
Farmaci intelligenti
Farmaci intelligenti
La locuzione farmaci intelligenti è stata introdotta nel linguaggio comune dal giornalismo scientifico italiano, e non se ne trova quasi traccia nella letteratura internazionale. Con essa si intende indicare l'insieme dei farmaci che hanno un bersaglio molecolare ben preciso responsabile di una determinata malattia. Questi farmaci, colpendo in modo specifico il bersaglio, indurrebbero giovamenti terapeutici importanti con modesti effetti collaterali. Lo scopo della farmacologia è sempre stato quello di produrre farmaci capaci di debellare la malattia senza un carico di tossicità eccessiva, ma spesso non è riuscita a raggiungere questo obiettivo per alcuni motivi fondamentali: a) la maggioranza delle patologie non è causata dalla disfunzione di una o poche molecole, ma assai sovente dal malfunzionamento di un complesso sistema dovuto alla disfunzione o perdita di molte molecole; b) in molte malattie sono compromesse molecole (recettori, canali, trasportatori, enzimi) che, pur avendo una moltitudine di effetti, sono alterate solo in alcuni tessuti o circuiti nervosi o cellule (si pensi, per es., alla schizofrenia, nella quale certamente vi è una disfunzione dei recettori dopaminergici nel sistema limbico, ma non nel sistema mesostriatale); è chiaro che molecole specifiche verso questo bersaglio possono produrre effetti benefici, ma, immancabilmente, anche effetti collaterali seri; c) talvolta, pur conoscendo il bersaglio da colpire, non si hanno ancora a disposizione farmaci specifici. I più recenti progressi della chimica sono stati così importanti da consentire di abbreviare molto il tempo tra la scoperta del bersaglio e l'ottenimento di molecole opportune. Un esempio tipico è la terapia dell'AIDS, malattia all'origine incurabile e che solo dopo vent'anni dalla scoperta del virus HIV (1983-84), è divenuta invece una malattia con prognosi molto meno drammatica. In un breve lasso di tempo dalla scoperta dei possibili bersagli utili per la terapia, la chimica e la farmacologia hanno saputo approntare strumenti terapeutici validi. Ma anche in questo caso, nel quale si hanno molti farmaci relativamente specifici a disposizione, la risposta terapeutica non è soddisfacente in quanto non si è ancora trovato il punto di attacco letale per il virus, capace di eradicarlo dall'organismo; inoltre, i farmaci continuano a possedere una loro alta tossicità, e in alcuni pazienti la terapia non è tollerata. La difficoltà di avere un farmaco che sia al contempo efficiente e selettivo, e quindi 'intelligente', non sta tanto nell'insufficienza della chimica e della farmacologia, quanto soprattutto nella nostra ignoranza del meccanismo patogenetico di quasi tutte le malattie, nella multifattorialità dei meccanismi che provocano molte delle malattie delle quali soffre l'uomo. L'analisi del genoma ha permesso di approfondire la complessità dell'organismo vivente, la diversità genetica degli individui, i motivi della loro diversa risposta ai farmaci e alle noxae patogene, e la natura di numerose malattie che affliggono gli esseri umani. Da queste ricerche risulta che il genoma umano è composto da circa 25.000-30.000 geni, che le proteine nel proteoma sono circa 21.600, e che di queste 1500-3000 sono potenziali bersagli dei farmaci, mentre i bersagli al momento aggredibili da farmaci sono 300-400. Inoltre, sono state messe a punto alcune metodologie di biologia molecolare e cellulare relativamente facili, riproducibili e di basso costo, che permettono di raffinare e rendere precoce la diagnosi molecolare delle malattie, affrontare la sintesi di nuovi tipi di farmaci e testarli su molti bersagli con metodi di alta efficienza e di basso costo. Contemporaneamente la chimica, attraverso la chimica combinatoriale e il disegno molecolare attraverso il computer (in silico drug design), ha aumentato molto la capacità di sintetizzare e analizzare nuove molecole. Si può calcolare che ogni chimico può produrre almeno 1000 analoghi di una sostanza per anno, costruiti con una logica che combini la struttura del bersaglio con quella del farmaco. La preparazione dei f. i. si basa su tre cardini: a) la farmacogenomica, che ricerca bersagli specifici e sempre più significativi che siano alla base delle malattie verso i quali indirizzare farmaci capaci di colpire solamente questi bersagli patologici; b) la farmacogenetica, per la quale il malato è qualcosa di più complesso che non un suo organo, e la pratica clinica ci insegna che non tutti i pazienti rispondono ai farmaci in modo uguale; è necessario, quindi, scoprire quali sono i motivi che rendono gli individui diversi tra loro rispetto alla risposta ai farmaci, con la speranza d'individuare per ogni paziente il farmaco più corretto per curare la sua malattia; c) drug delivery, ossia la disciplina che riguarda la ricerca di f. i. attraverso la somministrazione e il recapito dei farmaci stessi in modo che possano essere presenti nell'organismo solo alle concentrazioni volute, nel tempo utile per il loro effetto e nel sito dove essi devono agire.
