
Alzheimer, Alois
Alzheimer, Alois
(propr. Aloysius) Psichiatra tedesco (Marktbreit 1864 - Breslavia 1915). Prof. a Breslavia (dal 1912), contribuì in modo notevole all’affermazione dell’indirizzo anatomico in psichiatria. I suoi studi più importanti ebbero per oggetto la patologia della nevroglia e i processi di degenerazione del tessuto nervoso. Con la denominazione malattia di A. sono indicate le sindromi demenziali degenerative già distinte da E. Kräpelin in demenza presenile di A. (detta anche malattia di A.-Perusini, in quanto descritta da A. e G. Perusini) e demenza senile.
Morbo di Alzheimer Il morbo di A. è una forma di demenza (➔) che, a differenza di quanto si verifica nella demenza arteriosclerotica e in quella indotta da multipli focolai infartuali, è dovuta ad atrofia cerebrale diffusa (assottigliamento delle circonvoluzioni cerebrali, allargamento dei solchi, dilatazione dei ventricoli, riduzione dell’area occupata) e non a deficit circolatori cerebrali distrettuali.
Le fasi della malattia
Il morbo di A. può colpire le persone in modo diverso ma l’insieme di sintomi più comuni riguarda, nella fase iniziale, la progressiva difficoltà di ricordare nuove informazioni poiché, fra gli organi maggiormente colpiti prematuramente dal morbo, ci sono parti del cervello, quali l’ippocampo, che presiedono a questo tipo di funzioni. Con il progredire della malattia, l’individuo prova un aumentato senso di confusione e di pensiero disorganizzato, presenta alterata capacità di giudizio e di disorientamento nel tempo e nella propria localizzazione spaziale che lo può condurre a deambulare senza meta per le strade e ad avere comportamenti inappropriati. Negli stadi successivi, la persona affetta dal morbo necessita di assistenza per lavarsi, vestirsi e per compiere altre attività usuali. Negli stadi terminali il paziente perde la capacità di comunicare, di riconoscere le persone che gli sono care e diviene riluttante od ostile a ogni trattamento terapeutico. Sebbene i familiari preferiscano tenere la persona malata nella propria dimora il più a lungo possibile, la maggior parte dei pazienti necessita di cure più specifiche e specialistiche che inducono a un internamento in una casa di cura.
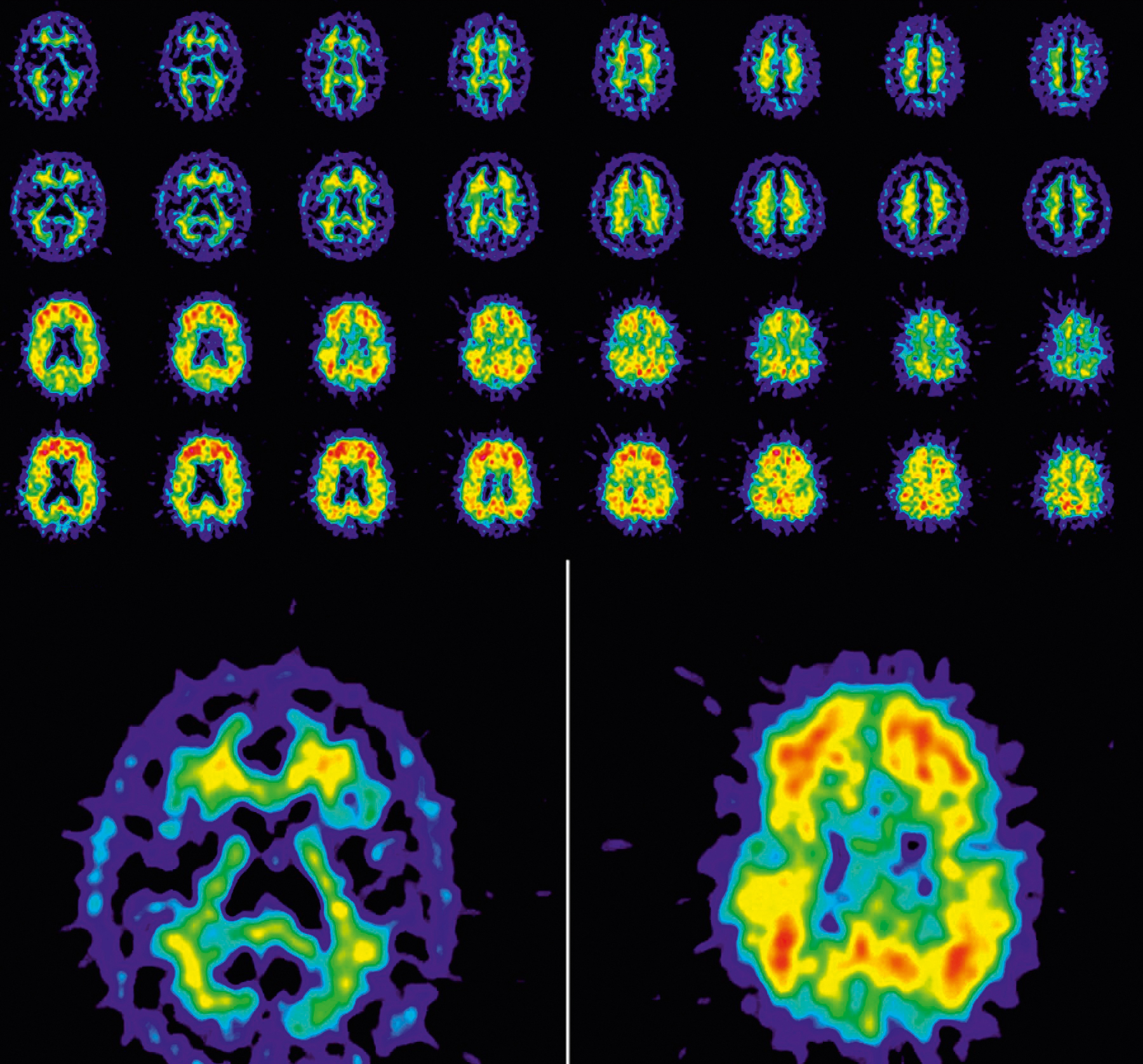
Incidenza e costi sociali
