
epilessia
epilessia
L’epilessia è una condizione clinica caratterizzata da eventi accessuali, le crisi epilettiche, dovuti a scariche ipersincrone che coinvolgono popolazioni neuronali della corteccia cerebrale. Si distinguono crisi parziali, che originano da una regione limitata, e crisi generalizzate, con coinvolgimento di tutta la corteccia. La diagnosi si formula essenzialmente su base clinica, con l’ausilio dell’EEG. Ai fini dell’inquadramento sindromico è importante stabilire la familiarità per epilessia, il tipo di crisi, il tipo di anomalie EEG, la presenza di lesioni cerebrali. Esistono epilessie idiopatiche, dovute a una predisposizione genetica, sintomatiche, causate da una lesione epilettogena, e criptogenetiche, dove la lesione, se pur sospetta, non è evidente. La terapia è per lo più di tipo farmacologico, con un controllo delle crisi in circa il 70% dei casi. Nelle epilessie parziali farmacoresistenti può essere indicata una terapia chirurgica. [➔ antiepilettico; convulsione; displasia; elettrofisiologia del sistema nervoso; eterotopie corticali; imaging cerebrale]
L’epilessia (dal greco ἐπιλαμβάνω, «sono colto di sorpresa, sono sopraffatto») è nota sin dall’antichità: una delle prime descrizioni di una crisi risale a circa 3.000 anni fa, in Mesopotamia. Le manifestazioni epilettiche, per il loro carattere improvviso e altamente drammatico, erano in genere interpretate come di origine divina (morbo sacro) o comunque soprannaturale. Spetta a Ippocrate il merito di aver per primo considerato, circa 2.500 anni fa, l’e. un’infermità con cause di tipo naturale e «non più divina né sacra di altre malattie». L’indagine scientifica moderna inizia con i lavori di Theodor Fritsch, Eduard Hitzig, David Ferrier e Richard Caton (seconda metà del 19° sec.), che riuscirono a registrare scariche epilettiche dalla corteccia cerebrale dell’animale. Hughling Jackson studiò invece accuratamente i sintomi di una crisi e li correlò con la sede anatomica della lesione cerebrale identificata post mortem; le sue descrizioni dei fenomeni epilettici rimangono ancora insuperate. La scoperta di Hans Berger (1929) che segnali elettrici provenienti dal cervello possono essere registrati mediante elettrodi dalla superficie del capo introdusse l’elettroencefalo;gramma (EEG) nello studio delle crisi epilettiche. Frederic Gibbs, William Lennox, Wilder Penfield, Herbert H. Jasper e successivamente Henri Gastaut hanno contribuito all’avanzamento delle conoscenze nel campo della classificazione delle crisi epilettiche.
Definizione ed epidemiologia
L’e. è una condizione clinica caratterizzata da manifestazioni motorie, sensoriali, psichiche e neurovegetative (crisi epilettiche) che insorgono e cessano bruscamente, in genere di durata breve, e che si ripetono nel tempo in maniera imprevedibile. Tali eventi accessuali sono la traduzione clinica di scariche improvvise, ipersincrone ed eccessive che coinvolgono popolazioni neuronali più o meno estese della corteccia cerebrale. L’e. è tra le più frequenti malattie neurologiche, colpendo quasi l’1% della popolazione: in Italia ne sono affetti circa 500.000 individui e ogni anno si verificano oltre 25.000 nuovi casi.
Classificazione delle crisi epilettiche
Da un punto di vista nosografico, si individuano due livelli di classificazione: uno relativo alle crisi epilettiche, che considera differenti tipi in base alle loro caratteristiche cliniche, l’altro relativo alle sindromi epilettiche, che invece identifica complessi sindromici sulla base della fenomenologia delle crisi, delle condizioni neurologiche generali, dell’EEG, della presenza o meno di una causa nota, ecc. Classificare le crisi è indispensabile ai fini dell’identificazione della sindrome epilettica, a sua volta importante per definire la terapia e la prognosi. La classificazione delle crisi epilettiche, elaborata dalla International league against epilepsy (ILAE, 1981) e in revisione dal 2001, distingue due gruppi, quello delle crisi parziali (o focali), dove la clinica e l’EEG indicano un’origine da una regione limitata della corteccia cerebrale, e quello delle crisi generalizzate, nelle quali invece la scarica epilettica coinvolge sin dall’inizio tutta la corteccia.
