
Chimica bioinorganica
Chimica bioinorganica
La chimica bioinorganica studia la funzione esercitata da alcuni elementi inorganici nei fenomeni chimici che avvengono negli esseri viventi e sono oggetto di studio della biochimica. Si tratta di una funzione fondamentale, nonostante la biochimica sia storicamente legata soprattutto alle reazioni delle sostanze organiche nei viventi. Il fatto che l'aggettivo bioinorganica richieda, a tutt'oggi, una giustificazione, testimonia quanto lentamente i progressi delle scienze chimiche si diffondano al di fuori della cerchia degli specialisti ed entrino a far parte della cultura generale. I termini minerali e sostanze organiche evocano immagini molto diverse fra loro: solidi puri e cristallini i primi, materiali impuri e amorfi le altre. Queste immagini, tuttavia, rappresentano un'eredità dei secoli scorsi: le sostanze organiche sono anch'esse composti puri, spesso ottenibili anche allo stato cristallino. Peraltro, i composti organici, così come sono presenti nella materia vivente, difficilmente potrebbero dar luogo a strutture con la robustezza e la solidità necessarie per costituire l'esoscheletro di un mollusco, o i denti e l'endoscheletro di un mammifero. Strutture di questo tipo sono realizzate in natura con i cosiddetti biominerali, ossia sostanze inorganiche, come carbonato di calcio, silice, fosfato di calcio, che si sviluppano in aggregati cristallini analoghi a quelli dei minerali sulla crosta terrestre ma il cui accrescimento è regolato dal metabolismo dell'essere vivente.
I principali elementi presenti nei sistemi biologici sono idrogeno (62,8%), ossigeno (25,4%), carbonio (9,4%), azoto (1,4%) e altri. Essi si trovano combinati in diversi composti, quali il carbonato di calcio, il fosfato di calcio, il solfato di calcio, gli ossidi e il solfato di ferro, la silice e il carbonato di magnesio. Sono diffusi negli animali e nelle piante con funzioni diversificate, con particolare riferimento all'apparato scheletrico. Tuttavia, la funzione dei biominerali non è solo strutturale. Piccoli cristalli di calcite si trovano in sospensione acquosa adagiati alla base dei canali semicircolari nel labirinto dell'orecchio interno, a livello delle macule e delle creste ampollari. Il loro contatto con queste ultime fornisce informazioni al cervello sulla direzione delle linee di forza del campo gravitazionale terrestre e permette il mantenimento della posizione eretta. Cristalli di magnetite all'interno di alcuni batteri forniscono informazioni sulla direzione delle linee di forza del campo magnetico terrestre e conferiscono un certo senso dell'orientamento a questi organismi non dotati di organi sensoriali sofisticati. Cristalli di idrossido di ferro vengono fatti crescere all'interno di strutture proteiche per immagazzinare il ferro in forma insolubile, fintantoché l'elemento non deve essere utilizzato per costituire il centro attivo di metalloproteine e metalloenzimi. Grazie alle moderne tecniche di diffrazione dei raggi X è ormai possibile analizzare la morfologia dei biominerali nei minimi dettagli. Rimane invece molto da capire sui meccanismi attraverso i quali l'organismo guida e controlla l'accrescimento dei biominerali. Si tratta di meccanismi molto complicati, come si può immaginare dalla complessità delle strutture cristalline risultanti e dalla loro varietà. La comprensione del controllo metabolico della formazione dei biominerali è indispensabile per elaborare le strategie di intervento nelle malattie a essi correlate.
I metalli negli esseri viventi
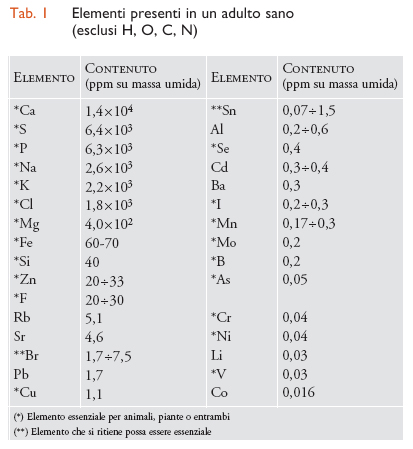
Nella tab. 1 sono elencati i dati relativi agli elementi presenti in un adulto sano di peso corporeo pari a 70 kg (non sono riportati, però, l'idrogeno, l'ossigeno, il carbonio e l'azoto). Molti di tali elementi sono metallici o semimetallici e alcuni sono notoriamente tossici, come il piombo, il rame o l'arsenico; eppure il rame e l'arsenico sono addirittura essenziali per gli organismi. Questo apparente paradosso si spiega molto semplicemente con le cosiddette curve di risposta alla dose. In questi diagrammi si indicano con 100 le condizioni ottimali di salute o di crescita e con 0 la morte dell'organismo. Ad alta concentrazione la curva della risposta scende a 0 per qualsiasi elemento, cioè una concentrazione eccessiva provoca sempre la morte. Per un elemento essenziale la curva parte da 0 a concentrazione 0 (cioè, anche la mancanza dell'elemento provoca la morte), mentre per un elemento non essenziale la curva parte da 100 (cioè, la mancanza dell'elemento non ha influenza sulla salute e la crescita). Si capisce, quindi, che anche elementi non essenziali possono essere normalmente presenti nell'organismo, se la loro concentrazione è abbastanza bassa da mantenere una risposta vicina a 100, mentre gli elementi essenziali devono mantenersi entro l'intervallo di concentrazione che dà una risposta vicina a 100. Per alcuni di questi elementi l'intervallo ottimale è abbastanza ristretto. Si noti anche che la massima concentrazione tollerata di un elemento non essenziale (per es., il rubidio) può essere molto più alta di quella di un elemento essenziale (per es., l'arsenico).
