
Corrosione
Corrosione
I materiali a contatto con ambienti aggressivi subiscono un degrado chimico e fisico che, per quanto riguarda in particolare i metalli, è denominato corrosione. La corrosione può essere dunque definita come la progressiva alterazione della superficie di un corpo da parte di agenti atmosferici o di altri mezzi aggressivi, che porta a una progressiva alterazione delle caratteristiche, spesso non soltanto superficiali, del materiale attaccato; o anche come la distruzione o il deterioramento di un materiale per reazione con l’ambiente; o ancora come la tendenza di un manufatto metallico a tornare al suo stato originale, nel quale si trova in natura. I processi di corrosione tendono infatti a portare spontaneamente i materiali metallici allo stato termodinamicamente più stabile, che è quello di combinazione con altri elementi, soprattutto ossigeno e zolfo. È in questo stato combinato che la grande maggioranza dei metalli si rinviene in natura nei corrispondenti minerali (fanno eccezione metalli molto nobili come oro e platino). Metalli quali il rame e il ferro si trovano in natura sotto forma metallica soltanto in piccole quantità (soprattutto il ferro), mentre molto più abbondanti sono, rispettivamente, gli ossidi e i solfuri. A partire da tale stato i materiali metallici sono ottenuti (estratti) tramite i processi metallurgici, che comportano una notevole somministrazione di energia. A seguito dei processi corrosivi, i metalli tornano a trasformarsi nei composti, più stabili, da cui provengono. Con riferimento a questo aspetto, la corrosione è detta anche antimetallurgia (metallurgy in reverse).
Un’idea dell’importanza della corrosione nelle attività industriali è data dall’impatto economico che essa esercita. Nei paesi industrializzati il costo della corrosione è stimato intorno al 3÷4% del prodotto interno lordo, valutato come somma dei costi dei danni diretti (per es., il costo dei materiali danneggiati che devono essere rimpiazzati e il costo dell’intervento di sostituzione) e di quelli indiretti (per es., il costo della perdita di produzione, il costo del disinquinamento ambientale, i danni a persone e cose, la perdita di immagine nella società). È stato stimato che il costo della corrosione può essere ridotto del 15÷20% mediante l’applicazione di tecniche che sfruttano le conoscenze di base della corrosione, quali la protezione catodica, i materiali inossidabili, l’uso di inibitori di corrosione, i rivestimenti.
La corrosione dei materiali metallici può essere classificata in due grandi categorie: la corrosione ad alta temperatura (o corrosione a caldo o anche corrosione a secco), tipica dei materiali che operano a elevata temperatura in presenza di gas caldi (per es., nelle caldaie lato fumi e nelle turbine a gas), e la corrosione a umido, caratteristica dei materiali che sono esposti a una soluzione elettrolitica (per es., l’acqua di mare, i terreni, il calcestruzzo inquinato da cloruri o carbonatato, i fluidi di processo). La distinzione tra corrosione a secco e corrosione a umido si deve al diverso meccanismo con cui il fenomeno ha luogo, nel primo caso di tipo chimico, basato su reazioni eterogenee, nel secondo di tipo elettrochimico.
Il processo corrosivo può colpire la superficie metallica in modo generalizzato, interessando l’intera superficie esposta all’ambiente, oppure in modo localizzato, cioè limitatamente ad alcune parti sensibili della superficie esposta.
Aspetti generali della corrosione
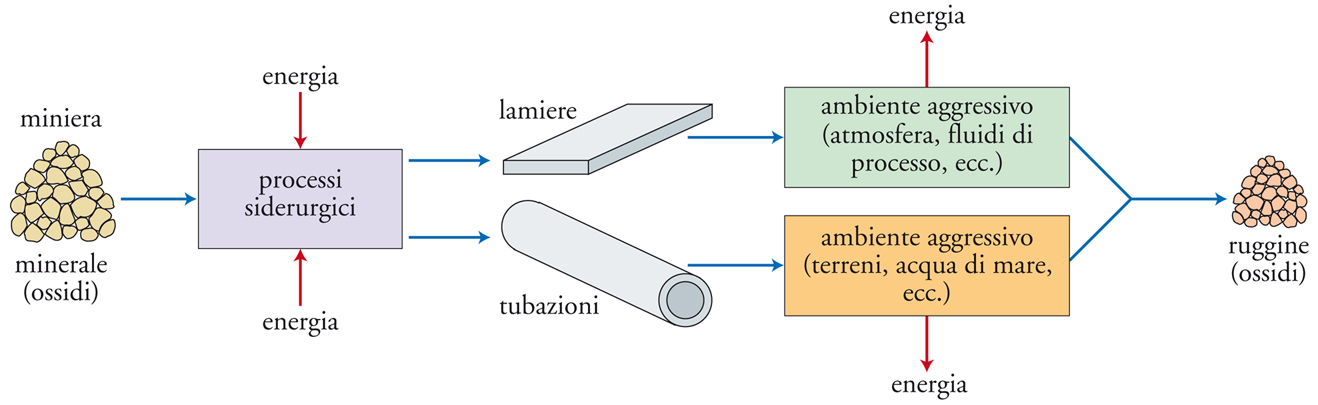
Dal punto di vista chimico-fisico, il risultato di un fenomeno corrosivo consiste nella trasformazione del metallo in un composto più stabile, che è generalmente la forma chimica nella quale il metallo è presente in natura come minerale, e dalla quale esso era stato ottenuto mediante un processo metallurgico. Di qui la denominazione di antimetallurgia talvolta usata per denotare il fenomeno della corrosione. La fig. 2 illustra schematicamente l’intero ciclo di trasformazione del materiale, a partire dal minerale originario fino ai prodotti del processo corrosivo.
Nel seguito, verranno illustrati alcuni aspetti fondamentali delle due grandi categorie dei fenomeni di corrosione: la corrosione a caldo e quella a umido.
Corrosione a caldo
La corrosione dei metalli a contatto con aria a temperature superiori ai 400 °C e fino a temperature di 1300°C è detta corrosione a caldo. La presenza di ossigeno provoca la formazione di una scaglia di ossido sulla superficie del metallo, mentre la presenza nei gas caldi di specie chimiche, quali zolfo, sodio e vanadio, porta alla formazione di sali con bassa temperatura di fusione che reagiscono con il metallo.
Per prevedere la formazione della scaglia e la sua crescita è necessario considerare le caratteristiche termodinamiche e la cinetica delle reazioni coinvolte. Le condizioni termodinamiche stabiliscono se la reazione di ossidazione procede spontaneamente alla temperatura di esercizio, mentre la cinetica determina la velocità con cui ha luogo il processo di crescita della scaglia.
Date le condizioni di esercizio piuttosto al limite, spesso con rischio di conseguenze catastrofiche a seguito di una rottura, la scelta dei materiali richiede in generale una maggiore attenzione rispetto alle applicazioni a basse temperature.
Cinetica della corrosione a caldo. - Poiché le condizioni termodinamiche mostrano che la corrosione a caldo è spontanea per i metalli e le leghe industriali, è necessario analizzare le condizioni cinetiche di crescita della scaglia. Il meccanismo di crescita comprende: (a) l’adsorbimento dell’ossigeno sulla superficie del metallo; (b) la formazione di un nucleo di ossido che si estende su tutta la superficie; (c) la crescita dello spessore del film.
La nucleazione dell’ossido è favorita nei siti a elevata energia quali i difetti di superficie (dislocazioni, bordi di grano, precipitati) ed è influenzata dal trattamento superficiale, dalla temperatura e dalla pressione parziale dell’ossigeno. Una volta formato il film su tutta la superficie, la sua crescita procede attraverso i processi di diffusione allo stato solido nella scaglia.
La valutazione della capacità protettiva di un ossido viene compiuta in modo qualitativo mediante il rapporto di Pilling-Bedworth, definito come il rapporto tra il volume dell’ossido e quello del metallo che l’ha prodotto. Se tale rapporto è inferiore a 1 o maggiore di 2,5 l’ossido non è protettivo; nel primo caso perché non è sufficiente per ricoprire il metallo, nel secondo caso perché esso tende a staccarsi a causa delle tensioni di compressione che si originano durante la crescita.
Quando lo strato di ossido è protettivo, la sua crescita dipende dai processi di diffusione degli ioni ossigeno O2- e degli ioni metallici. La velocità di crescita è proporzionale al flusso più lento degli ioni in gioco.