Il morbo di A. costituisce la settima di tutte le cause di decesso negli USA e nella maggior parte dei paesi della Unione Europea è la quinta causa dopo i 65 anni. Negli USA più di 5 milioni di persone ne sono affette e si stima che il numero sia destinato ad aumentare con il prolungamento dell’età media delle persone anziane, tanto che si calcola che nel 2050 la popolazione colpita si aggirerà intorno ai 10÷15 milioni. Il costo diretto e indiretto (spese correnti, diminuita produttività di colui che svilupperà definitivamente la malattia) per la cura delle persone ammalate del morbo di A. e di demenze correlate si aggira attualmente sui 148 miliardi di dollari l’anno. Si stimano in 10 milioni le persone che dedicano larga parte del loro tempo alla cura dei malati di A. e che non beneficiano di alcun guadagno da questa attività.
Influenza del genere e dei fattori culturali
Le donne dimostrano una maggiore predisposizione (16% delle persone tra i 65 e gli 85 anni) rispetto agli uomini (11%) a sviluppare il morbo di A:, ma ciò appare principalmente dovuto al fatto che l’incidenza della malattia progredisce con l’invecchiamento e la mortalità femminile, in rapporto all’età, è minore di quella maschile. Un dato quantitativamente più consistente è quello legato al livello di studi. Il fattore di rischio del morbo di A. risulta, infatti, molto più elevato in coloro che hanno avuto una ridotta scolarità rispetto a coloro che hanno seguito studi più avanzati. Questo dato è statisticamente valido anche se si pongono nella valutazione altri criteri, generalmente legati alla minore possibilità di studio, come quelli sociali o alimentari. Questo dato ha suscitato numerose ipotesi neurobiologiche, fra le quali quella attualmente più accreditata si basa sulla dimostrata utilità del mantenimento dell’attività nervosa per il buon funzionamento in vita delle cellule nervose. Studi in modelli animali che sviluppano sindromi analoghe da un punto di vista anatomopatologico e comportamentale, hanno dimostrato che la crescita in ambienti arricchiti e stimolanti le capacità cognitive riduce la produzione di sostanze tossiche, come per esempio il peptide beta-amiloide, responsabili della degenerazione neuronale. Analogamente, la sopravvivenza di neuroni in colture in vitro è maggiore in popolazioni coltivate in condizioni elettrofisiologiche che ne stimolano la continua attività elettrica rispetto a quelle in stato di riposo elettrofisiologico.
