
Chimica fisica dei sistemi non lineari
Chimica fisica dei sistemi non lineari
SOMMARIO: 1. Introduzione. 2. Definizioni e concetti elementari. 3. Tipi di fenomeni non lineari: a) sistemi chimici con stati stazionari multipli; b) reazioni chimiche oscillanti; c) altre variazioni temporali delle concentrazioni di specie chimiche. 4. Altri tipi di sistemi reattivi: a) reazioni eterogenee; b) la temperatura come variabile in sistemi reattivi; c) interazione della luce con le reazioni chimiche; d) reazioni chimiche in sistemi non ideali; e) interazioni delle onde sonore con le reazioni chimiche. 5. Strutture spaziali in sistemi reattivi. 6. Chimica della corrente alternata. 7. Funzioni logiche e calcolatori ottenuti mediante cinetiche chimiche macroscopiche. 8. La teoria termodinamica e stocastica di sistemi non lineari lontani dall'equilibrio. □ Bibliografia.
1. Introduzione
Negli ultimi quaranta anni l'interesse nei confronti della cinetica delle reazioni chimiche non lineari è cresciuto considerevolmente. Il termine ‛cinetica' definisce un vasto campo di ricerca, che comprende lo studio delle velocità e dei meccanismi di sistemi chimici non lineari, i tipi di fenomeni osservati e gli approcci concettuali impiegati per analizzare questi sistemi; l'interesse nei confronti di questo tipo di ricerche è dimostrato dai più di 5.000 articoli comparsi nella letteratura scientifica degli ultimi trent'anni. Dato che in questo articolo può essere presentata soltanto una breve rassegna di alcune delle attività svolte in questo campo, con un numero limitato di riferimenti bibliografici, rimandiamo il lettore interessato ad alcune recenti rassegne (v. Field e Burger, 1985; v. Gray e Scott, 1990; v. Marek e Schreiber, 1991; v. Perelson e altri, 1988; v. Glass e Mackey, 1988; v. Noyes, 1990).
2. Definizioni e concetti elementari
In un sistema isolato, in assenza di scambi di materia e di energia con l'ambiente circostante, possono aver luogo dei fenomeni transitori fino al raggiungimento di uno stato di equilibrio. Nello stato di equilibrio, l'entropia del sistema isolato, essendo costante l'energia, è massima. In un sistema chiuso, definito dal fatto che può scambiare energia ma non materia con l'ambiente circostante, in condizioni di pressione e temperatura costanti, lo stato di equilibrio è raggiunto quando è minima l'energia libera di Gibbs. Queste funzioni, cioè l'entropia per i sistemi isolati e l'energia libera di Gibbs per i sistemi chiusi a temperatura e pressione costanti, rappresentano le cosiddette ‛funzioni di Ljapunov', e sono impiegate per descrivere l'approccio dei sistemi all'equilibrio poiché indicano appunto la direzione dell'evoluzione del sistema verso l'equilibrio: un aumento di entropia per sistemi isolati e una diminuzione di energia libera di Gibbs per sistemi chiusi a temperatura e pressione costanti (v. Berry e altri, 1980).
È possibile impedire che sistemi chimici e fisici raggiungano l'equilibrio imponendo flussi di massa o di energia attraverso i loro confini. Si consideri il semplice sistema costituito da un reagente A, un prodotto intermedio X e un prodotto finale B nella configurazione mostrata in fig. 1, contenuto in un recipiente di volume costante, VII. Questo recipiente è separato dal serbatoio del reagente a volume variabile, VI, e dal serbatoio del prodotto a volume variabile, VIII, da membrane semimpermeabili tali da permettere soltanto ad A di passare da VI a VII e soltanto a B di passare da VII a VIII. Col procedere della reazione da A a B, il volume VI diminuisce poiché è stata applicata una pressione costante PA mediante un pistone, mentre il volume VIII cresce a pressione costante PB. In corrispondenza di uno stato stazionario, la concentrazione di X, come pure le concentrazioni di A e B nel volume VII, sono costanti, ma c'è un flusso costante di massa attraverso il sistema: A passa da VI a VII e B abbandona VII per andare in VIII. In uno stato stazionario le velocità di movimento dei pistoni nei due serbatoi sono le stesse. Al contrario, in uno stato transitorio, nel caso della semplice reazione A → B, la concentrazione di X varia col variare del tempo.
Una disposizione alternativa, più facile da realizzare in laboratorio, consiste in un recipiente di reazione (reattore) sottoposto ad agitazione continua (CSTR, Continuous Stirred Tank Reactor), oggetto di molti studi in chimica e in ingegneria chimica; tale disposizione è utilizzata per applicazioni industriali. I reagenti vengono pompati entro il CSTR attraverso i tubi che nella figura sono indicati come ‛flussi di alimentazione'; la reazione si svolge all'interno del reattore ben agitato; i prodotti e i reagenti che non hanno reagito escono attraverso lo ‛scarico'. Il reattore è circondato da una camicia d'acqua termostatata. Le concentrazioni delle specie chimiche sono misurate nel reattore, in questo caso, utilizzando un elettrodo ioni-selettivo.
3. Tipi di fenomeni non lineari
a) Sistemi chimici con stati stazionari multipli.
Si consideri, per esempio, il semplice modello (di Schlögl) costituito da due reazioni elementari successive:
A + 2X ⇋ 3X (1)
X ⇋ B. (2)
Per i gas ideali e le soluzioni ideali la velocità con la quale varia la concentrazione, x, della specie X è data dalla seguente equazione cinetica, derivata dalla legge dell'azione di massa:
dx/dt = k1ax2 + k4b - [k2x3 + k3x] ≡ (x) (3)
dove k1 e k2 sono le costanti di velocità della reazione (1) e della sua inversa e analogamente k3 e k4 per la reazione (2), x, a e b sono le concentrazioni di X, A e B, rispettivamente (v. cinetica chimica, vol. I). Questa equazione di velocità è non lineare per il fatto che vi appaiono potenze della concentrazione di X maggiori di 1. In questo caso la non linearità esercita una retroazione positiva (talvolta detta anche autocatalisi) sul primo stadio del meccanismo di reazione, poiché due molecole di X vengono trasformate in tre molecole di X. Finché la variazione di x nel tempo è diversa da zero, si verificherà un rilassamento transitorio verso uno stato stazionario definito dalla condizione dx/dt = 0. Poiché nello stato stazionario la (3) fornisce un'equazione cubica nella concentrazione di X, in corrispondenza di certe scelte della pressione, del reagente (A), del prodotto (B) e delle loro concentrazioni, potrebbero esistere tre stati stazionari. La stabilità di ciascuno stato stazionario può essere determinata considerando la variazione temporale di una piccola deviazione δx = x - xs di x rispetto al suo valore nello stato stazionario, indicato con xs. Poiché l'equazione cinetica per x = xs è dx/dt = 0, attraverso uno sviluppo in serie di Taylor si ottiene
F(xs) = 0 (4)
dδx/dt = [∂F(x)/∂x]xsδx = Ω (xs) δx. (5)
Si supponga che lo stato stazionario sia perturbato in modo tale da spostare il sistema da xs a x = xs + δx; in questo caso la variazione temporale della perturbazione δx è δx (t) = δx (t = 0) exp [Ω (xs) t]; pertanto, se la derivata Ω (xs) 〈 0, il sistema è stabile, poiché la deviazione rispetto allo stato stazionario decade a zero. Al contrario, se Ω (xs) > 0, allora il sistema è instabile in quello stato stazionario poiché la perturbazione rispetto allo stato stazionario cresce con il tempo. Date le concentrazioni di A e B e assegnati i valori delle costanti di velocità, si può risolvere la (4) rispetto a xs; un grafico di questi valori di xs è rappresentato in fig. 3. Per certi valori di a e b, indicati con il deponente eq (equilibrio), il rapporto aeq/beq diventa pari alla costante di equilibrio K della reazione. In corrispondenza di un certo campo di valori del rapporto a/b, esistono tre rami di stati stazionari: i rami 1 e 3 sono stabili, mentre il ramo 2 è instabile. I punti indicati con am e bm sono i cosiddetti ‛punti di stabilità marginale'. Se scegliamo il rapporto a/b in prossimità dell'equilibrio, allora un'analisi della stabilità lineare come quella eseguita nelle (4) e (5) dimostra che nel caso di sistemi ideali, in cui le concentrazioni di equilibrio sono univocamente determinate, lo stato stazionario è unico e stabile.