I metalli che formano ossidi dotati di crescita sufficientemente lenta e quindi idonei per contrastare la corrosione a caldo sono i seguenti: (a) il ferro, che forma tre ossidi stabili (ematite Fe2O3, magnetite Fe3O4 e wurstite FeO), protettivi solo a temperature inferiori a 570 °C; (b) il cromo, che forma l’ossido Cr2O3, protettivo fino a 900 °C; (c) il nichel, che forma l’ossido NiO, stabile a temperature oltre i 900 °C; (d) l’alluminio, che forma l’ossido Al2O3, molto stabile e protettivo fino a 1300 °C; (e) il silicio, che forma l’ossido SiO2, molto stabile e protettivo fino a 1200 °C.
Corrosione a umido
Una generica reazione di corrosione per un materiale metallico M può essere schematizzata con la seguente reazione:
[1] M+ambiente aggressivo ←→ prodotti di corrosione di M
dove M è un generico materiale metallico. Quando l’ambiente è una soluzione elettrolitica, la reazione globale di corrosione [1] implica un processo di ossidazione del metallo accompagnato dalla reazione di riduzione dell’ossigeno disciolto nella soluzione, secondo lo schema seguente, scritto per il caso del ferro:
[2] ferro + ossigeno + acqua ←→ prodotti di corrosione
oppure un secondo processo, tipico delle soluzioni acide, in cui ha luogo la riduzione dell’idrogenione, secondo la reazione, sempre nel caso del ferro:
[3] ferro + soluzione acida ←→ ioni di ferro + idrogeno.
Queste due reazioni procedono secondo un meccanismo elettrochimico in cui sono coinvolti gli elettroni del materiale metallico. La reazione è la somma di due processi elettrodici complementari: (a) un processo anodico che implica l’ossidazione del materiale metallico e rende disponibili elettroni nella fase metallica; (b) un processo catodico che consuma gli elettroni, resi disponibili dal processo anodico, mediante una reazione di riduzione (dell’ossigeno molecolare come gas disciolto nella soluzione o dell’idrogenione o di entrambi).
Poiché deve essere mantenuta l’elettroneutralità, le due reazioni devono prodursi simultaneamente e con la stessa velocità.
Le reazione catodica in una soluzione acida è la riduzione dello ione idrogeno con produzione di idrogeno molecolare, secondo lo schema:
[4] formula
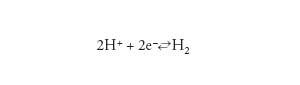
dove e− indica l’elettrone.
Negli ambienti naturali, la reazione catodica di gran lunga più importante è la reazione di riduzione dell’ossigeno, in ambiente neutro o basico:
[5] formula.
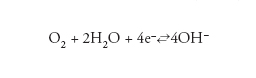
Poiché il meccanismo è elettrochimico, si possono applicare le leggi di Faraday che stabiliscono la relazione tra le masse e la carica elettrica circolante (numero di elettroni) attraverso gli equivalenti elettrochimici. L’equivalenza tra la velocità di corrosione espressa in mA/m2 e mm/a per metalli bivalenti con densità di circa 8 Mg/m3 (per es., Fe, Zn e Cu) è 1 mA/m2≈1 μm/a (per il ferro, il valore preciso è 1 mA/m2=1,17 μm/a).
Aspetti termodinamici. - Se indichiamo con Ea il potenziale della reazione anodica (dissoluzione del metallo) e con Ec il potenziale della reazione catodica (riduzione dell’ossigeno o sviluppo di idrogeno) la condizione termodinamica di spontaneità del processo di corrosione è:
[6] formula
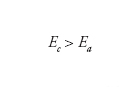
ossia la reazione di corrosione è spontanea se il potenziale della reazione catodica è più nobile del potenziale della reazione anodica.
Il potenziale della reazione anodica (dissoluzione del metallo) è dato dalla legge di Nernst:
[7] formula
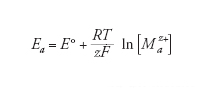
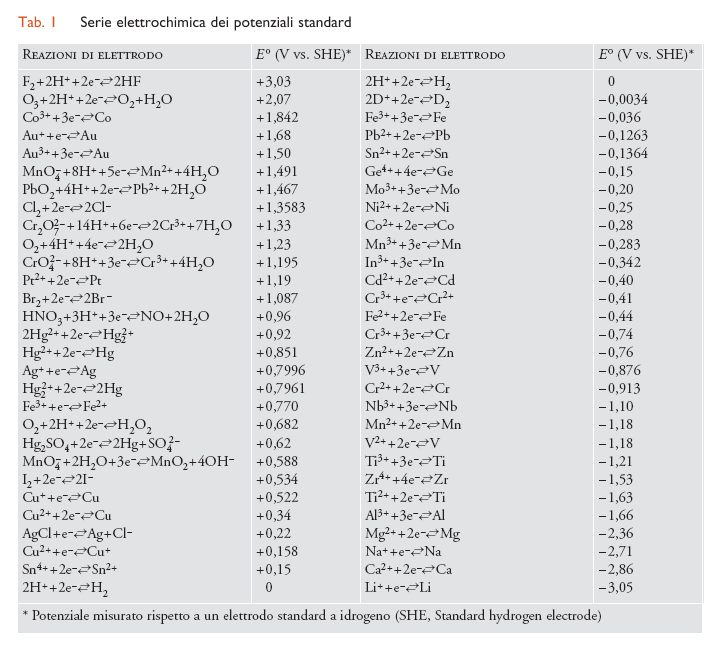
dove E° è il potenziale standard del metallo (tab.1) e [Maz+] è la concentrazione dei suoi ioni nell’elettrolita a contatto con la sua superficie. Convenzionalmente, la concentrazione oltre la quale si considera che il metallo subisca corrosione viene presa pari a 10−6 mol/L, come suggerito da Marcel Pourbaix (1973).
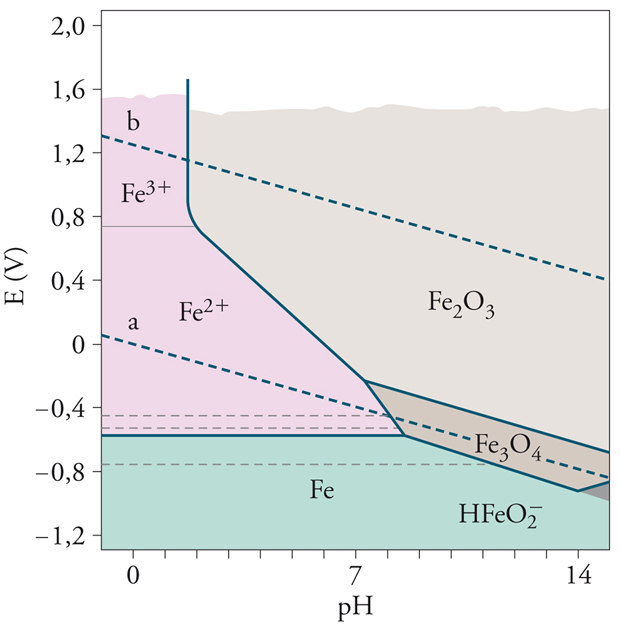
Le condizioni termodinamiche di corrosione sono illustrate efficacemente dai cosiddetti diagrammi di Pourbaix, ossia diagrammi potenziale-pH che forniscono i potenziali di equilibrio al variare del pH per i metalli e per le due reazioni catodiche di sviluppo di idrogeno [4] e di riduzione dell’ossigeno [5], rappresentate da due rette parallele (linee a e b tratteggiate in figura) aventi coefficiente angolare −0,059 V/decade e distanziate di 1,23 V. Nella fig. 3 è riportato il diagramma di Pourbaix semplificato del ferro.
I diagrammi di Pourbaix, costruiti sulla base dei dati termodinamici di equilibrio delle reazioni elettrochimiche che coinvolgono il metallo, mostrano le zone di stabilità delle specie chimiche coinvolte (zona di immunità del metallo; zona di stabilità degli ossidi, i quali potrebbero dare luogo a fenomeni di passivazione; zona di stabilità degli ioni metallici e delle forme complessate in ambienti fortemente alcalini) ma non possono fornire informazioni (se non qualitative) sulla cinetica dei processi, ossia sulla velocità di corrosione. È necessario inoltre osservare che in condizioni di non equilibrio, quali sono quelle che si verificano nella pratica, i campi di stabilità sono diversi, con ricadute importanti sulla cinetica. Nel caso del ferro, per esempio, il diagramma ottenuto sperimentalmente in acqua agitata evidenzia una zona di passività molto più estesa di quella prevista in teoria.