La chimica degli elementi metallici negli esseri viventi è essenzialmente quella delle loro soluzioni acquose, perché l'acqua è la sostanza (inorganica) in essi più abbondante (ca. il 70% in peso nei Mammiferi). Gli elementi metallici si trovano sempre sotto forma di ioni positivi, o cationi, coordinati da altri atomi che formano con essi legami covalenti più o meno polari. A seconda dell'elemento, gli atomi preferiti come leganti possono essere atomi di ossigeno, di azoto o di zolfo: se l'elemento preferisce essere coordinato da atomi di ossigeno, spesso questo si trova sotto forma di ione idrato, o aquaione, cioè legato esclusivamente da molecole d'acqua; altrimenti, l'elemento preferirà interagire con quelle sostanze organiche (amminoacidi, proteine, acidi nucleici, gruppi prostetici di varia natura, ecc.) che forniscono atomi leganti più forti dell'ossigeno dell'acqua. La forza del legame di coordinazione aumenta con la carica dello ione, M+〈M2+〈M3+, e quest'ultima dipende a sua volta dal numero di ossidazione (o stato di ossidazione) preferito dall'elemento.

La diversificazione dei ruoli biologici dei vari elementi metallici (tab. 2) trae origine essenzialmente dalla preferenza dell'elemento per certi atomi donatori, dalla forza dei legami di coordinazione che si formano e dalla possibilità per l'elemento di esistere in più stati di ossidazione. Gli ioni alcalini, come Na+ e K+, data la loro scarsa tendenza a formare composti di coordinazione stabili, sono principalmente usati come trasportatori di carica elettrica per regolare il potenziale elettrochimico attraverso le membrane cellulari. Gli ioni Mg2+ e Ca2+, che possono interagire con leganti organici formando composti discretamente stabili, sono spesso usati come attivatori, ossia per modificare temporaneamente la struttura, per esempio, di una proteina e per far sì che essa possa svolgere un certo compito quando richiesto. Gli ioni Mg2+ e Ca2+ possono anche avere un ruolo strutturale, possono cioè contribuire a determinare e a consolidare la struttura del composto organico (in genere della proteina) a cui sono legati; in questo caso ne diventano costituenti permanenti.
Metalloproteine
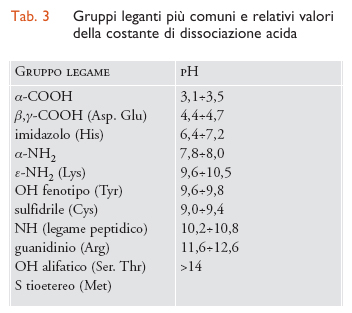
Le proteine sono fra i principali costituenti della materia vivente. Esse sono composte da unità monomeriche elementari, gli amminoacidi, polimerizzati per eliminazione di una molecola d'acqua e formazione di un legame peptidico. A volte gli atomi di azoto e di ossigeno del legame peptidico possono interagire con gli ioni metallici, ma più frequentemente l'interazione delle proteine con gli ioni metallici è a carico delle catene laterali degli amminoacidi: molte di queste contengono gruppi di atomi particolarmente adatti a funzionare come ligandi di ioni metallici. Nella tab. 3 sono riportati i più comuni gruppi leganti con le relative costanti di dissociazione acida. Infatti, gli atomi che coordinano gli ioni metallici devono usare una coppia di elettroni solitaria, o di non legame, e quest'ultima può anche legare uno ione idrogeno invece di uno ione metallico. Al diminuire del pH, ovvero all'aumentare della concentrazione dello ione idrogeno, questo può competere con lo ione metallico causandone la dissociazione dalla proteina che lo lega. Più alto è il pKa, ovvero più alta è l'affinità dell'atomo legante per lo ione idrogeno, più questo effetto può essere rilevante. I gruppi più importanti come ligandi sono: carbossilato, imidazolo, fenolato e tiolato.
Le tre funzioni principali degli ioni metallici nelle metalloproteine sono quelle di mantenere la struttura di queste ultime (ruolo strutturale), di legare l'ossigeno per la funzione respiratoria (ruolo di trasporto di ossigeno) e di cambiare numero di ossidazione accettando e cedendo elettroni. I metalli con ruolo strutturale sono il magnesio, il calcio e lo zinco. Gli altri due ruoli sono ricoperti quasi esclusivamente da metalli di transizione che presentano un riempimento incompleto degli orbitali d. I composti di coordinazione relativi hanno spesso la proprietà di poter variare facilmente il numero di coordinazione, vale a dire sono in grado di legare temporaneamente un ulteriore ligando oltre a quelli forniti dalla proteina e, inoltre, di cambiare il numero di ossidazione. Al ruolo di trasporto di ossigeno o di elettroni è sempre associato, ovviamente, anche un ruolo strutturale, perché la presenza di uno ione metallico legato alle catene laterali conferisce comunque maggiore stabilità alla struttura proteica.