Fattori ereditari
La causa del morbo di A. è ancora sconosciuta ma l’opinione più accreditata è che esso è la conseguenza di molti fattori più che di una singola causa. Ancora più dibattuto è se, una volta iniziati i primi danni ai neuroni e alle strutture sinaptiche, si verifichi una sorta di progressiva diffusione a macchia d’olio alle popolazioni neuronali vicine ancora indenni, come una sorta di contaminazione infettiva o, al contrario, se diversi focolai della malattia si ‘accendano’ indipendentemente uno dall’altro. Approssimativamente il 5% del morbo di A. è di natura indiscutibilmente genetica, causato da un alterato funzionamento di uno dei 3 geni coinvolti nel metabolismo della proteina precursore dell’amiloide, APP. In queste forme ereditarie i sintomi della malattia si manifestano molto prima, talvolta in individui dell’età di 30 anni. Un fattore genetico della malattia che si presenta dopo i 65 anni è l’apolipoproteina Ee4(APOEe4), una delle tre forme comuni espresse dal gene APOE che fornisce le istruzioni per la sintesi di una proteina coinvolta nel metabolismo del colesterolo. Ogni individuo eredita un gene per la proteina APOE da ciascun genitore. Coloro che ereditano un gene APOEe4 presentano un aumentato rischio di sviluppare la malattia e coloro che lo ereditano da entrambi i genitori presentano un rischio ancora maggiore.
Nel morbo di Alzheimer, così come in altri tipi di demenze molto meno frequenti, il numero di neuroni del cervello diminuisce progressivamente; la perdita più consistente e invalidante, riguarda tuttavia il numero di sinapsi progressivamente colpite. Le sinapsi sono strutture altamente specializzate che funzionano come giunzioni dei prolungamenti nervosi (dendriti) dei neuroni. Poiché il numero di dendriti di ciascun neurone varia da qualche centinaio a molte migliaia, si stima che nel cervello operino più di 100 mila miliardi di sinapsi. Studi recenti hanno dimostrato che, molto tempo prima della degenerazione e della morte di intere popolazioni di neuroni, la funzionalità sinaptica viene progressivamente e spesso irreversibilmente colpita. Questo fatto è alla base di uno dei primi sintomi clinici del morbo di Alzheimer, la progressiva perdita della memoria, e in particolare della cosiddetta memoria breve, cioè quella che si verifica a livello sinaptico senza sostanziale coinvolgimento dell’intero neurone.
I due marcatori principali della malattia
Anche se la descrizione clinica della malattia risale al neuropatologo Alois Alzheimer (1864-1915), per molto tempo questa patologia è stata confusa, e spesso identificata, con altre demenze senili dovute ad ischemie cerebrali e disturbi vascolari, o con demenze progressive di natura traumatica. Si deve, tuttavia, ad Alzheimer la descrizione dei due reperti istochimici che, in seguito, sono stati riconosciuti come patognomonici del morbo: le placche senili (SP) e i cosiddetti neurofibrillary tangles (NFT). Questi due ‘marcatori’ sono caratterizzati dalla presenza di due componenti di natura polipeptidica: il primo è principalmente costituito da peptidi di basso peso molecolare, composti da 40÷42 amminoacidi, chiamati beta-amiloide (bA). I secondi, al contrario, sono aggregati fibrillari principalmente costituiti da una proteina denominata tau. Grazie a una lunga e complessa serie di studi condotti su pazienti e su modelli animali, nell’ultimo decennio si sono compresi i meccanismi molecolari alla base della formazione delle SP e degli NFT e si sono avanzate numerose ipotesi sul significato patognomonico di questi due alterati depositi di aggregati proteici.
Se si induce un aumento di espressione del precursore di beta-amiloide (APP, Amyloid Precursor Protein) tramite opportune manipolazioni genetiche, si realizza una sindrome in tutto sovrapponibile al morbo di Alzheimer. Analoghe considerazioni valgono se topi transgenici sono indotti a produrre aumentate quantità della proteina tau.