Aspetti cinetici. - La disponibilità di un lavoro motore costituisce una condizione necessaria perché la reazione di corrosione possa avvenire, ma non sufficiente: l’intervento di resistenze di reazione (o attriti generalizzati) condiziona la velocità di corrosione fino anche ad annullarla e comunque non permette di prevedere, dalla sola conoscenza del lavoro motore, l’evoluzione temporale del processo di corrosione. In altre parole, come avviene in molti altri fenomeni chimici e fisici, intervengono i fattori cinetici che possono mutare radicalmente il comportamento di un materiale soggetto a corrosione dedotto da considerazioni termodinamiche. È il caso, per esempio, del titanio, che secondo la termodinamica è più reattivo del ferro avendo un potenziale standard più negativo di oltre 1 V, ma in pratica si comporta da metallo nobile e non subisce corrosione negli ambienti in cui invece il ferro si corrode, come l’acqua di mare. Questo comportamento è dovuto all’intervento di fenomeni di passivazione con formazione di film protettivi.
Per ridurre la velocità di corrosione si può agire sulle resistenze di reazione, dette sovratensioni, e in particolare: (a) sulla reazione anodica (dissoluzione del metallo) favorendo la formazione di film protettivi (passivazione); (b) sulle reazioni catodiche (riduzione di ossigeno o sviluppo di idrogeno) aumentandone le sovratensioni; (c) sulla resistenza al trasporto di corrente nell’elettrolita (aumento delle resistenze ohmiche).
La sovratensione della reazione anodica di dissoluzione dei metalli (comportamento attivo) passa da valori trascurabili per i metalli normali (cioè i metalli che hanno bassa temperatura di fusione, inferiore a 600 °C, come Cd, Hg, Sn, Pb, Mg, Al e Zn) a valori superiori a 100 mV per i metalli inerti (con alta temperatura di fusione, superiore a 1400 °C, come Fe, Co, Ni, Cr, Mo, Ti, i metalli del gruppo del Pt e i metalli di transizione). Infine i metalli intermedi (temperatura di fusione intorno a 1000°C, per es., Cu, Ag, Au) mostrano un comportamento compreso tra questi estremi.
Molti metalli e loro leghe ad alta affinità per l’ossigeno hanno la caratteristica di ricoprirsi di uno strato di ossido protettivo che li preserva dalla corrosione in ambienti corrosivi. Queste condizioni sono dette condizioni di passività e definiscono il comportamento attivo-passivo dei metalli. È il caso, ben noto, degli acciai inossidabili, che devono la loro condizione di passività alla formazione sulla superficie di uno strato di ossido di cromo molto protettivo che li rende, come indica lo stesso nome, resistenti alla ossidazione in molti ambienti dove il ferro o gli acciai al carbonio e bassolegati subiscono una corrosione anche molto forte. Un altro esempio di comportamento passivo, che spesso passa inosservato, è quello delle armature del calcestruzzo armato che risultano perfettamente passivate dall’alcalinità della pasta di cemento idratata; in altre parole, il ferro nel calcestruzzo (o in soluzioni molto alcaline) si comporta come l’acciaio inossidabile nelle acque dolci. Anche l’alluminio e il titanio sono resistenti alla corrosione grazie alla loro capacità di passivarsi.
Le sovratensioni di sviluppo di idrogeno, rappresentate dalla reazione [4], dipendono dalla natura del materiale metallico M su cui avviene la reazione, secondo una anticorrelazione con la sovratensione di dissoluzione del metallo, per cui i metalli normali caratterizzati da sovratensioni di dissoluzione molto basse mostrano elevate sovratensioni di sviluppo di idrogeno (Hg, Sn, Pb, Mg, Al, Zn), mentre i metalli inerti hanno sovratensioni di sviluppo di idrogeno molto basse (Fe, Co, Ni, Cr, Mo, Ti, Pt).
La reazione di riduzione dell’ossigeno disciolto nell’acqua è il principale processo catodico che ha luogo negli ambienti naturali e in soluzioni neutre o debolmente alcaline.
La solubilità dell’ossigeno nell’acqua diminuisce all’aumentare della temperatura (diviene praticamente nulla al di sopra di 60 °C) e all’aumentare del contenuto di sali disciolti nell’acqua. La massima solubilità dell’ossigeno è pari a 10 mL/L in acqua pura a 0 °C.
In condizioni stazionarie l’apporto di ossigeno è dato dalla prima legge di Fick, secondo la quale il flusso è direttamente proporzionale al gradiente di concentrazione e al coefficiente di diffusione, e inversamente proporzionale allo strato limite di diffusione. Il valore massimo del flusso di ossigeno che dalla soluzione arriva alla superficie del metallo, che corrisponde alla corrente limite di diffusione di ossigeno, jl, è dato dalla relazione:
[8] formula
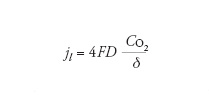
Il valore di jl dipende dunque da tre fattori: (a) dal coefficiente di diffusione D che aumenta all’aumentare della temperatura; (b) dalla concentrazione di ossigeno CO2 che diminuisce all’aumentare della temperatura; (c) dallo strato limite di diffusione δ, che è massimo in condizioni stagnanti e diminuisce all’aumentare della turbolenza. Un approccio semplificato porta a una relazione del tipo:
[9] formula
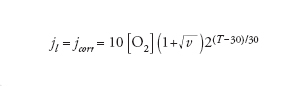
dove jcorr è la velocità di corrosione in μm/a, [O2] è la concentrazione di ossigeno in ppm, v è la velocità dell’acqua e T la temperatura in °C.
Forme di corrosione
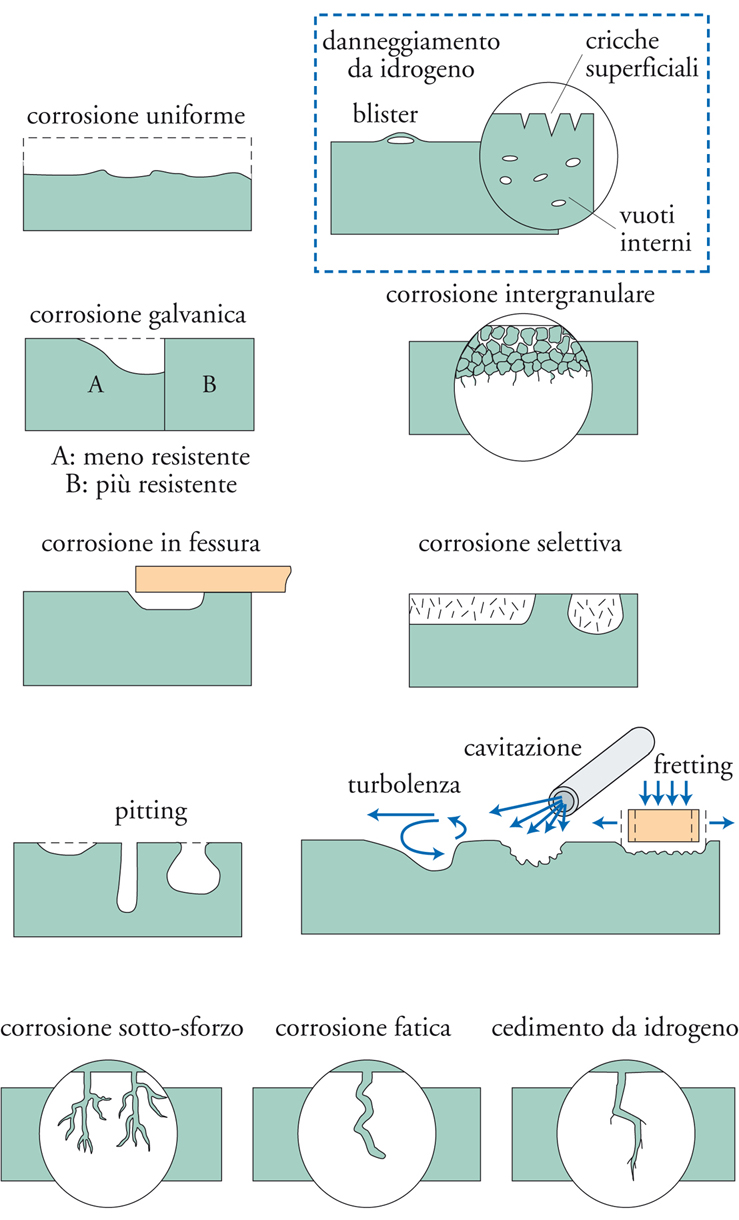
I fenomeni corrosivi si possono produrre alla superficie dei materiali metallici in modo diffuso oppure localizzato. Si ha corrosione generalizzata quando l’attacco interessa tutta la superficie del materiale esposta all’ambiente, e in particolare corrosione uniforme quando tale attacco generalizzato si produce in modo uniforme. Si ha corrosione localizzata, invece, quando l’attacco ha luogo solo su alcune parti della superficie del materiale esposta all’ambiente, con morfologia particolare, per esempio in forma di fenditure o di cricche, cavità, crateri, ulcere. Si parla poi di corrosione selettiva quando si verifica l’attacco di costituenti particolari del materiale, per esempio alcune fasi presenti come grani o al contorno di grani. Nella fig. 4 sono illustrate le principali forme di corrosione.
