
Coagulazione
Coagulazione
La coagulazione è la trasformazione di un liquido in una sostanza gelatinosa o solida per l'azione di agenti chimici o fisici. La coagulazione del sangue è il fenomeno che determina, attraverso fasi successive e con l'intervento di numerose sostanze chimiche (fattori della coagulazione) attivate a cascata, la formazione di un reticolo insolubile di fibrina o 'coagulo' (dal latino cogere, co-agere, "stringere insieme"). Il processo della coagulazione è normalmente seguito da quello della fibrinolisi. La concentrazione nel sangue dei fattori della coagulazione può essere aumentata o ridotta (con meccanismo congenito o acquisito), dando luogo a patologie di notevole impatto sociale.
Inquadramento introduttivo

Fuori dell'albero circolatorio, per es. in un contenitore di vetro, il sangue coagula in pochi minuti; successivamente il coagulo si contrae ed esprime il siero, liquido incoagulabile, ricco di proteine, ioni e altre sostanze biologiche; per evitare la coagulazione del sangue prelevato per motivi di studio (esami di laboratorio) o a scopo di trasfusione, è necessario aggiungere una sostanza anticoagulante (per es., citrato di sodio, eparina). In vivo il sangue può andare incontro a coagulazione all'interno di un vaso, che abbia perso la sua integrità anatomica o funzionale. Nel primo caso la coagulazione del sangue contribuisce al processo fisiologico dell'emostasi, con conseguente arresto dell'emorragia. Nel secondo caso, invece, la coagulazione contribuisce alla formazione patologica del trombo, con conseguente ostruzione totale o parziale del lume vascolare (trombosi). La base per lo sviluppo del processo emostatico e della coagulazione del sangue è costituita dall'interazione delle piastrine con la parete vascolare. Le piastrine (300.000/mm3 di sangue), le più piccole cellule ematiche circolanti, sono capaci di aderire rapidamente alle strutture sottoendoteliali della parete vascolare alterata o danneggiata (tab. 1). Il collageno e il fattore von Willebrand rappresentano le componenti vascolari critiche nel meccanismo dell'adesione piastrinica.I globuli rossi rivestono un ruolo importante nell'agevolazione del contatto fisico tra piastrine circolanti e parete vascolare, di contro i globuli bianchi (polimorfonucleati), che sono adesi alle cellule endoteliali, vengono a costituire un ulteriore elemento di attrazione per le piastrine, grazie a un complesso gioco di molecole adesive (selettine, integrine, immunoglobuline).
All'adesione alla parete vascolare alterata segue l'aggregazione delle piastrine, un fenomeno che porta alla formazione di una massa cellulare in grado di occludere (parzialmente o totalmente) il lume vascolare. Se la lesione vascolare è di tipo emorragico, la formazione del 'tappo' piastrinico è sufficiente per un primo effetto emostatico, che risulterebbe però provvisorio se nei minuti e nelle ore successivi non si attivasse il processo della coagulazione del sangue, con conseguente formazione e deposito di fibrina sulla superficie delle piastrine aggregate. Le piastrine non danno inizio alla coagulazione ma, accumulandosi rapidamente sulla lesione vascolare, permettono la deposizione preferenziale di fibrina a livello della lesione stessa, risparmiando invece le superfici vascolari rimaste integre. La formazione e il deposito locali di fibrina possono essere ulteriormente stimolati dalla presenza di globuli bianchi mononucleati (monociti) all'interno e alla periferia del tappo emostatico.
Meccanismo della coagulazione del sangue
Il processo della coagulazione è descritto tradizionalmente come una 'cascata', il cui inzio è attribuibile a due meccanismi alternativi confluenti in una via comune. Nella cosiddetta via intrinseca (tutti i fattori partecipanti sono presenti nel sangue), la coagulazione comincia con il contatto del fattore XII con superfici cariche negativamente (per es. il vetro). Nella via estrinseca invece, è la tromboplastina o fattore tissutale (TF) che viene resa disponibile al fattore VII circolante. La via comune, infine, parte dall'attivazione del fattore X in fattore X attivato (Xa) ottenuta da entrambe le vie alternative, e porta alla formazione di fibrina.
