
Difese contro le infezioni virali
Difese contro le infezioni virali
Come il sistema immunitario si è evoluto per combattere le infezioni, così gli agenti patogeni si sono evoluti per sfuggirlo. La forma e la struttura degli agenti patogeni e del sistema immunitario sono il prodotto di questa costante battaglia per la sopravvivenza. Durante questo processo di coevoluzione, sia gli agenti patogeni sia il sistema immunitario hanno sviluppato i propri punti di forza, ma anche le proprie debolezze. Questa competizione ha prodotto un sistema con molteplici livelli di difesa. Gli agenti patogeni si scontrano inizialmente con le difese innate non adatta bili, poi con quelle specifiche. Questi due meccanismi si fondono armoniosamente e diversi processi si svolgono parallelamente, in modo sia indipendente sia cooperativo. Molte delle tipologie di interazione tra sistema immunitario e agente patogeno sono attualmente conosciute a livello molecolare. Malgrado il raggiungimento di questi traguardi, le infezioni costituiscono ancora la maggiore minaccia per la salute nel mondo. La sfida per il futuro consisterà nell'applicare le nostre conoscenze in campo immunologico per debellare i continui attacchi dei germi patogeni.
Introduzione
Durante tutta la vita, il sistema immunitario ci difende dai continui attacchi di agenti patogeni potenzialmente letali, mentre tollera microbi che non rappresentano un'immediata minaccia. I microbi che si sviluppano sulla cute e nell'intestino superano in rapporto di circa 10:1 le nostre cellule. Si è stimato che il sistema immunitario affronti ed elimini, solo nel primo anno di vita, in media nove infezioni virali a livello respiratorio (Denny, 1995), ciascuna delle quali genera un nuovo insieme di antigeni che devono essere riconosciuti dallo stesso sistema. In ogni successivo anno di vita, il sistema immunitario incontra numerosi nuovi antigeni.

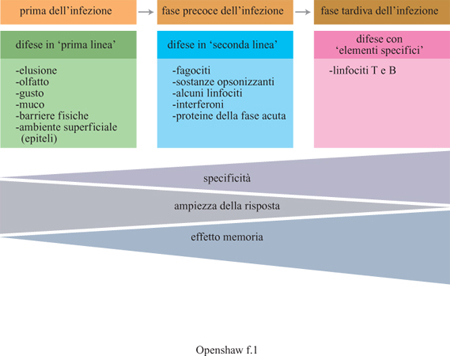
I meccanismi effettori che eliminano le infezioni costituiscono un'intricata rete, i cui singoli componenti sono tra loro interconnessi. In tabella (tab. I) sono indicate le principali componenti di questo sistema sequenziale di difese immunitarie (fig. 1). Sebbene la rete sia piuttosto complessa, si conoscono attualmente con notevole dettaglio le basi molecolari del funzionamento delle singole componenti.
La via preferenziale di ingresso della maggior parte degli agenti patogeni virali è costituita dalle mucose. La minaccia delle infezioni respiratorie e intestinali alla salute mondiale è in un certo senso inevitabile. Respirare, bere, mangiare e avere rapporti sessuali sono attività ineliminabili; queste funzioni necessitano di scambi di materiali con l'ambiente e con altri ospiti. Qualsiasi agente patogeno ben adattato è spinto a sfruttare queste funzioni per guadagnarsi l'accesso all'ambiente interno dell'organismo.
La cute e le mucose non sono solo i luoghi ideali di invasione, ma sono anche punti strategici per diffondere gli agenti patogeni ad altri ospiti. L'irritazione della mucosa nasale causata dal comune virus del raffreddore provoca la secrezione di muco, che contiene abbondante virus infettivo. Starnuti e tosse causano l'espulsione delle secrezioni. Particelle di muco si depositano sulle mucose dei nuovi ospiti. Il ciclo dell'infezione non richiede che il virus si diffonda in altri tessuti al di fuori del tratto respiratorio.
La trasmissione può in realtà essere più 'efficace' se le condizioni della persona infetta non sono così gravi da dover evitare i contatti sociali. Gli agenti che causano la diarrea e il vomito garantiscono la loro diffusione tramite questi effetti, che raramente sono letali per l'ospite. Si può sostenere che gli agenti patogeni che causano il comune raffreddore e forme acute gastrointestinali hanno raggiunto uno stadio ideale di evoluzione stabile. Aumentare o diminuire la patogenicità sarebbe uno svantaggio per l'aggressore. Gli agenti patogeni che causano elevata mortalità possono forse essere considerati 'intrusi' instabili, che ancora devono evolversi fino allo stato di equilibrio con l'ospite.
Le stime dell'Organizzazione mondiale della sanità indicano che circa 14 milioni di persone muoiono ogni anno di infezioni trasmesse attraverso le vie respiratorie, a confronto con i 5 milioni di decessi per malattie trasmesse attraverso la via gastrointestinale, che colpiscono per lo più i bambini. Le infezioni virali dei bambini sono di importanza fondamentale nel loro sviluppo e nell'andamento delle malattie nell'adulto. Si spera che ulteriori progressi nella comprensione delle difese contro le malattie infettive rendano tali patologie soggette a prevenzione e trattamento, riducendo in tal modo anche il loro costo per la salute umana.
Difese a livello di superficie
l virus hanno bisogno di cellule viventi per replicarsi ed è significativo che quasi tutte le cellule visibili sulla cute siano morte. l papillomavirus hanno bisogno di una lacerazione della cute per formare una verruca; anche l'HlV non riesce a infettare attraverso la cute intatta. Sebbene lo strato superficiale del tratto respiratorio e di quello gastrointestinaIe contengano cellule vive, la maggior parte di esse muore e viene eliminata dall' organismo in 24 ore. Questo 'muro di cadaveri' è senza dubbio la più importante singola difesa contro le infezioni invasive. Le cellule morte, tuttavia, non sono le sole difese di superficie. Batteri e funghi non patogeni, cioè la normale flora, ricoprono la cute, il tratto aereo superiore e il tratto digestivo, competendo con batteri potenzialmente nocivi per i siti di adesione e per le sostanze nutritive. Riducendo la densità di questa flora normale con gli antibiotici si può aumentare il potenziale invasivo degli agenti microbici non sensibili ai trattamenti. La competizione per una posizione offre una barriera importante alle infezioni invasive.
Nel tratto respiratorio vi è un costante flusso di muco dalle regioni profonde dei polmoni e dal naso verso la faringe, dove il muco è normalmente deglutito o in certi casi eliminato. Questo velo di muco comprende isole di spesso gel in un mare di sol. Il muco contiene enzimi antibatterici e antivirali, come illisozima, presente nella saliva, nel sudore e nelle lacrime. Inoltre le mucoproteine che contengono acidi sialici si adsorbono su virus come quello dell'influenza A, riducendo la capacità dei virus stessi di aderire ai recettori delle mucose. La cute, invece, produce secrezioni sebacee contenenti acidi grassi, che sono potenti agenti antimicrobici. Nell'intestino, l'alternanza di pH basso e alto e la peristalsi che smuove il materiale dallo stomaco all'intestino tenue, le proprietà detergenti ed enzimatiche della bile e delle altre peptidasi intestinali eliminano gran parte degli agenti infettivi che entrano nel tratto intestinale. Sulle mucose si è evoluto un sistema di difesa complesso, esteso e altamente specializzato. La presenza di materiali estranei sulla superficie di tali mucose necessita di un'attenta regolazione da parte delle risposte immunitarie per evitare danni ai tessuti vicini. La funzione di queste difese di superficie consiste nella protezione contro l'invasione da parte di potenziali agenti patogeni, tollerando però materiali non patogeni. Al contrario, il sistema immunitario interno funziona normalmente in un ambiente sterile e reagisce in modo più vigoroso alle sostanze antigeniche. Il successo dell'eliminazione degli antigeni dalle mucose, ottenuto senza danneggiare i tessuti vicini, dipende da risposte immunitarie bilanciate. Se i meccanismi immunitari non infiammatori falliscono nell' eliminazione dell' antigene, prevarranno meccanismi pro infiammatori che causeranno un danno. Questa alterazione immunitaria è responsabile di alcune manifestazioni cliniche che si verificano durante l'infezione virale.
Risposte immunitarie innate
Macrofagi
l macrofagi (letteralmente, grosse cellule mangiatrici) sono una componente chiave del sistema immunitario innato. Essi inglobano i virus e altri agenti patogeni che sono stati neutralizzati dagli anticorpi circolanti e che devono essere distrutti senza danneggiare i tessuti circostanti. l macrofagi derivano da quelle cellule del midollo osseo che diventano monociti del sangue. Questi si differenziano in macrofagi nei polmoni, nei linfonodi, nella milza, nella cute, nel sistema nervoso centrale (dove formano la microglia) e nel fegato (dove costituiscono le cellule di Kupffer). Sebbene la loro funzione primaria sia inglobare la materia particolata, l'ingestione è più efficace se la particella è ricoperta dagli anticorpi o dal complemento, che hanno entrambi la funzione di opsonizzare la particella. Si può paragonare questo processo di opsonizzazione all'aggiunta di salse o sughi ai cibi per renderli più gustosi e appetibili. Dopo aver inglobato la particella, il macrofago digerisce il contenuto della vescicola endocitata. l prodotti di digestione della particella vengono trasportati alla superficie cellulare in associazione con il complesso maggiore di istocompatibilità (MHC, Major Histocompatibility Complex). Se questo frammento peptidico non è riconosciuto come proprio dell' ospite, vengono attivati i linfociti T helper (v. oltre).