Corrosione generalizzata. - Interessa tutta la superficie di un metallo ed è tipica della corrosione in ambiente acido. Il parametro controllante è il pH: per il ferro e gli acciai in genere la corrosione acida diviene significativa per pH inferiori a 4.
La presenza di CO2 in fase acquosa porta alla formazione di acido carbonico che, nonostante sia un acido debole, è molto aggressivo nei confronti dell’acciaio al carbonio. Se si confronta la velocità di corrosione, per esempio, del ferro in acido carbonico e quella in un acido forte come l’acido cloridrico, HCl, entrambi a pH 4, la velocità di corrosione in acido carbonico è circa dieci volte più grande. La ragione di ciò sta nella diversa cinetica della reazione di riduzione dello ione idrogeno e di sviluppo di idrogeno molecolare: nel caso dell’acido carbonico anche lo ione bicarbonato partecipa direttamente alla reazione di riduzione.
Pur essendo un acido debole, l’H2S produce attacchi di corrosione particolarmente severi perché la formazione del solfuro di ferro, FeS, caratterizzato da un bassissimo prodotto di solubilità (dell’ordine di 10−24), provoca una diminuzione del potenziale anodico del ferro con conseguente disponibilità di lavoro motore anche in soluzioni neutre.
Per quanto riguarda la prevenzione di questa forma di corrosione – che rispetto alle altre forme è meno insidiosa perché si può prevedere sia come insorgenza sia come velocità media di perdita di spessore – essa si attua principalmente utilizzando un sovraspessore di corrosione, dimensionato valutando il prodotto della velocità di corrosione uniforme per la vita di progetto, o, più in generale, mediante i metodi tradizionali di controllo e prevenzione della corrosione, come la protezione catodica, l’uso di rivestimenti (organici, inorganici e metallici), le pitture e gli inibitori.
Corrosione localizzata. - Quando le reazioni anodica e catodica hanno luogo su superfici distinte si ha la corrosione localizzata, che interessa solo una parte limitata della superficie metallica esposta all’ambiente. La separazione delle aree instaura la circolazione di una corrente, detta corrente di macrocoppia, con circolazione di elettroni nel metallo, dall’area anodica a quella catodica, e circolazione di ioni nella soluzione: quelli positivi migrano dall’area anodica verso l’area catodica e quelli negativi nella direzione opposta.
Le cause che determinano l’instaurarsi di una corrente di macrocoppia sono molteplici: (a) la diversa nobiltà di metalli o leghe in contatto elettrico, che dà luogo alla corrosione per contatto galvanico; (b) la differente disponibilità di ossigeno che provoca (per es., sugli acciai), la separazione tra aree anodiche e aree catodiche; (c) la rottura locale del film di passività nei materiali attivo-passivi.
Corrosione per contatto galvanico. - È detta anche corrosione galvanica o bimetallica e avviene quando due metalli tra loro in contatto elettrico, entrambi esposti a un ambiente aggressivo, hanno diversa nobiltà: la corrosione ha luogo sul metallo meno nobile, mentre il metallo più nobile (o, se presente, un materiale non metallico dotato di conducibilità elettronica, come la grafite, o un film superficiale, costituito, per es., da magnetite o solfuro di ferro) funge da area catodica. In queste condizioni la velocità di corrosione del metallo meno nobile subisce un’accelerazione dipendente dal rapporto tra l’area del metallo più nobile (area catodica) e quella del metallo meno nobile che si corrode (area anodica). Un esempio tipico è l’attacco sugli acciai al carbonio bassolegati in acqua di mare o in soluzioni aerate quando sono accoppiati con materiali più nobili, come le leghe di rame, o di nobiltà pratica più alta, come gli acciai inossidabili e il titanio.
La velocità di corrosione per contatto galvanico è data dall’espressione generale:
[10] formula
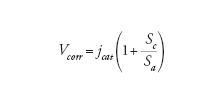
dove jcat è la velocità del processo catodico, e Sa e Sc sono rispettivamente le aree anodica e catodica. Nelle acque, la velocità del processo catodico è la densità di corrente limite di diffusione di ossigeno jcat=jl.

Per stabilire se possa instaurasi un contatto galvanico, prima di tutto si devono confrontare i potenziali dei due metalli nell’ambiente aggressivo al quale sono esposti. A questo scopo, è consuetudine far uso della scala dei potenziali pratici in acqua di mare, riportati nella tab. 2, anche per ambienti diversi quali terreni e acque.
La prevenzione della corrosione per contatto galvanico si può attuare in vari modi: (a) evitando l’accoppiamento di materiali con diversa nobiltà (per es., usando flange isolanti); (b) utilizzando aree anodiche grandi e aree catodiche piccole; (c) evitando elettroliti con bassa resistività; (d) applicando la protezione catodica. La pitturazione è un rimedio efficace se applicata sulle aree catodiche, cioè sul metallo più nobile; se la pitturazione (o il rivestimento isolante) interessa solo il metallo meno nobile, è invece pericolosa perché aumenta la velocità di corrosione in corrispondenza dei difetti del rivestimento.
Pitting. - Si presenta sotto forma di attacchi, detti pit o vaiolature, molto penetranti, ma che interessano una porzione di superficie metallica piccola rispetto alla superficie esposta. Le dimensioni lineari variano da poche decine di μm sino ad alcuni mm e le morfologie si presentano variabili, dal tipo a punta di spillo a quello cosiddetto a caverna.
È una forma di corrosione tipica dei materiali con comportamento attivo-passivo. Tra i metalli di impiego comune la corrosione per pitting interessa gli acciai inossidabili, il rame, l’alluminio. Nel meccanismo si distinguono due stadi: l’innesco e la propagazione.
Lo stadio di innesco ha una durata pari al tempo necessario per la rottura locale del film di passività a opera di specifiche specie chimiche presenti nell’ambiente corrosivo, come gli ioni cloruro, Cl−, nel caso degli acciai inossidabili e delle leghe di alluminio, qualora la loro concentrazione superi un valore di soglia che dipende dalla composizione (per es., contenuto di Cr e Mo per gli acciai inossidabili) e dai parametri ambientali (per es., temperatura e turbolenza: condizioni stagnanti riducono il tempo di innesco e in generale costituiscono un fattore aggravante per il pitting; la temperatura riduce il tempo di innesco). La presenza di inclusioni, la formazione di precipitati e il grado di incrudimento del metallo favoriscono l’innesco del pitting.
Nello stadio di propagazione si instaura una corrente di macrocoppia tra le aree dove l’ossido è stato danneggiato con dissoluzione del metallo (aree anodiche) e le aree passive con comportamento catodico. La velocità di corrosione risultante è assai elevata, in molti casi dell’ordine di alcuni millimetri per anno, dato il rapporto sfavorevole tra l’area anodica piccola e l’area catodica molto grande.
La prevenzione del pitting è attuata principalmente nello stadio di innesco, perché una volta che il processo è innescato è più difficile arrestarne la propagazione, e pertanto si basa sulla scelta di materiali resistenti all’innesco nelle condizioni di esercizio. Per gli acciai inossidabili austenitici e austeno-ferritici e per le leghe di nichel si impiega un indice, il Pitting resistance equivalent number (PREN), calcolato in base al contenuto di Cr, Mo, W e N nella lega, secondo la formula seguente:
[11] PREN = Cr% + 3,3(Mo% + 0,5W%) + 16N%.
Per valori di PREN superiori a 40 la resistenza al pitting è elevata, mentre per valori inferiori a 35 è bassa. La protezione catodica è efficace sia nella fase di innesco sia in quella di propagazione.
Corrosione interstiziale e sotto deposito. - La presenza di interstizi con spessori molto piccoli, dell’ordine del μm o inferiori, o di parti di superficie non liberamente esposte all’ambiente costituisce spesso un fattore aggravante per la corrosione di un metallo. Esempi di interstizi sono: l’accoppiamento mediante flange, la saldatura a punti, la presenza di depositi di varia natura.
Più articolato è invece il meccanismo di corrosione interstiziale degli acciai inossidabili in soluzioni contenenti ioni cloruro. Come per il pitting, si distinguono due stadi: uno di innesco e uno di propagazione.