Farmacogenomica
Le tecniche di biologia molecolare e cellulare e le loro applicazioni nel campo delle biotecnologie hanno reso possibile la descrizione quasi completa del genoma umano e delle modificazioni genomiche, proteomiche e trascrizionali a livello di molti tessuti patologici. Le ricerche più significative che sono approdate a f. i. già applicati in clinica sono quelle che riguardano i tumori. Questi ultimi sono stati il campo di battaglia preferenziale dei ricercatori in quanto costituiscono patologie molto gravi, con altissima mortalità e, quindi, con forte impatto sociale e affettivo.
I progressi tecnologici
Molti tumori umani derivano da singole cellule somatiche che hanno subito una serie di modificazioni geniche ed epigeniche con alterazioni nella espressione e nell'attività dei geni. Tra gli aspetti più importanti che caratterizzano un tumore vi sono: disregolazione dei segnali di crescita, aumento dell'invasività e dell'angiogenesi, mancata risposta ai segnali di differenziamento e di blocco della proliferazione e perdita dell'apoptosi. Le più importanti mutazioni, i riarrangiamenti genomici e le alterazioni trascrizionali responsabili sono in gran parte note, e ci si sta approssimando alla loro piena comprensione (Workman 2003). Notevoli progressi sono stati compiuti grazie alle nuove discipline della genomica e della proteomica. L'uso del sequenziamento automatico, il completamento delle conoscenze sul genoma umano e gli avanzamenti tecnologici più recenti hanno molto accelerato l'identificazione di mutazioni e alterazioni del genoma delle cellule tumorali come possibile bersaglio terapeutico. La tecnologia dei micro array ha permesso il confronto dell'espressione dei mRNA in tumori e tessuti normali, aprendo la strada a una classificazione dei tumori non solo su base istologica ma anche su base molecolare, rendendo così la diagnosi più facile e più legata a un possibile meccanismo eziopatologico. Infine, si sono molto sviluppate le tecniche per l'analisi delle proteine; questo ha permesso la nascita di una proteomica dei tumori, e la conseguente identificazione di alterazioni di proteine nei tessuti tumorali e di marcatori biologici per ogni tipo di tumore, e ancor più di caratterizzare dal punto di vista proteico il tumore per ogni singolo paziente.
L'associazione delle tecnologie genomiche e proteomiche ha comportato un approfondimento insperato nella patogenesi e biologia di diversi tumori, favorendo l'identificazione di bersagli specifici per diagnosticare e colpire i diversi tipi di tumore (Workman 2003).
I bersagli per i farmaci intelligenti
Dai dati che sono emersi a partire dagli anni Novanta del 20° sec., risulta che i bersagli più rilevanti per un possibile attacco specifico alla cellula tumorale sono i geni soppressori e gli oncogeni. In molti tumori vi è perdita della proteina soppressore p53 (che controlla il ciclo e che induce apoptosi) e presenza di oncogeni (geni che iniziano, stimolano e promuovono la crescita cellulare). Sia i geni soppressori sia gli oncogeni agiscono attraverso la regolazione del ciclo cellulare, intervenendo sui fattori di crescita e i loro recettori, sulla traduzione del segnale per i fattori di crescita e sull'apoptosi. La ricerca si è soprattutto concentrata sugli oncogeni, in quanto è più facile inibire un'eccessiva attività biologica che introdurre un'attività persa dalla cellula. I farmaci che hanno dato risultati clinici più brillanti sono stati quelli rivolti all'inibizione dell'attività dei recettori per i fattori di crescita e all'inattivazione delle tirosina chinasi (Dancey, Sausville 2003).
