
Enzimi
Enzimi
di Edwin C. Webb
SOMMARIO: 1. Premesse storiche. □ 2. Gli enzimi come catalizzatori: a) gli enzimi come parte di sistemi metabolici; b) gli enzimi come entità indipendenti; c) aspetti quantitativi: cinetica enzimatica. □ 3. Gli enzimi come sostanze: a) purificazione degli enzimi; b) composizione e struttura. □ 4. Relazione fra struttura e funzione: meccanismo d'azione degli enzimi. □ 5. Aspetti biologici dell'enzimologia: a) localizzazione degli enzimi; b) forme multiple e varianti genetiche; c) regolazione dell'attività enzimatica. □ 6. Prospettive future dell'enzimologia. □ Bibliografia.
1. Premesse storiche.
La conoscenza di reazioni enzimatiche è di molto anteriore al XX secolo, benché il fatto che esse fossero dei processi chimici catalizzati da agenti di origine biologica fosse compreso solo vagamente. Reazioni come la fermentazione di soluzioni zuccherine, l'inacidimento e la trasformazione in formaggio del latte, la putrefazione delle urine sono state di certo osservate per caso, come evenienze fortuite, ma l'uso di innesti da precedenti fermentazioni come catalizzatori è probabilmente molto antico. Il concetto di ‛fermentazione' si incontra nella Bibbia: ‟Un po' di lievito fa fermentare tutta la pasta" (1a Lettera ai Corinzi, 5, 6). Oltre al vino, la cui produzione dipende soltanto dai fermenti naturalmente presenti sulla buccia dell'uva, anche la birra (o un'analoga bevanda fermentata ricavata dall'orzo) è stata descritta da molti autori classici, fra i quali Senofonte, Erodoto, Plinio e Tacito. Essa era chiamata zythos in greco e xythum in latino e più tardi fu denominata con i termini latini cella o cervesia. Una particolareggiata ricetta per la produzione dello zythos egiziano ci è data da Zosimo nel III secolo d. C. Dopo la descrizione della germinazione dell'orzo a malto, essa prosegue: ‟Infine, tritarlo e farne dei pani, aggiungendo lievito come nel caso del pane ordinario. Quindi cuocere questi pani al forno, ma solo leggermente, e quando cominciano a gonfiarsi decantarvi acqua dolce e farla colare attraverso un filtro o un setaccio fine [...]". Il vocabolo latino fermentum, usato per esempio da Virgilio, si riferisce a ogni cosa che provochi un ribollio e quindi anche alla birra o al lievito stesso.
In ogni modo, il concetto fondamentale di enzimologia, nel senso di catalisi di reazioni chimiche da parte di agenti di origine biologica, cominciò a consolidarsi solo nel sec. XIX. L'idea di catalisi fu formulata per la prima volta dal chimico svedese J. J. Berzelius, come quella di ‟una forza differente dalle forze che finora conosciamo". Nella terza edizione del suo testo di chimica, pubblicata nel 1837, egli scrisse: ‟Questa è una forza probabilmente molto più diffusa di quanto si è finora pensato e la cui natura ci è ancora nascosta [...]. La capacità catalitica sembra consistere in realtà nel fatto che alcuni corpi, soltanto in virtù della loro presenza, e non della loro affinità, possono risvegliare affinità sopite a quella temperatura, e, come risultato, gli elementi di un corpo complesso si assestano in un nuovo ordine" (Berzelius, Lehrbuch der Chemie, vol. VI, Dresden-Leipzig 18373, pp. 19-25). Queste interpretazioni chimiche arrivarono al momento giusto, poiché diversi casi di catalisi biologiche erano stati descritti nei cinquant'anni precedenti. Nel 1783 L. Spallanzani, un fisiologo italiano, aveva mostrato che la carne veniva liquefatta dal succo gastrico dei corvi; nel 1810 Plancke faceva notare che estratti di radici vegetali potevano far diventare blu la tintura di guaiaco; nel 1830 R. Robiquet e A.-F. Boutron-Charard scoprivano l'idrolisi dell'amigdalina da parte delle mandorle amare, e nello stesso anno A.-P. Dubrunfaut mostrava che un estratto di malto trasformava l'amido in zucchero. In un articolo, che secondo molti rappresenta la nascita dell'enzimologia, A. Payen e J. Persoz (v., 1833) descrivevano la precipitazione con alcool, da estratti di malto, di una sostanza che poteva essere conservata allo stato secco mantenendo la capacità di idrolizzare l'amido e che chiamarono ‛diastasi'. La sinigrinasi e la pepsina furono descritte poco tempo dopo.
Nel mezzo secolo seguente sviluppi ulteriori furono ostacolati dalla disputa intorno alla possibilità che alcune di queste catalisi biologiche dipendessero, in qualche misura, dalla presenza di organismi viventi. La grande opera di Pasteur dimostrò (sulla scorta di precedenti contributi di Th. Schwann e di C. Cagniard de Latour) che il lievito era qualcosa di vivo e che molte fermentazioni, sia aerobiche sia anaerobiche, erano provocate da microrganismi viventi. Pur nella loro grande importanza per gli sviluppi generali della biologia, questi risultati incoraggiarono una tendenza ‛vitalistica' nello studio delle fermentazioni. Pasteur fece una distinzione fra ‛fermenti non organizzati o solubili', come la diastasi e la pepsina, e ‛fermenti organizzati', come il lievito, per quanto L. Traube suggerisse l'ipotesi che i fermenti organizzati dovessero le loro proprietà alla presenza in essi di fermenti solubili. D'altra parte, Liebig e altri chimici ridicolizzarono tutta l'ipotesi che reazioni chimiche dipendessero in qualche modo dalla presenza di vita; in un articolo satirico pubblicato anonimamente da Liebig nel 1839, questi fece l'ipotesi scherzosa che le cellule del lievito avessero ‟una ventosa facciale per divorare lo zucchero, un getto d'alcool che usciva dall'ano, mentre diossido di carbonio gorgogliava fuori da enormi organi genitali" (Das enträtselte Geheimnis der geistigen Gärung, in ‟Liebig's Annalen der Chemie", 1839, XXIX, pp. 100-104). La dimostrazione, fornita da E. Buchner (1897), che un succo privo di cellule estratto dal lievito poteva ancora fermentare lo zucchero, rivelò che Pasteur e Liebig avevano ambedue ragione, ciascuno dal proprio punto di vista. Il lievito è costituito da veri organismi viventi, ma i processi chimici della fermentazione sono provocati da un sistema di catalizzatori non viventi prodotti all'interno dalle cellule di lievito. L'espressione ‛enzima' fu suggerita da W. Kühne nel 1878 per indicare questo catalizzatore ‛nel lievito'; egli intendeva applicarla anche a organismi più complessi i quali, egli disse, ‟non sono così fondamentalmente diversi dagli organismi unicellulari come molti vorrebbero farci credere". La parola ‛enzima' sostituì ben presto (fuorché in tedesco) quella più ambigua di ‛fermento'.
Dopo la risoluzione della controversia fra Pasteur e Liebig, il XX secolo si apriva con la scena pronta per l'eccezionale sviluppo dell'enzimologia. L'inizio dell'interpretazione quantitativa e teorica del processo catalitico (di cui ci occuperemo nel cap. 2) può essere ravvisato nei contributi di C. O' Sullivan e A. J. Brown in Inghilterra e di V. Henri in Francia, apparsi tra il 1890 e il 1906, benché l'opera di L. Michaelis e M. L. Menten (v., 1913) sia spesso indicata come l'origine della cinetica enzimatica. Queste ricerche precedettero di poco i primi reali tentativi di studiare gli enzimi come sostanze (v. sotto, cap. 3, È a): in effetti la purificazione degli enzimi cominciò nei primi anni del decennio 1920-1930 con l'opera di R. Willstätter e collaboratori, cui seguirono la prima cristallizzazione di un enzima (l'ureasi), a opera di J. B. Sumner nel 1926, e i classici lavori di J. H. Northrop e del suo gruppo sugli enzimi digestivi, compiuti nel decennio seguente all'Istituto Rockefeller. Il primo serio lavoro di purificazione e studio di un enzima animale era stato quello di M. Dixon e collaboratori sulla xantinossidasi del latte (1924-1928).
Dal 1940 in poi numerosi enzimi, sia extracellulari che intracellulari, furono ottenuti allo stato puro e poterono essere sottoposti ai procedimenti analitici che i chimici delle proteine stavano sviluppando (v. proteine, struttura delle). A essi si è poi riconosciuta una tridimensionalità con l'uso della cristallografia a raggi X e perfino modelli tridimensionali completi di molecole enzimatiche sono stati proposti con discreta certezza in almeno una mezza dozzina di casi (v. sotto, cap. 3, È b). Solo quando si è ottenuto questo risultato si può tentare di spiegare con attendibilità il meccanismo dei processi catalitici in termini chimici (v. sotto, cap. 4).
Gli ultimi venticinque anni hanno visto un rinnovato interesse per gli aspetti più biologici dell'enzimologia, alcuni dei quali saranno discussi nel cap. 5. Sono stati studiati sistemi complessi comprendenti più di un enzima, per esempio nel mitocondrio, e si è anche rivolta l'attenzione al controllo dell'attività enzimatica all'interno della cellula. Dopo le prime osservazioni di E. S. Vessel e A. G. Bearn (v., 1957) e di C. L. Markert e F. Møller (v., 1959), lo studio delle forme multiple degli enzimi (‛isoenzimi') è divenuto un ramo fondamentale della materia.
Per un'informazione più particolareggiata sulla storia dell'enzimologia, il lettore è rimandato al testo di Haldane (v., 1930) e a quello di Dixon (v., 1970).
2. Gli enzimi come catalizzatori.
a) Gli enzimi come parte di sistemi metabolici.
Durante la prima metà del XX secolo i biochimici hanno gradualmente chiarito il funzionamento di quel complesso e articolato insieme di reazioni chimiche che ha luogo dentro ogni cellula e che prende il nome globale di ‛metabolismo'. Quasi tutte le sequenze di reazioni studiate si sono rivelate molto più complicate di quanto era sembrato a prima vista. Per esempio, la fermentazione del glucosio ad alcool e CO2, secondo l'equazione generale
C6H12O6 = 2 C2H5OH + 2 CO2
consta di 14 passaggi distinti, richiede l'intervento di adenosintrifosfato, nicotinammide-adenin-dinucleotide, tiamindifosfato, oltre a ioni magnesio e ortofosfato, ed è accoppiata con la trasformazione del fosfato inorganico in una forma esterificata.
Malgrado la loro complessità, appare chiara l'esistenza di certi caratteri comuni nei processi metabolici. In tutte le cellule viventi si svolgono continuamente sequenze metaboliche in cui substrati complessi sono degradati, o aerobicamente o anaerobicamente. La maggior parte di esse possono essere considerate come varianti di due processi fondamentali, l'ossidazione del glucosio a CO2 e acqua attraverso il sistema glicolisi-ciclo degli acidi tricarbossilici, e la demolizione ossidativa degli acidi grassi a catena lunga attraverso la β-ossidazione (v. metabolismo). Questi processi catabolici sono esoergonici e possono essere considerati analoghi alla combustione del carbone o della benzina nei motori convenzionali; essi costituiscono i meccanismi fondamentali di utilizzazione dell'energia dei combustibili biologici (come, nel caso degli animali, il cibo) per le manifestazioni che caratterizzano i sistemi viventi: crescita, riproduzione, movimento e, in casi speciali, produzione di altri tipi di energia (per es., la luce nelle lucciole e nei batteri luminescenti, o potenziali elettrici nel gimnoto). Gran parte dell'energia prodotta nel ‛catabolismo' è utilizzata per mezzo dell'accoppiamento diretto di reazioni cataboliche con un'ampia serie di processi biosintetici (‛anabolismo'). Anche se una cellula vivente non è in accrescimento e non mostra una variazione netta della sua composizione chimica, può avere luogo in essa una considerevole attività sintetica, poiché tutta la materia vivente è in equilibrio dinamico. Studi con metaboliti marcati mediante isotopi hanno dimostrato che la maggior parte dei componenti del corpo, anche in strutture apparentemente stabili come le ossa, si distrugge con grande rapidità, con vite medie di rinnovamento che vanno da pochi giorni ad alcuni mesi. Una discussione in extenso di tali sequenze metaboliche è al di fuori degli scopi di questo articolo (per un esame delle più importanti di esse v. Dixon e Webb, 19642, cap. XII). Ai nostri fini è importante ricordare che ogni passaggio metabolico, con poche eccezioni, è portato a termine da un singolo enzima e che in generale gli enzimi hanno un alto grado di specificità. Il numero complessivo degli enzimi che esistono come entità separate deve assommare a molte migliaia; la lista degli enzimi ben caratterizzati nella Enzyme nomenclature (v., 1965) conteneva 875 voci, che aumenteranno a quasi 2.000 in una nuova edizione di prossima pubblicazione.
In certi casi la sequenza metabolica può risultare semplicemente da una mescolanza di enzimi solubili coesistenti nel citoplasma della cellula; è l'alta specificità dei singoli enzimi ad assicurare in questo caso che le reazioni si succederanno in una sequenza costante. Così avviene per la glicolisi, la prima parte della demolizione del glucosio, fino al piruvato (che contiene tre atomi di carbonio). D'altronde molte sequenze ossidative (per es., il ciclo degli acidi tricarbossilici per l'ossidazione del piruvato e la β-ossidazione degli acidi grassi) sono portate a compimento da complessi multienzimatici organizzati in strutture disposte sulla membrana interna dei mitocondri animali.
b) Gli enzimi come entità indipendenti.
Reazioni enzimatiche. - A prima vista l'elenco delle reazioni catalizzate da enzimi fa pensare a una sconcertante gamma di attività; gamma forse non sorprendente se si considera la vasta serie di sostanze chimiche complesse che si trovano negli organismi viventi delle diverse specie. Tutti i prodotti naturali studiati dai chimici organici nell'ultimo secolo sono stati sintetizzati (in soluzione acquosa a temperatura normale!) da una delle sequenze di enzimi discusse nel paragrafo precedente. Tuttavia, uno studio approfondito rivela che gli enzimi catalizzano un numero di tipi di reazione relativamente piccolo.
La necessità di una classificazione sistematica delle reazioni enzimatiche si è presentata come requisito essenziale per creare un sistema razionale di nomenclatura, quale è discusso nel sottoparagrafo seguente. Sono stati proposti diversi schemi generali di classificazione: citeremo fra essi quelli di Oppenheimer (1925), Euler (1934), Baldwin (1947), Hoffmann-Ostenhof (1953), Dixon e Webb (1958). Nel 1956 l'Unione Internazionale di Biochimica insediò una Commissione internazionale, presieduta da M. Dixon, con lo scopo di formulare una classificazione e una nomenclatura degli enzimi. Il rapporto della Commissione per gli enzimi fu approvato dall'Unione nella sua sessione del 1961 a Mosca; per quanto questo rapporto sia stato in seguito riveduto, i principi fondamentali della classificazione sono rimasti gli stessi (v. Enzyme nomenclature, 1965). Le linee principali sono esposte qui di seguito.
La classificazione in uso è basata esclusivamente sul tipo di reazione chimica catalizzata, non sulla natura dell'enzima. In questo senso essa può essere considerata da molti biochimici come provvisoria, in quanto si può considerare possibile, in prospettiva, una definizione dei caratteri chimici e strutturali della molecola enzimatica che distinguono un tipo di enzima dall'altro. Finora, però, esistono solo pochissimi enzimi per i quali è stata ottenuta una descrizione chimica ragionevolmente completa della molecola.