Nel caso di corrosione sotto deposito dei materiali attivi, le misure di prevenzione riguardano innanzitutto l’eliminazione delle disomogeneità ambientali e in secondo luogo l’impiego di pitture e l’applicazione della protezione catodica. Per la prevenzione della corrosione interstiziale degli acciai inossidabili, devono essere adottate, in fase di progettazione, le soluzioni tecniche che escludono la presenza di interstizi; per esempio, sono preferibili le giunzioni saldate alla flangiatura. Come per il pitting, la protezione catodica è efficace sia nello stadio di innesco che in quello di propagazione.
Corrosione intergranulare. - Questa forma di corrosione si manifesta come attacco localizzato al bordo di grano per la presenza di precipitati. In effetti il bordo di grano è un sito preferenziale di processi di segregazione e precipitazione di composti (per es., carburi, solfuri, composti intermetallici). In linea di principio, tutte le leghe metalliche in cui sono presenti precipitati ai bordi di grano sono suscettibili di corrosione intergranulare. In pratica, quelle che hanno dimostrato maggiore suscettibilità sono gli acciai inossidabili e alcune leghe di nichel, cioè materiali ritenuti molto resistenti a corrosione.
Per impedire questa forma di corrosione è necessario evitare la precipitazione dei carburi, per esempio ripetendo il trattamento termico di solubilizzazione sull’intero manufatto saldato. Se questo non è fattibile, la precipitazione dei carburi di cromo è impedita mediante due strategie alternative: abbassando il contenuto di carbonio al di sotto di un valore soglia in genere inferiore allo 0,03%, in modo che la formazione dei carburi sia cineticamente più difficile (acciai di grado L, per es., AISI 304L); oppure aggiungendo elementi di lega che hanno maggiore affinità del cromo verso il carbonio, per esempio titanio o niobio, in modo che il carbonio sia già legato e quindi non disponibile per il cromo (acciai stabilizzati AISI 321 e AISI 347).
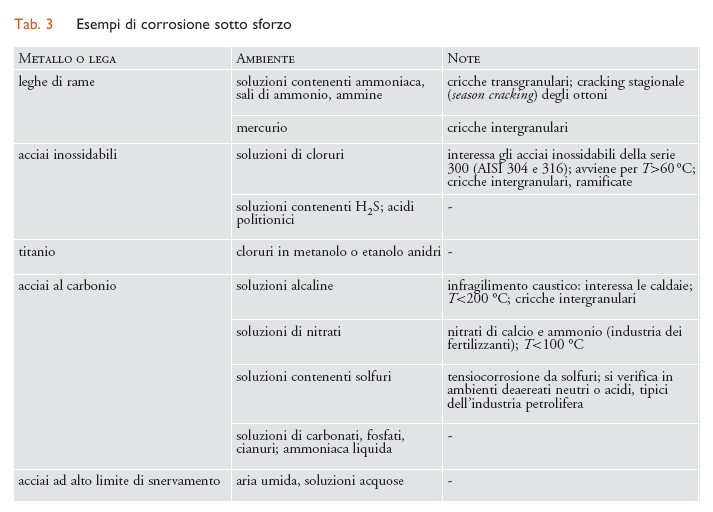
Corrosione sotto sforzo. - Con il termine corrosione sotto sforzo (o tensiocorrosione) (SCC, Stress corrosion cracking), si definiscono i fenomeni di innesco e propagazione di cricche in un metallo sotto l’azione combinata di sollecitazioni meccaniche di trazione (ma non di compressione) e di un ambiente corrosivo. È un fenomeno complesso che riguarda molteplici aspetti: per esempio, le condizioni di insorgenza sono caratteristiche di accoppiamenti molto specifici di un materiale metallico e di un ambiente; il fenomeno si innesca soltanto al di sopra di una soglia di sollecitazione meccanica; interessa soprattutto le leghe; la velocità di propagazione delle cricche, seppure elevata, è inferiore a quella relativa a cricche di natura puramente meccanica. Gli sforzi di trazione possono provenire da carichi esterni applicati al componente metallico, ma anche da stati di sollecitazione interni, quali per esempio sforzi residui da lavorazioni meccaniche, da operazioni di saldatura e da trattamenti termici. Le cricche possono essere di tipo intergranulare, con propagazione lungo i bordi dei grani, o di tipo transgranulare, quando la propagazione avviene attraverso i grani del metallo. Le cricche inoltre possono essere individuali o ramificate. Nella tab. 3 sono riportati i più comuni casi di SCC.
Corrosione sotto sforzo da cloruri. - Questo tipo di corrosione interessa soprattutto gli acciai inossidabili austenitici della serie 300, tipo AISI 304 e 316, e anche, in misura minore, gli acciai inossidabili austeno-ferritici. Ne sono praticamente immuni gli acciai inossidabili ferritici con tenori di Ni inferiori al 4%. Per manifestarsi richiede una temperatura superiore a 60 °C.
Infragilimento da idrogeno. - La reazione di riduzione di ioni idrogeno (2H++2e−←→H2), che ha luogo negli ambienti acidi, ha inizio attraverso un primo stadio (H++e−←→Hads) che porta alla formazione di idrogeno atomico adsorbito sulla superficie metallica (Hads). Questo può formare una molecola di idrogeno H2 (Hads+Hads←→H2), oppure penetrare all’interno del reticolo cristallino del metallo (Hass) se sulla superficie di questo sono presenti particolari veleni che inibiscono la formazione dell’idrogeno molecolare. Sono veleni i solfuri metallici e il solfuro di idrogeno (H2S), spesso presenti negli ambienti associati agli idrocarburi. La presenza di H2S dà luogo alla forma di corrosione sotto sforzo per infragilimento da idrogeno detta Sulphide stress cracking (SSC). Gli acciai suscettibili di SSC sono quelli ad alto limite di snervamento (carico di rottura superiore a 700 MPa) con una microstruttura che risulta particolarmente sensibile agli effetti dell’idrogeno. Le rotture sono caratterizzate da cricche transgranulari.
La prevenzione della corrosione sotto sforzo si attua innanzitutto in sede di progettazione attraverso la scelta di materiali non suscettibili nell’ambiente specificato.
Erosione-corrosione. - Questa forma di corrosione si manifesta quando il processo di corrosione è associato a condizioni fluidodinamiche spinte (per es., elevata velocità del fluido che provoca la rimozione dei prodotti di corrosione). La presenza di solidi sospesi accentua il fenomeno. Questo effetto sinergico tra corrosione e azione meccanica del fluido interessa gran parte dei materiali metallici quando si superano determinate condizioni di turbolenza espresse dalla velocità critica di erosione-corrosione.
Corrosione microbiologica. - Nelle acque e nei terreni ha luogo una forma di corrosione, detta MIC (Microbiologically induced corrosion), che è associata alla presenza e all’azione di batteri. Questa forma di corrosione coinvolge sia i batteri aerobici, che vivono e crescono in presenza di ossigeno, in genere producendo condizioni acide, sia i batteri anaerobici che vivono e crescono in condizioni di assenza di ossigeno. Questi ultimi, che sono ritenuti i più pericolosi, comprendono i batteri solfatoriduttori (SRB, Sulphate reducing bacteria).
Corrosione da correnti disperse. - Questa forma di corrosione, detta anche corrosione elettrolitica o da correnti vaganti, è provocata da fenomeni di interferenza elettrica. Il caso più frequente è quello in cui si manifesta come corrosione localizzata su tubazioni o serbatoi interrati che si trovano in vicinanza dei binari delle linee ferroviarie, tranviarie e metropolitane con trazione elettrica a corrente continua o in vicinanza di dispersori di corrente continua impiegati negli impianti di protezione catodica. Il meccanismo è il seguente: la corrente circolante nel terreno (corrente di ritorno nei sistemi di trazione elettrica a corrente continua o corrente di protezione negli impianti di protezione catodica) utilizza la struttura metallica interrata come conduttore elettrico, causando un attacco di corrosione nelle zone dove la corrente lascia la struttura. Le zone di ingresso della corrente si trovano, invece, in condizioni di protezione catodica e non subiscono corrosione. Gli attacchi sono spesso severi perché la zone di uscita della corrente si localizzano nei difetti del rivestimento isolante delle strutture, in vicinanza delle sottostazioni elettriche o delle strutture protette catodicamente; basti pensare che 1 mA rilasciato da 1 cm2 di superficie del metallo provoca una perdita di spessore di 10 mm/a.