Il processo di ingestione inizia con il legame del materiale opsonizzato al recettore del frammento cristallizzabile sulla membrana dei macrofagi e continua con l' endocitosi in una vescicola interna, chiamata fagosoma. Quest'ultimo si fonde con una vescicola, contenente lisozima e altri enzimi digestivi, chiamata liso soma; la vescicola che risulta dalla fusione si chiama fagolisosoma. Una combinazione di pH basso, mantenuto dalle pompe ioniche, di idrolasi acide, di perossido di idrogeno, di radicali reattivi dell'ossigeno e di ossido d'azoto è in grado di demolire il contenuto del fagoliso soma. Talvolta la particella da distruggere è troppo grossa per essere fagocitata, come nel caso di un parassita. Questo provoca il rilascio degli enzimi lisosomiali nel mezzo extracellulare, con la possibilità di causare danni localizzati alle cellule dell'ospite.
I macrofagi sono predominanti nei granulomi, processi infiammatori in cui vari tipi di cellule effettrici (monociti, linfociti, cellule polimorfonucleate), attratte dai fattori chemiotattici rilasciati dopo l'invasione del tessuto da parte di materiali estranei particolati (inclusi agenti patogeni), formano una barriera attorno alla particella estranea. Esempi tipici di questo fenomeno si osservano nella tubercolosi, dove micobatteri (gli agenti che causano la tubercolosi o la lebbra) possono sopravvivere per decenni all'interno di un granuloma.
Molti agenti microbici importanti hanno sviluppato sistemi per evitare gli effetti antimicrobici dei macrofagi. Alcuni virus, specialmente dengue e HIV, si sviluppano effettivamente dentro i macrofagi e possono usare anticorpi antivirali per attivare la loro capacità di infettare i macrofagi stessi, che divengono inefficaci e funzionano persino da 'cavallo di Troia', trasportando l'infezione in altre regioni del corpo. Alcuni agenti patogeni non virali (per esempio, Leishmania e molti micobatteri) sono ospiti intracellulari obbligati e riescono a replicarsi soltanto all'interno della cellula. I macrofagi sono efficienti non solo nel digerire antigeni e nel presentare componenti estranee, ma anche nel produrre alcune citochine, fra cui le interleuchine l e 6 (IL-l e IL-6). Un macrofago infetto allerta perciò le cellule circostanti, inclusi i linfociti B, della presenza di materiale estraneo potenzialmente infettivo.
Cellule natural killer e interleroni
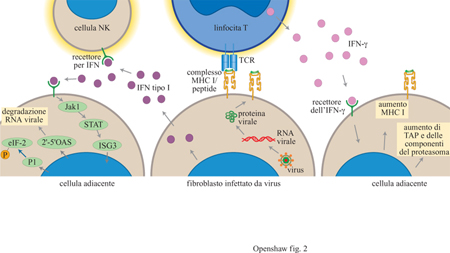
Una seconda componente della risposta immunitaria innata è costituita dal sistema delle cellule natural killer (NK) che di solito è collegato con l'azione degli interferoni (fig. 2).
L'interferone α/β, o interferone di tipo I, è prodotto da una grande varietà di cellule in seguito all'infezione virale e attiva la citotossicità mediata dalle cellule NK tramite il legame a specifici recettori. Alcuni virus sono in grado di produrre una molecola solubile analoga al recettore dell'interferone di tipo I che inibisce l'effetto antivirale dell'interferone stesso, competendo con i normali meccanismi di attivazione. L'esistenza di questo meccanismo di evasione virale suggerisce un ruolo importante dell'interferone nella resistenza ai virus. Oltre che dalle cellule NK, l'interferone γ è rilasciato dai linfociti T attivati; esso induce anche uno stato antivirale, per esempio inducendo nel topo il gene Mx che genera resistenza al virus influenzale, e stimola il processamento e la presentazione dell'antigene in modo simile all'interferone di tipo I. L'interferone γ è prodotto dalle cellule T che si infiltrano nei gangli neuronali infettati dal virus Herpes simplex e si ritiene possa essere coinvolto nel passaggio dalla fase litica al ciclo di replicazione latente del virus (v. oltre).
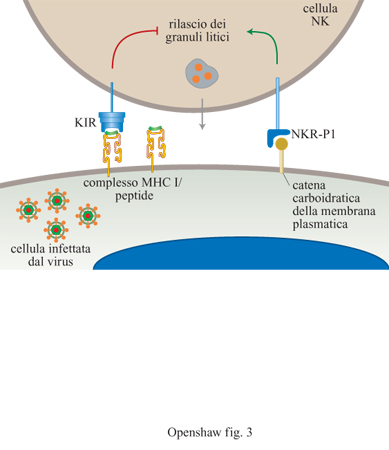
Le cellule NK differiscono dalle cellule T citotossiche in quanto non si differenziano nel timo. Il meccanismo attraverso cui le cellule NK riconoscono ed eliminano gli agenti patogeni è rimasto oscuro fino a tempi molto recenti (Biron, 1997). Le cellule NK sono reclutate entro i primi due giorni dall'infezione e si dividono e si differenziano localmente. Esse hanno importanti effetti antivirali, in parte perché uccidono le cellule infettate attraverso la formazione di granuli citolitici e in parte perché producono interferoni α, β e γ. Molti progressi in questo campo hanno riacceso l'interesse per le cellule NK, in particolare quelli riguardanti la descrizione di numerosi recettori esposti sulla superficie delle cellule NK e di potenziali cellule bersaglio che regolano l'attività delle cellule NK stesse. La prima interazione di questo tipo avviene tra l'MHC di classe I sulle normali cellule bersaglio e i recettori inibitori dei killer (KlR, Killer Inhibitory Receptor) sulle cellule NK (fig. 3). Il coinvolgimento delle molecole di classe I manda un segnale negativo alle cellule NK, inibendo la loro azione litica. L'assenza o l'alterazione di molecole di classe I previene la stimolazione di questo segnale inibitorio. La cellula NK viene quindi attivata da segnali trasmessi attraverso un secondo recettore e ciò provoca il rilascio del contenuto della cellula NK. Il rilascio direzionale di granuli provoca apoptosi nella cellula bersaglio. Qualsiasi cellula contenente un virus che diminuisca l'espressione delle molecole di classe I è soggetta a essere uccisa da questo meccanismo (v. il saggio di E.I.H.I. Wiertz, nel II volume).
Alcuni virus, come per esempio il citomegalovirus (CMV) umano e murino, cercano di sabotare questo meccanismo codificando molecole omologhe all'MHC di classe I. Inoltre, le cellule normali non infettate rispondono al rilascio locale di interferone aumentando l'espressione della classe I. Il fenomeno rende queste cellule, normalmente sensibili, resistenti all'uccisione da parte delle cellule NK. Se le cellule sono infettate, questa aumentata espressione può non avvenire, rendendo le cellule più suscettibili all'uccisione mediata da cellule NK. Questo è uno degli esempi dell'azione dei recettori inibitori dei killer, ma le cellule NK possono provare una varietà di KlR prima di raggiungere un punto definitivo e svuotare i granuli citotossici. Una volta che le risposte specifiche locali delle cellule T sono attivate, meccanismi aggiuntivi inibiscono le funzioni delle cellule NK. L'attività di queste cellule potenzialmente dannose è perciò di breve durata, ma copre l'intervallo tra l'inizio dell'infezione e lo sviluppo dell'immunità specifica.
Risposte immunitarie acquisite
Mentre le risposte immunitarie innate vengono innescate precocemente e hanno scarsa o nessuna specificità per il particolare agente patogeno, quelle acquisite si verificano più tardi nel tempo ma sono altamente specifiche. llinfociti specifici per l'antigene proliferano e si differenziano durante questo intervallo di tempo, raggiungendo valori che sono sufficienti a costituire una difesa efficace. Se si è incontrato un agente patogeno identico o simile in precedenza, il ritardo si riduce e la potenza della risposta immunitaria è fortemente rafforzata; questo fenomeno viene definito memoria immunitaria. Sulle mucose, le cellule linfoidi si concentrano in aggregati specializzati chiamati MALT (Mucosal Associated Lymphoid Tissue, tessuto linfoide associato alla mucosa). Quando queste cellule migrano, preferenzialmente tornano indietro, proprio nelle aree mucose. Questo processo di homing (localizzazione) è dovuto all'espressione di recettori di superficie che riconoscono in modo specifico le addressine vascolari, presenti sull'endotelio di venule specializzate dei tessuti linfoidi, le HEV (High Endothelial Venules, venule con endotelio alto).
Linfociti B
Uno dei principali tipi cellulari coinvolti nell'immunità acquisita è costituito dai linfociti B. Le cellule B sono così chiamate perché sono state studiate nella borsa di Fabrizio del pollo. In questa specie, l'asportazione della borsa provoca l'eliminazione delle cellule B e della produzione di anticorpi durante lo sviluppo. Le cellule B mature portano in superficie le immunoglobuline, che agiscono come recettori dell' antigene, e sono distribuite dal sangue e dalla linfa ai tessuti linfoidi, in particolare ai linfonodi e alla milza. Non esiste un equivalente anatomico della borsa di Fabrizio nei mammiferi, dove lo sviluppo delle cellule B inizia nel midollo osseo dalle cellule staminali che derivano originariamente dal fegato. Queste cellule formano un lignaggio attraverso le cellule pro-B, le cellule pre-B, fino alle cellule B vere e proprie e infine alle plasmacellule (v. il saggio di F. Melchers, Lo sviluppo dei linfociti). Per progredire in questo processo di maturazione, le cellule B rispondono a stimoli di sviluppo, in modo particolare a lL-7 prodotta dalle cellule stromali nel midollo osseo. Durante l'ontogenesi, i geni che codificano le molecole di anticorpi vengono ricombinati per produrre catene pesanti e leggere uniche che costituiscono il recettore specifico per l'antigene (v. il saggio di C. Rada, nel II volume). La plasmacellula rappresenta lo stadio finale di sviluppo, durante il quale sono se crete abbondanti quantità di anticorpi. Esistono due tipi principali di risposte da parte delle cellule B, quella dipendente dalle cellule T e quella indipendente dalle cellule T. È stato proposto che la via T-indipendente sia diretta soprattutto contro gli antigeni ripetitivi, come quelli presenti sulla superficie di molti agenti microbici, e che questa via sia responsabile dell'induzione di potenti risposte antivirali. Tali risposte sono più rapide, non richiedono aiuti, di preferenza inducono risposte neutralizzanti e molto durature, soprattutto se confrontate con le risposte T -dipendenti (Bachmann e Zinkemagel, 1996). Al contrario, le risposte T -dipendenti, come quelle verso le proteine coniugate agli apteni o agli eritrociti di pecora, dipendono dalla costimolazione delle cellule B di molecole accessorie, che avviene grazie alle interazioni che si stabiliscono tra gli anticorpi monoclonali B7.1 o B7.2, o entrambi, sui linfociti e il CD28 sulla superficie dei linfociti T helper, o tra CD40 e il suo ligando. L'interazione con i linfociti B induce inoltre i linfociti T helper a rilasciare citochine, che inducono la proliferazione, la produzione di anticorpi e lo switch (commutazione) isotipico dei linfociti B.