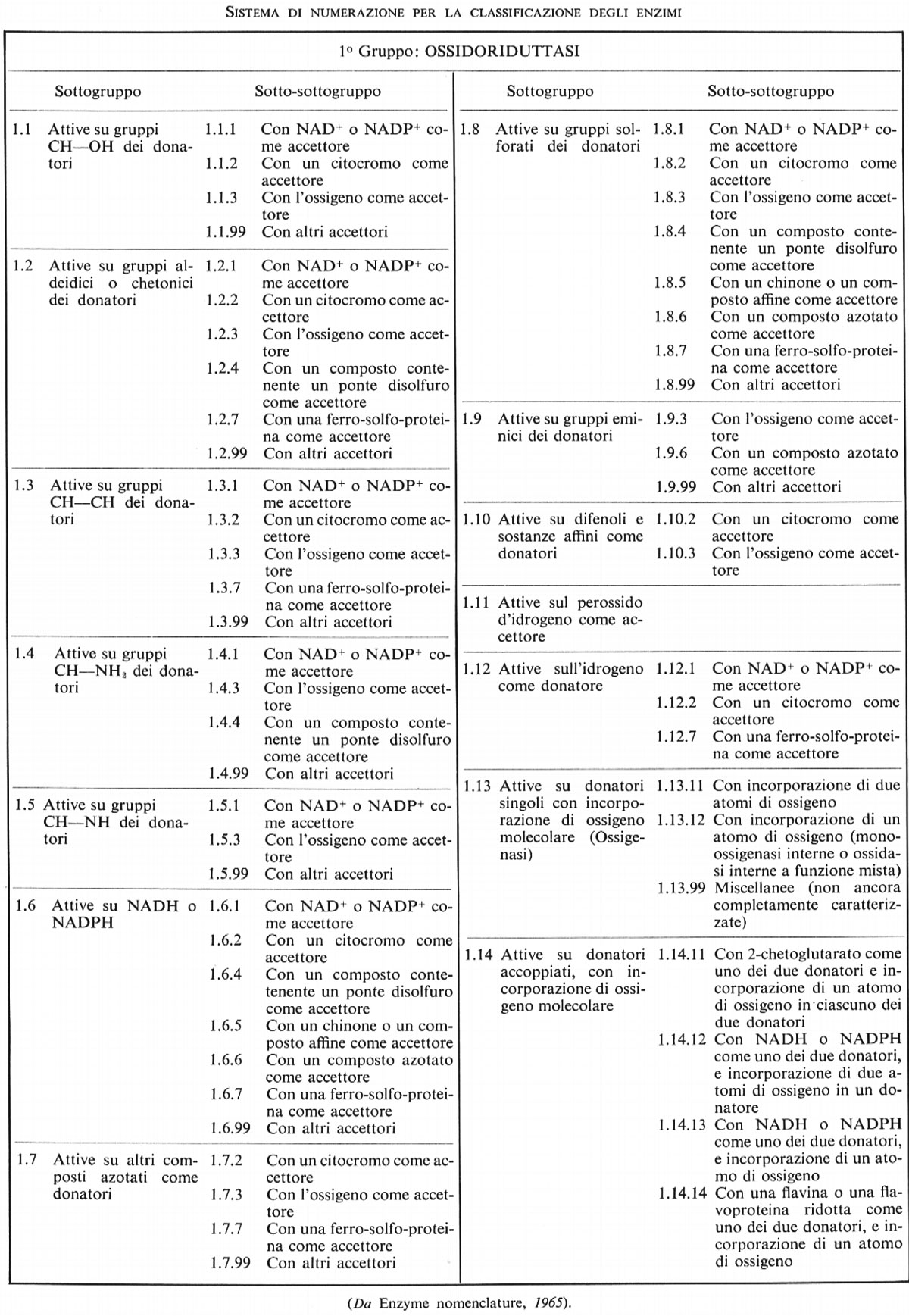
Nello schema approvato, gli enzimi sono disposti in sei gruppi concernenti la catalisi di processi simili, con sottogruppi diretti a specificare con maggior precisione la reazione effettivamente catalizzata e un'ulteriore suddivisione che o definisce ancor più precisamente la reazione o indica il tipo di substrato su cui ha luogo la reazione in oggetto. Per esempio, il gruppo delle idrolasi (o enzimi idrolitici, cioè che catalizzano la scissione del substrato con intervento di acqua) comprende, fra gli altri, un sottogruppo costituito da enzimi che agiscono su legami esterei; questo può essere suddiviso in più sotto-sottogruppi, secondo che al legame estereo partecipi un acido carbossilico, o un tiolo, o acido fosforico, o acido solforico, ecc. I sei gruppi sono:
1. Ossidoriduttasi
2. Transferasi
3. Idrolasi
4. Liasi
5. Isomerasi
6. Ligasi (o sintetasi).
La Commissione stabilì un sistema di numerazione a quattro numeri, basato sulla classificazione, che è stato ampiamente adottato. La classificazione generale, con la numerazione dei sottogruppi e dei sotto-sottogruppi, è presentata qui di seguito.
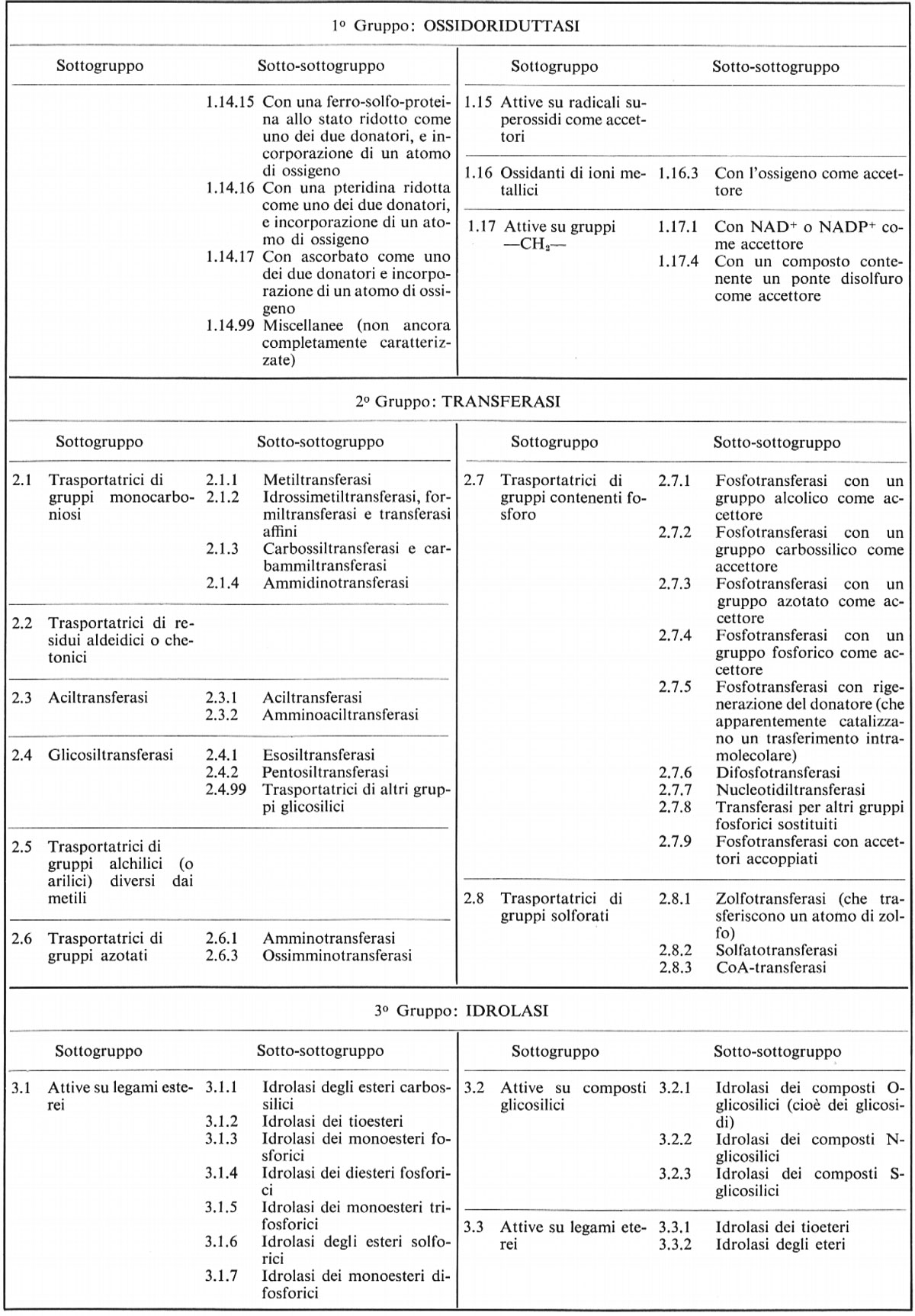
Per tutti gli enzimi del gruppo 1 (ossidoriduttasi), la reazione può essere scritta come trasporto di idrogeno:
AH2 + B ⇄ A + BH2.
È uno dei gruppi più ampi, all'interno del quale una grande quantità di enzimi catalizza un numero piuttosto piccolo di ossidazioni. Per esempio, nella più recente versione della lista degli enzimi, 186 voci concernono l'ossidazione di gruppi idrossilici a chetonici; complessivamente 279 enzimi catalizzano o l'ossidazione di
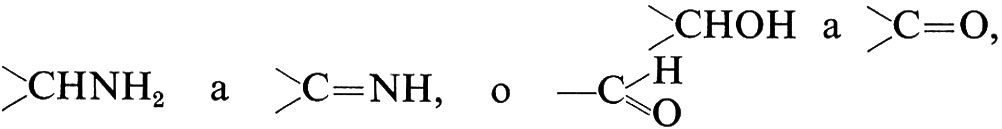
(equivalente a

Un numero di accettori di idrogeno relativamente piccolo è interessato in queste stesse reazioni: i più comuni sono i coenzimi derivati dalla nicotinammide, i citocromi e l'ossigeno. Il ruolo del nicotinammide-adenin-dinucleotide nella sfera anaerobica del metabolismo è interamente dovuto al fatto che molte deidrogenasi hanno in comune questo coenzima come accettore di idrogeno, cosicché esso può agire da trasportatore di idrogeno fra i substrati di deidrogenasi ‛accoppiate'.
Tre sottogruppi delle ossidoriduttasi meritano una menzione speciale. Nel sottogruppo 1.11 l'accettore è il perossido d'idrogeno; l'enzima ‛catalasi' è un caso speciale, in cui una molecola di H2O2 è ossidata da un'altra, secondo la reazione:
2 H2O2 = 2 H2O + O2.
Nei sottogruppi 1.13 e 1.14 l'ossigeno è incorporato nel substrato direttamente dall'ossigeno molecolare; nel caso delle idrossilasi, sono presenti due diverse molecole donatrici, in una delle quali è incorporato un atomo di ossigeno:
RH + AH2 + O2 = ROH + A + H2O.
Il gruppo 2 comprende le transferasi, che trasferiscono gruppi con un atomo di carbonio, gruppi acilici, gruppi glicosilici, gruppi amminici e altri gruppi azotati, gruppi contenenti fosforo o zolfo. In certo senso le ossidoriduttasi possono essere considerate come un tipo speciale di transferasi, che trasferiscono idrogeno, e tutte le reazioni catalizzate dai gruppi 1 e 2 possono essere scritte così:
AX + B ⇄ A + BX.
Questa equazione simboleggia, in effetti, le reazioni catalizzate da più di una buona metà degli enzimi finora scoperti.
Molte transferasi formano gruppi con un accettore comune, che così si comporta nelle vie metaboliche come coenzima, trasportando radicali attivi fra sistemi enzimatici accoppiati. Così l'ADP agisce come trasportatore di fosfato, il coenzima A di gruppi acilici, l'UDP di gruppi glicosilici, la S-adenosilmetionina di gruppi metilici e il tetraidrofolato di altri gruppi a un atomo di carbonio (idrossimetilici, formilici e formimminici).

Alcuni enzimi classificati in particolari gruppi di transferasi catalizzano reazioni che a prima vista sembrano differenti. Le fosfomutasi (sottogruppo 2.7.5) catalizzano apparentemente uno spostamento intramolecolare, ma in realtà hanno bisogno di un coenzima che è rigenerato nella reazione globale; così la reazione
glucosio-6-fosfato + glucosio-1,6-difostato =
= glucosio-1,6-difosfato + glucosio-1-fosfato
è un semplice trasporto di fosfato da una molecola a un'altra, ma la reazione netta è:
glucosio-6-fosfato = glucosio-1-fosfato.
Gli enzimi nei quali agisce come accettore o il fosfato o il pirofosfato possono essere considerati come ‛fosforilasi' o ‛pirofosforilasi', cioè enzimi che producono una scissione analoga all'idrolisi. Così la reazione
X-Y + H3PO4 = X-H2PO3 + YOH
può essere considerata come la fosforolisi di XY o come il trasporto di X da XY al fosfato. Per convenienza questi enzimi sono trattati come transferasi; ne sono esempi le transglicosilasi (come la classica fosforilasi dell'amido o del glicogeno) o le nucleotidiltransferasi, come gli enzimi, scoperti da A. Kornberg, responsabili della sintesi dei dinucleotidi:
ATP + flavin-mononucleotide = adenin-flavin-dinucleotide + pirofosfato
ATP + nicotinammide-mononucleotide = nicotinammide-
-adenin-dinucleotide + pirofosfato.
Questi due enzimi trasportano un gruppo adenina-ribosiofosfato dall'ATP all'altro reagente.
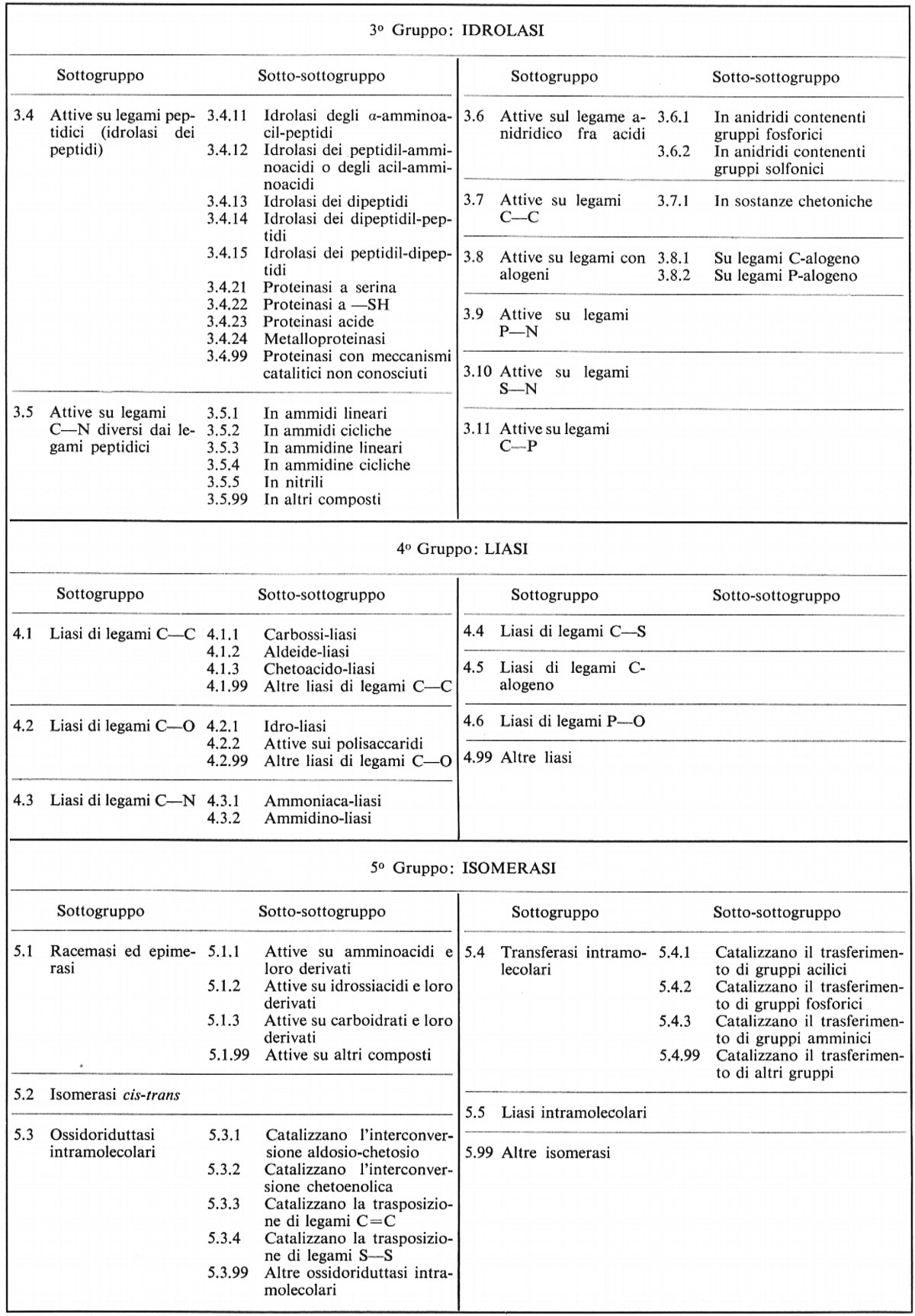
Le reazioni degli enzimi del gruppo 3 possono essere rappresentate come:
A-B + H2O → AH + BOH.
Il gruppo comprende esterasi, fosfatasi, peptidasi, deamminasi, glicosidasi ed enzimi che idrolizzano il legame anidridico (come il legame pirofosforico dell'ATP). Molte idrolasi, per quanto non tutte, catalizzano, in condizioni particolari, anche reazioni di trasferimento. Fra queste condizioni è compresa un'alta concentrazione dell'accettore, in quanto esso dovrà competere con l'acqua per il gruppo trasportato. Così le fosfatasi, che sono relativamente aspecifiche, possono portare a un accumulo di fosfato di etile se substrati come il fosfato di fenile o il fosfato di fenolftaleina sono idrolizzati in presenza di un'alta concentrazione di etanolo. In questi casi appare appropriato considerare un'idrolasi semplicemente come un caso speciale di transferasi in cui l'acqua è l'accettore permesso, cioè
A-X + B = A + BX
oppure
A-X + H2O = AH + XOH.
È spesso vero che queste idrolasi mostrano un grado di specificità molto più alto per il donatore che per l'accettore. Assieme ad altre prove, questo è indice di una reazione a due stadi, nella quale il gruppo X è trasferito sulla proteina enzimatica a formare un composto intermedio reattivo. Ritorneremo su questo argomento nel cap. 4.
Molti enzimi di questo gruppo entrano nei processi digestivi. Poiché essi sono extracellulari, furono scoperti e studiati molto prima di altri gruppi di enzimi. Essi rappresentano tuttora un materiale di studio preferito da molti enzimologi, in quanto si purificano facilmente in grandi quantità e danno reazioni che sono misurate e interpretate agevolmente. I soli enzimi di cui si conosce completamente la struttura tridimensionale sono idrolasi, come la ribonucleasi, la chimotripsina e il lisozima.
I tre gruppi rimanenti comprendono il 22% degli enzimi conosciuti. Mentre le reazioni finora descritte interessano due reagenti in ciascuno dei due sensi della reazione, il gruppo 4 ha due reagenti in una direzione e uno nell'altra; il gruppo 5 è monomolecolare in entrambe le direzioni e il gruppo 6 presenta tre molecole da ambedue le parti.