Per limitare i danni provocati dalle correnti disperse è necessario impedire che queste ultime entrino nelle strutture interferite; a questo riguardo si ricorre a varie strategie che comprendono il miglioramento del rivestimento isolante, il sezionamento elettrico delle strutture (per es., interposizione di giunti isolanti sulle tubazioni), il drenaggio elettrico e i collegamenti equipotenziali.
Anche la corrente alternata può causare danni sulle strutture oggetto di interferenza elettrica, ma è necessario che la densità della corrente scambiata tra metallo e terreno sia superiore a 30 A/m2, cioè di cinque ordini di grandezza superiore alle correnti di interferenza da corrente continua. Casi di interferenza da corrente alternata sono provocati dai parallelismi con le linee ad alta tensione, in genere superiore a 125 kV, e dalla dispersione nel terreno della linea ferroviaria ad alta velocità.
Controllo della corrosionee scelta dei materiali
Sulla base del meccanismo elettrochimico del processo di corrosione, è possibile individuare varie modalità di intervento mirato a bloccare o rallentare la corrosione.
Interventi sulla reazione anodica. - Possono essere basati sull’annullamento del lavoro motore, conseguito mediante l’uso di materiali di nobiltà superiore al processo catodico (Ea>Ec; per es., metalli nobili come l’oro e il platino in ambienti aerati o il rame in ambiente acido non ossidante), o realizzato portando il potenziale del materiale nella zona di immunità (E〈Eeq) mediante la protezione catodica; oppure per intervento di fenomeni di passivazione che portano il materiale in condizioni di passività (formazione di ossidi protettivi e stabili come nel caso degli acciai inossidabili e del titanio in ambienti aerati) o per applicazione della protezione catodica per passività o della protezione anodica sui materiali attivo-passivi; infine, mediante l’uso di inibitori passivanti aggiunti all’ambiente, che favoriscono la formazione di film di passivazione.
Interventi sulla reazione catodica. - Possono essere basati sull’eliminazione dell’ossigeno e sul mantenimento del pH alcalino; sull’aumento delle sovratensioni del processo di sviluppo di idrogeno nelle soluzioni acide mediante l’aggiunta di opportuni inibitori di corrosione filmanti; sulla diminuzione delle sovratensioni del processo catodico di sviluppo di idrogeno in ambienti acidi mediante alligazione catodica nel caso di leghe di Ti, di Cr e di Ta. In questi particolari casi l’aumento della velocità del processo catodico porta il materiale in condizioni di passività.
Interventi sull’ambiente. - Si realizzano mediante l’aumento della resistenza ohmica nell’elettrolita, per esempio mediante rivestimenti isolanti (pitture e rivestimenti plastici a elevato spessore).
Rivestimenti e pitture
Un metodo di prevenzione della corrosione efficace e molto usato è l’impiego di un rivestimento in grado di isolare la superficie del metallo dall’ambiente. La scelta è molto ampia in quanto i rivestimenti possono essere organici (pitture), inorganici (strati di conversione, smalti) o metallici.
Rivestimenti metallici. - Rappresentano uno dei metodi più comuni di prevenzione e sono costituiti da uno strato di un metallo o di una lega metallica resistente alla corrosione. Oltre alla resistenza a corrosione, tali strati devono possedere altre proprietà, quali: (a) resistenza meccanica; (b) durezza; (c) resistenza all’usura; (d) proprietà elettriche, ottiche e termiche; (e) aspetto esteticamente gradevole.
Dal punto di vista della corrosione sono particolarmente importanti le imperfezioni relative alla continuità e all’uniformità superficiali. Infatti, se il rivestimento è continuo e non poroso, la protezione nei confronti del metallo base è completa, mentre in presenza di imperfezioni si ha formazione di coppie galvaniche tra il metallo base e il rivestimento. Sono pericolosi i rivestimenti catodici che accelerano la corrosione del ferro, come rame, nichel, argento, piombo, cromo mentre sono benefici i rivestimenti anodici come lo zinco.
Strati di conversione. - Sono ottenuti sulla superficie di alcuni metalli, principalmente acciai, acciai zincati e alluminio, in seguito a reazioni chimiche o elettrochimiche. I processi di conversione più diffusi sono la fosfatazione, la cromatazione e l’ossidazione anodica. La fosfatazione si ottiene immergendo manufatti di acciaio o di acciaio zincato in soluzioni acide di fosfati di Zn e Mn che formano sulla superficie del metallo uno strato di fosfati aderenti e protettivi in grado di svolgere una blanda azione anticorrosiva e di costituire una mano di fondo per cicli di pitturazione (per es., nel caso di elettrodomestici e di scaffalature metalliche).
L’ossidazione anodica dell’alluminio e di altri metalli è un processo elettrolitico realizzato allo scopo di ispessire il film di ossido naturalmente presente, in modo da migliorarne la resistenza alla corrosione e all’abrasione e le caratteristiche estetiche, oppure al fine di ottenere un film di ossido con caratteristiche dielettriche speciali. Un tipico esempio di ossidazione anodica è quello che si effettua per la protezione dei serramenti di alluminio, detti appunto anodizzati. L’ossidazione anodica serve anche come pretrattamento per ancorare al metallo cicli di verniciatura o di pitturazione.
Rivestimenti cementizi. - Per la loro alcalinità (pH ca. 13) i materiali cementizi offrono una perfetta protezione all’acciaio al carbonio. Questa è una delle principali ragioni alla base del successo delle costruzioni in calcestruzzo armato, ma spiega anche la diffusione dei rivestimenti cementizi per la protezione interna di tubazioni per il trasporto dell’acqua (applicati per centrifugazione su tubazioni di acciaio o di ghisa), oppure per il ricoprimento esterno di tubazioni marine (anche con funzioni di appesantimento) o di strutture interrate, come per esempio l’esterno dei casing dei pozzi petroliferi.
Pitture e rivestimenti plastici. - Sono costituiti da un film sottile di natura organica di spessore 0,02÷3 mm circa, applicato sulla superficie del metallo mediante un processo di essiccazione di un solvente, per estrusione a caldo oppure per reticolazione chimica.
La mano di fondo anticorrosiva è costituita da pigmenti attivi di tipo anodico o catodico. I pigmenti anodici sono i composti che portano in condizioni di passivazione l’acciaio, come i fosfati, i cromati e il minio di piombo (cromati e minio sono stati aboliti per gli effetti tossici e cancerogeni). I pigmenti catodici sono costituiti da polvere di zinco metallico che agisce in modo duplice: contribuisce al rafforzamento dell’effetto barriera ed esplica un’azione di protezione catodica nei confronti dell’acciaio. La polvere di zinco metallico (oltre 90% in peso) può essere dispersa in due tipi di legante/solvente: il legante inorganico, costituito da silicato di sodio in solvente metanolo, oppure il legante organico, costituito da resine epossidiche bicomponenti e termoindurenti.
L’applicazione delle pitture prevede i cosiddetti cicli di pitturazione, caratterizzati dalle fasi di preparazione superficiale, mano di fondo, mano intermedia e mano di finitura. La preparazione superficiale consiste nella rimozione di ossidi, incrostazioni e sporcizia fino a portare il metallo al bianco lucente. Una buona preparazione superficiale è la premessa per il successo del ciclo di pittura.
Controllo dell’ambiente
La prevenzione della corrosione si può perseguire attraverso controlli o correzioni della composizione dell’ambiente.
Correzione del pH. - Tra le correzioni del pH più frequenti vi sono: (a) l’alcalinizzazione delle acque di caldaia per favorire la formazione di un film di magnetite; (b) le variazioni di pH per migliorare il potere incrostante delle acque; (c) l’aggiunta di sostanze alcaline per neutralizzare le condense acide.
Controllo dell’ossigeno. - L’eliminazione dell’ossigeno nelle acque neutre o alcaline riduce e in pratica annulla la corrosione dell’acciaio al carbonio. Si attua con metodi fisici mediante stripping con gas o trattamento sotto vuoto, raggiungendo tenori di ossigeno residui di 0,015 ppm (sufficienti per le acque di alimentazione di caldaie a bassa o media pressione), oppure con metodi chimici, realizzati spesso in serie a quelli fisici, che possono portare il tenore di ossigeno a qualche ppb. Le sostanze più frequentemente utilizzate a questo scopo sono il solfito o il bisolfito di sodio e l’idrazina, che agiscono rispettivamente secondo le reazioni: Na2SO3+1/2O2←→Na2SO4 e N2H4+O2←→2H2O+N2. L’idrazina si può in parte decomporre, dando ammoniaca secondo la reazione 3N2H4←→4NH3+N2, con il benefico effetto di produrre alcalinità che su l’acciaio favorisce la separazione di film protettivi di magnetite. Un altro vantaggio dell’idrazina rispetto al solfito è che essa non provoca un aumento della salinità dell’acqua. Tuttavia l’idrazina reagisce con l’ossigeno con velocità sensibile solo a partire da temperature superiori a 140 °C. Problemi di tossicità, inoltre, ne sconsiglierebbero l’impiego.