Inibitori delle tirosina chinasi
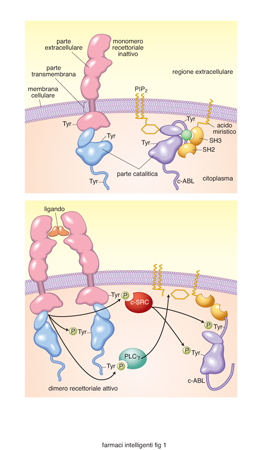
Le tirosina chinasi sono enzimi che catalizzano il trasferimento di fosforo dall'ATP alla tirosina di una proteina; sono molto importanti in quanto regolano la proliferazione, la sopravvivenza, il differenziamento, la motilità e molte altre funzioni della cellula. Nei tumori queste attività enzimatiche così importanti sono disregolate, e si è quindi pensato che potessero essere un bersaglio privilegiato per un intervento farmacologico specifico. La previsione si è rivelata corretta e gli inibitori delle tirosina chinasi tumorali hanno avuto un notevole successo terapeutico. Tra questi, l'imatinib mesilato (Glivec) che inibisce la Bcr-Abl tirosina chinasi, essenziale per la proliferazione cellulare nella leucemia mieloide cronica; il gefitinib (Iressa) e l'erlotinib (Tarceva) che inibiscono l'attività delle chinasi del recettore dell'EGF (Epidermal Growth Factor), importante nel controllare la crescita di alcuni tumori solidi. Le tirosina chinasi si possono dividere in due classi: la prima contiene i recettori a tirosina chinasi, proteine transmembrana che vengono attivate da fattori di crescita; la seconda classe è costituita da proteine che si trovano nel citoplasma della cellula, non hanno quindi la parte extracellulare e quella transmembrana, ma solo la parte catalitica, e sono legate alla membrana attraverso una catena di acido grasso, normalmente l'acido miristico (fig. 1). Questi enzimi sono fortemente regolati durante la vita cellulare e nelle cellule non proliferanti si hanno solo tracce di proteine fosforilate in tirosina. A riposo i recettori a tirosina chinasi non sono attivi in quanto, in assenza del ligando, sono monomeri incapaci di attivare la parte catalitica. Le tirosina chinasi citoplasmatiche sono invece tenute silenti da un legame intramolecolare tra due siti SH3 (Sarc homology-3) e dal legame con un lipide di membrana, il fosfatidil inositolo bisfosfato, PIP2 (Krause, Van Etten 2005). Quando i recettori a tirosina chinasi vengono attivati dal proprio ligando, i monomeri si avvicinano e formano dimeri, in modo che le parti catalitiche dei due recettori si trovino vicine e si attivino; così, ciascun recettore fosforila il recettore vicino (transfosforilazione). I recettori fosforilati vengono riconosciuti da una serie di proteine adattatrici presenti nel citoplasma che contengono un dominio SH2, come la fosfolipasi Cγ e il c-SRC, che in questo modo si attivano e danno origine a una cascata di eventi biochimici che portano tra l'altro alla proliferazione cellulare (fig. 1 A). Le tirosina chinasi citoplasmatiche sono attivate da una fosforilazione di due tirosine da parte di altre tirosina chinasi, da altri processi che attivano cSRC e dalla idrolisi del PIP2. I livelli di fosforilazione in tirosina delle proteine sono controllati da fosfatasi apposite che defosforilano le proteine fosforilate in tirosina, e che sono anch'esse modulate da processi di fosforilazione dipendenti dall'attività recettoriale. Nei tumori, le tirosina chinasi sono altamente disregolate, e questo avviene attraverso una serie di meccanismi, di cui citiamo i più rilevanti.
Sovra espressione. I recettori per l'EGF sono molto più espressi che nel normale, e questo porta a un'esagerata proliferazione di queste cellule e alla formazione del tumore (Herbst, Fukuoka, Baselga et al. 2004; Hynes, Lane 2005); questo avviene, per es., nel tumore della mammella.
Mutazioni puntiformi. Modificano la struttura del recettore rendendolo più attivo e/o indipendente dal legame con il ligando. Alcuni esempi: nella leucemia mieloide acuta, alcune cellule bianche del sangue hanno un recettore (FLT3) sempre attivo, e di conseguenza si riproducono molto velocemente; un recettore delle cellule epiteliali per il fattore di crescita delle cellule staminali (cKIT) è mutato e reso costituzionalmente iperattivo in alcuni tumori gastrointestinali, con conseguente proliferazione di queste cellule e, quindi, crescita del tumore intestinale.