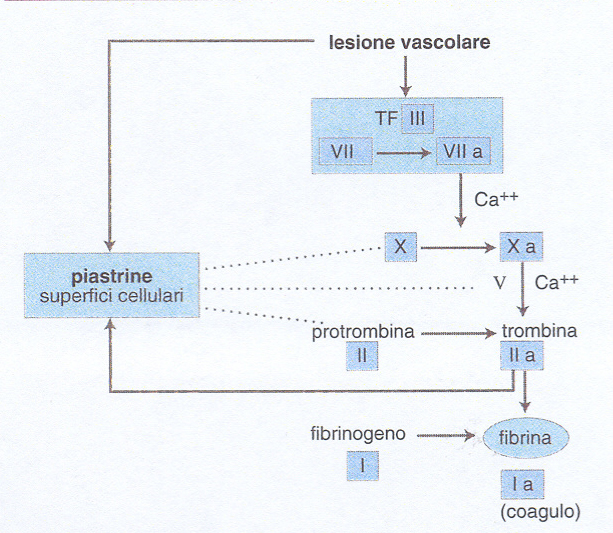

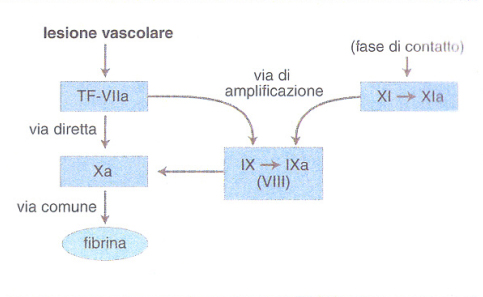

Un concetto fondamentale è quello della cascata che amplifica il processo della coagulazione, grazie a una sequenza di reazioni biochimiche che trasformano un precursore inattivo in un enzima attivo, il quale a sua volta attiva lo zimogeno successivo fino alla formazione della fibrina. L'identificazione dei fattori della coagulazione con numeri romani risale alla riunione di un Comitato internazionale svoltasi a Roma nel 1958, e non corrisponde alla sequenza di intervento dei fattori stessi nel processo di coagulazione. Più recentemente, lo schema della coagulazione è stato rielaborato tenendo in maggior conto i dati clinici rispetto a quelli ottenuti in provetta.Nell'organismo vivente, dunque, la coagulazione del sangue consegue a una lesione vascolare grave che, oltre a indurre i meccanismi cellulari dell'emostasi, rende disponibile il TF, complesso lipoproteico indispensabile per iniziare la cascata della coagulazione (fig. 1). Il TF (fattore III) è prodotto principalmente dalle cellule sottoendoteliali, ma anche da cellule endoteliali e monociti macrofagi attivati da numerose sostanze fisiologiche o patologiche (tab. 2). Il TF attiva il fattore VII (VIIa) presente nel plasma e forma con esso un complesso stechiometrico 1:1 che a sua volta attiva il fattore X in Xa: questo è il passaggio centrale della cascata che prosegue rapidamente in presenza di fattore V con l'attivazione della protrombina (II) in trombina (IIa), l'enzima specifico che trasforma il fibrinogeno (I) solubile in fibrina insolubile. Questa via diretta di attivazione della coagulazione è normalmente rafforzata da una via di amplificazione più lenta che coinvolge i due fattori antiemofilici (fig. 2): infatti il complesso TF-VIIa attiva anche il fattore IX →IXa, in presenza di fattore VIII che funge da cofattore della reazione (il fattore IX può essere anche attivato dal fattore XI, a sua volta attivato da meccanismi ancora mal definiti in vivo). Il fattore IXa è in grado di attivare il fattore X → Xa, amplificando così le reazioni ulteriori che determinano la formazione di fibrina. Questo schema della coagulazione, a differenza di quelli precedenti, integra tutti i fattori noti in un'unica via di attivazione - iniziata dal complesso TF-VIIa - e spiega perché un deficit grave di TF sia incompatibile con la vita e quello di fattore VII sia molto raro, mentre i numerosi pazienti affetti da emofilia A o B, pur avendo deficit severi di VIII o IX, soffrano di sindromi emorragiche gravi, ma non necessariamente fatali (v. emorragia). Le cellule ematiche quali piastrine e monociti - insieme alle cellule endoteliali della parete interna del vaso - forniscono ai fattori della coagulazione una superficie catalitica che contribuisce alla localizzazione e all'amplificazione del processo di coagulazione. È stato calcolato che, sulla superficie endoteliale, una molecola del complesso TF-VIIa può generare in un minuto 2,5∃105 molecole di trombina e 3∃109 molecole di fibrina. Alcuni fattori ritenuti favorire la coagulazione in vitro (tab. 3) non sono riportati nelle figg. 1 e 2, perché il loro significato clinico non appare al momento rilevante. Essi costituiscono la cosiddetta fase di contatto della coagulazione. Il processo della coagulazione è controllato da alcuni inibitori fisiologici: antitrombina III, cofattore