Nelle prove sperimentali è possibile ottenere soltanto stati stazionari stabili; infatti, anche la minima perturbazione provoca un allontanamento crescente da uno stato stazionario instabile: il sistema abbandona lo stato stazionario instabile e si porta in uno stato stazionario stabile. Si supponga quindi che il ramo 1 degli stati stazionari stabili venga attraversato lentamente per valori crescenti di a/b: in corrispondenza del punto di stabilità marginale am si verifica allora una netta transizione verticale verso il ramo 3 degli stati stazionari stabili e il sistema si mantiene su questo ramo anche per ulteriori aumenti di a/b. Al decrescere di a/b, il sistema si mantiene sul ramo 3 degli stati stazionari stabili fino al raggiungimento del punto di stabilità marginale bm, e a questo punto si verifica una netta transizione verticale fino al ramo 1 degli stati stazionari stabili. Questo tipo di transizione da un ramo degli stati stazionari stabili a un altro in corrispondenza di posizioni differenti dei punti di stabilità marginale am e bm è un esempio di isteresi chimica. L'esistenza di una bistabilità (due rami di stati stazionari stabili) rappresenta una sorta di commutatore chimico; su questo punto ritorneremo in seguito.
Esistono innumerevoli esempi di multistabilità degli stati stazionari nelle reazioni chimiche e biochimiche. La multistabilità nell'isteresi chimica ha luogo, ad esempio, nella reazione tra uno iodato e l'acido arsenioso, il cui comportamento è illustrato nella fig. 4, e nell'idrolisi degli esteri catalizzata dall'enzima papaina, rappresentata in fig. 5. Multistabilità e isteresi in un sistema chiuso, alimentato da un flusso di energia (luce) sono raffigurate in fig. 6. È possibile stabilizzare stati stazionari instabili tramite retroazioni esterne senza che ciò interferisca con la posizione dei rami degli stati stazionari di fig. 3. I rami instabili degli stati stazionari possono essere determinati sperimentalmente, come si può vedere in fig. 6.
La dimostrazione sperimentale di bistabilità e isteresi nei sistemi chimici fa sorgere alcune domande interessanti. Esistono criteri termodinamici che condizionano la stabilità di stati stazionari analoghi a quelli che condizionano sistemi all'equilibrio (v. cap. 2)? La stabilità relativa di due stati di equilibrio, ad esempio acqua allo stato liquido e acqua allo stato di vapore a una certa temperatura e pressione, è stabilita dal valore dell'energia libera di Gibbs in quelle condizioni: lo stato con energia libera di Gibbs più bassa è più stabile. Per una data condizione, per esempio una data velocità di flusso nel sistema iodato-acido arsenioso, quale dei due possibili stati stazionari stabili è il più stabile? Esistono criteri termodinamici di stabilità relativa in sistemi con stati stazionari multipli? Un altro problema da prendere in esame riguarda le fluttuazioni presenti in un sistema lontano dall'equilibrio, sia nel caso in cui sia vicino allo stato stazionario che quando subisca una fluttuazione più ampia. In un sistema all'equilibrio vi sono relazioni tra una funzione termodinamica come l'entropia o l'energia libera di Gibbs e la probabilità di una fluttuazione termica spontanea. Esistono relazioni di questo tipo per sistemi in stati stazionari lontani dall'equilibrio? Alcuni di questi problemi verranno discussi, almeno in parte, più avanti; ora soffermiamoci su un esperimento che riguarda la stabilità relativa.
Si consideri l'apparato sperimentale di un sistema dotato di una molteplicità di stati stazionari (1, 2), una parte del quale, dato un certo insieme di condizioni, si trova nello stato stazionario 1, e un'altra parte nello stato stazionario 2. Le condizioni imposte mantengono le parti del sistema nei loro rispettivi stati stazionari. Inizialmente le due parti del sistema sono separate, ma a un certo momento la separazione viene rimossa e si può allora osservare che o il sistema nello stato stazionario 1 si propaga e annulla il sistema nello stato 2, oppure avviene il contrario, a seconda di quale sia lo stato più stabile (v. Harding e Ross, 1990; v. Foerster e altri, 1993).
La descrizione dei sistemi cinetici, come ad esempio quella fornita dalla (3), viene detta macroscopica o deterministica: l'equazione fornisce la dipendenza dal tempo della media macroscopica delle concentrazioni delle specie. Comunque, poiché in effetti si verificano delle fluttuazioni termiche, per poterle considerare è necessario tenere conto delle deviazioni rispetto alle medie. L'approccio abituale a questo tipo di studi è basato su certe equazioni stocastiche, come la cosiddetta master equation (v. van Kampen, 19892; v. Gardiner, 19902), che, riferendoci per esempio al sistema contenuto nel volume VII di fig. 1, fornisce la probabilità P (X, t) che ci siano delle molecole di X al tempo t. La descrizione stocastica dipendente dal tempo vale per processi transitori; a tempo infinito, quando il sistema si trova in uno stato stazionario, la distribuzione stazionaria è indipendente dal tempo. Per un sistema all'equilibrio la distribuzione stazionaria è poissoniana, la nota distribuzione di forma semplice, con un picco pronunciato in corrispondenza del valore più probabile della concentrazione che rappresenta la media macroscopica. Per sistemi bistabili la probabilità di distribuzione stazionaria stocastica è bimodale: un picco è situato in corrispondenza del più probabile valore macroscopico della concentrazione di uno degli stati stazionari stabili e l'altro picco in corrispondenza del valore della concentrazione macroscopica dell'altro stato stazionario stabile. L'altezza relativa dei due picchi, almeno nel limite termodinamico di un numero elevato di particelle (cioè di un sistema macroscopico), è una misura della stabilità relativa dei due stati stazionari.
b) Reazioni chimiche oscillanti.
Nelle reazioni chimiche oscillanti le concentrazioni delle specie chimiche variano periodicamente; queste specie possono essere indifferentemente reagenti, intermedi o prodotti di reazione. Le oscillazioni possono verificarsi in sistemi con due o più variabili. Anche in questo caso, consideriamo un esempio semplice, quello di una reazione chimica caratterizzata da due variabili: un substrato (reagente), S, e un prodotto, P; la reazione è catalizzata da un enzima E attraverso la semplice stechiometria S + E → E + P. Il modello è solo un poco più semplice di quello dell'idrolisi degli esteri in cui il substrato genera due prodotti, uno dei quali è costituito da protoni; la reazione viene catalizzata dall'enzima papaina. L'enzima E (come nel caso della papaina) si ionizza in due stadi:
E ⇋ E′ + P; E′ ⇋ E″ + P, (6)
e solamente la forma E dell'enzima è attiva. Il sistema è confinato - ad esempio da una membrana, nel caso di una cellula, oppure all'interno di un CSTR - e le concentrazioni esterne del reagente, S0, e del prodotto, P0, sono mantenute costanti a valori tali che si svolga una reazione spontanea da S a P. In questo sistema le oscillazioni si verificano a causa della presenza di un meccanismo di controreazione. Supponiamo che l'enzima a un certo istante si trovi prevalentemente nelle forme E′ ed E″: la reazione sarà lenta poiché la concentrazione della forma attiva dell'enzima, E, è bassa. Ma via via che si svolge la reazione, costantemente alimentata dal flusso di S0, si ha formazione di prodotto, P, che può o reagire secondo la (6), oppure diffondere al di fuori della zona di reazione. Se le velocità delle reazioni (6) sono elevate, allora la produzione di P sposta queste reazioni verso la produzione di concentrazioni più elevate di E, con un conseguente aumento della velocità di reazione dovuta alla catalisi da parte di E. La velocità di reazione sarà alla fine così elevata che il flusso di S0 diviene insufficiente a mantenere la reazione. A questo punto le reazioni (6) si spostano rapidamente in senso inverso, cosicché aumenta la concentrazione di P nella zona di reazione; si produrrà quindi un rapido flusso di P fuori dal sistema, con un conseguente spostamento di E a E′ ed E″. A questo punto siamo tornati alle condizioni iniziali, dopo variazioni consistenti delle concentrazioni di S, P ed E. Può allora iniziare un altro ciclo di oscillazioni che si susseguono fintantoché vengono mantenute le condizioni esterne.