Le ‛liasi' (gruppo 4) catalizzano il tipo di reazione:
AX-BY ⇄ A = B + X-Y.
Questa reazione può essere considerata come l'addizione di molecole quali H2O, H2S, NH3 o aldeidi al doppio legame di una molecola insatura. Il nome del gruppo deriva dalla reazione in senso opposto, che può essere considerata come una scissione non idrolitica di una molecola complessa. Le decarbossilasi vengono classificate come liasi (sottogruppo 4.1.1) piuttosto che come idrolasi producenti acido carbonico. Così la classica ‛carbossilasi' del lievito, che trasforma il piruvato in aldeide acetica, catalizza la reazione:
CH3COCOOH = CH3CHO + CO2
che può essere scritta:
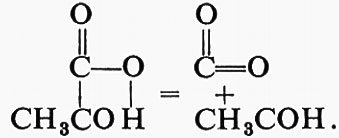
Il gruppo 5 (‛isomerasi') catalizza reazioni intramolecolari, cioè:
A ⇄ B.
Le isomerasi includono enzimi che catalizzano reazioni simili a quelle dei gruppi 1, 2 e 4, ma in cui l'intera reazione si svolge nella stessa molecola. Così ci sono ossidoriduttasi, transferasi e liasi intramolecolari. Inoltre, ci sono alcuni enzimi che catalizzano semplicemente una trasformazione stereochimica (racemasi, epimerasi e isomerasi cis-trans).
Le reazioni prodotte dagli enzimi del gruppo 6 sono più complesse di tutte quelle già descritte. Esse portano a termine l'attacco reciproco fra due molecole, cioè una reazione di sintesi, collegata alla scissione simultanea di un nucleoside-polifosfato, che è normalmente l'ATP. L'energia liberata dalla scissione dell'ATP può essere considerata come la sorgente dell'energia necessaria per la sintesi; questi enzimi formano così uno dei principali tipi di collegamento fra catabolismo e anabolismo. La reazione completa è:
X + Y + ATP = X-Y + ADP + ortofosfato
oppure
X + Y + ATP = X-Y + AMP + pirofosfato.
Queste reazioni non possono avvenire in un solo stadio e in molti casi si hanno indicazioni di due o tre stadi che compongono l'intera sequenza di reazioni. Questi stadi sono essenzialmente di tipo transferasico.
L'esame rapido e superficiale delle reazioni enzimatiche che abbiamo qui presentato serve a mettere in risalto la fondamentale somiglianza, nelle linee essenziali, di quello che a prima vista sembra un complicato panorama di tipi di reazione. La grande maggioranza delle reazioni prodotte dagli enzimi può essere descritta semplicemente come il trasferimento di un gruppo chimico da una molecola all'altra o come una sequenza di tali trasferimenti, che coinvolge una o più molecole di substrato, o eventualmente la stessa molecola dell'enzima.
Nomenclatura degli enzimi. - Come si è detto nell'introduzione, Payen e Persoz proposero il nome di diastasi per l'enzima che idrolizza l'amido. Agli enzimi proteolitici digestivi furono dati nomi terminanti in ‛ina', come pepsina, ma nel 1883 E. Duclaux propose che gli enzimi dovessero essere denominati aggiungendo il suffisso ‛asi' a una radice indicante la natura del substrato (cioè la sostanza su cui agisce l'enzima). Quasi tutti i nomi proposti in questo secolo hanno usato tale sistema come punto di partenza.
Il sistema della Commissione per gli enzimi (v. Enzyme nomenclature, 1965) dà due nomi per ogni enzima: un nome ‛sistematico', che è fondato su principî strettamente logici ma che è spesso lungo e scomodo, e un nome ‛ordinario', abbastanza breve per un uso generale ma non necessariamente molto esatto o sistematico. Il sistema dà anche un numero a quattro membri, che definisce univocamente ogni enzima. I nomi sistematici sono molto meno usati rispetto ai nomi ordinari seguiti dai numeri, che permettono un riferimento rapido alla lista ufficiale. Così, 2.7.1.6 è la galattochinasi, il cui nome sistematico è ATP: D-galattosio-1-fosfotransferasi, che riflette chiaramente la reazione catalizzata:
ATP + D-galattosio ⇄ ADP + D-galattosio-1-fosfato.
I nomi ordinari, costituiti da una radice derivata dal nome del substrato più il suffisso -asi, sono attualmente limitati a enzimi producenti l'idrolisi del substrato in oggetto, per esempio acetilcolinesterasi, glucosio-6-fosfatasi. Nei gruppi diversi dalle idrolasi i nomi sono tipicamente composti da due parole, la prima indicante il substrato e la seconda, che termina in -asi, denotante il tipo di reazione che ha luogo. La seconda parola è spesso comune a un intero gruppo o sottogruppo di enzimi. Per esempio la maggioranza delle ossidoriduttasi sono deidrogenasi, riduttasi o ossidasi, come la lattatodeidrogenasi o la xantinossidasi. La Commissione per gli enzimi ha proposto che alcuni nomi, finora usati piuttosto indiscriminatamente, siano limitati a certi significati definiti; per esempio, ‛sintetasi' è limitato a quegli enzimi del gruppo 6 che catalizzano sintesi accoppiate alla scissione di nucleosidi-polifosfati. Un termine nuovo, ‛sintasi', è usato nei casi di enzimi di altri gruppi che prendono il nome ordinario da una reazione di sintesi, per esempio citratosintasi, una liasi che catalizza la reazione:
acetilcoenzima A + ossalacetato = citrato + coenzima A.
Per evitare confusione e ambiguità, sono state poste regole precise per la formulazione di nuovi nomi d'enzimi (v. Enzyme nomenclature, 1965).
c) Aspetti quantitativi: cinetica enzimatica.
L'osservazione quantitativa delle reazioni enzimatiche ha fornito un gran numero di informazioni sul processo della catalisi enzimatica (v. catalisi enzimatica). Il procedimento sperimentale che è fondamento degli studi cinetici consiste essenzialmente nel misurare il prodotto formato a tempi diversi in un sistema di composizione nota. La fig. 1 mostra un tipico grafico del prodotto in funzione del tempo, o ‛curva di cinetica' della reazione. Questa è una curva complessa, che tende verso una curva di reazione monomolecolare alla fine della reazione e mostra una velocità costante, equivalente a una reazione di ‛ordine zero', all'inizio, purché la concentrazione iniziale del substrato sia sufficientemente alta. Fra questi due estremi, l'‛ordine' della reazione varia fra 0 e 1. Curve di questo tipo furono pubblicate per la prima volta da O'Sullivan e Tompson nel 1890, benché questi autori pensassero di poterle considerare approssimativamente curve del primo ordine; ma A. J. Brown nel 1902 confermò le anomalie e suggerì l'ipotesi che potessero essere dovute a una combinazione fra enzima e substrato. V. Henri pubblicò un'equazione empirica adatta a spiegare queste curve e nel 1903 mostrò che questa equazione poteva essere dedotta supponendo una fase rapida reversibile per la formazione del complesso enzima-substrato, seguita da una fase, la cui velocità era invece limitante della velocità complessiva della reazione, rappresentata dalla decomposizione del complesso con formazione del prodotto.
Le curve di cinetica della reazione sono complicate da numerosi fattori, come l'instabilità dell'enzima stesso, l'inattivazione dell'enzima da parte dei prodotti di reazione, cambiamenti di pH, ecc. Gli enzimi sono molto sensibili ai cambiamenti di pH e quindi il progresso degli studi sperimentali fu lento finché la classica opera di S. P. L. Sørensen non permise l'uso di soluzioni tampone per controllare il pH. Le altre complicazioni possono essere eliminate misurando la velocità iniziale di reazione in una serie di differenti miscele di reazione. La fig. 2 mostra tre rappresentazioni dell'andamento della velocità iniziale v in funzione della concentrazione del substrato s. Dati di questo tipo furono pubblicati da Michaelis e Menten (v., 1913) per l'idrolisi del saccarosio a opera dell'invertasi del lievito, insieme con un'elaborazione matematica. Curve come quelle della fig. 2 sono spesso descritte come ‛curve di Michaelis'. La teoria di Michaelis e Menten è molto semplice. Ammette che si stabilisca rapidamente un equilibrio fra enzima, substrato e un complesso ES e che questo complesso si decomponga a una velocità che è limitante della velocità dell'intero processo:
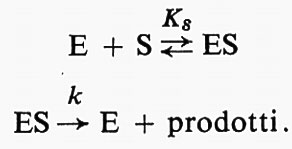
Se e è la concentrazione dell'enzima e s la concentrazione iniziale del substrato, si può facilmente dimostrare che la velocità di formazione del prodotto (all'inizio della reazione) è data da:
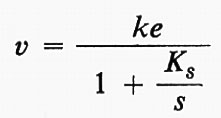
Questa è l'equazione di un'iperbole che, per valori molto grandi di s, tende a una velocità massima ke, che può essere indicata con V. Allora,
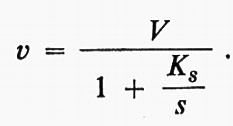
Nel grafico in forma diretta, usato nella fig. 2A, il valore di Ks è dato dalla concentrazione del substrato corrispondente a metà della velocità massima. V e Ks, i parametri dell'equazione di Michaelis e Menten, possono essere ricavati in maniera più conveniente da grafici d'altro tipo e, in particolare, mediante il grafico detto ‛dei doppi reciproci' mostrato nella fig. 2C, proposto da H. Lineweaver e D. Burk. Essi possono essere ottenuti anche con procedimenti statistici che possono essere posti alla base di programmi per calcolatori elettronici (v. Cleland, 1967).
V e Ks non sono soltanto i valori numerici che definiscono l'effetto della concentrazione del substrato per un dato enzima; essi forniscono anche informazioni sul meccanismo enzimatico stesso, se la teoria è valida. Ks misura sotto forma di reciproco l'affinità dell'enzima per il suo substrato, e V indica la velocità costante di decomposizione del complesso enzima-substrato, vale a dire l'efficienza catalitica dell'enzima. Sfortunatamente la teoria può essere interpretata in molti modi. Briggs e Haldane (v., 1925), in un importante articolo teorico, mostrarono che un'equazione della stessa forma di quella di Michaelis e Menten poteva essere ottenuta senza ipotizzare una fase di equilibrio rapido. Al contrario essi ammisero che si stabilisse uno stadio stazionario, un ‛dinamico', in cui la concentrazione del complesso cambiava molto più lentamente di quella del substrato o del prodotto. Definendo le costanti di velocità come appare nelle seguenti equazioni:

essi mostrarono che la velocità complessiva era data da:
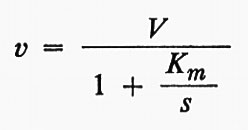
dove
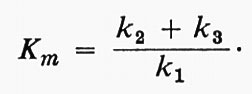
In questo modo Km, cioè la Ks apparente ricavata dalle curve sperimentali, sarà in realtà:
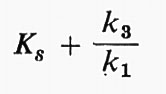
vale a dire la costante d'affinità più un fattore di correzione che rappresenta l'effetto di deformazione operato sull'equilibrio dal complesso di tutte le costanti di reazione.
Queste considerazioni ebbero solamente un interesse teorico finché B. Chance non elaborò, nel 1943, un metodo ingegnoso per misurare le singole costanti cinetiche per un enzima particolare, la perossidasi, il cui spettro di assorbimento cambia in presenza del substrato. La Km sperimentale, lungi dall'essere anche approssimativamente simile alla Ks, si rivelò in gran parte dipendente dal rapporto k3/k1; quindi non misurava in nessun modo l'affinità dell'enzima per il substrato. L'opera di Chance segnò l'inizio di un copioso lavoro, sia sperimentale sia teorico, basato su questo tipo di elaborazione, comunemente noto come ‛cinetica di stato stazionario'. Buone trattazioni se ne possono trovare in molti manuali classici: Laidler (v., 1958); Webb (v., 1963); Reiner (v., 1959); Boyer, Lardy e Myrbäck (v., 19592); Alberty (v., 1956 e 1962) e il IV capitolo del libro di Dixon e Webb (v., 19642).
La semplice esposizione qui presentata presuppone, ovviamente, una reazione enzimatica in cui un'unica molecola di substrato si trasforma nei prodotti. La maggior parte delle reazioni enzimatiche è più complessa, con due o più substrati. Per ogni substrato può essere ottenuta una Km, ma la formulazione di stato stazionario mostra che questo parametro è una funzione complessa in cui compaiono non solo le costanti di velocità relative al substrato variabile, ma anche quelle della combinazione con l'altro substrato. Le equazioni diventano ancora più complicate nel caso che nella reazione intervenga un attivatore (v. sotto).
Le curve sperimentali della velocità in funzione della concentrazione del substrato non sempre seguono la curva teorica di Michaelis-Menten. Una frequente anomalia, specialmente nel caso di enzimi idrolitici, è che la velocità diminuisce nuovamente ad alte concentrazioni di substrato. Ciò può essere dovuto a cause svariate, fra cui la formazione di un complesso inattivo ES2, con una seconda molecola di substrato, che si comporta come un inibitore.
La fig. 3 mostra il caso tipico dell'effetto del pH sulla velocità della reazione enzimatica, benché vi siano molte eccezioni. Queste curve si riferiscono a effetti reversibili; ma esiste anche una distruzione irreversibile dell'enzima per esposizione a valori di pH al di fuori dell'intervallo di stabilità, che spesso è molto stretto. La curva reversibile tipica, che è illustrata nella fig. 3, mostra un optimum di pH, con una diminuzione della velocità da una parte e dall'altra dell'optimum, e somiglia a una curva di dissociazione semplice. In altri termini, la tipica curva velocità-pH sembra riflettere, come Michaelis e Davidsohn avevano suggerito fin dal principio, la curva di dissociazione di un anfolita semplice, quale un amminoacido, più che quella di una molecola proteica, con molti gruppi ionizzabili. Questo fatto, insieme con la teoria di Michaelis e Menten e l'alto grado di specificità di molti enzimi, ha portato al concetto della presenza sull'enzima di un sito di combinazione limitato e specifico o ‛centro attivo'. Si potrebbe allora ammettere che i due gruppi ionizzabili responsabili della curva attività-pH siano contenuti in questo centro attivo.
Lo scopo di gran parte degli studi di cinetica enzimatica è stato quello di chiarire i particolari del meccanismo dei processi enzimatici, sia nel senso di conoscere le varie tappe della reazione, sia in quello di identificare i gruppi chimici interessati. Dalla discussione precedente risulta chiaro che l'effetto del pH sull'attività potrebbe permettere la determinazione dei pK dei gruppi ionizzabili del centro attivo e così portare a ragionevoli congetture sulla natura di questi gruppi. A prima vista il problema non appare difficile, specie se i valori di pK sono ben separati, ma la teoria completa non è così semplice. Contributi importanti le sono stati arrecati da Michaelis, Alberty, Dixon e Laidler (v. Dixon e Webb, 19642; v. Laidler, 1958; v. Alberty, 1956). Queste teorie distinguono gli effetti del pH su V, la velocità limite o massima, da quelli su Km: i primi dipendono dai gruppi ionizzabili del centro attivo che prendono parte al passaggio finale del processo catalitico, la decomposizione di ES; gli effetti su Km dipendono dai gruppi coinvolti in ogni passaggio le cui costanti di velocità sono comprese nell'espressione che definisce Km. Nel caso ideale ‛tipo Michaelis', dove Km = Ks, gli effetti del pH su Ks dipenderanno solamente dai gruppi dell'enzima che partecipano alla combinazione con il substrato.
Un'elaborazione grafica particolarmente semplice di dati in funzione del pH è stata descritta da Dixon (v., 1953). Se V, o Km, o la costante di velocità monomolecolare a basse concentrazioni di substrato (v0) sono riportate su una scala logaritmica in funzione del pH, le curve che si ottengono hanno dei segmenti rettilinei con inclinazioni che sono zero o numeri interi, congiunti da brevi porzioni curvilinee. Estrapolando i segmenti rettilinei, i punti d'intersezione rappresentano valori di pH che corrispondono ognuno al pK di un gruppo implicato nel processo enzimatico o in E,o in ES, o in S. Combinando i dati ottenuti da V e da Km si ha un quadro soddisfacente del centro attivo. La fig. 4 mostra una curva teorica con l'intepretazione, in base alla teoria di Dixon, dei vari tipi di curve pKm/pH che possono essere ottenute. La fig. 5 mostra poi alcuni dati sperimentali ricavati con la xantinossidasi, disposti in grafico da Dixon; essi indicano pKs uguali a 5,3 e 7,9 per i gruppi dell'enzima (E) implicati nella combinazione con il substrato, e un pK di 4,4 per un gruppo in ES che partecipa al processo catalitico (in quanto è presente anche nelle curve V/Km). In genere, tali gruppi possono essere identificati solo in via ipotetica; tuttavia un caso generale è la frequente ricorrenza di gruppi con pK fra 6 e 7, che vengono di solito identificati come istidine. Spesso altre prove, del tutto indipendenti, confermano l'importanza di un residuo di istidina nel centro attivo.
Un altro capitolo, tra i più importanti, della cinetica enzimatica è quello dedicato allo studio dell'effetto della temperatura. Esso è più complicato e meno fruttuoso dello studio degli effetti della concentrazione del substrato e del pH. Le reazioni enzimatiche hanno un optimum di temperatura, per quanto non si tratti di un valore costante, bensì dipendente dalle condizioni dell'esperimento. Infatti esso è la risultante di due fenomeni contrapposti, l'accresciuta velocità catalitica e l'accresciuta velocità di denaturazione, ambedue prodotti dall'aumento di temperatura. Il coefficiente di temperatura per l'inattivazione degli enzimi è abitualmente molto alto, corrispondente a quello per la denaturazione proteica. Studi sull'effetto della temperatura sulla velocità iniziale possono perciò essere compiuti in maniera soddisfacente solo in un intervallo di temperature relativamente ristretto; si possono distinguere, come nel caso del pH, effetti su V ed effetti su Km.
