
Argille e origine della vita
Argille e origine della vita
L'origine della vita è materia interdisciplinare dagli ampi confini. Uno di questi è con la geochimica, e in modo particolare con la mineralogia delle argille. Si assume generalmente che la vita si sia originata su una Terra o ve, molto probabilmente, le argille erano minerali diffusi come lo sono oggi. È noto da molto tempo che le molecole organiche hanno interagito con le argille: ciò ha condotto all'idea che le argille, o altri materiali simili, possano aver contribuito alla produzione delle molecole chiave necessarie all'origine della vita. Un punto di vista più radicale è che alcuni di questi minerali possano essere stati i veri componenti dei primi sistemi capaci di evolvere attraverso la selezione naturale. In questo saggio verranno discussi aspetti acclarati e controversi di queste idee e saranno presi in esame alcuni modelli ed esperimenti recenti.
Introduzione
Non c'è nulla di veramente nuovo nell'idea che l'argilla possa aver avuto a che fare con l'origine della vita sulla Terra. Nella mitologia tradizionale è stato immaginato che un creatore avesse dato forma alla prima creatura vivente con questo materiale comune e malleabile. Tali ipotesi appaiono ingenue alla scienza moderna, anche perché le forme e le configurazioni cruciali degli organismi viventi sono minutamente inscritte nelle molecole e non appartengono alla scala delle sculture o delle ceramiche. Inoltre, anche il materiale sembra inadatto. La vita sulla Terra è oggi di natura essenzialmente organica, basata sul carbonio e non sul sistema silicio-ossigeno. l suoi atomi, per la maggior parte, derivano dall'aria e dall'acqua, soltanto pochi dalle rocce. In ogni caso, le strutture più importanti, quali i geni, gli enzimi, le membrane, sono costituite quasi esclusivamente da molecole organiche. C'è tuttavia qualcosa di strano. Queste importanti strutture di regolazione, come quelle che si trovano in tutte le forme di vita sulla Terra, sono sempre costruite partendo da un numero sorprendentemente limitato di piccoli componenti molecolari: amminoacidi, nucleotidi e così via. Può sembrare ovvio che la spiegazione di questa uniformità biochimica debba risiedere nell'origine della vita, nell'origine di un unico processo evolutivo, di un singolo albero ereditario. Può anche sembrare ovvio che le particolari molecole che ora sono universali lo siano perché esse, o molecole simili, erano facilmente disponibili sulla Terra primitiva. Però, questa uniformità biochimica è troppo bella per essere vera: potremmo dire troppo esatta per essere un indicatore plausibile di quanto stesse accadendo nel momento preciso dell'inizio dell'evoluzione. Da una così perfetta concordanza tra, per esempio, la scelta degli amminoacidi necessari per fabbricare le proteine e le procedure e le regole generali della sintesi proteica, deriva in modo evidente - e ciò è accettato generalmente da tutti - che le creature sulla Terra siano biochimicamente simili perché condividono un ultimo antenato comune. Più precisamente, perché è esistito un antenato comune a tutte le forme di vita sulla Terra le cui caratteristiche biochimiche comprendevano quelle che sono ora universali. È anche chiaro che questo antenato comune doveva essere già evoluto: a monte deve essersi verificato un processo evolutivo più importante e non si può certo escludere che durante tale periodo la scelta dei materiali principali, ora così fissati, non facesse parte del processo evolutivo che stava avvenendo. N.W. Pirie (1957) ha discusso a lungo questo punto, sottolineando recentemente (Pirie, 1994) che "non si dà più per scontato che le forme esistite in origine fossero caricature dei più semplici organismi attuali". Si può dire che i primi organismi erano costituiti di materiale simile agli acidi nucleici, proteine e così via? Ciò potrebbe essere vero, e una tale opinione ha un suo valore pragmatico, ma il processo evolutivo che ha preceduto l'u1timo antenato comune della vita sulla Terra deve essere stato difficile e tortuoso, a giudicare dalla complessità 'ad alta tecnologia' del suo prodotto. La somiglianza dei processi biochimici tra le forme di vita originarie e quelle attuali è un'ipotesi che necessita ancora di altre prove e argomentazioni. A sostegno di questa ipotesi va detto che alcuni amminoacidi sono facili da sintetizzare, mentre gli zuccheri e i nucleotidi creano maggiori problemi (Shapiro, 1988; Joyce e Orgel, 1993; Shapiro, 1995). L'idea del 'mondo a RNA' (Gilbert, 1986; Wilson e Szostak, 1995) presuppone una buona risorsa di nucleotidi. Altri materiali però possono essere considerati quali possibili coadiuvanti o componenti dei primi sistemi capaci di evolvere: le argille sono tra questi.
Inizieremo questo saggio con una breve rassegna sulla struttura e sulle condizioni di formazione dei minerali argillosi, quindi divideremo in tre parti la discussione sul possibile ruolo delle argille nell'origine della vita. Nella prima parte (l'opzione conservatrice) seguiremo il punto di vista della maggioranza, e assumeremo che le molecole fondamentali siano sempre state più o meno quali sono ora. Con questo approccio le argille, ammesso che abbiano avuto un ruolo, sarebbero state semplici assistenti nell'allestimento dello scenario: forse punti di raccolta di molecole organiche, catalizzatori, cioè agenti involontari di una evoluzione chimica. L'opinione seguita nella parte successiva (l'opzione radicale) sarà più estrema. Supporremo che tipi diversi di minerali argillosi siano stati i reali componenti dei primi organismi (cioè dei primi sistemi capaci di prendere parte a una lunga evoluzione attraverso la selezione naturale). Essi avrebbero fornito i materiali per i primi geni, o per i primi catalizzatori interni, o per le membrane o altre strutture di ordine superiore o per tutti questi sistemi insieme: in breve, essi sarebbero stati tra i principali componenti regolativi dei primi sistemi capaci di evolvere. Sarà discusso come le argille posseggano strutture, caratteristiche e condizioni di formazione tali da poter essere considerate seriamente per questo ruolo. Nell'ultima parte (territori di frontiera) riconsidereremo i problemi derivanti sia dal punto di vista conservatore (che è troppo ristretto) sia da quello radicale (che è troppo ampio) e cercheremo di tracciare gli orientamenti futuri.
Formazione e struttura dei minerali argillosi
La definizione di argilla prevede che le particelle di questi minerali siano molto piccole (inferiori a 2 μm), ma è un errore immaginare questi materiali semplicemente come rocce dure sminuzzate o disintegrate. A volte può essere così, ma la maggior parte delle argille sono prodotti di ricristalizzazione di materiali ero si da fenomeni atmosferici. Un pezzo di granito, per esempio, si forma sotto l'azione delle alte temperature e pressioni presenti all'interno della Terra. Quando, attraverso processi tettonici o altro, tale roccia emerge alla superficie, è metastabile e su lunghi periodi di tempo tenderà a trasformarsi in altri materiali quali, in presenza di acqua, i minerali argillosi. In poche parole, le rocce dure si dissolvono gradualmente nell' acqua e ricristallizzano come minerali argillosi. Per esempio, la caolinite è un tipico prodotto di erosione atmosferica del granito.
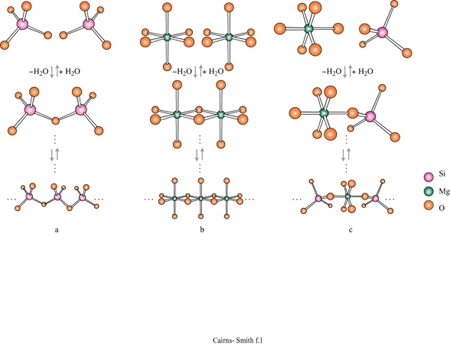
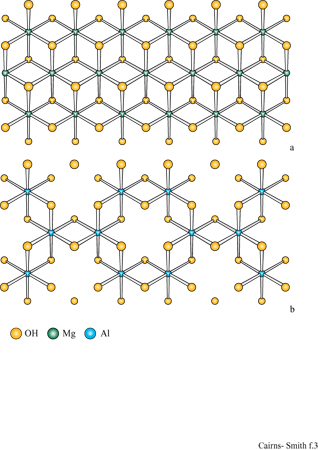
Per la sintesi delle argille sono necessari due tipi di unità monomeriche presenti generalmente nelle acque naturali. Una è l'acido silicico, Si(OH)4 Queste unità polimerizzano velocemente da sole con eliminazione di acqua e poi depolimerizzano nuovamente (fig. 1); il processo generale della polimerizzazione dipende dal pH (un pH basso lo favorisce) e, ovviamente, dalla concentrazione. A temperature ordinarie, una polimerizzazione rapida in un primo tempo darà luogo immancabilmente a una forma non cristallina di silice di tipo polimerico tridimensionale, come il gel di silice, mentre forme cristalline comuni come il quarzo (fig. 2) si possono formare dopo lunghi periodi di tempo, oppure a temperature elevate. Le altre unità richieste per la sintesi delle argille sono cationi quali Mg(H2O)62+. Anche questi tendono a polimerizzare, specialmente nelle condizioni di pH moderatamente basico e di alta concentrazione. L'inizio di tale processo è mostrato in figura 1b. Il magnesio, per esempio, se polimerizza completamente, produce un polimero bidimensionale, la brucite, con una struttura a strati di idrossido come mostrato in figura (fig. 3). Tale struttura è elettricamente neutra, con le parti anioniche e cationiche perfettamente bilanciate. In realtà, cariche locali si possono originare da vari tipi di difetti. Per esempio, possono essere mancanti cationi magnesio e si avrà una carica netta negativa. La sostituzione di qualche ione magnesio con litio monovalente potrebbe avere un effetto simile, mentre l'alluminio trivalente potrebbe creare cariche locali positive. Un cristallo di brucite è costituito da molti strati come quello mostrato in figura 3a, impilati uno sull'altro.
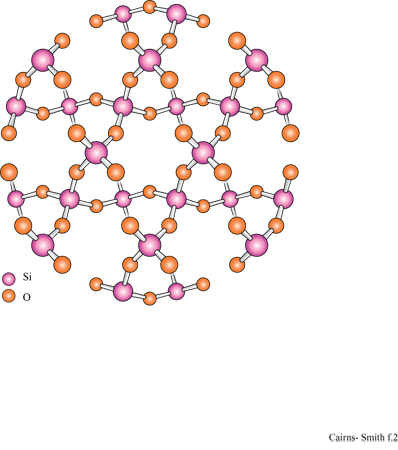
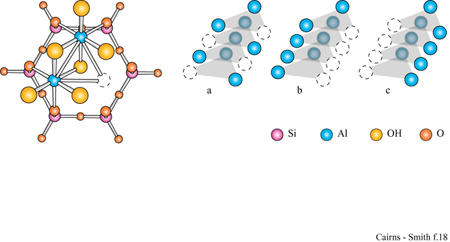
La figura 3b mostra la struttura analoga dello strato di idrossido di alluminio della gibbsite. Anche questa struttura ideale è neutra, nonostante l'alluminio abbia tre cariche invece di due, poiché solo i due terzi dei possibili siti cationici sono occupati. Le sostituzioni sono comuni e in questo caso alcuni siti formalmente vuoti possono essere in realtà occupati e disturbare il bilanciamento delle cariche. Quando in soluzione acquosa sono presenti sia l'acido silicico che i cationi metallici, diventa possibile la polimerizzazione incrociata (v. figura 1 c); se ciò avviene per un periodo di tempo sufficientemente lungo, si possono formare strutture di minerali argillosi meravigliosamente elaborate. In figura (fig. 4) è mostrata una di queste strutture: uno strato di caolinite costituito da un sotto strato o foglio di gibbsite, con una rete polimerica di unità di acido silicico fusa su un lato (lo strato sottostante nella figura).
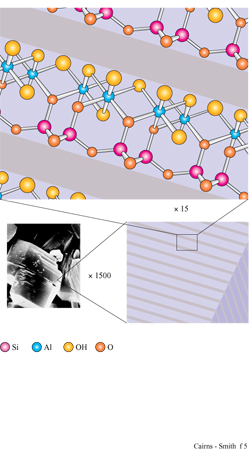
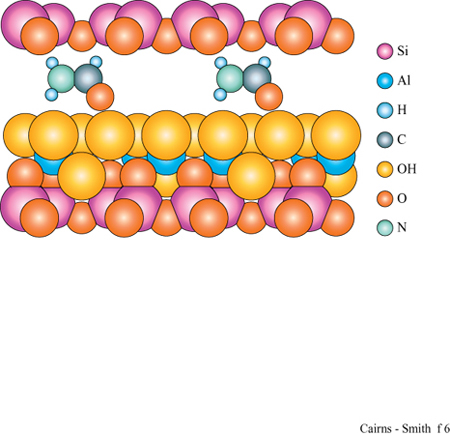
Un tipico minerale argilloso contiene pile di questi strati di silice ed è classificato come l: l se, come nella caolinite, una rete di silossano è fusa su un solo lato, oppure come 1:2 se, più simmetricamente, c'è una rete su entrambi i lati di un sottostrato di idrossido. In figura (fig. 5) è illustrata la struttura l: l della caolinite, con gli strati visti di lato. In questo caso i lati sono tenuti uniti piuttosto saldamente da legami idrogeno. Tra gli strati di caolinite possono anche essere intercalate alcune molecole organiche, quali la formammide (fig. 6).