Sono noti più di 120 sistemi chimici oscillanti (reazioni inorganiche e organiche; v. anche termodinamica irreversibile e sinergetica, vol. VII) e circa 150 reazioni biologiche oscillanti in vivo (v. Rapp, 1979). Un esempio di reazione inorganica è quello della reazione clorito-ioduro, di cui vengono mostrate nella fig. 7 le oscillazioni temporali per alcune delle specie chimiche implicate. Sia le ampiezze che il periodo delle oscillazioni sono ben riproducibili e stabili rispetto a piccole perturbazioni che possono però alterare la fase delle oscillazioni. La reazione esaminata, come molte altre reazioni chimiche oscillanti, rappresenta un esempio dei cosiddetti ‛cicli limite stabili'. Mentre uno stato stazionario stabile è un singolo punto che serve da attrattore entro un certo dominio dello spazio delle concentrazioni, un ciclo limite è un attrattore costituito da un circuito chiuso nello spazio delle concentrazioni. Interessanti esempi di sistemi biochimici oscillanti sono le oscillazioni delle reazioni glicolitiche (v. fig. 8), le oscillazioni di volume dei mitocondri (v. fig. 9) e le oscillazioni di potenziale del cuore umano (v. fig. 10). I meccanismi delle reazioni oscillanti possono essere abbastanza complessi per il fatto che sono costituiti da parecchi stadi elementari, in ciascuno dei quali avviene una reazione chimica attraverso una singola collisione tra reagenti (di solito non più di due). Un meccanismo di reazione ipotizzato per la reazione clorito-ioduro è mostrato in tab. I; ciò che era stato teoricamente previsto in relazione a questo meccanismo è - per la gran parte, ma non completamente - in accordo con le osservazioni sperimentali. Proporre e verificare i meccanismi di reazione è un compito difficile. Vi sono stati diversi contributi intesi a individuare una metodologia che determini i meccanismi delle reazioni oscillanti e possibilmente quelli di altri tipi di reazione attraverso la messa a punto di esperimenti dai quali sia possibile dedurre almeno la parte essenziale del meccanismo di reazione (v. Tyson, 1975; v. Luo e Epstein, 1990; v. Hynne e altri, 1993; v. Chevalier e altri, 1993; v. Noyes, 1990).

c) Altre variazioni temporali delle concentrazioni di specie chimiche.
Oltre alle variazioni periodiche delle concentrazioni di specie chimiche, se ne possono verificare diverse altre. Esistono vari tipi di attrattori periodici, caratterizzati o da una singola frequenza fondamentale, come mostrato negli esempi delle figg. 7-10, oppure da cicli limite con due o più frequenze fondamentali correlate razionalmente. Vi sono anche variazioni quasi periodiche delle concentrazioni di specie chimiche, come esemplificato in fig. 11, in cui sono presenti due frequenze fondamentali non correlate razionalmente. Pertanto il moto nello spazio delle concentrazioni non si svolge su un anello chiuso, ma su un toro, cioè un tubo curvato a formare un anello chiuso; una frequenza descrive il moto attorno al tubo e l'altra frequenza il moto attorno all'anello. Se immaginiamo una sezione del tubo, cioè quello che viene detto ‛piano di Poincaré', le traiettorie del moto che intersecano quel piano costituiscono un cerchio (v. fig. 11).
Oscillazioni composte di vari tipi possono aver luogo in modo tale che, ad esempio, le concentrazioni variano regolarmente con una grande ampiezza per un determinato periodo di tempo al quale segue un intervallo di oscillazioni di piccola ampiezza. Un esempio interessante è costituito dalle oscillazioni improvvise nella reazione tra diossido di cloro e ioduro, simili ai fenomeni improvvisi che si osservano quando gli impulsi nervosi vengono trasmessi lungo un neurone.
Un tipo di variazione oscillatoria non regolare delle concentrazioni in reazioni chimiche viene detto ‛caos chimico'. Il moto caotico in sistemi fisici, meccanici e idrodinamici, e chimici, così come in molti altri campi della scienza, della medicina e delle scienze sociali, è stato oggetto di grande attenzione (v. Marek e Schreiber, 1991; v. Schneider e Muenster, 1991; v. Vidal, 1986). Le equazioni che descrivono la cinetica chimica sono deterministiche, cioè, date le concentrazioni delle specie chimiche a un certo istante, le soluzioni delle equazioni cinetiche differenziali determinano in maniera completa le concentrazioni delle specie in ogni istante futuro. Per i sistemi caotici, però, la precisione necessaria per ottenere tali soluzioni aumenta esponenzialmente con il tempo. Due sistemi infinitamente vicini nello spazio delle concentrazioni a un certo istante iniziale divergono esponenzialmente in quello spazio al crescere del tempo. I sistemi caotici hanno effettivamente un attrattore stabile verso il quale le traiettorie decadono dopo un periodo transitorio di rilassamento. Il moto in direzione di quell'attrattore non è un anello chiuso, come in un ciclo limite; un attrattore caotico è una superficie di dimensione frattale maggiore di 2. La superficie di un tavolo è bidimensionale, una stanza è tridimensionale; le misure della circonferenza del tavolo, o del volume della stanza, non dipendono dalle dimensioni dello strumento usato per effettuarle, che può essere indifferentemente, ad esempio, di 1 metro oppure di 10 cm. Un oggetto di dimensioni frattali pari, per esempio, a 2,23 ha invece l'interessante proprietà, tra le altre, che la lunghezza del perimetro di quell'oggetto (per es. la linea costiera di un'isola) dipende dalle dimensioni dello strumento che la misura. Esempi di variazioni caotiche temporali di una concentrazione in un sistema chimico sono stati illustrati in altri articoli (v. termodinamica irreversibile e sinergetica, vol. VII, fig. 23); una diversa rappresentazione di una tale variazione temporale viene fornita nella fig. 13; per confronto, la rappresentazione nello spazio delle fasi di un sistema oscillante è un singolo anello chiuso. La traiettoria caotica è più complessa di quella oscillante, ma mostra in realtà una regolarità che smentisce l'immagine comunemente associata al termine ‛caos'. Il caos non deve essere confuso con il ‛rumore', che in una rappresentazione grafica riempirebbe l'intero spazio delle fasi diversamente da un sistema caotico. La prova della presenza del caos richiede parecchi test severi che solitamente, perché siano efficaci, richiedono un gran numero di misurazioni. Le pretese di aver scoperto un sistema caotico non sono spesso sostenute da prove concrete.
4. Altri tipi di sistemi reattivi.
a) Reazioni eterogenee.