Le immunoglobuline 'saggiano' tutte le proteine con le quali vengono in contatto e formano legami stabili solo con molecole la cui superficie mostra caratteristiche chimicofisiche, come forma, distribuzione di cariche e idrofobicità, che esprimono la maggiore complementarità per l'immunoglobulina (v. il saggio di R.A. Mariuzza, Riconoscimento dell'antigene da parte dei recettori del sistema immunitario). Quando la corrispondenza spaziale è esatta, l'immunoglobulina resta attaccata sufficientemente a lungo per innescare altre risposte. Ciò significa che devono esistere tante immunoglobuline sufficientemente differenti per riconoscere quasi tutte le proteine patogene esistenti e potenziali. Nello stesso tempo, si deve evitare il riconoscimento delle molecole proprie dell'organismo. Individuare il modo in cui sono prodotte le diverse immunoglobuline è stato uno dei principali enigmi dell'immunologia, risolto solo pochi anni or sono. La generazione della diversità è raggiunta combinando molti diversi segmenti genici codificanti polipeptidi che riarrangiano durante lo sviluppo per produrre catene ibride leggere e pesanti (v. i saggi di G.J.V. Nossal e di C. Rada, nel II volume). Nella linea germinale esistono molti elementi variabili che possono ricombinarsi con una notevole percentuale di 'imprecisione'. Le mutazioni puntiformi sono frequenti e diverse catene leggere e pesanti possono associarsi per produrre almeno 10¹⁶ forme diverse. La variabilità si concentra nei domini di legame per l'antigene delle immunoglobuline. Sebbene questa variabilità innata non sia elevata come quella del recettore delle cellule T, le cellule B possiedono un'altra via molto utile per raggiungere la specificità: la mutazione somatica. Questa è raggiunta permettendo l'introduzione di errori durante la trascrizione del DNA, caso che si verifica circa una volta ogni appaiamento di mille coppie di basi. Questa frequenza è 100 volte maggiore della frequenza di mutazione dell'RNA virale e suggerisce un interessante parallelo tra la capacità di microevolvere del sistema immunitario e quella degli agenti patogeni. Le cellule B contenenti errori che migliorano il legame con l'antigene sono ricompensate da un aumento di stimolazione, crescita e divisione cellulare. Le cellule che producono anticorpi che legano fortemente particelle estranee proliferano, producendo una maggior quantità di anticorpi che trovano così un bersaglio opportuno.

La seconda componente delle immunoglobuline è chiamata frammento cristallizzabile (Fc, Fragment crystallizable). Per una data sotto classe di anticorpi questa componente è costante e non è rivolta verso l'antigene (fig. 4). Essa interagisce con il complemento o con i propri recettori sulle cellule, determinando così l'effetto del legame dell'antigene alla porzione Fab (Fragment antigen-binding, frammento che lega l'antigene). l differenti tipi di immunoglobuline sono identificati in relazione alla componente Fc e ciascuna ha un diverso ruolo. Così come la risposta immunitaria si modifica nel tempo, anche le cellule B maturano e vanno incontro allo switch isotipico. Lo stesso Fab codificato dai geni V H si unirà successivamente con diversi prodotti dei geni CH, in modo da produrre IgM, IgG, ecc. Le IgG costituiscono le più abbondanti immunoglobuline del siero e i principali anticorpi presenti dopo l'immunizzazione con la maggior parte degli antigeni. Non sono attivamente secrete nel tratto respiratorio ma travasano dal circolo sanguigno quando la mucosa è infiammata. Le cellule B che producono IgG di solito giacciono negli strati più profondi della mucosa. Le IgG sono molto potenti nell'opsonizzare l'antigene che è inglobato dalle cellule polimorfonucleate (PMN) e dai macrofagi. L'adesione è seguita dall'ingestione (fagocitosi) e dalla fusione del fagosoma internalizzato con i lisosomi. Gli enzimi proteolitici, i radicali dell' ossigeno e l' acidificazione del fagolisosoma contribuiscono attivamente alla demolizione del contenuto del fagolisosoma stesso.
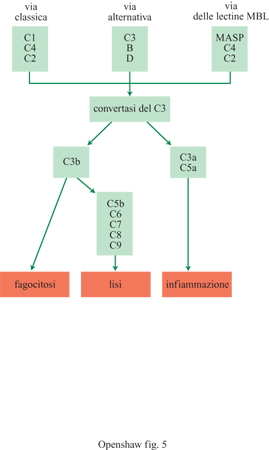
l monomeri di IgM possono unirsi con le catene J che legano le componenti secretrici sulla superficie basolaterale degli enterociti. Questi anticorpi rilasciati complessano l'antigene che è quindi eliminato dall'interno dell'organismo (esclusione immunitaria). Le IgM sono anticorpi opsonizzanti assai potenti e sono presenti nel momento in cui le cellule B incontrano l'antigene per la prima volta. Sono anche forti attivatori delle componenti del complemento, che formano pori nelle cellule (lisi). La componente inattiva del complemento, chiamata Clqrs (dove q, re s sono le tre subunità del complesso Cl) può essere attivata dal legame a una singola molecola IgM (o ad almeno due molecole IgG), che è andata incontro a un cambio conformazionale dopo il legame con l'antigene. Questa a sua volta attiva Clq, che attiva Clr, che attiva CIs (v. fig. 4c), iniziando la via classica di attivazione del complemento (fig. 5).
Le IgA sono immunoglobuline specializzate presenti a basse concentrazioni nel siero ma molto abbondanti nelle secrezioni mucose. Sono attivamente trasportate attraverso le cellule epiteliali, dove si legano alla componente secretrice, e quindi rilasciate nelle secrezioni (latte, saliva, ecc.). La loro azione antivirale si esplica, per esempio, proteggendo la mucosa intestinale dall' infezione da parte di poliovirus e il tratto respiratorio dai comuni virus influenzali, inclusi il virus respiratorio sinciziale e quello dell'influenza A. Le IgD sono presenti a livelli bassissimi nel siero, ma si trovano sulla superficie di molte cellule B nelle fasi precoci del differenziamento. Anche le IgE sono presenti a concentrazioni bassissime nel siero, ma sono importanti perché la componente Fc si lega molto fortemente ai mastociti, ai baso fili e agli eosinofili. Un antigene che si leghi alle IgE fissate alla superficie di queste cellule provoca il rilascio di istamina, bradichina, enzimi, leucotrieni, fattore attivante le piastrine, ecc. Si ritiene che siano particolarmente importanti nella protezione contro infezioni da parte di vermi, ma che inneschino anche reazioni allergiche, come, per esempio, febbre da fieno o asma. Gli anticorpi contro i virus possono essere neutralizzanti; in questo caso il legame dell'anticorpo blocca la capacità infettiva del virus ostruendo fisicamente i siti critici per l'adesione e l'ingresso del virus sulle proteine di superficie. In alternativa, l'anticorpo può legare altri siti che non interferiscono con il legame del virus. Questo anticorpo non neutralizzante può opsonizzare il virus direttamente o lasciarlo legare dal complemento. Infine, alcuni virus sembrano infettare meglio in presenza di un anticorpo, usando l'opsonizzazione per penetrare in cellule che non sono capaci di distruggere il virus. Questi sono anticorpi che aumentano l'infezione.
L'anticorpo, di qualsiasi tipo sia, è spesso inefficace nel prevenire le infezioni virali delle superfici mucose; ciò dipende dal fatto che esso non è specifico o è presente nel posto o nel momento sbagliati. Se le cellule B non hanno ancora incontrato un determinato antigene virale, gli anticorpi con la corretta specificità impiegano tempo per espandersi. Per esempio, se il sistema immunitario non ha mai incontrato il ceppo di influenza diffuso in un particolare anno, gli anticorpi contro altri ceppi sono inefficaci contro le proteine di superficie del nuovo ceppo. Un caso recente è stato l'esplosione dell'influenza aviaria H5Nl nel 1997 a Hong Kong. Alcune altre specie di virus del raffreddore, per esempio i rhinovirus, sono in grado di assumere un centinaio di rivestimenti diversi, costringendo il sistema immunitario a trattare ogni infezione come se fosse nuova. Nel caso del virus respiratorio sinciziale, inoltre, anticorpi presenti nel siero ai normali livelli sono inefficaci contro la reinfezione per via nasale. Al contrario, IgA localizzate nel naso sono efficaci, ma persistono solo per 9 mesi dopo l'infezione. Purtroppo, dato che le esplosioni virali si succedono circa una volta all'anno, questi anticorpi sono di poca utilità nella protezione contro le ondate successive.