eparinico II e il sistema della trombomodulina-proteina C-proteina S. L'azione di tali inibitori si esplica in modo diverso: l'antitrombina III forma complessi con la trombina e il fattore Xa, reazioni amplificate dall'eparina e da altre sostanze anticoagulanti eparino-simili. Il cofattore eparinico II potenzia invece l'effetto anticoagulante dell'eparina come inibitore della trombina, ma non del fattore Xa. La trombina che si forma durante la coagulazione si lega alla trombomodulina, proteina sintetizzata dalle cellule endoteliali, alla quale è legata anche la proteina C, che risulta così attivata. La proteina C attivata, che utilizza la proteina S come cofattore di amplificazione, blocca l'attivazione dei due cofattori della coagulazione, il fattore VIII e il fattore V. In assenza di inibitori, la trombina che si genera in 1 ml di sangue in meno di 15 s potrebbe coagulare tutto il fibrinogeno circolante (3 g/l).
Più recentemente è stato identificato un inibitore del complesso TF/VIIa/Xa (TFPI, Tissue factor pathway inhibitor). A differenza degli altri inibitori fisiologici della coagulazione, il TFPI si ritrova in gran parte legato alla parete vascolare. È stato finora impossibile individuare pazienti con un'alterazione plasmatica misurabile di questo inibitore, la cui funzione fisiopatologica resta perciò tutta da chiarire. Quando il complesso TF/VIIa attiva il X, il Xa che si forma è capace di indurre l'attività inibente del TFPI: così la generazione di Xa è autolimitata e il processo di coagulazione si rallenterebbe o si arresterebbe se il TF/VIIa non avesse nel frattempo attivato la via di amplificazione dei fattori antiemofilici IX e VIII e se questa via non fosse attivata indipendentemente anche dal fattore XI (fig. 2). I livelli plasmatici dei fattori della coagulazione (e della fibrinolisi) sono regolati in gran parte dall'interazione di determinanti genetiche e di fattori ambientali (alimentazione, fumo di sigaretta, infezioni ecc.). All'interno di una popolazione, la variabilità genetica legata a polimorfismi allelici può spiegare, almeno in parte, la variabilità e la distribuzione (normalmente di tipo gaussiano o a campana) dei livelli plasmatici dei fattori della coagulazione (e della fibrinolisi). Recentemente, alcuni polimorfismi dei geni del fibrinogeno, del fattore VII e del PAI-1 (Plasminogen activator inhibitor, un inibitore della fibrinolisi; v. oltre) sono stati associati a livelli plasmatici aumentati o ridotti dei corrispondenti fattori. Anche la risposta a situazioni ambientali (per es. l'aumento di fibrinogeno plasmatico nei pazienti affetti da infezione da Helicobacter pylori) sembra essere geneticamente determinata. La fibrina depositata sulle cellule accumulate nella zona della lesione vascolare diventa a sua volta superficie catalitica e cofattore del sistema fibrinolitico. Il sistema fibrinolitico comprende un insieme complesso di enzimi e di inibitori che porta alla formazione di plasmina dal precursore plasminogeno. Il termine fibrinolisi deriva dalle prime osservazioni che limitavano l'effetto della plasmina alla degradazione o lisi della fibrina. Oggi sappiamo che la plasmina e l'intero sistema fibrinolitico sono coinvolti in diversi processi fisiopatologici, quali la migrazione dei macrofagi, l'ovulazione e l'impianto della blastocisti, l'involuzione della ghiandola mammaria e la disseminazione dei tumori.