Fin qui abbiamo discusso soltanto le reazioni omogenee, che hanno luogo in un'unica fase, quale una soluzione gassosa oppure liquida. Le reazioni eterogenee si svolgono all'interfaccia tra due o più fasi. Un esempio è rappresentato dall'ossidazione catalitica del monossido di carbonio su una superficie di platino che agisce da catalizzatore. La miscela gassosa di CO, O2 e del prodotto CO2 costituisce una fase, mentre il platino solido con i reagenti e i prodotti assorbiti è la seconda fase. Tutti i diversi tipi di reazioni non lineari, stati stazionari multipli, oscillazioni, oscillazioni composte e caos sono stati osservati anche in sistemi eterogenei (v. Krischer e altri, 1992 e 1993).
b) La temperatura come variabile in sistemi reattivi.
Gli esempi citati fin qui si riferivano a reazioni chimiche condotte a temperatura costante, cioè ai cosiddetti sistemi isotermi. La temperatura, tuttavia, può essere una variabile nei sistemi reattivi non lineari. Si consideri l'ossidazione di un composto organico, come l'acetaldeide, e si conduca la reazione in un CSTR termostatato a una certa temperatura T0, diciamo 250 °C. In queste condizioni si svolge un'ossidazione a fiamma fredda, nella quale sono stati osservati stati stazionari multipli e oscillazioni di vario tipo sia delle specie chimiche che della temperatura (v. Gray e altri, 1981; v. Pugh e altri, 1986; v. Tsujimoto e altri, 1991). Via via che i reagenti chimici O2 e CH3CHO entrano nel reattore, i due componenti gassosi reagiscono dando luogo a un processo di combustione esotermico: viene liberato calore e la temperatura cresce. Al crescere della temperatura la velocità di reazione cresce a sua volta, finché diventa così alta da esaurire tutti i reagenti presenti nel CSTR. La velocità di combustione allora decresce, i prodotti della reazione abbandonano il CSTR e il processo appena descritto si ripete.
c) Interazione della luce con le reazioni chimiche.
Abbiamo già citato l'esempio di una reazione chimica condotta in un sistema chiuso che interagisce con la luce e che potrebbe dar vita a una multistabilità sia rispetto alle concentrazioni sia rispetto alla temperatura (v. fig. 6). La luce può interagire con reazioni chimiche anche per dar vita a oscillazioni e altre variazioni temporali sia in condizioni isotermiche che non isotermiche (v. Borderie e altri, 1992; v. Li e Ross, 1991).
d) Reazioni chimiche in sistemi non ideali.
Per reazioni chimiche tra gas ideali o in soluzioni ideali vi è un unico stato di equilibrio. In queste condizioni, i tipi di fenomeni non lineari fin qui descritti, come stati stazionari multipli e oscillazioni, non possono aver luogo in prossimità dell'equilibrio, ma solo lontano da esso. Nel caso di sistemi non ideali lo stato di equilibrio potrebbe non essere unico, e in questo caso, per esempio, potrebbero verificarsi oscillazioni perfino in prossimità dell'equilibrio (v. Chu e Ross, 1990; v. Hjelmfelt e Ross, 1992).
e) Interazioni delle onde sonore con le reazioni chimiche.
Nelle reazioni chimiche in cui la pressione rappresenta una variabile, come ad esempio nelle reazioni di combustione, si possono verificare interazioni con le onde sonore. Un esempio interessante è rappresentato dall'amplificazione delle onde sonore attraverso fiamme (reazioni di combustione) (v. Gilbert e altri, 1972).
5. Strutture spaziali in sistemi reattivi.
In un sistema reattivo ben agitato, le concentrazioni delle specie chimiche sono funzioni del tempo negli stati transitori, indipendenti dal tempo negli stati stazionari e dipendenti dal tempo in maniera periodica in presenza di attrattori tipo i cicli limite. Consideriamo ora il caso di concentrazioni di specie chimiche che dipendono sia dal tempo che dallo spazio. Abbiamo già visto un caso, discusso in relazione agli esperimenti sulla stabilità relativa, in cui il fronte di uno stato stazionario più stabile si sposta verso uno stato stazionario meno stabile in sistemi che, in date condizioni, presentano stati stazionari multipli. Per descrivere lo spostamento del fronte - che è costituito da una variazione di concentrazione nello spazio, in una certa direzione - è necessario prendere in considerazione le equazioni di diffusione con reazione; ad esempio, per il modello di Schlögl, descritto dalle reazioni (1)-(2), l'equazione di diffusione con reazione in una dimensione spaziale, z, ha la forma
∂x/∂t = F(x) + DX(∂2x/∂z2) (7)
dove x è la concentrazione della specie X e DX è il coefficiente di diffusione di X nella reazione.
Oltre alla propagazione del fronte, in sistemi oscillanti ed eccitabili sono stati osservati molti altri fenomeni dipendenti dal tempo e dallo spazio, quali onde chimiche, pulsazioni e fronti (v. Ross e altri, 1988; v. Winfree, 1973; v. Field e Burger, 1985; v. Cross e Hohenberg, 1993). Se in un sistema eccitabile, che ha un solo stato stazionario, in corrispondenza di quello stato si verifica una perturbazione di una certa specie che supera un certo valore limite, si determinano ampie variazioni delle concentrazioni di tutte le specie variabili prima che si ripristini lo stato stazionario. La fig. 14 mostra la propagazione di un'onda innescata dalla reazione oscillante di Belousov-Chabotinsky in uno stato stazionario eccitabile (per altre raffigurazioni di onde chimiche, v. termodinamica irreversibile e sinergetica, vol. VII, figg. 24-25). L'ampiezza e la struttura del fronte della variazione di concentrazione (di Fe2+, misurata tramite spettroscopia di assorbimento-trasmissione) sono invarianti, poiché i due ripidi gradienti di concentrazione si propagano nello spazio, uno a sinistra e uno a destra. Via via che i fronti si propagano, si ha un aumento di concentrazione causato dalla diffusione delle specie chimiche determinata dal brusco gradiente di concentrazione; il flusso entrante di materia scatena allora l'eccitazione della regione che si trova immediatamente prima del fronte. Ne risulta una consistente variazione delle concentrazioni delle specie reagenti prima del ritorno allo stato stazionario eccitabile e tale variazione determina la creazione di un marcato gradiente di concentrazione nella nuova regione e pertanto causa la propagazione del fronte. Questo tipo di fenomeno d'onda non lineare è simile alla propagazione di un impulso nervoso (v. FitzHugh, 1961), durante la quale l'ampiezza e la struttura del fronte si mantengono inalterate. Queste cosiddette ‛onde scatenanti' che si formano in mezzi che si trovino lontani dall'equilibrio in sistemi eccitabili, inclusi i nervi, sono fondamentalmente diverse dall'eccitazione di un'onda idrodinamica ottenuta, per esempio, lasciando cadere un sasso in un laghetto. In quest'ultimo caso viene creata un'onda che si propaga dal punto d'impatto verso l'esterno con ampiezza decrescente in ciascuna oscillazione dell'onda a causa della dissipazione di energia nel mezzo viscoso costituito dall'acqua. Analoga dissipazione ha luogo nella propagazione di onde chimiche dovuta alla reazione e alla diffusione, ma l'ampiezza e la struttura del fronte dell'onda sono mantenute inalterate dalla forza trainante, cioè dalla variazione di energia libera di Gibbs, via via che si svolge la reazione.
Vi sono diversi tipi di onde (v. Ross e altri, 1988): per esempio le onde cinematiche, che non implicano diffusioni di massa e sono in realtà un'illusione ottica, cioè sembra che si propaghino in un mezzo a causa di un gradiente del periodo di oscillazione (forse dovuto a un piccolo gradiente di temperatura) oppure di un gradiente nella fase delle oscillazioni. Se esiste un gradiente di fase, dopo un po' di tempo avrà luogo un trasferimento di massa tra elementi contigui nella soluzione e quindi si verificherà una diffusione di fase. Le onde chimiche si possono formare in una, due, tre dimensioni. Un fenomeno interessante è quello delle onde a spirale (v. fig. 15). L'onda lentamente si avvolge a spirale nello spazio e l'origine della spirale può dar luogo a figure geometriche. Sono stati compiuti progressi sostanziali nella comprensione della propagazione delle onde nel tessuto cardiaco e della sua correlazione con i moti di contrazione del cuore in condizioni normali e in certe aritmie (v. Winfree, 1987).