Le crisi parziali. Queste crisi a loro volta sono classificate come semplici se non vi è alterazione dello stato di coscienza, complesse quando al contrario la coscienza è compromessa, e tonico-cloniche secondariamente generalizzate quando le iniziali manifestazioni focali esitano in un episodio convulsivo generalizzato. Le crisi parziali semplici si possono presentare con manifestazioni motorie, versive (deviazione di capo e occhi verso un lato), sensitive, vegetative e psichiche. Le crisi parziali complesse, prototipo delle quali sono le crisi a origine dal lobo temporale, si caratterizzano per l’alterazione dello stato di coscienza e per comportamenti automatici (automatismi), che possono essere di tipo alimentare (masticazione o deglutizione), gestuali semplici (movimenti afinalistici auto- o eterodiretti) e complessi (esecuzione di gesti significativi quali per es. abbottonarsi la camicia, svestirsi, ecc.), genitali, verbali (pronuncia di parole o frasi fuori contesto). Tali crisi sono anche note come automotorie, in accordo a una recente (1998) classificazione semeiologica. Altre forme di crisi parziali complesse possono essere di tipo ipermotorio e spesso originano dal lobo frontale. Esse si caratterizzano per automatismi complessi bizzarri e spesso violenti, con scalciamento/pedalamento, movimenti ritmici del bacino a volte mimanti l’atto sessuale, vocalizzazioni.
Le crisi generalizzate. Le crisi tonico-cloniche secondariamente generalizzate si presentano con convulsioni generalizzate precedute da un’aura o da una crisi parziale complessa. Le crisi generalizzate comprendono 6 sottotipi differenti: crisi tipo assenza, cloniche, miocloniche, toniche, atoniche e tonico-cloniche. Le assenze si manifestano come episodi di alterazione della coscienza (incantamento) di durata inferiore ai 20 s, a esordio e fine bruschi, talora accompagnati da ammiccamenti palpebrali. Si accompagnano a un caratteristico pattern EEG rappresentato da complessi punta-onda a 3 cicli al secondo. Le crisi cloniche si caratterizzano per scosse muscolari ritmiche diffuse a tutta la muscolatura corporea, correlate a un pattern EEG di punta o polipunta-onda. Le crisi miocloniche sono caratterizzate da brevi e bruschi scatti muscolari, aritmici, con correlato EEG di polipunta-onda. Le crisi toniche spesso colpiscono pazienti con altre alterazioni neurologiche e si caratterizzano per una estensione o flessione della testa, del tronco o delle estremità, a esordio brusco e con durata di qualche secondo, spesso in corso di sonno. Il correlato EEG tipico è di tipo elettrodecrementale (attività rapida di basso voltaggio diffusa che evolve talora in complessi di punta– o polipunta-onda). La crisi tonico-clonica (convulsiva) generalizzata (grande male) si caratterizza per improvvisa perdita della coscienza con caduta a terra, emissione di un urlo dovuto alla violenta contrazione dei muscoli respiratori, sospensione del respiro e conseguente cianosi del volto, ipertonia muscolare diffusa e, in fase terminale, scosse ritmiche di contrazione e rilasciamento. Fenomeni collaterali, espressione della compartecipazione del sistema nervoso vegetativo, sono emissione di bava, morsicatura della lingua, incontinenza degli sfinteri con perdita di urina o di feci. Segue uno stato similcomatoso di durata variabile e nessun ricordo resta di quanto è accaduto. Il correlato EEG prevede punte ritmiche diffuse seguite da polipunta-onda e quindi da silenzio elettrico di durata variabile. Le crisi atoniche si presentano in genere in persone con disturbi neurologici e si caratterizzano per atonia muscolare diffusa, spesso con caduta a terra traumatica. Il correlato EEG è simile a quello delle crisi toniche.