Un diagramma in cui log V sia riportato in funzione del reciproco della temperatura assoluta dà normalmente segmenti rettilinei; in altre parole, il processo catalitico segue l'equazione di Arrhenius. Dall'inclinazione di queste curve possono essere ottenute le energie di attivazione per il passaggio che porta alla formazione dei prodotti. Si può affermare in generale che i valori che si ottengono sono molto bassi in confronto a quelli delle corrispondenti reazioni non enzimatiche. Per esempio, le energie di attivazione per l'idrolisi del saccarosio sono 26.000 calorie per mole nel caso della catalisi da H+, 13.000 calorie per mole nel caso della saccarasi del malto e 11.000 calorie per mole per la saccarasi del lievito. Per la decomposizione del perossido di idrogeno, i valori (in calorie per mole) sono 18.000, 11.000 e 5.500 rispettivamente per la reazione in assenza di catalizzatori, per quella catalizzata dal platino colloidale e per quella catalizzata dalla catalasi. Così, l'effetto dell'enzima è quello di ridurre la ‛barriera di energia' che il substrato deve oltrepassare per essere trasformato nel prodotto.
In alcuni casi, le reazioni enzimatiche mostrano discontinuità nei diagrammi di Arrhenius. Questi cambiamenti dell'energia di attivazione a determinate temperature indicano presumibilmente trasformazioni dell'enzima, ma non sono stati mai spiegati in maniera adeguata.
Nei casi in cui Km può essere considerata una costante di equilibrio per la combinazione enzima-substrato, l'effetto della temperatura su di essa dipenderà dal calore di combinazione fra enzima e substrato e il ΔH per questa reazione può essere ricavato da un grafico lineare in cui è riportato log Km contro il reciproco della temperatura assoluta. Anche in questi casi, le curve di alcuni enzimi presentano delle discontinuità. In un caso, quello della fumaratoidratasi, Massey (v., 1953) ha ottenuto una buona coincidenza fra i dati dell'effetto della temperatura su V e Km per ambedue i sensi della reazione e quelli relativi all'effetto sull'equilibrio complessivo; in questo modo è riuscito a dare un quadro soddisfacente degli scambi di calore nella sequenza di reazione a due differenti temperature (v. fig. 6). I substrati fumarato e malato sono rappresentati da F e da M e i relativi complessi da EF e EM. EX* rappresenta lo stato attivato che permette la trasformazione del substrato nel prodotto, o viceversa. Così la sequenza complessiva è:
E + F ⇄ EF ⇄ EX* ⇄ EM ⇄ E + M.
(+ H2O)
Nella fig. 6, EF1 è il livello ricavato dai diagrammi lineari al di sopra di 18 °C, ed EF2 quello dedotto dai dati ottenuti al di sotto di 18 °C. La differenza nei livelli delle curve al punto EX* mostra il buon accordo dei dati ottenuti partendo da tre basi differenti.
Ricorderemo qui anche un altro aspetto degli studi cinetici, vale a dire lo studio degli inibitori. Molte sostanze che non sono substrati di un enzima possono avere un effetto sulla velocità di reazione, o come ‛inibitori' o come ‛attivatori'. Talvolta questi effetti sono abbastanza aspecifici, come nel caso di agenti denaturanti. D'altra parte, l'alta specificità di molti inibitori fa supporre che essi agiscano a livello dello stesso centro attivo dove agisce il substrato; questi inibitori sono stati utilizzati per investigare la natura del centro attivo stesso, cioè come un'estensione degli studi di specificità. Gli inibitori specifici, capaci di bloccare particolari reazioni enzimatiche, sono stati anche ampiamente impiegati per chiarire le sequenze metaboliche.
Anche in questo caso, possono essere distinti effetti su V e su Km. Fin dal principio gli inibitori furono distinti in inibitori competitivi, che agiscono combinandosi con il centro attivo e perciò ‛competono' con il substrato e formano un complesso stabile, e inibitori non competitivi che, pur non combinandosi nello stesso punto in cui si combina il substrato, influenzano la natura del complesso che si forma, in modo da impedire l'attivazione catalitica. Le reazioni ipotizzate nei vari casi possono essere rappresentate in questa maniera:
E + S ⇄ ES
E + Ic ⇄ EIc
EIc + S ↛
oppure
E + S ⇄ ES
E + Inc ⇄ EInc
EInc + S ⇄ EIncS
EIncS ↛
dove Ic rappresenta un inibitore competitivo e Inc uno non competitivo.
Si può dimostrare che, nel caso di un equilibrio semplice, gli inibitori competitivi hanno effetto solamente su Km e i non competitivi solamente su V. Alcuni inibitori hanno un effetto misto.
Il tipo di inibitore può essere determinato studiando o l'effetto della concentrazione del substrato in presenza e in assenza di una quantità fissa di inibitore, o viceversa l'effetto di diverse concentrazioni di inibitore a due differenti e costanti concentrazioni di substrato. La fig. 7 mostra i risultati che si ottengono da vari tipi di grafici ricavati con il primo procedimento. (Per maggiori particolari, si veda Dixon e Webb, 19642, e Webb, 1963).
Abbiamo qui presentato una rapida esposizione dei principali metodi usati in cinetica enzimatica. Lo studio degli aspetti quantitativi dell'attività enzimatica è progredito in maniera assai rapida dalla scoperta dell'esistenza degli enzimi, dato che non dipende dal loro isolamento allo stato puro. Una volta che si sia ottenuto un estratto di tessuto provvisto di un ragionevole livello di attività enzimatica e privo di altri enzimi che agiscano o sul substrato o sul prodotto dell'enzima in oggetto, e che sia stato elaborato un metodo per misurare la quantità del prodotto, allora è possibile compiere gli studi in questione. I risultati così ottenuti hanno grandemente contribuito alle teorie sul meccanismo d'azione degli enzimi che saranno discusse nel cap. 4; in certi casi è stato possibile ottenere una spiegazione plausibile del processo enzimatico già prima di qualsiasi studio sull'enzima come sostanza.
Molti manuali offrono trattazioni generali degli argomenti discussi in questa sezione (v. in particolare Dixon e Webb, 19642; Boyer e altri, 19592; Florkin e Stotz, 1964).
3. Gli enzimi come sostanze.
a) Purificazione degli enzimi.
Per purificare con successo un enzima occorrono alcune condizioni fondamentali. Deve essere disponibile una buona fonte di enzima, preferibilmente provvista di un'attività relativamente alta già nel materiale di partenza. Deve essere disponibile, per la determinazione dell'attività enzimatica, un processo conveniente e riproducibile che dovrebbe, possibilmente, richiedere solo piccole quantità di enzima. Bisogna adottare un metodo che fornisca un indice cui riferire, per paragone, le variazioni di attività; esso può essere il peso secco, il carbonio totale o (dato che l'enzima è una proteina) la quantità totale di proteine o di azoto. Bisogna quindi elaborare tecniche di frazionamento che non inattivino in grado apprezzabile l'enzima. Esse, ovviamente, devono escludere alte concentrazioni di acidi o alcali e temperature elevate, o l'aggiunta di agenti denaturanti come gli ioni di metalli pesanti.
La purificazione degli enzimi non fu intrapresa se non dopo il 1920 e i primi tentativi furono del tutto empirici. Anche nel periodo aureo della purificazione degli enzimi intracellulari, e cioè verso la fine degli anni trenta e negli anni quaranta, ogni purificazione enzimatica era una successione di stadi unica nel suo genere, per quanto di solito comprendesse alcuni procedimenti convenzionali. I recenti sviluppi delle tecniche di scambio ionico e di filtrazione molecolare hanno trasformato la purificazione di un nuovo enzima in un'operazione quasi automatica.
Per valutare se un determinato metodo è un passaggio che contribuisce utilmente alla purificazione, il materiale ad attività enzimatica è suddiviso in frazioni, in ognuna delle quali si determina l'attività e la quantità di materiale (peso secco, carbonio o azoto totale). Da questi dati (e dal volume delle frazioni) si possono calcolare due indici: l'‛attività totale' (comunemente espressa in unità di enzima) e un rapporto che indica il ‛grado di purezza', per esempio le unità/mg di peso secco o le unità/mg di azoto. In questo modo possono essere scelte particolari frazioni, che sono poi combinate e usate come materiale di partenza per un altro passaggio. Lo scopo della scelta delle frazioni è quello di avere il massimo aumento di purezza e, per quanto possibile, il recupero totale delle unità di enzima, cioè ‛purificazione' e ‛rendimento'.
Due metodi sono stati di fondamentale importanza per le prime purificazioni, cioè l'adsorbimento su gel colloidali di fosfato di calcio o di allumina (introdotto da Willstätter) e la precipitazione salina, in genere con solfato d'ammonio (metodo usato con enorme successo da Northrop e dal suo gruppo). Questi procedimenti non apportano drastici aumenti di purezza: un aumento di circa cinque volte con un recupero dell'80% in un singolo passaggio è considerato abbastanza soddisfacente. Tuttavia, ripetendo più volte l'uno o l'altro di questi frazionamenti o meglio ancora applicando l'uno e l'altro in maniera alternata, si riuscì, fra il 1920 e il 1950, a purificare molti enzimi.
Fra i più recenti metodi di frazionamento vi sono la precipitazione per aggiunta graduale, a bassa temperatura, di solventi organici come alcool etilico, acetone o butanolo (aggiunta che deve essere fatta raffreddando adeguatamente la miscela), l'adsorbimento su colonne di resine a scambio ionico e l'uso di colonne di materiali noti come ‛setacci molecolari'. I metodi su colonna sono ora i più usati; le tecniche sono simili, sia che utilizzino scambiatori di ioni come la DEAE-cellulosa e la CM-cellulosa, sia setacci molecolari come il Sephadex (un polimero sintetico del gruppo dei carboidrati, che può essere ottenuto con porosità di varie dimensioni), sia materiali a funzione mista come il DEAE-Sephadex. La preparazione enzimatica è aggiunta alla colonna sciolta in un tampone diluito con il quale la colonna stessa è stata equilibrata; la colonna viene quindi lavata con il tampone e poi sottoposta a un ‛gradiente salino' o a tamponi di differenti pH. La soluzione effluente è suddivisa, mediante un raccoglitore di frazioni, in piccoli campioni, sui quali si determinano l'attività enzimatica e la concentrazione proteica.
Alcuni metodi di purificazione risultano più utili quando la purificazione è già abbastanza avanzata. L'elettroforesi zonale serve sia per purificare sia per controllare la purezza. Analogamente la cristallizzazione spesso conferma che la purificazione è quasi completa e nello stesso tempo è a pieno diritto un mezzo di purificazione essa stessa, specie se ripetuta due o tre volte.
L'accresciuta conoscenza della localizzazione biologica degli enzimi ha facilitato in alcuni casi la purificazione per mezzo dell'isolamento del materiale di partenza. Così D. E. Green e collaboratori, dell'Università del Wisconsin, hanno usato mitocondri isolati da omogenati di tessuti animali come fonte principale per gli enzimi ossidativi localizzati unicamente nei mitocondri.
Prima dello sviluppo di adatte tecniche biofisiche, era difficile giudicare con certezza se la purificazione di un enzima fosse completa. La cristallizzazione è senza dubbio una prova molto indicativa che spesso è stata la meta dell'enzimologo, ma molte proteine possono cristallizzare anche in presenza di una considerevole percentuale di proteine estranee nei cristalli. Migliori criteri di purezza sono la presenza di un'unica banda all'ultracentrifuga analitica e all'elettroforesi in fase libera, o una solubilità costante in presenza di quantità crescenti di fase solida. Bisogna tuttavia tener presente che preparazioni omogenee in base a questi criteri possono mostrare, all'elettroforesi zonale, più bande di attività, dovute alla presenza di ‛isoenzimi'.
b) Composizione e struttura.
Benché intorno al 1925 numerosi enzimi fossero stati purificati di centinaia di volte rispetto agli estratti grezzi, sir W. Bayliss, in una monografia intitolata The nature of enzyme action, pubblicata in quell'anno, affermava che gli enzimi probabilmente non erano proteine, ma forse carboidrati. Anche Haldane (v., 1930, p. 174), nel suo classico testo, sollevava dubbi in proposito e citava una lista di enzimi che non davano le reazioni delle proteine. Egli scriveva: ‟Se gli enzimi sono tutti delle proteine, come molti sperimentatori credono, è certamente degno di nota che la maggioranza dei tentativi riusciti di purificazione abbia portato a ottenere sostanze che, almeno in gran parte, non sono proteine, benché il materiale di origine fosse costituito soprattutto da proteine. Bisogna tener presente che queste preparazioni, che non danno le reazioni delle proteine, non sono dializzabili e all'analisi mostrano di contenere C, H, O e N". Haldane ammetteva che l'ureasi cristallina di Sumner sembrava una proteina pura, ma formulava l'ipotesi, seguendo le opinioni di Willstätter, che si potesse trattare di una proteina che agiva come colloide protettore su di un enzima a essa adsorbito.
Queste difficoltà nascevano soprattutto dall'altissima attività catalitica degli enzimi, a causa della quale si possono preparare soluzioni purificate in cui l'attività enzimatica è facilmente misurabile ma la proteina enzimatica è presente in quantità così piccola da non essere rivelabile con molte tecniche chimiche. Malgrado che dal 1930 in poi sia stata ogni tanto annunciata la scoperta di ‛enzimi privi di proteine', la natura proteica di tutti gli enzimi conosciuti è al giorno d'oggi accettata come un assioma. Dixon e Webb (v., 19642, p. 5) definiscono un enzima come ‟una proteina con proprietà catalitiche dovute alla sua capacità di attivazione specifica".
Dopo la scoperta che gli enzimi non erano che particolari proteine, lo studio chimico degli enzimi purificati divenne parte del problema generale della chimica delle proteine. L'eccezionale progresso compiuto in questo campo negli ultimi venticinque anni, dovuto all'introduzione di metodi completamente nuovi d'indagine per queste molecole di grandi dimensioni e prive di una struttura regolare, è stato forse il più importante fattore del rapido sviluppo delle nostre conoscenze sugli enzimi. L'esposizione delle tecniche della chimica delle proteine esula dagli scopi di questo articolo (v. proteine, struttura delle); saranno brevemente trattati solo alcuni risultati concernenti le proteine enzimatiche. A ogni modo, su circa 2.000 enzimi conosciuti, meno di 100 sono stati esaminati dal punto di vista chimico come proteine pure, meno di 20 in ogni particolare e solo in 4 o 5 casi si dispone di qualcosa di simile a una rappresentazione completa della molecola. Inoltre, gli enzimi idrolitici sono molto meglio conosciuti di quelli di altri tipi.
I primi enzimi purificati furono sottoposti ad analisi fisico-chimiche e in alcuni casi all'analisi degli amminoacidi. Non furono trovati caratteri particolari. Gli enzimi hanno pesi molecolari assai diversi, dal più piccolo (la ribonucleasi) che è appena superiore a 10.000, alla glutammatodeidrogenasi e alla piruvatodecarbossilasi per le quali sono riportati pesi molecolari di un milione e più. Gli enzimi più grandi, tuttavia, sono spesso chiaramente costituiti da sub-unità. Molte proteine enzimatiche sono globulari, ma altre (per es. la miosina, l'ATP-asi) sono notevolmente asimmetriche. La composizione in amminoacidi varia ampiamente e non mostra alcuna particolarità rispetto alle altre proteine; in genere, sono presenti tutti i venti comuni amminoacidi di configurazione L.
Una pietra miliare nella chimica delle proteine fu il metodo di F. Sanger, applicato per la prima volta all'insulina, che permise di chiarire la vera e propria sequenza degli amminoacidi nella struttura primaria di una catena peptidica di una proteina. La prima sequenza primaria stabilita per un enzima fu quella della ribonucleasi, in seguito al lavoro dei gruppi guidati da C. B. Anfinsen e da S. Moore e W. H. Stein. Essa è mostrata schematicamente nella fig. 8. Anche la posizione dei ponti − S − S − , oltre alla sequenza degli amminoacidi dell'unica catena di 124 residui, può essere determinata chimicamente. Essi sono formati da residui di cistina composti da due ‛mezze cistine' situate in differenti posizioni della catena peptidica, cioè:
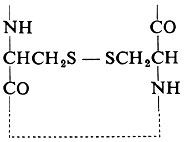
Questi ponti pongono un limite al possibile ripiegamento della molecola: per esempio, il ponte fra i residui 65 e 72 implica che in questo punto esista una voluta relativamente piccola. Queste restrizioni sono indicate nel disegno della catena nella fig. 8, ma bisogna ricordare che queste tecniche puramente chimiche permettono solamente una previsione molto vaga della vera struttura tridimensionale. La fig. 9 mostra la struttura primaria del lisozima. Informazioni di questo tipo sono state ottenute anche per la chimotripsina, la tripsina, la papaina, la proteasi del B. subtilis e la carbossipeptidasi.