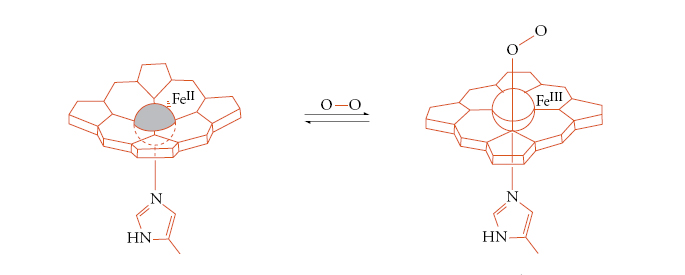
I principali gruppi metallo-leganti responsabili del trasporto di ossigeno si trovano nell'emoglobina (fig. 2) e nella mioglobina, nell'emocianina e nell'emoeritrina. L'emoglobina e la mioglobina usano il gruppo prostetico eme, che, a sua volta, lega uno ione Fe2+, mentre l'emocianina usa una coppia di ioni Cu+ e l'emoeritrina una coppia di ioni Fe2+, in entrambi i casi coordinati da catene laterali imidazoliche dell'amminoacido istidina. Queste strutture piuttosto complicate si sono evolute per risolvere il problema di trasportare l'ossigeno, che è il responsabile di tutte le reazioni di ossidazione negli esseri viventi aerobici, senza farlo reagire prima del tempo. Gli ioni metallici (ferro o rame) sono indispensabili per questo scopo, perché possono cambiare il proprio stato di ossidazione (da +2 a +3 il ferro, e da +1 a +2 il rame), riducendo temporaneamente l'ossigeno a superossido o a perossido, per aumentarne le capacità leganti e quindi impedirgli di dissociarsi prima di essere giunto al luogo di utilizzo. L'ossigeno viene poi liberato all'interfaccia della membrana mitocondriale, dove viene ridotto ad acqua ricevendo gli elettroni da una serie di proteine di trasporto che formano la cosiddetta catena respiratoria. Molte di queste proteine sono anch'esse metalloproteine.
Numerose metalloproteine di trasporto elettronico sfruttano la stessa struttura ferro-eme già vista per l'emoglobina e la mioglobina, con la differenza che le due posizioni di coordinazione assiale sono entrambe occupate da ligandi della proteina stessa, e quindi il ferro non può interagire con ligandi esterni come l'ossigeno. Può, però, cambiare il proprio stato di ossidazione, per esempio acquistando un elettrone da una proteina per passare da Fe3+ a Fe2+ e cedendo l'elettrone a un'altra proteina in un diverso comparto biologico per tornare a Fe3+. Oltre alle emoproteine, altre tipiche metalloproteine di trasporto elettronico sono le cosiddette proteine a ferro-zolfo. La proteina a ferro-zolfo lega 4 atomi di ferro tramite i gruppi tiolati delle catene laterali dell'amminoacido cisteina; gli atomi di ferro sono ulteriormente legati da ioni solfuro per formare un aggregato, o cluster. Il numero di ossidazione di tutto il cluster Fe4S4 può variare di un'unità, permettendo alla proteina di esercitare la sua funzione di trasporto elettronico. Le proteine a ferro-zolfo si trovano in un gran numero di organismi, dalle forme di vita primordiali precedenti i batteri (Archaea) all'uomo. Alcune recenti teorie circa l'evoluzione della vita sulla Terra ipotizzano addirittura che i primi sistemi chimici dotati della capacità di autoriprodursi siano originati dall'interazione di alcune semplici sostanze organiche con la pirite (FeS2), per formare dei cluster ferro-zolfo.
Si potrebbe, a buon diritto, definire una quarta classe di metalloproteine: quella costituita dalle proteine adibite al trasporto o all'immagazzinamento degli ioni metallici stessi. La disponibilità biologica degli ioni metallici per l'organismo dipende da molti fattori, fra i quali la relativa abbondanza nell'ambiente esterno e nel cibo, ma anche la speciazione, ovvero la forma chimica sotto la quale gli ioni metallici si trovano. Per esempio, lo ione Fe3+ è abbondante, ma è assai poco solubile al pH fisiologico; occorre quindi una proteina (la transferrina) che sia capace di legarlo con alta affinità per solubilizzarlo nel plasma e trasportarlo ai siti di utilizzazione (sintesi dell'emoglobina, ecc.) o di accumulo. Nei batteri il trasporto del ferro è affidato a piccole molecole dette siderofori, anch'esse dotate di alta affinità per il ferro, sebbene non così alta come quella della transferrina. Una varietà di quest'ultima, detta lattoferrina, è presente nel latte dei Mammiferi con funzioni batteriostatiche: la sua presenza riduce drasticamente la biodisponibilità del ferro ai batteri occasionalmente presenti e ne impedisce la crescita. Il trasporto del rame è prevalentemente a carico della ceruloplasmina, mentre il trasporto dello zinco, e probabilmente l'accumulo del cadmio (che è un metallo non essenziale), è a carico delle tioneine. È stato rilevato che la biosintesi di queste ultime è stimolata nei casi di avvelenamento da cadmio o da altri metalli pesanti.
Metalloenzimi
L'importanza degli elementi metallici nei sistemi biologici è esemplificata ancora meglio dai metalloenzimi. Gli enzimi sono catalizzatori biologici e quindi, come tali, accelerano il procedere di una reazione chimica verso l'equilibrio finale, senza essere consumati nella reazione stessa. La vita non potrebbe esistere senza gli enzimi perché, alle temperature ottimali per la vita degli organismi, molte reazioni necessarie al metabolismo procederebbero a velocità troppo bassa. Sono noti migliaia di enzimi suddivisi in 6 classi, a seconda del tipo di reazione che catalizzano: ossidoreduttasi, transferasi, idrolasi, liasi, isomerasi e ligasi. In molti casi, la catalisi richiede la presenza di uno ione metallico: quasi la metà degli enzimi finora scoperti sono metalloenzimi. Un importantissimo metalloenzima della classe delle ossidoreduttasi è la citocromo-c-ossidasi. La riduzione dell'ossigeno ad acqua, che avviene alla superficie della membrana mitocondriale, è il passaggio chiave per l'utilizzazione della capacità ossidante dell'ossigeno nel metabolismo degli aerobi:
[1] O2 + 4H+ + 4e− → 2H2O.