Fisiopatologia
In termini fisiopatologici una crisi epilettica è il risultato di uno squilibrio tra sistemi eccitatori e inibitori nell’ambito di un network di neuroni corticali a favore di una parossistica eccitazione. I sintomi critici esprimono la disorganizzazione delle aree corticali sede del network interessato dalla scarica, riflettendone, in modo spesso grossolano e caricaturale, la funzione. Lo squilibrio sopraddetto può dipendere o da meccanismi che portano a una diminuita inibizione (per es., riduzione del GABA, principale neurotrasmettitore cerebrale inibitorio), oppure da meccanismi che portano a un aumento di eccitazione (per es., aumento del glutammato, principale neurotrasmettitore eccitatorio).
Diagnosi
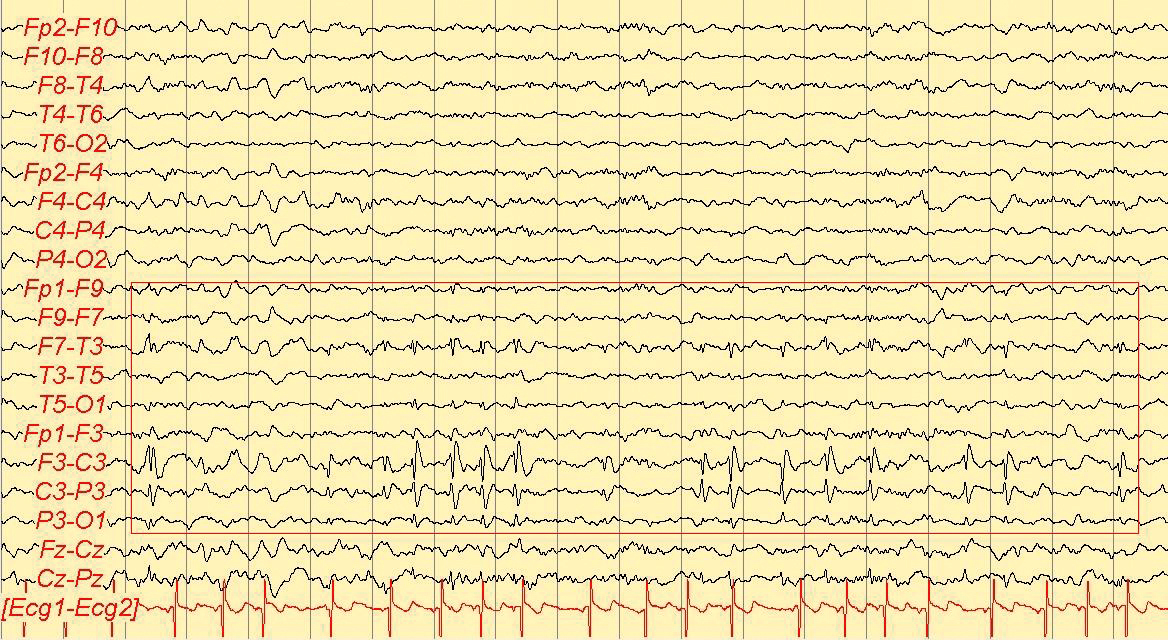
La diagnosi di crisi epilettica si formula essenzialmente su base clinica, analizzando le manifestazioni soggettive eventualmente esperite dal paziente e interrogando eventuali osservatori che abbiano assistito all’evento critico. In genere le manifestazioni epilettiche hanno un carattere stereotipato. Ai fini dell’inquadramento sindromico è importante la ricognizione su un’eventuale familiarità per e., sulla presenza di fattori di rischio quali una sofferenza perinatale anossico-ischemica, convulsioni febbrili complesse, traumi cranici significativi o esiti di infezioni dell’SNC. A tal fine, anche l’esame obiettivo, generale e neurologico sono importanti poiché la presenza di particolari alterazioni somatiche o di deficit neurologici si associa spesso all’e. in specifiche sindromi. L’EEG, nelle sue varianti (in veglia, in sonno o dopo deprivazione di sonno, dinamico ambulatoriale, video-EEG), è un test utile per la diagnosi (diagnosi differenziale con eventi di natura non epilettica) e per la caratterizzazione sindromica. Da ricordare che un EEG negativo non esclude la diagnosi di e. e che uno positivo non necessariamente la consente senza una clinica adeguata. Altri test, come quelli ematochimici, metabolici, genetici, neurofisiologici e neuroradiologici, sono importanti essenzialmente per l’inquadramento eziologico e sindromico.
