
dolore
dolore
Sensazione sgradevole o di sofferenza, susseguente alla stimolazione di recettori sensoriali presenti nell’organismo, dovuta all’azione di un agente che compromette l’integrità somatica, o suscitata dallo stato patologico di un organo.
La trasmissione dell’impulso doloroso
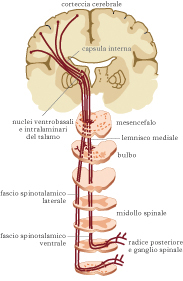
La sensazione dolorosa si basa su due elementi collegati e distinti: la nocicezione, che rappresenta il collegamento tra i recettori sensoriali e le vie nervose che presiedono i meccanismi neurologici di risposta allo stimolo doloroso; la percezione del d., rappresentante un meccanismo più complesso che coinvolge aspetti emozionali e cognitivi e organizza quella che è la sgradevole sensazione che al d. si associa. Per ciò che riguarda il meccanismo nocicettivo il d. viene percepito da recettori specifici che si trovano a livello cutaneo, a livello muscolare, articolare e viscerale. La stimolazione di uno di questi recettori si traduce fondamentalmente in tre momenti: la trasduzione (lo stimolo doloroso viene tradotto in impulsi elettrici), la trasmissione (l’impulso elettrico viene trasmesso alle corna posteriori del midollo spinale e di qui risale verso l’encefalo), la modulazione (nella quale l’impulso stesso viene amplificato o inibito durante il suo tragitto). Lo stimolo doloroso si trasmette con diversa velocità e intensità a seconda del fascio nervoso che lo trasporta, che può avere diverse caratteristiche anatomiche. Il meccanismo in generale relativo alla trasmissione dello stimolo doloroso non avviene esclusivamente attraverso lo spostamento di potenziali elettrici, ma vi contribuiscono anche sostanze chimiche che influiscono sulla modulazione del d. associando fenomeni infiammatori (bradichinine, citochine e prostaglandine) e condizioni di iperalgesia (per diminuzione della soglia di eccitazione dei recettori periferici). Le vie del d. non arrivano direttamente in sede centrale, ma hanno due stazioni: la prima a livello midollare e la seconda a livello del talamo. Il talamo trasmette le informazioni ricevute alla corteccia cerebrale dove si realizza il meccanismo della percezione, dell’analisi e della memorizzazione, ossia della coscienza dell’evento doloroso. Una parte degli stimoli dolorifici dal talamo viene condotta all’ipotalamo, che diventa responsabile dell’evocazione delle reazioni endocrine e neurovegetative che possono associarsi al d.: nausea, vomito, bradicardia o tachicardia e ipertensione. Così come esistono mediatori chimici che hanno la capacità di accentuare il d., esistono mediatori capaci di attenuarne gli effetti. Gli studi su questo particolare settore hanno portato a identificare sostanze ad azione analgesica. Il nostro organismo è capace di produrre elementi attenuatori del d. quali le endorfine e, più in generale, un’ampia gamma di oppioidi endogeni quale l’anandammide, un cannabinoide endogeno (mediatore lipidico del cervello capace di legare i recettori cannabinoidi).
Terapia del dolore
Il primo irrinunciabile passo in una terapia del d., e quindi la base di ogni strategia antalgica, è la valutazione qualitativa e quantitativa del sintomo. I farmaci antalgici più usati appartengono a tre categorie fondamentali: i farmaci antinfiammatori non steroidei (FANS); gli oppioidi, naturali o di sintesi; gli anestetici locali. A questi gruppi fondamentali si affiancano farmaci adiuvanti quali gli antidepressivi o i cortisonici. L’OMS ha elaborato una scala sequenziale di impiego di sostanze che, nata per il d. neoplastico, è stata successivamente adottata anche per d. diversi. In questo schema di trattamento si prevede l’impiego, in successione, di FANS, di oppioidi inizialmente deboli e poi forti, e di farmaci adiuvanti. La scala farmacologica può essere progressivamente integrata, caso per caso, con l’uso di tecniche invasive o seminvasive, di derivazione anestesiologica, che consentono di portare i farmaci, sia analgesici locali sia oppioidi, a contatto con le radici spinali (analgesia epidurale) o con il sistema nervoso centrale (tecniche subaracnoidee spinali o intraventricolari centrali), modulando l’entrata di impulsi dolorosi (neuromodulazione spinale). Infine, se le associazioni farmacologiche precedentemente impiegate si sono dimostrate o sono diventate inefficaci, possono essere utilizzate sostanze neurotossiche quali il fenolo o l’alcol le quali, portate a diretto contatto con il tessuto nervoso o con i gangli simpatici e i plessi parasimpatici, causano un certo grado di lesione più o meno duratura (neurolisi spinale o gangliare) in grado di interrompere la trasmissione dello stimolo doloroso. Le tecniche neurolesive vengono impiegate quasi esclusivamente nelle fasi finali del d. causato dal cancro. Infine, possono fare parte della terapia del d. sistemi di elettrostimolazione, come quelli utilizzati dall’agopuntura.