In certi casi è stata determinata gran parte della sequenza di amminoacidi intorno a un gruppo del centro attivo che può essere ‛marcato' in maniera stabile, come per esempio la serina attiva nella chimotripsina, nella tripsina, nella carbossilesterasi, ecc., che può reagire irreversibilmente col gas nervino diisopropilfluorofosfato (DFP). Se l'enzima si fa reagire con DFP contenente 32P radioattivo, i prodotti della sua parziale scissione contengono fosfato di serina radioattivo. Si è visto che parecchi enzimi hanno sequenze piuttosto simili intorno al centro attivo; nella tab. I si riporta per esempio la disposizione degli amminoacidi intorno alla serina marcata del centro attivo di dieci enzimi.

I primi sette sono inibiti dal DFP; la fosfoglucomutasi e la fosfatasi alcalina vengono fosforilate, nella serina marcata, durante l'azione catalitica; l'ultimo enzima è trasformato nella sua forma attiva dalla fosforilazione della serina indicata.
Nell'ultimo decennio l'uso della cristallografia a raggi X ha aggiunto in maniera sensazionale una nuova dimensione alla nostra conoscenza della molecola enzimatica. Alla base di questo progresso c'è l'opera svolta da W. Th. Astbury sulle proteine fibrose negli anni trenta, insieme alla paziente e accurata analisi della mioglobina e dell'emoglobina da parte di M. F. Perutz e J. C. Kendrew dal 1947 al 1967, che fruttò loro il premio Nobel. Una buona esposizione della struttura tridimensionale delle molecole proteiche e dei metodi per determinarla hanno fornito H. Neurath (v., 1964) e R. E. Dickerson e I. Geis (v., 1969). L'analisi cristallografica ai raggi X è ora attivamente in corso per numerosi enzimi cristallini e in due o tre casi sono stati già ottenuti risultati definitivi, che saranno brevemente discussi.
In generale si può affermare che, sorprendentemente, la struttura delle molecole proteiche manca di regolarità. Abbiamo già visto che la sequenza degli amminoacidi e la distribuzione dei legami trasversali è più o meno casuale. Ci sono alcuni tipi di struttura secondaria della catena peptidica, fra i quali particolarmente l'α-elica destrogira (come nella lana e nella cheratina dei capelli) e la struttura β ‛a fogli pieghettati' (come nella fibroina della seta), che si rinvengono nelle molecole enzimatiche globulari, ma queste strutture spesso si estendono solo per brevi tratti e gran parte della catena è in forma di avvolgimento irregolare e complesso, comunemente detto random coil (letteralmente ‛avvolgimento casuale'), per quanto si tratti di una definizione ambigua. Tuttavia, una regolarità strutturale è rappresentata dalla distribuzione degli amminoacidi con catena laterale idrofobica, come la leucina e la valina, in opposizione a quelli con catene laterali idrofile, sia elettricamente cariche, come gli acidi glutammico e aspartico e la lisina, sia non cariche, come la serina e la treonina. I residui idrofobici sono spesso stipati da un lato dell'α-elica e quelli idrofili dall'altro; nella distribuzione generale gli amminoacidi idrofobici sono spesso sepolti nell'interno di molecole più o meno globulari, mentre le catene laterali idrofile sono esposte sulla superficie. Questa distribuzione è assai evidente nel lisozima, che ora descriveremo.
Il lisozima è un enzima ampiamente diffuso, segnalato per la prima volta da A. Fleming nel muco nasale nel 1922, il quale ‛lisa' molte cellule batteriche dissolvendo un polisaccaride della loro parete cellulare; agisce anche sulla chitina, l'esoscheletro degli Artropodi. La sua funzione è quella di idrolizzare specificamente i legami glicosidici di questi polisaccaridi e precisamente, nel caso dei complessi della parete cellulare batterica, il legame fra il C-1 di un residuo di acido N-acetilmurammico e il C-4 di un residuo di N-acetilglucosammina. Possono essere idrolizzati i polimeri dell'N-acetilglucosammina con più di tre residui, ma non i polimeri dell'acido N-acetilmurammico. L'enzima è stato purificato a partire da diversi materiali, ma gran parte del lavoro chimico si riferisce all'enzima dell'albume d'uovo di gallina. La sequenza primaria degli amminoacidi, determinata da R. E. Canfield e J. Jollès, è mostrata nella fig. 9, che indica anche, schematicamente, in raffigurazione bidimensionale, l'avvolgimento determinato dalla presenza di quattro ponti − S − S − .
La struttura tridimensionale completa di questo enzima fu ottenuta per mezzo della cristallografia a raggi X e pubblicata da D. C. Phillips e collaboratori nel 1965-1967 (v. Phillips, 1966). La fig. 10 mostra un disegno da un modello in scala della molecola costruito da Phillips e collaboratori, e la fig. 11 l'avvolgimento della catena peptidica principale, da cui sono state tolte per chiarezza le catene laterali, ma in cui è mostrata una molecola di substrato collocata nella fessura che attraversa la proteina (v. sotto, cap. 4). La fig. 12A mostra un modello a spazi pieni, che mette in evidenza la profonda fessura sulla superficie della molecola. Questa fessura divide la molecola in due parti; una, che contiene i residui 1-40 e 101-129, è in gran parte a elica, mentre l'altra, che contiene i residui 41-87, è molto più irregolare, per quanto fondamentalmente sia una struttura β. I residui 88-100 formano un'α-elica che tiene insieme le due parti come un cardine; un lato di essa, sepolto nel fondo della fessura, è idrofobico. La superficie interna della fessura è rivestita dalle poche catene laterali idrofobiche presenti all'esterno della molecola, ma contiene anche altri gruppi. Phillips e collaboratori hanno studiato, sempre con la cristallografia a raggi X, la struttura dell'enzima combinato con il trimero della N-acetilglucosammina, che è un inibitore, e hanno concluso che esso si adatta a un'estremità della fessura. La combinazione con questo inibitore (e quindi, per analogia, con il substrato) avviene per mezzo di legami a idrogeno con la catena peptidica principale e coinvolge anche i triptofani in posizione 62, 63 e 108 e l'acido aspartico in posizione 101. I suddetti autori suppongono che la combinazione con un'intera molecola di substrato dipenda anche da una glutammina, due asparagine e un'arginina all'altra estremità della fessura e che i principali gruppi catalitici (v. sotto, cap. 4) siano l'acido aspartico in posizione 52 e l'acido glutammico in posizione 35. L'ultimo gruppo si trova in una zona notevolmente idrofobica della fessura e probabilmente rimane non ionizzato, mentre l'acido aspartico è in una zona polare.
Un'immagine sufficientemente particolareggiata si ha anche per la ribonucleasi, la cui struttura primaria è mostrata nella fig. 8. L'analisi cristallografica della ribonucleasi S, una forma modificata per scissione di un legame peptidico, è stata eseguita da Harker e Katha e mostra anche in questo caso una molecola divisa in due parti da un'accentuata fessura lineare. Una contiene due brevi segmenti di α-elica con una zona interna idrofobica che riveste la fessura, l'altra contiene una struttura a fogli pieghettati molto più estesa che nel lisozima. Tre residui, una lisina e due istidine, rispettivamente in posizione 41, 12 e 119, che altri metodi hanno dimostrato essere implicati nel processo catalitico, si trovano nella fessura vicini fra di loro.
Si hanno descrizioni ancora incomplete per altri enzimi proteolitici. La papaina, dal frutto di Carica papaya, è fondamentalmente simile, nella forma, al lisozima e alla ribonucleasi, con due residui importanti per la catalisi, la cisteina in posizione 25 e l'istidina in posizione 158, posti uno di fronte all'altro sui due lati della fessura; tuttavia, questa molecola è solo in piccola parte ad α-elica. Al contrario, la chimotripsina e la tripsina, proteinasi del tipo contenente serina nel centro attivo, sembrano essere molecole più o meno sferiche costituite da parecchie catene distese, disposte parallelamente, con la serina attiva in una depressione profonda piuttosto che in una fessura.
W. N. Lipscomb e collaboratori (v., 1970) hanno descritto diffusamente la carbossipeptidasi A, che è una proteina contenente zinco e che sembra differire notevolmente dagli enzimi discussi precedentemente. Il centro della molecola è costituito da un grande foglio pieghettato fatto da 8 catene parallele, che rappresenta più del 20% dell'intera molecola. Esternamente ad esso vi sono otto sezioni a elica; questa disposizione è l'opposto di quanto è stato descritto nei casi precedenti. L'atomo di zinco giace in una larga depressione sulla superficie della molecola e sotto di esso c'è una tasca idrofobica nell'interno della molecola. Un acido glutammico e una tirosina prendono parte, nella depressione, al sito attivo, che ha certe somiglianze con il sito attivo della chimotripsina e della tripsina.
Prima di lasciare questi argomenti di carattere strutturale, va ricordata la tecnica della microscopia elettronica (v. Haschmeyer, 1970). Essa non ha ancora acquistato quel grado di risoluzione che le permetterebbe di competere con la cristallografia a raggi X nel determinare nei particolari la geometria delle più piccole molecole enzimatiche, benché gli strumenti commerciali possano ora arrivare a risoluzioni di 5 Å o anche meno. Il fattore limitante è il ‛contrasto' in densità elettronica fra differenti parti della molecola, per quanto esso possa essere migliorato con le tecniche delle ombre e della colorazione negativa. La microscopia elettronica ha avuto particolare successo nel dimostrare al di fuori di ogni dubbio l'organizzazione in subunità di alcune delle più grandi molecole enzimatiche. Così si è visto che la piruvatocarbossilasi e l'aldolasi contengono 4 subunità, la glutamminasintetasi dell'Escherichia coli ha 12 subunità identiche ordinate in due strati esagonali e la glutammatodeidrogenasi ha due piani sovrapposti, ciascuno con 3 subunità.
4. Relazione fra struttura e funzione: meccanismo d'azione degli enzimi.
Lo scopo finale di uno studio enzimologico è riuscire a dare una rappresentazione chimica precisa di come agisce un enzima. Se si possiede una sufficiente comprensione del meccanismo chimico di reazione e una completa conoscenza della struttura chimica tridimensionale di un determinato enzima, l'esame del modello molecolare dovrebbe consentire la predizione della sua attività catalitica. Questo traguardo è naturalmente abbastanza lontano, ma in certi casi è stato effettivamente possibile correlare, con sufficiente sicurezza, il meccanismo proposto con la conoscenza chimica dell'enzima.
Molte teorie sul modo di agire degli enzimi, sia in generale sia in casi particolari, sono state proposte senza chiare prove in base alle quali potessero essere accettate o rifiutate. Le considerazioni cinetiche discusse più sopra (v. cap. 2, È c) portarono a immaginare che l'enzima si combinasse con il substrato per formare un complesso in cui il substrato reagisse più agevolmente e ciò ha indotto a formulare l'ipotesi che il legame chimico che viene attaccato nel substrato sia ‛teso' in qualche modo, meccanico o elettronico, per la vicinanza di gruppi polari o di altri gruppi reattivi. Haldane (v., 1930) propose, nel caso degli enzimi idrolitici, che la specificità di combinazione con parti del substrato ai due lati del legame scisso potesse produrre la ‛tensione' appropriata.
La formulazione di teorie più realistiche dipende dalla possibilità di sapere quali gruppi chimici, fra i molti presenti in una proteina enzimatica, prendono effettivamente parte al processo catalitico; perciò gran parte della ricerca, specie dopo il 1945, è stata rivolta all'identificazione di gruppi importanti nel centro attivo. I gruppi che partecipano al legame fra enzima e substrato possono essere dedotti da considerazioni di specificità; per esempio, il fatto che l'acetilcolinesterasi richieda la presenza nel substrato di un gruppo carico positivamente suggerisce l'esistenza del carbossile di un acido glutammico o aspartico nel centro attivo. Gli studi sull'effetto del pH e della temperatura sull'attività enzimatica forniscono parametri dai quali si può risalire ai gruppi cataliticamente significativi; abbiamo già ricordato (v. sopra, cap. 2) la frequenza con cui questi metodi sembrano indicare l'istidina. Anche metodi chimici e la perdita d'attività in seguito a fotossidazione sono serviti a mostrare l'importanza della catena laterale imidazolica dell'istidina nelle reazioni enzimatiche (v. Barnard e Stein, 1958). Indicazioni della presenza di istidine essenziali per l'attività enzimatica si hanno per la chimotripsina, la tripsina, la colinesterasi e altre esterasi, la ribonucleasi, ecc. Un altro amminoacido considerato importante in numerosi enzimi è la cisteina. Un gruppo di reattivi, fra i quali certi gas lacrimogeni, attacca i gruppi tiolici in maniera più o meno specifica, per ossidazione o per alchilazione, e inibisce i cosiddetti ‛enzimi a − SH' causando la perdita di uno o di pochi gruppi tiolici dell'enzima. Questi enzimi comprendono molte chinasi e parecchie deidrogenasi. Tuttavia occorre sottolineare che la perdita di attività in seguito all'attacco di un gruppo tiolico non prova che questo partecipi direttamente alla catalisi; esso potrebbe, per esempio, essere essenziale per il mantenimento della giusta configurazione della proteina nel suo insieme.
Uno sviluppo molto importante fu rappresentato dalla scoperta che un residuo, che normalmente non è considerato particolarmente reattivo, può far parte del centro attivo e in questa situazione acquistare una reattività speciale. Abbiamo ricordato (v. sopra, cap. 3, È b) l'inibizione di certi enzimi a opera del DFP. Il DFP è una sostanza relativamente stabile; quando la chimotripsina è inibita da quantità stechiometriche di DFP, si trova che soltanto una serina è stata fosforilata. Le altre 24 molecole di serina non reagiscono, neanche ad alte concentrazioni di DFP. Se la chimotripsina è denaturata, nessuna serina reagisce col DFP. Si può concludere che nella chimotripsina nativa c'è una serina dotata di una reattività particolare, tale da dar vita a proprietà catalitiche; la sua reattività è in qualche modo dovuta alla sua collocazione, nella configurazione proteica nativa, in vicinanza di qualche punto particolare della catena peptidica. Un'ipotesi attendibile è che l'istidina, considerata importante in base ad altre tecniche, possa essere posta in vicinanza della serina in seguito all'avvolgimento della molecola proteica, per quanto non si trovi a essa adiacente nella sequenza primaria. La cristallografia a raggi X ha confermato che la serina 195 e l'istidina 57 sono in effetti molto vicine fra di loro.
Non si può dare una chiara descrizione di un meccanismo enzimatico se non si conoscono, in maniera particolareggiata, i cambiamenti chimici che avvengono nel substrato, compresi quelli concernenti gli stadi intermedi della reazione. Oltre agli studi cinetici, è stato molto utile l'uso degli isotopi, sia in esperimenti di incorporazione, sia in esperimenti di scambio. Per esempio la collocazione dell'isotopo nel prodotto, dopo idrolisi in H218O, rivela quale legame è stato scisso durante l'idrolisi stessa. Per mezzo di questa tecnica, M. Cohn dimostrò che la fosforilasi e la saccarosioglucosiltransferasi attaccano il legame C − O del glucosio-1-fosfato, e non il legame O − P, mentre le fosfatasi, sia acide che alcaline, attaccano il legame O − P degli esteri fosforici. In genere, se si considera un atomo che scambia due legami in direzioni opposte, la scissione avverrà da quella parte che è più vicina alla parte del substrato per cui l'enzima ha più specificità, cioè avverrà il più vicino possibile al gruppo che deve essere trasferito.
L'incorporazione di un isotopo può anche essere usata per scegliere fra due possibili tappe intermedie di reazione. La reazione catalizzata dalla metilmalonil-CoA-mutasi è:
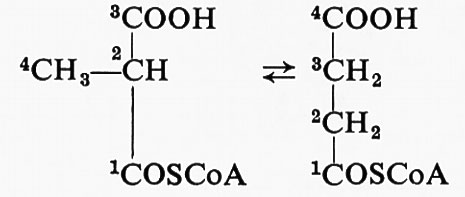
Il meccanismo più semplice sarebbe il trasferimento del − COOH sul − CH3. Invece, F. Lynen ha mostrato che, se il metilmalonil-coenzima A è marcato in posizione 2, l'isotopo si ritrova nella posizione 3 del succinil-coenzima A. Questo risultato può essere spiegato con il trasferimento del gruppo − COSCoA, ma non con quello del − COOH. La letteratura è ricca di esempi di questo tipo di analisi.
Gli esperimenti di scambio di un isotopo sono quelli in cui l'enzima catalizza lo scambio di una porzione marcata con isotopi fra due molecole, in assenza di catalisi della reazione totale. D. Rittenberg ha dimostrato, per esempio, che la chimotripsina, in H218O, causava l'incorporazione di 18O in molecole del tipo acilfenilalanina, che possono formarsi come prodotto di reazioni catalizzate dalla chimotripsina. Questo fatto si può spiegare facilmente ammettendo che si formi un enzima acilato, o acil-enzima, come composto intermedio dell'idrolisi, cioè, chiamando Ac-Phe-COX uno specifico substrato peptidico:
Ac-Phe-CO • X + EH ⇄ Ac-Phe-CO • E + HX
Ac-Phe-CO • E + H2O → Ac-Phe-COOH + EH.
Lo scambio isotopico è allora dovuto a:
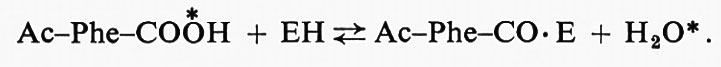
Oltre a indicare la probabile partecipazione di composti intermedi in cui un radicale del substrato si lega in maniera covalente all'enzima, gli studi di scambio possono essere usati per decidere fra differenti meccanismi di reazioni enzimatiche di trasferimento. Essi sono stati particolarmente utili nello studio degli enzimi del gruppo 6, le ligasi, la cui reazione totale è rappresentata schematicamente da:
A + B + XY ⇄ AB + X + Y.