Classificazione delle sindromi epilettiche
Nel 1989 è stata adottata una nuova classificazione delle sindromi epilettiche che valuta anche i criteri eziologici, così da individuare, per ciascuna delle due categorie principali, generalizzate o focali, tre sottocategorie: forme idiopatiche, dipendenti dall’età e la cui causa è una predisposizione genetica; forme sintomatiche, in cui è documentabile una lesione epilettogena; forme criptogenetiche, in cui, pur non essendo dimostrata una lesione, questa è verosimilmente presente. Inoltre vengono adottati criteri topografici per la diagnosi di sede delle e. parziali (lobo temporale, parietale, frontale, occipitale). Tale classificazione è al momento sotto revisione da parte di una task force dell’ILAE che sta introducendo nuove sindromi su cui si è creato consenso, con elaborazione di un sistema diagnostico multiassiale (descrizione della crisi, tipo di crisi, sindrome epilettica, eziologia e grado di compromissione del paziente).
Epilessie generalizzate idiopatiche. Sono forme a elevata predisposizione genetica che colpiscono soggetti neurologicamente normali, con esordio legato all’età, quindi condizionato dal diverso grado di maturazione cerebrale: si distinguono forme dell’infanzia, dell’adolescenza e della giovinezza. In tali casi il ritmo di fondo (ritmo di base o fisiologico) dell’EEG e le neuroimmagini (TC e RM) sono normali. Tale categoria di e. include diverse sindromi, caratterizzate da tre tipi di crisi che possono manifestarsi in modo variabile anche nello stesso soggetto: crisi tonico-cloniche generalizzate, assenze tipiche e mioclonie. Svolgono spesso un ruolo fattori scatenanti quali l’alterazione del ritmo sonno-veglia e la stimolazione luminosa intermittente. In genere hanno una buona risposta alla terapia medica. Le caratteristiche cliniche delle sindromi più frequenti sono quelle di seguito elencate. L’e. a tipo assenza dell’infanzia è una delle forme più frequenti: insorge in bambini di età compresa tra 2 e 10 anni, con un picco tra i 5 e i 6 anni. Le assenze sono molto frequenti, a volte fino a 200 al giorno (picnolessia), con evidenti ripercussioni sul livello di attenzione e sul rendimento scolastico. L’e. mioclonica giovanile si manifesta intorno alla pubertà, con mioclonie bilaterali agli arti, soprattutto superiori; l’intensità delle mioclonie può essere elevata e provocare la caduta di oggetti dalle mani o la caduta a terra del soggetto; non vi è disturbo della coscienza. Le crisi compaiono in genere dopo il risveglio; sovente la privazione di sonno o i risvegli precoci costituiscono fattori scatenanti. Spesso il paziente presenta crisi di grande male, meno frequentemente assenze. L’e. con crisi di grande male al risveglio insorge generalmente tra i 10 e i 20 anni. Le crisi, di tipo tonico-clonico generalizzato, si verificano poco dopo il risveglio, con un secondo picco nel periodo serale di relax, e sono precipitate da privazione di sonno.