Fusioni con altre proteine. Consiste nella fusione di due diversi geni normalmente localizzati su cromosomi lontani tra loro. Per es., nella leucemia mieloide cronica, una fusione dei cromosomi 9 e 22 provoca la formazione di un nuovo gene composto da una parte del gene BCR e una parte del gene ABL. Da ciò risulta una proteina dimerizzata (Bcr-Abl), con attività tirosina chinasica costituzionalmente sempre attiva (P) che darà origine a un segnale intracellulare molto intenso e persistente (fig. 1 B). La disregolazione delle attività tirosina chinasiche e una loro conseguente iperattivazione porta le cellule a proliferare, a una maggiore sopravvivenza e, nei tumori, a crescita, invasività e neoproduzione di vasi sanguigni. Le tirosina chinasi tumorali sono state identificate come possibile bersaglio specifico per la terapia utilizzando come farmaci inibitori specifici delle forme tumorali di tirosina chinasi, in modo da uccidere il tumore senza recare troppi danni alle cellule normali. In alcuni casi, come con l'imatinib nella leucemia mieloide cronica, il successo è stato strabiliante, e si è ottenuta la guarigione dei pazienti senza gravi effetti collaterali. Se si tiene conto del carico doloroso che una normale chemioterapia antitumorale comporta e della sua scarsa efficienza terapeutica, ci si può immaginare le aspettative che questo successo ha comportato. Come si possono aggredire le tirosina chinasi tumorali? Esistono diversi punti di attacco: a) si può inattivare la parte extracellulare del recettore attraverso anticorpi specifici e diminuire così il numero e l'attività dei recettori; b) si possono inattivare i ligandi specifici per i recettori e impedire così che i recettori vengano attivati; c) si può inibire l'attività delle tirosina chinasi sia recettoriali sia citoplasmatiche; d) si possono inibire le tirosina chinasi citoplasmatiche impedendo la traduzione del mRNA per la proteina, oppure impedendo la sua corretta traslocazione nel citoplasma interferendo con l'associazione con la Heat Shock Protein 90. Risultati significativi hanno riguardato i punti a e c; per quanto riguarda il punto a, il 20-25% dei tumori della mammella sovraesprime alcuni recettori per il Fattore di Crescita dell'Epidermide (EGFR, Epidermal Growth Factor Receptor), chiamati ERB2 o Neu, che conferiscono loro un'alta aggressività e un'alta invasività.
L'anticorpo monoclonale Transtuzumab (Herceptin) si lega al recettore e ne favorisce l'internalizzazione e la sua rimozione dalla superficie cellulare. Esso induce nelle pazienti portatrici di questo tipo di tumore un consistente aumento di sopravvivenza e una forte riduzione delle metastasi, quando esso sia unito alla chemioterapia tradizionale. In seguito a questo successo altri anticorpi verso questi recettori sono allo studio e anticorpi contro altri recettori, soprattutto verso il recettore per l'EGF, sono stati sperimentati in clinica con un certo successo come coadiuvanti nella terapia del tumore del colon (Dancey, Sausville 2003; Hynes, Lane 2005). Per quanto riguarda il punto c, invece, sono stati ottenuti brillanti risultati inibendo l'attività catalitica delle tirosina chinasi citoplasmatiche attraverso molecole di piccole dimensioni che bloccano il sito di legame dell'ATP all'enzima. Tra questi il più noto è l'imatinib (Glivec) che inibisce la chinasi Bcr-Abl che è la causa diretta della leucemia mieloide cronica. In questa malattia il farmaco induce una guarigione in un'alta percentuale dei pazienti con pochissimi effetti collaterali. Questo farmaco produce buoni risultati clinici anche in circa il 50% dei pazienti portatori di tumori gastrointestinali nei quali sia presente un recettore a tirosina chinasi mutato (cKIT), che è sempre attivato (vedi sopra). Anche in questo caso gli effetti collaterali sono scarsi. Altri farmaci sono stati trovati attivi soprattutto verso