
Atmosfera, chimica della
Atmosfera, chimica della
Sommario: 1. Storia ed evoluzione dell'atmosfera terrestre. 2. Gli strati dell'atmosfera. 3. Tempo di permanenza (tempo di vita) dei composti atmosferici. a) Processi di eliminazione atmosferica. b) Tempo di permanenza. 4. Chimica della stratosfera. a) Ozono stratosferico. b) Clorofluorocarburi e ozono stratosferico. c) Buco dell'ozono. 5. Chimica della troposfera in condizioni naturali. a) Ossidi dell'azoto. b) Chimica del metano. c) Chimica dello zolfo. 6. Chimica della troposfera inquinata. a) Composti organici diversi dal metano. b) Chimica dello zolfo. 7. Chimica dell'atmosfera e clima globale. a) Gas responsabili dell'effetto serra. b) Aerosol e clima. □ Bibliografia.
1. Storia ed evoluzione dell'atmosfera terrestre
È opinione comune che il sistema solare si sia formato per effetto della condensazione di una nuvola interstellare di gas e polvere, nota come ‛nebulosa solare primordiale', circa 4,6 miliardi di anni fa. Si pensa che l'atmosfera della Terra e quella degli altri pianeti simili a essa, Venere e Marte, si sia formata in seguito a liberazione di composti volatili intrappolati nelle componenti solide del pianeta. Si ritiene che inizialmente l'atmosfera terrestre fosse una miscela di anidride carbonica (più propriamente, diossido di carbonio, CO2), azoto (N2) e vapor d'acqua (H2O) con tracce di idrogeno (H2), una miscela simile a quella emessa ora dai vulcani.
La composizione dell'atmosfera attuale (v. tab. I) è molto diversa da quella dell'atmosfera primitiva. La maggior parte del vapor d'acqua emesso in origine dall'interno della Terra si è condensata abbandonando l'atmosfera e formando gli oceani, mentre gran parte del CO2 si è prima disciolto negli oceani e quindi ha sedimentato formando rocce di carbonato. Si stima che per ogni molecola di CO2 presente oggi nell'atmosfera, ve ne siano circa 105 incorporate in rocce sedimentarie sotto forma di carbonati. Poiché l'N2 è chimicamente inerte, non solubile in acqua e non condensabile, nel corso del tempo geologico si è per la gran parte accumulato nell'atmosfera divenendone il componente più abbondante.

L'atmosfera primitiva della Terra era una miscela leggermente riducente, mentre attualmente è fortemente ossidante. L'enorme aumento di ossigeno (O2) è dovuto alla sua emissione come prodotto secondario dell'azione di fotosintesi. È stato calcolato che l'O2 atmosferico abbia raggiunto il livello odierno circa 400 milioni di anni fa (v. Cloud, 1983), e che tale livello si mantenga grazie all'equilibrio tra la sua produzione per fotosintesi e la sua eliminazione dovuta alla respirazione e all'ossidazione del carbonio organico. Se tutte le attività di fotosintesi cessassero, la riserva di carbonio organico in superficie verrebbe completamente esaurita in circa 20 anni, ma la quantità di O2 nell'atmosfera diminuirebbe di meno dell'1% (v. Walker, 1977). In assenza di carbonio organico di superficie, l'azione meteorologica sulle rocce sedimentarie consumerebbe tutto l'O2 dell'atmosfera in un periodo di circa 4 milioni di anni.
2. Gli strati dell'atmosfera
L'atmosfera viene genericamente suddivisa in regioni inferiore e superiore. Di solito si considera atmosfera inferiore quella regione che arriva alla zona superiore della stratosfera, a un'altitudine di circa 50 km; il suo studio viene detto ‛meteorologia', mentre quello dell'atmosfera superiore è chiamato ‛aeronomia'. L'atmosfera terrestre è caratterizzata da variazioni di temperatura e pressione a seconda dell'altezza. Anzi, la variazione dell'andamento medio della temperatura al variare dell'altezza è il criterio per distinguere le diverse regioni dell'atmosfera qui di seguito brevemente descritte.
Troposfera. - È lo strato inferiore dell'atmosfera: si estende dalla superficie terrestre fino alla tropopausa, che si trova a un'altitudine compresa tra gli 8 e i 16 km a seconda della latitudine e del periodo dell'anno; è caratterizzata da una diminuzione della temperatura con l'altezza e da una rapida miscelazione verticale.
Stratosfera. - Si estende dalla tropopausa alla stratopausa (da 45 a 55 km di altitudine circa); la temperatura cresce con l'altitudine, fino ad arrivare a uno strato in cui la miscelazione verticale è lenta.
Mesosfera. - Si estende dalla stratopausa alla mesopausa (da 80 a 90 km di altitudine circa); la temperatura decresce con la quota fino alla mesopausa, che è il punto più freddo dell'atmosfera; la miscelazione verticale è rapida.
Termosfera. - È la regione al di sopra della mesopausa, caratterizzata da temperature elevate per effetto dell'assorbimento di radiazioni a basse lunghezze d'onda da parte di N2 e O2 e da un rapido miscelamento verticale. La ‛ionosfera' è una regione della mesosfera superiore e della termosfera inferiore nella quale vengono prodotti ioni per fotoionizzazione.
Esosfera. - È la regione più esterna dell'atmosfera (quota > 500 km) in cui le molecole di gas dotate di sufficiente energia possono sfuggire all'attrazione gravitazionale terrestre.
La troposfera si estende fino a un'altezza media di circa 11 km: sopra l'equatore l'altezza media è di circa 16 km, sopra i poli, attorno agli 8 km. Il nome, coniato dal meteorologo inglese sir Napier Shaw, deriva dalla parola greca tropos, che significa rivolgimento, e in effetti la troposfera è un'area di continua turbolenza e rimescolamento. La troposfera, da cui hanno origine i mutamenti climatici, contiene quasi tutto il vapor d'acqua dell'atmosfera. Attualmente sappiamo che, sebbene costituisca solamente una piccola frazione dell'altezza complessiva dell'atmosfera, essa contiene circa l'80% della sua massa totale. Nella troposfera la temperatura decresce quasi linearmente con l'altezza, con un andamento di 6,5 °C/km. La ragione di questo progressivo raffreddamento è la crescente distanza dalla Terra riscaldata dal Sole. Nella tropopausa la temperatura scende a un valore medio di circa 217 K (-56 °C).
La stratosfera, che si estende da circa 11 a 50 km di altitudine, fu individuata all'inizio del XX secolo dal meteorologo francese Léon-Philippe Teisserenc de Bort. Utilizzando dei palloni sonda per effettuare misurazioni della temperatura, egli scoprì che, contrariamente alla comune credenza dei suoi tempi, la temperatura non diminuiva costantemente con l'altitudine fino ad arrivare allo zero assoluto, ma cessava di diminuire a una quota di circa 11 km. Egli chiamò questa regione ‛stratosfera' dalla parola latina stratum. Sebbene esista effettivamente una regione isoterma che va, a latitudini medie, da 11 a 20 km, la temperatura cresce progressivamente dai 20 ai 50 km, fino a raggiungere 271 K in corrispondenza della stratopausa, una temperatura che non è poi molto diversa da quella media di 288 K della superficie terrestre.
Sono necessari da 1 a 2 mesi perché una specie chimica si disperda per tutto un emisfero ma, sorprendentemente, ci vogliono da 1 a 2 anni perché si disperda nell'intera atmosfera inferiore della Terra. Il tempo relativamente lungo necessario per la miscelazione tra gli emisferi nord e sud deriva dalla presenza all'equatore della cosiddetta ‛zona di convergenza intertropicale'. Poiché l'aria qui tende ad ascendere, questa zona è caratterizzata da notevole nuvolosità e piogge, mentre non esistono forti venti in direzione nord-sud che aiuterebbero a miscelare i gas tra i due emisferi.
La miscelazione verticale nella troposfera è relativamente vigorosa. Una specie chimica emessa in corrispondenza della superficie terrestre può essere miscelata fino alla parte superiore della troposfera in pochi giorni. Salendo nella stratosfera, tuttavia, i moti verticali d'aria si riducono drasticamente e il processo di miscelazione diventa assai più lento che nella troposfera. Di conseguenza, una specie potrebbe impiegare parecchi anni, dal momento in cui attraversa la tropopausa, per raggiungere i livelli più elevati della stratosfera.
Le scale di moto nell'atmosfera vanno da piccolissimi vortici di 1 cm o meno a enormi movimenti di masse d'aria delle dimensioni di un continente. Per comodità sono state utilizzate tre categorie per classificare le scale del moto atmosferico: 1) microscala (scala locale): fenomeni che hanno luogo su una scala dell'ordine di 1 km, quali il flusso e la dispersione del fumo di un camino e il complicato regime di flusso che caratterizza le correnti d'aria che entrano in collisione con palazzi di grandi dimensioni; 2) mesoscala (scala regionale): fenomeni che hanno luogo sulla scala delle centinaia di chilometri, quali la brezza tra terraferma e mare, venti tra monti e valli, e fronti migratori tra alte e basse pressioni; 3) macroscala (scala globale): fenomeni che hanno luogo sulla scala delle migliaia di chilometri, quali le aree di alta e bassa pressione continentali e oceaniche.
Mentre si trova nell'aria, una sostanza può essere alterata chimicamente secondo due modalità. In primo luogo, la stessa luce solare può contenere energia sufficiente per disgregare la molecola: si tratta, cioè, di una reazione fotochimica. Il secondo tipo di alterazione chimica, che è il più comune, ha luogo quando due molecole, urtandosi, reagiscono producendo nuove specie.
Le trasformazioni chimiche nell'atmosfera possono avvenire per via omogenea o eterogenea. Sono omogenee quelle reazioni chimiche che si svolgono interamente in un'unica fase; le reazioni chimiche eterogenee, invece, implicano più di una fase, come ad esempio nel caso di un gas che interagisce con un liquido o con una superficie solida. Durante il trasporto attraverso l'atmosfera, tutte le sostanze, salvo quelle più inerti, partecipano con tutta probabilità a una qualche reazione chimica. Tale processo può trasformare lo stato originario di una sostanza chimica - ossia la forma fisica (gassosa, liquida o solida) e chimica con cui entra nell'atmosfera - in uno stato differente, che può avere caratteristiche simili ma anche molto diverse. I prodotti di trasformazione possono essere molto diversi da quelli da cui hanno origine, dal punto di vista delle proprietà chimiche, della tossicità e di altre caratteristiche. Le modalità con cui questi prodotti vengono eliminati dall'atmosfera possono essere molto differenti da quelle operanti nel caso dei loro precursori: per esempio, quando una sostanza originariamente emessa allo stato gassoso viene trasformata in una particella (v. cap. 3, § a), la velocità complessiva di eliminazione solitamente aumenta poiché le particelle tendono a essere rimosse velocemente dall'aria.
Nonostante il fatto che l'atmosfera sia composta soprattutto da molecole relativamente inerti quali N2 e O2, essa è un mezzo ossidante piuttosto efficace. Una ragione della capacità ossidante dell'atmosfera deriva dal fatto che essa contiene piccole quantità di frammenti molecolari estremamente reattivi, detti ‛radicali liberi'. Inoltre l'atmosfera contiene tracce di specie che, pur essendo meno reattive dei radicali liberi, lo sono abbastanza da attaccare numerosi componenti dell'aria (v. anche radicali liberi: Chimica).
Dopo la loro emissione, le specie chimiche vengono convertite con velocità diverse in sostanze generalmente caratterizzate da stati di ossidazione chimica più elevati di quelli delle specie originarie. Di frequente questa trasformazione ossidativa è accompagnata da un aumento di polarità (e quindi di solubilità in acqua) o da altre variazioni fisiche o chimiche rispetto alla molecola progenitrice. Un esempio è fornito dalla conversione dell'anidride solforosa (o diossido di zolfo, SO2) in acido solforico (H2SO4). L'SO2 è moderatamente solubile in acqua, ma il prodotto di ossidazione, l'acido solforico, è così solubile che perfino molecole isolate di acido solforico nell'aria vengono immediatamente circondate da molecole d'acqua. La scomparsa di una sostanza attraverso una trasformazione chimica può divenire la fonte in situ di un'altra specie. In generale, quindi, una specie emessa nell'aria può essere trasformata tramite un processo chimico in un prodotto dalle proprietà chimico-fisiche molto differenti e con un suo destino particolare. Nel capitolo seguente illustreremo in particolare la chimica dell'atmosfera inferiore, cioè della troposfera e della stratosfera, che sono gli strati in cui hanno luogo le reazioni chimiche essenziali alla vita dell'uomo.
3. Tempo di permanenza (tempo di vita) dei composti atmosferici
Immaginiamo che sia possibile seguire tutte le singole molecole di una certa sostanza emessa nell'aria. Alcune di queste potrebbero venire eliminate vicino al punto di emissione per contatto con le goccioline d'acqua presenti nell'aria o con la superficie terrestre. Altre potrebbero invece venire trascinate verso l'alto nell'atmosfera e trasportate a grande distanza prima di essere alla fine eliminate. Facendo la media dei tempi di vita di tutte le molecole di una sostanza, è possibile calcolare un tempo di vita medio, o ‛tempo di permanenza' per quella sostanza, cioè quanto in media una molecola di quella sostanza rimarrà nell'atmosfera prima di venire eliminata. Tale tempo è definito convenzionalmente come il tempo occorrente per arrivare dalla concentrazione iniziale C0 a C0/e, cioè C0/2,7183; esso è perciò superiore al tempo di dimezzamento. Le possibilità di eliminazione dall'atmosfera sono rappresentate dalla precipitazione e dalla superficie terrestre stessa. Le specie emesse nell'aria devono prima o poi essere eliminate attraverso una di queste due vie.
a) Processi di eliminazione atmosferica
I processi di eliminazione delle specie atmosferiche possono per comodità essere raggruppati in due categorie: deposizione secca e deposizione umida. La deposizione secca è un trasferimento diretto di specie, sia gassose sia sotto forma di particelle, sulla superficie della Terra e procede senza l'aiuto di precipitazione. La deposizione umida, invece, comprende tutti i processi in cui le specie presenti nell'aria sono trasferite sulla superficie terrestre in forma acquosa (cioè pioggia, neve o nebbia).
I fenomeni di deposizione umida comprendono i seguenti processi: 1) dissoluzione di gas atmosferici in goccioline presenti nell'aria, come gocce di nuvole, pioggia o nebbia; 2) rimozione di particelle atmosferiche che vanno a costituire i nuclei di condensazione dell'acqua atmosferica e vengono incorporate nelle goccioline di una nuvola o della nebbia; 3) rimozione di particelle atmosferiche attraverso la collisione con una goccia all'interno o al di sotto delle nuvole. È importante notare che un gas o una particella, anche se incorporati in una goccia, potrebbero non essere rimossi definitivamente dall'aria qualora la goccia evaporasse invece di cadere sulla Terra.
Con l'espressione di ‛materia particellare' (o ‛particella') indichiamo qualunque sostanza, eccetto l'acqua pura, esistente in forma liquida o solida nell'atmosfera in condizioni normali e avente dimensioni microscopiche o submicroscopiche, ma comunque più grandi delle dimensioni molecolari. Tra tutti i componenti dell'atmosfera la materia particellare è unica per la sua complessità. Essa deriva non solo dall'emissione diretta di particelle, ma anche dall'emissione di certi gas che condensano sotto forma di particelle direttamente o dopo aver subito una trasformazione chimica. Per descrivere in modo completo le particelle atmosferiche è necessario specificare non solo la loro concentrazione, ma anche le loro dimensioni, composizione chimica, fase (liquida o solida) e morfologia.
Le dimensioni di una particella sono espresse convenzionalmente come se la particella fosse una sfera con un certo diametro. L'unità abituale è il micron o micrometro (µmm), pari a 10-6 m. Particelle di diametro inferiore a 2,5 µmm sono dette ‛fini' e quelle di diametro superiore a 2,5 µmm, ‛grossolane'.
Gli aerosol atmosferici sono costituiti da particelle le cui dimensioni vanno da poche decine di ångström (1 Ao;= =10-10 m) a parecchie centinaia di micron. Le fonti di aerosol possono essere classificate come primarie e secondarie. Gli aerosol primari sono quelli emessi direttamente sotto forma di particelle, come ad esempio la polvere sollevata dal vento oppure le particelle contenute nel fumo che esce da una ciminiera. Gli aerosol secondari sono costituti da particelle che vengono generate nell'atmosfera, per esempio attraverso reazioni in fase gassosa che producono specie condensabili. Mentre le sorgenti primarie possono dar vita a particelle di ogni dimensione, le sorgenti secondarie producono soprattutto particelle di dimensioni inferiori al micron.
Una volta che gli aerosol si trovano nell'atmosfera, dimensione, numero e composizione chimica delle particelle che li compongono vengono modificati attraverso diversi meccanismi, finché vengono eliminati mediante processi naturali. Alcuni dei processi chimici e fisici che influenzano l'‛invecchiamento' degli aerosol atmosferici sono più efficaci in un certo ambito di dimensioni delle particelle piuttosto che in un altro. Sebbene l'invecchiamento delle particelle sia influenzato da processi specifici, il loro tempo di permanenza nell'atmosfera inferiore di solito è dell'ordine di alcune settimane. Molto vicino al suolo, i meccanismi principali per la rimozione delle particelle sono la deposizione o l'impatto su superfici; al di sopra di altezze di 100 metri circa, invece, il meccanismo principale di rimozione è l'incorporamento per fenomeni di precipitazione.
Allorché l'aria sale attraverso una nuvola e diviene leggermente soprasatura di vapor d'acqua (la sua umidità relativa supera cioè il 100%), si formano goccioline di acqua su nuclei di condensazione - solitamente particelle solubili di aerosol (ad esempio particelle microscopiche di alcuni sali) che esistono nell'atmosfera a concentrazioni che vanno da 100 a 3.000/cm3 - che aumentano di dimensioni per condensazione di vapor d'acqua. Al processo di condensazione è associato un rilascio di energia, e questo dà ulteriore impulso all'ascesa dell'aria. La crescita delle goccioline e gli urti tra di esse creano gocce di pioggia, che crescono rapidamente incorporando, mentre cadono, altre goccioline delle nuvole ed evaporano quando incontrano aria non satura (con umidità relativa inferiore al 100%); l'evaporazione raffredda l'aria e, unitamente all'effetto del peso dell'acqua liquida, causa spinte verso il basso nei flussi d'aria.
La condensazione del vapor d'acqua in un'atmosfera completamente priva di particelle richiederebbe che il vapor d'acqua fosse estremamente soprasaturo, mentre il valore di soprasaturazione dell'atmosfera in genere non oltrepassa l'1% (equivalente a un'umidità relativa compresa fra 100 e 101%). Ciò è dovuto al fatto che nell'atmosfera le goccioline delle nuvole e della nebbia si formano per nucleazione eterogenea, ovvero per condensazione d'acqua su particelle di aerosol, i cosiddetti ‛nuclei di condensazione delle nuvole'. A differenza dei nuclei di ghiaccio, che sono distribuiti in modo abbastanza rarefatto, i nuclei di condensazione delle nuvole, sia d'origine naturale sia antropogenici, sono presenti ovunque nell'atmosfera. La velocità di crescita di una singola gocciolina dipende non solo dall'affinità del suo nucleo per l'acqua (ossia dalle sue proprietà igroscopiche), ma anche dai processi di diffusione e conduzione termica che determinano la velocità a cui il vapor d'acqua si trasferisce alla goccia e quella a cui il calore se ne allontana. La velocità di crescita è estremamente rapida per una goccia di piccole dimensioni, ma diminuisce al crescere della goccia. Per esempio, un nucleo di sale del diametro di 1 µmm a una temperatura di 274 K (cioè appena al di sopra di 0 °C) in condizioni di soprasaturazione costante dello 0,05% (tipica delle condizioni base di una nuvola) darà origine a gocce di diametro via via crescente di 10, 20, 40 e 80 µmm in tempi rispettivamente di 1 minuto, 14,5 minuti, 1,64 ore, e 11,5 ore. In condizioni atmosferiche normali, gocce con diametro di 80 µmm sono ancora tropppo piccole per raggiungere la Terra prima di evaporare; di solito, soltanto gocce con diametro superiore ai 200 µmm possono essere considerate gocce di pioggia. Pertanto, la crescita per condensazione da sola non potrebbe produrre gocce in grado di precipitare entro il tempo di vita tipico di una nube convettiva (cioè tra 30 minuti e 1 ora); la formazione di gocce di pioggia deve quindi aver luogo fondamentalmente per coalescenza di goccioline o per fenomeni di congelamento.
b) Tempo di permanenza
Il principio fisico fondamentale che regola il comportamento delle specie chimiche nell'atmosfera è quello di conservazione della massa. In qualsiasi ipotetico cubo d'aria deve sussistere il seguente equilibrio tra le specie chimiche:
velocità di ingresso - velocità di uscita + velocità di introduzione - velocità di eliminazione = velocità di accumulo nell'ipotetico volume.
Questo equilibrio deve valere nel caso del più piccolo volume d'aria così come per l'intera atmosfera.
Se indichiamo con Q la massa totale della sostanza contenuta nel volume d'aria, con Fi e Fu i flussi di ingresso e uscita, rispettivamente, di quella sostanza dal volume d'aria, con P la velocità di introduzione di una specie da una sorgente e con R la velocità di rimozione delle specie, il principio di conservazione della materia può essere espresso in forma matematica come segue:
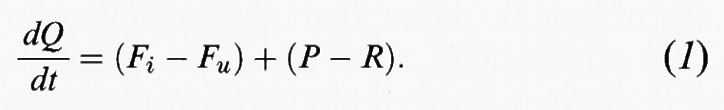
Che cosa succede se la quantità Q della sostanza nel volume considerato non varia con il tempo? In questo caso Q è una costante e dQ/dt=0. Affinché il valore di Q possa rimanere immutato, qualsiasi quantità di sostanza generata deve essere bilanciata da una analoga scomparsa nel volume d'aria. Ciò significa che:
Fi + P = Fu + R. (2)
In questo caso si dice che ci si trova in condizioni di ‛stato stazionario'.
Se il volume a cui ci si riferisce è l'intera atmosfera, allora Fi = 0 e Fu = 0, e nel caso di una sostanza in condizioni di stato stazionario la velocità di immissione da parte delle fonti deve essere uguale alla sua velocità di eliminazione, cioè P=R. Il tempo di permanenza medio, τ, nei termini delle quantità introdotte in precedenza, è:

Poiché in condizioni di stato stazionario è R + Fu = P + +Fi, il tempo di permanenza è dato anche da:

Se il volume d'aria considerato coincide con l'intera atmosfera, in condizioni di stato stazionario si avrà:

Per illustrare il concetto di tempo di permanenza, si considerino tutti i composti contenenti zolfo presenti nella troposfera. Se la concentrazione media di questi composti è di una parte su un miliardo (1 ppb), valutata sulla massa, e si suppone l'esistenza di uno stato stazionario, allora, dato che la massa della troposfera è di circa 4 × 1021 g, la massa totale dei composti contenenti zolfo nella troposfera è Q = 4 × 1012 g. Si calcola che le fonti naturali e antropogeniche di zolfo contribuiscano a dare un valore totale di P di circa 200 × 1012 g/anno, probabilmente con un fattore di precisione di circa 2. Quindi il tempo di permanenza dei composti solforati nell'atmosfera può essere valutato pari a:
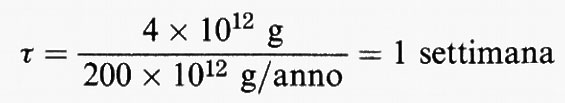
con un fattore di precisione compreso tra 2 e 4.
Il calcolo dei tempi di permanenza può essere utile per valutare a quale distanza dalla sua fonte è possibile che una sostanza rimanga nell'aria prima di essere eliminata dall'atmosfera.
Se consideriamo una particolare regione dell'atmosfera, diciamo il volume d'aria sopra una città oppure sopra l'emisfero nord, o anche l'intera stratosfera, possiamo definire un tempo di miscelazione caratteristico per quel volume come il tempo necessario per miscelare completamente all'interno di tale volume un certo composto chimico. Indichiamo il tempo caratteristico di miscelazione con τM. Una specie risulta poco miscelata all'interno di un volume se almeno uno dei tempi di miscelazione caratteristici τM non è piccolo rispetto al tempo di permanenza τ di quella specie. Si noti che questo significa che un particolare volume può presentare una buona miscelazione rispetto ad alcune specie e cattiva rispetto ad altre, a seconda del tempo di permanenza di ciascuna specie. Inoltre, i tempi di miscelazione nell'atmosfera sono tipicamente diversi per diverse direzioni. Ad esempio, come abbiamo appena notato, il tempo caratteristico di miscelazione verticale nella troposfera, cioè il tempo richiesto per miscelare una specie uniformemente dal suolo fino alla tropopausa, è di circa una settimana, mentre il tempo di miscelazione orizzontale della troposfera, cioè il tempo richiesto per miscelare completamente un componente attorno al globo nella troposfera, è di un anno circa. Quindi la troposfera può essere considerata ben miscelata rispetto al 85Kr (uno degli isotopi radioattivi del kripton), che ha una semivita di 10 anni, mentre per i composti dello zolfo, il cui tempo di permanenza è di circa una settimana, non c'è una buona miscelazione neppure verticalmente.
La miscelazione della stratosfera è considerata cattiva rispetto a tutti i componenti atmosferici presenti in tracce. Il tempo di miscelazione verticale caratteristico può essere valutato in base al rapporto tra il quadrato della profondità verticale della stratosfera, che è di circa 40 km, e un coefficiente di diffusione verticale caratteristico, pari a 1 m2/s circa:
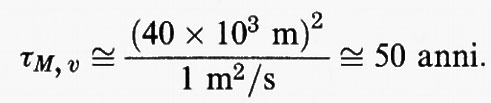
Di conseguenza la stratosfera può essere considerata ben miscelata soltanto rispetto a specie che hanno tempi di permanenza ben superiori ai 50 anni. In effetti, uno dei pochi esempi di specie dalla vita così lunga è l'elio, la cui fonte è la superficie terrestre, che viene eliminato in quanto fugge nello spazio attraverso gli strati più alti dell'atmosfera.
Spesso la velocità di eliminazione di una specie chimica dall'atmosfera è proporzionale alla sua concentrazione (eliminazione del primo ordine): tanto più alta la concentrazione, tanto maggiore la sua velocità di eliminazione. Questa è solitamente la situazione sia nel caso di deposizioni asciutte sulla superficie terrestre, sia dell'incorporamento da parte delle goccioline delle nuvole. Si consideri una specie per la quale valgano le condizioni di stato stazionario e che venga eliminata a una velocità proporzionale alla sua concentrazione con una costante di proporzionalità λ. Una specie di questo tipo è il 85Kr, per il quale l'unico processo di rimozione significativo è il decadimento radioattivo. Per il 85Kr, quindi:

Perciò, per calcolare il tempo di permanenza del 85Kr non è neppure necessario conoscere la quantità in cui esso è presente nell'atmosfera, Q, ma solamente la costante di decadimento radioattivo, λ, che è legata alla semivita, t1/2, dalla relazione
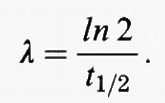
Di conseguenza non ha alcuna importanza se il 85Kr è miscelato uniformemente nell'intera atmosfera. Nel caso in cui il processo di eliminazione sia del primo ordine, è possibile valutare in modo semplice e accurato il tempo di permanenza anche per una specie poco miscelata purché si possa stimare accuratamente la sua velocità di eliminazione.
Consideriamo ora il caso di una specie, avente una massa Q nell'atmosfera, che venga rimossa da due processi indipendenti, il primo a una velocità λ1Q e il secondo a una velocità λ2Q, dove λ1 e λ2 sono i due coefficienti di eliminazione del primo ordine. Il suo tempo di permanenza complessivo è dato da:
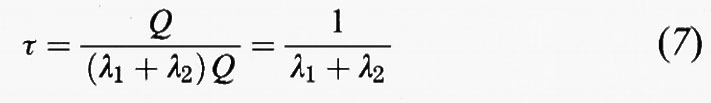
ovvero

Il primo processo potrebbe essere quello della deposizione asciutta e il secondo quello dell'incorporamento entro una nuvola. È possibile associare delle costanti di tempo ai due singoli processi di eliminazione:

in cui τ1 può essere considerato come il tempo di permanenza che la specie avrebbe se l'unico processo di eliminazione fosse il processo 1; analoga definizione per τ2. Sulla base delle equazioni (8) e (9) possiamo esprimere il tempo di permanenza complessivo nei termini dei due singoli tempi di eliminazione τ1 e τ2 come:

L'equazione (10) mostra che le diverse modalità di eliminazione possono essere sommate tra di loro per dare un tempo di permanenza totale, così come due resistenze elettriche in parallelo si sommano per dare una resistenza totale che è perfino più piccola della più piccola delle due resistenze. Dall'equazione (10) si ricava:

Se τ1 ≫ τ2, se cioè il tempo di permanenza associato con l'eliminazione attraverso il processo 1 è molto più lungo di quello associato con il processo 2, quest'ultimo è il meccanismo di rimozione più efficace e τ ≅ τ2. Quindi, quando esistono più modalità di eliminazione in competizione tra di loro, al fine di stimare il tempo di permanenza complessivo di una specie, bisognerebbe sempre cercare di ottenere delle valutazioni più accurate per la velocità di eliminazione più elevata.
Si mostrerà tra breve che il meccanismo di eliminazione primario nella troposfera per quei composti organici che contengono atomi di H labili o doppi legami C=C è la reazione con il radicale ossidrile (• OH). Nella troposfera l'eliminazione di quei composti che reagiscono con i radicali • OH ha luogo secondo la reazione chimica:
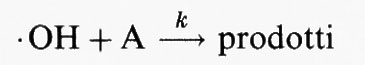
Il parametro k è la costante cinetica della reazione, per cui la velocità della reazione è data da k[• OH][A], in cui le parentesi quadrate indicano la concentrazione delle specie in esse contenute.
Dall'analisi dei tempi di permanenza atmosferici, se Q è la quantità totale della specie A nella troposfera e la sua velocità di rimozione è R = k[• OH]Q, allora il tempo di permanenza del composto, in base all'equazione (6), è:
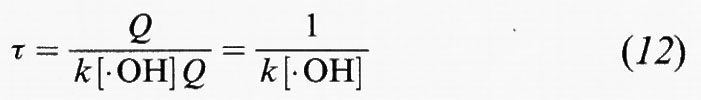
in cui [• OH] è una media globale appropriata della concentrazione dei radicali • OH nella troposfera.
4. Chimica della stratosfera
A causa della rapida miscelazione che ha luogo tra la superficie terrestre e la tropopausa, nella troposfera la composizione chimica dell'aria in condizioni normali, non contaminata, è essenzialmente indipendente dall'altitudine. Come già sottolineato, l'andamento crescente della temperatura nella stratosfera fa sì che si generi uno strato dinamicamente stabile e privo di miscelazione verticale di aria più calda sopra ad aria più fredda. Di conseguenza, il tempo medio necessario a una molecola che si trova nella tropopausa per miscelarsi nella parte centrale della stratosfera è di circa 10 anni. A causa di questa lenta miscelazione, le specie nella stratosfera, come già notato, non sono distribuite uniformemente nelle diverse quote. Il componente chimico qualitativamente più importante della stratosfera è l'ozono (O3).
a) Ozono stratosferico
L'ozono è entrato a far parte dei componenti dell'atmosfera nel periodo durante il quale l'O2 ne è divenuto il costituente più importante. In realtà la quantità di O3 nell'atmosfera è estremamente piccola. Nella troposfera originaria e non inquinata le frazioni, in volume, di ozono, o i cosiddetti ‛rapporti di miscelazione', sono compresi tra le 10 e le 40 ppb, con valori leggermente più alti nella troposfera superiore. L'ozono raggiunge un rapporto di miscelazione massimo di circa 10 parti per milione (ppm), in volume, a un'altitudine compresa tra i 25 e i 30 km nella stratosfera. Si può avere un'idea di quanto piccole siano le quantità di ozono presenti nell'atmosfera considerando che se tutto l'ozono atmosferico fosse portato sulla superficie terrestre a temperatura e pressione normali, esso formerebbe uno strato dello spessore di 0,5 cm soltanto. Il 90% dell'ozono atmosferico si trova nella stratosfera.
Nonostante si tratti di un elemento presente solo in tracce, l'ozono esercita un'influenza profonda sull'atmosfera e sulla vita sulla Terra. Esso assorbe le radiazioni solari ultraviolette (UV) con lunghezze d'onda comprese tra i 210 e i 290 nanometri (nm); se così non fosse, molte forme di vita cesserebbero di esistere. Assorbendo radiazioni ultraviolette, l'ozono riscalda la stratosfera ed è una delle principali cause dell'aumento della temperatura della stratosfera al crescere dell'altitudine.
Nella stratosfera, la presenza dell'ozono è dovuta all'assorbimento della luce da parte di O2, seguito da dissociazione in atomi - la cosiddetta fotolisi. Nella troposfera, invece, l'ozono è prodotto attraverso una serie complessa di reazioni che coinvolgono gli ossidi di azoto - monossido (NO) e diossido (NO2) - e gli idrocarburi. È ragionevole prendere la stratosfera come punto di partenza di una discussione sulla chimica atmosferica poiché le reazioni chimiche che vi hanno luogo sono meno complicate rispetto a quelle che si svolgono nella troposfera.
La formazione dell'ozono ha luogo nella stratosfera a un'altitudine superiore ai 30 km circa, dove le radiazioni solari ultraviolette con lunghezze d'onda, λ, inferiori ai 242 nm dissociano lentamente l'ossigeno molecolare:
O2 + hν → O + O,
dove hν indica un fotone. L'atomo d'ossigeno reagisce rapidamente con l'O2 in presenza di una terza molecola che indichiamo con M (M di solito è un altro O2 o N2) per formare ozono:
O + O2 + M → O3 + M. (13)
Dal punto di vista pratico, la reazione (13) è l'unica reazione che produce ozono nell'atmosfera. Lo stesso O3 formato nella reazione (13) assorbe fortemente radiazioni in un campo di lunghezze d'onda comprese tra i 240 e i 320 nm per decomporsi di nuovo in O2 e O:
O3 + hν → O2 + O. (14)
Inoltre l'O3 può reagire con l'ossigeno atomico per generare nuovamente due molecole di O2:
O3 + O → O2 + O2. (15)
Questo meccanismo per la produzione dell'ozono nella stratosfera è stato proposto nel 1930 dal meteorologo inglese Sydney Chapman, ed è stato considerato l'insieme principale di reazioni che determinano la concentrazione di O3 nella stratosfera fino all'inizio degli anni settanta, quando il lavoro pionieristico di Paul Crutzen, di Harold Johnston, e di Mario Molina e F. Sherwood Rowland chiarì la intricata chimica della stratosfera. È ora noto che il meccanismo di Chapman darebbe origine a una quantità di O3 cinque volte maggiore di quella realmente presente nella stratosfera (v. Rowland, 1991). Questa differenza è dovuta al fatto che esistono altri processi, oltre alle reazioni (14) e (15), attraverso i quali viene distrutto O3 nella stratosfera. Membri di altre famiglie chimiche possono distruggere l'ozono attraverso una serie di cicli catalitici, costituiti da catene di reazioni le quali rigenerano la molecola che inizialmente reagisce con O3, in modo tale che il ciclo può essere ripetuto in media parecchie volte per una singola molecola reattiva. Sono state identificate tre famiglie chimiche che partecipano a quei cicli catalitici che contribuiscono a riportare la quantità di ozono predetta dalle reazioni di Chapman al valore effettivamente osservato nella stratosfera:

Tre dei più importanti cicli catalitici che riconvertono l'O3 nella stratosfera in O2 sono descritti qui sotto (da qui in poi con NOx verrà indicata la somma di NO e NO2).
Nel ciclo HOx • il radicale ossidrile (• OH) reagisce con l'O3 per formare il radicale idroperossido (HO2 •) che successivamente reagisce con l'ossigeno atomico per rigenerare •OH:

Nel ciclo NOx il monossido di azoto reagisce con O3 per innescare il ciclo:
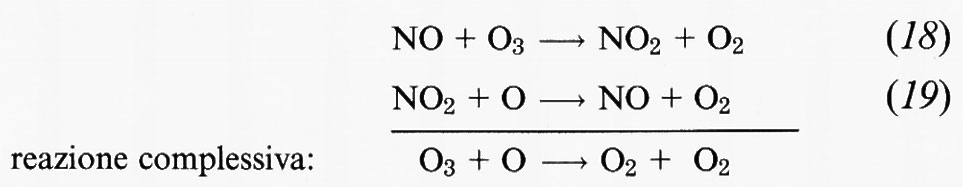
che riveste un ruolo particolarmente importante tra la stratosfera media e quella superiore a quote comprese tra i 30 e i 45 km di altitudine. L'effetto complessivo di ciascuno di questi cicli è quello di convertire 1 molecola di ozono e 1 atomo di ossigeno in 2 molecole di ossigeno. Nell'atmosfera naturale, non alterata, si deve a questi due cicli la maggior parte dell'eliminazione dell'ozono relativo al ciclo di Chapman.
Il radicale ossidrile è una delle specie reattive più importanti della chimica atmosferica e, come si vedrà più avanti, ha un ruolo centrale nella chimica della troposfera. Il radicale • OH risulta dalla fotolisi dell'O3 a lunghezze d'onda inferiori a 319 nm, e produce l'atomo di ossigeno in stato eccitato O(1D):
O3 + hν → O (1D) + O2. (20)
La maggior parte dell'O(1D), che è estremamente reattivo, viene riportato al suo stato base di ossigeno atomico da una collisione con N2 o O2, ma una piccola frazione reagisce chimicamente con altre specie, in particolare con vapor d'acqua e metano:
O (1D) + H2O → 2 •OH (21)
O (1D) + CH4 → •OH + •CH3. (22)
La reazione (21) è la principale fonte di radicali • OH nella troposfera a causa dell'elevata concentrazione di vapor d'acqua che vi si riscontra. Nella stratosfera, vapor d'acqua e metano hanno concentrazioni grosso modo simili, e pertanto entrambe le reazioni (21) e (22) sono importanti fonti di • OH.
Il monossido di azoto emesso dalla superficie terrestre ha un tempo di vita troppo breve nella troposfera per sopravvivere abbastanza a lungo da essere trasportato nella stratosfera. L'ossido di diazoto (N2O) emesso sulla superficie terrestre da sorgenti biologiche presenti nel suolo e nell'acqua è invece abbastanza poco reattivo nella troposfera e, dato il suo lungo tempo di vita atmosferico (120 anni circa), sopravvive e si accumula nella stratosfera. L'N2O viene distrutto nella stratosfera principalmente per fotolisi,
N2O + hν → N2 + O (1D)
e, in misura minore, da una reazione con O(1D) per formare due molecole di NO:
N2O + O (1D) → NO + NO.
Quest'ultima reazione è la principale fonte di NOx nella stratosfera. L'N2O viene emesso in piccole quantità da un gran numero di fonti e di conseguenza le stime sulla sua emissione globale sono piuttosto imprecise. Le valutazioni relative alle fonti di N2O, espresse in Tg/anno (1 Tg = 1012 g) sono le seguenti (v. IPCC, 1992):