Processamento e presentazione degli antigeni virali alle cellule T
Le cellule T riconoscono esclusivamente gli antigeni che sono stati processati, cioè ridotti a livello di frammenti peptidici. Questi frammenti sono presentati sulla superficie delle normali cellule legati a specifici siti di proteine di superficie, chiamate MHC. Se le cellule sono infettate da un virus, i nuovi peptidi endogeni, compresi alcuni peptidi di origine virale, vengono campionati dall'MHC di classe I e portati sulla superficie cellulare. Al contrario, alcune cellule specializzate che presentano l'antigene (APC, Antigen Presenting Cells) prelevano proteine dal mezzo extracellulare e le digeriscono in frammenti peptidici per la presentazione da parte dell'MHC di classe II. Quando le cellule T incontrano questi nuovi complessi MHC/peptide, vi si legano attraverso il recettore delle cellule T (TCR, T Cell Receptor); il legame è stabilizzato da CD4, molecola attraverso la quale le cellule T coadiuvanti legano la classe II, o da CD8, molecola attraverso la quale i linfociti T citotossici (CTL, Cytotoxic T Lymphocytes) legano la classe I. Singoli linfociti T maturi riconoscono un numero molto limitato di antigeni. Il riconoscimento avviene attraverso l'appaiamento della struttura della regione apicale del recettore delle cellule T, posto sulla superficie della cellula T, con una struttura complementare dell'antigene presentato da altre cellule ospiti.
Le più importanti cellule APC sono i macrofagi, le cellule dendritiche e i linfociti B. La modalità con cui l' antigene entra nelle APC è rilevante per determinare quale tipo di cellula T verrà stimolata dall'antigene. Antigeni esogeni (solubili) vengono inglobati dalle APC, in modo particolare quando sono opsonizzati dagli anticorpi o dal complemento, o quando sono legati all' anticorpo sulla superficie delle cellule B. Dopo essere stato intemalizzato, l'antigene viene demolito nei fagolisosomi. l peptidi derivati dalla digestione si incontrano con le proteine dell'MHC di classe II neo sintetizzate (o riciclate) che sono accompagnate, fino a questo punto, da una proteina chiamata catena invariante (v. il saggio di G.J. Hämmerling e collaboratori, nel II volume). La catena invariante si lega strettamente all'MHC di classe II e si infila nel solco sull' apice della proteina di classe II, dove si è formato il sito di legame per i peptidi prodotti dalla digestione dell'antigene. Dopo che la catena invariante viene rimossa, il sito di legame per il peptide cattura un qualsiasi peptide disponibile che si adatti al solco. Se non sono disponibili peptidi estranei, l'MHC II cattura peptidi normali della cellula ospite prodotti dal naturale riciclo delle proteine in questo comparto della cellula. Ogni proteina MHC II può potenzialmente trattenere diverse migliaia di peptidi differenti. Sebbene sembri un numero enorme, appare piccolo se confrontato con il numero potenziale di peptidi presenti in ogni cellula. Questi peptidi sono di solito lunghi 15 ÷18 amminoacidi e sporgono per un breve tratto a una delle estremità del solco dell'MHC II. Si pensa che circa 100 o 200 proteine MHC debbano essere occupate dal peptide appropriato perché avvenga il riconoscimento. Questa quota rappresenta 0,1 ÷ 10% delle proteine MHC presenti sulla superficie di una APC.
La via di presentazione degli antigeni endogeni mostra alcune importanti differenze rispetto alla presentazione di antigeni esogeni. Gli antigeni endogeni sono sintetizzati dentro la cellula e quindi demoliti in peptidi di 8 ÷ 10 amminoacidi. Questi peptidi più corti incontrano la proteina MHC di classe I mentre essa viene prodotta nel reticolo endoplasmatico rugoso e l'associazione del peptide con l'MHC l aiuta a stabilizzare il legame con la β2-microglobulina. Il complesso trimolecolare si muove attraverso l' apparato del Golgi, dove le proteine vengono glicosilate prima di essere esposte in superficie. l peptidi si adattano al solco all'apice dell'MHC l in modo quasi analogo ai peptidi che si adattano all'MHC 11, tranne per il fatto che il solco dell'MHC l è chiuso a entrambe le estremità. Ciò implica che ciascun MHC l ha una scelta più limitata di peptidi che vi si adattano in modo adeguato: per questa ragione, vengono presentate solo poche centinaia di peptidi diversi entro il solco degli MHC l sulla superficie di cellule caratteristiche. Questo repertorio limitato è compensato, in parte, da una gamma più vasta di cellule che esprimono l'MHC I. La maggior parte delle cellule nucleate porta l'MHC l, incluse tutte le cellule dell' epitelio delle mucose, le cellule che presentano l'antigene, i fibroblasti e l'endotelio dei vasi. Al contrario, l'MHC II è presente in quantità importanti solo su cellule APC 'professioniste'. Le due proteine MHC funzionano perciò in due modi fondamentalmente diversi. L 'MHC di classe I mostra quali proteine la cellula sta sintetizzando e invita i linfociti T citotossici a uccidere la cellula che mostra complessi MHC/peptide anomali. L'MHC di classe II è invece presente solamente sulle cellule che sono specializzate nel 'diffondere notizie', che campionano le proteine presenti nel mezzo extracellulare e innescano una risposta immunitaria coordinata mediata dalle cellule T helper.
La capacità delle cellule T di discriminare tra il self e il non self, cioè tra ciò che è proprio dell' organismo e ciò che è estraneo, si sviluppa nel timo. Dopo aver lasciato il midollo osseo, le cellule destinate a diventare cellule T migrano nel timo; qui vanno incontro a una serie di cambiamenti nel passaggio dallo strato corticale a quello midollare. Il timo è effettivamente una 'stazione di esame', progettata per selezionare le cellule T con bassa reattività verso gli MHC, che presentano antigeni del self ma sono peraltro capaci di riconoscere peptidi estranei presentati da MHC propri. Appena dopo il loro arrivo, le cellule migrate dal midollo osseo mostrano sulla propria superficie sia CD4 sia CD8. Quando entrambe queste proteine sono presenti con un complesso TCR, la forza di interazione con le cellule che presentano l'antigene nello stroma del timo determina il destino della cellula T in via di sviluppo. Quelle che si legano troppo fortemente o troppo debolmente vengono eliminate o entrano in uno stato di inattività semipermanente, denominata anergia. Questo processo di selezione timica è molto rigoroso e la maggior parte delle cellule T in sviluppo muore nel timo per apoptosi. Le cellule T selezionate per essere di potenziale utilità futura per l'ospite si moltiplicano ed escono dal timo; circolano poi attraverso i linfonodi, la milza, il fegato, l'intestino, la cute e altre regioni, cercando nuovi complessi MHC/peptide sulla superficie delle cellule ospiti. Una volta in periferia, le opportunità di ulteriori cambiamenti nelle proteine di superficie delle cellule T sono estremamente scarse. Queste cellule T hanno un destino ben stabilito e difficilmente mutabile: infatti, hanno perso la capacità di esprimere altri corecettori e non possono modificare il loro TCR. Esse restano quiescenti fino a quando vengono in contatto con una cellula che possiede le caratteristiche per il cui riconoscimento sono programmate e solo allora si attivano. Il processo attraverso il quale le cellule T raggiungono le cellule che presentano l' antigene non è soltanto casuale, sebbene la frequenza giochi una parte quando vi sono solo pochissimi linfociti con un TCR che si adatta intimamente con qualsiasi complesso MHC/peptide. Nelle zone in cui avviene l'infezione, l'infiammazione provoca un aumento del flusso sanguigno, un travaso più attivo di linfociti nei tessuti e un'attivazione delle APC, che aumenta la quantità di proteine MHC sulla superficie. Quando una cellula che presenta l'antigene incontra il TCR complementare, la cellula T è portata in uno stato di attivazione metabolica, che culmina con la divisione cellulare. Il processo di sviluppo dei linfociti è perciò un equilibrio finemente controllato tra crescita, inattività, attività e morte. Tutte queste caratteristiche sono essenziali per un corretto funzionamento del sistema immunitario. Lo sviluppo dei linfociti corrisponde, in un certo senso, allo sviluppo e alla capacità adattativa degli organismi patogeni, contro cui il sistema immunitario si è evoluto. La diversità potenziale dei TCR è immensa. Si è calcolato che i geni per i TCR sono capaci di unirsi in modi diversi per generare circa 10¹⁶ specificità differenti, assicurando che uno o più TCR saranno quasi perfettamente adatti a ogni proteina estranea. Molte di queste possibilità saranno rigettate dal timo perché avranno una interazione troppo forte o troppo debole con le normali cellule ospiti. In ciascuno di noi vi è stata una corsa evolutiva effettuata dai nostri linfociti, già prima della nascita, attraverso la morte per apoptosi di circa il 95% delle cellule che hanno iniziato il programma differenziativo. Durante tutta la vita, i linfociti continuano a essere selezionati, resi tolleranti o eliminati. Una quota relativamente esigua di cellule T sopravvive negli anziani e questo spiega forse in parte le capacità limitate del sistema immunitario di rispondere alle infezioni in età avanzata.
Cellule T helper
La presenza di CD4 sulla superficie di una cellula T provoca un grande rafforzamento del legame del recettore delle cellule T all'MHC di classe II che porta il peptide 'giusto'. Quando le cellule vergini uscenti dal timo entrano in contatto con una cellula che presenta l'antigene, iniziano a proliferare e a produrre segnali chimici sotto forma di citochine. Esempi importanti di citochine prodotte da queste cellule T helper sono IL-l, lL-3, lL-4, IL-5, IL-10 e interferone γ. Queste sostanze chimiche segnalano ad altre cellule di prepararsi ad attaccare l'agente patogeno invasore. lL-3 è un fattore chiave che provoca la crescita e la divisione delle cellule staminali, producendo più cellule B e T. lL-2 ha un ruolo chiave nell'attivazione delle cellule T, mentre lL-4 e lL-6 attivano le cellule B. lL-3, in combinazione con GM-CSF (Growth Macrophage Colony Stimulating Factor, fattore che stimola la crescita della colonia di macrofagi), è un potente promotore della crescita e della proliferazione delle cellule fagocitiche, compresi eosinofili, polimorfonucleati, baso fili e macrofagi. Le cellule T helper giocano un ruolo centrale nell'identificare in modo preciso l'agente patogeno invasivo e nell'accendere il sistema d'allarme dell'ospite che coinvolge altri giocatori-chiave della difesa immunitaria.