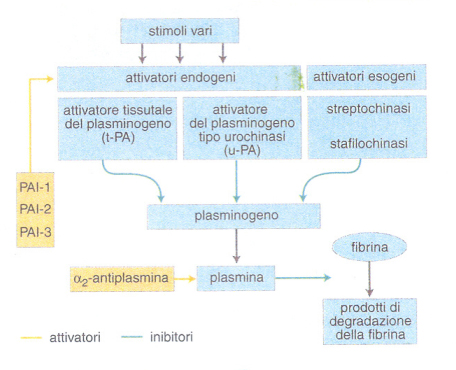

La fibrinolisi è il maggiore sistema di difesa contro la deposizione di fibrina nell'albero vascolare e quindi riveste un ruolo fondamentale nella protezione contro un eccesso di coagulazione o un evento coagulativo che si potrebbe verificare nel momento o nel posto sbagliato (trombosi). La coagulazione e la fibrinolisi esistono fisiologicamente in uno stato di reciproco equilibrio compensato. Se questo equilibrio viene disturbato, può svilupparsi una trombosi o un'emorragia.Il sistema fibrinolitico è innescato in vivo dall'attivazione della coagulazione e dalla conseguente deposizione di fibrina. Quest'ultima è capace di concentrare sulla sua superficie i componenti del sistema fibrinolitico quali il plasminogeno, e l'attivatore tissutale del plasminogeno. La plasmina che si forma dal plasminogeno attivato scinde la fibrina nei suoi prodotti di degradazione.L'attivatore tissutale del plasminogeno è presente sull'endotelio di tutti i vasi sanguigni, dal quale viene rilasciato nel sangue in seguito a stimoli vari (per es., esercizio fisico, attivazione della coagulazione, farmaci; fig. 3). L'urochinasi, attivatore del plasminogeno, è presente nel plasma come precursore inattivo prourochinasi, finché non sia attivato dalla plasmina. La streptochinasi, invece, è un attivatore esogeno del plasminogeno, isolato da culture di streptococco β-emolitico. La streptochinasi forma un complesso stechiometrico 1:1 con il plasminogeno circolante, che diviene in tal modo capace di autoformare plasmina. Al contrario della streptochinasi, gli attivatori endogeni sono altamente specifici per la fibrina. Più recentemente è stato identificato un altro attivatore esogeno, derivante da colture di stafilococco aureo (stafilochinasi), che eserciterebbe in vivo un effetto relativamente fibrino-specifico. Tra gli inibitori, l'α₂-antiplasmina neutralizza rapidamente l'enzima attivo, mentre diversi 'inibitori dell'attivatore del plasminogeno' (PAI-1, PAI-2, PAI-3, nexinproteasi) possono regolare il sistema fibrinolitico e contribuire all'insorgenza di sindromi emorragiche o trombotiche. Un ulteriore meccanismo di controllo è esercitato da recettori o siti di legame per i vari fattori della fibrinolisi che appaiono distribuiti su cellule del sangue e della parete vascolare.In sintesi, le reazioni dei processi di coagulazione e di fibrinolisi avvengono tutte secondo uno schema costante: enzima-substrato-cofattore-superficie catalitica-prodotto (tab. 4).
Patologia della coagulazione
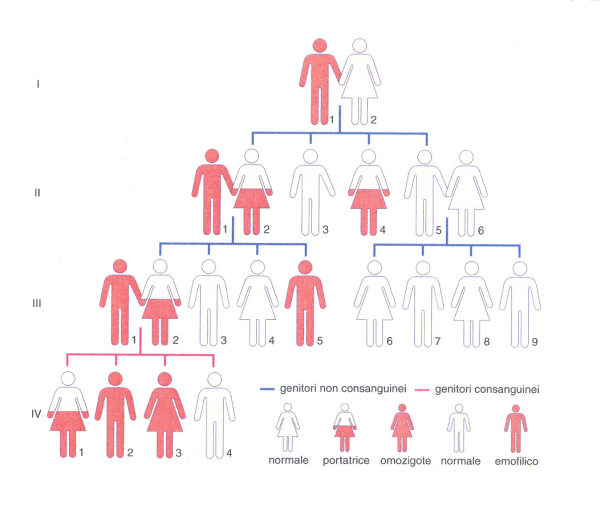
a) Emofilia A ed emofilia B. Le alterazioni della coagulazione possono essere congenite o acquisite. Tra le prime, la più nota sin dai tempi biblici è l'emofilia A, dovuta al difetto di un cofattore della coagulazione, il fattore VIII che interviene nell'attivazione del fattore IX (o fattore antiemofilico B; fig. 4). L'emofilia A (emofilia classica) e l'emofilia B (malattia di Christmas) costituiscono le malattie ereditarie della coagulazione più frequenti e importanti. Si calcola che in Italia i pazienti affetti da emofilia A siano più di 5000 e quelli affetti da emofilia B più di 1000. L'emofilia (A e B) colpisce non solo la specie umana, ma anche cani, gatti e cavalli. Le due malattie emorragiche sono indistinguibili dal punto di vista clinico e sono entrambe condizioni ereditarie recessive, con trasmissione legata al sesso e manifestazione (quasi) esclusivamente nei maschi (fig. 4). I figli maschi di un emofilico che sposi una donna normale sono sani, mentre le figlie sono portatrici sane. Se una portatrice sana sposa un uomo normale, i suoi figli maschi