Il ruolo di APP e di tau
I peptidi di amiloide sembrano essere la probabile causa scatenante della complessa sequenza di eventi che, coinvolgendo anche le numerose cruciali funzioni di tau, porta alla degenerazione e alla morte neuronale. In seguito a un abnorme produzione o accumulo di bA, tau subisce una serie di modificazioni postraduzionali e un attacco da parte di enzimi appartenenti alla classe delle calpaine e delle caspasi; a causa di ciò essa si aggrega a formare gli NFT e la sua funzione fondamentale, collegata con il funzionamento dei microtubuli, viene progressivamente a cessare. Di conseguenza, gli alterati metabolismi di APP e di tau provocano il duplice effetto negativo di cessata funzione fisiologica e di produzione di derivati altamente tossici. Mentre la funzione di APP non è ancora del tutto nota, si sa che quella della proteina tau è fondamentale per il funzionamento di diverse attività neuronali, fra le quali il trasporto assonale sia anterogrado (dal corpo della cellula alle terminazioni sinaptiche), sia retrogrado (dalle sinapsi al corpo neuronale). Tale trasporto svolge un ruolo essenziale nell’economia neuronale poiché garantisce il rifornimento di materiale prezioso, insostituibile, per la vita e il funzionamento della cellula nervosa.
Il probabile ruolo dell’apoptosi
Se, dunque, gli eventi molecolari alla base del morbo di Alzheimer sono ormai chiariti, rimane il quesito di fondo: qual è la causa o quali sono le cause che provocano l’attivazione della proteina amiloide e la degradazione di tau? Tra le varie ipotesi avanzate, si ritiene che il processo apoptotico (suicidio cellulare) possa costituire un effetto scatenante. L’apoptosi svolge un ruolo fondamentale nel corso dello sviluppo del cervello e della formazione dei circuiti nervosi, provvedendo a eliminare tutti i neuroni che non hanno formato circuiti appropriati e ben funzionanti. In altri termini, tutte le cellule che non hanno formato i circuiti funzionanti, seguendo ‘direttive’, sia dovute a particolari geni sia al contesto ambientale (quali stimoli sensoriali di varia natura), si autoeliminano e i loro dendriti vengono rapidamente digeriti da apposite cellule per evitare ingombro dannoso ed eccessivo consumo energetico. Ma se questi fenomeni di autoeliminazione venissero comunque ‘accesi’, nel cervello dell’adulto si verificherebbe una progressiva e irreversibile perdita di intere popolazioni neuronali che, nell’arco di qualche anno, condurrebbe l’individuo ai sintomi devastanti del morbo. Al contrario dei tumori, che non obbediscono all’ordine di attivare i propri geni apoptotici, i neuroni compirebbero l’errore di attivarli ‘a sproposito’ in quanto verrebbero a mancare loro i segnali che, di norma, li tengono repressi. In queste cellule, come è comprensibile, il congegno ‘a orologeria’ che attiva i geni dell’apoptosi deve essere bloccato per tutta la vita dell’organismo. Quando questo blocco viene a mancare, le cellule nervose si autoeliminano e, a seconda delle funzioni che esse svolgono nel cervello, vengono a essere gravemente compromesse memoria, intelligenza, movimento. Un processo apoptotico anomalo può essere attivato da diverse cause e fra queste si annoverano il diminuito apporto di fattori neurotrofici, fra i quali ricordiamo l’NGF, o la riduzione di condizioni o di fattori che mantengono sempre attive le funzioni cerebrali cognitive.
Biografia
1864 Nasce a Marktbreit in Baviera
1887 Si laurea in medicina all’Univ. di Würzburg
1888 Inizia la specializzazione presso l’ospedale psichiatrico di Francoforte sul Meno
1889 Pubblica con F. Nissl il trattato Histologische und histopathologische Arbeiten über die Grosshirnrindeder
1895 Diventa direttore dell’ospedale psichiatrico di Francoforte e si dedica allo studio della depressione e della schizofrenia
1902 Si trasferisce presso la clinica psichiatrica dell’Univ. di Heidelberg, invitato da E. Kräpelin
1904 Pubblica un trattato sui cambiamenti neurologici nei pazienti affetti da sifilide
1906 Descrive il caso clinico di «una particolare malattia della corteccia cerebrale», cui verrà poi attribuito il suo nome nel 1910
1908 Diventa professore all’istituto di psichiatria di Monaco e direttore del laboratorio di anatomia clinica
1912 Diventa professore di psichiatria all’Univ. di Breslavia
1915 Muore a Breslavia