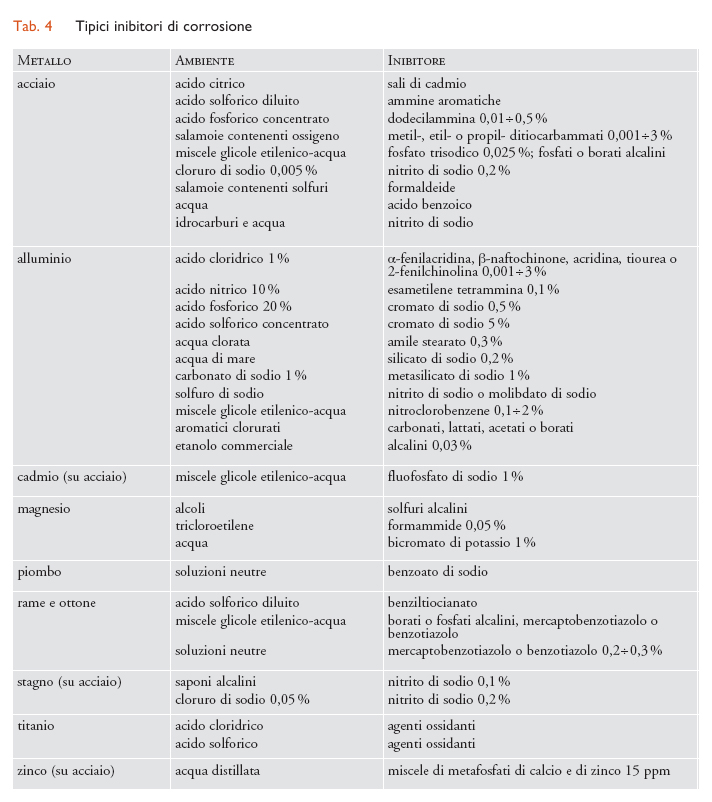
Inibitori di corrosione. - Gli inibitori sono sostanze che, aggiunte in piccole quantità agli ambienti aggressivi, possono rallentare fino anche ad annullare i processi di corrosione. Le sostanze in possesso di queste caratteristiche sono molto numerose e di natura assai diversa. Il meccanismo con cui operano in molti casi non è ancora chiarito. In generale comunque agiscono modificando lo stato di superficie del materiale metallico da proteggere in seguito a processi di adsorbimento o di reazione che portano alla separazione di strati protettivi. L’aumento della resistenza alla reazione di corrosione ha luogo a volte attraverso un’inibizione del processo catodico (per es., aumentando la sovratensione di idrogeno o impedendo all’ossigeno di raggiungere la superficie metallica) e/o di quello anodico; altre volte in seguito all’instaurazione di condizioni di passività. Nella tab. 4 sono riportati gli inibitori usualmente impiegati in alcuni ambienti. La stragrande maggioranza degli inibitori trova impiego essenzialmente nei seguenti casi: (a) in ambienti neutri o leggermente alcalini, costituiti principalmente da acque naturali o industriali di raffreddamento o comunque da soluzioni con un pH compreso tra 5 e 9; (b) in ambienti acidi (decapaggio, trattamenti acidi di disincrostazione); (c) nei processi di estrazione e di raffinazione del greggio.
Gli inibitori possono essere classificati in relazione alla loro natura chimica (inibitori organici e inorganici), al tipo di impiego (inibitori per acqua di alimentazione di caldaia, per decapaggio, disincrostazione, imballaggi) e alle condizioni di impiego (inibitori in soluzione o in fase a vapore), oppure secondo il meccanismo con cui operano (inibitori catodici, inibitori anodici ossidanti o non ossidanti e inibitori misti o ad azione multipla).
Per svolgere la loro azione, gli inibitori devono essere presenti in tenori superiori a una data concentrazione, detta efficace, in genere più bassa per gli inibitori ossidanti (indicativamente 10−3 mol/L) e più elevata per gli altri, che spesso devono agire anche da correttori di pH. La concentrazione efficace dipende dalle condizioni superficiali (per superfici lisce e pulite sono richiesti tenori molto più bassi che per quelle porose o ricoperte da prodotti di corrosione o da scaglie varie) e dalla composizione dell’ambiente (la concentrazione degli inibitori va aumentata in presenza di specie che tendono a contrastarne l’azione, come per es., i cloruri o le sostanze riducenti nel caso di inibitori ossidanti).
Protezione elettrica catodica e anodica
La cosiddetta protezione elettrica si basa su due principi: uno termodinamico di annullamento del lavoro motore (protezione catodica per immunità o quasi-immunità termodinamica) e uno cinetico che consiste nel realizzare e mantenere le condizioni di passività (protezione catodica per passività e protezione anodica).

Protezione catodica. - La protezione catodica si realizza inviando una corrente alla superficie del metallo (corrente catodica) mediante un anodo galvanico o un sistema a corrente impressa (fig. 5). La corrente provoca un abbassamento del potenziale del metallo e una riduzione della velocità di corrosione. Se la corrente (o meglio la densità di corrente) è sufficiente per portare il potenziale del metallo al di sotto del suo potenziale di equilibrio, dato dall’equazione di Nernst [7], si instaurano le condizioni di immunità termodinamica o quasi-immunità termodinamica. I potenziali di protezione di immunità e di quasi-immunità dei principali materiali metallici sono riportati nella tab. 5.
Per quanto riguarda i materiali con comportamento attivo-passivo che operano in condizioni di passività (per es., gli acciai inossidabili e il ferro nel calcestruzzo), per prevenire o bloccare la corrosione localizzata promossa dai cloruri si possono seguire due strade: la protezione catodica per immunità, applicabile a tutti i materiali, e la protezione catodica per passività, che ha lo scopo di mantenere il materiale passivo.
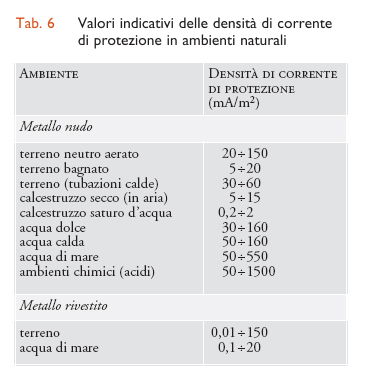
Per realizzare le condizioni di protezione occorre far giungere sulla superficie del metallo un’adeguata densità di corrente di protezione. Nella tab. 6 sono riportati i valori delle densità di corrente di protezione nei principali ambienti. La densità di corrente di protezione jprot dipende dalla velocità Ic con cui, al potenziale di protezione, i processi catodici si producono alla superficie della struttura e pertanto dipende, come Ic, dalle condizioni ambientali. Se il processo di corrosione, per esempio, ha luogo, come avviene nei terreni e nelle acque, in condizioni di controllo di diffusione di ossigeno, la densità di corrente di protezione dipende da tutte le condizioni che definiscono la velocità limite di apporto di ossigeno e cioè dal tenore di ossigeno disciolto, dallo stato di agitazione dell’ambiente, dalla temperatura, dalla presenza di depositi o di rivestimenti. In acqua di mare, per esempio, la densità di corrente di protezione, al variare dell’agitazione, della temperatura e del tenore di ossigeno, può passare dai 20 mA/m2, richiesti per proteggere strutture immerse nel fango del fondo marino, a 1 A/m2 e più, necessari per proteggere parti scoperte di navi vicino alle eliche, cioè nelle condizioni di massima agitazione e ossigenazione. Un effetto benefico, che si riscontra sia nei terreni sia nelle acque naturali, è dato dalla precipitazione sulla superficie metallica, in seguito a processi di alcalinizzazione locale, di carbonati e di prodotti di corrosione che riducono la superficie esposta del metallo.
Gli ambienti naturali e tutti quelli in cui la reazione catodica è la riduzione di ossigeno si dimostrano particolarmente adatti per l’applicazione della protezione catodica, perché la corrente di protezione è uguale alla corrente limite di diffusione dell’ossigeno, che non è mai troppo elevata. Altri ambienti, proprio per le ragioni opposte, risultano poco adatti all’applicazione del metodo; per esempio gli ambienti acidi, nei quali la reazione catodica è quella di sviluppo di idrogeno e le correnti di protezione sono di due o tre ordini di grandezza superiori alla corrente limite di diffusione dell’ossigeno.