La teoria matematica su cui si fonda la soluzione delle equazioni differenziali parziali di reazione e diffusione è un argomento complesso che non può essere trattato in questa sede. Sono disponibili vari tipi di approssimazione, come lo sviluppo in piccole ampiezze, lo sviluppo per piccoli vettori d'onda, lo sviluppo in scala multipla temporale e la teoria della biforcazione (v. Ross e altri, 1988). Una grande quantità di informazioni è stata ottenuta attraverso le soluzioni numeriche delle equazioni di reazione e diffusione, ricavate mediante computer ad alta velocità. Un altro approccio per la soluzione di queste equazioni differenziali è quello dei metodi ad automi cellulari (v. Kapral e altri, 1992).
Le disomogeneità spaziali che si stabiliscono in sistemi lontani dall'equilibrio per l'interazione tra reazioni e diffusione, e in assenza di qualunque gradiente imposto di concentrazione o temperatura, sono note col nome di ‛strutture di Turing' (v. Turing, 1952). Le prime osservazioni di una struttura di Turing, nel suo sviluppo temporale, sono state riportate in una serie di articoli (v. Flicker e Ross, 1974; v. Feinn e altri, 1978; v. Müller e altri, Periodic precipitation..., e Mesoscopic structure..., 1982; v. Lovett e altri, 1978), mentre l'osservazione di una struttura di Turing indipendente dal tempo è stata riportata per la prima volta da Castets e altri (v., 1990). Un esempio di modello di Turing quasi bidimensionale nelle reazioni tra clorito-ioduro e acido malonico è mostrato in fig. 16. Strutture di Turing sono state notate in sistemi omogenei ed eterogenei e la teoria è stata applicata al problema della formazione di strutture nei sistemi biologici, quale può essere, ad esempio, quella delle macchie del leopardo (v. Murray, 1988).
6. Chimica della corrente alternata.
La chimica della corrente alternata studia le modalità di funzionamento dei dispositivi di trasformazione dell'energia chimica e biologica, quali possono essere, ad esempio, le batterie, le pompe a protoni e i processi che avvengono durante la fotosintesi. Si consideri una reazione chimica che si svolge in una batteria: le semicelle della reazione generano una differenza di potenziale V, in corrispondenza di un certo carico sulla batteria con una forza contro-elettromotrice applicata, Ea. La batteria produce una potenza pari a Ea × I, dove I è l'intensità della corrente; (V - Ea) × I è la potenza dissipata, e in condizioni stazionarie tutte queste quantità sono costanti. Nel caso di una pompa a protoni, la potenza prodotta è pari alla differenza di potenziale tra i due lati della membrana moltiplicata per l'intensità della corrente di protoni, la potenza assorbita è data dalla velocità d'idrolisi dell'adenosintrifosfato (ATP) moltiplicata per la variazione di energia libera di Gibbs di quell'idrolisi, e la dissipazione è pari alla differenza tra queste due quantità. Nel funzionamento in condizioni stazionarie di una pompa a protoni, tutte queste quantità sono costanti. Nel caso di sistemi non lineari possono verificarsi stati dinamici che danno vita a oscillazioni delle concentrazioni dei prodotti intermedi, caos, oppure a uno stato stazionario, che viene chiamato ‛fuoco', in cui una perturbazione decade in modo oscillatorio nello stesso stato. Si confronti l'efficienza termodinamica di una pompa a protoni (ed esistono prove sperimentali che alcune pompe a protoni si trovano in un fuoco stabile oppure in uno stato oscillante) nel caso di due diversi modi di alimentazione: una concentrazione costante oppure una concentrazione oscillante di ATP, rispettivamente. Un'analisi teorica mostra che l'efficienza, definita come il rapporto tra la potenza prodotta dalla pompa e quella immessa, può cambiare, nel caso in cui l'alimentazione avvenga con una concentrazione oscillante di ATP, in relazione sia alla frequenza che all'ampiezza delle oscillazioni di concentrazione di ATP (v. Ross e Schell, 1987). In condizioni oscillanti per l'ATP, il rapporto di efficienza mostra variazioni significative, in particolar modo in corrispondenza della metà e del doppio della frequenza propria del fuoco stabile, rispetto a quello che si otterrebbe in corrispondenza di un'alimentazione stazionaria di ATP alla stessa velocità complessiva di consumo del carburante, cioè dell'ATP. Esistono verifiche sperimentali di queste previsioni teoriche nel caso della reazione oscillante dell'ossidazione dell'NADH catalizzata dalla perossidasi del rafano (v. Lazar e Ross, Changes in..., ed Experiments on..., 1990). Nel caso in cui si abbia un'immissione costante di ossigeno nel sistema, è possibile osservare oscillazioni dell'NADH e dell'ossigeno in soluzione. La reazione avviene e si mantiene lontano dall'equilibrio, un processo che, da un punto di vista termodinamico, è analogo a ciò che accade in una pompa biochimica, cioè a stabilire e mantenere un gradiente di concentrazione ai due lati di una membrana. La variazione di energia libera di Gibbs può essere calcolata sulla base delle concentrazioni dei reagenti e dei prodotti (la concentrazione di NADH sommata a quella del NAD+ è costante), mentre la velocità di reazione può essere calcolata sulla base delle variazioni nel tempo delle concentrazioni dei reagenti. Nella chimica della corrente alternata nasce un nuovo concetto, quello di spostamento di fase tra la variazione dell'energia libera di Gibbs e la velocità di reazione; questa quantità non si osserva nel caso di cinetiche ordinarie che si svolgono in condizioni stazionarie oppure in una situazione di rilassamento monotono verso l'equilibrio. Poiché lo spostamento di fase determinato sperimentalmente è diverso a seconda che l'alimentazione di O2 (gas) sia costante o oscillante, è possibile stabilire l'energia prodotta e quella dissipata per ogni determinata energia immessa. Nelle reazioni in cui non si ha produzione d'energia, la dissipazione è data dal prodotto tra la variazione di energia libera di Gibbs e la velocità di reazione in condizioni isoterme. In una reazione oscillante lo spostamento di fase tra ΔG e la velocità determina il valore della dissipazione. In reazioni con produzione di energia, sia la dissipazione che l'energia prodotta dipendono dalla frequenza e dall'ampiezza della perturbazione oscillante del reagente.
La variazione di energia libera di Gibbs di una reazione, ad esempio in una pompa a protoni per il trasferimento di un protone da un potenziale chimico basso a uno più alto, è proporzionale alla differenza di potenziale ai due lati della membrana, secondo quanto stabilito dall'equazione di Nernst. La velocità di pompaggio dei protoni attraverso la membrana si identifica con una corrente e l'energia prodotta dalla membrana è il prodotto del voltaggio per la corrente, ovvero l'energia libera di Gibbs che corrisponde a quel voltaggio moltiplicata per la velocità di una pompa a protoni. Per una certa energia immessa, quella prodotta potrebbe essere alterata nei sistemi non lineari attraverso periodiche variazioni esterne delle condizioni, come ad esempio una concentrazione variabile, anziché costante, di ATP. Questo è un esempio di chimica della corrente alternata. Esiste pertanto la possibilità di variare l'efficienza, le concentrazioni nello stato stazionario, la dissipazione e la produzione di energia in sistemi non lineari di reazioni chimiche mediante perturbazioni delle condizioni esterne, come le concentrazioni dei reagenti. La comparsa di reazioni oscillanti in sistemi viventi potrebbe essere stata favorita dai vantaggi evolutivi rappresentati dalle variazioni di efficienza e concentrazione nelle specie al variare delle condizioni esterne. Nei sistemi in stato stazionario con un decadimento non oscillante verso questo stato (nodo stabile), le efficienze, così come le altre grandezze citate, diminuiscono se le condizioni esterne, anziché essere costanti, variano. La capacità di variare la concentrazione media nello stato stazionario senza variare l'immissione media di carburante consente ai sistemi biologici di adattarsi a richieste variabili anche in presenza di gradienti di concentrazione fissi e senza variare radicalmente la quantità di carburante necessario. Il sistema può quindi adattarsi velocemente a una variazione della concentrazione necessaria a formare un determinato prodotto biologico senza cambiare la quantità media di carburante consumata.