La halloysite è una forma di caolinite in cui gli strati sono separati da molecole di acqua e non sono così saldamente attaccati. In questo caso gli strati, asimmetrici, della caolinite tendono a curvarsi e a formare scanalature e protuberanze.
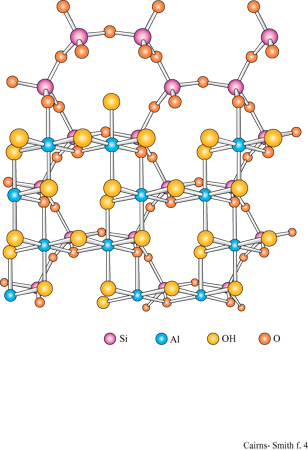
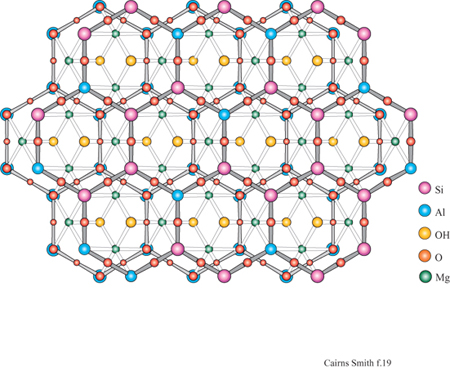
La montmorillonite è un minerale argilloso con strati 2:1. Una caratteristica tipica degli strati di silicato 2:1 è di avere un certo numero di atomi di sostituzione che conferiscono loro una carica netta negativa. Per esempio, lungo il piano centrale, gli atomi di magnesio, che possiedono solo due cariche formali positive, possono sostituire alcuni atomi di alluminio che ne hanno tre. Oppure, nelle reti di silossano, gli atomi di alluminio con tre cariche positive possono trovarsi al posto di quelli di silicio che hanno quattro cariche positive formali. Per compensare la carica negativa di questi strati di silicato devono essere incorporati cationi aggiuntivi. Questi ioni di compensazione giacciono tra uno strato e l'altro, dove, nel caso di argille quali la montmorillonite, si trovano anche molecole di acqua. Tipici ioni di compensazione sono il sodio e il calcio, ma anche altri, come il catione ammonio, possono interporsi.
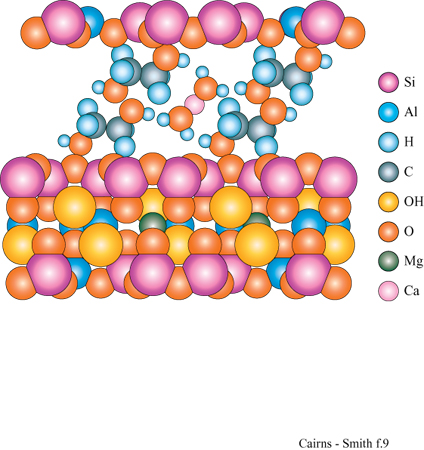
In questo contesto è particolarmente interessante che tra gli strati dei minerali argillosi si interpongano facilmente alcuni tipi di molecole organiche (Theng, 1974; Raussell-Colom e Serrato sa, 1987). Per esempio, i cationi delle basi organiche come piridina o adenina si inseriscono nelle zone interstrato della montmorillonite. Inoltre, alcune molecole organiche neutre si possono interporre in parte o totalmente al posto dell' acqua. Il glicole etilenico lo fa così bene (fig. 9) da essere usato nei test di identificazione delle smectiti, o 'argille espansibili' (argille 2:1 che tra gli strati hanno quantità variabili di acqua: la montmorillonite appartiene a questa classe). Quando il glicole si inserisce nelle zone interstrato, gli strati vengono allontanati tra di loro, creando un effetto facilmente visibile mediante diffrazione di raggi X.
I minerali argillosi 2:1 sono strutturalmente correlati alle miche che si presentano in forma macrocristallina. In un tipo di mica, la muscovite, gli strati somigliano a quelli di montmorillonite tranne per il fatto che portano più cariche dovute alle numerose sostituzioni di alluminio, circa 1 su 4, nelle reti di silossano e che gli ioni di compensazione sono esclusivamente ioni potassio. Questi ultimi sono ben collocati in alveoli trigonali delimitati sotto e sopra da reti di silossano; con questa precisa disposizione non c'è spazio per l'acqua. Il minerale argilloso illite è quello più strettamente collegato alla muscovite, con un alto contenuto in potassio e con molti strati privi d'acqua; l'illite è molto comune nei sedimenti marini.
Altri silicati 2:1 (per esempio, la mica flogopite) hanno strati i cui piani centrali sono più simili alla brucite che alla gibbsite, anche se, invariabilmente, nei campioni naturali alcuni atomi di magnesio sono sostituiti con altri ioni come il ferro (Il). La mica biotite ha una struttura simile, generalmente con più ferro che magnesio. Esistono inoltre materiali microcristallini correlati a questi e con struttura simile, per esempio minerali argillosi ricchi di ferro.

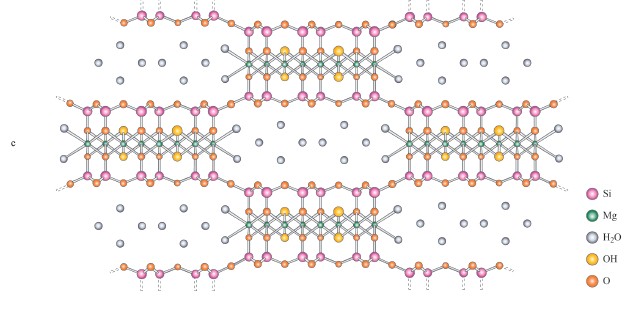
La sepiolite (fig. 11 a-b, c) è tra i minerali argillosi più singolari. Anch'essa è basata su strati simili a quelli della brucite e con struttura fondamentale 2:1, ma questa volta con inversioni periodiche tali da formare microcanali. La figura 11 b mostra microcanali più ampi e più complessi e anche scanalature della superficie che originano da irregolarità della struttura cristallina. Molecole organiche di forma appropriata, come quelle degli eterocicli aromatici, per esempio l'arancio di acridina (Ridler e Jennings, 1980) e l'indaco (van Olphen, 1966), si possono legare alle superfici scanalate della sepiolite e della paligorskite, un'argilla simile a questa.
L'imogolite è una comune argilla tubulare. Allo stesso modo della caolinite, la sua struttura è basata su uno strato di gibbsite con unità di acido silicico attaccate su un lato. Qui, tuttavia, finiscono le somiglianze. Nell'imogolite le unità di acido silicico sono attaccate allo strato di gibbsite attraverso tre ossigeni, pertanto le unità non possono essere connesse lateralmente le une alle altre a formare il tipo di rete a silossano peculiare della caolinite e di molti altri minerali argillosi.
Materiali simili all'argilla
Secondo una definizione più ampia, le argille possono essere considerate "materiali a cui sono interessati gli esperti di mineralogia delle argille", come si può leggere nella rivista Clay and Clay Minerals. In questa definizione sarebbero inclusi vari minerali importanti per la questione dell' origine della vita. Certamente le zeoliti sarebbero incluse nella categoria, come anche alcuni idrossidi (non quelli di silicio), ossidi-idrossidi, ossidi e solfuri, specialmente di ferro e manganese (Cairns-Smith, 1982; Arrhenius, 1986; Costanzo e Laszlo, 1988; Cairns-Smith et al., 1992; Holm et al., 1993). Di straordinario e recente interesse è il gruppo di minerali della piroaurite studiati da G. Arrhenius e collaboratori, e da loro discussi in relazione alle origini della vita (Kuma et al., 1989; Pitsch et al., 1995). Questi materiali si sintetizzano facilmente in laboratorio in condizioni non drastiche. Le loro strutture derivano da elaborazioni della brucite. Consideriamo uno strato di brucite (v. figura 3a) con un'ampia proporzione di cationi trivalenti (per es., alluminio) al posto dei normali bivalenti (magnesio). Invece di essere neutro, questo strato avrebbe una forte carica positiva e potrebbe impilarsi sugli altri a esso simili soltanto se le cariche degli strati fossero compensate da anioni posti tra essi. Nei diversi minerali della piroaurite cationi metallici di tipo diverso creano strati di idrossido a valenza mista che si alternano con allineamenti di anioni, tipicamente CO32-, Cl- o SO4-2. Altri anioni, quali, per esempio, gli ioni cianuro o fosfato, di particolare interesse per l'origine della vita, possono anch'essi interporsi (Kuma et al., 1989).
Anche i ferrocianuri sono stati presi in considerazione come materiali che potrebbero aver avuto un ruolo in qualche modo simile a quello dell'argilla (Arrhenius, 1990; Kamaluddin et al., 1990; 1994). Più di recente è stato proposto che il ferrocianuro di zinco potrebbe aver avuto importanza per l'adsorbimento di ioni ammonio, concentrando e proteggendo l'ammoniaca che nelle condizioni dell'atmosfera primitiva sarebbe stata tendenzialmente instabile (Braterman et al., 1995).
L'opzione conservatrice
In questo paragrafo seguiamo il punto di vista convenzionale e consideriamo il modo in cui i minerali d'argilla potrebbero aver coadiuvato l'evoluzione chimica delle molecole sulla Terra primitiva verso i tipi di molecole che attualmente costituiscono il perno della biochimica.
Nel famoso esperimento condotto nel 1953 nel laboratorio di S. Miller venne simulato un temporale in un'atmosfera primitiva e vennero prodotte molecole organiche tra cui alcuni amminoacidi (Miller, 1953). L'atmosfera, nel modello originale di Miller, era fortemente riducente contenendo, per esempio, il carbonio sotto forma di metano e l' azoto sotto forma di ammoniaca, in linea con l'idea dell'atmosfera primitiva della Terra che A.I. Oparin, H.C. Ureye altri avevano sostenuto. Tuttavia i geologi non hanno mai appoggiato con entusiasmo l'idea di un'atmosfera primitiva fortemente riducente (Rubey, 1951; Abelson, 1966), preferendo quella di un'atmosfera più o meno neutra, né troppo riducente né troppo ossidante. Questa posizione si è nel tempo consolidata (Kerr, 1980; Holm, 1992). Considerazioni di natura fotochimica aumentano i dubbi. Un'atmosfera fortemente riducente non sarebbe durata a lungo con l'intensità della radiazione ultravioletta che il Sole le avrebbe fornito (Wayne, 1991). È più verosimile che l'atmosfera primitiva fosse dominata da diossido di carbonio e azoto, più simile quindi a quella di Marte e Venere che a quella di Giove: qualcosa di analogo all'attuale atmosfera terrestre senza l'ossigeno (Walker, 1977). Il problema è che tali atmosfere elettrochimicamente neutre, o soltanto moderatamente riducenti, non avrebbero facilmente prodotto molecole organiche (Chang et al., 1983). Sono state prese in considerazione altre possibili fonti di molecole organiche sulla Terra primitiva. Per esempio, la luce ultravioletta avrebbe potuto portare alla formazione di alcoli, aldeidi e acidi in un'atmosfera contenente monossido di carbonio (Bar-Nun e Hartman, 1978) o sulla superficie di minerali esposti a tale atmosfera (Hubbard et al., 1973). Piccole quantità di acido formico e ossalico si possono formare dalla riduzione di anidride carbonica attraverso l'irradiazione con luce ultravioletta di soluzioni contenenti ferro (Il) (Getoff, 1962; 1963; Akermark et al., 1980). I sistemi geologici idrotermali, se non sono troppo caldi, sono un'altra possibile fonte di materiale organico (Russell et al., 1988; Holm, 1992; Shock et al., 1995).
Un problema di diluizione
Sulla Terra primitiva saranno esistite senza alcun dubbio fonti di materiale organico. Tuttavia, l'idea originale di un brodo oceanico ricco di molecole biologiche, prodotte in maniera sistematica prima che si sviluppasse la vita sulla Terra, non appare più così convincente come nel passato. Oggi, infatti, c'è meno ottimismo riguardo all'esistenza di fonti di molecole organiche; al contrario, la discussione si è incentrata piuttosto sul ruolo dei cosiddetti 'pozzi' (sink), cioè di quelle situazioni capaci di favorire la sottrazione di materia organica; la radiazione ultravioletta, per esempio, serve più a distruggere le molecole che a costruirle (Hull, 1960; Hulett, 1969; Dose, 1975). Inoltre, i minerali avrebbero potuto ripulire gli oceani dalle molecole organiche, agendo da 'pozzi', come fanno oggi (Nissenbaum, 1976), e il riciclo delle acque oceaniche attraverso sistemi idrotermali a elevata temperatura probabilmente avrebbe distrutto molecole organiche complesse (Nisbet, 1986a; Miller e Bada, 1988). È chiaro che ora dobbiamo pensare in termini di stati stazionari e fonti locali piuttosto che di un semplice accumulo di brodo primordiale. In una Terra che rispetto al passato è considerata molto più dinamica, la chimica è basata più su stati che cambiano lentamente che su accumuli. Ora, sembrerebbe che, in presenza di 'pozzi' così attivi, la concentrazione dei materiali organici allo stato stazionario fosse scarsa nei mari e negli oceani della Terra primitiva (Nisbet, 1986a). In che modo, in una soluzione così diluita, piccole molecole organiche hanno potuto aggregarsi per costruire strutture di ordine superiore? Y.M. Goldschmidt (1952) ha fornito una possibile soluzione a questo problema. Nel 1945 egli suggerì che la superficie dei minerali avrebbe avuto la tendenza ad adsorbire qualsiasi molecola organica presente sulla Terra primitiva. Inoltre, tali minerali potrebbero aver imposto un certo ordine ai materiali organici, selezionando particolari tipi di molecole, allineando le e forse perfino catalizzando le loro reazioni. Anche J.D. Bernal (1949; 1951; 1967) si è mosso secondo la stessa linea di pensiero, sottolineando l'importanza dei minerali argillosi. Egli ha posto in evidenza la tendenza delle argille a intercalare molecole organiche in maniera ordinata (v. figure 6, 9). Ancora oggi molta materia organica della Terra presente nel suolo è associata ad argille.