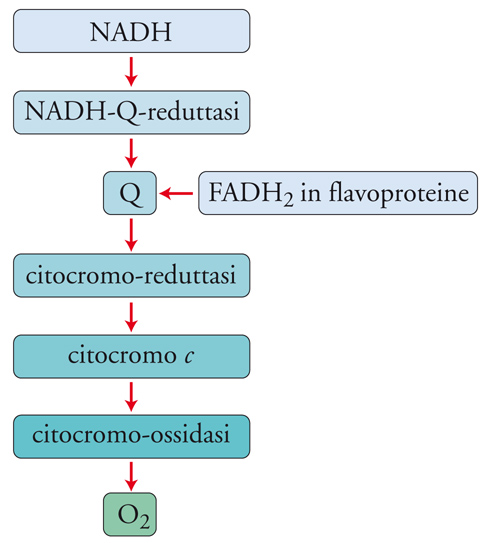
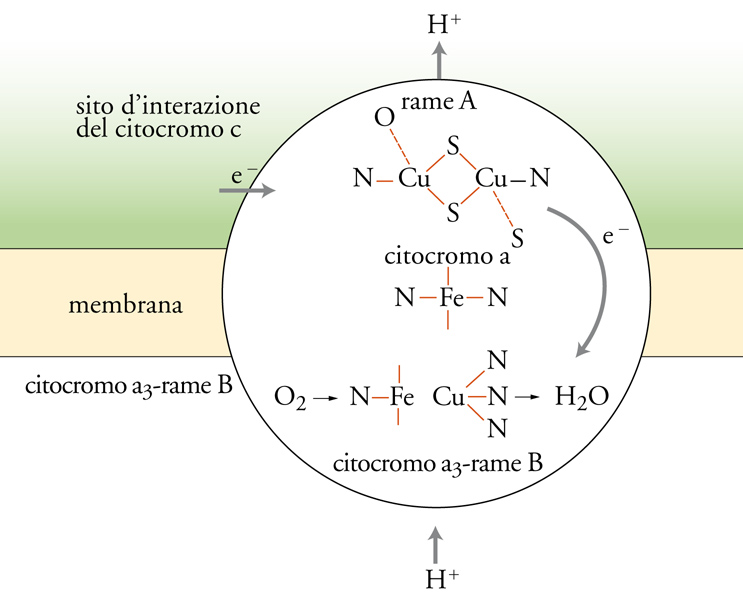
Questa è la reazione finale della catena respiratoria (fig.3). La riduzione della molecola di O2 ad acqua richiede 4 elettroni che, quando sono ceduti uno alla volta alla molecola, producono intermedi come il superossido, il perossido o il radicale ossidrile, che sono molto tossici se liberati nei fluidi biologici. La natura si è cautelata affidando la riduzione dell'ossigeno alla citocromo-c-ossidasi, un enzima inglobato nella membrana mitocondriale; l'enzima ha un sito di attacco per la metalloproteina di trasporto elettronico (citocromo c) all'interno del mitocondrio, e un sito di attacco per l'ossigeno all'esterno. L'enzima contiene 4 centri metallici, costituiti da 2 gruppi ferro-eme, 1 ione rame e 2 ioni rame raggruppati in un cluster, che possono acquistare un totale di 4 elettroni da 4 molecole di citocromo c (fig. 4). L'enzima, quando è totalmente ridotto, può legare l'ossigeno e ridurlo completamente ad acqua, cedendogli in rapida sequenza i 4 elettroni, senza liberare nessuno degli intermedi tossici.
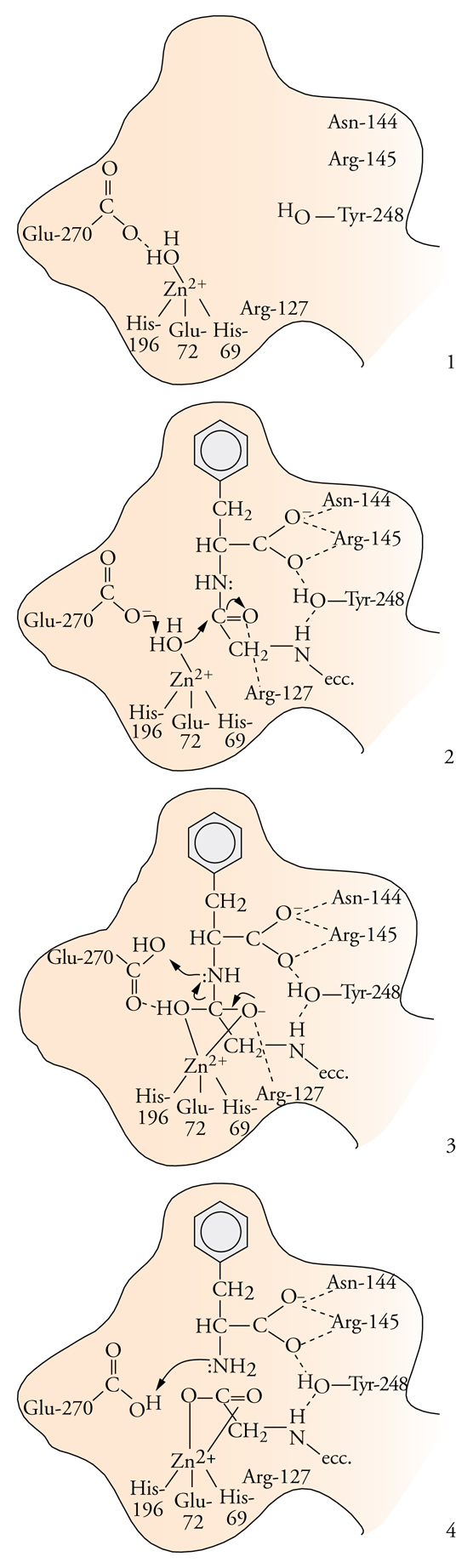
Un'altra classe in cui i metalloenzimi sono particolarmente numerosi è quella delle idrolasi. Spesso le idrolasi contengono zinco, un metallo che non può svolgere funzioni di ossidoriduzione, perché ha gli orbitali d completamente riempiti e un solo numero di ossidazione accessibile, il 2+. La completezza del sottostrato d conferisce però allo zinco una grande flessibilità nel numero di coordinazione, che può passare da 4 a 5 e a 6 senza grandi variazioni di energia del sistema. Lo zinco, quindi, risulta molto adatto a legare transitoriamente il substrato che deve subire la reazione e, nel caso delle idrolasi, ad attivare per coordinazione la molecola d'acqua che deve attaccare il substrato stesso. La fig. 5 mostra uno schema del meccanismo catalitico della carbossipeptidasi: il substrato è un peptide, cioè un poliamminoacido, e l'enzima catalizza l'idrolisi del legame peptidico dell'amminoacido carbossi-terminale. Il peptide si lega in una cavità della molecola dell'enzima, posizionandosi in modo tale che l'atomo di ossigeno della molecola d'acqua coordinata allo zinco possa formare un legame con il carbonio del legame peptidico e provocare la rottura del legame C−N. Il gruppo carbossilato della catena laterale di un amminoacido adiacente serve da accettore dello ione idrogeno della molecola d'acqua, che diventa uno ione ossidrile e risulta così attivata per l'attacco al carbonio peptidico.