le chinasi dei recettori per l'EGF. Tra questi, gefitibib (Iressa) ed erlotinib (Tarceva) sono già entrati in clinica e hanno dimostrato di avere buone attività terapeutiche nei tumori del polmone non a piccole cellule, soprattutto in quei tumori che esprimono più copie dei recettori per l'EGF (Dancey, Sausville 2003; Krause, Van Etten 2005). Perché non tutti gli inibitori delle tirosina chinasi sono così attivi come l'Imatinib nella leucemia linfatica cronica e nei tumori gastrointestinali? I risultati hanno evidenziato che il tumore può essere dovuto a un'alterazione di un solo gene, come è il caso della leucemia mieloide cronica, dovuta all'attivazione di Abl, ma più spesso all'attivazione sequenziale di più geni che producono tumore solo se sono presenti due o più alterazioni (Workman 2003). Questo concetto di oncogenesi sequenziale ha cambiato l'approccio terapeutico, che insiste non più su un unico bersaglio, ma su più bersagli specifici. La difficoltà sta nell'individuare in ogni tumore quale siano i bersagli essenziali che portano all'oncogenesi, e quando sia il tempo e momento opportuno per intervenire. Lo straordinario successo di questi f. i. ha stimolato molto la ricerca di altri possibili bersagli selettivi (Herbst, Fukuoka, Baselga 2004). I geni che sono oggetto di studio riguardano tanto la cascata della trasmissione del segnale recettoriale, e in paricolare le MAP chinasi (Mitogen Activate Protein Kinases) e la PI3 chinasi (inositol fosfato chinasi), le molecole di membrana responsabili dell'adesione cellulare, quanto eventi più a valle come le istone deacetilasi, molecole fondamentali nella regolazione dell'espressione genica, e che spesso sono alterate in molti tumori. Molti farmaci sono stati trovati verso questi bersagli e alcuni di questi sono già in fase di sperimentazione clinica. La lezione appresa con i farmaci antitumorali si sta estendendo anche ad altre patologie, ma la difficoltà consiste nello scoprire quali siano le alterazioni specifiche che stanno all'origine delle diverse malattie. Le difficoltà nella scoperta di f. i. aumentano inoltre quando la patologia da curare è una patologia cronica, che si instaura lentamente e di conseguenza dovuta ad alterazioni magari piccole e poco significanti di molte molecole, le quali diventano significative soltanto se presenti contemporaneamente e per un lungo periodo.
Farmacogenetica
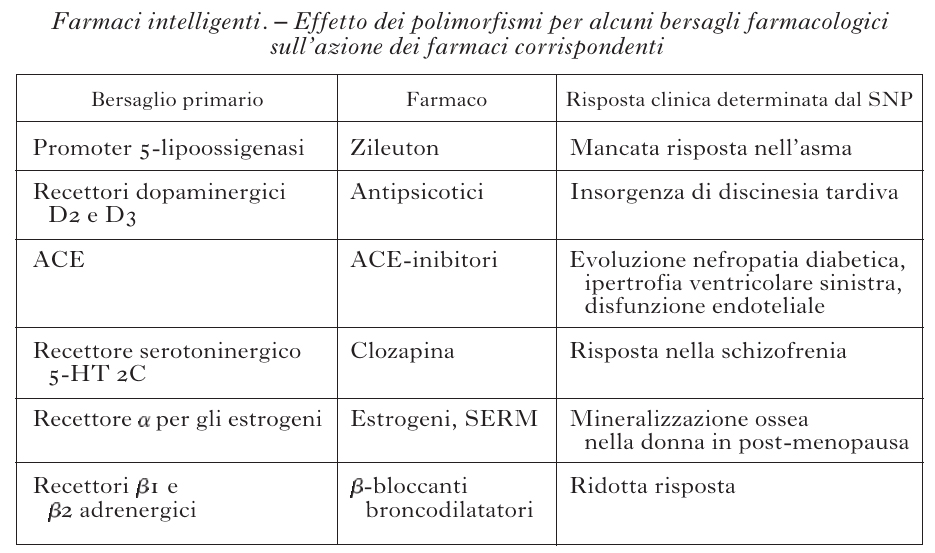
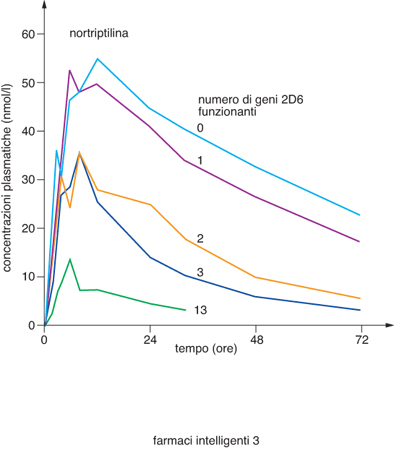
È esperienza comune nella pratica medica che lo stesso farmaco somministrato alla stessa dose può essere efficace nella maggioranza dei pazienti, scarsamente efficace o inefficace in un numero significativo di pazienti e indurre effetti collaterali indesiderati, a volte anche gravi, in alcuni di essi. La variabilità individuale rispetto a un determinato trattamento farmacologico può essere influenzata da fattori legati alla fisiologia (età, sesso, peso corporeo), alla fisiopatologia del paziente (funzionalità epatica e renale, presenza di patologie concomitanti), a fattori ambientali (nutrizione, consumo di alcool, di fumo, di trattamenti farmacologici concomitanti), ma, soprattutto, dall'assetto genetico del singolo soggetto. Esiti terapeutici diversi sono in parte causati da differenze interindividuali nella farmacocinetica (assorbimento, distribuzione, metabolismo ed eliminazione del farmaco assunto) e/o da differenze interindividuali di tipo farmacodinamico, per es., diversa interazione del farmaco con il bersaglio terapeutico (recettore, canale ionico, enzima), in quanto esso può risultare leggermente diverso. La comprensione della risposta individuale a un farmaco permette di selezionare, tra tutti, i pazienti che rispondono, escludere dal trattamento quelli che poco rispondono o non rispondono affatto e quelli che hanno effetti collaterali eccessivi. In questo modo si ottiene