
Fosforilazione ossidativa
Fosforilazione ossidativa
di Edward Ch. Slater
SOMMARIO: 1. Premessa storica. □ 2. Reversibilità della fosforilazione ossidativa. □ 3. Stato attuale delle ricerche sul meccanismo della fosforilazione ossidativa: a) catena trasportatrice di elettroni; b) meccanismo per la sintesi dell'ATP; c) meccanismo di accoppiamento tra catena trasportatrice di elettroni e apparato per la sintesi di ATP. □ 4. Ipotesi chemiosmotica. □ 5. Ipotesi conformazionale. □ 6. Aspetti comuni delle ipotesi proposte circa il meccanismo della fosforilazione ossidativa. □ 7. Nota finale. Un'ipotesi unitaria sul meccanismo della fosforilazione ossidativa. □ Bibliografia.
1. Premessa storica.
Con il termine ‛fosforilazione ossidativa' si indica una reazione di fosforilazione legata a un'ossidazione. In tutti i casi studiati la reazione di fosforilazione è, o può essere, connessa alla fosforilazione dell'ADP con fosfato inorganico (Pi). Formalmente la fosforilazione ossidativa può esprimersi con l'equazione
AH2 + B + ADP + Pi⇄A + BH2 + ATP + H2O.
che rappresenta la somma di due reazioni essenzialmente irreversibili:
AH2 + B → A + BH2
ADP + Pi Á ATP + H2O.
La prima reazione deve liberare energia sufficiente per spostare la seconda verso destra.
Le prime informazioni sulla fosforilazione ossidativa ci sono venute dallo studio della trasformazione del glucosio in alcool etilico da parte del lievito o in acido lattico da parte del muscolo. La presenza indispensabile di quantità stechiometriche di fosfato per la fermentazione da parte di estratti di lievito fu dimostrata nel 1906 da A. Harden e W. J. Young, che identificarono il composto intermedio formatosi come fruttosiodifosfato e stabilirono che la reazione nel suo complesso poteva essere descritta dall'equazione
2 glucosio + 2 Pi → 2 etanolo + 2 C02 + 1 fruttosiodifosfato + 2 H2O.
In altre parole, una molecola di glucosio verrebbe fosforilata dal fosfato inorganico a fruttosiodifosfato, mentre una seconda molecola sarebbe demolita con formazione di etanolo e CO2.
Gli studi di C Neuberg, G. Embden, J. Parnas, O. Meyerhof, J. Needham e O. Warburg negli anni venti e trenta consentirono di scoprire che l'energia necessaria per la reazione di fosforilazione deriva dalla reazione ossidativa che avviene durante la fermentazione e la glicolisi, e precisamente dalla ossidazione della fosfogliceraldeide da parte del piruvato o dell'acetaldeide
fosfogliceraldeide + piruvato (o acetaldeide) + H2O → fosfoglicerato + lattato (o etanolo).
e che l'energia di questa reazione è immagazzinata sotto forma di composti fosforilati ad alto contenuto di energia. Nel 1939 Warburg dimostrò che il primo a formarsi di tali composti altamente energetici è l'acido 1,3-difosfoglicerico che, in presenza di un enzima specifico, reagisce con l'ADP per formare ATP. L'equazi9ne su riportata, con la parallela fosforilazione di ADP, può essere ora formulata come la somma di tre equazioni e cioè:
fosfogliceraldeide + NAD+ + Pi ⇄ acido 1,3-difosfoglicerico + NADH + H+
acido 1,3-difosfoglicerico + ADP ⇄ acido 3-fosfoglicerico + ATP
NADH + H+ + piruvato (o acetaldeide) ⇄ NAD+ + lattato (o etanolo).
L'ATP, scoperto da K. Lohmann, occupa una posizione centrale nel metabolismo cellulare, poiché il suo gruppo fosforico terminale può essere rimosso in modo da conservare l'energia libera che sarebbe dissipata nella semplice idrolisi dell'ATP. Dobbiamo principalmente a F. Lipmann lo sviluppo delle conoscenze circa il ruolo dei composti fosforici ad alta energia.
Che la respirazione potesse anche fornire l'energia per la sintesi dell'ATP era stato suggerito dalla scoperta di Meyerhof, nel 1920, secondo cui l'ossigeno consumato durante il periodo di recupero dopo la contrazione muscolare corrisponde solo a un quinto dell'acido lattico formatosi durante la contrazione e inoltre l'energia prodotta dall'ossidazione di questo quinto può essere usata per convertire il rimanente acido lattico in glicogeno. Negli anni venti P. Eggleton e G. P. Eggleton da un lato e C. H. Fiske e Y. Subbarov dall'altro dimostrarono che durante il recupero in presenza di ossigeno viene risintetizzata rapidamente la fosfocreatina.