Le cellule T helper possono essere classificate in tipi diversi, che producono combinazioni caratteristiche di citochine: alcune combinazioni dirigono le risposte immunitarie, mentre altre attivano la produzione di anticorpi da parte delle cellule B (Fiorentino et al., 1989; Mosmann e Sad, 1996; Street et al., 1990).
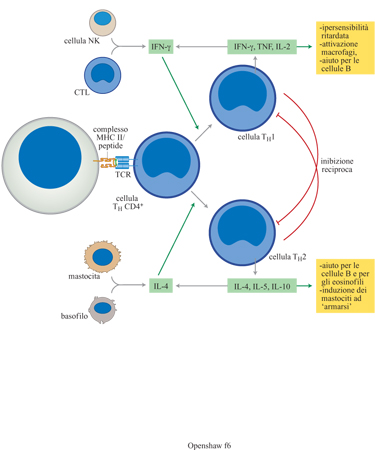
Alcuni antigeni e diverse modalità di immunizzazione inducono in modo differenziale due principali sotto gruppi funzionali, THl e TH2 (fig. 6). Le cellule THl producono cito chine di tipo l, che includono TNF-α (Tumor Necrosis Factor α, fattore di necrosi tumorale α) e lFN-γ; queste hanno un potente effetto antivirale. Le cellule TH2 producono cito chine di tipo 2 che comprendono lL-4, lL-5 e IL-10 e sono predominanti nelle risposte antielmintiche e allergiche. Popolazioni di cellule che producono citochine di tipo l e 2 sono mutualmente inibitorie. Sebbene sia stata osservata produzione polarizzata di citochine in cloni di cellule T provenienti da parecchie specie (sia di primati che di roditori), il grado di polarizzazione nelle cellule T naturali si sta chiarendo solo ora.
Cellule T citotossiche
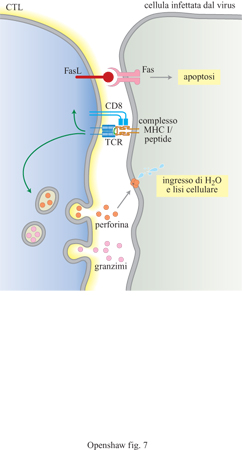
La via di processamento endogena porta dapprima all'attivazione delle cellule T con CD8, che funzionano per lo più come cellule T citotossiche. Di nuovo, il processo di coinvolgimento della cellula T 'giusta' consiste essenzialmente nel trovare un TCR con la struttura adeguata per adattarsi al complesso MHC/peptide. Una volta avvenuto il contatto, la presenza di CD8 sulla superficie della cellula T blocca la cellula T stessa sull' APC che porta il peptide appropriato. La risposta delle cellule T CD8+ (cellule T che esprimono CD8) è di nuovo l'attivazione (fig. 7); sebbene tali cellule producano anche citochine, soprattutto di tipo l, l'effetto principale dell'attivazione è che le cellule diventano citotossiche. l CTL attivati riorientano il cito scheletro e l'apparato secretore verso la cellula bersaglio e rilasciano granuli contenenti perforina e granzimi nello spazio formatosi tra le due cellule. La perforina forma polimeri in presenza di Ca²+ e si inserisce nel doppio strato lipidico della membrana della cellula bersaglio, formando pori delimitati da polimeri di perforina. Il conseguente ingresso di acqua e ioni provoca lo scoppio della cellula ospite. La cellula allora frammenta il suo DNA e il suo RNA e muore. A questo punto, la cellula T si stacca dalla cellula morente, si divide e si sposta per cercare altre cellule da contattare e uccidere. È importante che le cellule T citotossiche siano altamente selettive nella scelta dell'antigene; l'uccisione incontrollata di cellule erroneamente riconosciute come infettate sarebbe infatti disastrosa.
Un altro meccanismo citotossico, il principale utilizzato dai CTL che non possiedono granuli, consiste nell' espressione del ligando per Fas (FasL) indotta dall'attivazione. FasL si lega al suo recettore Fas presente sulla membrana della cellula bersaglio, portando bersaglio e CTL molto vicini. Il legame innesca l'apoptosi, che conduce alla frammentazione del DNA e alla morte cellulare. l CTL si staccano dal bersaglio dopo aver scagliato il colpo mortale e riescono così a uccidere più cellule in rapida successione.
Esempi di risposte immunitarie a particolari agenti patogeni
Virus respiratorio sinciziale
Le infezioni polmonari sono la causa dominante di morbilità e mortalità infantile. Il virus respiratorio sinciziale (RSV, Respiratory Syncytial Virus) è il principale agente patogeno dell'apparato respiratorio infantile a livello mondiale e rappresenta una sfida irrisolta per lo sviluppo di un vaccino (Heilman, 1990). Dal punto di vista clinico, l'infezione è caratterizzata da sintomi e segni di restringimento bronchiale e molti bambini che si sono ristabiliti dalla bronchiolite sono stati successivamente diagnosticati asmatici. Le difese immunitarie contro RSV sono interessanti per molti aspetti. Innanzi tutto, si verificano nuove infezioni durante la vita malgrado risposte immunitarie apparentemente forti alla prima infezione. Inoltre, la vaccinazione può aumentare la gravità della malattia. Infine, vi sono legami apparenti tra le infezioni infantili e il successivo sviluppo dell'asma. RSV appartiene al genere Pneumovirus della famiglia Paramyxoviridae ed è simile al virus Marburg, al virus del morbillo, al virus del cimurro canino e ai virus della parotite e della parainfluenza. Il microscopio elettronico mostra virioni di forma irregolare e spesso aggregati, con un involucro lipidico che porta le glicoproteine di superficie G, F e SH (v. oltre). Il nucleocapside contiene un genoma a RNA a singolo filamento negativo (vengono designati a filamento negativo quei virus che trascrivono l'RNA messaggero necessario per la sintesi delle proteine a partire dall'RNA genomico e replicano quest'ultimo tramite un RNA complementare di senso positivo), di massa molecolare 5 ∙ 10⁶ kDa, non segmentato. Sono presenti 10 geni con 12 potenziali prodotti. La trascrizione procede sequenzialmente da 3' a 5'; i primi geni trascritti sono lc e lb, che codificano proteine non strutturali con funzione sconosciuta. Viene poi trascritta la nucleoproteina (N), che è relativamente ben conservata tra ceppi isolati naturali. La fosfoproteina (P) e la piccola proteina idrofobica (SH) sono trascritte di seguito, seguite dalla proteina di adesione (G) e dalla proteina di fusione (F). Queste ultime due proteine sono le principali glicoproteine superficiali, contro cui sono diretti gli anticorpi neutralizzanti, e mostrano la maggiore variabilità naturale tra diverse forme isolate naturali di RSV. Infine vi sono M2, seconda proteina di matrice, di 22 kDa, e la proteina large (L) che è la RNA-polimerasi. Si pensa che entrambe queste proteine siano relativamente ben conservate. Studi sull'uomo e su animali modello hanno mostrato il doppio ruolo delle cellule T antivirali nell'eliminare il virus e nel provocare malattie più acute. Questo paradosso immunopatologico è ora compreso in modo più chiaro sia per la malattia provocata da RSV sia per quelle provocate da qualsiasi altra comune infezione umana.
La prima prova che l'immunità specifica potesse essere pericolosa si è avuta negli anni Sessanta, quando dei bambini furono vaccinati con RSV inattivato dalla formalina. l bambini riceventi il vaccino svilupparono forti risposte sierologiche, ma non furono protetti dall'infezione. Molti di questi individui, che vennero in seguito in contatto con RSV, svilupparono una grave affezione del tratto respiratorio inferiore e alcuni ne morirono. L'esame post mortem dei soggetti vaccinati col virus inattivato mostrava un'infiltrazione di eosinofili. Si sono a lungo studiate le cause dell'affezione aggravata dal vaccino inattivato, ma purtroppo non si è ancora prodotto un vaccino sicuro ed efficace. Recentemente sono state riesaminate le risposte immunitarie a RSV (Openshaw, 1995). L'immunità umana naturale a seguito di un'infezione ha durata breve e durante la vita si possono verificare nuove infezioni. Dopo i deludenti risultati dei primi tentativi con RSV inattivato dalla formalina, si sono proposti vaccini alternativi nel tentativo di indurre risposte protettive nella mucosa respiratoria. Tra questi sono stati testati ceppi attenuati di RSV, vaccini peptidici o derivati costituiti da singole subunità incorporati in sostanze adiuvanti in grado di potenziare le risposte delle mucose, adenovirus ricombinanti o virus vaccini che esprimono le proteine F o G, o entrambe, di RSV. A tutt'oggi, l'efficacia di questi protocolli di vaccinazione non è ancora stata determinata nell'uomo. Nel modello murino per l'affezione da RSV, la maggior parte dei linfociti recuperati durante i primi 5 giorni dall'infezione primaria è di fenotipo CD4- e CD8- e presenta le caratteristiche delle cellule NK, la cui attività mostra un picco circa nello stesso momento. Durante l'eliminazione del virus dai polmoni (6 - 9 giorni), il principale sottogruppo di linfociti è di tipo CD8+, sebbene si trovino anche cellule CD4+. In questo modello, l'induzione di forme distinte di immunopatologie può essere spiegata dalla produzione di citochine di tipo 2 dalle cellule T, che riconoscono la principale glicoproteina di superficie G, e di citochine di tipo l da parte di cellule T specifiche per la proteina di fusione F e per le proteine della matrice M2. La prova più convincente che l'aggravarsi della malattia sia causato dalle cellule T viene da studi sul trasferimento passivo dell'immunità. Topi infettati solo con RSV mostrano una lieve infermità, ma la gravità della malattia aumenta drammaticamente con l'iniezione di cellule T che riconoscono RSV. Le cellule T H l o i CTL CD8 possono provocare emorragie polmonari (che ricordano il polmone da shock), mentre le cellule CD4, producendo citochine di tipo 2, provocano eosinofilia nei polmoni (Alwan et al., 1994). Al contrario, il trasferimento di anticorpi non aggrava mai la malattia in vivo e può talvolta proteggere dall'infezione.