una terapia efficace, selettiva e quindi 'intelligente'. Gli effetti farmacocinetici e farmacodinamici sono mediati da numerose proteine ed enzimi, ciascuno dei quali è il prodotto di uno specifico gene. Molti di questi geni sono polimorfici, presentano cioè in individui diversi variazioni di sequenza che frequentemente sono a carico di singole basi della catena polinucleotidica; questo tipo di polimorfismo viene perciò indicato con la sigla SNP (Single Nucleotide Polymorphism, Polimorfismo a singolo nucleotide). La presenza di SNP in un determinato gene può modificare la struttura, il livello di espressione o la funzionalità della proteina codificata. Qualora la proteina stessa sia implicata nella catena di eventi metabolici legati alla somministrazione di un farmaco, si potranno di conseguenza manifestare degli effetti sull'efficacia del trattamento terapeutico o sulla eventuale comparsa di reazioni indesiderate; qualora la proteina sia invece il bersaglio terapeutico del farmaco si può avere una diversa interazione con il farmaco stesso. La farmacogenetica è la disciplina che studia le relazioni tra la costituzione genetica di un individuo e la sua risposta a un trattamento farmacologico, con l'obiettivo di favorire un utilizzo più razionale e personalizzato dei farmaci, attraverso l'ottimizzazione della loro efficacia e la minimizzazione degli effetti avversi. Schematicamente, le differenze genetiche possono riguardare le molecole responsabili della cinetica dei farmaci nell'organismo e/o le molecole bersaglio dei farmaci. Tra i vari complessi processi che regolano sia la vita sia la permanenza di un farmaco all'interno dell'organismo (assorbimento, distribuzione, metabolismo ed escrezione), il metabolismo è quello più rilevante e più geneticamente determinato. Il metabolismo può trasformare il farmaco in una sostanza non più attiva o in una sostanza tossica, oppure trasformare una sostanza inattiva (profarmaco) in un farmaco. Il metabolismo avviene prevalentemente a livello epatico da parte di una serie di enzimi, tra i quali sono molto importanti i citocromi P450 (CYP450). Sono state descritte almeno 60 isoforme del CYP450, classificate in 17 famiglie, e per tutti questi citocromi sono state trovate varianti alleliche che correlano con la risposta individuale ai farmaci. Ogni isoforma del CYP450 ha una affinità per un dato tipo di farmaco e ciascuno di noi ha una particolare dotazione di CYP450, e di loro varianti alleliche (tab.). Si può affermare che ogni individuo ha una sua capacità metabolica che condiziona la risposta ai farmaci. Questo fatto risulta tanto più importante quanto più mista da un punto di vista genetico diviene la nostra società che è aperta a immigrazioni e complementazioni genetiche. I risultati di un esperimento, riportati nella fig. 2, rappresentano un esempio paradigmatico di questo fatto: è stata misurata la concentrazione plasmatica di nortriptilina, un farmaco antidepressivo e del suo metabolita inattivo in individui con diversi polimorfismi per il CYP2D6 (2D6), CYP che metabolizza i farmaci psicotropi; si è osservato che gli individui con poche copie di geni hanno una concentrazione di farmaco nel plasma molto più elevata, e per un tempo più lungo, di quella degli individui con molte copie. L'effetto del farmaco, quindi, che è quasi sempre proporzionale alla sua concentrazione plasmatica, sarà molto differente nei diversi individui. Mentre negli individui con una copia dell'enzima si può avere la piena efficacia o addirittura una tossicità da dose eccessiva di farmaco, nell'individuo con più copie dell'enzima il farmaco non produrrà alcun effetto. La conoscenza del patrimonio dei geni che codificano per il CYP450 porterà quindi a individuare meglio per ogni individuo la scelta del farmaco da somministrare, tra quelli aventi le stesse proprietà farmacologiche, e la dose utile per