b) Clorofluorocarburi e ozono stratosferico
Nel 1974 Molina e Rowland scoprirono che la classe di molecole dette clorofluorocarburi (CFC), prodotti e utilizzati dall'uomo in diverse applicazioni tecnologiche che vanno dagli impianti frigoriferi ai propellenti per i vari tipi di spray, non vengono distrutti nella troposfera ma sopravvivono nell'atmosfera fino a diffondersi negli strati più elevati della stratosfera, dove la potente luce UV li fotolizza (v. Rowland, 1991). Le due molecole di CFC più comuni sono il CFCl3 (detto CFC-11) e il CF2Cl2 (detto CFC-12). Le reazioni di fotolisi rilasciano un atomo di cloro (Cl), ad esempio,
CFCl3 + hν → CFCl2 + Cl.
L'atomo di cloro è estremamente reattivo nei confronti dell'O3 e innesca un rapido ciclo di distruzione dell'O3 che coinvolge un radicale molto reattivo, il monossido di cloro (ClO •):
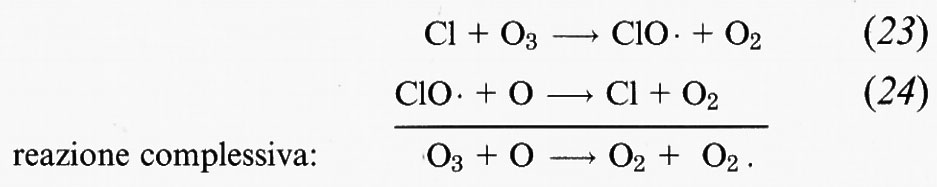
Alle condizioni presenti nella stratosfera, la reazione (23) ha una costante di tempo di circa 1 secondo, mentre quella della reazione (24) è di circa 1 minuto.
Apparentemente le coppie di reazioni (16) e (17), (18) e (19), e (23) e (24) dovrebbero portare alla distruzione definitiva dell'O3. In realtà il ciclo si interrompe quando le specie reattive • OH, NO2, Cl e ClO • vengono coinvolte in altre reazioni. Conoscendo quali sono le reazioni che possono interrompere questo ciclo, è possibile calcolare quante volte in media si ripeterà ogni ciclo prima che uno dei composti di tali reazioni prenda parte a una delle altre possibili reazioni, ponendo fine al ciclo. Per esempio, alle attuali concentrazioni nella stratosfera, il ciclo costituito dalle reazioni (23) e (24) si ripete, in media, 105 volte prima di interrompersi. Ciò significa che in media un atomo di cloro può distruggere 100.000 molecole di ozono attraverso il ciclo delle reazioni (23) e (24). L'eliminazione di una specie reattiva può essere permanente se il prodotto viene effettivamente eliminato dall'atmosfera, e ciò di solito avviene per migrazione nella troposfera e successiva dispersione mediante precipitazioni. Esempi di reazioni che possono portare a un'eliminazione definitiva dall'atmosfera sono:
•OH + NO2 + M → HNO3 + M (25)
Cl + CH4 → HCl + •CH3 (26)
e poiché sia l'acido nitrico (HNO3) che l'acido cloridrico (HCl) sono estremamente solubili in acqua, possono nuovamente migrare verso la troposfera ed esserne eliminati tramite precipitazione.
Una specie reattiva può anche essere eliminata temporaneamente attraverso un ciclo catalitico ed essere conservata in una ‛specie serbatoio' di per sé relativamente poco reattiva, ma senza essere effettivamente eliminata dall'atmosfera. Un esempio tipico di specie serbatoio è il nitrato di cloro (ClONO2), che si forma in questo modo:
ClO• + NO2 + M → ClONO2 + M.
Il nitrato di cloro può andare incontro a fotolisi rigenerando cloro atomico:
ClONO2 + hν → Cl + NO•3;
esso è una specie serbatoio particolarmente importante che mantiene sia l'NO2 sia il ClO •. Insieme HCl e ClONO2 immagazzinano più del 99% del cloro presente nella stratosfera, lasciando libera di partecipare alla distruzione catalitica dell'ozono una quantità inferiore all'1%.
Le reazioni descritte sopra hanno massima efficienza alle medie latitudini terrestri e particolarmente ai tropici, dove la luce ultravioletta è più abbondante, l'ossigeno atomico è più facilmente disponibile e vi è una continua formazione di ozono.
La chimica della stratosfera è in realtà più complessa di quanto abbiamo descritto. Intervengono altri cicli catalitici, ad esempio quelli che coinvolgono i composti del bromo (Br); alcuni hanno importanza alle alte latitudini, altri a latitudini più basse, a seconda della distribuzione dei reagenti e delle lunghezze d'onda disponibili per la fotolisi. Tutti i cicli sono accoppiati l'uno con l'altro, e un'analisi dettagliata della chimica stratosferica deve tener conto delle loro interrelazioni. Ad esempio, un aumento delle emissioni di N2O porterebbe a un aumento delle concentrazioni stratosferiche di NO e quindi a un maggior consumo di ozono attraverso il ciclo catalitico NOx. Analogamente, un aumento dei livelli di CFC porterebbe a un maggior consumo di ozono tramite il ciclo ClOx. Tuttavia, presi insieme, l'aumento di NOx prodotto da N2O porterebbe a un innalzamento del livello del serbatoio ClONO2 e a una riduzione del ciclo cloro. Pertanto, l'effetto complessivo sull'ozono di aumenti simultanei di N2O e CFC è inferiore alla somma dei loro effetti separati. Anche aumenti del CH4 o dell'H2O nella stratosfera ridurrebbero l'efficienza dei cicli di distruzione dell'ozono catalizzati da NOx e ClOx. Bisogna osservare a questo proposito che una quantità molto limitata del vapor d'acqua della troposfera penetra nella stratosfera, che è pertanto relativamente asciutta - 3 ÷ 4 ppm di H2O - se confrontata con la troposfera. Il rapporto di miscelazione dell'H2O cresce con l'altitudine, fino ad arrivare a 6 ppm nella stratosfera superiore a causa dell'ossidazione del CH4 nella stratosfera. Come vedremo nel prossimo paragrafo, ogni molecola di CH4 ossidata dal radicale • OH produce due molecole d'acqua.
c) Buco dell'ozono
Fino a metà degli anni ottanta, si pensava che la chimica della stratosfera precedentemente illustrata fosse relativamente completa, particolarmente per ciò che riguardava la distruzione catalitica dell'ozono iniziata dai CFC. Tuttavia, nel 1985 un gruppo di scienziati guidato dall'inglese J. Farman sconvolse la comunità degli studiosi di chimica atmosferica con dati relativi a massicce diminuzioni annuali dell'ozono stratosferico sopra l'Antartide durante la primavera polare (settembre-ottobre), un'osservazione che le nozioni relative alla chimica del cloro nella stratosfera allora comunemente accettate non riuscivano a spiegare. Questo fenomeno è stato chiamato dai mass media ‛buco dell'ozono'.
Sebbene sull'Antartide si riscontrino concentrazioni di ozono tra le più elevate della Terra per gran parte dell'anno, l'ozono viene in realtà prodotto in prevalenza ai tropici e trasferito in Antartide, insieme alle riserve molecolari di cloro, da movimenti d'aria su grande scala. La situazione dell'ozono artico è analoga. La carenza di ossigeno atomico che si riscontra nella stratosfera antartica è dovuta all'assenza di una intensa radiazione UV. Il vortice polare antartico, un modello di circolazione tipico del polo sud, mantiene intrappolato ad alti livelli di concentrazione l'ozono sopra l'Antartide per parecchi mesi all'anno. Normalmente la quantità di ozono nel vortice polare comincia a diminuire allorché, a fine agosto o inizio settembre, l'Antartide emerge dalla notte australe - che dura molti mesi -, si mantiene costante in ottobre e cresce in novembre. La comparsa del buco dell'ozono antartico ha determinato un mutamento significativo degli andamenti finora riscontrati: i livelli primaverili sono diminuiti a valori mai riscontrati in precedenza e continuano a diminuire di anno in anno.
La stratosfera è molto asciutta e generalmente priva di nubi. La lunga notte polare causa temperature di circa - 90 °C ad altezze comprese tra 15 e 20 km, abbastanza basse da condensare anche le piccole quantità di vapor d'acqua presenti nella stratosfera che danno origine alle nubi stratosferiche polari. Nell'Antartide, dove il vortice polare è più stabile che non al polo artico, si registrano così le temperature più basse. L'eccezionale stabilità del vortice al polo sud è probabilmente un effetto della distribuzione praticamente simmetrica dell'oceano attorno all'Antartide. Il vortice polare artico, meno stabile, tende a miscelarsi maggiormente con l'aria che lo circonda e pertanto non raggiunge l'isolamento e le basse temperature tipici del polo sud.
Le nubi stratosferiche polari sono formate principalmente da ghiaccio o da cristalli di acido nitrico triidrato (HNO3 • 3H2O). Esse favoriscono la conversione dei più importanti serbatoi di cloro - acido cloridrico (HCl) e nitrato di cloro (ClONO2) - a una forma fotoliticamente attiva. Ricerche di laboratorio hanno dimostrato che il primo passo del processo, cioè l'assorbimento di HCl gassoso da parte delle particelle di ghiaccio, ha luogo in maniera molto efficiente. Questo passo è seguito dalla reazione eterogenea della particella con il ClONO2 gassoso:
HCl (ghiaccio) + ClONO2 → Cl2 + HNO3 (ghiaccio).
Il Cl2 prodotto dalla reazione fotolizza rapidamente generando atomi liberi di cloro, mentre l'altro prodotto, HNO3, rimane nel ghiaccio, dando luogo a un'eliminazione globale degli ossidi di azoto dalla fase gassosa. Ciò favorisce ulteriormente la distruzione dell'ozono da parte dei radicali liberi a base di cloro, che altrimenti verrebbero incorporati efficacemente da NO e NO2.
Gli atomi di cloro reagiscono con O3 per produrre ClO •. A causa dell'assenza di atomi d'ossigeno nella stratosfera polare, il ClO • reagisce con se stesso per produrre il dimero del monossido di cloro Cl2O2:
ClO• + ClO• + M → Cl2O2 + M
che può fotolizzare per rigenerare l'atomo di cloro e ClO2 il quale a sua volta può essere decomposto termicamente per rilasciare l'altro atomo di cloro:
Cl2O2 + hν → Cl + ClO2
ClO2 + M → Cl + O2 + M.
Finché gli ossidi d'azoto restano legati in HNO3 solido e la quantità di NO2 gassoso è bassa, l'effetto mitigante prodotto usualmente quando si forma nitrato di cloro è assente. In mancanza di NOx che possano reagire con ClO •, le reazioni a catena si svolgono senza impedimenti per 5 o 6 settimane, provocando una diminuzione del 95% o più dell'O3 all'altezza delle nubi. Quando l'aria polare si riscalda per mescolamento con masse d'aria più calde e per l'aumento dell'intensità del calore solare, le nuvole stratosferiche polari evaporano, rilasciando HNO3, il quale fotolizza e reagisce con • OH rigenerando NOx gassosi.
Molti aspetti della chimica eterogenea del buco dell'ozono promossa dal ghiaccio rappresentano un'attiva area di ricerca della chimica dell'atmosfera. È probabile che ulteriori dettagli sulle reali velocità e sui meccanismi delle reazioni che causano il buco dell'ozono possano emergere via via che progrediranno gli studi di base.
5. Chimica della troposfera in condizioni naturali
La chimica della troposfera è qualitativamente molto diversa da quella della stratosfera. Questa differenza è dovuta in primo luogo alla gran quantità di composti chimici di origine antropica e naturale emessi a livello della superficie terrestre, e poi anche alla minor energia della radiazione UV che raggiunge la troposfera, e all'alta concentrazione del vapor d'acqua che determina la presenza di nubi e precipitazioni. La luce con lunghezze d'onda minori di 290 nm circa è assorbita dalle molecole presenti nella stratosfera. Affinché possa aver luogo la fotolisi di un composto della troposfera, esso deve essere in grado di assorbire radiazioni e di subire una trasformazione chimica per lunghezze d'onda al di sopra dei 290 nm.
Il radicale • OH, che è il principale agente ossidante di praticamente tutti i composti della troposfera e in particolare dei composti organici, è il componente più importante nella chimica della troposfera. Ogni composto anche moderatamente reattivo con il radicale • OH verrà eliminato nella troposfera e non avrà un tempo di vita sufficientemente lungo per essere trasportato nella stratosfera. I radicali ossidrile sono generati nella troposfera dalle reazioni (20) e (21): ad esempio, a temperatura ambiente e con umidità relativa del 50%, si formano 0,2 radicali • OH per ogni atomo O(1D) generato dalla fotolisi dell'ozono. La chimica della troposfera in condizioni naturali, non inquinata, è governata dal metano - l'idrocarburo più abbondante dell'atmosfera, emesso da fonti sia antropiche che naturali - e dagli ossidi d'azoto, NO e NO2. L'ozono viene prodotto nella stratosfera come risultato della fotolisi di O2; una parte dell'ozono viene trasportato in basso dalla stratosfera attraverso la tropopausa, ma una porzione almeno equivalente dell'ozono troposferico viene generato in situ per via chimica dall'ossidazione del metano.
Mentre i livelli d'ozono nella stratosfera sono in diminuzione a causa delle reazioni chimiche che distruggono l'ozono provocate dai CFC, i livelli di O3 nella troposfera sono in aumento. Gli unici dati attendibili relativi all'ozono risalenti al XX secolo sono quelli dell'osservatorio di Montsouris, vicino a Parigi, che si riferiscono al periodo compreso tra il 1895 e il 1910. Questi dati indicano che a livello della superficie terrestre, alle medie latitudini, l'ozono troposferico è aumentato di circa il 100% rispetto all'inizio del secolo (v. Volz e Kley, 1988; v. Staehelin e Schmid, 1991).
a) Ossidi dell'azoto
La presenza di ossidi di azoto (NOx) è fondamentale per la chimica della troposfera (come abbiamo già detto, con NOx si indica la somma di NO e NO2). Le emissioni globali di NOx all'anno nella troposfera sono state calcolate come segue:

È opportuno iniziare la descrizione della chimica della troposfera partendo dalla chimica degli NOx.
Nella troposfera NO reagisce con O3 per formare NO2 attraverso la reazione (18),
NO + O3 → NO2 + O2.
NO2, a sua volta, subisce una rapida fotolisi durante le ore diurne, con una durata di fotolisi di 2 minuti circa in condizioni di sole a picco,
NO2 + hν → NO + O. (27)
Questa reazione è seguita, in pratica istantaneamente, dalla reazione (13) che, come notato, è l'unica reazione nell'atmosfera che formi ozono (il tempo di vita di un atomo di ossigeno a 298 K e pressione atmosferica consumato dalla reazione (13) è di circa 10-5 s). In conseguenza della rapidità delle reazioni (18), (27) e (13), si produce O3 in una concentrazione, allo stato stazionario, espressa da:

dove le parentesi quadre indicano le concentrazioni, J è la velocità di fotolisi di NO2, e k è la costante di velocità per la reazione (18). Questa equazione è talvolta chiamata ‛relazione di stato fotostazionario' poiché esprime la concentrazione in stato stazionario dell'ozono generato dal ciclo NO−NO2−O3, che è alimentato dalla luce. Si noti che la concentrazione di O3 raggiunta è direttamente correlata al rapporto tra le concentrazioni di NO2 e NO.
Vi è una serie di altre reazioni che coinvolgono gli ossidi di azoto. Fra queste, il principale processo di eliminazione di NOx durante le ore diurne è la reazione di NO2 con il radicale • OH per formare acido nitrico, attraverso la reazione (25). Di notte, quando i livelli di • OH si avvicinano allo zero, le reazioni di NO2 con O3 e il radicale nitrato (NO3 •) sono i principali processi di eliminazione. Il tempo di vita degli NOx nella troposfera è breve e varia da meno di 1 giorno in prossimità della superficie terrestre a 1 settimana circa nella tropopausa. A causa della complessa distribuzione geografica delle loro fonti e del loro breve tempo di vita, la distribuzione spaziale e temporale degli NOx è altamente variabile. I rapporti di miscelazione degli NOx possono variare da 10 parti per trilione (ppt) sopra le zone più remote degli oceani a 100 parti per miliardo in aree urbane ricche di fonti, presentando quindi una variazione di 1:10.000.
b) Chimica del metano
Il metano (CH4) è l'idrocarburo più abbondante dell'atmosfera. Si stima che le emissioni di CH4 da parte di fonti sia naturali che antropogeniche ammontino a circa 510 Tg/anno. Le principali fonti sono rappresentate dalle risaie, dai ruminanti domestici e dalle paludi. Il metano viene eliminato dall'atmosfera prevalentemente attraverso la reazione con il radicale • OH, che va a produrre il radicale metile (• CH3) e una molecola d'acqua:
CH4 + •OH → •CH3 + H2O. (28)
Negli anni ottanta, le concentrazioni di CH4 sono aumentate di circa l'1% annuo (v. Blake e Rowland, 1988; v. Khalil e Rasmussen, 1990). È stato possibile risalire alle concentrazioni di metano atmosferico negli ultimi 150.000 anni, grazie all'analisi della composizione dell'aria intrappolata nelle calotte polari (v. Stauffer e altri, 1985; v. Khalil e Rasmussen, 1987), dimostrando che durante questo periodo le concentrazioni di CH4 non hanno mai superato la metà dei livelli attuali. Il recente aumento di CH4 nell'atmosfera è verosimilmente il risultato dell'aumento delle emissioni, e solo parte dell'aumento potrebbe dipendere dalla diminuzione dei livelli di • OH. Una volta rilasciato nell'atmosfera, il CH4 ha un tempo di vita, dovuto alla reazione (28), che va dagli 8 ai 10 anni. Poiché la maggior parte delle fonti di CH4 si trova sulla terraferma e molte di queste sono legate ad attività umane, le concentrazioni di CH4 sono più elevate al di sopra dei continenti e sull'emisfero nord in generale. Il rapporto di miscelazione attuale è di circa 1,7 ppm.
Nelle attuali condizioni della troposfera, il radicale • CH3 formato nella reazione (28) reagisce esclusivamente con O2 producendo il radicale metilperossido (CH3O2 • ),
•CH3 + O2 + M → CH3O2• + M.
Il radicale CH3O2 • può reagire con i radicali NO, NO2, idroperossido (HO2 •) e altri radicali perossidi (indicati con RO2•); le reazioni con NO e HO2 • sono le più importanti. La reazione con NO porta alla formazione di NO2 e di radicale metossido (CH3O •):
CH3O2• + NO → CH3O• + NO2 (29)
mentre quella con il radicale HO2 • porta alla formazione di metilidroperossido (CH3OOH):
CH3O2• + HO2• → CH3OOH + O2. (30)
CH3OOH può a sua volta essere sottoposto a fotolisi o reagire con radicali • OH per rigenerare radicali CH3O • o CH3O2 •
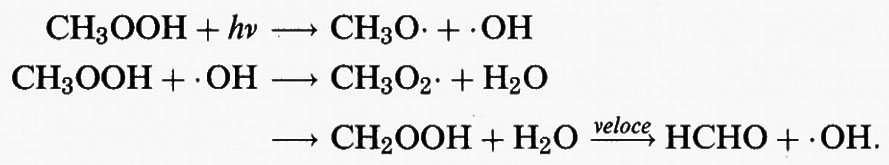
Il tempo di vita atmosferico del CH3OOH risultante dalla fotolisi e dalla reazione con il radicale • OH è di 2 giorni circa, e pertanto rappresenta un serbatoio temporaneo di radicali liberi. Può essere eliminato dall'atmosfera mediante assorbimento in una nube d'acqua.
L'unica reazione atmosferica importante per il radicale metossido (CH3O •) nelle condizioni della troposfera è quella con O2 per produrre formaldeide (HCHO) e il radicale HO2 •:
CH3O• + O2 → HCHO + HO2•.
La formaldeide, un importante gas presente in tracce nella troposfera, può essere soggetta a sua volta a fotolisi:
HCHO + hν → H2 + CO (55%) (31a)
→ H + HCO• (45%) (31b)
dove le percentuali riportate si riferiscono a condizioni di sole a picco. La formaldeide può anche reagire con • OH:
HCHO + •OH → HCO• + H2O. (32)
Nella troposfera l'atomo H e il radicale formile (HCO •) reagiscono esclusivamente con O2 per formare il radicale HO2 •:
H + O2 + M → HO2• + M (33)
HCO• + O2 → HO2• + CO•. (34)
I tempi di vita di HCHO che risultano dalla fotolisi e dalla reazione con il radicale • OH sono rispettivamente di 4 ore e 1,5 giorni circa, che portano a un tempo di vita complessivo di circa 3 ore in condizioni di sole a picco.
Le reazioni (31) e (34) rappresentano fonti importanti di monossido di carbonio (CO) nell'atmosfera (l'ossidazione del CH4 produce circa i 2/3 del CO presente nell'atmosfera). Il passo conclusivo nella catena di ossidazione del metano è l'ossidazione del CO per reazione con il radicale • OH (l'unica reazione del CO nella troposfera):
CO + •OH → CO2 + H. (35)
Il tempo di vita del CO prodotto da questa reazione è di 3 mesi circa. Le concentrazioni di CO nella troposfera sono più elevate nell'emisfero nord (attorno alle 120 ppb) che nell'emisfero sud (80 ppb) e più elevate in inverno che in estate. Queste osservazioni sono coerenti con il fatto che nell'emisfero settentrionale esistono importanti fonti industriali che emettono CO e con il fatto che la reazione di eliminazione dominante è la (35). Le valutazioni relative alle fonti globali di CO sono le seguenti (v. IPCC, 1992):

Poiché buona parte del metano che produce CO proviene da fonti di origine umana, si stima che 2/3 circa delle emissioni di CO siano riconducibili ad attività antropogeniche.
Il radicale HO2 • può reagire con NO, O3, oppure con se stesso, e ciò dipende soprattutto dalla concentrazione di NO. La reazione con NO porta alla rigenerazione del radicale • OH:
HO2• + NO → NO2 + •OH (36)
mentre le reazioni con HO2 • e O3 sono:
HO2• + HO2• → H2O2 + O2 (37a)
HO2• + O3 → •OH + 2O2. (37b)
Le reazioni (37a) e (37b) portano a una perdita netta di O3 troposferico. La produzione del radicale • OH da O3 mediante la reazione (20), attraverso l'intermediazione dell'atomo O(1D), rappresenta un processo di perdita di O3 troposferico. Pertanto, l'assenza di una fase in cui si forma O3 nel ciclo di ossidazione del CH4 equivale a una perdita netta di O3. Quando il radicale HO2 • reagisce con NO attraverso la reazione (36), l'NO2 prodotto partecipa immediatamente al ciclo fotolitico NOx-O3 delle reazioni (27), (13) e (18) per generare O3. Tuttavia, se l'HO2 • partecipa a una delle reazioni (37a) o (37b) non viene prodotto O3. Si stima che in genere prevalga la reazione (36) se il rapporto di miscelazione dell'NO supera le 10 parti per trilione (ppt).
Se le concentrazioni di NO sono elevate, cioè se il rapporto di miscelazione dell'NO è superiore a circa 1 ppb, il risultato netto della catena di ossidazione del CH4 è la formazione di due molecole di O3, una molecola di HCHO e la rigenerazione del radicale • OH:
CH4 + •OH + 4O2 → H2O + 2O3 + HCHO + •OH.
Analogamente, se le concentrazioni di NO sono elevate, l'effetto della fotolisi della formaldeide e della successiva reazione con • OH è:
Formula
L'ossidazione dell'HCHO è quindi una sorgente di radicali • OH. Nelle regioni della troposfera che si trovano al di sopra di remote zone marine, dove le concentrazioni di NO sono dell'ordine delle 20 ppt o meno, l'ossidazione del metano porta a una distruzione netta di O3 a causa del prevalere delle reazioni (37a) e (37b) rispetto alla (38). Nella troposfera superiore e al di sopra di aree continentali dove predominano fonti di NO da combustione, le concentrazioni di NO sono sufficientemente elevate da far sì che l'ossidazione del CH4 porti alla formazione netta di O3.
Sulla base di una media approssimata globale annua, è stato stimato che per ogni molecola di CH4 distrutta vengano consumate 0,22 molecole di • OH e vengano prodotte 0,82 molecole di CO e 1,15 molecole di O3 (v. Tie e altri, 1992).
c) Chimica dello zolfo
I composti dello zolfo hanno un ruolo importante nella chimica della troposfera. L'ossidazione dei composti gassosi dello zolfo con formazione di particelle di solfati è una fonte importantissima di nuclei di condensazione delle nuvole e può influenzare direttamente il bilancio della radiazione terrestre attraverso la dispersione della radiazione solare a basse lunghezze d'onda. Nelle regioni industrializzate del mondo l'acido solforico che deriva da fonti antropogeniche di diossido di zolfo (SO2) è una delle cause principali dell'acidità delle piogge.
I composti dello zolfo emessi nell'atmosfera comprendono il solfuro di carbonile (COS), il solfuro di metile (CH3SCH3), il solfuro di carbonio (CS2), l'idrogeno solforato (H2S) e SO2. Il COS è il composto dello zolfo più abbondante nell'atmosfera. Esso viene emesso da fonti situate sia sulla terraferma che negli oceani e dall'ossidazione del CS2, e si stima che la sua quantità atmosferica totale sia di circa 2,5 Tg riferita al solo zolfo. Il COS reagisce con • OH nella troposfera, ma a una velocità piuttosto bassa, cosicché la sua più importante causa di distruzione, a parte la deposizione sulla superficie terrestre, è la fotolisi nella stratosfera, con formazione di SO2; esso rappresenta quindi una delle fonti più importanti dello strato di solfati presente nella stratosfera. Il COS ha un tempo di vita atmosferico di un anno circa.
Il dimetilsolfuro (DMS) è emesso nell'atmosfera dal fitoplancton oceanico (v. Andreae, 1990). Esso viene ossidato nell'atmosfera marina da • OH e NO3 • con produzione di SO2 e acido metansolfonico (CH3SO3H). La successiva ossidazione dell'SO2 a particelle di acido solforico è stata proposta come uno dei meccanismi principali della formazione dei nuclei di condensazione delle nuvole al di sopra degli oceani (v. Charlson e altri, 1987). La concentrazione media del DMS nell'atmosfera marina è di 100 ppt circa. Con una tale concentrazione sopra tutti gli oceani del mondo nel primo chilometro inferiore dell'atmosfera e una concentrazione di 10 ppt a quote superiori, si calcola che l'atmosfera contenga circa 0,8 Tg di zolfo sotto forma di DMS.
Gli spruzzi del mare sono ricchi di solfati. Tuttavia, parecchie misure effettuate sugli aerosol nello strato a immediato contatto con la superficie marina indicano la presenza di una quantità di solfati doppia rispetto a ciò che si potrebbe presumere sulla base dell'Na e del Cl presenti nelle particelle. Tale quantità ‛in più' o ‛solfato non-marino' è dovuta all'ossidazione del DMS e di altri gas solforati e al trasporto a grandi distanze di solfati antropogenici dai continenti.
6. Chimica della troposfera inquinata
a) Composti organici diversi dal metano
Sebbene l'idrocarburo prevalente nell'atmosfera sia il metano, composti organici diversi dal metano vengono emessi da fonti antropogeniche nelle regioni urbane e continentali, e inoltre grandi quantità di composti organici di origine biologica vengono emessi dalla vegetazione. In totale si calcola che vengano emesse le seguenti quantità di composti organici diversi dal metano:

Le attività umane portano anche a emissioni di NOx, le cui concentrazioni sono di parecchi ordini di grandezza superiori sulle aree urbane che non in aree remote della troposfera. I composti organici diversi dal metano reagiscono in presenza di NOx e luce solare in maniera simile a quella del metano; sostanzialmente la chimica della troposfera inquinata è simile a quella della troposfera originaria, il cui componente predominante era il metano, ma anche significativamente più complessa a causa della grande varietà di composti organici (v. Atkinson, 1990).

Le reazioni dei composti organici con gli NOx portano alla formazione di ozono, proprio come il ciclo globale di ossidazione del metano. Il CH4 reagisce soltanto con il radicale • OH; anche nel caso degli altri composti organici la reazione prevalente è quella con il radicale • OH, sebbene alcuni possano reagire con il radicale nitrato (NO3 •) e con O3. Le classi principali di tali composti organici sono gli alcani, gli alcheni, gli aromatici e i composti carbonilici. La tab. II elenca le concentrazioni medie dei 25 composti organici più abbondanti misurati nell'estate del 1987 nell'ambito del Southern California Air Quality Study.
Nell'aria delle città è presente un gran numero di alcani (v. per es. tab. II). L'unico processo di rimozione degli alcani che abbia rilevanza è quello attraverso una reazione con il radicale • OH. Come nel caso del metano, che è l'alcano a molecola più breve, la reazione radicalica con • OH procede attraverso l'estrazione di un atomo di H dai vari legami C-H. Indicando un alcano con R-H, dove R rappresenta un gruppo alchilico, la reazione con • OH è:
RH + • OH → R + H2O.
Il radicale alchilico, così come il radicale metilico nel caso del metano, addiziona rapidamente O2 per formare un radicale alchilperossidico (RO2 •):
R + O2 + M → RO2 • + M.
Il primo elemento della serie degli RO2 • è il radicale metilperossidico (CH3O2 •). Così come avviene con il radicale CH3O2 •, i radicali alchilperossidici possono reagire con i radicali NO, NO2, HO2 • e altri radicali RO2 •, sebbene quest'ultima classe di reazioni sia meno importante delle altre alle condizioni in cui si trova la troposfera. Nelle aree urbane e in altre regioni della troposfera dove le emissioni degli NOx sono molto elevate, la reazione più importante a cui partecipa il radicale RO2 • è con NO (si ricordi la reazione 29):
RO2 • + NO → NO2 + RO •.
Come nel caso del radicale metossilico (CH3O •), i radicali alcossidici (RO •) che hanno un atomo di H disponibile possono reagire con O2 per formare HO2 • e un composto carbonilico come avviene, ad esempio, per il radicale alcossidico che si forma da n-butano:
CH3CH(O •)CH2CH3 + O2 → CH3C(O)CH2CH3 + HO2 •
Gli alcheni sono molecole organiche che contengono legami C=C. Tra gli alcheni più abbondanti nell'atmosfera vi sono l'etilene (H2C=CH2), il propene (CH3CH=CH2) e i buteni. I principali idrocarburi di origine vegetale sono gli alcheni: isoprene (2-metil-1,3-butadiene) e i monoterpeni C10H16. Oltre che col radicale • OH, gli alcheni reagiscono nell'atmosfera con O3 e il radicale NO3 •. Tutte e tre le reazioni hanno inizio con una addizione sui doppi legami C=C. La reazione tra propene e • OH, ad esempio, è:
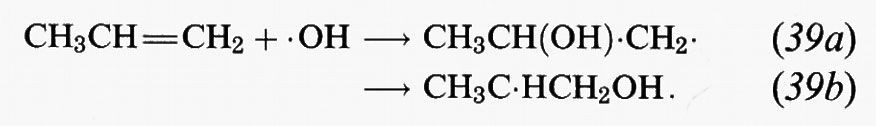
Le reazioni successive di questi radicali sono simili a quelle dei radicali alchilici che si formano dalla reazione di • OH con gli alcani. Pertanto i radicali che si formano nelle reazioni (39a) e (39b) addizionano O2; successivamente il radicale perossidico risultante, in condizioni di elevate concentrazioni di NOx, reagisce con NO per produrre NO2 e un radicale idrossialcossidico. Questi radicali contenenti OH appaiono essere soggetti soprattutto a decomposizione. Se, per esempio, consideriamo il radicale CH3C • HCH2OH che deriva dal propene, il percorso di reazione è:
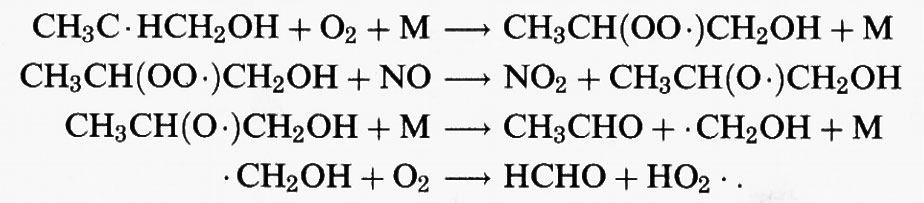
Pertanto, i prodotti stabili che derivano dall'attacco dell'• OH sul propene sono l'acetaldeide (CH3CHO) e la formaldeide (HCHO).
Il benzene, il toluene e gli xileni sono gli idrocarburi aromatici più abbondanti nell'atmosfera. Come nel caso degli alcani, l'unico processo importante in base al quale gli idrocarburi aromatici vengono eliminati dall'atmosfera è la reazione con il radicale • OH. Per i benzeni alchil-sostituiti, le reazioni con • OH seguono due modalità: l'estrazione di un atomo di H dai legami C−H dei gruppi (o del gruppo) alchilici sostituenti e l'addizione del radicale • OH all'anello aromatico. Per il toluene, ad esempio,
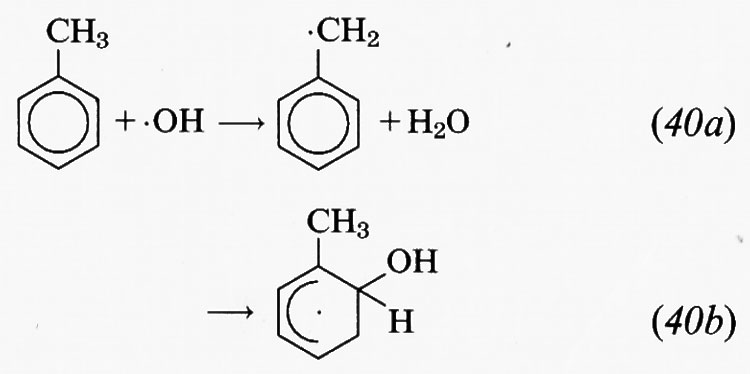
Nel 90% circa dei casi, la reazione procede secondo la modalità (40b) alle condizioni in cui si trova la troposfera. Successivamente, la reazione più probabile del radicale contenente un OH che si forma nella reazione (40b) è con l'O2, e molti dati sembrano indicare che alla fine della catena di reazioni l'anello aromatico si rompe per dar vita, insieme ad altri prodotti, a composti α-dicarbonilici, cioè gliossale (CH(O)CHO) e metilgliossale (CH3C(O)CHO).
L'ossidazione atmosferica di alcani, alcheni e aromatici porta, spesso con rese significative, alla produzione di composti carbonilici. L'ossidazione del metano dà formaldeide come prodotto principale. Nell'atmosfera i composti carbonilici predominanti sono: formaldeide, acetaldeide e aldeidi alifatiche superiori, acetone (CH3C(O)CH3) e alcuni chetoni superiori; infine i composti dicarbonilici semplici gliossale e metilgliossale. I composti carbonilici che non contengono legami C=C vengono eliminati dall'atmosfera principalmente attraverso la reazione con il radicale • OH e la fotolisi (si ricordino le reazioni 31 e 32); quest'ultimo processo è il principale responsabile dell'eliminazione dell'aldeide più semplice, HCHO, e del chetone più semplice, CH3C(O)CH3, come pure dei composti dicarbonilici. Per le aldeidi superiori o i chetoni la reazione con il radicale • OH è il processo di eliminazione dominante in fase gassosa.
Dopo la formaldeide, l'acetaldeide è il più importante composto carbonilico nell'atmosfera. La sua reazione con l'• OH procede per estrazione di un atomo di H dal gruppo -CHO per formare il radicale acetilico (CH3C • O):
CH3CHO + • OH → CH3C • O + H2O
che addiziona rapidamente O2 per formare il radicale acetilperossidico:
CH3C • O + O2 + M → CH3C(O)OO • + M.
Alle condizioni della troposfera, il radicale acetilperossidico reagisce con NO e NO2:
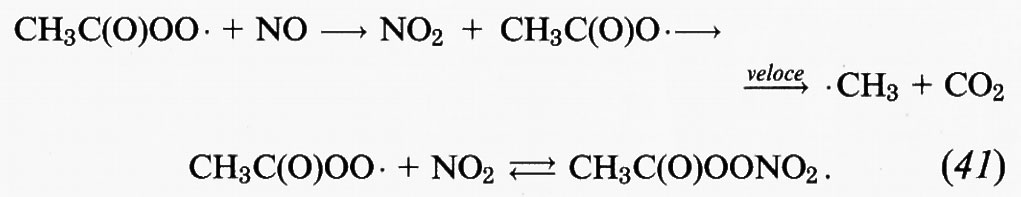
Il prodotto della reazione (41) è il perossiacetilnitrato, comunemente indicato per semplicità con PAN; il suo tempo di vita rispetto alla decomposizione termica attraverso la reazione (41) è di circa 30 minuti a 298 K nella troposfera inferiore. Nella troposfera superiore, dove le temperature sono molto più basse, le decomposizione termica diventa estremamente lenta e sono la reazione con • OH oppure la fotolisi che determinano alla fine la decomposizione del PAN. A causa della natura reversibile della sua formazione, il PAN funge da serbatoio temporaneo di NOx. Benché il tempo di vita abituale degli NOx nella troposfera sia di un giorno o due soltanto, nel caso in cui vengano sequestrati in PAN a temperature sufficientemente basse, gli NOx possono essere trasportati a grandi distanze in aree meno inquinate.
La catena di ossidazione globale del metano è la principale fonte di ozono nella troposfera remota. Nelle aree urbane e continentali, dove le quantità di idrocarburi e ossidi di azoto superano nettamente i livelli di fondo, l'ozono viene prodotto anche dall'ossidazione di composti organici diversi dal metano in presenza di NOx e luce solare. La catena di ossidazione dei composti organici diversi dal metano, sebbene notevolmente più complessa di quella del metano, ha essenzialmente le stesse caratteristiche. Il ciclo fotochimico NO−NO2−O3, cioè le reazioni (27), (13) e (18), che producono un livello stazionario di O3, è spostato sensibilmente in direzione dell'NO2 invece che dell'NO a causa di reazioni con radicali perossidici che convertono NO in NO2, per esempio:
HO2 • + NO → NO2 + • OH
RO2 • + NO → NO2 + RO •.
In accordo con il livello stazionario di O3 generato dalle reazioni (27), (13) e (18), la concentrazione di O3 cresce al crescere del rapporto NO2/NO. La conversione diminuisce via via che l'NO2 viene rimosso attraverso la reazione con • OH con formazione di acido nitrico,
NO2 + • OH + M → HNO3 + M
e via via che gli NOx vengono eliminati nel processo che dà origine al PAN attraverso la reazione (41). A temperatura ambiente il PAN si decompone gradualmente per via termica rilasciando NO2, cosicché il destino finale degli NOx è quello di formare acido nitrico e nitrati organici (questi ultimi meccanismi non vengono qui descritti).
Un'analisi della chimica dei composti organici diversi dal metano e degli NOx rivela l'esistenza nella chimica troposferica di due tipi di comportamento qualitativamente diversi a seconda dei livelli di NOx (v. Zimmerman e Poppe, 1993; v. Stewart, 1993). In un ambiente ad alte concentrazioni di NOx, come quello urbano, un aumento delle emissioni degli NOx inibisce la produzione di O3, che è invece favorita se tale circostanza si verifica in un ambiente a basse concentrazioni di NOx, come l'atmosfera di una remota zona marina. Analogamente, i livelli dei radicali ossidrilici, così come quelli di O3, sono favoriti da immissioni di NOx se le concentrazioni di NOx sono basse, e inibiti se sono invece elevate. I due regimi chimici a basse ed elevate concentrazioni di NOx non rappresentano i limiti di un sistema che varia in maniera continua al variare dei livelli di NOx, ma in realtà riflettono un cambiamento strutturale nel meccanismo di reazione (v. Kleinman, 1991). I radicali vengono prodotti soprattutto da reazioni di fotolisi, come le reazioni (20) e (21). Tutti i radicali formati nell'atmosfera vengono alla fine eliminati, solitamente in pochi minuti, determinando una concentrazione stazionaria. Come descritto sopra, esistono tre percorsi fondamentali per l'eliminazione dei radicali: 1) reazioni che coinvolgono NOx, come le reazioni (25) e (41); 2) processi di distruzione bimolecolare come:
• OH + HO2 • → H2O + O2;
3) reazioni di combinazione tra radicali perossidici, come le reazioni (30) e (37a).
Nell'eliminazione di radicali attraverso la reazione con gli NOx, ogni molecola di NO o NO2 può reagire con un solo radicale al massimo. La velocità massima di eliminazione dei radicali mediante reazione con gli NOx è quindi data dalla velocità di emissione degli NOx nell'atmosfera.