Epilessie generalizzate sintomatiche e criptogenetiche. Queste e. includono forme gravi con crisi resistenti alla terapia medica, in pazienti con danni neurologici. Il tracciato EEG presenta un ritmo di fondo rallentato e disorganizzato e le neuroimmagini presentano spesso alterazioni. La sindrome di West insorge entro il primo anno di vita ed è contraddistinta da una tipica triade: spasmi simultanei della muscolatura degli arti e del tronco (che si presentano soprattutto dopo il risveglio), arresto dello sviluppo psicomotorio e ipsaritmia (onde lente e punte di alto voltaggio caotiche e multifocali) all’EEG. Le forme sintomatiche possono avere cause prenatali (complicanze durante la gravidanza, malformazioni cerebrali congenite), perinatali (traumi da parto, anossia), postnatali (meningiti, encefaliti, malattie metaboliche). La sindrome di Lennox-Gastaut insorge tra 1 e 8 anni, unita a ritardo mentale, con crisi toniche nel sonno, crisi atoniche ad assenze atipiche. L’EEG mostra complessi punta-onda a 2 cicli al secondo e scariche di polipunte a 10 cicli al secondo nel sonno.
Epilessie parziali o focali. Queste e. riguardano quelle forme in cui la scarica che le sottende interessa una popolazione di cellule cerebrali circoscritta. Le forme idiopatiche, che hanno un esordio legato all’età e una predisposizione genetica, sono prive di lesioni anatomiche e spesso soggette a remissione spontanea. A questa categoria appartiene l’e. benigna dell’infanzia con punte centrotemporali, così detta per l’EEG tipico, che presenta punte di ampio voltaggio localizzate nelle regioni centrotemporali di un emisfero, più evidenti nelle fasi di addormentamento. La sindrome, frequente in età scolare, è caratterizzata da crisi parziali semplici motorie coinvolgenti un lato del volto o l’arto superiore, che nel 75% dei casi si verificano durante il sonno, determinando il risveglio del paziente; le crisi sono talvolta associate a crisi generalizzate tonico-cloniche. La prognosi è favorevole in quanto le crisi tendono a scomparire nell’adolescenza, indipendentemente dall’impiego di farmaci. Le forme criptogenetiche e quelle sintomatiche, che differiscono tra loro unicamente per l’evidenza eziologica, assente nelle prime e presente nelle seconde, costituiscono la maggior parte delle e. parziali, e possono comparire a qualsiasi età, anche se per lo più iniziano nell’adolescenza o nella gioventù. Le cause sono numerose e possono verificarsi in epoca prenatale, perinatale o postnatale. I fattori prenatali sono spesso legati a un’inadeguata ossigenazione intrauterina (per es., per traumi, distacco di placenta, ecc.), ma possono riguardare l’alterazione dell’organizzazione dello sviluppo corticale (per es., displasie focali, ➔ eterotopie corticali). I fattori perinatali comprendono le emorragie intracraniche su base traumatica, oppure la sofferenza ipossica, che può derivare da un parto difficoltoso. I fattori postnatali includono le patologie infettive dell’SNC, quelle vascolari (più spesso ischemie che emorragie), i traumi cranici e i tumori, più frequentemente nel caso delle neoplasie a lento sviluppo (tumori di basso grado, quali disembrioneuroepiteliomi e gangliogliomi). L’e. del lobo temporale mesiale si caratterizza per crisi automotorie, della durata di qualche minuto, spesso precedute da aure, come una sensazione epigastrica ascendente o un déjà-vu (➔). L’EEG fra una crisi e l’altra mostra anomalie lente e irritative sulle derivazioni anteriori, la risonanza magnetica può evidenziare un’eventuale lesione epilettogena (sclerosi temporomesiale, displasia corticale, neoplasia, angioma cavernoso). Spesso è resistente alla terapia medica, ma può avere un’ottima prognosi in seguito al trattamento chirurgico. L’e. del lobo frontale, più rara della precedente, si caratterizza per crisi ricche di segni motori, spesso configurando pattern di tipo ipermotorio. Le crisi sono di breve durata, in sonno e in grappoli, con frequente generalizzazione o stati di male. Le e. parieto-occipitali sono forme più rare e risultano caratterizzate per una sintomatologia sensoriale (parestesie), un’alterata percezione spaziale, talvolta vertigine (parietale), manifestazioni visive, che possono essere rappresentate da fenomeni sia positivi, per es., lampi, scintille, sia negativi, quali scotomi (nel caso di e. occipitale). Possono verificarsi anche illusioni visive, per cui si vedono gli oggetti più grandi, più piccoli, deformati. Le forme occipitali possono essere anche idiopatiche.