La prima dimostrazione della sintesi di ATP accoppiata alla respirazione fu ottenuta da W. A. Engelhardt nel 1930: egli scopri che negli eritrociti di coniglio, che non sono nucleati e non respirano, si forma fosfato inorganico se si inibisce la glicolisi con il fluoruro, mentre non ha effetto l'inibizione col cianuro, che blocca la respirazione. Invece negli eritrociti di piccione gli inibitori della respirazione (cianuro, uretano o monossido di carbonio) danno luogo alla produzione di fosfato. La quantità di fosforo inorganico trovata da Engelhardt corrispondeva precisamente alla quantità di ‛fosforo a 7 minuti' (ATP) scomparsa. Egli poté dimostrare che dopo l'allontanamento del cianuro per lavaggio avveniva nuovamente la sintesi di fosforo acido- labile. Da ciò concluse che la scissione dell'ATP può avvenire sia in assenza sia in presenza di inibitori della respirazione, ma che in loro assenza la scissione è compensata da risintesi.
Le ricerche di Engelhardt non ebbero grande risonanza quando furono pubblicate e l'interesse per la ‛fosforilazione ossidativa', come si chiamò da allora tale processo, cominciò dalle ricerche di H. Kalckar negli anni 1937-1939, condotte con omogenati acellulari di rene e di altri tessuti. Kalckar dimostrò che quando tali omogenati catalizzano l'ossidazione degli acidi citrico, glutammico, fumarico o malico, avviene la fosforilazione del glucosio, del glicerolo o dell'AMP.
Circa nello stesso periodo Lipmann vide che l'ossidazione del piruvato da parte del Bacterium delbruckii è accoppiata con la fosforilazione.
Harden e Young nei loro primi studi sul ruolo del fosfato nella fermentazione stabilirono che l'eccesso di CO2 liberato per aggiunta di fosfato a un estratto di lievito è equimolare al fosfato aggiuntò; la velocità di evoluzione del C02 diminuisce bruscamente quando tutto il fosfato è esterificato, e una seconda aggiunta di fosfato ristabilisce la fermentazione. Con questi esperimenti, nel dimostrare per la prima volta il controllo del metabolismo da parte del fosfato, essi stabilivano la stechiometria della fosforilazione associata alla glicolisi e alla fermentazione. Molto più difficile si rivelò lo stabilire la stechiometria della fosforilazione associata alla respirazione.
Nel 1939-1940 due gruppi di ricercatori, guidati da S. Ochoa e da V. A. Belitser e T. E. Tsibakova, fecero l'importante osservazione che più di un atomo di P veniva esterificato per ogni atomo di ossigeno (cioè due equivalenti di ossidazione) consumato da preparati che respiravano, in condizioni di assenza di glicolisi. Entrambi i gruppi ne conclusero che la fosforilazione debba avvenire non solo quando il substrato è deidrogenato - come nella reazione ossidativa della glicolisi o della fermentazione ma anche durante l'ulteriore passaggio di atomi di idrogeno (o di elettroni) lungo la catena respiratoria fino all'ossigeno. Quindi questi autori per primi ipotizzarono il processo ora noto come fosforilazione a livello della catena respiratoria. Tuttavia non risultò subito chiaro che il nuovo tipo di fosforilazione era differente da quella implicata nella glicolisi, in quanto era allora in auge l'ipotesi di A. Szent-Györgyi secondo cui sarebbero state partecipi della catena respiratoria le coppie succinato-fumarato e malato-ossalacetato. La scoperta che il NADH formato dalla deidrogenazione della maggior parte dei substrati veniva ossidato dalla catena respiratoria formata da flavoproteine e dal sistema dei citocromi, in assenza di componenti a basso peso molecolare, portò alla convinzione che i coenzimi nicotinammidici, le flavine o gli emi dovevano essere più intimamente connessi con il processo fosforilativo. Tuttavia una dimostrazione diretta della fosforilazione a livello della catena respiratoria non si ebbe fino al 1949, quando A. L. Lehninger scoprì che la sintesi di ATP era connessa con l'ossidazione del NADH da parte dei mitocondri, in assenza di altri substrati.