Esistevano le argille sulla Terra primitiva?
l minerali argillosi non rimangono inalterati per periodi molto lunghi, per cui non abbiamo una prova diretta della loro presenza sulla Terra primitiva. Sembra probabile, comunque, che essi vi fossero, almeno durante il periodo critico in cui apparve per la prima volta la vita, perché si può assumere che l'acqua sia stata un pre-requisito essenziale per l'origine della vita sulla Terra, ed essa è l'unico fattore realmente importante per la formazione di argille nella crosta terrestre ricca di silicati. Dal momento in cui la Terra fu sufficientemente fredda da permettere la vita abbiamo buone prove che su di essa ci fosse molta acqua. Per esempio, a lsua in Groenlandia esistono rocce antiche 3,8 miliardi di anni costituite da sedimenti alterati (Allaart, 1976; Ernst, 1983). L'enorme quantità di acqua circolante che in quel caso (e presumibilmente altrove sulla Terra a quel tempo) sarebbe stata necessaria per la deposizione di tali sedimenti, avrebbe sicuramente anche favorito la formazione di argille.
Una complicazione è rappresentata dal fatto che oggi sulla Terra la materia organica di derivazione biologica è spesso un fattore importante nella sintesi delle argille. La ragione principale è che i metalli a valenza elevata tendono a essere insolubili in condizioni di pH prossimo alla neutralità, necessario per la sintesi delle argille. Argille ricche di alluminio, come la caolinite, sono particolarmente difficili da produrre in laboratorio a temperature moderate senza additivi organici, come gli acidi di- e tricarbossilici, che servono a solubilizzare e mobilizzare l'alluminio chelandolo. L'acido fulvico, una miscela complessa di acidi carbossilici derivata dal suolo, è efficace per questo scopo (Linares e Huertas, 1971), ma lo è anche l'ossalato (Siffert, 1986), un materiale molto più semplice che, come notato in precedenza, potrebbe benissimo essere stato presente sulla Terra primitiva come prodotto dell'irradiazione ultravioletta di soluzioni di ferro (II) (Getoff, 1962; 1963; Åkermark et al., 1980). D'altro canto, anche la caolinite può essere sintetizzata in laboratorio in assenza di molecole organiche se le soluzioni sono sufficientemente diluite e la scala dei tempi sufficientemente lunga (Siffert, 1986). Molta caolinite naturale si forma, inoltre, a elevate temperature e pressioni in condizioni idrotermali dove, probabilmente, non sono coinvolte le molecole organiche. In ogni caso la sintesi di argille in condizioni idrotermali senza materiale organico aggiuntivo è effettuata di routine in laboratorio (Eberl, 1986). La sintesi di argille di magnesio può essere relativamente facile (Baird et al., 1971). Per esempio, la hectorite, una argilla di tipo 2:1, si può formare semplicemente facendo bollire per una settimana in un recipiente di vetro una fanghiglia contenente idrossido di magnesio e una piccola quantità di fluoruro di litio. Il Fe (II), relativamente solubile, potrebbe essere stato una specie comune quando l'atmosfera sulla Terra conteneva poco o per nulla ossigeno. Perciò potremmo supporre che le argille ricche in ferro fossero anche più diffuse sulla Terra primitiva di quanto lo siano oggi (Odin et al., 1986).
La realizzazione di reazioni termodinamicamente sfavorite
Un problema particolare del punto di vista conservatore sull' origine della vita è che i più importanti polimeri biologici - DNA, RNA, proteine e polisaccaridi - sono tutti termodinamicamente instabili (giacciono su un massimo di energia) rispetto ai loro monomeri in ambiente acquoso: infatti in acqua essi tendono spontaneamente a depolimerizzare e non a polimerizzare. Gli organismi che si trovano oggi sulla Terra sono dotati di elaborati sistemi che, consumando energia, alterano le strutture di appropriate unità monomeriche così da 'attivarle', cioè da renderle capaci di polimerizzare in maniera termodinamicamente favorita.
M. Paecht-Horowitz e collaboratori (1970) riuscirono a polimerizzare amminoacidi attivati (amminoaciladenilati) in presenza di montmorillonite (Warden et al., 1974; Paecht-Horowitz e Eirich, 1988). Questi sono esperimenti molto interessanti su cui ritorneremo, ma dal punto di vista della evoluzione chimica resta il problema fondamentale di come tali molecole, complesse e ricche di energia, quali gli amminoaciladenilati, possano essersi formate in quantità significative sulla Terra primitiva. Minerali appropriati e condizioni ambientali fluttuanti potrebbero fornire un'alternativa alla necessità di monomeri attivati. Supponiamo che l'assemblaggio di monomeri sulla superficie di un'argilla avesse creato una situazione per cui i polimeri fossero più stabili dei loro costituenti monomerici; ciò è abbastanza verosimile, in quanto la stabilità relativa dipende dalla differenza tra le energie di legame e di solvatazione delle specie molecolari coinvolte. Se i polimeri avessero formato legami più forti dei monomeri, questi, una volta adsorbiti, avrebbero mostrato tendenza a polimerizzare, soprattutto se opportunamente orientati e catalizzati, per esempio attraverso gruppi donatori di protoni (acidi) posti sugli strati superficiali di argilla. Ciò non infrange alcuna legge della termodinamica. Sarebbe stata comunque necessaria energia per mettere in atto un ciclo efficiente di operazioni al fine di desorbire il polimero, ma questa energia sarebbe potuta provenire da fluttuazioni ambientali. In un esperimento classico N. Lahav, D. White e S. Chang (1978) dimostrarono che le molecole di glicina potevano essere assemblate con legami peptidici in presenza di montmorillonite, attraverso un ciclo in cui si alternavano assenza e presenza d'acqua. Oligomeri quali la di- e la triglicina possono essere assemblati nella stessa maniera (Bujdak et al., 1995). Presumibilmente la polimerizzazione è favorita dall' assenza d'acqua, dopodiché il polimero reidratato è in grado di solubilizzarsi. Discutendo tali effetti in termini più generali, A.W.J. Muller (1983; 1985) ha suggerito che il ciclo termale è usato probabilmente anche oggi da alcuni organismi e che può aver costituito una risorsa primaria di energia per l'origine della vita.
Cicli locali di pH potrebbero, in modo simile, essere stati capaci di modificare la forza relativa di adsorbimento dei monomeri e dei polimeri sui minerali, per esempio alterando le cariche delle molecole organiche o dei minerali o di entrambi. Possiamo anche immaginare dispositivi costituiti da minerali in grado di accoppiare l'idrolisi termodinamicamente favorita del polifosfato con reazioni termodinamicamente sfavorite (Cairns-Smith, 1982).
Specificità
Potremmo dire che minerali come le argille, in grado di adsorbire molecole organiche, si sono rivelati utili nel risolvere un importante problema entropico (un problema di diluizione) e un importante problema energetico: come costruire polimeri che siano metastabili rispetto ai loro monomeri. È anche una questione di tipo probabilistico. Ci si potrebbe chiedere perché dovrebbe esserci una concordanza tra il tipo di molecole e i processi che possono essere adatti, diciamo, al materiale genetico, e il tipo di molecole con cui i minerali tendono a interagire e i modi in cui lo fanno. Chiaramente alcune coincidenze fortunate saranno necessarie, ma a favore delle teorie sull'origine della vita basate su interazioni con i minerali si può dire che esistono migliaia di tipi diversi di minerali (e migliaia di situazioni diverse in cui essi si trovano); questo aumenta le possibilità che alcuni di essi abbiano facilitato qualche fase cruciale della chimica organica. In effetti, il fatto che migliaia di singoli minerali di argilla siano ancora abbastanza comuni individualmente rende plausibile una loro comparsa sulla Terra primitiva, forse in maniera tale e in quantità sufficiente da aver creato in modo fortuito localizzazioni persistenti. C'è tuttavia un limite alla fortuna in cui possiamo ragionevolmente sperare! L'affinità della montrnorillonite per le basi eterocicliche è già stata menzionata, così come la naturale tendenza delle argille ad associarsi all'acqua. Possiamo ora approfondire queste nozioni: la montmorillonite, nelle zone interstrato, contiene sodio o calcio, ma negli ambienti marini tende a trasformarsi in illite; questa, più simile alle miche, nelle zone interstrato contiene potassio come catione dominante. Ciò risulta strano in quanto l'acqua del mare contiene molto più sodio che potassio. La spiegazione sembra essere che il potassio non legato a molecole d'acqua si adatta particolarmente bene tra gli strati dei silicati 2:1 (Eberl, 1980; 1986). Potremmo dire che questi minerali argillosi 'preferiscono' il potassio, come le cellule viventi. Inoltre i fosfati, specialmente i polifosfati, hanno un'elevata affinità per gli spigoli degli strati delle argille carichi positivamente a causa dei cationi esposti (van Olphen, 1977). Tutto ciò è abbastanza indicativo, ma non ci sono dubbi che la vita sulla Terra sia oggi in condizioni svantaggio se dipendendo dal potassio e dal fosfato, nutrienti spesso limitanti. Forse questi inconvenienti derivano dal fatto che la vita si è originata ed evoluta per un certo periodo di tempo all'interno di un ambiente in cui vi era abbondanza di potassio e di fosfato.
Argille per un mondo a RNA?
E.G. Nisbet (1986a; 1986b) ha proposto l'ipotesi di sistemi idrotermali a bassa temperatura come ambienti naturali di un primitivo mondo a RNA. Per prima cosa, si deve considerare il fatto che doveva esserci una buona scorta di fosfato. Ma Nisbet va oltre: "Un pH idoneo (oscillante intorno a 8) e temperature intorno ai 40°C sono caratteristici dei sistemi idrotermali sulla Terra. Inoltre, lave modificate nelle facies metamorfiche ricche di zeoliti, argille e solfuri di metalli pesanti, avrebbero fornito superfici catalitiche, pori e setacci molecolari in cui le molecole di RNA sarebbero state contenute e avrebbero potuto assemblarsi". Così, continua Nisbet, "l'occasione per creare la vita diventa non impossibile ma semplicemente altamente improbabile".
La cautela è necessaria, specialmente quando pensiamo più approfonditamente a cosa sarebbe stato necessario per dare inizio a un mondo a RNA. Forse, come suggerisce J.P. Ferris (1993), semplicemente non conosciamo abbastanza la catalisi minerale, tuttavia il problema ha a che fare più con la specificità che con la formazione e la rottura di legami covalenti. Ci sarebbe voluta una specificità più netta di quella in cui ci siamo imbattuti finora con le argille (o con le zeoliti, preferite da Nisbet) proprio per fornire il requisito essenziale: un appropriato insieme di nucleotidi opportunamente attivati, prodotti continuativamente in maniera idonea e allo stato puro, forse per migliaia di anni. Possono esserci stati minerali in grado di ridurre il diossido di carbonio a formaldeide usando, per guidare il processo, la cristallizzazione della pirite da soluzioni di solfuri (Wächtershäuser, 1988), oppure fluttuazioni termiche o di pH, o qualche altra fonte di energia. La formaldeide, in condizioni moderatamente alcaline, può produrre una miscela di zuccheri assai complessa, il formosio, ma ciò non costituisce una fonte adeguata di ribosio, e ancor meno di un particolare enantiomero del ribosio (Shapiro, 1988). Muller ha descritto una sintesi con una resa molto più elevata di zuccherofosfati a partire da glicolaldeide fosfato (Muller et al., 1990). È molto interessante che tale reazione possa aver luogo in maniera controllata in condizioni piuttosto moderate con i minerali della piroaurite (Pitsch et al., 1995), discussi in precedenza. La sintesi di glicolaldeide fosfato nelle condizioni della Terra primitiva resta comunque un problema.
Anche la produzione di adenina presenta alcune difficoltà (Shapiro, 1995), pur se essa può essere sintetizzata molto facilmente da soluzioni di cianuro moderatamente concentrate (Oró, 1960). Si potrebbe proprio parlare di una felice congiunzione di circostanze se sulla Terra primitiva si fosse formato e accumulato cianuro, concentrandosi abbastanza da permettere la formazione di adenina in condizioni di sufficiente purezza. Comunque, saremmo ancora lontani da una situazione in cui l'RNA si sarebbe potuto formare e replicare. Anche altre basi si sarebbero dovute sintetizzare allo stato puro e avrebbero dovuto essere presenti fosfati e ribosio o i loro precursori; tali pezzi avrebbero dovuto assemblarsi correttamente per formare i nucleotidi, pronti per essere attivati, pronti per una polimerizzazione guidata ...