7. Funzioni logiche e calcolatori ottenuti mediante cinetiche chimiche macroscopiche
Si consideri un meccanismo di reazione astratto (v. fig. 17) in cui Aj e Bj sono, per esempio, la forma attiva e quella inattiva di un enzima, come accade in molti meccanismi di reazioni biochimiche. Se un tale meccanismo viene portato a operare in condizioni lontane dall'equilibrio, allora la concentrazione di stato stazionario, poniamo, di Aj si sposta nettamente da un valore basso a un valore elevato via via che varia la concentrazione dell'enzima, Cj. Questo tipo di variazione - che può verificarsi solo se il sistema di reazione è lontano dall'equilibrio - è analogo a quello che si riscontra in un neurone, il quale può essere attivo, allorché c'è passaggio di segnale, oppure inattivo, quando non passa alcun segnale. Pertanto, meccanismi di reazione di questo tipo sono detti ‛neuroni chimici' (v. Okamoto e altri, 1987). L'accoppiamento di neuroni chimici può aver luogo attraverso connessioni di natura sia eccitatoria che inibitoria stabilite attraverso un coenzima, Eij. Considerando connessioni di questo tipo tra due neuroni chimici e un terzo neurone, possiamo costruire delle porte logiche (AND, OR, NOR e altre); ad esempio in una porta AND la concentrazione di A nel neurone efferente risulterà elevata se, e solo se, lo sarà anche nei neuroni afferenti. Con porte logiche chimiche costruite sulla base di cinetiche macroscopiche è possibile progettare un decodificatore binario e un sommatore binario; la loro combinazione costituisce una macchina a stati finiti, nota anche come calcolatore universale di Turing, che può effettuare qualunque calcolo. Il calcolatore di Turing viene completato con nastri di ingresso e di uscita, detti anche cataste; questi apparecchi possono essere costruiti anche usando neuroni chimici (v. Hjelmfelt e altri, 1992).
In una macchina parallela a rete neuronale costituita da neuroni chimici accoppiati in maniera inibitoria, gli stati stazionari del sistema chimico vengono determinati da forze di accoppiamento tra neuroni scelte in precedenza. In corrispondenza di una certa condizione iniziale, per esempio un modello vicino a uno degli stati stazionari, il sistema riconosce il modello preselezionato più vicino, avvicinandosi allo stato stazionario di quel modello. Questo approccio costituisce l'attuazione chimica delle reti neuronali (v. Hjelmfelt e Ross, 1992). Reattori ad agitazione continua tra i quali è possibile scambio di massa, con una reazione chimica bistabile in ciascuno di essi (cioè due possibili stati stazionari stabili), possono servire come congegni per il riconoscimento di modelli. I due reattori sono connessi da tubi per il trasferimento di massa e i coefficienti della velocità di trasferimento vengono determinati attraverso l'applicazione di una regola di Hebb, che viene derivata dai modelli che devono essere riconosciuti dal sistema. Sia stati stazionari multipli che trasferimenti di materia sono di facile riscontro nei sistemi biologici. Riconoscimento di modelli, caos e multistabilità possono aver luogo nei sistemi eccitabili accoppiati da scambio di massa (v. Hjelmfelt e Ross, 1993). In questo caso le funzioni di calcolo e il riconoscimento di modelli vengono effettuati durante la transitoria escursione del sistema nello spazio delle concentrazioni piuttosto che quando viene raggiunto uno stato stazionario. Pertanto questo sistema effettua i calcoli in maniera ben diversa rispetto alle reti neuronali usuali.
L'attuazione di funzioni di calcolo attraverso cinetiche macroscopiche può essere utile per molti scopi. Il primo riguarda il problema dei meccanismi di calcolo nei sistemi viventi, incluso il cervello umano. Qualunque sia il meccanismo, esso può essere soltanto di natura chimica; in tale ambito, il meccanismo può essere basato essenzialmente su caratteristiche molecolari, come ad esempio le diverse configurazioni di una proteina, oppure può essere dovuto a cinetiche macroscopiche. Gli studi descritti aprono la possibilità che le funzioni di calcolo siano effettuate sulla base di cinetiche macroscopiche. Il secondo scopo è collegato con lo studio dei meccanismi delle reazioni chimiche. Se facendo funzionare i meccanismi di reazione in condizioni lontane dall'equilibrio è possibile eseguire funzioni di calcolo, ci si può chiedere se in meccanismi di reazione noti, e in particolare nei meccanismi delle reazioni biochimiche, possano svolgersi funzioni di calcolo, e in questo caso che ruolo abbiano e a cosa servano. Terzo, se meccanismi di reazioni chimiche contengono componenti di calcolo, allora la difficile analisi di meccanismi complessi potrebbe essere portata avanti attraverso l'applicazione di esperimenti che stabiliscano tavole di verità e funzioni logiche per il meccanismo e correlazioni tra specie reagenti.
8. La teoria termodinamica e stocastica di sistemi non lineari lontani dall'equilibrio.
Questo complesso argomento è ancora in fase di sviluppo e per esso sono stati suggeriti diversi approcci (v. Glansdorff e Prigogine, 1971; v. Keizer, 1978; v. Stratanovich, 1992; v. Graham e Tèl, 1985). Una rassegna completa, in particolar modo una rassegna comparativa, esula dallo scopo di questo articolo. Comunque, per sistemi a una sola variabile, tipo il modello di Schlögl descritto dalle reazioni (1) e (2), sono stati compiuti dei progressi sostanziali (v., ad esempio, Chu e altri, 1993) Una teoria dei sistemi lontani dall'equilibrio può essere basata sul concetto di affinità specie-specifica per la variabile intermedia X, così come nel modello di Schlögl:
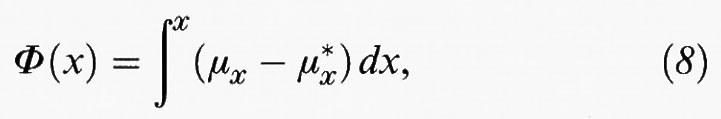
dove μx indica il potenziale chimico e μx* il suo valore in uno stato di riferimento corrispondente allo stato stazionario dell'equivalente sistema lineare con lo specifico valore x* di x.
Questa quantità rappresenta anche un lavoro in eccesso espresso in termini della cinetica della reazione diretta e dell'inversa
μx - μx* = - kT ln (tx+/tx-) (9)
essendo tx+ e tx- i flussi chimici che rispettivamente aumentano e diminuiscono il valore di x in un sistema a volume costante. Si può interpretare x* come la concentrazione di X in uno stato stazionario di un sistema lineare equivalente al sistema non lineare, sia termodinamicamente che cineticamente, per il dato valore, x*, di x.