Diversi studi hanno suggerito una relazione tra la bronchiolite da RSV e l'asma. N. Sigurs e collaboratori (1995) hanno realizzato uno studio prospettivo di gruppo sulla bronchiolite da RSV infantile e hanno scoperto che essa è uno dei maggiori fattori di rischio per lo sviluppo di asma e malattie atopiche. Da questi studi non emerge una chiara indicazione che la bronchiolite operi come fattore di rischio indipendente, ma sembra sia un fattore aggiuntivo che agisca indipendentemente dalla storia familiare dell' atopia. Il tipo di risposta immunitaria che si verifica nel contatto iniziale con RSV dipende da parecchi fattori; tra questi le influenze genetiche, lo stato immunologico, la presenza di risposte immunitarie in atto contro altri antigeni e la presenza di memoria di antigeni simili.
HIV e AIDS
L'infezione provocata dal virus dell' immunodeficienza umana (HlV, Human lmmunodefieieney Virus) non è in genere eliminata dal sistema immunitario. Dopo esser penetrato nell'organismo, HlV costituisce rapidamente delle riserve di virus al di fuori della portata dei meccanismi immunitari di eliminazione. Queste riserve sono:
1) infezione virale latente di cellule T CD4+ quiescenti; nell'organismo si genereranno nuovi virus dopo l'attivazione delle cellule T.
2) Virioni ricoperti dagli anticorpi e dal complemento intrappolati nella rete delle cellule dendritiche follicolari nei linfonodi; questi virioni non sono neutralizzati ma, al contrario, sono altamente infettivi e sono concentrati nei siti obbligati di ricircolazione delle cellule T CD4+.
3) Varianti che hanno perso gli epitopi antigenici e sfuggono al controllo immunitario. Inoltre, l'instabilità del genoma virale è alta e varianti del virus si sviluppano per tutta la durata dell'infezione in diverse regioni anatomiche, costituendo un bersaglio in continuo cambiamento per meccanismi di controllo effettori. Ne consegue che molti processi immunitari vengono attivati durante l'infezione, ma non riescono a estirpare il virus.
Dopo l'ingresso di HlV, l'infezione si diffonde rapidamente alle cellule che esprimono CD4 e i recettori per le chemochine CCR5 e CXCR4 (Hill et al., 1997). Le risposte cellulari e anticorpali seguono la prima massiva ondata di viremia che scaturisce dalla replicazione virale. La quantità di cellule T CD4+ circolanti crolla contemporaneamente al picco della viremia; questo è in relazione al fatto che i CD4+ sono gli ospiti principali per la replicazione di HlV. Il picco della risposta dei CTL è seguito da un crollo nella viremia e dal ritorno a livelli normali delle cellule CD4+; ciò suggerisce che almeno temporaneamente i CTL sono capaci di controllare HIV. La risposta degli anticorpi neutralizzanti compare più tardi e arriva lentamente a plateau. Questa fase acuta dell'infezione dura circa 3 mesi ed è seguita da una fase cronica, durante la quale le risposte dei CTL restano molto attive, il livello degli anticorpi è alto, quello dei virus circolanti è basso e le cellule CD4+ scendono lentamente. La fase cronica può durare parecchi anni ed è caratterizzata da un veloce e costante riciclo dei virus, che fa diminuire progressivamente il numero delle cellule CD4+ circolanti (Ho et al., 1995). Con il tempo si verifica, nella gran parte degli individui infettati, una catastrofica transizione alla sindrome da immunodeficienza acquisita (AIDS, Acquired lmmunodeficiency Syndrome): il numero delle cellule CD4+ circolanti crolla sotto la soglia critica (<200/μl), i CTL e gli anticorpi scompaiono e l'ospite alla fine soccombe a causa di infezioni opportunistiche o di tumori correlati all'AIDS.
Individui con varianti genetiche delle molecole dei corecettori virali mostrano resistenza all'infezione e alla progressione verso l'AIDS (Liu et al., 1996). Questo non è dovuto a meccanismi immunitari effettori, anche se i recettori delle chemochine coinvolti hanno un ruolo nelle risposte immunitarie, ma riflette semplicemente il debole legame alle cellule. Altri fattori coinvolti nella progressione lenta verso l'AIDS potrebbero essere risposte protettive finora non identificate, inoculi scarsamente infettivi o potrebbero semplicemente rappresentare l'estremo limite di una gamma di velocità di progressione.
Cellule CD4+. - Il decremento delle cellule CD4+ è probabilmente dovuto a una combinazione tra gli effetti citopatici virali e l'uccisione da parte di HIV dei CTL che riconoscono i peptidi di HIV espressi in associazione a molecole MHC di classe I sulla membrana delle cellule CD4+ infettate. Anche prima che un calo nel numero delle cellule CD4+ divenga evidente, si riscontra una riduzione delle risposte immunitarie ad alcuni altri stimoli. Con il tempo, si perde la capacità di rispondere agli alloantigeni e alla fine anche quella ai mitogeni. Si sono osservate la perdita precoce delle risposte di tipo THl (produzione di IFN-γ e IL-2) e la ritenzione preferenziale delle risposte di tipo T H2 (produzione di IL-4 e IL-10) durante il progredire verso l'AIDS. Ciò può essere dovuto al fatto che le cellule T H l sono più suscettibili all'uccisione da parte di HIV delle cellule TH2, o all'induzione positiva delle cellule TH2 mediata da HIV. Si è osservato anche il coinvolgimento di cellule CD4+ citotossiche, che riconoscono i peptidi derivati da gpl20 presentati dalle molecole MHC di classe II, ma non è chiara la loro importanza.
Cellule CD8+. - I livelli di CTL circolanti specifici per HIV sono estremamente alti (circa l'1% dei linfociti) durante tutta la fase cronica (Moss et al., 1995), al punto che è possibile saggiare direttamente la loro attività in saggi citotossici ex vivo. Di solito sono presenti più cloni di CTL che riconoscono differenti antigeni virali e si sono osservate dinamiche complesse di espansione clonale delle cellule CDS+ e del carico di virus mutanti (Nowak et al., 1995). Oltre all'attività citotossica, i CTL possono rilasciare IFN-γ (effetto antivirale diretto) e chemochine come RANTES, MIP-l α e MIP-lβ, che possono competere con HIV per i corecettori usati dal virus per penetrare nelle cellule CD4+ e infettarle. Questa competizione impedisce l'ingresso di virus libero in nuove cellule ospiti, anche se questo non è un effettore diretto e non è in grado di eliminare l'infezione.
Anticorpi. - Le risposte anticorpali si sviluppano più lentamente di quelle dei CTL, forse perché necessitano dell'aiuto da parte delle cellule CD4+. I principali anticorpi neutralizzanti sono diretti contro la glicoproteina (gp) virale di rivestimento gp120, in particolare contro un'ansa molto variabile chiamata V3. Il rapporto tra mutazioni codificanti e non codificanti dell'ansa V3 suggerisce che essa sia sotto una pressione selettiva, forse una combinazione tra la neutralizzazione da parte degli anticorpi e la disponibilità di molti corecettori alternativi. Non è chiaro quale sia l'esatto meccanismo di neutralizzazione. Non tutti gli anticorpi antiansa V3 sono neutralizzanti; gli anticorpi neutralizzanti che riconoscono l'ansa V3 non impediscono il legame a CD4, ma potrebbero bloccare la fase di ingresso del virus dopo il legame. Un altro meccanismo possibile è l'inibizione del taglio della glicoproteina dell'involucro, un passaggio richiesto per l'infettività mediato da una proteasi che trasforma il precursore gpl60 env in gpl20 e gp41. Altri anticorpi neutralizzanti riconoscono il dominio di legame per CD4 su gpl20 e permettono la neutralizzazione crociata di ceppi virali con diverse anse V3. Anche altre regioni di gpl20 sono bersaglio degli anticorpi neutralizzanti che possono funzionare alterando la conformazione del virione o interferendo con il legame ai corecettori. In sistemi sperimentali che utilizzano gli scimpanzé, alti livelli di anticorpi neutralizzanti che riconoscono la glicoproteina dell'involucro possono proteggere contro l'infezione da HIV, ma tali livelli non possono essere sostenuti per lungo tempo. Inoltre, variazioni nell'involucro virale possono impedire tale protezione. Nelle prove cliniche di vaccini basati sull'involucro virale, gli anticorpi prodotti non riescono a neutralizzare i ceppi isolati primari, mostrando le limitazioni della neutralizzazione ceppo-specifica.
ADCC. - È stata anche descritta una forma di citotossicità cellulare (ADCC, Antibody Directed Cellular Cytitoxicity) mediata dagli anticorpi che riconoscono le proteine dell'involucro gpl20 e gp41 e che prevede il coinvolgimento delle cellule NK. Il ruolo di questo meccanismo nel controllo della replicazione di HIV dipende dall' attività delle cellule NK, poiché gli anticorpi sono presenti ad alta concentrazione durante gran parte della fase cronica. Questa attività può essere meno utile dei meccanismi di citotossicità mediati dai CTL, o addirittura dannosa, perché potrebbe causare la lisi delle cellule T CD4+ non infettate che hanno legato la gpl20 rilasciata, o delle cellule B che esprimono anticorpi di membrana che riconoscono gp120.