Terapia
La terapia delle e. è per lo più di tipo farmacologico. Nelle e. parziali resistenti ai farmaci, in partic. se connesse a un sito cerebrale per il quale può essere indicata l’asportazione, è possibile intervenire chirurgicamente con completa guarigione del paziente. In caso di controindicazione alla terapia resettiva si può avviare il paziente farmacoresistente a interventi di tipo palliativo. Formulata la diagnosi di e., la terapia farmacologica va iniziata con un unico farmaco di prima scelta per il tipo di crisi diagnosticato (monoterapia). Se non si ottiene un completo controllo è necessario aumentare la posologia sino alla dose massima tollerata. In caso di insuccesso si procede a un cambio della terapia in atto con un secondo farmaco o a una bioterapia. Nel caso in cui la diagnosi formulata sia esatta, e l’iter terapeutico sia stato correttamente seguito, si ottiene un controllo delle crisi in circa il 70% dei casi. Nell’e. parziale non idiopatica, in caso di insuccesso con un secondo o un terzo farmaco, va verificata l’ipotesi neurochirurgica. I farmaci (➔ antiepilettico) attualmente disponibili sono: benzodiazepine, fenobarbitale, fenitoina, primidone, acido valproico, carbamazepina, oxcarbazepina, vigabatrin, felbamato, tiagabina, lamotrigina, topiramato, levetiracetam, zonisamide, rufinamide. Giancarlo Di Gennaro
La chirurgia dell’epilessia
La terapia chirurgica dell’epilessia riguarda qualsiasi intervento neurochirurgico che abbia come obiettivo la cura delle epilessie focali non controllate dalla terapia medica. Lo scopo è di ottenere il massimo controllo dell’epilessia con il minimo di effetti collaterali, evitando così l’impatto negativo delle crisi e della terapia medica sullo stato di salute, sugli aspetti cognitivi, socio-lavorativi e affettivorelazionali dei pazienti, migliorando la loro qualità della vita.