La funzione Φ ha le seguenti proprietà: 1) è un estremante in corrispondenza di ogni stato stazionario e il segno della sua derivata seconda determina la stabilità dello stato stazionario corrispondente; queste sono condizioni necessarie e sufficienti per la stabilità globale di tutti gli stati stazionari; per ogni variazione rispetto a uno stato stazionario, δΦ > 0 e pertanto Φ fornisce un criterio termodinamico per ogni stato stazionario; 2) la funzione Φ rappresenta un lavoro in eccesso, una forza termodinamica che spinge verso stati stazionari stabili, e pertanto essa costituisce una funzione di Ljapunov nella forma compatibile con il teorema H di Boltzmann; 3) Φ è un componente della dissipazione totale ed è nullo in corrispondenza di stati stazionari stabili; 4) dΦ rappresenta un lavoro in eccesso misurabile e per sistemi lineari è un lavoro negativo, diverso dal lavoro pressione-volume, che si può ricavare dal rilassamento spontaneo dX a un punto X lungo la traiettoria deterministica meno il lavoro necessario a spostare di una pari quantità il sistema a partire da uno stato stazionario stabile; per un sistema non lineare è possibile fornire un'interpretazione simile; per un sistema che si avvicina allo stato stazionario di equilibrio, la funzione Φ è una variazione di energia libera di Gibbs o di Helmholtz, a seconda delle condizioni del sistema; 5) la funzione Φ fornisce un criterio per la stabilità relativa di due stati stazionari stabili; 6) la funzione Φ fornisce la distribuzione di probabilità stazionaria di un'equazione stocastica (la master equation che contiene la nascita e la morte nella cinetica chimica) corrispondente alla cinetica deterministica; la distribuzione stazionaria di quella master equation, data dalla funzione Φ, è una generalizzazione della formula delle fluttuazioni di Einstein per uno stato stazionario in condizioni lontane dall'equilibrio; per sistemi a una sola variabile la funzione Φ è una funzione di stato; 7) si possono derivare le correlazioni fluttuazione-dissipazione (v. Keizer, 1978; v. Vlad e Ross, 1994). Le sopra elencate proprietà forniscono una teoria termodinamica e stocastica per i sistemi a una variabile lontani dall'equilibrio.
BIBLIOGRAFIA
Berry, R. S., Rice, S. A., Ross, J., Physical chemistry, New York 1980.
Boiteux, A., Hess, B., Oscillations in glycolysis, cellular respiration and communication, in Physical chemistry of oscillatory phenomena. 9th Faraday symposium of the Chemical Society, London 1974, pp. 202-214.
Borderie, B., Lavabre, D., Micheau, J. C., Laplante, S. P., Nonlinear dynamics, multiple steady states, and oscillations in photochemistry, in ‟Journal of physical chemistry", 1992, XCVI, pp. 2953-2961.
Caplan, S. R., Naparstek, A., Zabusky, N. J., Chemical oscillations in a membrane, in ‟Nature", 1973, CCXLV, pp. 364-366.
Castets, V., Dulos, E., Boissonade, J., De Kepper, P., Experimental evidence of a sustained standing Turing-type nonequilibrium chemical pattern, in ‟Physical review letters", 1990, LXIV, p. 2953.
Chevalier, T., Schreiber, I., Ross, J., Towards a systematic determination of complex reaction mechanism, in ‟Journal of physical chemistry", 1993, XCVII, pp. 6776-6787.
Chu, X., Hunt, K.L.C., Hunt, P. M., Ross, J., Thermodynamic and stochastic theory of reaction-diffusion systems with multiple stationary states, in ‟Journal of chemical physics", 1993, XCIX, pp. 3444-3454.
Chu, X., Ross, J., Complex kinetics of systems with multiple stationary states, in ‟Journal of chemical physics", 1990, XCIII, pp. 1613-1625.
Cross, M. C., Hohenberg, P. C., Pattern formation outside of equilibrium, in ‟Reviews of modern physics", 1993, LXV, pp. 855-1112.
De Kepper, P. Boissonade, J., Epstein, I. R., Chlorite-iodide reaction: a versatile system for the study of nonlinear dynamical behavior, in ‟Journal of physical chemistry", 1990, XCIV, pp. 6525-6536.
Dolnik, M., Epstein, I. R., A coupled chemical burster: the chlorine dioxide-iodide reaction in two flow reactors, in ‟Journal of chemical physics", 1993, XCVIII, pp. 1149-1155.
Feinn, D., Ortoleva, P. J., Scalff, W., Schmidt, S., Wolff, M., Spontaneous pattern formation in precipitating systems, in ‟Journal of chemical physics", 1978, LXIX, pp. 27-39.
Field, R. J., Burger, M., Oscillations and traveling waves in chemical systems, New York 1985.
Fitz Hugh, R., Impulses and physiological states in models of nerve membrane, in ‟Biophysics", 1961, I, pp. 445-466.
Flicker, F., Ross, J., Mechanisms of chemical instability for periodic precipitation phenomena, in ‟Journal of chemical physics", 1974, LX, pp. 3458-3465.
Foerster, P., Zhang, Y.-X., Ross, J., Experiments on relative stability in the bistable multivariable bromate-ferroin reaction, in ‟Journal of physical chemistry", 1993, XCVII, pp. 4708-4713.
Ganapathisubramanian, N., Showalter, K., Bistability, mushrooms, and isolas, in ‟Journal of chemical physics", 1984, LXXX, pp. 4177-4184.
Gardiner, C. W., Handbook of stochastic methods, New York 19902.
Gilbert, R. G., Hahn, H.-S., Ortoleva, P. J., Ross, J., Interaction of sound with gas phase reactions, in ‟Journal of chemical physics", 1972, LVII, pp. 2672-2679.
Glansdorff, P., Prigogine, I., Thermodynamic theory of structure, stability and fluctuations, New York 1971.
Glass, L., Mackey, M. C., From clocks to chaos, Princeton, N. J., 1988.
Graham, R., Tèl, T., Weak-noise limit of Fokker-Planck models and nondifferentiable potentials for dissipative dynamic systems, in ‟Physical review. A", 1985, XXXI, pp. 1109-1122.
Gray, P., Griffiths, J. F., Hasko, S. M., Lignola, P. G., Oscillatory ignitions and cool flames accompanying the non-isothermal oxidation of acetaldehyde in a well stirred flow reactor, in ‟Proceedings of the Royal Society" (Series A), 1981, CCCLXXIV, pp. 313-339.
Gray, P., Scott, S. K., Chemical oscillations and instabilities: non-linear chemical kinetics, Oxford 1990.
Harding, R. H., Ross, J., Experimental measurement of the relative stability of two stationary states in optically bistable ZnSe interference filters, in ‟Journal of chemical physics", 1990, XCII, pp. 3579-3589.
Hjelmfelt, A., Ross, J., Chemical implementation and thermodynamics of collective neural networks, in ‟Proceedings of the National Academy of Sciences", 1992, LXXXIX, pp. 388-391.
Hjelmfelt, A., Ross, J., Mass-coupled chemical systems with computational properties, in ‟Journal of physical chemistry", 1993, XCVII, pp. 7988-7992.
Hjelmfelt, A., Ross, J., Pattern recognition, chaos, and multiplicity in neural networks of excitable systems, in ‟Proceedings of the National Academy of Sciences", 1994, XCI, pp. 63-67.
Hjelmfelt, A., Ross, J., Weinberger, E. D., Chemical implementation of neural networks and Turing machines, in ‟Proceedings of the National Academy of Sciences", 1991, LXXXVIII, pp. 10983-10987.
Hjelmfelt, A., Ross, J., Weinberger, E. D., Chemical implementation of finite-state machines, in ‟Proceedings of the National Academy of Sciences", 1992, LXXXIX, pp. 383-387.
Hynne, F., Møller, T., Sørensen, P. G., Complete optimization of models of the Belousov-Zhabotinski reaction at a Hopf bifurcation, in ‟Journal of chemical physics", 1993, XCVIII, pp. 219-230.