I meccanismi possibili sono molteplici. Anzitutto vi sono i ‛meccanismi di trasferimento triplice' del tipo:
A-B + E = A + B-E
B-E + X = B + X-E
X-E + Y = X-Y + E
(dove E rappresenta la molecola di enzima). Un altro possibile tipo di reazione implica la formazione di un composto intermedio del tipo A − X, per esempio:
A-B + X = A-X + B
A-X + Y = A + X-Y.
Ognuno di questi passaggi, in qualsiasi meccanismo, dovrebbe poter essere dimostrato da un corrispondente scambio isotopico. Per esempio, se avviene il passaggio
A-B + E = A + B-E
un campione di A marcato dovrebbe scambiare con AB in assenza di XY, X o Y e lo scambio sarà inibito da X, che tende a ridurre la concentrazione di B − E (per una discussione completa di tutte le possibilità, v. Dixon e Webb, 19642, cap. 7). Per mezzo di questa tecnica è stato dimostrato che, mentre la glutammilcisteinasintetasi agirebbe mediante un meccanismo di trasferimento triplice con un glutammilenzima come composto intermedio, l'acetilcoenzima A-sintetasi e molte altre ligasi operano mediante il secondo meccanismo, con la formazione di adenilil-acetato (o acetil-AMP) come composto intermedio.
Il problema di determinare le tappe della reazione, anche nel senso più ristretto di determinare l'ordine in cui i reagenti si legano all'enzima che catalizza la reazione e i prodotti si staccano da esso, diviene particolarmente complesso se l'enzima non solo coinvolge due substrati e due prodotti (è questo il caso più comune), ma ha anche bisogno di un attivatore. Per esempio, la maggior parte delle ‛chinasi', che catalizzano il trasferimento di fosfato dall'ATP a un'altra molecola, necessitano di uno ione metallico bivalente (il più delle volte Mg2+) che si combina reversibilmente con l'enzima. Se si considera l'enzima creatinachinasi, che catalizza la reazione:
ATP + C ⇄ ADP + CP,
dove C e CP rappresentano rispettivamente la creatina e il creatinfosfato, si deve ammettere la presenza nella miscela di reazione delle specie molecolari seguenti: E, ATP, C, ADP, CP, E − ATP, E − C, E − ADP, E − CP, E − Mg, E − Mg − ATP, E − Mg − ADP, E − Mg − C, E − Mg − CP, E − C − ATP, E − CP − ADP, E − Mg − C − ATP, E − Mg − CP − ADP, E − CP − ATP, E − C − ADP, E − Mg − CP − ATP, E − Mg − C − ADP, Mg − ATP, Mg − ADP, e Mg − CP. In più, c'è la possibilità di composti intermedi rappresentati dalla temporanea fosforilazione di gruppi dell'enzima stesso da parte del gruppo fosforico che viene trasferito. Da ciò, ovviamente, non consegue di necessità che tutti questi composti siano effettivamente presenti in ogni determinata miscela di reazione; solo pochi di essi possono avere importanza funzionale nella sequenza catalitica. Alcune delle specie elencate, per esempio E − C − ADP e E − CP − ATP, sono complessi ‛abortivi' e non possono portare a buon termine la reazione. In casi come questo, individuare le tappe della reazione è il preliminare necessario per elaborare una teoria attendibile sul meccanismo chimico della catalisi.
Numerosi gruppi di ricercatori hanno contribuito negli ultimi dieci anni alla soluzione di alcuni di questi problemi di sequenza, ma forse il contributo più importante è stato quello di W. W. Cleland (v., 1963), il quale fra l'altro ha introdotto una terminologia nuova che ha facilitato la discussione di questi problemi. Possiamo brevemente sintetizzare prendendo come esempio un enzima simbolico che, non considerando l'attivatore, catalizza la reazione:
A + B ⇄ P + Q.
Le diverse tappe possibili possono essere rappresentate nella seguente maniera schematica:

Vengono così rappresentate tutte le vie che portano dall'enzima libero, a sinistra, all'enzima libero, a destra. I più importanti meccanismi possibili distinti da Cleland sono i seguenti.
1. Successione ordinata. Si tratta di quella successione in cui i substrati e i prodotti reagiscono in un ordine definito, per esempio:
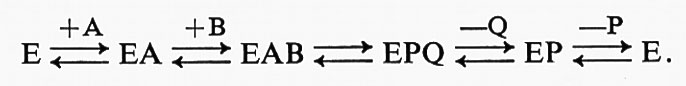
I complessi EB ed EQ non esistono.
2. Successione casuale. Si tratta di quella successione nella quale i siti di combinazione con A, B, P e Q partecipano contemporaneamente. Le reazioni saranno quelle mostrate nello schema precedente, eccetto quelle che contengono EX. Esse possono costituire un sistema a equilibrio rapido, in cui tutte le reazioni sono rapide rispetto a EAB ⇄ EPQ, oppure alcune velocità di combinazione o di distacco possono essere lente e contribuire alla cinetica della reazione complessiva.
3. Cosiddetto ‛meccanismo di Theorell-Chance'. Si tratta del caso in cui la reazione EAB → EPQ è così rapida che non contribuisce allo schema cinetico, cosicché si ha

4. Meccanismo ‛non in successione' (o ‛a ping-pong'). In questo caso si ha:

Naturalmente EX deve essere un composto intermedio in cui l'enzima contiene ancora parte del reagente originale, per esempio un gruppo fosforico che deve essere trasferito da A a B.
La scelta fra queste possibilità può essere fatta in vari modi. L'analisi cinetica, sebbene complicata, è uno strumento efficacissimo. Molte di queste possibilità non riconducono alla normale ‛cinetica di Michaelis' (di cui abbiamo già parlato) quando la velocità di reazione è misurata in funzione della concentrazione di uno dei reagenti; anche l'effetto degli inibitori e la cinetica dell'inibizione da prodotti di reazione possono essere usati come mezzi diagnostici. (Per una trattazione dei relativi procedimenti matematici, v. Cleland, 1963; v. Dixon e Webb, 19642; v. Wong e Hanes, 1962; v. Cleland, 1967). P. D. Boyer e altri hanno anche elaborato a questo scopo dei metodi di scambio isotopico: essenzialmente, si lascia andare a equilibrio il sistema, quindi lo si sposta leggermente introducendo un reagente altamente marcato e si misura la velocità iniziale di scambio isotopico. Con questi mezzi Morrison e Cleland (v., 1966) riuscirono a confermare che la creatinfosfochinasi, discussa precedentemente, agisce con un meccanismo di ‛successione casuale' a equilibrio rapido.
Finora sono stati descritti metodi che investono il meccanismo della catalisi enzimatica indipendentemente da una completa e intima conoscenza della chimica della proteina enzimatica. In molti casi, essi si sono rivelati sufficienti a stabilire un certo meccanismo con notevole sicurezza. Un buon esempio di successo nell'uso di vari metodi è il caso dell'aldolasi, della transaldolasi e di enzimi affini, discusso da Morse e Horecker (v., 1968). Lo studio di sistemi modello, della specificità, degli inibitori, di scambi isotopici in acqua pesante o triziata, o con substrati marcati con 14C, di cambiamenti nello spettro ultravioletto dipendenti dalla cinetica generale della reazione e infine di un composto di addizione enzima-substrato, di cui è possibile l'isolamento, ha dimostrato che la fase essenziale della catalisi è la formazione di una ‛base di Schiff' con un gruppo ε-amminico di un residuo di lisina dell'enzima. La reazione catalizzata dall'aldolasi di muscolo di coniglio, in cui il fruttosio-1,6-difosfato è scisso in gliceraldeide-3-fosfato e diidrossiacetonfosfato, è mostrata nella fig. 13. Si ammette che il substrato sia attirato dentro una fessura apolare dell'enzima, dove è trattenuto da gruppi positivi di questo. Quindi si forma, per eliminazione di acqua, una base di Schiff con il residuo di lisina. Un gruppo dell'enzima fortemente basico (B − ) accetta un protone dell'idrossile in C-4, iniziando così la reazione di dealdolizzazione (II). Si liberano gliceraldeide-3-fosfato e, successivamente, il protone (III). Il carbanione della base di Schiff che si produce è neutralizzato da un protone proveniente da un'istidina dell'enzima (IV); esso è idratato e si stacca come diidrossiacetonfosfato, rigenerando la configurazione originaria dell'enzima (I). Molte altre reazioni enzimatiche, per cui sono state proposte attendibili spiegazioni chimiche, sono discusse da Jencks (v., 1969).
La completa chiarificazione di alcuni casi di struttura tridimensionale di enzimi, che è stata descritta più sopra, ha aperto nuove possibilità per spiegarne il modo di azione. Nel caso del lisozima, come si è visto, la caratteristica più singolare è la profonda fessura, essenzialmente non polare, che attraversa la molecola. Phillips (v., 1966) ha mostrato che un analogo del substrato, un trimero della N-acetilglucosammina, entrava perfettamente in un'estremità di questa fessura. Non fu difficile supporre, per estrapolazione, che un'intera molecola di substrato avrebbe riempito la fessura in tutta la sua lunghezza; la fig. 11 mostra una molecola di substrato nella posizione presunta. La fig. 12B mostra un modello a spazi pieni del complesso enzima-substrato: si vede come questi si adattino in maniera perfetta l'uno all'altro, realizzando l'analogia con il sistema ‛chiave-serratura' proposta molto tempo fa da E. Fischer. L'esame dei residui contenuti nella fessura ha permesso una plausibile formulazione del processo catalitico, rappresentata nella fig. 14. La scissione idrolitica del substrato avviene fra l'anello D, che è tenuto nella fessura in posizione distorta, e l'anello E. L'acido aspartico in posizione 52, che è in un ambiente polare, ha normalmente una carica negativa, mentre l'acido glutammico in posizione 35 non è ionizzato, a causa del suo intorno non polare. Si pensa che l'acido glutammico possa cedere uno ione idrogeno all'ossigeno glicosidico fra D ed E: il legame glicosidico viene allora scisso e sull'anello D resta uno ione carbonio. Quest'ultimo è reso stabile dalla carica dell'acido aspartico in posizione 52. Una molecola d'acqua può quindi fornire un OH- che si combina con lo ione carbonio e un H+ per il residuo 35, completando così la reazione.
Descrizioni abbastanza simili possono essere proposte per l'azione della ribonucleasi e degli enzimi proteolitici dei quali è stata analizzata la struttura chimica. Per la discussione di questi casi, v. Goodwin e altri, 1964; v. Perutz, 1969; v. Koshland e Neet, 1968; v. Dickerson e Geis, 1969.
5. Aspetti biologici dell'enzimologia.
Uno degli aspetti più significativi della ricerca moderna è il fatto che gli enzimologi s'interessano sempre più agli enzimi nel loro ambiente biologico, piuttosto che come componenti isolati. Dopo aver separato la cellula vivente e averne esaminato chimicamente le singole parti, ora i biochimici stanno studiando come queste parti siano in relazione l'una con l'altra e come funzionino nella cellula intatta. Questo tipo di ricerca confluisce in altri campi biologici quali la genetica, la biologia dello sviluppo, la fisiologia e la morfologia, e perciò nel nostro articolo se ne tratterà solo brevemente.
a) Localizzazione degli enzimi.
Nel periodo d'oro della purificazione degli enzimi, e specialmente negli anni trenta, gli enzimi erano in genere ricavati da ogni materiale che fosse facilmente ottenibile e che avesse un'attività enzimatica relativamente alta. La fonte non era generalmente considerata di grande importanza: molti fra gli esperimenti destinati a ricostruire, per esempio, le sequenze nelle ossidazioni biologiche hanno utilizzato nella stessa miscela di reazione componenti ottenuti dal lievito, dal muscolo di coniglio, dal cuore di maiale e dal cuore di cavallo. Più recentemente, la questione della localizzazione di un enzima si è rivelata per se stessa di grande interesse.
Distribuzione nelle specie e nei tessuti. - Gli schemi metabolici fondamentali di tutti gli animali sono simili, cosicché è prevedibile che gli stessi gruppi di enzimi si trovino nei corrispondenti tessuti di tutte le specie. Vi sono naturalmente alcune limitazioni, quando si paragonano i tessuti fra di loro, corrispondenti alle specializzazioni fisiologiche dell'organo. Così gli enzimi del ciclo dell'urea si trovano nel tessuto epatico e non in altri. Abbiamo già ricordato (v. sopra, cap. 2, È a) che molti importanti processi metabolici, e specialmente quelli catabolici, sono comuni praticamente a tutti gli organismi viventi e di conseguenza molti enzimi sono presenti, oltre che in tutti i tessuti animali, nelle piante, nei lieviti e nei Batteri. D'altra parte, nelle piante e nei miceti (e, in una certa misura, anche nei Batteri) hanno luogo molte sequenze biosintetiche specializzate, come per esempio quelle responsabili della sintesi di alcaloidi complessi e di altri composti eterociclici, ed esse dipendono da enzimi con distribuzione di specie relativamente ristretta.
Quando si esamina la quantità di enzima effettivamente presente in varie sedi cellulari, si riscontrano variazioni abbastanza sorprendenti, che non sembrano potersi spiegare con la diversa attività metabolica delle cellule in oggetto (v. Dixon e Webb, 19642). Si possono ricordare tre esempi, scelti fra enzimi della sequenza principale della demolizione dei carboidrati. La citratosintasi è presente in concentrazioni pressappoco eguali nel muscolo cardiaco di maiale, cane, coniglio, ratto e piccione, in concentrazione fra un terzo e un quarto di questa nel rene delle stesse specie e pari a un terzo di essa nel fegato. Invece nel muscolo scheletrico abbiamo, ponendo uguale a 100 la concentrazione nel piccione, 34 nel cane, 6 nel ratto e 2 nel coniglio. Le concentrazioni relative in tre tessuti del piccione sono 100 per il muscolo scheletrico, 63 per il cuore, 2 per il fegato. Il secondo esempio riguarda l'attività aldolasica (cioè l'attività nei confronti del fruttosio-1,6-difosfato, alla quale possono contribuire enzimi diversi). Nel ratto le attività relative sono 100 per il muscolo scheletrico, 6 per il cuore, 6 per il fegato. Nel caso della malatodeidrogenasi, le attività relative nel ratto sono 100 per il cuore, 38 per il muscolo scheletrico e 19 per il fegato. Le differenze di specie per il muscolo cardiaco risultano 100 per il piccione, 116 per il ratto e 30 per il coniglio. È chiaro che queste attività non possono essere del tutto correlate col livello funzionale del metabolismo dei carboidrati nelle sedi citate e che alcune di esse esistono certamente in un notevole eccesso rispetto alle esigenze fisiologiche.
Durante le prime fasi dello sviluppo ci possono essere variazioni rilevanti nella composizione enzimatica dei vari tessuti, dato che diversi enzimi compaiono a diversi stadi della morfogenesi. Il metabolismo ossidativo dei carboidrati appare nell'embrione di pollo prima della capacità di ossidare il β-idrossibutirrato, e gli enzimi della β-ossidazione dei grassi appaiono anche più tardi. Gli enzimi del ciclo dell'urea appaiono negli Anfibi durante la metamorfosi.
In questa sezione abbiamo discusso enzimi provenienti da specie diverse che catalizzano la stessa reazione, come se si trattasse dello stesso enzima. Questo non significa che essi siano proteine identiche. Infatti, in molti casi ci possono essere precise differenze nelle proprietà chimiche di enzimi di specie differenti: differenze in solubilità, peso molecolare, punto isoelettrico o forma cristallina. Anche in specie strettamente affini, i metodi immunologici possono di solito rivelare in un enzima differenze specie-specifiche; per esempio, possono distinguere fra le catalasi epatiche di cavallo e di bue, fra le alcooldeidrogenasi dei lieviti di pane e di birra, fra le ureasi di soia e di un'altra leguminosa, la Canavalia ensiformis. In alcuni casi la determinazione della sequenza di amminoacidi ha indicato che si sono verificate regolarmente, durante l'evoluzione, sostituzioni di amminoacidi in varie posizioni nella struttura proteica; queste mutazioni devono o essere cambiamenti che influiscono poco sulle proprietà della proteina, come una valina al posto di una leucina o un acido glutammico al posto di un acido aspartico, oppure avvenire in una parte della molecola poco importante per l'attività funzionale. La maggior parte delle informazioni su queste variazioni chimiche delle proteine durante l'evoluzione riguarda certe proteine non enzimatiche come l'insulina, l'emoglobina, la mioglobina, il citocromo c; ma sono stati raccolti alcuni dati anche per gli enzimi idrolitici esaminati in precedenza (v. sopra, cap. 3, È b). Il numero di posizioni di amminoacidi che sono differenti nelle proteine corrispondenti di due specie diverse dipende ovviamente dal periodo di tempo intercorso dalla separazione delle specie da un antenato comune durante la loro evoluzione. Nel caso del citocromo c, che nell'uomo ha 104 amminoacidi, il numero di posizioni in cui gli amminoacidi differiscono rispetto all'uomo è 0 nello scimpanzé, 1 nel macaco, 10 nel canguro, 11 nel cane, 12 nel cavallo, 13 nel pollo, 14 nel serpente a sonagli, 23 nel pescecane, 31 in una specie di farfalla, 35 nel frumento, 43 nella Neurospora, 44 nel lievito.