avranno la stessa possibilità statistica di essere sani o emofilici e le sue figlie di essere normali o portatrici sane. Il figlio sano di un emofilico che sposi una donna normale avrà tutti figli sani. Nei rari casi di un matrimonio (consanguineo) tra un emofilico e una portatrice sana potrà nascere anche una donna emofilica (omozigote). È da notare che il 30-40% di pazienti emofilici non presentano anamnesi familiare della malattia: ciò può essere dovuto sia a mutazioni genetiche (relativamente rare), sia al fatto che il difetto può essere trasmesso da donna portatrice a donna portatrice per varie generazioni finché, quando nasce un maschio ammalato, l'evento viene erroneamente attribuito a una mutazione.
b) Malattia di von Willebrand. È la malattia emorragica ereditaria più comune, ma non è primariamente un'alterazione coagulativa: il fattore von Willebrand, infatti, carente nella malattia, facilita l'adesione piastrinica alla parete vascolare alterata in condizioni di flusso relativamente veloce. Esso serve anche come vettore plasmatico del fattore VIII (antiemofilico), il livello del quale risulta perciò ridotto nel sangue dei pazienti affetti dalla malattia. Per questo essa venne descritta originariamente come pseudo-emofilia. La malattia di von Willebrand si trasmette in modo autosomico dominante e colpisce ugualmente maschi e femmine. Dal punto di vista laboratoristico e clinico, sono state distinte 3 forme varianti della malattia e numerosi sottotipi.
c) Altre alterazioni congenite della coagulazione. Altre rare affezioni congenite della coagulazione derivano dalla carenza più o meno grave di uno o più fattori della coagulazione. Da ricordare l'afibrinogenemia (carenza di fibrinogeno) e numerose forme di disfibrinogenemia caratterizzate da varie alterazioni funzionali della complessa molecola proteica. Sebbene il fibrinogeno sia necessario per l'aggregazione piastrinica e per la formazione di fibrina, paradossalmente i difetti di questa proteina non causano emorragie spontanee importanti.Come già ricordato, non sono stati osservati difetti congeniti del TF; animali sperimentalmente privati del gene del TF (knock-out) muoiono allo stato di embrione per gravi anomalie del tessuto vascolare ed emorragia. Anche il deficit congenito severo di fattore VII è una condizione talmente grave da essere stata riscontrata soltanto in un centinaio di soggetti (con grado variabile di livelli plasmatici di fattore VII). Il deficit di fattore X è ugualmente raro, soprattutto nella forma omozigote, così come quello di protrombina (fattore II). Il deficit di fattore XI è praticamente limitato agli ebrei ashkenazi e non comporta un difetto emorragico grave. Il fattore XIII, che rende il coagulo di fibrina più stabile e resistente alla lisi, è una condizione rara (autosomica recessiva) che non può essere diagnosticata con i comuni test di laboratorio: si richiede pertanto un forte sospetto clinico per giungere alla diagnosi del difetto. Tra i difetti congeniti combinati dei fattori della coagulazione, il più comune è quello dell'VIII e del V, entrambi cofattori rispettivamente della via di amplificazione e della via comune del processo coagulativo (fig. 2). Per tutti i fattori della coagulazione sono stati identificati polimorfismi e varianti genetiche relativamente frequenti e con distribuzione geografica non sempre uniforme, associati o meno a difetti funzionali (abitualmente di tipo emorragico). Polimorfismi del fibrinogeno e del PAI-1 sono stati associati ad aumentato rischio trombotico, mentre alcuni polimorfismi del fattore VII potrebbero essere protettivi contro la trombosi e l'infarto coronarico.