Kai, S., Müller, S. C., Ross, J., Periodic precipitation patterns in the presence of concentration gradients. 2. Spatial bifurcation of precipitation bands and stochastic pattern formation, in ‟Journal of physical chemistry", LXXXVII, pp. 806-813.
Kampen, N. G. van, Stochastic processes in physics and chemistry, Amsterdam 19892.
Kapral, R., Lawniczak, A., Masiar, P., Reactive dynamics in a multispecies lattice-gas automation, in ‟Journal of chemical physics", 1992, XCVI, pp. 2762-2776.
Keizer, J., Statistical thermodynamics of nonequilibrium processes, New York 1978.
Krischer, K., Eiswirth, M., Ertl, G., Periodic perturbations of the oscillatory CO oxidation on Pt (110): model calculations, in ‟Journal of chemical physics", 1992, XCVII, pp. 307-319.
Krischer, K., Lubke, M., Eiswirth, M., Wolf, W., Hudson, J. L., Ertl, G., A hierarchy of transitions to mixed mode oscillations in an electrochemical system, in ‟Physica D: nonlinear phenomena", 1993, LXII, pp. 123-133.
Lazar, J. G., Ross, J., Changes in mean concentration, phase shifts, and dissipation in a forced oscillatory reaction, in ‟Science", 1990, CCXLVII, pp. 189-192.
Lazar, J. G., Ross, J., Experiments on the effects of external periodic variation of constraints on the thermodynamics of an oscillatory system, in ‟Journal of chemical physics", 1990, XCII, pp. 3579-3589.
Lengyel, I., Epstein, I. R., Turing structures in simple chemical reactions, in ‟Accounts of chemical research", 1993, XXVI, pp. 235-240.
Li, R.-S., Ross, J., Chemical instabilities in closed systems with illumination, in ‟Journal of physical chemistry", 1991, XCV, pp. 2426-2430.
Lovett, R., Ortoleva, P. J., Ross, J., Kinetic instabilities in first order phase transitions, in ‟Journal of chemical physics", 1978, LXIX, pp. 947-955.
Luo, Y., Epstein, I. R., Feedback analysis for chemical oscillators, in ‟Advances in chemical physics", 1990, LXXIX, pp. 269-299.
Marek, M., Schreiber, I., Chaotic behavior of deterministic dissipative systems, Praha 1991.
Markus, M., Müller, S. C., Hess, B., Observation of entrainment, quasiperiodicity and chaos in glycolyzing yeast extracts under periodic glucose input, in ‟Berichte der Bunsengesellschaft für physikalische Chemie", 1985, LXXXIX, pp. 650-654.
Müller, S. C., Kai, S., Ross, J., Mesoscopic structure of pattern formation in initially uniform colloids, in ‟Journal of physical chemistry", 1982, LXXXVI, pp. 4294-4297.
Müller, S. C., Kai, S., Ross, J., Periodic precipitation patterns in the presence of concentration gradients. 1. Dependence of ion product and concentration differences, in ‟Journal of physical chemistry", 1982, LXXXVI, pp. 4078-4087.
Murray, J. D., Mathematical biology, Berlin 1988.
Nitzan, A., Ortoleva, P. J., Ross, J., Nucleation in systems with multiple stationary states, in Physical chemistry of oscillatory phenomena. 9th Faraday symposium of the Chemical Society, London 1974, pp. 241-253.
Noyes, R. M., The current state of chemical oscillators, in ‟Reaction kinetics and catalysis letters", 1990, XLII, pp. 169-180.
Okamoto, M., Sakai, T., Hayashi, K., Switching mechanism of a cyclic enzyme system: role as a ‟chemical diode", in ‟BioSystems", 1987, XXI, pp. 1-11.
Olsen, R. J., Epstein, I. R., Bifurcation analysis of chemical reaction mechanisms. I. Steady state bifurcation structure, in ‟Journal of chemical physics", 1991, XCIV, pp. 3083-3095.
Ortoleva, P. J., Nonlinear chemical waves, New York 1992.
Perelson, A. S., Goldstein, B., Dembo, M., Jacquez, J. A. (a cura di), Nonlinearity in biology and medicine: proceedings of the seventh annual international conference of the Center for Nonlinear Studies, Los Alamos, N. M., 1987, Amsterdam 1988.
Pugh, S., Schell, M., Ross, J., Effects of two periodic perturbations on the oscillatory combustion of acetaldehyde, in ‟Journal of chemical physics", 1986, LXXXV, pp. 868-878.
Rapp, P. E., An atlas of cellular oscillators, in ‟Journal of experimental biology", 1979, LXXXI, pp. 81-281.
Ross, J., Müller, S. C., Vidal, C., Chemical waves, in ‟Science", 1988, CCXL, pp. 460-465.
Ross, J., Schell, M., Thermodynamic efficiency in nonlinear biochemical reactions, in ‟Annual review of biophysics and biophysical chemistry", 1987, XVI, pp. 401-422.
Samples, M. S., Hung, Y. F., Ross, J., Further experimental studies on the horseradish peroxidase-oxidase reaction, in ‟Journal of physical chemistry", 1992, XCVII, pp. 7338-7342.
Sander, M., Imbihl, R., Ertl, G., Kinetic oscillations in catalytic CO oxidation on a cylindrical Pt single crystal surface, in ‟Journal of chemical physics", 1992, XCVIII, pp. 5193-5204.
Schneider, F. W., Muenster, A. F., Chemical oscillations, chaos, and fluctuations in flow reactors, in ‟Journal of physical chemistry", 1991, XCV, pp. 2130-2138 .
Sevickova, H., Marek, M., Waves and patterns in chemical and biological media, in ‟Physica D: non linear phenomena", 1991, pp. 114-124.
Steinbock, O., Zykov, V., Müller, S. C., Control of spiral-wave dynamics in active media by periodic modulation of excitability, in ‟Nature", 1993, CCCLXVI, pp. 322-324.
Stratanovich, R. L., Nonlinear nonequilibrium thermodynamics, vol. I, Berlin 1992.
Tsujimoto, K. K., Hjelmfelt, A., Ross, J., Measurements and calculations of oscillations and phase relations in the driven gas-phase combustion of acetaldehyde, in ‟Journal of chemical physics", 1991, XCV, pp. 3113-3223.
Turing, A. M., Chemical basis of morphogenesis, in ‟Philosophical transactions of the Royal Society" (Series B), 1952, CCXXXVII, pp. 37-72.
Tyson, J. J., Classification of instabilities in chemical reaction systems, in ‟Journal of chemical physics", 1975, LXII, pp. 1010-1015.
Vidal, C., Order within chaos, New York 1986.
Vidal, C., Lemarchand, H., La réaction créatrice. Dynamique des systèmes chimiques, Paris 1988.
Vlad, M. O., Ross, J., Fluctuation-dissipation relations for chemical systems far from equilibrium, in ‟Journal of chemical physics", 1994, C, pp. 7268-7278.
Winfree, A. T., Scroll-shaped waves of chemical activity in three dimensions, in ‟Science", 1973, CLXXXI, pp. 937-939.
Winfree, A. T., When time breaks down. The three-dimensional dynamics of electrochemical waves and cardiac arrhythmias, Princeton, N. J., 1987.
Wood, P. M., Ross, J., A quantitative study of chemical waves in the Belousov-Zhabotinsky reaction, in ‟Journal of chemical physics", 1985, LXXXII, pp. 1924-1936.
Zeeman, E. Z., Oscillations in potential in the normal human heart, in Towards a theoretical biology; an IUBS symposium (a cura di C. H. Waddington), Edinburgh 1972, pp. 8-67.
Zimmerman, E. C., Schell, M., Ross, J., Stabilization of unstable states and oscillatory phenomena in an illuminated thermochemical system: theory and experiment, in ‟Journal of chemical physics", 1984, LXXXI, pp. 1327-1336.