Kleinman (v., 1991) ha notato come in base alle grandezze relative della velocità di formazione dei radicali liberi (F) e della velocità di emissione degli NOx (ENOx) si possano definire due stati atmosferici: nello stato a bassa concentrazione di NOx, dove F>;ENOx, reazioni quale quella di formazione dell'acido nitrico (reazione 25) eliminano rapidamente gli NOx. Poiché l'eliminazione degli NOx per via radicalica è limitata dalla quantità di NOx emessa, l'eccesso di radicali liberi viene eliminato per via biomolecolare e attraverso reazioni di combinazione tra radicali perossidici. Nello stato a elevata concentrazione di NOx, dove F〈;ENOx, il numero dei radicali liberi prodotti è insufficiente a eliminare tutti gli NOx. La velocità di produzione dei radicali dipende dalle concentrazioni di O3, HCHO e altri precursori che, a loro volta, dipendono dalla velocità di emissione dei composti organici diversi dal metano e da altre condizioni.
b) Chimica dello zolfo
Il più importante composto dello zolfo emesso da fonti antropogeniche è l'SO2. Nella troposfera, SO2 reagisce in fase gassosa con il radicale • OH, viene assorbito entro le goccioline delle nuvole dove viene ossidato in fase acquosa, e viene eliminato attraverso deposizione sulla superficie terrestre. La costante di velocità della reazione tra SO2 e • OH è tale che il tempo di vita dell'SO2 rispetto a questa reazione è di 15 giorni circa. Il meccanismo della reazione tra SO2 e • OH è il seguente:
Formula
La dissoluzione dell'SO2 in nubi, pioggia o nebbia procede in questo modo:
SO2(g) ⇄ SO2 • H2O
SO2 • H2O ⇄ H+ + HSO−3
HSO−3 ⇄ H+ + SO32-.
Gli ioni idrogenosolfito (HSO−3) e solfito (SO32-) possono partecipare a reazioni che li convertono in ioni solfato SO42-. Due tra le più importanti di tali reazioni coinvolgono acqua ossigenata (H2O2) e ozono:
HSO−3 + H2O2 → H + SO42- + H2O
SO32-+ O3 → SO42- + O2.
Si ritiene che la reazione tra SO2 disciolto e H2O2 disciolto sia il processo di ossidazione dell'SO2 atmosferico dominante in fase acquosa. Quando sono presenti nuvole o nebbia, l'ossidazione in fase acquosa dell'SO2 può procedere molto più rapidamente dell'ossidazione in fase gassosa da parte dei radicali • OH.
I processi chimici che hanno luogo all'interno delle goccioline di nubi o nebbia, benché si ritenessero dominati da quelli dei composti dello zolfo in esse disciolti, sono in pratica tanto complicati quanto la chimica in fase gassosa della troposfera (v. Pandis e Seinfeld, 1989). L'assorbimento dei gas in acqua:
A(g) + H2O ⇄ A • H2O
viene descritto da una relazione di equilibrio, detta legge di Henry,
[A • H2O] = HApA
dove HA è il coefficiente di Henry, espresso in mol/m3 Pa e pA è la pressione parziale della specie A in fase gassosa, espressa in Pascal. I coefficienti di Henry per alcuni gas atmosferici in condizioni standard sono:
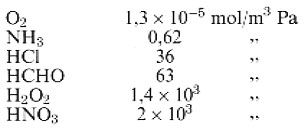
Più è solubile un gas, più elevato è il suo valore di HA. Gas molto solubili, cioè con coefficienti di Henry maggiori di 10 circa, possono venir concentrati in fase acquosa, ad esempio le goccioline di una nuvola, e raggiungere concentrazioni molto più elevate di quelle che avrebbero se rimanessero in fase gassosa. Due di queste specie molto solubili sono l'acqua ossigenata e la formaldeide. I valori forniti sopra riflettono solamente la solubilità del gas a prescindere dal destino successivo di A • H2O. Una buona parte delle specie elencate, una volta disciolte, sono soggette a ulteriori reazioni chimiche che accrescono la loro solubilità al di sopra di quanto indicato dal coefficiente di Henry. Il diossido di zolfo, l'acido nitrico e l'acido solforico hanno ricevuto una grande attenzione a causa della loro solubilità nelle precipitazioni atmosferiche, ma anche numerosi composti organici, se presenti sotto forma di gas nell'atmosfera, possono sciogliersi in acqua liquida atmosferica.
7. Chimica dell'atmosfera e clima globale
a) Gas responsabili dell'effetto serra
Le concentrazioni dei gas presenti in tracce nella troposfera sono in continuo aumento (v. Ramanathan e altri, 1985). La tab. III riassume le concentrazioni in superficie, le velocità con cui aumentano e le valutazioni relative ai tempi di vita nell'atmosfera per un certo numero di specie presenti in tracce. Molti di questi gas hanno effetti sul clima, in quanto assorbono radiazioni infrarosse emesse dalla superficie terrestre e dirette verso l'esterno dell'atmosfera; tale fenomeno è noto come ‛effetto serra' (v. Ramanathan, 1988). La crescente concentrazione di CO2, dovuta in gran parte alla combustione di combustibili fossili, ha ricevuto una grandissima attenzione. Secondo le attuali valutazioni, un raddoppio della concentrazione di CO2 rispetto all'epoca precedente la rivoluzione industriale, cioè il passaggio da un valore di 280 ppm a uno di 560 ppm, aumenterebbe la temperatura media globale della superficie terrestre di 2,5 ÷ 3 °C. La somma degli effetti esercitati sulla radiazione da altri gas, quali CH4, N2O e i CFC, potrebbe effettivamente raddoppiare l'impatto climatico degli aumenti previsti della concentrazione di CO2, nel caso in cui le tendenze attuali dei livelli di questi gas nell'ambiente dovessero persistere. Il ‛forcing radiativo' del sistema superficie-troposfera, dovuto a una variazione, per esempio, nelle concentrazioni di questi gas, è la variazione di irradianza netta espressa in watt al metro quadrato (W/m2) a livello della tropopausa. Le variazioni della temperatura della superficie terrestre sono quindi direttamente connesse alle variazioni del forcing. (Viene scelta la tropopausa perché è più facile calcolare il forcing sulla tropopausa e perché, da un punto di vista globale annuale, superficie terrestre e tropopausa sono accoppiate così strettamente che esse si comportano come un unico sistema termodinamico). Il forcing radiativo risultante da un qualunque livello di CO2 e altri gas è ben compreso da un punto di vista generale e non è oggetto di discussione. Ciononostante, a causa di complessi fenomeni di retroazione, il livello delle reali variazioni climatiche che potrebbero avere luogo è estremamente incerto.
Il tempo di risposta di un composto atmosferico, che è l'intervallo di tempo entro cui il carico atmosferico d'eccesso associato a una singola perturbazione decade a 1/e del suo valore iniziale, non corrisponde necessariamente al suo tempo di vita. Si consideri il metano, per esempio, il cui tempo di vita è legato al tempo caratteristico della reazione fra CH4 e • OH. Se nell'atmosfera viene introdotta una quantità in eccesso di CH4, l'aumento della concentrazione di questo gas ha come effetto una produzione ulteriore di CO. Entrambi eliminano • OH, che è l'agente che distrugge il CH4; pertanto il tempo caratteristico per il decadimento della perturbazione si estende oltre quello che si avrebbe in assenza della perturbazione. Nel caso del metilcloroformio (CH3CCl3), il cui processo di distruzione è legato alla reazione con • OH nella troposfera (v. tab. III), il contributo delle molecole di CH3CCl3 alla reazione complessiva degli • OH è così piccolo che il tempo di risposta del CH3CCl3 è uguale al suo tempo di vita. Invece, un composto che probabilmente ha tempo di vita e tempo di risposta differenti è N2O. Il principale mezzo di controllo sulla concentrazione di O3 nella stratosfera superiore è rappresentato dagli NOx derivati da N2O. Variazioni delle concentrazioni di O3 portano a variazioni nel campo delle radiazioni UV e quindi nella velocità di eliminazione dell'N2O stesso. Contrariamente a quanto avviene per il CH4, si presume che il tempo di risposta per l'N2O sia più breve del suo tempo di vita, dato che aumenti nella concentrazione di N2O provocano diminuzioni delle concentrazioni di O3 e quindi un aumento della velocità di ossidazione per via fotolitica di N2O.
Tabella 3
Mentre il radicale • OH di per sé non è un gas responsabile dell'effetto serra, esso è, come risulta da quanto scritto finora, il più importante agente chimico che elimina le impurità della troposfera (v. tab. III). Una variazione dei livelli globali di • OH può avere effetti sui tempi di vita di molti dei ‛gas-serra' dell'atmosfera e quindi influenzare le loro quantità e di conseguenza il clima stesso. Inoltre, gli • OH e le specie ad esso strettamente correlate, come HO2 •, svolgono un ruolo centrale nella generazione dell'ozono troposferico. Le vie di eliminazione più importanti per il CH4 e il CO sono le loro reazioni con • OH. Come abbiamo visto, aumenti delle quantità di CH4 e CO possono causare diminuzioni degli • OH, con conseguenti aumenti del tempo di vita di questi gas nell'atmosfera. Poiché il vapor d'acqua è la fonte degli • OH, variazioni della sua concentrazione, che sono legate alla temperatura, dovrebbero condurre a variazioni dei livelli di • OH. Un aumento di 2 °C della temperatura potrebbe determinare un aumento dal 10 al 30% dei livelli di H2O nella troposfera, implicando un piccolo aumento percentuale di • OH e altri membri della famiglia HOx •. Aumenti delle concentrazioni di NOx e O3 nella troposfera possono provocare un incremento di • OH poiché favoriscono le reazioni cicliche che convertono HO2 • in • OH. Un aumento di O3 di un fattore 2 potrebbe provocare aumenti di • OH forse del 10% al di sopra delle aree oceaniche e anche superiori al 10% sopra i continenti (v. Thompson, 1991). Variazioni delle temperature troposferiche stesse provocano variazioni delle velocità di reazione: ad esempio, l'energia di attivazione della reazione fra CH4 e • OH è pari a 1.710 K (assumendo pari a 1 la costante di Boltzmann). Un aumento di 5 K provocherebbe un incremento della velocità di reazione del 10%. Diminuzioni dell'ozono stratosferico possono influenzare la chimica della troposfera in due modi: 1) diminuzione del flusso di O3 nella troposfera; 2) maggiore radiazione UV nella troposfera, che provoca una maggiore produzione primaria di • OH attraverso la formazione di O(1D).
L'ozono esercita un duplice effetto sul clima. Assorbendo le radiazioni ultraviolette con lunghezze d'onda superiori ai 200 nm provenienti dal Sole, l'ozono governa la struttura termica della stratosfera; ma poiché assorbe anche le radiazioni infrarosse emesse dalla Terra, esso è anche un gas-serra. Pertanto, l'effetto sul clima di variazioni della concentrazione di O3 atmosferico dipende dalla differente distribuzione di O3 con l'altezza e la latitudine. Variazioni delle quantità di O3 nella troposfera superiore e nella stratosfera inferiore sono molto efficaci nel causare una variazione di temperatura alla superficie terrestre; un aumento di O3 in questa regione porta a temperature più elevate come risultato dell'effetto serra. L'effetto serra prodotto da un certo incremento nella quantità di un gas presente in tracce è direttamente proporzionale alla differenza tra la temperatura a cui la radiazione viene assorbita e quella alla quale essa viene riemessa. Questa differenza è maggiore vicino alla tropopausa. Al di sopra di circa 30 km, un aumento di O3 provoca in realtà una diminuzione della temperatura superficiale, poiché l'O3 in eccesso assorbe un'ulteriore quantità di radiazioni solari UV e visibili, impedendo a quell'energia di raggiungere la troposfera.