Studio prechirurgico
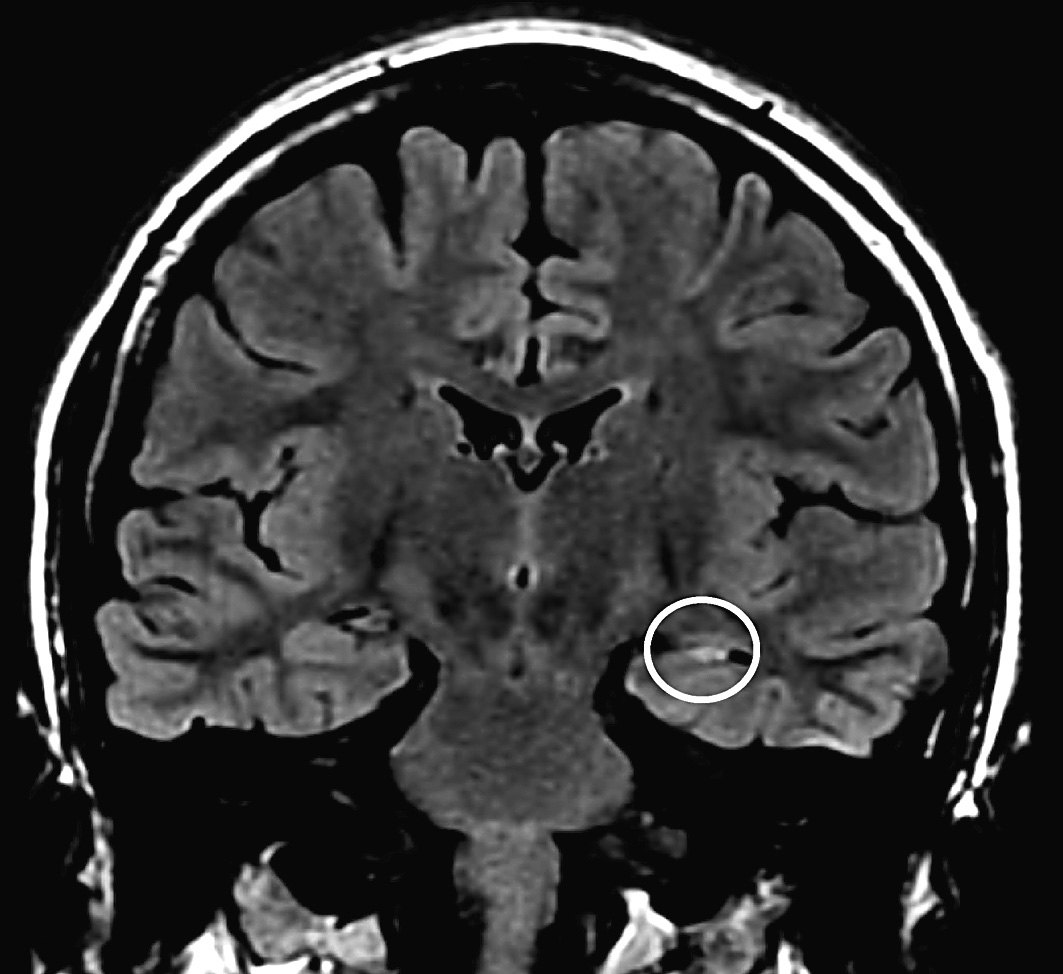
Prima dell’intervento è necessaria una valutazione multidisciplinare del paziente, finalizzata principalmente alla definizione della zona epilettogena (la regione corticale da cui originano le crisi e che è necessario rimuovere o disconnettere per consentirne la guarigione). Tale studio inizia con l’anamnesi epilettologica, che analizza le caratteristiche cliniche delle crisi e la loro stabilita nel tempo. La loro semeiologia può essere infatti considerata il risultato della disorganizzazione funzionale delle regioni corticali sequenzialmente interessate dalla scarica epilettica e fornisce, pertanto, i primi elementi per una ipotesi di localizzazione della zona epilettogena. Le caratteristiche cliniche anamnestiche delle crisi sono verificate dalla video- EEG, esame cardine dello studio prechirurgico, che accoppia la ripresa in video del paziente in corso di crisi con le eventuali modificazioni dell’EEG, in modo da rinforzare l’ipotesi localizzatoria. Il paziente è monitorato 24 ore su 24 ed è anche costantemente osservato da personale specializzato che, interagendo con lui durante una crisi, valuta la presenza di sintomi o segni non altrimenti desumibili dal solo esame del video (perdita di coscienza, disturbi del linguaggio, sottili modificazioni del tono muscolare, ecc.). La risonanza magnetica cerebrale ad alta risoluzione riveste un ruolo fondamentale in quanto consente di svelare un’eventuale lesione strutturale alla base dell’epilessia del paziente (lesione epilettogena). Per ottenere il massimo rendimento da questo esame occorre che venga guidato da un’ipotesi di localizzazione, formulata sulla base dei dati elettroclinici a disposizione. In questo senso la collaborazione tra epilettologo e neuroradiologo permette di effettuare un esame individualizzato. Altre tecniche di neuroimaging cosiddette funzionali (RM funzionale, PET e SPECT) sono oggi disponibili, ma il loro ambito di utilizzo nelle varie epilessie e la loro attendibilità non riscuotono un consenso unanime e sono considerate, pertanto, ancillari. Un aspetto importante è rappresentato dalla valutazione neuropsicologica e da quella psichiatrica. In generale, la valutazione delle funzioni cognitive del paziente mediante una serie di test neuropsicologici contribuisce alla identificazione della corteccia sede di eventuale disfunzione, alla valutazione dei rischi di deficit di memoria (come conseguenza della resezione del lobo temporale) e, nei casi dubbi, alla determinazione della dominanza emisferica. La valutazione psichiatrica esplora invece principalmente la presenza di componenti psicopatologiche e di eventuali disturbi della personalità, a rischio di scompenso postchirurgico, nonché la tenuta della