I difetti ereditari degli inibitori naturali della coagulazione, così come alcune alterazioni del sistema fibrinolitico, possono indurre uno stato di coagulazione non controllata (ipercoagulabilità) e predisporre a eventi trombotici (circa 10% dei casi di tromboembolia ricorrenti). I casi finora descritti sono tutti ereditati come tratti autosomici dominanti: mentre è abitualmente necessario un difetto grave dei fattori della coagulazione per indurre una sindrome emorragica clinicamente rilevante, la riduzione anche solo del 50% di un inibitore comporta un aumentato rischio di trombosi.
Più recentemente è stato introdotto il concetto di 'resistenza alla proteina C attivata' (APC), definita come una ridotta risposta anticoagulante del plasma a questo inibitore. La resistenza all'APC è un fenotipo relativamente frequente ed è stato riscontrato in circa il 40% dei casi di trombosi venosa (particolarmente nelle forme familiari). La base molecolare della resistenza risiede, nella maggior parte dei casi (80-95%), in una mutazione del gene del fattore V (fattore V Leiden, FVR506Q, dalla sostituzione dell'arginina-506 con acido glutammico). Il fattore V Leiden è più lentamente inattivato dall'APC, inducendo così un'elevata tendenza trombotica (rischio aumentato 3-8 volte negli eterozigoti e 30-140 volte negli omozigoti).

d) Coagulazione intravascolare disseminata. Tra i disordini emorragici acquisiti, il più drammatico è certamente la sindrome di coagulazione intravascolare disseminata (CID), nota anche come coagulopatia da consumo: liberazione di fattori tissutali (per es. da tumori necrotici o da incidenti ostetrici) o espressione di attività tissutale sulla superficie di cellule endoteliali e/o monociti da parte di endotossine (in corso di sepsi da batteri gram-negativi), scatenano il processo di coagulazione con deposizione diffusa di fibrina nella microcircolazione (tab. 5) e attivazione secondaria della fibrinolisi. Se la causa scatenante persiste, si osserva deplezione (consumo) dei fattori della coagulazione e delle piastrine con conseguente sintomatologia emorragica. Essendo una sindrome associata ad altre condizioni patologiche, la presentazione clinica della CID può variare molto, ma è caratterizzata da gravi sanguinamenti cutanei e mucosi che possono esporre il paziente al rischio di morte. Le alterazioni che si riscontrano più frequentemente sono: piastrinopenia, riduzione del fibrinogeno plasmatico e aumento dei prodotti di degradazione della fibrina (PDF) per l'intensa fibrinolisi secondaria. La terapia della CID deve tendere innanzitutto a rimuovere la causa scatenante; si deve inoltre controllare la sintomatologia emorragica con terapia sostitutiva ed eventualmente ridurre il consumo mediante farmaci anticoagulanti, in particolare eparina.
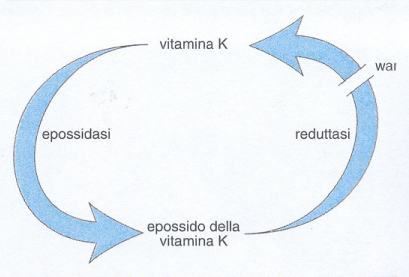
e) Altre alterazioni acquisite della coagulazione. La carenza di vitamina K comporta la riduzione di quattro fattori della coagulazione (VII, IX, X e II) e di due inibitori (proteine C e S). La vitamina K è infatti un cofattore della formazione di residui γ-carbossi-glutammici che rendono funzionali le proteine della coagulazione sopra menzionate. Cause principali di carenza di questa vitamina sono malassorbimento intestinale, dieta inadeguata e danno epatico. La somministrazione sistematica di vitamina K a tutti i neonati ha praticamente fatto scomparire nel nostro paese i frequenti deficit di vitamina K alla nascita e la conseguente sindrome emorragica. La terapia anticoagulante orale permanente con warfarin e altri farmaci cumarinici induce una condizione analoga a quella da deficit di vitamina K, in quanto previene la riduzione degli epossidi di questa vitamina nei microsomi epatici (fig. 5). Malgrado risultino inibiti dal warfarin fattori sia procoagulanti sia anticoagulanti, il sistema viene sbilanciato in senso emorragico/antitrombotico.