I metalloenzimi più interessanti e importanti per la loro funzione sono l'enzima nitrogenasi e gli enzimi coinvolti nel fotosistema II. Il primo catalizza la reazione di fissazione dell'azoto atmosferico, molecola particolarmente inerte, in alcuni batteri (detti azoto-fissatori):
[2] N2 + 10H+ + 8e− → 2NH4+ + H2 .
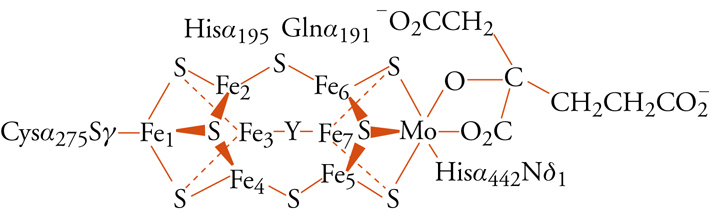
La reazione di fissazione dell'azoto, con produzione di ammonio o ammoniaca, è fondamentale per l'immissione dell'elemento nella biosfera, ed è anche molto importante dal punto di vista industriale, perché quasi tutti i composti chimici che contengono azoto vengono sintetizzati a partire dall'ammoniaca; tuttavia, è molto difficile da realizzare proprio per l'inerzia della molecola di N2. Il meccanismo della nitrogenasi non è ancora del tutto chiarito, ma è ormai evidente, anche grazie alla recente risoluzione della struttura ai raggi X della parte del sistema enzimatico contenente il cosiddetto cofattore ferro-molibdeno, che gli ioni metallici sono essenziali. La fig. 6 mostra il cluster metallico ferro-molibdeno come risulta dalla risoluzione della struttura ai raggi X. Sembra che sia cruciale, perché la reazione proceda a velocità accettabile, che il sistema sia in grado di fornire in rapida sequenza i 6 elettroni che servono per ridurre la molecola di N2 (o gli 8 elettroni, se la reazione coinvolge anche, come sembra, una riduzione di ioni idrogeno). Riduzioni parziali dell'azoto sono molto sfavorite dal punto di vista energetico e producono composti instabili, come N2H2 o N2H4; proprio a questo è dovuta l'inerzia chimica della molecola.
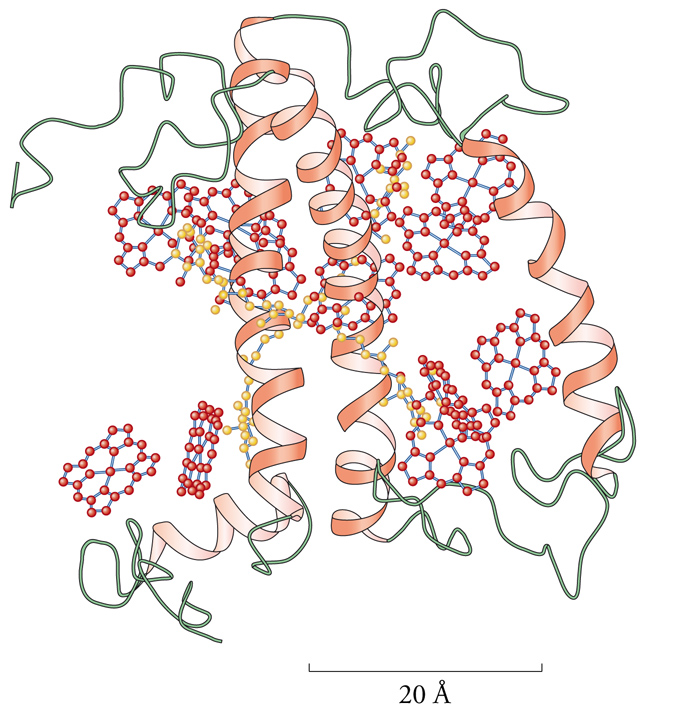
Altrettanto affascinante è la sfida posta agli scienziati dalla comprensione del sistema fotosintetico, che è responsabile della produzione di ossigeno da parte delle piante e di alcuni batteri. La luce è trasformata in energia chimica durante il ciclo fotosintetico nei cosiddetti centri di reazione, che contengono ioni magnesio legati a gruppi eme. L'assorbimento di fotoni si traduce in ossidazione di un altro componente del fotosistema II, il centro di svolgimento di ossigeno (fig. 7). Di quest'ultimo fanno parte 4 ioni manganese, che sono quindi ossidati allo stato di ossidazione 3+ dall'azione indiretta (tramite i centri di reazione già visti) della radiazione solare ultravioletta. La struttura del complesso è ancora ignota e costituisce una delle grandi sfide tuttora aperte per la chimica bioinorganica. Probabilmente, gli ioni manganese passano dallo stato di ossidazione 3+ allo stato 2+, sottraendo all'acqua i 4 elettroni necessari per la formazione di ossigeno, in una reazione inversa a quella catalizzata dalla citocromo-c-ossidasi:
[3] 2H2O → O2 + 4H+ + 4e−.