Anticorpi potenzianti. - I complessi circolanti tra HIV e anticorpi sono infettanti e non vengono distrutti dai macrofagi. Infatti, gli anticorpi possono aumentare l'infettività di HIV. Questo si verifica per legame diretto dei complessi immunitari o alle cellule che esprimono il recettore per Fc, soprattutto CDl6 sulle cellule NK e il recettore FcRI sui monociti, oppure alle cellule che esprimono CR2 per il potenziamento mediato dal complemento. Tale potenziamento si dimostra con sieri neutralizzanti che sono stati diluiti oltre la concentrazione richiesta per la neutralizzazione. Così i livelli decrescenti di anticorpi nel decorso della malattia possono facilitare il progredire verso l' AIDS. Inoltre, anticorpi che neutralizzano un ceppo di HIV possono potenziarne un altro. Questo effetto può contribuire alla selezione di varianti resistenti ed evidenzia un possibile problema per le vaccinazioni indirizzate all'induzione dell'immunità mediata dagli anticorpi.
Cellule NK. - L'attività delle cellule NK sembra diminuire con il progredire verso l'AIDS, ma l'aggiunta di IL-2 può far recuperare l'attività nei saggi in vitro di funzione delle NK stesse. Anche se un sistema immunitario senza aiuti sembra essere tutt'al più capace solo di rallentare la progressione della malattia, potrebbe ancora essere possibile contrattaccare la variabilità e il vantaggio cinetico di HIV mediante l'immunizzazione preventiva. Parecchi lavori hanno descritto risposte cellulari specifiche all'RIV in individui che, pur essendo stati esposti a tale virus, rimangono sieronegativi, anche se è stato difficile mostrare l'effettiva eliminazione di un inoculo virulento di RIV. Si è raggiunta protezione contro le infezioni da HIV in scimpanzé vaccinati e contro le infezioni con il virus correlato dell'immunodeficienza di scimmia nei macachi (Daniel et al, 1992). Il recente successo della terapia antivirale combinata nella diminuzione della carica virale negli individui infetti suggerisce che un trattamento antivirale precoce ed aggressivo dopo l'infezione potrebbe prevenire l'esaurimento delle forze immunitarie, permettere lo sviluppo di un'immunità protettiva solida e infine controllare la restante replicazione virale (Autran et al., 1997).
Infezioni da Herpes simplex
Il virus Herpes simplex (HSV) ha un genoma a DNA lineare a doppio filamento che codifica 70 o più proteine virali. Il genoma è contenuto in un core (capside) circondato da un materiale amorfo, il tegumento, ricoperto a sua volta da un involucro lipidico costellato di glicoproteine. Vi sono due tipi di questo virus: HSV-l e HSV-2. Il secondo è di solito associato con infezioni genitali.
HSV provoca una vasta gamma di malattie, dalle più comuni, come herpes labiale, gengivostomatite, herpes genitale, cheratocongiuntivite, alle infezioni viscerali di ospiti immunocompromessi e alle rare encefaliti erpetiche. Negli USA, il 60 ÷ 80% della popolazione entro i 50 anni è stato infettato da HVS-l e il 20% da HSV-2. HSV infetta per contatto attraverso le mucose o le abrasioni cutanee. L'infezione è seguita dalla replicazione nelle cellule epiteliali del sito di ingresso. A questo punto HSV entra nelle terminazioni periferiche dei neuroni sensitivi e viene trasportato dal flusso assonale retrogrado alle radici dei gangli dorsali, dove va incontro a una replicazione limitata e transeunte. L'infezione neurale è di solito controllata, anche se si verificano infezioni letali a carico del sistema nervoso centrale in individui immunocompromessi, e il virus resta latente, cioè il genoma si mantiene in forma episomica, non vengono sintetizzate proteine virali e l'espressione genica virale è limitata ai trascritti associati alla fase latente. Fattori di stress, come l'esposizione ai raggi ultravioletti, stress emotivi, febbre, altre infezioni (per esempio, polmonite pneumococcica), immunosoppressione, taglio di un nervo periferico, possono essere seguiti dalla ricomparsa del virus nei siti innervati dagli stessi neuroni infetti. Qui il virus entra nel ciclo litico replicativo a livello delle cellule epiteliali, producendo vescicole e ulcere. In questo stadio si verifica la trasmissione ad altri individui. Sono frequenti variazioni individuali della frequenza di riattivazione, da una volta ogni pochi anni a 10 ÷ 20 volte all'anno. La frequenza di infezioni ricorrenti (riattivate) potrebbe dipendere dalla carica virale iniziale così come da fattori dell'ospite, compresa la risposta immunitaria. HSV potrebbe diffondersi dal sito mucocutaneo di ingresso attraverso i neuroni sensitivi per causare un'encefalite letale, ma la maggior parte degli ospiti oppone resistenza alle affezioni diffuse provocate da HSV. Le infezioni in ospiti che non sono mai stati esposti al virus precedentemente provocano risposte infiammatorie locali nei siti mucocutanei di replicazione. Per il precoce contenimento della diffusione del virus nell'epitelio sono richieste la produzione locale di interferone di tipo I (IFN-α e IFN-β) e le cellule NK. Oltre a dirigere gli effetti antivirali, gli interferoni aumentano l'espressione dell'MHC di classe I e la presentazione antigenica nei cheratinociti, fornendo un migliore bersaglio per i CTL. Tuttavia, questi meccanismi effettori iniziali limitano ma non prevengono né l'infezione dei neuroni né lo stabilirsi della latenza. L' eliminazione dell'infezione locale richiede l'intervento dei linfociti T.
Esperimenti in modelli murini di infezione da HSV hanno rivelato la complessità delle interazioni tra fattori genetici nell'ospite e risposte delle cellule CD4+ e CD8+. In generale, l'eliminazione di HSV dal sito primario di infezione dipende dalle cellule T CD4+, mentre le cellule T CD8+ ne limitano la diffusione nei neuroni. Lo sviluppo di cellule T CD8+ specifiche per HSV dipende dalle cellule CD4+ in alcuni, ma non in tutti, ceppi di topi. La deplezione selettiva delle cellule CD4+ o CD8+ nell'animale produce effetti limitati sull'eliminazione di HSV dalle lesioni primarie, mentre il trattamento dei topi con anticorpi che neutralizzano IFN-γ, secreto da entrambi i sottotipi di cellule T, prolunga la durata dell'infezione locale.
Le cellule T CD8+ si infiltrano nei gangli neuronali con HSVe sembrano essenziali per controllare l'infezione neuronale, poiché il trattamento con anticorpi anti-CD8 provoca una maggiore distruzione dei neuroni (Simmons e T scharke , 1992). Il meccanismo di azione delle cellule T CD8+ specifiche per HSV nei gangli infetti è di tipo non litico: in realtà esse aumentano il numero di neuroni che contengono il virus. Questo può essere dovuto al fatto che l'espressione delle molecole MHC di classe I, normalmente assenti nel tessuto nervoso, è bloccata a livello postrascrizionale nei neuroni. Tuttavia, le cellule CD8+ riconoscono i peptidi virali prodotti in una cellula infettata e presentati dalle molecole MHC di classe I di quella cellula. L'infezione incrementa l'espressione della classe I nei gangli, ma solo nelle cellule 'satellite' e in quelle di Schwann (Pere ira et al., 1994). Diversamente dai neuroni, le cellule 'satellite' non sono permissive per la replicazione di HSV, ma possono essere sede di un'infezione abortiva che permette la presentazione dei prodotti virali sintetizzati nella fase iniziale del ciclo replicativo. La sequenza di eventi dovrebbe quindi essere la seguente: l'infezione litica dei neuroni rilascia il virus che infetta le cellule 'satellite' senza successo, ma stimola da parte loro l'espressione delle molecole MHC di classe I. Esse presentano i peptidi virali alle cellule CDS+ infiltranti attratte dalle chemochine rilasciate durante le fasi iniziali dell'infezione. Le cellule T CDS+ attivate rilasciano poi IFN-γ, che agisce sui neuroni neo infettati e modula il ciclo replicativo del virus dalla fase litica alla latenza. L' espressione dell'MHC di classe I cessa durante la fase latente, quando si verifica solo la minima trascrizione dei geni virali e diminuisce lo stato di attivazione immunitaria.
La latenza può durare per mesi o anni, finché stimoli idonei provocano la riattivazione del virus. A questo punto la sintesi delle proteine virali riprende e il virus infettivo ritorna indietro lungo le fibre sensitive periferiche, fino ai siti mucocutanei innervati da tali fibre. Qui la replicazione virale causa effetti citopatici evidenti a un esame clinico, cioè vescicole trasparenti e ulcere. In circa il 50% dei casi di riattivazione, tuttavia, la diffusione del virus è asintomatica, favorendo così la sua trasmissione. In questa fase vengono innescati vari meccanismi dell'immunità acquisita: le riattivazioni hanno durata più breve delle lesioni primarie. Gli anticorpi neutralizzanti contro le glicoproteine dell'involucro (soprattutto gD e gB) limitano la diffusione dell'infezione mediata da virus libero. HSV ha evoluto misure contro gli anticorpi e il complemento. Le glicoproteine dell'involucro gE e gl sono in effetti recettori dell'Fc che legano IgG monomeriche, mentre gE da sola può legare aggregati di IgG. Tutto ciò sembra proteggere i virioni dalla neutralizzazione. In modo simile, gC ha una parziale omologia con il recettore del complemento CRI e lega C3b. Questo può aiutare HSV ad attaccarsi a certi tipi cellulari.
Cellule mononucleate, compresi i linfociti CD4+ e CDS+, si infiltrano nelle lesioni dovute all'infezione ricorrente. Le cellule CD4+ predominano nelle lesioni precoci. I linfociti T CD4+ e CDS+ specifici per HSV sono presenti a bassa frequenza nelle PBMC (Peripheral Blood Mononuclear Cells, cellule mononucleate del sangue periferico). Il clonaggio di cellule T isolate da pazienti affetti da lesioni ricorrenti ha mostrato che i CTL riconoscono parecchi antigeni diversi, derivati da proteine virali sia strutturali sia non strutturali. In questi studi si sono usate linee di cellule B linfoblastoidi infettate da HSV come cellule bersaglio. Quando sono stati utilizzati come bersaglio i fibroblasti, la lisi mediata dai CTL è apparsa meno efficace. Questo ha portato alla scoperta che HSV contiene un gene, denominato rx47, che codifica il prodotto ICP4 7, la cui espressione può far trattenere l'MHC di classe I nella cellula mediante l'interazione con il trasportatore associato al processamento dell'antigene e l'inibizione del trasporto dei peptidi nel reticolo endoplasmatico (York et al.,1994). In tal modo HSV può evitare o limitare l'eliminazione da parte dei CTL (v. il saggio di E.J.H.J. Wiertz, nel II volume).