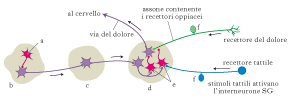
Le droghe presenti nel nostro cervello
Per spiegare come il pensiero e le emozioni influenzino la nostra percezione del dolore lo psicologo canadese Ronald Melzack (1929) e il neurofisiologo britannico Patrick Wall (1925-2001) proposero, negli anni Sessanta del secolo scorso, l’esistenza di un ‘cancello’ nelle corna posteriori del midollo spinale. In questa zona, fibre nervose di piccolo calibro (dette fibre nocicettive) e fibre nervose di grande calibro (dette non nocicettive) stabiliscono sinapsi con i neuroni di proiezione che formano il tratto spino-talamico e con interneuroni inibitori localizzati nella sostanza gelatinosa di Rolando (SG). Le afferenze nocicettive aprono il cancello, perché inibiscono l’interneurone inibitorio SG: le informazioni raggiungono pertanto i centri di riconoscimento. L’attivazione del neurone inibitorio SG da parte di uno stimolo non nocicettivo, tattile inibisce i neuroni di proiezione, chiude il cancello e il segnale viene trasmesso in forma attenuata o non trasmesso del tutto ai centri superiori. Perciò, una modalità per chiudere il cancello è di stimolare i recettori tattili della cute nell’area del dolore. Questo risultato si può ottenere attraverso la stimolazione elettrica transcutanea del nervo o tramite l’agopuntura. L’interneurone inibitorio SG può essere attivato anche da fibre discendenti che provengono dal sistema di modulazione centrale formato da strutture coinvolte nei processi cognitivi, affettivi e legati allo stato di vigilanza del soggetto.
Le droghe endogene
Nei circuiti nervosi di modulazione del dolore vengono prodotte sostanze molto simili a quelle che per millenni gli esseri umani hanno assunto per sedare il dolore. Le proprietà analgesiche dell’oppio (Papaver somniferum) e dei suoi derivati, quali la morfina, e della marijuana (Cannabis sativa), erano note sin dall’antichità.
Negli anni Settanta del secolo scorso una clamorosa scoperta ha provato che gli oppiacei agiscono legandosi strettamente e selettivamente con vari tipi di recettori specifici nel cervello, detti recettori oppiodi. Questi sono di tre tipi: μ, δ e κ. La loro scoperta ha indotto a formulare l’ipotesi dell’esistenza di sostanze di natura endogena in grado di legarsi a tali recettori. Queste sostanze sono state successivamente isolate e identificate come peptidi. Esse sono simili alla morfina e sono chiamate genericamente endorfine o peptidi oppiodi endogeni. Esistono tre classi di endorfine: le encefaline, la β-endorfina e le dinorfine. Tutte contengono una sequenza comune di quattro amminoacidi (Tyr-Gly-Gly-Phe) e tutte derivano da precursori di dimensioni maggiori codificati da tre geni distinti. Per esempio, la β-endorfina deriva dal POMC (propiomelanocortina), un precursore che contiene anche le sequenze per l’ormone MSH, che stimola i melanociti, e per l’ormone ACTH, l’ormone dello stress. La β-endorfina condivide con l’ACTH gli stessi ritmi circadiani, con un picco massimo di produzione al risveglio. Ciò spiega perché la tolleranza al dolore è maggiore al mattino. Le endorfine hanno di regola effetti inibitori che si realizzano con l’apertura dei canali per il potassio o con la chiusura dei canali del calcio nelle cellule bersaglio. Ciò provoca inibizione del rilascio di neurotrasmettitori, quali il glutammato e la sostanza P, da parte dei terminali delle fibre afferenti nocicettive, che pertanto non attivano le fibre di proiezione. Il principio attivo della Cannabis, isolato e identificato nel 1964, è il Δ-tetraidrocannabinoide. È stata dimostrata l’esistenza, nel sistema nervoso centrale e periferico, di recettori specifici per queste sostanze. Ne sono stati clonati due sottotipi (CB1 e CB2) e sono stati generati topi carenti dei geni sia per il recettore CB1 sia per il recettore CB2. L’attivazione del recettore CB1 ha un effetto inibitorio sui neuroni bersaglio e riduce la liberazione dei neurotrasmettori. L’attivazione del recettore CB2 inibisce nelle cellule bersaglio la liberazione di ossido nitrico (NO), un altro mediatore della comunicazione tra neuroni.
Nel 1992 è stato isolato dal cervello di maiale il primo endocannabinoide: l’anandammide, un ecasonide costituito dall’acido arachidonico legato all’etanolammina. Successivamente, dall’intestino di cane è stato isolato un altro endocannabinoide, il 2-arachidonilglicerolo. Il sistema cannabinoide controlla il dolore in modo simile a quello oppiode. Viene attivato nelle regioni di rilevanza per la percezione del dolore come la sostanza grigia periacqueduttale e nei tessuti periferici come la pelle. Avrebbe cioè un ruolo, in parte ancora da decifrare, sia nella trasduzione dell’impulso doloroso a livello periferico sia nell’elaborazione dell’informazione a livello centrale.
Significato biologico dell’analgesia
Non sempre gli stimoli che provocano dolore tendono a scatenare delle risposte di fuga e di evitamento. In molte specie animali la soppressione del dolore è necessaria durante comportamenti importanti come il combattimento o l’accoppiamento. I maschi che lottano per la conquista delle femmine durante la stagione degli accoppiamenti non potranno assicurare la trasmissione dei loro geni se il dolore scatenerà risposte di fuga che interferiscano con la lotta.