Fantasticando si può immaginare ogni singolo passo, ma per i nucleotidi sarebbero stati necessari troppi passi (Cairns-Smith, 1982). Soltanto gruppi di enzimi, ognuno dei quali adattato dalla selezione naturale a una specificità estrema, o gruppi di chimici, in laboratori ben attrezzati e con tecniche raffrnate, possono fare realmente questo tipo di cose, per lo meno per quanto ne sappiamo noi. Per esempio, come abbiamo visto, si può affermare che le argille presentano una naturale affinità per le basi eterocicliche, per i fosfati e i polifosfati, ma non in modo particolare per l'adenina o per gli zuccherofosfati, tanto meno per i fosfati di qualche zucchero specifico come il ribosio, e ancor meno per un particolare enantiomero del ribosio. Considerando una più ampia gamma di minerali, senza dubbio aumentano le possibilità di trovarne qualcuno adatto alla sintesi di quelle molecole a cui siamo interessati; comunque, tale gamma sarebbe più ampia di quella già considerata, nel migliore dei casi, di uno o due ordini di grandezza, non abbastanza da fare molta differenza a fronte della enorme improbabilità che caratterizza a priori le sequenze dei processi non guidati che devono realizzarsi secondo un ordine appropriato (Cairns-Smith, 1985). Malgrado ciò, sembra essere esistita qualche specificità nelle interazioni dell'argilla e di altri minerali simili con le molecole organiche, che avrebbe potuto fornire un qualche ruolo a questi minerali, forse in un'era precedente la comparsa delle proteine; per esempio, se i nucleotidi sono associati a cationi bivalenti di metalli di transizione, come Zn²+, essi vengono adsorbiti in maniera ordinata nelle regioni interstrato delle argille smectiti, mentre senza questi cationi, che compensano la loro carica negativa, i nucleotidi si legano alle argille solo piuttosto debolmente (Odom et al., 1979; Lawless, 1986). Viceversa essi si legano direttamente alle superfici dell'apatite che sono cariche positivamente (Winter e Zubay, 1995). Inoltre, come gli amminoaciladenilati, che sono nucleotidi modificati, possono formare polimeri di amminoacidi, anche gli oligomeri di RNA possono derivare da nucleotidi attivati legati alla montmorillonite (Ferris e Ertem, 1992; 1993; Ferris 1993).
Una nota sulle argille e la chiralità

A. Yamagishi (1987) ha suggerito il possibile ruolo svolto dai minerali argillosi nell' origine della chiralità. Egli ha trovato che miscele racemiche di chelati tris(1. 10- fenantrolina)metallo (11), per esempio ioni [Fe(phen)3]2+, vanno incontro ad adsorbimento racemico nelle zone interstrato della montmorillonite contenente sodio, in ragione di due volte la capacità di scambio dei cationi propria dell'argilla. Ciò equivale a dire che, nelle zone interstrato, gli ioni chelati porterebbero il doppio della carica positiva che sarebbe necessaria a neutralizzare le cariche negative degli strati dell'argilla, così che anioni interstrato dovrebbero essere incorporati per dare una struttura complessivamente neutra. Da ciò risulta che non è soltanto la carica degli strati a permettere l'ingresso dei chelati, ma c'è anche un effetto cooperativo, originato si da forze tra i chelati organici stessi, che favorisce il riempimento delle zone interstrato. Tutto ciò è però vero solo per le miscele racemiche. Quando viene usata una soluzione di uno solo degli enantiomeri, l'adsorbimento è commisurato alla capacità di scambio dei cationi, cioè è pari soltanto alla metà di quello precedente. Questo complesso argilla-enantiomero avrebbe un'affinità specifica per l'altro enantiomero (fig. 14), e, in molti casi, una preferenza per uno degli enantiomeri di altre molecole chirali. Curiosamente lo ione [Ru(bpy)3]2+, strutturalmente simile al precedente, mostra un adsorbimento enantiomerico esattamente opposto, così come pure effetti opposti di selettività chirale (Yamagishi, 1993). Complessi argilla-enantiomero sono stati usati per la separazione cromato grafica degli enantiomeri (Yamagishi, 1985). Riguardo l'origine dell'attività ottica del materiale biologico, Yamagishi suggerisce che la predisposizione alla chiralità possa essere apparsa (in qualche modo) nei chelati metallici e poi trasferita ai composti del carbonio asimmetrico attraverso tali interposizioni. Due questioni principali sono associate alla diffusa omochiralità delle molecole biologiche. La prima riguarda l'esistenza di L-amminoacidi e D-zuccheri malgrado una biochimica 'speculare' potesse funzionare ugualmente. Sono state fatte molte proposte, ma la scelta fatta dalla biochimica terrestre può essere attribuita all'effetto di un comune progenitore, così come per molte altre cose comuni alla biochimica di tutti gli organismi (Cairns-Smith, 1986). La seconda e più significativa questione è precedente a queste considerazioni e riguarda l'origine della selettività chirale. Prima della comparsa delle proteine appropriate, quali erano i sistemi che potevano gestire le molecole chirali e che potevano anche riconoscere le differenze tra gli enantiomeri? Generalmente le reazioni organiche tra piccole molecole, in soluzioni omogenee, non sono selettive dal punto di vista della chiralità.
Come ha dimostrato Yamagishi, le zone interstrato delle argille possono produrre una disposizione delle molecole in grado di distinguere la chiralità; ciò è una conseguenza delle limitazioni spaziali di queste zone. Anche i processi di cristallizzazione possono essere selettivi dal punto di vista della chiralità, e per lo stesso motivo: i vincoli dovuti all'impacchettamento. Un esempio classico è l'acido D,L-tartarico che può essere ottenuto sotto forma di cristalli racemici, oppure, in condizioni diverse, come miscela di cristalli di cui alcuni sono formati solo dal D-enantiomero e altri soltanto dal L-enantiomero. Mediante la cernita di cristalli, come fece L. Pasteur, o mediante la crescita selettiva, si possono ottenere enantiomeri puri con relativa facilità. Spesso è stato suggerito che se questo tipo di fenomeni si fosse verificato in condizioni naturali, si sarebbe avuta una disponibilità locale di particolari enantiomeri. In particolare, è stato studiato il quarzo (Bernal, 1951), perché le unità di acido silicico di cui è costituito non sono chirali: la chiralità scaturisce totalmente dalla struttura cristallina stessa. Per quanto i cristalli di quarzo, in linea di principio, possano adsorbire enantiomeri in vario grado, in pratica questi effetti, in soluzione acquosa, sono pressoché inesistenti (Bonner, 1995). Simili contraddizioni riguardano anche i minerali argillosi (Youatt e Brown, 1981; Bonner, 1995). Per esempio, la struttura della caolinite è chirale anche se, come il quarzo, è costituita da unità non chirali. In linea di principio i cristalli di caolinite potrebbero distinguere le diversità chirali, in pratica sembra che non possano fado; in questo caso il problema è che tali cristalli sono costituiti da minuti domini (Mansfield e Bailey, 1972) che fanno sì che i cristalli siano di fatto 'racemici'.
L'adsorbimento racemico di Yamagishi non dipende da alcuna chiralità dell'argilla adsorbente sebbene dipenda, in gran parte, dal fenomeno dell'interposizione visto nella smectite; né esso di per sé origina chiralità. Il suo interesse risiede nell'essere un modello di elevata capacità di selezione chirale, che è un prerequisito essenziale. Possono essere prodotti un grande numero di 'involucri' capaci di discriminare le differenze chirali, e in un mondo pre-proteico queste cose non sarebbero state facili da trovare.
Terminiamo questo paragrafo con un altro esempio in cui le argille potrebbero aver contribuito a sostituire le proteine in modo molto semplice.
Le argille come catalizzatori e gli pseudoenzimi di Mortland
Le argille sono state usate come catalizzatori a secco e in condizioni di elevata temperatura nel cracking del petrolio e in altri processi simili che procedono tipicamente attraverso ioni carbonio come intermedi (Rupert et al., 1987). A temperature moderate, in ambiente acquoso a un pH vicino alla neutralità - condizioni per noi più interessanti - le argille sono meno attive come catalizzatori e possono addirittura impedire che le reazioni avvengano agendo non come catalizzatori ma come inibitori, protettori di molecole organiche (van Olphen, 1966). Ciò non è necessariamente negativo: l'attività regolatrice è di estrema importanza, bloccare un fenomeno può essere fondamentale come permettere che avvenga. Le argille, in condizioni normali, possono catalizzare le reazioni sulla loro superficie per mezzo dei siti acidi di Bronsted o, più comunemente, attraverso la dissociazione delle molecole d'acqua legate ai cationi metallici di scambio (Pinnavaia e Mortland, 1986). I cationi idratati (v. figura 1b) sono acidi per natura: più elevata è la carica del catione, più forte è l'acido e maggiore sarà la sua tendenza a perdere un protone. L'argilla smectite, per esempio, con ioni Zn²+ o Cu²+ situati nelle regioni interstrato, somiglia vagamente a un enzima. Infatti le molecole d'acqua, situate negli interstrati e vicine a questi ioni, sono siti acidi fissi che possono donare protoni a molecole organiche adiacenti, come fanno i gruppi donatori di protoni che si trovano intorno al sito attivo di un enzima (Fersht, 1985). Un'altra analogia riguarda il potenziale della catalisi elettrostatica mediata dagli ioni metallici. Per esempio, se una reazione deve passare attraverso un intermedio, in cui una carica negativa si sviluppa su un ossigeno di un gruppo carbonile, la disponibilità di un catione metallico può facilitare il verificarsi di questo passaggio (Fersht, 1985).
M.M. Mortland nel 1984 andò oltre (Mortland, 1984): incorporò il coenzima piridossalfosfato in una smectite 'caricata' con Cu²+ e mostrò che questa combinazione poteva deamminare selettivamente la glutammina. Dimostrò inoltre che la reazione procedeva tramite la formazione di una base di Schiff tra il gruppo amminico dell' acido glutammico e quello aldeidico del piridossalfosfato, seguita dalla formazione di un composto di coordinazione con il rame che si dimostrò essenziale per il processo. Sebbene i candidati più promettenti a essere gli agenti primitivi del trasferimento di elettroni siano, forse, i solfuri di ferro (Hall et al., 1971; Hartman, 1975; de Duve, 1991; Kaschke et al., 1994), altrettanto efficaci potrebbero essere state le smectiti contenenti atomi di ferro; le smectiti, per esempio, reagiscono in modo caratteristico con la benzidina per trasferimento di un singolo elettrone, dando il colore blu caratteristico del radicale cationico della benzidina. L' accettore sembra essere il ferro (III) ottaedrico, cioè il ferro dello strato centrale idrossilico (Solomon, 1968; Solomon et al., 1968; Theng, 1971).
Quanti passi avanti abbiamo fatto immaginando le condizioni in cui si resero disponibili i pilastri centrali della nostra biochimica? Il problema non è come si siano formati, per esempio, i nucleotidi sulla Terra primitiva, ma piuttosto come si siano sviluppati i mezzi necessari alla loro produzione. Questo è ancora un quesito scoraggiante; sembra di avere solo alcuni pezzi della risposta, pezzi che dovrebbero sistemarsi in un puzzle. Ma in quale maniera questi pezzi vanno fatti combaciare? In che modo gli eventi si sono avvicendati in maniera appropriata? Come si sono stabilite le linee di produzione? Forse è il momento di essere più radicali.
L'opzione radicale
Stiamo supponendo che la vita sulla Terra si sia realmente originata per cause naturali. Tuttavia iniziamo a sospettare che non esista alcuno scenario geologico plausibile per un'ingegneria chimica così avanzata quale sembra essere richiesta, in particolare, per la sintesi regolare dei nucleotidi. Se comunque questo evento non si è verificato in uno scenario geologico, esso deve essere avvenuto in uno scenario biologico, e la tecnologia richiesta deve in qualche modo essere stata prodotta dalla selezione naturale. Senza dubbio la selezione naturale imita un progettista intelligente. Quando essa è al lavoro diviene possibile che sequenze di processi gradualmente si riuniscano, si accumulino e si organizzino adeguatamente per uno scopo. Ciò che distingue la sottoclasse dei sistemi chimico-fisici che chiamiamo viventi è proprio il fatto che essi sono stati prodotti in questo modo, cioè attraverso la selezione naturale operante su lunghi periodi di tempo. Abbiamo visto come i minerali argillosi possono aver coadiuvato l'organizzazione delle molecole organiche in situazioni cruciali. Ciò che mancava loro però era un'attività regolatrice globale. Le funzioni delle argille, che abbiamo preso in esame, rientrano nella categoria del chimicamente e fisicamente possibile come, nella maggior parte dei casi, è stato messo in evidenza dagli esperimenti. Esse si avvicinerebbero al biologicamente plausibile se soltanto potessimo immaginare un modo in cui tali processi sono stati organizzati e adattati dalla selezione naturale per cooperare nel dare esiti funzionalmente specifici.