Cloni di cellule T CD4+ isolati dalle lesioni da HSV sono specifici per molti antigeni virali, comprese l'abbondante componente del tegumento VPl6 e le glicoproteine dell'involucro gB, gC e gD. I ruoli diversi delle cellule THl e TH2 sono stati studiati (Thomas e Rouse, 1997) nei modelli murini della cheratite stromale erpetica (HSK, Herpetic Stromal Keratitis). L'infezione oculare da HSV è la principale causa infettiva di cecità nei paesi sviluppati; si pensa che HSK sia un'immunopatologia mediata dalle cellule T per due ragioni: animali deficitari di cellule T risultano protetti dalla malattia; HSK si sviluppa dopo che il titolo virale sta già diminuendo, quando le risposte T -specifiche contro HSV si stanno sviluppando nella cornea.
Poiché fattori genetici correlati al ceppo murino e al ceppo virale sono coinvolti in HSK, non sorprende che differenti cellule effettrici sembrino causare HSK in diversi sistemi modello. Nei topi BALB/c infettati con i ceppi KOS o RE di HSV-l, HSK è mediata dalle cellule CD4+ che secernono TNF e IFN-γ, ma in topi AlJ gli effettori di HSK sono i CTL CDS+. Anticorpi anti-IFN-γ bloccano HSK, mentre l'infiltrazione della cornea da parte delle cellule di Langerhans (come effetto dell 'infiammazione) è richiesta per HSK, poiché MHC di classe II non è espresso dalle cellule corneali. Sono stati descritti altri meccanismi che coinvolgono danni prodotti dagli anticorpi e dal complemento e una forma di citotossicità cellulare mediata dagli anticorpi e dalle cellule NK. Per evidenziare la complessità di questi meccanismi, HSK può essere indotta anche da un clone CD4+, mantenuto a lungo in coltura, specifico per gD che produce IL-4, cioè un clone TH2.
Non esiste nessun vaccino efficace contro HSV. Gli anticorpi neutralizzanti specifici per le glicoproteine dell'involucro gD e gB possono proteggere e migliorare l'affezione ricorrente nel modello della cavia e queste glicoproteine sono candidate per lo sviluppo di un vaccino. Altri approcci promettenti comprendono i virus vivi attenuati o geneticamente resi meno efficienti e l'immunizzazione mediante DNA. Un vaccino che prevenga completamente le infezioni da HSV potrebbe essere irraggiungibile, ma l'induzione combinata di anticorpi e immunità cellulare a livello delle mucose potrebbe limitare la replicazione iniziale di HSV e l'instaurarsi dell'infezione latente, portando a una forma più lieve o asintomatica dell'affezione ricorrente.
Conclusioni
La risposta immunitaria è essenziale nella difesa contro le infezioni virali, ma è anche in grado di danneggiare l'ospite. Senza risposte immunitarie, i virus si diffonderebbero in modo incontrollato il che, si assume, sarebbe dannoso per l'ospite. Quando vengono sintetizzate le proteine virali, queste riducono la capacità della cellula di sintetizzare le sue normali proteine. È da notare tuttavia che alcuni virus non danneggiano particolarmente le cellule in assenza di risposta immunitaria. Si può sostenere che questa possa essere buona o cattiva non solo per l'ospite, ma anche per il virus. L'infiammazione che si genera dalla risposta immunitaria è spesso d'aiuto al virus per raggiungere un nuovo ospite e, poiché i virus sopravvivono solo se trasmessi a nuovi ospiti, la risposta è un aiuto essenziale per molti virus.
Bibliografia citata
ALWAN, W.H., KOZLOWSKA, W.l, OPENSHAW, P.l.M. (1994) Distinct types of lung disease caused by functional subsets of antiviral T cells. J. Exp. Med., 179, 81-89.
AUTRAN, B., CARCELAIN, G., LI, T.S., BLANC, C., MATHEz, D., TUBIANA, R., KATLAMA, C., DEBRE, P., LEIBOWITCH, J. (1997) Positive effects of combined antiretroviral therapy on CD4+ T cell homeostasis and function in advanced HIV disease. Science, 277, 112-116.
BACHMANN, M.F., ZINKERNAGEL, R.M. (1996) The influence of virus structure on antibody responses and virus serotype formation. Immunol. Today, 17, 553-558.
BIRON, C.A. (1997) Activation and function of natural killer cell responses during viraI infections. Curr. Opin. Immunol., 9, 24-34.
DANIEL, M.D., KIRCHHOFF, F., CZAJAK, S.C., SEHGAL, P.K., DEsROSIERS, R.C. (1992) Protective effects of a live attenuated SIV vaccine with a deletion in the nef gene. Science, 258, 1938-1941.
DENNY, F.W. J.R. (1995) The clinical impact ofhuman respiratory virus infections. Am. J. Respir. Crit. Care. Med., 152 (4) Suppl., S4-S12.
FIORENTINO, D.F., BOND, M.W., MOSMANN, T.R. (1989) Two types of mouse T helper cell (IV): T H2 clones secrete a factor that inhibits cytokine production by THl clones. J. Exp. Med., 170, 2081-2095.
HEILMAN, C.A. (1990) Respiratory syncytial and parainfluenza viruses. J. Infect. Dis., 161, 402-406.
HILL, C.M., DENG, H., UNUTMAZ, D., KEWALRAMANI, V.N., BASTIANI, L., GORNY, M.K., ZOLLA PAZNER, P.S., LITTMAN, D.R. (1997) Envelope glycoproteins from human immunodeficiency virus types 1 and 2 and simian immunodeficiency virus can use human CCR5 as a coreceptor for viraI entry and make direct CD4-dependent interactions with this chemokine receptor. J. Virol., 71, 6296-6304.
HO, D.D., NEUMANN, A.U., PERELSON, A.S., CHEN, W., LEONARD, lM., MARKOWITZ, M. (1995) Rapid tumover of plasma virions and CD4 Iymphocytes in HIV-l infection. Nature, 373, 123-126.
LU, R., PAXTON, W.A., CHOE, S., CERADINI, D., MARTIN, S.R., HORUK, R., McDoNALD, M.E., STUHLMANN, H., KOUP, R.A., LANDAU, N.R. (1996) Homozygous defect in HIV-l coreceptor accounts for resistance of some multiply-exposed individuals to HIV-l infection. CelI, 86, 367-377.
MOSMANN, T.R., SAD, S. (1996) The expanding universe of T cell subsets: Thl, Th2 and more. Immunol. Today, 17, 138-146.
MOSS, P.A., ROWLAN-JONES, S.L., FRODSHAM, P.M., McADAM, S., GIANGRANDE, P., McMICHAEL, AJ., BELL, lI. (1995) Persistent high frequency of human immunodeficiency virus-specific cytotoxic T cells in peripheral blood of infected donors. Proc. Natl. Acad. Sci. USA, 92, 5773-5777.
NOWAK, M.A et al. (1995) Antigenic oscillations and shifting immunodominance in HIV-l infections. Nature, 375, 606-611.
OPENSHAW, P.J.M. (1995) Immunopathological mechanisms in respiratory syncytial virus disease. Seminars in immunopathol., 17, 187-201.
PEREIRA, R.A, TSCHARKE, D.C., SIMMONS, A (1994) Upregulation of class I major histocompatibility complex gene expression in primary sensory neurons, satellite cells, and Schwaun cells of mice in response to acute but not latent herpes simplex virus infection in vivo. J. Exp. Med., 180, 841-850.
SIGURS, N., BJARNASON, R., SIGURBERGSSON, F., KIELLMAN, B., BJÖRKSTÉN, B. (1995) Asthma and immunoglobulin E antibodies after respiratory syncytial virus bronchiolitis: a prospective cohort study with matched controls. Pediatrics, 95, 500-505.
SIMMONS, A., TSCHARKE, D.C. (1992) Anti-CD8 impairs clearance of herpes simplex virus from the nervous system: implications for the fate of virally infected neurons. J. Exp. Med., 175, 1337-1344.
STREET, N.E., SCHUMACHER, J.H., FONG, T.A., BASS, H., FIORENTINO, D.F., LEVERAH, J.A, MOSMANN, T.R. (1990). Heterogeneity ofmouse helper T cells. Evidence from bulk cultures and limiting dilution cloning for precursors of T H 1 and T H2 cells. J. Immunol., 144, 1629-1639.
THOMAS, J., ROUSE, B.T. (1997) Immunopathogenesis of herpetic ocular disease. Immunol. Res., 16, 375-386.
YORK, L.A., ROOP, C., ANDREWS, D.W., RIDDELL, S.R., GRAHAM, F.L., JOHNSON, D.C. (1994) A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T Iymphocytes. CelI, 77, 525-535.
Bibliografia generale
FIELDS, B.N., KNIPE, D.M., HOWLEY, P.M., CHANOCK, R.M., MELNICK, lL., MONATH, T.P., ROIZMAN, B., STRAUS, S.E., a c. di, FieIds viroIogy, 3' ed., Filadelfia, Lippincott-Raven, 1996.
JANEWAY, C.A. JR., TRAVERS, P. Immunobiology, 3' ed., Londra-S. Francisco, Current Biology, e New Y ork, Garland Publishing Inc., 1997.
MIMS, C.A. Mims' pathogenesis of infectious disease, 4' ed., Londra, Academic Press, 1995.
PAUL, W.E., a c. di, Fundamental immunology, 3' ed., New York, Raven Press, 1993