b) Aerosol e clima
Le particelle di aerosol, sebbene costituiscano soltanto un miliardesimo circa dell'atmosfera, possono influenzare il clima mediante il loro effetto sul trasferimento delle radiazioni attraverso l'atmosfera e sulla nucleazione delle nubi (v. Charlson e altri, 1992). Gli aerosol possono disperdere la radiazione in arrivo, aumentando notevolmente l'albedo planetaria, e agire come nuclei di condensazione delle nuvole, aumentando la concentrazione numerica delle goccioline delle nuvole ed eventualmente aumentando l'albedo delle nuvole. Contrariamente ai gas serra (v. tab. III), gli aerosol atmosferici hanno un breve tempo di vita nell'atmosfera (una settimana circa), e quindi, visto anche che la distribuzione delle fonti di aerosol è assai poco uniforme, risultano diffusi in modo geograficamente molto poco omogeneo. Vi sono, ad esempio, differenze significative nella distribuzione degli aerosol antropogenici e naturali tra gli emisferi nord e sud, dato che le fonti industriali si trovano in prevalenza nell'emisfero settentrionale e gli oceani in quello meridionale.
Gli aerosol atmosferici possono essere di origine primaria, cioè emessi dalle fonti come particelle, o secondaria, cioè formati nell'atmosfera dalla trasformazione di gas. Andreae (v., 1995) ha stimato il flusso globale annuale di aerosol nel modo seguente:
Tabella
Le caratteristiche degli aerosol che sono rilevanti per quanto riguarda le proprietà di irradiamento dell'atmosfera includono: 1) numero, area della superficie, e concentrazione; 2) indice di rifrazione (funzione della composizione chimica); 3) solubilità in acqua e igroscopicità. La dimensione delle particelle è la proprietà che ha più importanza nel condizionare il ruolo potenziale degli aerosol nei riguardi del clima. La distribuzione delle dimensioni di un aerosol atmosferico è spesso rappresentata come trimodale, con un gruppo cosiddetto dei nuclei (di diametro inferiore a 0,1 µmm), un gruppo di accumulazione (diametro compreso tra 0,1 e 1,0 µmm), e un gruppo di particelle più grosse (diametro superiore a 1 µmm). Le particelle del gruppo dei nuclei sono quelle appena prodotte da processi di combustione o nucleazione. Il gruppo di accumulazione si chiama così perché le particelle tendono ad accumularsi in questo campo di dimensioni per coagulazione di particelle più piccole, oppure per crescita di particelle più piccole dovuta a condensazione di specie gassose. Le particelle che appartengono al gruppo dei nuclei o a quello di accumulazione sono dette ‛particelle fini'. La concentrazione numerica delle particelle fini è compresa tra 10 e 105/cm3, mentre quella delle particelle più grosse è solitamente inferiore a 1/cm3. In genere, prevalgono numericamente le particelle che hanno diametro inferiore a 0,1 µmm, anche se le particelle più grosse possono essere dominanti rispetto alla massa dell'aerosol. Particelle aventi diametro compreso tra 0,1 e 1,0 µmm hanno la massima efficienza per unità di massa per quanto riguarda la dispersione della radiazione solare.
Come notato in precedenza in questo articolo, le goccioline atmosferiche si formano e crescono per condensazione del vapor d'acqua su particelle che agiscono da nuclei di condensazione per le nuvole. La soprasaturazione critica alla quale una particella agisce da nucleo di condensazione dipende dalle dimensioni e dalla composizione della particella. Quando l'aria viene raffreddata e la soprasaturazione cresce, viene attivato un numero maggiore di nuclei finché la rimozione dell'acqua per condensazione sui nuclei viene bilanciata da quella formatasi per raffreddamento dell'aria. Le soprasaturazioni massime nelle nuvole raramente superano l'1%. Di solito, particelle di dimensioni superiori a 0,1 µmm di diametro vengono attivate per formare delle goccioline. L'effetto di un aumento della concentrazione di aerosol sulle proprietà microfisiche e di irradiazione delle nuvole viene evidenziato dalla formazione delle cosiddette ‛rotte navali' che si formano nelle nuvole marine stratiformi per effetto delle emissioni dalle ciminiere delle navi (v. Radke e altri, 1989). Rispetto alla massa totale delle particelle, l'ossidazione dell'SO2 e degli NOx per formare solfati e nitrati sotto forma di aerosol rappresenta una fonte modesta; tuttavia le particelle che vengono prodotte per questa via, a causa della loro igroscopicità e delle loro piccole dimensioni, possono essere molto efficaci come nuclei di condensazione. Le particelle di fumo derivanti da fenomeni di combustione sono ricche di carbonio, ma possono contenere altri prodotti della combustione. L'efficacia degli aerosol da combustione come nuclei di condensazione è estremamente variabile e dipende dal materiale bruciato e dalla natura del processo di combustione.
BIBLIOGRAFIA
Andreae, M. O., Ocean-atmospheric interactions in the global biogeochemical sulfur cycle, in ‟Marine chemistry", 1990, XXX, pp. 1-29.
Andreae, M. O., Climate effects of changing atmospheric aerosol levels, in World survey of climatology, vol. XVI, Future climates of the world (a cura di A. Henderson-Sellers), Amsterdam 1995, pp. 341-392.
Atkinson, R., Gas-phase tropospheric chemistry of organic compounds: a review, in ‟Atmospheric environment" (Part A), 1990, XXIV, pp. 1-41.
Barker, J. R., A brief introduction to atmospheric chemistry, in Progress and problems in atmospheric chemistry, (a cura di J. R. Barker), Singapore 1995, pp. 1-33.
Barnes, R. A., Holland, A. C., Kirchoff, V. W. J. H., Equatorial ozone profiles from the ground to 52 km during the southern hemisphere autumn, in ‟Journal of geophysical research", 1987, XCII, pp. 5573-5583.
Blake, D. R., Rowland, F. S., Continuing worldwide increase in tropospheric methane, 1978 to 1987, in ‟Science", 1988, CCXXXIX, pp. 1129-1131.
Charlson, R. J., Lovelock, J. E., Andreae, M. O., Warren, S. G., Oceanic phytoplankton, atmospheric sulphur, cloud albedo and climate, in ‟Nature", 1987, CCCXXVI, pp. 655-661.
Charlson, R. J., Schwartz, S. E., Hales, J. M., Cess, R. D., Coakley, J. A. Jr., Hansen, J. E., Hofmann, D. J., Climate forcing by anthropogenic aerosols, in ‟Science", 1992, CCLV, pp. 423-430.
Cloud, P., The biosphere, in ‟Scientific American", 1983, CCXLIX, pp. 176-189 (tr. it.: La biosfera, in ‟Le scienze", 1983, XXXI, 183, pp. 132-146).
IPCC (Intergovernmental Panel on Climate Change), Climate change 1992: the supplementary report to the IPCC Scientific assessment (a cura di J. T. Houghton, B. A. Callander, S. K. Varney), Cambridge 1992.
Khalil, M. A. K., Rasmussen, R. A., Atmospheric methane: trends over the last 10,000 years, in ‟Atmospheric environment", 1987, XXI, pp. 2445-2452.
Khalil, M. A. K., Rasmussen, R. A., Atmospheric methane: recent global trends, in ‟Environmental science and technology", 1990, XXIV, pp. 549-553.
Kleinman, L. I., Seasonal dependence of boundary layer peroxide concentration: the low and high NOx regimes, in ‟Journal of geophysical research", 1991, XCVI, pp. 20721-20733.
Logan, J. A., Tropospheric ozone: seasonal behavior, trends, and anthropogenic influence, in ‟Journal of geophysical research", 1985, XC, pp. 10463-10482.
Logan, J. A., Prather, M. J., Wofsy, S. C., McElroy, M. B., Tropospheric chemistry: a global perspective, in ‟Journal of geophysical research", 1981, LXXXVI, pp. 7210-7254.
Lurmann, F. W., Main, H. H., Analysis of ambient VOC data collected in the Southern California air quality study, Sacramento, Cal., 1992.
McElroy, M. B., Salawitch, R. J., Changing composition of the global stratosphere, in ‟Science", 1989, CCXLIII, pp. 763-770.
National Research Council, Rethinking the ozone problem in urban and regional air pollution, Washington 1991.
Pandis, S., Seinfeld, J. H., Sensitivity analysis of a chemical mechanism for aqueous-phase atmospheric chemistry, in ‟Journal of geophysical research", 1989, XCIV, pp. 1105-1126.
Radke, L. F., Coakely, J. A. Jr., King, M. D., Direct and remote sensing observations of the effects of ships on clouds, in ‟Science", 1989, CCXLVI, pp. 1146-1149.
Ramanathan, V., The greenhouse theory of climate change: a test by an inadvertent global experiment, in ‟Science", 1988, CCXL, pp. 293-299.
Ramanathan, V., Cicerone, R. J., Singh, H. B., Kiehl, J. T., Trace gas trends and their potential role in climate change, in ‟Journal of geophysical research", 1985, XC, pp. 5547-5566.
Rowland, F. S., Chlorofluorocarbons and the depletion of stratospheric ozone, in ‟American scientist" 1989, LXXVII, pp. 36-45.
Rowland, F. S., Stratospheric ozone depletion, in ‟Annual review of physical chemistry", 1991, XLII, pp. 731-768.
Seinfeld, J. H., Atmospheric chemistry and physics of air pollution, New York 1986.
Seinfeld, J. H., Urban air pollution: state of the science, in ‟Science", 1989, CCXLIII, pp. 745-752.
Staehelin, J., Schmid, W., Trend analysis of tropospheric ozone concentrations utilizing the 20-year data set of ozone balloon soundings over Payerne (Switzerland), in ‟Atmospheric environment" (Part A), 1991, XXV, pp. 1739-1749.
Stauffer, B., Fischer, G., Neftel, A., Oeschger, H., Increase of atmospheric methane recorded in Antarctic ice core, in ‟Science", 1985, CCXXIX, pp. 1386-1388.
Stewart, R. W., Multiple steady states in atmospheric chemistry, in ‟Journal of geophysical research", 1993, XCVIII, pp. 20601-20611.
Thompson, A. M., Interaction of atmospheric chemical and climate change: implications for tropospheric ozone, in Atmospheric chemistry: models and predictions for climate and air quality (a cura di C. S. Sloane e T. W. Tesche), Boca Raton, Flo., 1991, pp. 47-61.
Tie, X., Kao, C.-Y., Mroz, E. J., Net yield of OH, CO, and O3 from the oxidation of atmospheric methane, in ‟Atmospheric environment" (Part A), 1992, XXVI, pp. 125-136.
Volz, A., Kley, D., Evaluation of the Montsouris series of ozone measurements made in the nineteenth century, in ‟Nature", 1988, CCCXXXII, pp. 240-242.
Walker, J. C. G., Evolution of the atmosphere, New York 1977.
Warneck, P., Chemistry of the natural atmosphere, San Diego, Cal., 1988.
Wayne, R. P., Chemistry of atmospheres: an introduction to the chemistry of the atmospheres of earth, the planets, and their satellites, Oxford 19912.
Zimmerman, J., Poppe, D., Nonlinear couplings in the tropospheric NOx-HOx gas phase chemistry, in ‟Journal of atmospheric chemistry", 1993, XVII, pp. 141-155.