Localizzazione intracellulare. - Gli enzimi diffusi in modo uniforme all'interno di una cellula vivente sono in realtà molto pochi. Indicazioni sulla presenza di enzimi negli organelli endocellulari erano state ottenute per mezzo di tecniche istochimiche. Tuttavia lo studio quantitativo è incominciato solo alla fine degli anni quaranta, con lo sviluppo dei metodi di separazione delle particelle intracellulari, fondati sull'invenzione dell'omogeneizzatore di vetro di V. R. Potter. Nell'omogeneizzatore un tessuto, per esempio il fegato, sminuzzato e sospeso in una soluzione tampone, viene forzato fra la parete interna di un provettone e la superficie di un pistone di vetro rapidamente rotante. Le pareti cellulari sono rotte dall'attrito, mentre il nucleo, i mitocondri, ecc. rimangono abbastanza integri e possono essere isolati per mezzo della centrifugazione differenziale. Questi metodi furono elaborati e sviluppati da Hogeboom, Schneider, Claude, de Duve e altri ancora (v. Roodyn, 1967; v. de Duve e altri, 1962).
Le prime prove d'attività enzimatica nelle varie frazioni dettero luogo a interessantissimi risultati e a generalizzazioni talora avventate. Molte ossidazioni cataboliche e le fosforilazioni a esse associate sembrarono essere localizzate unicamente nel ‛mitocondrio', che A. L. Lehninger chiamò ‟la centrale elettrica della cellula"; molti sistemi idrolitici furono localizzati nella frazione dei ‛microsomi', costituita da piccole particelle che sedimentano, centrifugando a velocità elevata, dopo la separazione dei mitocondri. Si pensa che i microsomi si producano durante l'omogeneizzazione dal ‛reticolo endoplasmatico', che si osserva al microscopio elettronico. Lavori successivi hanno modificato i particolari di queste conclusioni, ma non le grandi linee. Il problema maggiore è stato quello di accertare se i componenti delle frazioni isolate erano strettamente correlati con quelli della cellula integra. L'aspetto morfologico può essere usato come guida per scegliere le soluzioni tampone, le condizioni di omogeneizzazione, ecc., che provochino il minor danno possibile, ma anche in questo caso c'è la possibilità che enzimi o altri componenti passino da una frazione cellulare all'altra durante le fasi della separazione. Le frazioni nucleari sono generalmente contaminate con mitocondri e possono adsorbire enzimi dal citoplasma diluito; mitocondri distrutti possono formare piccole particelle che contaminano la frazione microsomiale, e così via. Inoltre frazionamenti più accurati hanno mostrato che le quattro frazioni ottenute originariamente da W. C. Schneider e da altri (nuclei, mitocondri, microsomi e succo cellulare) non sono esse stesse omogenee. Per mezzo di un'accurata centrifugazione differenziale, utilizzando soprattutto un gradiente di densità, C. de Duve riuscì a separare i ‛lisosomi' da frazioni mitocondriali grezze.
L'ossidazione terminale dei carboidrati, la β-ossidazione dei grassi e altri processi ossidativi hanno luogo, presi nel loro complesso, prevalentemente nei mitocondri della cellula animale. Tuttavia, solo alcuni degli enzimi che vi prendono parte, per esempio la succinatodeidrogenasi e la citocromossidasi, sono localizzati esclusivamente nei mitocondri. Infatti, altri enzimi essenziali delle sequenze cataboliche, come l'aconitatoidratasi e l'aldolasi, si trovano prevalentemente nel succo cellulare, insieme con una gran parte degli enzimi della glicolisi. Studi su enzimi singoli possono ovviamente portare a conclusioni errate; la velocità del processo metabolico nel suo complesso può dipendere dalla concentrazione di un enzima singolo che è presente in quantità limitanti.
L'interesse verso la localizzazione intracellulare degli enzimi è stato molto accresciuto dallo sviluppo della microscopia elettronica, che ha rivelato la presenza nella cellula di un'infrastruttura altamente organizzata (v. cellula). G. E. Palade ha dimostrato che il mitocondrio ha una membrana esterna, una membrana interna, delle cristae (membrane interne sinuose rivestite di particelle dense) e un ‛succo mitocondriale' che compenetra e circonda queste strutture. D. E. Green e la sua scuola dell'Università del Wisconsin, in particolare, hanno messo in relazione con questa disposizione strutturale le attività riscontrate in frazioni provenienti da mitocondri disgregati. Essi pensano che nelle particelle dense dei mitocondri ci sia un'organizzazione strutturale di enzimi che corrisponde all'organizzazione schematica delle reazioni enzimatiche nelle catene metaboliche. Per esempio, porzioni della catena respiratoria sono situate sulle cristae in differenti particelle. Si ritiene che il processo della fosforilazione ossidativa sia intimamente collegato con queste strutture di membrane (v. fosforilazione ossidativa).
Uno dei capitoli più controversi in questo campo è stato quello concernente l'enzimologia del nucleo, studiata ed esposta esaurientemente da Siebert e Humphrey (v., 1965). Ovviamente il nucleo contiene gli enzimi connessi con la conservazione e la trasmissione dell'informazione genetica, come il sistema della DNA-polimerasi; ma il suo contenuto in enzimi è in genere molto basso. D. B. Roodyn ha dimostrato che molti enzimi che erano stati trovati in piccola quantità in frazioni nucleari erano dovuti a contaminazioni con mitocondri o a un adsorbimento sulla superficie nucleare. Nuovi metodi per la preparazione di nuclei puri sono stati elaborati da Behrens, Mirsky e Siebert; essi si basano sul congelamento rapido del tessuto, in modo da impedire processi di decomposizione; alcuni utilizzano centrifugazioni in mezzi non acquosi, come cicloesano e bromobenzene, o tetracloruro di carbonio e benzene. Ora è stabilito che il nucleo contiene in basse concentrazioni molti enzimi, compresi quelli della glicolisi e del ciclo dell'acido citrico, alcuni sistemi di fosforilazione ossidativa ed enzimi implicati nella biosintesi di coenzimi. Non è stato chiarito se tutti questi enzimi siano sintetizzati nel nucleo stesso o se molti di essi vi arrivino passando dal citoplasma attraverso la membrana nucleare.
b) Forme multiple e varianti genetiche.
Al principio degli anni cinquanta A. Meister e J. B. Neilands dimostrarono che la lattatodeidrogenasi preparata dal cuore di bue in forma apparentemente pura poteva essere separata in due componenti attive. Allora non si dette a questa osservazione molta importanza, ma nel 1957 Vessel e Bearn (v., 1957) mostrarono che, nell'elettroforesi del siero, la lattatodeidrogenasi si trova in tre bande distinte. Un'ampia serie di dati elettroforetici è stata pubblicata da Markert e Møller (v., 1959), i quali mostrarono che, se si sottoponevano a elettroforesi siero o estratti di tessuto, si potevano osservare fino a cinque bande di lattatodeidrogenasi, due di malatodeidrogenasi, tre di fosfatasi alcalina, e dieci ciascuna di esterasi e di perossidasi. Inoltre, la distribuzione era differente in tessuti differenti e cambiava durante lo sviluppo dell'organismo. Essi avanzarono l'ipotesi che questo fosse un fenomeno generale e proposero il nome ‛isozima' (più tardi cambiato in ‛isoenzima') per le diverse forme molecolari di un enzima esistenti in una singola specie. Dal 1960 in poi è apparsa sugli isoenzimi una vasta letteratura (v. Kaplan, 1968; v. Markert e Whitt, 1968; v. Wilkinson, 1970; v. Masters e Holmes, 1972).
Le tecniche per rivelare gli isoenzimi sono relativamente semplici. Quelle usate più diffusamente sono stati certi tipi di elettroforesi zonale, in blocchi di gel d'amido, strati sottili di gel d'agar, colonne di cellulosa o amido, o colonne cilindriche di gel di poliacrilammide sintetica. I gel possono essere tagliati in strisce o dischi per il saggio dell'attività enzimatica o, in certi casi, l'attività può essere osservata sullo stesso supporto elettroforetico per mezzo di adatte miscele di substrato. In alcuni casi certi isoenzimi sono stati separati con la cromatografia a scambio ionico.
Attualmente si sa che molti enzimi esistono in forme multiple non solo nei tessuti animali, ma anche nelle piante e nei microrganismi. Tuttavia il primo sistema scoperto, quello della lattatodeidrogenasi, resta ancora il più studiato. La specificità di tessuto della distribuzione degli isoenzimi è stata usata come mezzo diagnostico in clinica, specialmente da F. Wroblewski. Il siero normale ha una bassa attività lattatodeidrogenasica, distribuita in cinque bande isoenzimatiche, indicate con i numeri da 1 a 5, in ordine decrescente di velocità di migrazione all'anodo. L'estratto di muscolo cardiaco contiene solo due forme, gli isoenzimi 1 e 2, mentre l'estratto di fegato contiene in misura molto ridotta tutte le forme all'infuori dell'isoenzima 5. In certe condizioni patologiche la lattatodeidrogenasi del siero aumenta in seguito alla sua fuoriuscita dai tessuti danneggiati. L'elettroforesi del siero in questi casi mostrerà agevolmente se, per esempio, l'enzima proviene dal fegato o dal rene.
Malgrado vi sia più di una spiegazione chimica possibile per le differenze fra gli isoenzimi, è un dato di fatto che la maggioranza degli enzimi che mostrano forme multiple sono costituiti da più di una catena peptidica. La lattatodeidrogenasi è un tetramero e possiede due specie differenti di catene, codificate da geni diversi. Queste forme furono chiamate da C. L. Markert A e B e da N. O. Kaplan M e H per indicare le forme del muscolo e del cuore (in inglese, rispettivamente, muscle e heart). I cinque isoenzimi sono i cinque possibili tetrameri: A4, A3B, A2B2, AB3 e B4. Se si congela e scongela in NaCl molare una miscela di isoenzimi 1 e 5 (A4 e B4, si producono fenomeni di dissociazione e riassociazione delle subunità, le quali si possono così ridistribuire fra di loro, con il risultato finale di una miscela dei cinque isoenzimi nella prevista distribuzione ‛binomiale':
In questo modo, la differenza nella distribuzione isoenzimatica nei vari tessuti potrebbe essere spiegata in base a differenti gradi di espressione dei due geni A e B.
Alcuni autori ritengono che quasi tutti i sistemi di isoenzimi potranno essere spiegati sulla base di subunità multiple del tipo di quelle della lattatodeidrogenasi. Tuttavia esistono altre possibilità, come differenti forme polimeriche che contengono un numero diverso di una stessa subunità, la combinazione con differenti molecole a basso peso molecolare, una serie di forme prodotte dalla degradazione di un'unica catena peptidica, e isomeri conformazionali, cioè differenti organizzazioni tridimensionali della stessa catena peptidica.
C'è anche la possibilità che gli enzimi siano il prodotto completo di geni del tutto separati. Sembra questo il caso dell'anidrasi carbonica degli eritrociti umani, che esiste in due forme con proprietà cinetiche assai differenti e con diversa composizione in amminoacidi.
Abbiamo visto che enzimi corrispondenti in specie differenti sono proteine differenti prodotte da cambiamenti nel codice genetico durante l'evoluzione, e non sorprende la scoperta che questo accade talvolta anche all'interno di una medesima specie. Naturalmente una mutazione può anche portare a cambiamenti che producono la perdita totale di un'attività enzimatica. Questo fenomeno è ben noto nei microrganismi ed è stato sfruttato da O. W. Beadle e altri per chiarire alcune sequenze metaboliche. Quando un enzima è assente in seguito a mutazione, si può accumulare un metabolita intermedio fino ad allora ignorato, e il prodotto formato dall'enzima può diventare un fattore d'accrescimento. In altri casi la mutazione può portare alla produzione di una proteina le cui proprietà enzimatiche sono notevolmente cambiate. Nell'uomo, per esempio, c'è un ceppo genetico, presente tra la popolazione nella misura di 1 su 4.000, che ha un livello di attività colinesterasica del siero pari a circa un quarto del normale. Livelli intermedi si riscontrano negli individui eterozigoti. L'enzima posseduto dagli individui con bassa attività colinesterasica differisce dall'enzima normale in vari aspetti, per esempio è relativamente insensibile alla dibucaina, un inibitore della colinesterasi. Ovviamente la mutazione genetica ha modificato il centro attivo: ciò spiega l'attività più bassa. Alcuni casi di resistenza agli insetticidi negli Insetti sono stati attribuiti alla selezione di mutanti i quali modificano gli enzimi normali in modo da renderli insensibili all'insetticida o capaci di distruggerlo. (Per una discussione su queste variazioni genetiche negli animali v. Harris, 1959; per il controllo genetico degli enzimi nelle piante v. Scandalios, 1969).
Tornando agli isoenzimi classici, tipo lattatodeidrogenasi, bisogna ricordare brevemente la loro possibile funzione. La forma B4, che predomina nel tessuto cardiaco, è fortemente inibita da alte concentrazioni di piruvato e perciò sarebbe adatta a un tessuto altamente aerobio, nel quale normalmente il livello di piruvato rimane basso. Al contrario, la forma A4 funziona bene in presenza di alte concentrazioni di piruvato e perciò sarebbe capace di produrre lattato nel muscolo scheletrico, che ha bisogno di energia in maniera sporadica e perciò necessita di un meccanismo che permetta esplosioni intermittenti di metabolismo anaerobico. È indicativo il fatto che mentre il muscolo scheletrico dei Mammiferi e del pollo contiene soprattutto lattatodeidrogenasi di tipo A4, quello di Uccelli come il colibrì e la procellaria, che hanno necessariamente lunghi periodi di attività, possiede in gran parte il tipo B4, come il cuore.
In altri casi gli isoenzimi possono avere un ruolo nei sistemi regolatori dell'attività enzimatica (v. sotto, È c).
Numerosi ricercatori hanno cominciato a usare sistemi isoenzimatici come ‛traccianti' per studiare uno dei problemi biologici non risolti, cioè quello della differenziazione dei tessuti (v. Masters e Holmes, 1972). L'uovo fecondato contiene geni per la produzione di tutti gli enzimi del corpo, eppure, come abbiamo visto, alcuni di questi enzimi si trovano nell'adulto solamente in determinati tessuti. Allo stesso modo, l'embrione deve contenere sia il gene A sia il gene B della lattatodeidrogenasi, malgrado l'espressione di questi geni nei diversi tessuti vari ampiamente. Se si riportano su un grafico, per una particolare specie animale, le curve dei cambiamenti della distribuzione degli isoenzimi nei tessuti in sviluppo, esse possono essere estrapolate a un comune punto di origine, ma le proporzioni iniziali sono notevolmente diverse nelle varie specie. Esprimendo i risultati per la lattatodeidrogenasi come percentuale delle subunità totali di tipo B, il punto iniziale è meno di 10 nel ratto e nel topo, di 40 per il coniglio, circa 50 per il gatto, superiore a 50 per i Ruminanti e infine 100 per l'uomo. Tuttavia, risultati recenti fanno pensare che alcuni enzimi possano derivare da fonti esterne anche prima dell'impianto dell'uovo fecondato, quando si trova ancora nella tuba.
Poco si conosce sui processi che fanno divenire le cellule embrionali in parte cellule epatiche, in parte cellule muscolari, e così via, in seguito all'espressione di alcuni geni e alla soppressione di altri. Lo studio di condizioni che alterano la distribuzione degli isoenzimi può contribuire alla comprensione di questo importante problema.
c) Regolazione dell'attività enzimatica.
Abbiamo rilevato come la quantità di un enzima sia molto diversa nei vari tessuti. In effetti, alcuni tessuti possono esserne totalmente privi, benché il gene responsabile della sua produzione sia ovviamente presente. In molti casi la quantità di enzima può variare notevolmente anche in uno stesso tessuto, a seconda della domanda metabolica nei riguardi della reazione catalizzata dall'enzima. La quantità di enzima sintetizzato è influenzata dall'accumulo sia del substrato sia del prodotto; i processi relativi, noti da tempo, sono detti ‛induzione' e ‛repressione'.
All'incirca nell'ultimo decennio si è giunti alla convinzione che, in molti casi, l'attività funzionale di una sequenza metabolica è modificata dall'effetto che un metabolita, che si produce negli ultimi stadi della sequenza (eventualmente lo stesso prodotto finale), esercita su un enzima chiave, in genere uno di quelli che catalizzano i primi passaggi della sequenza e spesso proprio l'enzima del primo passaggio. Questo enzima chiave, la cui attività è posta sotto controllo metabolico, è detto ‛enzima regolatore' e il processo di controllo ‛inibizione a retroazione' (feedback). (Per le osservazioni che hanno portato a questi concetti v. Stadtman, 1966; v. Atkinson, 1966; v. Koshland e Neet, 1968). Gli enzimi di questo tipo hanno in comune molte proprietà, che si considerano connesse con la loro funzione regolatrice.
Molti enzimi regolatori mostrano una dipendenza della velocità di reazione dalla concentrazione del substrato che non è quella tipica, in quanto è espressa dalla curva sigmoide presentata nella fig. 15 e non dalla curva iperbolica precedentemente descritta (v. fig. 2A). J. Monod, J. P. Changeux e F. Jacob (v., 1963) misero in evidenza la somiglianza fra queste curve sigmoidi e la curva di ossigenazione dell'emoglobina, essa stessa contrastante con la curva iperbolica di ossigenazione della mioglobina, e proposero che questi enzimi, che denominarono ‛allosterici', fossero costituiti da due o più subunità, ognuna con un sito catalitico. Infatti la curva sigmoide è appunto espressione del fatto che la combinazione di uno dei siti con il substrato (o con l'ossigeno nel caso dell'emoglobina) ha effetto sull'affinità degli altri siti. La maggior parte degli enzimi allosterici si sono dimostrati effettivamente a struttura polimerica e alcuni mostrano altre proprietà che senza dubbio dipendono da questa struttura, come la sensibilità alle basse temperature e l'accresciuta stabilità al calore in presenza di ‛effettori allosterici' (siano essi inibitori o attivatori). Le espressioni ‛enzima allosterico' e ‛sito allosterico' sono diventate di uso corrente: un sito allosterico è un sito che si combina con piccole molecole che hanno un effetto sull'attività enzimatica e che esiste sull'enzima in aggiunta ai siti normali cataliticamente attivi. La molecola del substrato può essere essa stessa capace di combinarsi con il sito allosterico e allora l'enzima regolatore è detto ‛omotropico'; l'enzima è ‛eterotropico' se gli unici effettori sono molecole diverse dal substrato.