Perché la selezione naturale possa operare è necessaria la riproduzione; più precisamente, deve esistere un'eredità a lungo termine, e ancora più precisamente un'eredità a lungo termine dei mezzi che producono caratteristiche specifiche. Ciò implica un materiale che sia dotato di memoria, un materiale genetico che possa conservare l'informazione su lunghi periodi di tempo, attraverso copie di copie di copie ... Questa non è l'unica cosa di cui deve essere capace un materiale genetico, ma è la più caratteristica, la più ardua, la conditio sine qua non.
Se sosteniamo che i mezzi per produrre molecole così sofisticate come l'RNA debbano essersi evoluti attraverso la selezione naturale, dobbiamo iniziare a pensare a un altro materiale genetico venuto prima. Si deve cercare un materiale genetico veramente primitivo che, a differenza dell'RNA, possa essere verosimilmente apparso sulla Terra senza selezione naturale in uno scenario geologico. Inoltre, dobbiamo pensare a come il materiale genetico primitivo possa essersi evoluto, forse attraverso molti stadi, per produrre sistemi di controllo genetico sorprendentemente sofisticati che sono ora condivisi da tutte le forme di vita sulla Terra. Potrebbe sembrare un'ipotesi azzardata: e se un'argilla, o un minerale microcristallino simile, potesse essere un materiale genetico? Questa è l'essenza dell' opzione radicale per quanto concerne il ruolo delle argille nell' origine della vita.
Modificazione genetica
Prima di tutto dobbiamo chiarire cosa intendiamo per modificazione genetica (Cairns-Smith, 1966). Potremmo supporre che prima dell'RNA ci fossero versioni di molecole di RNA che avevano basi in qualche modo differenti o scheletri più semplici (Schwartz e Orgel, 1985; Joyce et al., 1987; Schwartz, 1993; E schenmoser, 1994), o che, forse, avevano scheletri di glucosio piuttosto che di ribosio (Eschenmoser e Dobler, 1992), o scheletri di tipo peptidico (Wittung et al., 1994; B6hler et al., 1995). Queste sono idee che oggi vengono discusse ampiamente. Nel seguito della storia del mondo a RNA, quando si arriva a immaginare il DNA che sostituisce l'RNA come materiale genetico principale, si immagina una modificazione genetica: un'ipotetica evoluzione di versioni di materiale genetico abbastanza simili da essere capaci, come in effetti erano, di leggere i messaggi l'una dell'altra. È senz'altro plausibile, comunque, che l'evoluzione dei nostri sistemi centrali di controllo sia passata attraverso varie modificazioni di questo tipo e che questo sia stato il modo principale in cui il nostro materiale genetico si è evoluto in quella lontana era, prima dell'ultimo antenato comune a tutta la vita sulla Terra, quando l'evoluzione fondamentale dei sistemi centrali di controllo era ancora possibile. Presumibilmente, in quella situazione, ci sono stati organismi intermedi che possedevano sia geni a RNA che geni a DNA. Da questo punto di vista non ci sono grosse difficoltà. Per esempio, non sarebbe una grande rivoluzione se, in un organismo odierno, si trovassero molecole di RNA in grado di replicarsi ed essere in qualche modo utili. Tuttavia tale organismo avrebbe due materiali genetici diversi. Potremmo chiamare organismi siffatti eterogenetici.
Organismi radicalmente eterogenetici
Questo porta a una possibilità più radicale: ci potrebbero essere tipi di materiali genetici del tutto diversi che collaborano all'interno di un singolo organismo; materiali che, in generale, non potrebbero leggere i messaggi l'uno dell'altro, che immagazzinerebbero e replicherebbero informazioni in modi differenti e con differenti tecniche di espressione e, ciononostante, in grado di collaborare e di essere vincolati da un'utilità reciproca. Anche a questa idea non si può porre alcun veto. Sebbene tutte le forme di vita siano costituite dallo stesso materiale, anche i geni attuali hanno una certa indipendenza d'azione. Essi, per esempio, si scambiano durante la riproduzione sessuale. Per regola, un gene rappresenta un certo tipo di informazione scritta in modo tale da poter essere replicata e da avere l'effetto di migliorare le prospettive di sopravvivenza e riproduzione della comunità di geni alla quale esso appartiene. Niente, in queste regole, impone che differenti geni debbano operare nello stesso modo o essere fatti dello stesso materiale, sebbene l'essere omogenetici renda i sistemi avanzati più efficienti. È probabile che ciò non fosse così efficiente all'inizio, quando i geni dovevano agire direttamente e un dato tipo di gene poteva fare soltanto poche cose, prima che una proteina factotum entrasse in scena. In quel momento sarà stato meglio avere materiali specializzati predisposti per differenti funzioni. Facciamo un parallelo sociale. È probabile che il macellaio abbia imparato il suo mestiere guardando suo padre e ascoltando ciò che gli diceva, il pasticciere francese leggendo libri scritti nella sua lingua, il fabbricante di candele studiando attentamente i disegni nel museo locale. Perciò ciascuno ha usato un'informazione, ereditata, di tipo diverso. Tuttavia, non è stato necessario che essi fossero in grado di approfondire le fonti di informazione di ciascuno per poter beneficiare delle reciproche attività. È tuttora comune nella nostra società avere a disposizione un misto di tecniche, riserve, trasmettitori ed effettori di informazione. l dischetti magnetici costituiscono una delle ultime innovazioni in questo senso, ma sono ancora in uso altri sistemi informativi del tutto incompatibili con essi, come carta e penna.
'Usurpazione' genetica
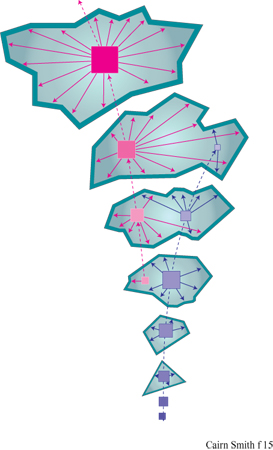
L' 'usurpazione' genetica è stata intesa originariamente come la graduale sostituzione di un certo materiale genetico a opera di un altro, incompatibile, notevolmente differente, attraverso organismi intermedi radicalmente eterogenetici (Cairns-Smith, 1977). Tuttavia, la locuzione è talvolta usata nel significato di modificazione genetica (B6hler et al., 1995). L'usurpazione genetica è un'ipotesi generale che non riguarda gli stadi fmali dell' evoluzione del nostro sistema genetico ma quelli iniziali. Si potrebbe definirla prima origine della vita, in contrapposizione all'origine della nostra attuale macchina biochimica che F. Dyson ha definito seconda origine della vita (Dyson, 1985). Secondo la nostra ipotesi, quest'ultima è il frutto di una fase precedente dell'evoluzione. La figura (fig. 15) illustra la più semplice versione (a uno stadio) di questa idea. Possiamo immaginare che, all'inizio, un materiale genetico primitivo fosse costituito da unità monomeriche facilmente reperibili. Questo materiale si può replicare senza alcun particolare aiuto esterno, ma ci sono situazioni nelle quali lo sviluppo di un fenotipo indipendente costituisce un vantaggio. Per rendere la nostra ipotesi più specifica, supponiamo che il primo materiale genetico fosse un minerale microcristallino, come un'argilla che contiene informazione sotto forma o di un certo assetto nella distribuzione delle cariche, o di una particolare configurazione delle scanalature della superficie, o di un particolare impilamento di strati replicanti si attraverso la crescita e la sfaldatura dei cristalli (fig. 16). Tali caratteristiche possono influenzare il tipo di molecole organiche adsorbite dall' ambiente circostante e le trasformazioni catalitiche di tali molecole che potrebbero essere prodotte dai cristalliti del minerale. Il fenotipo di questo organismo molto primitivo potrebbe, così, essere una sorta di caotico guazzabuglio di molecole organiche, che potrebbero aver funzionato in vari modi: come una gelatina o una colla che impedisce alle particelle di argilla di scivolar via; oppure come stabilizzatori del pH in un arco di valori adatti alla sintesi replicativa, o adsorbendosi sulle facce o sugli spigoli dei cristalli per prevenire la crescita verticale (figura 16a) o laterale (figura 16b), oppure chelando l'alluminio (si ricordi l'acido fulvico) in modo da facilitarne la mobilità e catalizzando così la sintesi delle argille, o ancora svolgendo altri ruoli in circostanze più complesse. Se immaginiamo che un mezzo poroso sia stato il luogo ove si sono svolte queste attività, non sono più necessarie membrane esterne che tengano insieme tutte le parti. Queste molecole costituirebbero l'organismo più semplice a cui si può pensare: geni che creano perturbazioni locali nel loro ambiente il quale, per il fatto di essere il loro ambiente, avrebbe potuto fornire loro un vantaggio selettivo qualora queste perturbazioni locali avessero determinato la sintesi o la sopravvivenza delle argille. Si può notare che i fenotipi in gran parte organici che qui si ipotizzano non sono costituiti dallo stesso materiale dei supposti sistemi di controllo centrale che, al contrario, sono dei minerali microcristallini. Questa sarebbe stata una soluzione particolarmente buona per sistemi veramente primitivi, con ancora uno scarso controllo sulla chimica organica, in quanto sarebbe servita a evitare la confusione tra sistemi di controllo e sistemi controllati. Non è necessario che il controllo gene → fenotipo sia particolarmente accurato. Abbiamo proprio ora immaginato come perfino rudimentali miscele di molecole organiche potrebbero essere servite allo scopo. Ma la replicazione dei geni è tutta un'altra cosa. Qui l'accuratezza è essenziale affinché esista qualcosa al di là di una banale evoluzione. Certamente alcuni 'errori' devono verificarsi affinché la selezione naturale possa lavorarci sopra, ma essi avvengono troppo facilmente. L'accuratezza è di gran lunga il problema più importante ed è, forse, la ragione principale per considerare come primissimi geni un tipo particolare di polimeri cristallini inorganici, piuttosto che dei polimeri organici.
Tipi di geni cristallini
Molto tempo prima dell'identificazione del materiale genetico sono state ipotizzate analogie tra geni e cristalli (Troland, 1917). Nel 1944 E. Schr6dinger usò la famosa dizione "cristallo aperiodico" per definire quella che pensava essere una caratteristica essenziale del gene. Il gene sarebbe stato un cristallo con irregolarità, più simile a un arazzo che a una tappezzeria. Un'irregolarità fisica fondamentale fu presto trovata nella disposizione delle basi nelle molecole di DNA. Ma il DNA si rivelò essere piuttosto diverso da un cristallo. In particolare, la replicazione del DNA (o dell'RNA) non ha niente della spontaneità della crescita dei cristalli, in quanto richiede una complicata assistenza enzimatica e un consumo di energia per avvenire e procedere in modo regolare. Ciononostante, se assumiamo che non fossero necessarie relazioni strutturali tra il materiale genetico primitivo e quello successivo, possiamo riprendere in considerazione i cristalli aperiodici, questa volta nell' ambito dell'origine della vita. Essi, probabilmente, non erano molto efficienti, tuttavia i geni cristallini, poiché usano i processi di crescita dei cristalli per replicare l'informazione, sembrerebbero essere più semplici nelle loro operazioni, più adatti a un inizio. Il ciclo tettonico, indotto dal riscaldamento radioattivo all'interno della Terra, trasforma continuamente i minerali. Insieme al ciclo dell'acqua (a cui il Sole fornisce energia), esso produce innumerevoli sistemi aperti quali fiumi, ruscelli, flussi di acque sotterranee, all'interno dei quali i minerali microcristallini, come le argille, cristallizzano continuamente. Queste condizioni sembrerebbero adatte per la formazione di geni cristallini, ammesso che ciò sia possibile. E lo è?
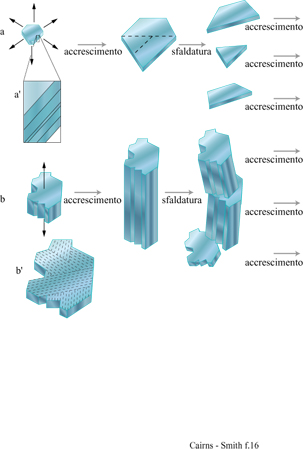
l cristalli aperiodici sono comuni. Tutti i veri cristalli hanno caratteristiche aperiodiche o, come sono anche chiamate, difetti. l cristalli d'argilla ne hanno più degli altri e noi ne abbiamo già considerati alcuni. È importante capire se alcune di queste aperiodicità nella configurazione delle scanalature, nella disposizione dei cationi o in qualche altro aspetto si possano replicare attraverso la crescita del cristallo. La figura 16a è lo schema formale di replicazione di una configurazione unidimensionale di un cristallo (una sequenza di strati con impilamento aperiodico) attraverso un'appropriata combinazione di crescita e di sfaldatura del cristallo. La figura 16b è uno schema simile della replicazione di una specifica configurazione bidimensionale (un 'arazzo'). Per la maggior parte, le caratteristiche aperiodiche di un cristallo non vengono replicate, o perché sono 'accidenti' casuali, tipici della crescita dei cristalli, o perché non passano attraverso le sequenze di eventi necessari alla formazione di ciascuno dei tipi ideali di geni cristallini mostrati in figura 16. Per esempio, la configurazione delle scanalature nel cristallo di sepiolite della figura 11 si sarà certamente estesa durante la crescita dei cristalli. Quanto allo schema della figura l6b, la direzione di crescita sarà stata sufficientemente determinata? Le scanalature avranno, forse, cambiato leggermente dimensione e forma durante l'allungamento dell' asse. Tutte queste possibilità sono in contrasto con la straordinaria uniformità della configurazione di scanalature per tutta la lunghezza del cristallo, anche se non possiamo esserne certi. E ancora, non è chiaro se questo materiale abbastanza fibroso potrebbe rompersi in maniera sufficientemente netta. La caolinite vermiforme sembra più promettente da questo punto di vista, ma ancora non possiamo esserne certi.