ogni individuo. In questo modo si avrà una terapia più efficace e con minori effetti collaterali. Anche nei geni che codificano per i bersagli dei farmaci (recettori, canali, trasportatori, enzimi) possono essere presenti polimorfismi che interferiscono con la risposta di queste proteine ai farmaci, ampliandola o diminuendola. Questi polimorfismi sono quasi sempre i responsabili della mancata azione dei farmaci in alcuni individui. Alcuni esempi di polimorfismi dei geni che codificano per alcuni bersagli farmacologici e del loro possibile effetto in clinica sono riportati nella tabella. È evidente che l'individuazione, prima del trattamento, dei portatori di polimorfismi che rendono un bersaglio farmacologico non responsivo oppure iper-responsivo a un farmaco eviterebbe il fallimento o le complicanze del trattamento. I progressi nella tecnologia molecolare hanno permesso di rendere l'analisi dei polimorfismi relativamente facile ed economica. Il conoscere questi dati permette di applicare ai singoli pazienti i farmaci che meglio corrispondono alla composizione molecolare dei loro enzimi metabolici e dei loro bersagli terapeutici, e attuare così una terapia personalizzata e individuale specifica, efficace e con il minimo di effetti collaterali.
Drug delivery
Il terzo aspetto che può rendere una terapia farmacologica 'intelligente' è quello che riguarda la possibilità di recapitare il farmaco nella sede nella quale agire, al tempo opportuno nonché con il dosaggio desiderato. Fino a qui abbiamo considerato che il trattamento con farmaci, anche i più selettivi, avvenga somministrando il farmaco per via generale. Questo, dopo essere stato assorbito, si distribuirà a tutti i tessuti per via ematica e raggiungerà i siti nei quali agire. È intuitivo pensare che se noi potessimo concentrare il farmaco nei punti dove deve agire (per es., nella cellula tumorale invece che in quella sana, in alcune cellule nervose rispetto ad altre, nel fegato piuttosto che nel rene ecc.), otterremmo una maggior efficacia e specificità. La ricerca in questo settore ha fatto notevoli passi avanti, cercando di incapsulare il farmaco in micro- o nanostrutture che possano avere al loro interno il farmaco e alla loro superficie molecole (anticorpi o ligandi specifici) capaci di riconoscere certi tipi cellulari. In questo modo il farmaco è portato in vicinanza del bersaglio dove potrà essere liberato, e raggiungere concentrazioni elevate rispetto ai tessuti vicini. I materiali utilizzati possono essere lipidi più o meno complessi (liposomi) (Meyer 2004), polimeri di polianidridi, poliesteri, poliuretani, eteropolimeri tra questi composti e zuccheri, idrogel (Torchilin 2005; Hughes 2005). Oltre a fornire un trasporto all'indirizzo preciso, molti di questi materiali si prestano anche a rilasciare in modo controllato, nel tempo e nello spazio, il farmaco contenuto. Si può pensare a materiali che vengono assorbiti e si disgregano con tempi diversi, o a materiali che rilasciano il contenuto in base alle condizioni esterne nelle quali si trovano (per es., cariche elettriche, campi magnetici, pH). Consideriamo, a titolo di esempio, il trattamento con un farmaco antinfiammatorio. Se esso si trova incapsulato in un sistema che si disintegra in ambiente acido oppure a una temperatura superiore a quella media corporea (situazioni ambientali tipiche di un tessuto infiammato) il suo rilascio avverrà prevalentemente in questi tessuti, nei quali raggiungerà una buona concentrazione. Oppure si può cercare di veicolare farmaci attivi contro batteri e parassiti nelle cellule infettate attraverso la loro inclusione in particolari nanostrutture, come è stato fatto per l'anfotericina B nella leishmaniosi viscerale. Con questi sistemi si possono rendere vecchi farmaci, non troppo efficienti e specifici, più attivi, più selettivi e diminuire i loro effetti collaterali.