In presenza di un rivestimento isolante, la corrente di protezione può diventare una percentuale anche bassissima di quella richiesta dal metallo nudo, perché lo scambio di corrente avviene sulle zone non ricoperte dal rivestimento, cioè in corrispondenza di pori, difetti o danneggiamenti del rivestimento.
Le strutture metalliche protette catodicamente in acqua di mare si ricoprono di uno strato protettivo costituito in gran parte da carbonato di calcio e idrossido di magnesio, comunemente chiamato deposito calcareo. La sua formazione, soprattutto sulle strutture nude, è quanto mai provvidenziale, poiché riduce di un ordine di grandezza la corrente di protezione. Il deposito calcareo esplica una duplice azione protettiva: costituisce una barriera che limita la diffusione di ossigeno e mantiene il pH alcalino sulla superficie metallica che perciò tende a passivarsi.
Le applicazioni più comuni della protezione catodica riguardano strutture poste negli ambienti naturali. Gli anodi galvanici sono impiegati negli ambienti a elevata conducibilità, per esempio in acqua di mare, e possono essere convenienti quando sono richieste piccole correnti anche negli ambienti con bassa conducibilità, come nei terreni e nella prevenzione catodica del calcestruzzo armato. I sistemi a corrente impressa sono necessari negli ambienti resistivi, come i terreni e il calcestruzzo, e sono preferiti per la protezione di strutture estese. Un notevole vantaggio è dato dal fatto che il sistema presenta una grande flessibilità di esercizio, potendo variare e regolare la corrente erogata.
Come menzionato in precedenza, la protezione catodica può essere realizzata mediante l’impiego di anodi galvanici o mediante l’erogazione di corrente impressa (fig. 4).
La prima applicazione della protezione catodica con anodi galvanici, realizzata da Humphry Davy nel 1824, riguardava la protezione del rame ottenuta con anodi di ferro e di zinco, cioè con materiali meno nobili. Per le strutture in acciaio al carbonio, molti sono i materiali che verificano questa condizione, come risulta dall’esame della serie elettrochimica degli elementi; tuttavia le realizzazioni pratiche sono effettuate ricorrendo a leghe a base di soli tre elementi: alluminio, magnesio e zinco.
Nei sistemi a corrente impressa, la corrente è fornita da un generatore esterno di corrente continua, mediante un dispersore che è in grado di erogare corrente nell’ambiente. Lo scambio di corrente tra dispersore e ambiente avviene attraverso una reazione anodica, che dipende dal materiale anodico e dall’ambiente. Nel caso di anodi di acciaio al carbonio, per esempio, la reazione anodica è quella di dissoluzione del ferro, con consumo dell’anodo; per gli anodi cosiddetti insolubili, realizzati con il titanio platinato, la grafite o altri materiali, le reazioni possono sviluppare ossigeno o cloro a seconda dell’ambiente e della densità di corrente di erogazione.
Protezione anodica. - Si applica solo ai materiali con comportamento attivo-passivo e si realizza inviando una corrente dal metallo alla soluzione (corrente anodica) mediante un catodo con un sistema a corrente impressa che mantiene il potenziale costante (condizioni potenziostatiche). La corrente provoca un aumento del potenziale del metallo e un iniziale aumento della velocità di corrosione finché, superata la densità di corrente di passivazione, il materiale si passiva e la corrente anodica crolla al valore della densità di corrente di passività. In queste condizioni il materiale è passivo e tale rimane finché il potenziale è mantenuto all’interno dell’intervallo di passività. Tipiche applicazioni della protezione anodica sono la protezione dell’acciaio al carbonio in acido solforico e in soluzioni ammoniacali, degli acciai inossidabili in acido solforico, miscele di acido solforico e nitrico, acido fosforico, soda, e del titanio in soluzioni di acido cloridrico. A differenza della protezione catodica, la protezione anodica è realizzata con uno speciale alimentatore, detto potenziostato, in grado di mantenere costante il potenziale del metallo.
Monitoraggio della corrosione
Negli impianti in cui i materiali subiscono corrosione sono richieste attività di verifica della velocità di corrosione e dell’entità del danno causato (perdita di spessore, presenza di cricche, corrosione localizzata) che possono essere distinte in ispezioni e controlli periodici e monitoraggio in continuo della velocità di corrosione.
Ispezioni
Le ispezioni hanno lo scopo di verificare l’integrità dei componenti di un impianto mediante tecniche dirette, quali: (a) l’ispezione visiva; (b) la misura degli spessori; (c) la ricerca di cricche mediante varie tecniche basate sull’uso di particelle magnetiche; (d) i liquidi penetranti; (e) le correnti indotte; (f) i campi magnetici; (g) gli ultrasuoni. Queste attività sono svolte durante gli arresti periodici degli impianti, in punti prescelti ritenuti rappresentativi delle condizioni operative del materiale nell’impianto.
Misura della velocità di corrosione
Durante l’esercizio di un impianto è spesso opportuno, se non necessario, conoscere in tempo reale la velocità di corrosione a cui è sottoposto il materiale per ottimizzare i trattamenti anticorrosivi, come l’iniezione di inibitori di corrosione o la rimozione dell’ossigeno nei circuiti di trattamento delle acque.
A tale scopo sono appositamente inseriti nell’impianto i provini di corrosione (corrosion coupons) che consistono in una lamina dello stesso materiale dell’impianto esposta all’ambiente corrosivo per un tempo prefissato (per es., da 1 a 6 mesi). Una variante è rappresentata dal tronchetto di corrosione (corrosion spool), consistente in un tratto di tubazione delimitato da due valvole che può essere periodicamente ispezionato. La misura della perdita di peso rapportata al periodo di esposizione permette il calcolo della velocità di corrosione media. Questo metodo è affidabile ed economico ma fornisce un risultato ritardato, in quanto la corrosione viene rilevata solo dopo che si è verificata.
Per ovviare a tale inconveniente, si può ricorrere alle sonde elettriche, costituite in pratica da un provino di corrosione a forma di filo o di spirale di cui viene misurata in continuo la resistenza elettrica, che, a causa della riduzione della sezione, aumenta in proporzione alla velocità di corrosione. La sonda elettrica ha il vantaggio di funzionare anche in ambienti con bassa conducibilità, come in caso di esposizione diretta a fluidi multifase (idrocarburi liquidi e gassosi e fase acquosa).
Negli elettroliti si può applicare una tecnica elettrochimica detta resistenza di polarizzazione lineare, che fornisce la misura della velocità di corrosione istantanea. Queste sonde sono usate nei circuiti d’acqua.
Infine si fa cenno alle sonde a idrogeno che misurano la quantità di idrogeno prodotta dal processo di corrosione in ambiente acido quando è presente l’idrogeno solforato. La velocità di corrosione è proporzionale alla quantità di idrogeno, valutata indirettamente dalla misura della pressione nella sonda. Dispositivi questi che sono molto usati nell’industria petrolchimica.
bibliografia
Bertolini 2004: Bertolini, Luca e altri, Corrosion of steel in concrete, Weinheim, Wiley-VCH, 2004.
Bianchi, Mazza 1989: Bianchi, Giuseppe - Mazza, Francesco, Corrosione e protezione dei metalli, 3. ed., Milano, Masson Italia, 1989.
Corrosion, in: Metals handbook, 9. ed., Metals Park (Ohio), American Society for Metals, XIII, 1987.
Dexter 1986: Biological induced corrosion, edited by Stephen C. Dexter, Houston, National Association of Corrosion Engineering, 1986.
Fontana 1986: Fontana, Mars G., Corrosion engineering, 3. ed., New York-London, McGraw-Hill, 1986.
Hoar 1971: Hoar, Thomas P., Report of the Committee onCorrosion and Protection, London, HMSO, 1971.
Jones 1991: Jones, Denny A., Principles and prevention ofcorrosion, New York, Macmillan, 1991.
LaQue 1975: LaQue, Francis L., Marine corrosion, New York, Wiley Interscience, 1975.
Lazzari, Pedeferri 2006: Lazzari, Luciano - Pedeferri, Pietro, Cathodic protection, Milano, Polipress, 2006.
Pedeferri 2007: Pedeferri, Pietro, Corrosione e protezione dei materiali metallici, Milano, Polipress, 2007.
Pourbaix 1973: Pourbaix, Marcel, Lectures on electrochemical corrosion, New York, Plenum, 1973.
Revie 2000: Uhlig’s corrosion handbook, 2. ed., edited by Robert W. Revie, New York, Wiley, 2000.
Shreir 1994: Corrosion, edited by Lionel L. Shreir, London, Newnes, 1994.