Nei geni cristallini unidimensionali illustrati nella figura 16a l'informazione sarebbe contenuta in una dimensione, come una sequenza di strati impilati. In questi geni l'unità di informazione che corrisponde, per esempio, a un appaiamento di basi in un acido nucleico, è nella scala di un intero strato. Le permutazioni di impilamento sono caratteristiche comuni delle argille e dei minerali simili organizzati in strati. Sono possibili innumerevoli permutazioni sia perché strati successivi possono essere orientati in maniera diversa (politipismo), sia perché gli strati che formano il cristallo possono essere di differenti tipi (stratificazione mista) sia, come accade molto spesso, per entrambe le ragioni. Di nuovo, le caratteristiche più importanti hanno a che fare con le direzioni di crescita. In questo caso le pile di strati devono crescere soltanto attraverso l'aggiunta laterale delle unità, e mai attraverso la formazione di nuovi strati alla sommità o al fondo delle pile.
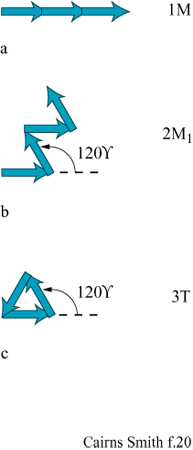
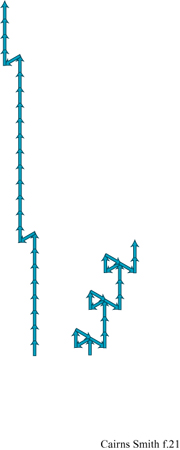
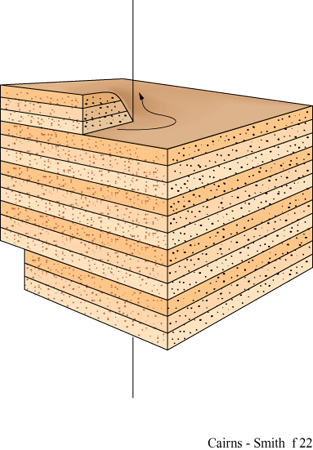
Si ricordi che lo strato unitario della caolinite (v. figura 4) ha un'asimmetria dovuta al fatto che soltanto due terzi dei siti ottaedrici sono occupati dall'alluminio (v. figure 4, 5). Strati successivi non si impilano regolarmente uno sull'altro ma subiscono una deviazione in una direzione, a seconda che il sito vacante dell'alluminio si trovi in una posizione equivalente a quella dello strato successivo oppure no. Due sono le alternative per i siti vacanti: trovarsi alla destra o alla sinistra della direzione di deviazione dell'impilamento. Nella struttura ideale della caolinite il sito vacante si trova regolarmente o a destra (un enantiomero) o a sinistra (l'altro enantiomero). Nella dickite, un politipo di caolinite, i siti vacanti sono alternati (fig. 18). Tuttavia, i cristalli reali si uniformano raramente a queste regole; generalmente le sequenze di impilamento sono, in certo qual modo, aperiodiche. Le caoliniti con struttura altamente disordinata sono comuni. Esse, in linea di principio, potrebbero contenere l'informazione sotto forma di tale sequenza disordinata. Le miche forniscono un altro esempio, ben studiato, di politipismo degli strati di silicati basato su un'asimmetria di tipo diverso. All'interno degli strati di silicato 2:1 delle miche le reti superiori e inferiori non giacciono regolarmente una sull'altra. C'è una deviazione in una direzione (fig. 19) poiché uno strato di mica ha, in effetti, un verso che dipende dal modo in cui la rete superiore di silossano è dislocata rispetto a quella inferiore. Consideriamo ora l'impilamento di tali strati uno sull'altro. l 'versi' degli strati successivi potrebbero tutti giacere nella stessa direzione. Questa è la caratteristica che defrnisce un politipo 1M. In alternativa, le direzioni della deviazione possono oscillare tra + 120° e -120° come nel politipo 2M1, oppure possono mantenere lo stesso cambiamento di direzione di 120° (o + o -) negli strati successivi, determinando il politipo 3T (fig. 20). Questi esempi costituiscono le più comuni sequenze di impilamento che si trovano nelle miche. Tuttavia, come per la caolinite, anche nelle miche sono comuni politipi più o meno disordinati. È ancora più sorprendente che 'errori' isolati o sequenze abbastanza disordinate si trovino spesso ripetuti più e più volte all'interno di un singolo cristallo, specialmente nelle miche biotiti (Baronnet e Kang, 1989). La figura a lato (fig. 21) ne illustra alcuni esempi usando la stessa rappresentazione a freccia della figura 20. Sono stati riportati periodi ripetuti di oltre 100 strati (Czank, 1986). A. Baronnet ha studiato il politipismo delle miche flogopiti, correlate alle precedenti, cristallizzando le in laboratorio in condizioni idrotermali ed esaminando le con un microscopio elettronico per cercare di capire come possa essersi originato un polimorfismo così complesso (Baronnet, 1980; Baronnet e Kang, 1989). Furono trovate prove di un meccanismo di dislocazione a vite, in linea con quanto suggerito da F.C. Frank (1951) perl'origine dei politipi in generale (fig. 22). Da diverso tempo si conoscono politipi dotati di periodi lunghi presenti in altri materiali, come nel carburo di silicio per il quale sono stati riportati periodi superiori a 1000 Å (Verma e Krishna, 1966). È difficile credere che queste ripetizioni esatte, complesse, con ampio spettro, siano da spiegarsi in termini termodinamici, cioè che una particolare disposizione sia semplicemente stata la più stabile nelle condizioni di crescita. Il punto di vista più generale è che in questi casi le ripetizioni originino dai processi di crescita dei cristalli, che esse siano il prodotto di effetti cinetici e che qualsiasi configurazione capiti nel primo stadio di crescita verrà poi copiata più e più volte, come può accadere, per esempio, nella crescita attraverso dislocazione a vite (Verma e Krishna, 1966; Savage e Tauber, 1967; Cairns-Smith, 1988; Heine e Cheng, 1990). Materiali a strati misti forniscono esempi analoghi. In questo caso tipi diversi di strati sono impilati uno sull'altro in una disposizione regolare o irregolare. Gli strati misti sono comuni nelle argille, per esempio possono esserci permutazioni di impilamento tra illite e smectite o, anche, tra caolinite e smectite (Reynolds, 1986). Anche in questi casi, in linea di principio, l'informazione potrebbe essere stata contenuta in questa struttura, poiché sono possibili sequenze irregolari.
Territori di frontiera: modelli e difficoltà
L'idea di un mondo a RNA è in accordo con ciò che sappiamo dell'RNA e delle sue funzioni negli organismi moderni. Ma come teoria sulla natura dei primi organismi viventi, essa dipende fortemente dalla presenza di una costante disponibilità di nucleotidi attivati sulla Terra primitiva. È questo il problema principale se si cerca di unire l'idea del mondo a RNA con una visione strettamente conservatrice dell' origine della vita, basata sull' evoluzione chimica dei nucleotidi. Il problema si semplifica se non dobbiamo assumere che i primi sistemi capaci di evolvere abbiano usato un precursore dell'RNA, piuttosto che l'RNA stesso. Se consideriamo le usurpazioni genetiche come una parte della storia, l'idea del mondo a RNA diventa, piuttosto, l'idea di un 'quartiere a RNA'. Possiamo immaginare che i precursori dell'RNA si siano evoluti come parte del metabolismo secondario, 'opzionale', di alcuni tipi di forma di vita primitiva. Questa forma primitiva non doveva necessariamente basarsi su un materiale genetico che fosse anche vagamente simile all'RNA; in questo modo la prospettiva di trovare qualcosa di adeguato ai primi organismi diviene più ampia, ma sorgono altri problemi. La visione conservatrice può risultare troppo ristretta, ma questa visione radicale può sembrare troppo ampia. Se durante l'evoluzione primitiva le basi materiali della vita cambiavano, come potremo mai sapere quali siano stati i primi materiali? Non esistono risposte certe a questa domanda, ma in laboratorio dovrebbero essere disponibili prove di fattibilità, in parti colar modo per il materiale genetico primitivo che, per definizione, è capace di evolvere in un ambiente non evoluto. Studi recenti (Rotello et al., 1991; Terfort e von Kiedrowski, 1992; Sievers e von Kiedrowski, 1994) consolidano la conoscenza generale dei problemi di replicazione (Orgel, 1992) e questa è, senza dubbio, la strada da cui partire per la ricerca del primo materiale genetico. Inoltre, è giusto che il campo di interesse si sia ampliato fino a includere sistemi completamente artificiali che hanno la potenzialità di evolvere attraverso la selezione naturale. Finora, tuttavia, al di fuori dei campi di ricerca dominati dall'attuale biochimica (o dall'informatica) non sono stati fatti esperimenti significativi sull' evoluzione, basati cioè su polimeri organici di acidi non nucleici, su argille o su qualsiasi altro materiale concreto, naturale, in grado di replicarsi.
La replicazione dei cristalli può sembrare più facile e geochimicamente più fattibile di quella dei polimeri organici. In realtà, per molti aspetti, è ancora di difficile attuazione entro i binari canonici. La maggior parte dei problemi riguarda l'inibizione. Come indicato nella figura 16, la crescita dei cristalli deve aver luogo soltanto in alcune direzioni e in modo ordinato, senza l'introduzione di nuove caratteristiche aperiodiche durante la crescita. Questa è forse parte della risposta a una domanda che viene posta spesso: se il primo materiale genetico era costituito da minerali comuni, perché la vita non si origina continuamente? Un'altra parte della risposta a tali questioni proviene dall' effetto più interessante che ci si può aspettare dal primo materiale genetico. Anche considerando che questo materiale sia in grado di replicarsi, sarebbe stata necessaria una pressione selettiva continua perché qualcosa d'interessante potesse realmente accadere. Non ci sarebbe stato nessun 'incentivo' per le argille, per esempio, a continuare a replicare configurazioni complesse se esse potevano crescere in qualsiasi modo, come generalmente avveniva, qualunque configurazione stessero replicando. Infatti, in qualsiasi momento si fossero semplificate le condizioni, esse avrebbero mostrato la tendenza a perdere l'informazione acquisita precedentemente. Una caratteristica particolare dei materiali genetici primitivi sarebbe la mancanza dell'effetto di irreversibilità che permette a tutti i moderni organismi di essere complessi (una volta persi i sotto sistemi primitivi non è facile tornare indietro). Tutti gli organismi oggi devono essere complessi per sopravvivere comunque e dovunque, ma è probabile che ciò non fosse vero al principio dell'evoluzione (Cairns-Smith, 1982; 1995).
La domanda seguente riguarda il modo in cui la informazione 'secca' contenuta nella sequenza di strati impilati di un cristallo potrebbe aver influenzato le possibilità di sopravvivenza e replicazione del cristallo stesso. Che cosa avrebbe potuto significare tale informazione? Possiamo soltanto fare congetture al riguardo. In modo particolare nelle strutture a strati misti, in cui i diversi tipi di strati quando non sono impacchettati hanno quasi certamente leggere differenze dimensionali, la sequenza di impacchettamento sarà spesso associata a piccole tensioni locali; in questo modo, una pila con una sequenza asimmetrica di strati tenderà a piegarsi e, forse, a formare strutture di ordine superiore; oppure i cristalli con differenti sequenze di impilamento tenderanno a rompersi in modo da rivelare diversi tipi di superfici che tendono ad adsorbire differenti gruppi di molecole organiche e inorganiche e di ioni e che, forse, hanno diverse proprietà catalitiche. Più specificamente, se ci fosse adsorbimento differenziale di monomeri organici sui margini dei diversi tipi di strati, si eserciterebbe un controllo sulle sequenze delle molecole prodotte dalla loro polimerizzazione, molecole che potrebbero ripiegarsi secondo il modo adatto alla sequenza acquisita. Forse quest'ultima congettura è influenzata troppo dall'analogia con le proteine. Tuttavia, per un momento, può essere interessante insistere con un'analogia con la biochimica moderna. Le proteine costituiscono la via principale attraverso cui la sequenza di informazione contenuta nel DNA acquista un significato. Il momento critico dell' espressione di questo significato è quando una molecola proteica si ripiega, trasformando il messaggio in un minuscolo pezzo di macchinario molecolare. La flessibilità di una catena proteica è parte essenziale di questo evento e può essere messa a confronto con la relativa maggiore rigidità del DNA. La rigidità, forse, contribuisce a un'accurata replicazione dell'informazione mentre la flessibilità è necessaria all'espressione di questa informazione.