L'ultimo aspetto da considerare è quello che concerne la possibilità di somministrare il farmaco alle dosi e alle concentrazioni necessarie. Esistono dei farmaci che per la loro ottimale azione debbono essere somministrati solo nel momento giusto e nelle concentrazioni necessarie. Si prenda in esame, per es., l'insulina: essa fisiologicamente viene secreta in concentrazioni opportune solo quando vi sia una certa concentrazione plasmatica di glucosio. Nel diabetico incapace di secernere insulina il trattamento con insulina esogena dovrebbe mimare ciò che avviene fisiologicamente, mentre solitamente l'insulina viene somministrata in modo molto approssimato, con limitata efficacia nel controllo del diabete. Per questo si è pensato a una somministrazione che venga fornita da un sistema capace di 'sentire' i livelli di glucosio nel plasma e di erogare l'opportuna quantità di insulina.
Queste piccole macchine, altamente miniaturizzate ed efficienti, permettono un trattamento molto simile al fisiologico e, quindi, teoricamente molto più efficace a lungo termine rispetto ai trattamenti insulinici praticati. Procedimenti analoghi sono allo studio per altri farmaci che debbano essere somministrati al bisogno. Quest'ultimo aspetto della farmacologia, che si è potuto sviluppare grazie ai progressi della chimica, dei nanomateriali e delle nanomacchine, permette di trasformare vecchi farmaci in farmaci più selettivi e 'intelligenti', conservando quindi nell'armamentario terapeutico farmaci di comprovata attività e dei quali si conoscono pregi e difetti, con innovazioni relativamente poco costose.In conclusione, la scoperta di f. i. non è che la logica conseguenza della maggiore conoscenza che abbiamo dell'organismo umano, delle più importanti malattie che ci affliggono, delle scoperte nel campo delle biotecnologie e delle nanotecnologie e dei progressi della chimica farmaceutica. Queste conoscenze ci permettono di selezionare i bersagli essenziali e specifici contro cui indirizzare i farmaci, selezionare i pazienti in relazione alle caratteristiche della malattia e della loro costituzione genetica, indirizzare i farmaci nel sito specifico dove devono agire alla concentrazione giusta, ma anche nel tempo e per il tempo opportuno. Combinando tutti questi accorgimenti si tende a ottenere una terapia personalizzata per ogni individuo, in modo da sfruttare al massimo le caratteristiche terapeutiche del nostro armamentario farmacologico.
bibliografia
J. Dancey, E.A. Sausville, Issues and progress with protein kinase inhibitors for cancer treatment, in Nature reviews. Drug discovery, 2003, 2, pp. 296-313.
O. Kayse, C. Olbrich, S.L. Croft, A.F. Kiderlen, Formulation and biopharmaceutical issues in the development of drug delivery systems for antiparasitic drugs, in Parasitology research, 2003, 90, suppl. 2, pp. 63-70.
P. Workman, The opportunities and challenges of personalized genome-based molecular therapies for cancer: targets, technologies, and molecular chaperones, in Cancer chemotherapy and pharmacology, 2003, 52, suppl. 1, pp. 45-56.
R.S. Herbst, M. Fukuoka, J. Baselga et al., Gefitinib-a novel targeted approach to treating cancer, in Nature reviews. Cancer, 2004, 4, pp. 956-65.
U.A. Meyer, Pharmacogenetics - five decades of therapeutic lessons from genetic diversity, in Nature reviews. Genetics, 2004, 5, pp. 669-76.
A.D. Roses, Pharmacogenetics and drug development: the path to safer and more effective drugs, in Nature reviews. genetics, 2004, 5, pp. 645-56.
G.A Hughes, Nanostructure-mediated drug delivery, in Disease-a-month: DM, 2005, 51, 6, pp. 342-61.
N.E. Hynes, H.A. Lane, ERBB receptors and cancer: the complexity of targeted inhibitors, in Nature reviews. Cancer, 2005, 5, pp. 341-54.
D.S. Krause, R.A. Van Etten, Mechanisms of Disease: tyrosine kinases as targets for cancer therapy, in The New England journal of medicine, 2005, 353, pp. 172-87.
V.P. Torchilin, Recent advances with liposomes as pharmaceutical carriers, in Nature reviews. Drug discovery, 2005, 4, pp. 145-59.