Si chiude così il ciclo: la radiazione solare fornisce l'energia necessaria per ossidare gli ioni manganese al numero di ossidazione 3+; questi sono quindi in grado di ossidare l'acqua a ossigeno, processo che sarebbe sfavorito dal punto di vista energetico. L'ossigeno viene utilizzato dagli organismi aerobi per liberarsi degli elettroni che si producono nel ciclo metabolico, e così viene nuovamente ridotto ad acqua. Il ciclo metabolico produce, fra gli altri, composti organici ad alta energia come l'ATP (adenosintrifosfato). In questo modo l'energia solare viene immagazzinata negli esseri viventi in una forma conveniente per essere utilizzata quando opportuno.
Metalli e acidi nucleici
Gli ioni metallici possono interagire direttamente con gli acidi nucleici (RNA e DNA), che sono polimeri aventi come unità monomeriche costituenti, rispettivamente, ribonucleotidi e desossiribonucleotidi, polimerizzati tramite la formazione di legami fosfodiesterici. Gli acidi nucleici assumono le ben note strutture a doppia elica, a loro volta organizzate in superstrutture in cui le doppie eliche formano anse o si avvolgono ulteriormente formando doppie eliche di doppie eliche. Alcune delle interazioni dei metalli con gli acidi nucleici sono piuttosto aspecifiche, come, per esempio, la stabilizzazione di alcune superstrutture da parte di ioni Na+ e Mg2+ attraverso interazioni elettrostatiche che schermano i gruppi fosfato, carichi negativamente, gli uni dagli altri. Recentemente, però, si è scoperto che gli ioni Mg2+ possono anche funzionare da attivatori specifici di certe attività catalitiche dell'RNA, e che gli ioni K+ sembrano stabilizzare in particolare la struttura delle unità terminali delle doppie eliche del DNA alla fine dei cromosomi. Un'altra relazione importante fra ioni metallici e acidi nucleici è data dal ruolo che molte proteine contenenti zinco svolgono nell'esplicare le loro funzioni di attivatori o regolatori del DNA. In particolare, una classe di zinco-proteine scoperte di recente, le cosiddette zincfingers (dita di zinco), sembra utilizzare un certo numero di ioni zinco per creare strutture peptidiche che si protendono all'infuori della proteina (come le dita) e vengono usate per intercalarsi tra le spire della doppia elica del DNA. In questo caso il metallo sembra risultare indispensabile per costringere la catena peptidica ad assumere la struttura adatta per l'interazione con il DNA.
Prospettive della disciplina
Occorre premettere che è difficile parlare di prospettive a lunga o anche a media scadenza per una disciplina giovane e in tumultuoso sviluppo come la chimica bioinorganica. Fra le principali componenti che hanno permesso lo sviluppo delle scienze biochimiche, in generale, e della bioinorganica, in particolare, abbiamo cercato di mettere in luce l'importanza dei progressi verificatisi nel campo della strumentazione, dell'informatica, dei nuovi materiali e dei prodotti chimici, delle biotecnologie. Non a caso, questi campi sono oggetto, in Italia come in tutti i paesi industrializzati, di particolare attenzione e sono considerati temi strategici da parte degli organi di finanziamento della ricerca. Per ciò che riguarda la strumentazione, si possono prevedere ulteriori progressi a breve termine nelle tecniche di raccolta dei dati di diffrazione di raggi X. È prevedibile che il numero annuo di risoluzioni di strutture di metalloproteine, metalloenzimi e, soprattutto, di addotti metalloproteine-DNA aumenti in modo ancor più significativo di quanto avvenuto nel recentissimo passato. La tecnica dovrebbe anche potersi avvantaggiare della crescente automazione delle metodiche di crescita di cristalli di macromolecole biologiche. Progressi altrettanto rapidi si attendono nelle tecniche di risonanza magnetica, grazie sia alla diffusione di strumenti EPR (Electron paramagnetic resonance) pulsati, sia al continuo innalzamento dei campi magnetici (fino a oltre 20 T) per l'NMR (Nuclear magnetic resonance). Piccoli produttori di apparecchi scientifici stanno già offrendo sul mercato strumenti per la rilassometria, tecnica finora ristretta a ricercatori che disponevano di strumenti autoprodotti, data la crescente importanza che essa riveste come tecnica di supporto per la diagnostica medica per immagini.