Altre affezioni della coagulazione si riscontrano in corso di malattie epatiche, poiché il fegato gioca un ruolo centrale nella sintesi e nel metabolismo della maggior parte dei fattori della coagulazione e dei suoi inibitori. Si aggiunga inoltre il ridotto assorbimento della vitamina K da colestasi e la piastrinopenia da splenomegalia conseguente a ipertensione portale. La terapia è essenzialmente sostitutiva con plasma fresco congelato.

In alcuni soggetti, più frequentemente in pazienti con deficit congenito di fattori della coagulazione sottoposti a trasfusioni ripetute, si riscontrano anticorpi che interferiscono con il processo coagulativo (anticoagulanti circolanti). Altri inibitori non-specifici possono alterare dei test di laboratorio, ma non il processo coagulativo in vivo: è il caso dell'anticoagulante lupico (malgrado il nome, si tratta di anticorpi antifosfolipidi, anticardiolipina, associati a condizioni di vasculite e rischio trombotico).Numerose patologie di frequente riscontro clinico possono essere associate a un aumento di rischio trombotico per uno squilibrio dei meccanismi di controllo della coagulazione (stato di ipercoagulabilità; tab. 6).
Sintomatologia clinica, diagnosi e trattamento
La componente plasmatica della coagulazione può essere valutata essenzialmente mediante due test di laboratorio (il tempo di protrombina, PT, e il tempo di tromboplastina parziale, PTT), ai quali si può aggiungere il tempo di trombina e il dosaggio del fibrinogeno. Nel PT si aggiunge al plasma anticoagulato con citrato di sodio una miscela di tromboplastina tissutale e calcio, riproducendo così le condizioni di attivazione della via diretta della coagulazione. Nel PTT, invece, si aggiunge una miscela di fosfolipidi, calcio e un fattore di contatto capace di attivare in vitro la via di amplificazione della coagulazione. Entrambi i test esplorano anche la via comune della coagulazione (fig. 2). Il PT risulta selettivamente prolungato quando sono carenti i fattori del complesso protrombinico vitamina K-dipendenti (VII, X, II), mentre un allungamento del PTT riflette un deficit di fattore VIII o IX (emofilia A o B). Quando entrambi i test sono patologici, si deve pensare a un difetto della via comune (fattori X, V, II e fibrinogeno). Sulla base dei risultati del PT e del PTT, si possono dosare i singoli fattori plasmatici allo scopo di giungere a una diagnosi specifica del difetto di coagulazione. Non sono attualmente disponibili esami di laboratorio di routine attraverso i quali identificare i pazienti con sospetta ipercoagulabilità o stati protrombotici acquisiti. Le sindromi da anticoagulanti circolanti vengono evidenziate quando l'aggiunta di plasma normale a quello del paziente non è sufficiente a riportare alla norma gli esami alterati.La raccolta di un'anamnesi accurata permette facilmente di sospettare un disordine ereditario della coagulazione. A differenza dei sanguinamenti legati a difetti vasopiastrinici, le emorragie da difetto della coagulazione si manifestano diverse ore o giorni dopo la lesione (estrazione dentaria, parto, piccola chirurgia), non sono sensibili alla terapia locale e si verificano soprattutto in tessuti sottocutanei, muscoli, articolazioni e cavità sierose (v. emorragia).
La terapia dei difetti congeniti della coagulazione è sostitutiva (plasma fresco congelato o concentrati di fattori specifici), quella dei difetti acquisiti deve tendere innanzitutto alla risoluzione della patologia sottostante e al controllo della sintomatologia emorragica predominante.
Bibliografia
a.l. bloom et al., Haemostasis and thrombosis, 2 voll., Edinburgh, Churchill Livingstone, 1994.
g. de gaetano, Piastrine, trombosi e aterosclerosi, Milano, Masson, 1983.
g. de gaetano, v. bertelé, Fisiopatologia dell'emostasi primaria, in Trattato italiano di medicina interna, diretto da P. Introzzi, 5° vol., Firenze, USES Edizioni Scientifiche, 1988, pp. 4253-69.
State of the art 1997, ed. G. de Gaetano, "Thrombosis and Haemostasis", 1997, 78, pp. 1-784.