Possiamo pensare i cristalli sia come oggetti rigidi che come oggetti facilmente frantumabili. Tuttavia non è sempre cosÌ. Molti minerali argillosi sono molto flessibili e resistenti. Questo, senza dubbio, deriva dal loro carattere covalente. Come abbiamo visto (v. figure 1, 4) uno strato d'argilla oltre a essere un cristallo è anche un polimero bidimensionale. Queste caratteristiche consentono ai minerali argillosi di formare strutture di ordine superiore (v. figura 11) alcune delle quali potrebbero essere servite da contenitori più grandi e più complessi degli spazi interstrato delle argille, luoghi ove le molecole organiche avrebbero potuto reagire tra loro in maniera controllata. Giungiamo quindi alla domanda sul perché, sollevata dalla teoria dell'usurpazione genetica. Supponendo, per amore di discussione, che, come nella figura 15, ci fosse stato un solo avvicendamento, per esempio tra i geni minerali e l'RNA, in questo caso la produzione dei nucleotidi sarebbe stata una conseguenza dell'evoluzione delle prime forme di vita basate su geni minerali. Ma perché ciò sarebbe successo? G.F. Joyce e L.E. Orgel (1993), e in seguito A. Eschenmoser (1994), si chiedono: "Quale vantaggio selettivo potrebbe ottenere un piccolo sistema metabolicamente adeguato dalla sintesi di oligonucleotidi?".
Non è possibile rispondere esaurientemente a questa domanda (non conosciamo nemmeno la causa dell'evoluzione di molti metaboliti secondari presenti nelle piante moderne), ma possiamo comunque provarci. Ci sono esempi di questo fenomeno nell'evoluzione successiva. Esso è chiamato preadattamento ed è la fonte principale della capacità inventiva dell'evoluzione. Una struttura, sia essa un osso, un tipo cellulare o una molecola, evolve in un primo tempo con un certo ventaglio di funzioni, sotto un ben definito insieme di pressioni selettive. Ma poi le accade di avere altri usi e quindi di evolvere sotto la spinta di nuove pressioni selettive. In origine le ossa del nostro orecchio interno, meravigliosamente articolate, avevano usi diversi nelle mascelle dei rettili. Le cellule eucariotiche non furono inizialmente 'progettate' per essere neuroni. La maggior parte delle molecole proteiche si sono evo Iute da tipi che spesso avevano usi completamente diversi.
Per quanto si sa attualmente sulle molecole di RNA, le loro funzioni strettamente replicative sono confinate ai virus. Siamo propensi a pensare all'RNA soprattutto come alla molecola che rilascia l'informazione dal DNA alla proteina. Ma la maggior parte dell'RNA delle cellule serve a costruire i pezzi del macchinario, esso è un materiale microstrutturale, così come lo è un nastro portatore di messaggi, e presumibilmente ciò è più vicino alla funzione che doveva avere in origine. La grande idea progettuale a questo riguardo è che sequenze complementari di RNA siano reciprocamente e perfettamente coesive. Questa caratteristica è di per sé importante, indipendentemente da qualsiasi collegamento essa possa avere con la replicazione. l polisaccaridi come gli alginati, che non sono in grado di replicarsi, sono un buon esempio di questo fenomeno. Essi sono progettati per attorcigliarsi l'uno con l'altro, cosa che facilita la produzione di gel.
Una storia perfetta
Poche cose possono essere più importanti per qualsiasi settore avanzato della biologia molecolare della disponibilità di mezzi per assemblare, in maniera precisa, le molecole. Se assumiamo che sia esistito un sistema primitivo di vita in grado di evolvere la capacità metabolica di costruire particolari monosaccaridi e i loro fosfati, allora potremmo immaginare che, in qualche specie, le pressioni selettive avrebbero determinato la produzione di zuccheri 'autocoesivi', cioè in grado di legarsi tra loro, e polimeri di zucchero fosfati. Una molecola simile all'RNA capace di generare una struttura, con gruppi laterali in grado di combaciare tra loro, sarebbe un'elaborazione di questa idea di 'autocoesione', che non ha nulla a che fare, però, con la replicazione. Potremmo immaginare che all'inizio vi fossero soltanto sequenze di qualcosa come poli-A e poli-U attorcigliati insieme a formare un gel ben organizzato. Ma poi accadde che tali sequenze doppie potessero usare la replicazione come mezzo alternativo di produzione. E qui siamo arrivati al punto critico. Questi polimeri ora avrebbero potuto evolversi da soli, attraverso mutazione e selezione, producendo strutture maggiormente intricate basate su sequenze più interessanti di solo poli-A o poli-D. Questo nuovo materiale genetico sarebbe stato dipendente dal vecchio per lungo tempo, per esempio per il rifornimento di nucleotidi. L'informazione contenuta in questo proto-RNA sarebbe stata di un nuovo tipo: non avrebbe avuto mai la necessità di essere trascritta o tradotta dal primitivo materiale genetico. Più probabilmente i due sistemi sarebbero stati incompatibili, incapaci di leggere i reciproci messaggi. Come abbiamo già discusso, ciò non avrebbe impedito una reciproca utilità. Soltanto molto tempo dopo, secondo questa storia, sarebbe diventato possibile un sistema funzionante interamente sugli acidi nucleici, forse soltanto dopo che i mezzi per produrre le proteine si erano perfezionati. Così come il problema del perché, riguardante la pressione selettiva per formare i precursori dell'RNA, anche il problema del come riguardante i meccanismi, è arduo, ma lo è nel senso che è sempre difficile conoscere la storia dettagliata dell' evoluzione, e ancor di più quella dell'era oscura dell'ultimo antenato comune. Comunque, non ci si deve scoraggiare.
Un altro modello per i geni cristallini
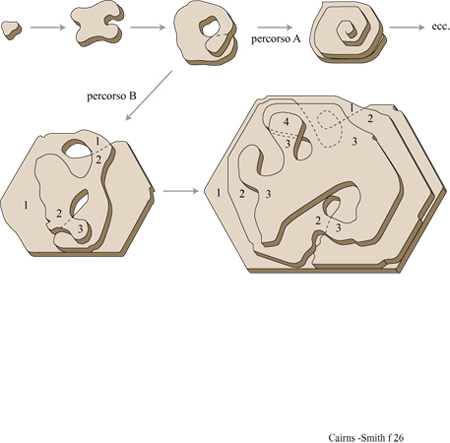
Cristalli argillosi ben formati si trovano più spesso in natura, ove possono crescere da soluzioni molto diluite, che in laboratorio. Le miche sono parenti strette dei minerali argillosi e, come detto precedentemente, forniscono utili modelli da laboratorio per studiare il politipismo caratterizzato da periodi lunghi. Le ferriti esagonali di bario sono modelli di argille particolarmente interessanti, regolarmente interstratificate nonostante siano prodotte a più di 1000 cC: mostrano ripetizioni con un arco di possibilità straordinariamente lungo. Esistono due classi di ferriti di bario costituite da due tipi di strati, designati come M e Y. Esse hanno, rispettivamente, uno e due piani centrali contenenti bario in quella che altrimenti è principalmente una struttura a ossigeno e ferro (lll) (fig. 24). l cristalli M e Y puri sono comuni ma ci sono cristalli costituiti da complesse sequenze impilate, confermate tramite la microscopia elettronica che ha messo in evidenza ripetizioni di lungo raggio, fino a 1500 Å con indizi di sequenze ripetute di 4000 Å e più (Kohn et al., 1971). In uno studio recente sulle caratteristiche morfologiche di queste ferriti (Tumer et al., 1997), è stato proposto che ripetizioni con periodi sorprendentemente lunghi potrebbero essere derivate da una crescita laterale con ramificazioni e autosovrapposizioni (fig. 26).
Una nota sulle crescite laterali
La sintesi degli strati di silicati sembra verificarsi spesso, se non sempre, attraverso l'unione di unità di acido silicico e di ioni metallici ai margini degli strati o delle pile di strati (Cairns-Smith, 1988). La cristallizzazione della mica sintetica flogopite, che avviene attraverso un meccanismo di dislocazione a vite (v. figura 22) è in accordo con questo modello. L'idea alternativa per cui piccole unità verrebbero aggiunte alle superfici principali degli strati è molto meno verosimile. Ciò richiederebbe, infatti, un ciclo di aggiunte in cui, nel caso per esempio della flogopite, le unità di acido silicico, alluminio, magnesio e potassio, conoscano la specifica sequenza di assemblaggio, attraverso strati, sintetizzati solo in parte, probabilmente instabili. Anche la hectorite sintetica sembra formarsi attraverso un tipo di polimerizzazione laterale che, in un primo tempo, produce lamine di materiale con struttura molto appiattita, le quali solo più tardi iniziano a impilarsi (Baird et al., 1971; 1973). Anche i cristalli sottili dell'illite marina come quelli mostrati nella figura 10 (McHardy et al., 1982; McHardy, 1986) suggeriscono una crescita principalmente laterale, così come le strisce incredibilmente sottili di un minerale simile, la rectorite. Queste indicazioni sul modo in cui crescono gli strati di silicato delle argille, insieme ai modelli costituiti dalle ferriti e dalle miche, fanno concentrare l'interesse sui geni cristallini unidimensionali (v. figura 16a). Il disegno della figura 26 si collega alla figura 16a, poiché ciascuna rappresenta l'idea di una crescita esclusivamente laterale. La differenza sta nel fatto che nel disegno la pila di strati non si separa per completare il processo di replicazione, bensì si sovrappone su se stessa. Ciò è apparentemente conveniente per ottenere copie multiple (questo processo mantiene vicini gruppi di strati correlati). Ma per realizzare cicli successivi di replicazioni e quindi per effettuare esperimenti sull' evoluzione, sarebbe necessario che i cristalli si spezzino adeguatamente, o che, per lo meno, si separino alcuni pezzi contenenti almeno una copia della sequenza di impilamento. Essi potrebbero funzionare come germi per la futura crescita di altri cristalli. Questo non sembra proprio un traguardo impossibile ma, tuttavia, deve essere ancora raggiunto.
Bibliografia citata
ABELSON, P.H. (1966) Proc. Natl. Acad. Sci. USA, 55, 1365-1372.
AKERMARK, B., EKLUND-WESTLIN, D., BAECKSTROM, P., LOF, R. (1980) Acta Chem. Scand., Ser. B, 34, 27-30.
ALLAART, J.H. (1976) In The early history of the Earth, a c. di Windley B.F., Londra, John Wiley & Sons, pp. 177-189.
ARRHENIUS, G. (1986) In Clay minerals and the origin of life, a c. di Cairns-Smith A.G., Hartman H., Cambridge, Cambridge Dniversity Press, pp. 97-104.
ARRHENIUS, G. (1990) Fourth Symposium on Chemical Evolution. NASA Ames Research Center.
BAIRD, T., CAIRNS-SMITH, AG., MACKENZIE, D.W. (1973) CIay Minerals, 10, 17-26.
BAIRD, T., CAIRNS-SMITH, A.G., MACKENZIE, D.W., SNELL, D.S. (1971) CIay Minerals, 9, 250-252.
BAR-NuN, A, HARTMAN, H. (1978) Synthesis of organic compounds from carbon monoxide and water by UV photolysis. Origins of Life, 9, 93-101.
BARONNET, A. (1980) Current Topics in Materials Science, 5, 547-548.
BARONNET, A., KANG, Z.C. (1989) Phase Transitions, 16/17, 477-493.
BERNAL, I.D. (1949) Proc. Roy. Soc., 62A, 537.
BERNAL, I.D. (1951) The physical basis of life. Londra, Routledge and Kegan PauI.
BERNAL, I.D. (1967) The origin of Iife. Londra, Weidenfeld and Nicholson.
BOHLER, C., NIELSEN, P.E., ORGEL, L.E. (1995) Template switching between PNA and RNA oligonucleotides. Nature, 376, 578-58l.
BONNER, W.A. (1995) Origins of Life, 25, 175-190.
BRATERMAN, P.S., ARRHENIUS, G., Hm, S., PAPLAWSKY, W. (1995) Origins of Life, 25, 531-538.
BUJDAK, l, FAYBIKOVA, K. EDER, A., YONGYAI, Y., RODE, B.M. (1995) Origins of Life, 25, 431-44l.
CAIRNS-SMITH, AG. (1966) In Towards a theoretical biology, I, Prolegomena, a c. di Waddington C.H., Edimburgo, Edinburgh University Press, pp. 57-66.
CAIRNS-SMITH, AG. (1977) Takeover mechanisms and early biochemical evolution. BioSystems, 9, 105-109.
CAIRNS-SMITH, AG. (1982) Genetic takeover and the mineraI origins of Iife, Cambridge, Cambridge University Press.
CAIRNS-SMITH, AG. (1985) Seven clues to the origin of life, Cambridge, Cambridge University Press, cap. 6.
CAIRNS-SMITH, AG. (1986) Chemistry in Britain, 22, 559-561.
CAIRNS-SMITH, AG. (1988) Intl. Revs. Phys. Chem., 7, 209-250.
CAIRNS-SMITH, AG. (1995) Astron. Soc. Pacific Conf. Series, 74, 31-36.
CAIRNS-SMITH, AG., HALL, A.J., RUSSELL, M.l (1992) Origins of Life, 22, 161-180.