Nel campo dell'informatica, le previsioni di un rapido sviluppo sono tanto facili quanto, spesso, errate per difetto. I progressi dell'informatica hanno contribuito notevolmente ‒ ed è logico attendersi che continueranno a farlo ‒ al crescente successo delle determinazioni strutturali, sia tramite raggi X sia per mezzo dell'NMR. Se poi nel prossimo decennio la potenza di calcolo continuasse ad aumentare anche solo al ritmo attuale, potrebbe non essere azzardato prevedere la risoluzione di un problema centrale della biochimica: quello della previsione a tavolino dell'avvolgimento (folding) di una qualunque proteina, data la sua sequenza primaria. Gli algoritmi esistenti sono già basati su buone previsioni teoriche dei potenziali per le interazioni atomo-atomo per tutti i residui amminoacidici. Invece mancano ancora, in gran parte, buoni potenziali per gli ioni metallici; la loro messa a punto costituirà probabilmente un campo di ricerca molto attivo nel prossimo futuro.
In relazione ai nuovi materiali e prodotti chimici, va rilevato che l'isolamento e la purificazione di sostanze biologiche da estratti cellulari hanno fatto enormi progressi, grazie allo sviluppo di tecniche di laboratorio, quali le tecniche cromatografiche, elettroforetiche, e così via. Queste ultime continueranno a trarre vantaggio dalle tecnologie chimiche sviluppate per la produzione di resine, comprese quelle per affinity chromatography che permettono la separazione immediata della proteina voluta. Usi molto promettenti di nuovi materiali e prodotti chimici si dovrebbero avere nel campo delle zeoliti e in quello dei kit analitici di riconoscimento o di misura dell'attività biologica. Non è questa la sede per illustrare l'importanza delle biotecnologie nel progresso della scienza di questi ultimi anni. Dal punto di vista della caratterizzazione strutturale e funzionale di metalloproteine e metalloenzimi, soprattutto quando si usano metodi fisici d'avanguardia, spesso il fattore limitante è dato dalla quantità di sostanza disponibile. La semplice possibilità di superesprimere una certa proteina, isolabile in piccolissime quantità da un certo organismo, in un organismo diverso, sta già cambiando radicalmente il modo di concepire la ricerca.
Le tecniche del DNA ricombinante stanno progredendo e standardizzandosi così rapidamente che già ora è impensabile affrontare lo studio di una nuova metalloproteina o di un nuovo metalloenzima senza programmare, in parallelo, l'isolamento o la sintesi chimica del gene relativo e la sua espressione in un microrganismo compiacente. Disporre di una proteina espressa significa, poi, non solo poter superare il problema della quantità di sostanza, ma anche poter progettare e caratterizzare un gran numero di mutanti ottenuti tramite mutagenesi sito-specifica e, attraverso di essi, formulare ipotesi sui dettagli del meccanismo d'azione. Infine, è doveroso accennare ai sempre più numerosi programmi di ricerca che si pongono l'obiettivo ambizioso di progettare e realizzare, attraverso tecniche genetiche, enzimi artificiali cone il cosiddetto de novo design. La progettazione di nuovi metalloenzimi è particolarmente promettente, perché già in natura esistono molti esempi di uso dello stesso gruppo prostetico (per es. l'eme) per funzioni che vanno dal trasporto elettronico a quello dell'ossigeno, alla sua stessa attivazione. Le differenze funzionali del gruppo eme devono essere ascritte principalmente alla natura dei leganti assiali: tramite mutagenesi sito-specifica si sta già realizzando la trasformazione, per esempio, della mioglobina o di un citocromo in una perossidasi. Si può prevedere che il de novo design diverrà uno dei maggiori temi della chimica bioinorganica dei prossimi anni.
L'importanza che questa avrà nel prossimo futuro, però, non sarà tanto legata al progresso delle metodiche sperimentali, quanto e soprattutto alla capacità della disciplina di porsi in posizione centrale nei riguardi dei grandi problemi del nostro tempo: la durata e la qualità della vita, la salute, l'ambiente. Per i primi, le prospettive della disciplina dovrebbero risultare ormai chiare. Occorre, invece, soffermarsi sulla collocazione che la bioinorganica potrebbe avere in futuro nelle problematiche ambientali, dato il ruolo fondamentale che rivestono i microrganismi del suolo nella cosiddetta bonifica biologica (bioremediation) di siti inquinati dalla presen-za di composti organici in gran parte derivati dalla petrolchimica. La presenza di questi nuovi inquinanti sulla superficie del nostro pianeta ha stimolato l'evoluzione di nuovi cammini (pathway) metabolici da parte dei batteri del suolo. Attraverso lo sviluppo di plasmidi, questi batteri hanno codificato enzimi che permettono, per esempio, la diossigenazione di composti aromatici o poliaromatici condensati, o la monoossigenazione degli alcani, seguite da ulteriori ossigenazioni che portano, dopo vari passaggi, a intermedi del ciclo di Krebs, e quindi metabolizzabili facilmente da tutti gli organismi. La maggior parte di queste nuove diossigenasi e monoossigenasi sono metalloenzimi, in genere contenenti ferro, il cui meccanismo di azione deve ancora essere chiarito. In questi studi la chimica bioinorganica avrà un ruolo preminente. È anche plausibile che gli enzimi più interessanti dal punto di vista ambientale non siano ancora perfetti dal punto di vista evolutivo, come la maggior parte degli enzimi del corredo genetico stabile. Si può quindi ipotizzare che la bioinorganica possa aiutare l'evoluzione dei microrganismi benefici, contribuendo a progettare plasmidi modificati che migliorino l'efficienza catalitica o lo spettro d'azione dei loro enzimi.