rete di supporto socio-familiare. Qualora le indagini non invasive non riescano a individuare la zona epilettogena, si può ricorrere a quelle invasive. La stereoelettroencefalografia (SEEG) è una metodologia di registrazione dell’attività elettrica cerebrale, che utilizza elettrodi intracerebrali impiantati con tecnica stereotassica (➔ stereotassico, apparato). La strategia d’impianto si basa sulla formulazione di un’ipotesi di localizzazione della zona epilettogena e va pertanto individualizzata in base alle esigenze del singolo caso. Vanno esplorate le strutture da cui si ipotizza che origini la scarica critica e quelle verosimilmente sede della sua successiva organizzazione. Vengono utilizzati elettrodi ad ago con contatti registranti lungo il loro decorso. Un’altra metodica di esplorazione è quella con elettrodi subdurali, organizzati in griglie di silicone di varia estensione, applicati mediante craniotomia direttamente a contatto della corteccia cerebrale. Lo scopo, al pari della SEEG, è la registrazione diretta dell’attività elettrica cerebrale, ma è anche consentita l’elaborazione di una mappa delle funzioni corticali tramite stimolazione elettrica.
Chirurgia
La chirurgia curativa si propone l’obiettivo dell’abolizione completa delle crisi. Esistono resezioni individualizzate, come la lesionectomia (rimozione di una lesione strutturale), la resezione sublobare, lobare e multilobare (a seconda dell’estensione dell’ablazione), o standardizzate, come la lobectomia temporale anteromesiale, l’amigdalo-ippocampectomia selettiva (interventi eseguibili nell’epilessia del lobo temporale mesiale), l’emisferectomia (per es., nell’epilessia associata all’encefalite cronica di Rasmussen). Vi è inoltre la possibilità di praticare la disconnessione, ossia la separazione anatomico-funzionale, anziché l’asportazione, del tessuto epilettogeno, per il quale viene preservata la vascolarizzazione. Lo scopo è quello di ridurre le complicanze postoperatorie, per es. in interventi molto estesi. La chirurgia palliativa è invece tesa alla riduzione dell’intensità e della frequenza delle crisi epilettiche e non alla loro soppressione. Lo scopo è quello di limitare o impedire la propagazione della scarica elettrica e di diminuire l’eccitabilità neuronale modulando circuiti inibitori o facilitatori. Possono essere considerati interventi palliativi la callosotomia (separazione delle vie intercommissurali che passano attraverso il corpo calloso; ➔ commissurotomia), le transezioni subpiali multiple (selettiva interruzione delle fibre intracorticali a orientamento orizzontale, preservando l’architettura colonnare) e le varie tecniche di neuromodulazione, quali la stimolazione del nervo vago e di nuclei cerebrali profondi (DBS, Deep Brain Stimulation).