CHANG, S., DESMARAIS, D., MACK, R., MILLER, S.L., STRATHEARN, G.E. (1983) In Earth's earliest biosphere, a c. di Schopf I.W., Princeton, Princeton University Press, cap. 4.
COSTANZO, P.M., LASZLO. P. (1988) Origins of Life, 18, 327-345.
CZANK, M. (1986) Proc. IMA Meeting 86, Stanford, CA, 85.
De Duve, C. (1991) Blueprint for a cel!l. Burlington, North Carolina, Neil Patterson.
DOSE, K. (1975) Peptides and amino acids in the primordial hydrosphere. BioSystems, 6, 224-228.
DYSON, F. (1985) Origins of life. Cambridge, Cambridge University Press.
EBERL, D.D. (1980) CIays and CIay Minerals, 28, 161-172.
EBERL, D.D. (1986) In CIay minerals and the origin of Iife, a c. di Cairns-Smith A.G., Hartman H., Cambridge, Cambridge University Press, pp. 65-71.
ERNST, W.G. (1983) In Earth's earliest biosphere, a c. di Schopf lW., Princeton, Princeton University Press, cap. 3.
ESCHENMOSER, A (1994) Origins of Life, 24, 389-423.
ESCHENMOSER, A, DOBLER, M. (1992) Relv. Chim. Acta, 75, 218-259.
FERRIS, J.P. (1993) Catalysis and prebiotic RNA synthesis. Orig. Life Evol. Biosph., 23, 307-315.
FERRIS, J.P., ERTEM, G. (1992) Origins of Life, 22, 369-381.
FERRIS, J.P., ERTEM, G. (1993) Origins of Life, 23, 229-241.
FERSHT, A (1985) Enzyme structure and mechanism. 2a ed., New York, W.H. Freeman & Co.
FRANK, F.C. (1951) Phil. Mag., 42, 1014-1021.
GETOFF. N. (1962) Zeit. Naturforsch., 17B, 87-90.
GETOFF, N. (1963) Zeit. Naturforsch., 18B, 169-170.
GILBERT, W. (1986) The RNA world. Nature, 319, 618.
GOLDSCHMIDT, V.M. (1952) New Biology, 12, 97-105.
HALL, D.O., CAMMACK, R., RAO, K.K. (1971) Nature, 233, 136-138.
HARTMAN. H. (1975) Speculations on the origin and evolution of metabolismo J. Mol. Evol., 4, 359-370.
HEINE, V., CHENG, C. (1990) In Geometry and thermodynamics, a C. di Tolédano I.C., New York, Plenum, pp. 311-322.
HOLM, N.G. (1992) Marine hydrothermal systems and the origin of Iife. Dordrecht, Kluwer Academic Publishers.
HOLM, N.G., ERTEM, G., FERRIS, I.P. (1993) The binding and reactions of nucleotides and polynucleotides on iron oxide hydroxide polymorphs. Orig. Life Evol. Biosph., 23, 195-215.
HUBBARD, lS., HARDY, lP., VOECKS, G.E., GOLUB, E.E. (1973) Photocatalytic synthesis of organic compounds from CO and water: involvement of surfaces in the formation and stabilization ofproducts. J. Mol. Evol., 2, 149-166.
HULETT, H.R. (1969) Limitations on prebiological synthesis. J. Theor. Biol., 24, 56-72.
HULL, D.E. (1960) Nature, 186, 693-694.
JOYCE, G.F., ORGEL, L.E. (1993) In The RNA world, a c. di Gesteland R.F., Atkins I.F., Cold Spring Harbor, Cold Spring Harbor Laboratory Press, pp. 1-25.
JOYCE, G.F., SCHWARTZ, AW., MILLER, S.L., ORGEL, L.E. (1987) The case for an ancestral genetic system involving simple analogues of the nucleotides. Proc. Natl. Acad. Sci. USA, 84, 4398-4402.
KAMALUDDIN, N.M., DEOPUJARI, S.W., SHARMA, A. (1990) Origins of Life, 20, 259-268.
KAMALUDDIN, N.M., SHARMA, A. (1994) Origins of Life, 24, 469-477.
KASCHKE, M., RUSSELL, M.J., COLE, W.J. (1994) Origins of Life, 24, 43-56.
KERR, R.A (1980) Origin of life: new ingredients suggested. Science, 210, 42-43.
KOHN, J.A, ECKART, D.W., COOK, C.F. (1971) Crystallography of the hexagonal ferrites. Science, 172, 519-525.
KUMA, K., PAPLAWSKY, W., GEDULIN, B., ARRHENIUS, G. (1989) Origins of Life, 19, 573-602.
LAHAV, N., WHITE, D., CHANG, S. (1978) Peptide formation in the prebiotic era: thermal condensation of glycine in fluctuating clay envirouments. Science, 201, 67-69.
LAWLESS, J.G. (1986) In CIay minerals and the origin of life, a c. di Cairns-Smith AG., Hartman H., Cambridge, Cambridge University Press, pp. 135-137.
LINARES, J., HUERTAS, F. (1971) Science, 171, 896-897.
MANSFIELD, C.F., BAILEY, S.W. (1972) Amer. Mineral., 57, 411-425.
McHARDY, W.J. (1986) In CIay minerals and the origin of life, a c. di Cairns-Smith AG., Hartman H., Cambridge, Cambridge University Press, pp. 52-57.
McHARDY, W.J., WILSON, M.J., TAIT, I.M. (1982) CIay Minerals, 17, 23-39.
MILLER, S.L. (1953) Science, 117, 528-529.
MILLER, S.L., BADA, I.L. (1988) Submarine hot springs and the origin of life. Nature, 334, 609-611.
MORTLAND, M.M. (1984) J. Mol. CataIysis, 27, 143-155.
MULLER, D., PITSCH, S., KITTAKA, A., WAGNER, E., WINTER, C.E., ESCHENMOSER, A (1990) Relv. Chim. Acta, 73, 1410-1468.
MULLER, A.W.J. (1983) Physics Letters, 96A, 319-32l.
MULLER, A.W.J. (1985) J. Theor. Biol., 115, 429-453.
NISBET, E.G. (1986a) RNA and hot-water springs. Nature, 322, 206.
NISBET, E.G. (1986b) Episodes, 9, 83-90.
NISSENBAUM, A. (1976) Scavenging of soluble organic matter from the prebiotic oceans. Origins of Life, 7, 413-416.
ODIN, G.S., REYNOLDS, R.C., HARDER, H., ARRHENIUS, G. (1986) In Clay minerals and the origin of life, a c. di Cairns-Smith AG., Hartman H., Cambridge, Cambridge University Press, pp. 81-104.
ODOM, D.G., RAO, M., LAWLESS, J.G., ORO, J. (1979) Association of nucleotides with homoionic clays. J. Mol. Evol., 12, 365-367.
OLPHEN, H. VAN (1966) Science, 154, 645-646.
OLPHEN, H. VAN (1977) An introduction to clay colloid chemistry. 2a ed., New Y ork, John Wi1ey & Sons.
ORGEL, L.E. (1992) Nature, 358, 203-209.
ORÓ, J. (1960) Biochem. Biophys. Res. Comm., 2, 407-412.
PAECHT-HOROWITZ, M., BERGER, J., KATCHALSKY, A. (1970) Prebiotic synthesis of po1ypeptides by heterogeneous polycondensation of amino-acid adenylates. Nature, 228, 636-639.
PAECHT-HOROWITZ, M., EIRICH, F. R. (1988) The polymerization of amino acid adenylates on sodium-montmorillonite with preadsorbed polypeptides. Orig. Life Evol. Biosph., 18, 359-387.
PINNA VAIA, T.1., MORTLAND, M.M. (1986) In Clay minerals and the origin of life, a c. di Cairns-Smith AG., Hartman H., Cambridge, Cambridge University Press, pp. 131-135.
PIRIE, N.W. (1957) Nature, 180, 886.
PIRIE, N.W. (1994) Interdisciplinary Science Reviews, 19, 13-21.
PITSCH, S., ESCHENMOSER, A., GEDULIN, B., HUI, S., ARRHENIUS, G. (1995) Origins of Life, 25, 297-334.
RAUSSELL-COLOM, lA, SERRATOSA, lM. (1987) In Chemistry of clays and clay minerals, a c. di Newman A.C.D., Har10w, Essex, Longman, cap. 8.
REYNOLDS, R.C. (1986) In Clay minerals and the origin of life, a c. di Cairns-Smith AG., Hartman H., Cambridge, Cambridge University Press, pp. 46-52.
RIDLER, P.S., JENNINGS, B.R. (1980) Clay Minerals, 15, 121-133.
ROTELLO, V., HONG, J.I., REBEK J. Jr. (1991) J. Am. Chem. Soc., 113, 9422-9423.
RUBEY, W.W. (1951) Bull. Geol. Soc. Amer., 62, 1111-1147.
RUPERT, lP., GRANQUlST, W.T., PINNA VAI A, T.1. (1987) In Chemistry of clays and clay minerals, a c. di Newman AC.D., Harlow, Essex, Longman, cap. 6.
RUSSELL, M.J., HALL, A.J., CAIRNS-SMITH, A.G., BRATERMAN, P.S. (1988) Submarine hot springs and the origin of life. Nature, 336, 117.
SAVAGE, R.O., TAUBER, A (1967) Materials Res. Bull., 2, 469-478.
SCHWARTZ, AW. (1993) Origins of Life, 23, 185-194.
SCHWARTZ, AW., ORGEL, L.E. (1985) Template-directed synthesis of novel, nucleic acid-like structures. Science, 228, 585-587.
SHAPIRO, R. (1988) Prebiotic ribose synthesis: a criticaI analysis. Orig. Life Evol. Biosph., 18,71-85.
SHAPIRO, R. (1995) Origins of Life, 25, 83-98.
SHOCK, E.L., McCoLLOM, T., SCHULTE, M.D. (1995) Origins of Life, 25, 141-159.
SIEVERS, D., VON KIEDROWSKI, G. (1994) Self-replication of complementary nucleotide-based oligomers. Nature, 369, 221-224.
SIFFERT, B. (1986) In Clay minerals and the origin of life, a c. di Cairns-Smith AG., Hartman H., Cambridge, Cambridge University Press, pp. 75-78.
SOLOMON, D.H. (1968) Clays and Clay Minerals, 16, 31-39.
SOLOMON, D.H., LOFT, B.C., SWIFT, lD. (1968) Clay Minerals, 7, 389-408.
TERFORT, A, VON KIEDROWSKI, G. (1992) Angew. Chem. Int. ed., 31, 654-655.
THENG, B.K.G. (1971) Clays and Clay Minerals, 19, 383-390.
THENG, B.K.G. (1974) The chemistry of clay-organic reactions. New York, John Wiley.
TROLAND, L.T. (1917) American Naturalist, 51, 321-350.
TURNER, G., STEWART, B., BAIRD, T., PEACOCK, R.D., CAIRNS-SMITH, A.G. (1997) J. Crystal Growth, in corso di stampa.
VERMA, A.R., KRISHNA, P. (1966) Polymorphism and polytypism in crystals. New York, John Wiley.
WÄCHTERSHÄUSER, G. (1988) System. Appl. Microbiol., 10, 207-210.
WALKER, J.C.G. (1977) Evolution of the Atmosphere. New York, Macmillan.
WARDEN, J.T., McCULLOUGH, J.J., LEMMON, R.M., CALVIN, M. (1974) Letter to the editor: a re-examination of the zeolitepromoted, clay-mediated peptide synthesis. J Mol. Evol., 4, 189-194.
WAYNE, R.P. ( 1991) Chemistry of atmospheres. 2a ed., Oxford, Clarendon Press, pp. 398-400.
WILSON, C., SZOSTAK, J.W. (1995) In vitro evolution of a self-alkylating rybozyme. Nature, 374, 777-782.
WINTER, D., ZUBAY, G. (1995) Origins of Life, 25, 61-81.
WITTUNG, P., NIELSEN, P.E., BUCHARDT, O., EGHOLM, M., NORDÉN, B. (1994) DNA-like double helix formed by peptide nucleic acido Nature, 368, 561-563.
YAMAGISHI, A (1985) J. Amer. Chem. Soc., 107, 732-734.
YAMAGISHI, A (1987) J. Coord. Chem., 16, 131-211.
YAMAGISHI, A (1993) Dynamic processes on solid surfaces, a c. di Tamaru, K., New York, Plenum, pp. 307-347. YOUATT, J.B., BROWN, R.D. (1981) Origins of chirality in nature: a reassessment of the postulated role of bentonite. Science, 212, 1145-1146.
Bibliografia generale
CAIRNS-SMITH, A.G. Genetic takeover and the mineraI origins of life. Cambridge, Cambridge University Press, 1982.
CAIRNS-SMITH, A.G., HARTMAN, H. Clay minerals and the origin of life. Cambridge, Cambridge University Press, 1986.
DAWKINS, R. The Blind Watchmaker. Harlow, Essex, Longman, 1986.
HOLM, N.G. Marine hydrothermal systems and the origin of life. Dordrecht, Kluwer Academic Publishers, 1982.
NEWMAN, AC.D. Chemistry of clays and clay minerals. Mineralogical Society Monograph No 6. Harlow, Essex, Longman, 1987.
WELLS, A.F. Structural inorganic chemistry. Oxford, Clarendon Press, 1997