Vari modelli sono stati proposti per spiegare questi fenomeni. Secondo il modello di Monod, Wyman e Changeux (v., 1965), un enzima allosterico sarebbe costituito da due o più subunità identiche, ognuna con un solo sito catalitico e con siti allosterici indipendenti, uno per ogni effettore. Si ammette che le subunità possano esistere in due configurazioni e che, in ogni data molecola polimerica, tutte le subunità debbano essere presenti nella stessa configurazione. Gli effetti allosterici sono allora dovuti a variazioni dell'equilibrio tra le due forme, in conseguenza delle differenti affinità che le due forme hanno per substrato ed effettore. In un'altra formulazione, descritta diffusamente da Koshland e Neet (v., 1968), si suppone che la combinazione di una subunità con l'effettore provochi un cambiamento di conformazione nella subunità stessa e che tale cambiamento influenzi la forma e la stabilità delle subunità adiacenti. Si pensa che possano formarsi degli stati conformazionali ibridi. Gran parte dell'abbondante letteratura sui singoli sistemi allosterici è stata diretta a discriminare, sulla base di analisi cinetiche o d'altro tipo, fra i vari meccanismi possibili.
Bisogna ricordare che solo in alcuni casi l'esistenza di siti allosterici è stata dimostrata direttamente. Un esempio interessante è l'enzima aspartatotranscarbammilasi che, quando viene estratto dall'Escherichia coli, ha un peso molecolare di 300.000 e 12 subunità. La dipendenza della velocità dalla concentrazione di aspartato è sigmoide e l'enzima è inibito dal citidintrifosfato (CTP), che è il prodotto terminale di una serie di reazioni cui partecipa l'enzima. L'ATP compete con il CTP per il sito allosterico, riducendo l'effetto inibitorio. L'aggiunta di sostanze come il p-idrossimercuribenzoato provoca la dissociazione dell'enzima, e allora la cinetica torna al classico tipo di Michaelis-Menten. L'enzima dissociato può essere separato in due tipi di subunità. Un tipo è cataliticamente attivo, ma non è inibito dal CTP; l'altro è inattivo e capace di combinarsi con il CTP. Sembra che l'enzima sia costituito da sei subunità, provviste di un sito catalitico ciascuna, e da altre sei, differenti, ognuna con un sito allosterico.
È necessario menzionare brevemente un altro tipo di controllo metabolico. C. F. Cori e A. A. Green, nel 1943, dimostrarono che la glicogenofosforilasi del muscolo esiste in due forme, fosforilasi a e fosforilasi b. Esse differiscono nella composizione primaria, in quanto la forma b contiene su ogni subunità un residuo di fosfato legato in maniera covalente. La fosforilasi a può essere trasformata nella forma b per fosforilazione a opera dell'ATP, sotto l'influenza di un enzima speciale (fosforilasichinasi); la trasformazione inversa è catalizzata da una fosfatasi, che stacca il fosfato legato alla fosforilasi b. I due enzimi differiscono inoltre per proprietà cinetiche, sicché questo sistema può dar luogo a un controllo metabolico in risposta al grado di fosforilazione dell'ATP nel sistema stesso. I meccanismi regolatori per questo enzima sono particolarmente complessi, perché ambedue le forme sono esse stesse enzimi allosterici. La fosforilasi a è un tetramero, la b è un dimero, e la fosforilazione operata dalla fosforilasichinasi ha anche l'effetto di dissociare l'enzima in due metà. La fosforilasichinasi esiste nel muscolo in una forma inattiva, che si trasforma nella forma attiva per azione della chinasi della fosforilasichinasi. Anche quest'ultimo enzima è allosterico, essendo attivato dall'adenosin-3′-5′-monofosfato ciclico. Altri esempi di enzimi regolatori in cui l'attività è modificata da variazioni chimiche prodotte da ‛enzimi modificanti' sono stati discussi da Holzer (v., 1969).
Per finire, ricordiamo che una possibile spiegazione dell'esistenza di isoenzimi è la presenza nella stessa cellula di enzimi che catalizzano la stessa reazione ma sono sotto un diverso controllo metabolico, vale a dire proteine che hanno lo stesso sito catalitico ma differenti siti allosterici. Per esempio, l'Escherichia coli ha tre aspartatochinasi sottoposte a feedback da parte, rispettivamente, della lisina, della treonina e dell'omoserina.
6. Prospettive future dell'enzimologia.
Durante l'ultimo mezzo secolo la scoperta di nuovi enzimi ha avuto luogo con un incremento che può dirsi logaritmico, con un tempo di raddoppiamento di circa otto anni. È probabile che presto questo ritmo comincerà a rallentare, dato che oramai molte vie metaboliche sono state completamente chiarite per quanto riguarda i singoli passaggi enzimatici, ma bisogna riconoscere che molto rimane da fare nel caso delle vie biosintetiche più esoteriche, specie nelle piante. Si può ragionevolmente sperare che alla fine del XX secolo saranno conosciuti praticamente tutti gli enzimi esistenti in natura (almeno nel senso che saremo in grado di scrivere le reazioni da essi catalizzate).
La descrizione completa di una molecola enzimatica come sostanza chimica, con la spiegazione della sua attività catalitica come conseguenza della sua composizione e della sua struttura chimica, è appena agli inizi, e ci si può attendere per i prossimi decenni un aumento enorme delle ricerche in questo campo. L'automazione dei metodi d'analisi degli amminoacidi e di determinazione della loro sequenza rende quasi meccanica l'esecuzione di tali studi su piccole quantità di enzimi purificati; è inoltre probabile che la cristallografia a raggi X sarà ampiamente utilizzata adesso che è chiaro, in seguito agli studi precorritori compiuti su alcuni enzimi idrolitici, che questa tecnica (benché niente affatto semplice e corrente) può rivelare particolari straordinariamente sottili della struttura terziaria che non possono essere ottenuti in nessun altro modo. La descrizione del meccanismo intimo della catalisi enzimatica diverrà sempre più materia di concreta realtà chimica piuttosto che di teoria non dimostrabile.
È anche probabile che continui la tendenza dei biochimici a interessarsi sempre più degli enzimi nella loro disposizione funzionale all'interno della cellula, piuttosto che come prodotti naturali isolati. Nei dieci anni trascorsi da quando per la prima volta s'incentrò l'interesse su questo problema, è venuto alla luce un grandissimo numero di sistemi regolatori dell'attività enzimatica, ma sembra probabile che molti ne restino ancora da scoprire. A mano a mano che diventa più completa la comprensione dei processi della sintesi proteica, della replicazione e dell'espressione dei geni, è probabile che molti problemi connessi con la presenza di particolari enzimi in particolari cellule vengano alla luce e siano risolti. In questo campo di ricerca sulla biologia degli enzimi può essere trovata la chiave per la soluzione di alcuni dei più vasti problemi biologici, come lo sviluppo, il differenziamento, la senescenza, il controllo ormonale e forse il cancro.
Tornando alla questione dei singoli enzimi, si deve ricordare un traguardo che non è stato ancora raggiunto: la sintesi completa di un enzima. Benché sia dubbio che l'effettiva sintesi possa aggiungere alla conoscenza o alla comprensione degli enzimi qualcosa che non sia stato già raggiunto per altra via, come nel caso del lisozima o della ribonucleasi, nondimeno la produzione, senza l'aiuto di una cellula vivente, dei catalizzatori fondamentali dai quali dipende il processo vitale rappresenterà per l'uomo un'enorme conquista. Probabilmente questa meta non è remota. Essa potrà portare anche alla produzione di enzimi non esistenti in natura, catalizzatori di reazioni diverse da quelle delle vie metaboliche naturali. Si può immaginare che questi enzimi sintetici avranno applicazioni nell'ingegneria chimica, nella sintesi di nuovi composti e forse anche nella pratica clinica.
Un risultato della ricerca futura, che è alquanto più lontano (ma, in prospettiva, più importante), è la possibilità d'interferire nei sistemi che, nella cellula vivente, producono gli enzimi. Se si potesse modificare il materiale genetico o i sistemi che controllano l'espressione dei geni, si potrebbe cambiare non solo la quantità, ma anche la natura degli enzimi dentro i tessuti, con la conseguenza d'influenzare l'attività metabolica generale dell'organismo. La capacità di alterare in questo modo le attività chimiche fondamentali di un individuo vivente ha evidentemente enormi possibilità pratiche, tanto nel bene quanto nel male.
Si può ricavare, da quanto abbiamo fin qui detto, una considerazione conclusiva: l'enzimologia ha percorso, nel XX secolo, un lungo tratto di strada, ma ne ha davanti ancora molta da percorrere.
Bibliografia.
Alberty, R. A., Enzyme kinetics, in Advances in enzymology (a cura di F. F. Nord), vol. XVII, New York 1956, pp. 1-64.
Alberty, R. A., The interpretation of steady state kinetic data on enzymatic reactions, in ‟Brookhaven symposia in biology", 1962, XV, pp. 18-31.
Atkinson, D. E., Regulation of enzyme activity, in ‟Annual review of biochemistry", 1966, XXXV, pp. 85-124.
Barnard, E. A., Stein, W. D., The roles of imidazole in biological systems, in Advances in enzymology (a cura di F. F. Nord), vol. XX, New York 1958, pp. 51-110.
Boyer, P. D., Lardy, H., Myrbäck, K. (a cura di), The enzymes, vol. I, New York 19592.
Briggs, G. E., Haldane, J. B. S., A note on the kinetics of enzyme action, in ‟Biochemical journal", 1925, XIX, pp. 338-339.
Canfield, R. E., Liu, A. K., The disulphide bonds of egg white lysozyme (muramidase), in ‟Journal of biological chemistry", 1965, CCXL, pp. 1997-2002.
Cleland, W. W., The kinetics of enzyme catalyzed reactions with two or more substrates or products. I. II. III, in ‟Biochimica et biophysica acta", 1963, LXVII, pp. 104-137, 173-187, 188-196.
Cleland, W. W., The statistical analysis of enzyme kinetic data, in Advances in enzymology (a cura di F. F. Nord), vol. XXIX, New York 1967, pp. 1-32.
Dayhoff, M. O., Eck, R. V., Atlas of protein sequence and structure 1967-1968, Washington 1968.
Dickerson, R. E., Geis, I., The structure and action of proteins, New York 1969.
Dixon, M., The effect of pH on the affinities of enzymes for substrates and inhibitors, in ‟Biochemical journal", 1953, LV, pp. 161-170.
Dixon, M., The history of enzymes and of biological oxidations, in The chemistry of life (a cura di J. Needham), Cambridge 1970, pp. 15-37.
Dixon, M., Webb, E. C., Enzymes, London-New York 19642 (tr. it.: Enzimi, Milano 1972).
Duclaux, E., Microbiologie, Paris 1883.
Duve, C. de, Wattiaux, R., Baudhuin, P., Distribution of enzymes between subcellular fractions in animal tissues, in Advances in enzymology (a cura di F. F. Nord), vol. XXIV, New York 1962, pp. 291-358.
??? Enzyme nomenclature, Recommendations (1964) of the International Union of Biochemistry on the nomenclature and classification of enzymes, Amsterdam-London-New York 1965.
Florkin, M., Stotz, E. H. (a cura di), Comprehensive biochemistry, vol. XII, Enzymes. General considerations, Amsterdam-London-New York 1964.
Goodwin, T. W., Harris, J. I., Hartley, B. S. (a cura di), Structure and activity of enzymes, New York 1964.
Haldane, J. B. S., Enzymes, London 1930.
Harris, H., Human biochemical genetics, Cambridge 1959.
Haschmeyer, R. H., Electron microscopy of enzymes, in Advances in enzymology (a cura di F. F. Nord), vol. XXXIII, New York 1970, pp. 71-118.
Holzer, H., Regulation of enzymes by enzyme-catalyzed chemical modification, in Advances in enzymology (a cura di F. F. Nord), vol. XXXII, New York 1969, pp. 297-326.
Jencks, W. P., Catalysis in chemistry and enzymology, New York 1969.
Jollès, J., Jauregui-Adell, J., Bernier, I., Jollès, P., La structure chimique du lysozyme de blanc d'oeuf de poule. Etude détaillée, in ‟Biochimica et biophysica acta", 1963, LXXVIII, pp. 668-669.
Kaplan, N. O., Nature of multiple molecular forms of enzymes, in ‟Annals of the New York Academy of Sciences", 1968, CLI, pp. 382-399.
Karlson, P., Kurzes Lehrbuch der Biochemie für Mediziner und Naturwissenschaftler, Stuttgart 19644 (tr. it.: Biochimica, Milano 1966).
Koshland, D. E., Neet, K. E., The catalytic and regulatory properties of enzymes, in ‟Annual review of biochemistry", 1968, XXXVII, pp. 359-410.
Laidler, K. J., The chemical kinetics of enzyme action, Oxford 1958.
Lehninger, A. L., Biochemistry, New York 1970.
Lipscomb, W. N., Reeke, G. N., Hartsuck, J. A., Quiocho, F. A., Bethge, P. H., The structure of carboxypeptidase A, in ‟Philosophical transactions of the Royal Society" (Series B), 1970, CCLVII, pp. 177-214.
Mahler, H. R., Cordes, E. H., Biological chemistry, New York 1966.
Markert, C. L., Møller, F., Multiple forms of enzymes, in ‟Proceedings of the National Academy of Sciences", 1959, XLV, pp. 753-763.
Markert, C. L., Whitt, G. S., Molecular varieties of isoenzymes, in ‟Experientia", 1968, XXIV, pp. 977-991.
Massey, V., Studies on fumarase. III. The effect of temperature, in ‟Biochemical journal", 1953, LIII, pp. 72-79.
Masters, C. J., Holmes, R. S., Isoenzymes and ontogeny, in ‟Biological reviews", 1972, XLVII, pp. 309-361.
Michaelis, L., Menten, M. L., Die Kinetik der Invertinwirkung, in ‟Biochemische Zeitschrift", 1913, XLIX, pp. 333-351.
Monod, J., Changeux, J. P., Jacob, F., Allosteric proteins and cellular control systems, in ‟Journal of molecular biology", 1963, VI, pp. 306-329.
Monod, J., Wyman, J., Changeux, J. P., On the nature of allosteric transitions. A plausible model, in ‟Journal of molecular biology", 1965, XII, pp. 88-118.
Morrison, J. F., Cleland, W. W., Isotope exchange studies of the mechanism of the reaction catalyzed by adenosine triphosphate: creatine phosphotransferase, in ‟Journal of biological chemistry", 1966, CCXLI, pp. 673-683.
Morse, D. E., Horecker, B. L., The mechanism of action of aldolases, in Advanecs in enzymology (a cura di F. F. Nord), vol. XXXI, New York 1968, pp. 125-181.
Neurath, H. (a cura di), The proteins, vol. II, New York 1964.
Payen, A., Persoz, J., Mémoire sur la diastase, in ‟Annales de chimie et de physique", 1833, LIII, pp. 73-92.
Perutz, M. F., X-ray analysis, structure and function of enzyme molecules, in ‟European journal of biochemistry", 1969, VIII, pp. 455-466.
Phillips, D. C., The three-dimensional structure of an enzyme molecule, in ‟Scientific American", 1966, CCXV, 5, pp. 78-90.
Reiner, J. M., Behavior of enzyme systems, Minneapolis 1959.
Roodyn, D. B. (a cura di), Enzyme cytology, London-New York 1967.
Scandalios, J., Genetic control of multiple molecular forms of enzymes in plants. A review, in ‟Biochemical genetics", 1969, III, pp. 37-79.
Siebert, G., Humphrey, G. B., Enzymology of the nucleus, in Advances in enzymology (a cura di F. F. Nord), vol. XXVII, New York 1965, pp. 239-288.
Smyth, D. G., Stein, W. H., Moore, S., The sequence of amino acid residues in bovine pancreatic ribonuclease. Revisions and confirmations, in ‟Journal of biological chemistry", 1963, CCXXXVIII, pp. 227-234.
Stadtman, E. R., Allosteric regulation of enzyme activity, in Advances in enzymology (a cura di F. F. Nord), vol. XXVIII, New York 1966, pp. 41-154.
Vessel, E. S., Bearn, A. G., Localization of lactic acid dehydrogenase in serum fractions, in ‟Proceedings of the Society for Experimental Biology and Medicine", 1957, XCIV, pp. 96-99.
Webb, J. F., Enzyme and metabolic inhibitors, vol. I, New York 1963.
White, A., Handler, P., Smith, E. L., Principles of biochemistry, New York 19684.
Wilkinson, J. H., Isoenzymes, London 1970.
Wong, J. T., Hanes, C. S., Kinetic formulations for enzymic reactions involving two substrates, in ‟Canadian journal of biochemistry and physiology", 1962, XL, pp. 763-804.