Bibliografia
Baker, Edward N. e altri, Structure, function and flexibility of human lactoferrin, ‟International journal of biological macromolecules", 13, 1991, pp. 122-129.
Banci, Lucia - Bertini, Ivano - Luchinat, Claudio, Nuclear and electron relaxation, Weinheim, VCH, 1991.
Banci, Lucia e altri, The three-dimensional structure in solution of the paramagnetic high-potential iron-sulfur protein I from Ectothiorhodospira halophila through nuclear magnetic resonance, ‟European journal of biochemistry", 225, 1994, pp. 715-725.
Beinert, Helmut, Recent developments in the field of iron-sulfur proteins, ‟FASEB journal", 4, 1990, pp. 2483-2491.
Berg, Jeremy M., Zinc fingers and other metal binding domains, ‟Journal of biological chemistry", 265, 1990, pp. 6513-6516.
The coordination chemistry of metalloenzymes, edited by Ivano Bertini, Russell S. Drago, Claudio Luchinat, Dordrecht-London, Reidel/NATO Scientific Affairs Division, 1983.
Zinc enzymes, edited by Ivano Bertini e altri, Boston, Birkhäuser, 1986.
Bioinorganic chemistry, edited by Ivano Bertini e altri, Mill Valley (Cal.), University Science Books, 1994.
Bertini, Ivano - Luchinat, Claudio, NMR of paramagnetic molecules in biological systems, Menlo Park (Cal.), Benjamin/Cummings, 1986.
Bertini, Ivano - Luchinat, Claudio, NMR of paramagnetic substances, Amsterdam, Elsevier, 1996.
Cammack, Richard - Sykes, A. Geoffrey, in: Iron-sulfur proteins, San Diego (Cal.), Academic Press, 1992.
Christianson, David W., Structural biology of zinc, in: Advances in protein chemistry, edited by Christian B. Anfinsen e altri, San Diego (Cal.), Academic Press, 1991, XLII, pp. 281-355.
Cowan, James A., Inorganic biochemistry: an introduction, Weinheim, VCH, 1993.
Origin, evolution and modern aspects of biomineralization in plants and animals, edited by Rex E. Crick, New York-London, Plenum, 1989.
Dickerson, Richard E., A little ancient history, "Protein science", 1, 1992, pp. 182-186.
Mössbauer spectroscopy, edited by Dominic P.E. Dickson, Frank J. Berry, Cambridge, Cambridge University Press, 1986.
Drago, Russell S., Physical methods for chemists, 2. ed., Fort Worth (Tex.)-London, Saunders College, 1992.
Fraústo da Silva, João José R. - Williams, Robert J., The biological chemistry of the elements: the inorganic chemistry of life, Oxford, Clarendon, 1991.
Metal ion homeostasis: molecular biology and chemistry, edited by Dean H. Hamer, Dennis R. Winge, New York, Liss, 1989.
Hill, Hugh A.O., Bio-electrochemistry, ‟Pure and applied chemistry", 59, 1987, pp. 743-748.
Hoff, Arnold J., Advanced EPR-application in biology and biochemistry, Amsterdam, Elsevier, 1989.
Hoffman, Brian M., Electron nuclear double resonance (ENDOR) of metalloenzymes, ‟Accounts of chemical research", 24, 1991, pp. 164-170.
Kim, Jongsun - Rees, Douglas C., Structural models for the metal centers in the nitrogenase molybdenum-iron protein, ‟Science", 257, 1992, pp. 1677-1682.
Lippard, Stephen J. - Berg, Jeremy M., Principles of bioinorganic chemistry, Mill Valley (Cal.), University Science Books, 1994.
Lowe, David J., ENDOR and EPR of metalloproteins, ‟Progress in biophysics & molecular biology", 57, 1992, pp. 1-22.
Luchinat, Claudio - Sola, Marco, Zinc enzymes, in: Encyclopedia of inorganic chemistry, New York, Wiley, 1994, VIII, pp. 4406-4434.
Okamura, Mel Y. - Feher, Georg, Proton transfer in reaction centers from photosynthetic bacteria, ‟Annual review of biochemistry", 61, 1992, pp. 861-896.
Renger, Gernot, On the mechanism of photosynthetic water oxidation to dioxygen, ‟Chemica scripta A", 28, 1988, pp. 105-109.
Metal ions in biological systems, XXI: Applications of NMR to paramagnetic species, edited by Helmut Sigel, New York, Dekker, 1987.
Stryer, Lubert, Biochemistry, 4. ed., New York, Freeman, 1995 (trad. it.: Biochimica, 4. ed., Bologna, Zanichelli, 1996).
Advances in inorganic and bioinorganic mechanisms, edited by Alfred G. Sykes, New York, Academic Press, 1985.
Theil, Elizabeth C. - Raymond, Kenneth N., Transition metal storage, transport and biomineralization, in: Bioinorganic chemistry, edited by Ivano Bertini e altri, Mill Valley (Cal.), University Science Books, 1994, pp. 1-35.
Metal-DNA chemistry, edited by Thomas D. Tullius, Washington D.C., American Chemical Society, 1989.
Wächtershäuser, Günter, The origin of evolution in an iron-sulfur world, ‟Journal of inorganic biochemistry", 56, 1994, p. 59.
Biomineralization and biological metal accumulation, edited by Pieter Westbroeck, Elizabeth W. De Jong, Dordrecht-London, Reidel, 1983.
Wüthrich, Kurt, NMR of proteins and nuc-leic acids, New York,Wiley, 1986.