Divenne allora importante stabilire più precisamente la stechiometria del processo, cioè determinare esattamente quanti atomi di fosforo sono esterificati per ogni molecola di substrato ossidato. Il primo tentativo di Ochoa in questo senso, nel 1943, apri un capitolo molto controverso in tale campo.
L'origine della controversia risiede nelle difficoltà tecniche: il problema è costituito dalla misura di n nella reazione

dove SH2 è un substrato ossidabile.
Le preparazioni enzimatiche usate dai primi ricercatori contenevano esochinasi e fosfofruttochinasi, che catalizza- no la fosforilazione da parte dell'ATP del glucosio o del fruttosio contenuto nella miscela di reazione:
n ATP + n glucosio → n ADP + n glucosio-6-fosfato.
La reazione complessiva che ne risulta è
SH2 + O + n glucosio + n Pi → S + n EMP + (n+1) H2O.
in cui EMP è l'abbreviazione di esosomonofosfato, di solito una miscela di glucosio-6-fosfato e fruttosio-6-fosfato formata per azione della glucosio-6-fosfatoisomerasi presente; in questa reazione non appaiono più i nucleotidi adenosinici, cioè essi si comportano da catalizzatori della reazione stessa. Tuttavia le preparazioni usate contenevano anche ATP-asi, che idrolizza l'estere fosforico formatosi rigenerando, nel mezzo di reazione, il fosfato inorganico:
n ATP + n H2O → n ADP + n Pi.
Da ciò consegue che la misura della scomparsa del Pi non sempre riflette accuratamente il valore di n. Infatti
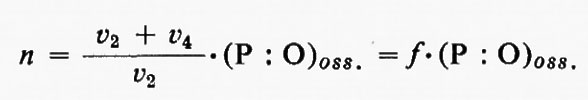
in cui v2 e v4 sono le velocità delle reazioni catalizzate rispettivamente dalla esochinasi e dalla ATP-asi.
Ochoa si rese conto di questa difficoltà e determinò il valore di f misurando il rapporto Pilattato nella reazione di fosforilazione glicolitica condotta nelle stesse condizioni della reazione di fosforilazione ossidativa. Egli suppose che l'ATP formatosi nella glicolisi possa suddividersi tra esochinasi e ATP-asi nello stesso modo in cui si suddivide l'ATP formato nella fosforilazione ossidativa a livello della catena respiratoria. In tal modo egli calcolò che il vero rapporto P : O (cioè n) per l'ossidazione dell'acido piruvico in omogenati di cuore o di cervello è uguale a 3. Pur incontrando questo risultato, a quel tempo, considerevoli resistenze, esso costituiva un elegante e obiettivo tentativo di superare una difficoltà reale e diede risultati molto vicini a quelli ora generalmente accettati.
Nel 1948-1949 furono introdotte nella tecnica sperimentale due innovazioni che hanno grandemente diminuito l'importanza ditale causa di errore. La prima era costituita dall'aggiunta alla miscela di reazione di un grande eccesso di esochinasi purificata dal lievito, di cui si poté allora disporre. In tal modo la reazione dell'ATP con il glucosio è talmente favorita da rendere insignificanti le perdite dovute all'ATP-asi. La seconda consisteva nell'uso di ‛particelle di tessuto lavate', composte prevalentemente di mitocondri, invece che di omogenati grezzi, come sistema enzimatico catalizzante la fosforilazione ossidativa. In tal modo veniva allontanata dal sistema una certa quantità di ATP-asi; ulteriori progressi nella tecnica di isolamento dei mitocondri, specialmente quelli dovuti a H. A. Lardy e J. W. Wellman, portarono alla virtuale abolizione dell'idrolisi dell'ATP come causa di errore nella misura del rapporto P : O.
Tuttavia nello stesso tempo si constatò che il rapporto P : O determinato sperimentalmente variava alquanto da preparazione a preparazione e che esso era tanto più alto quanto maggiore era la cura nel preparare i mitocondri. Ciò è dovuto alle reazioni da ‛perdita', anteriori alla formazione di ATP, determinate dal danneggiamento dei mitocondri. Per un certo periodo divenne abituale accettare come significativi solo i rapporti ‛migliori' (cioè i più alti). Inevitabilmente ciò condusse ad accettare acriticamente le condizioni sperimentali che davano alti rapporti P : O, per cui vennero pubblicati rapporti perfino di 6 per l'ossidazione di substrati connessi col NAD. Oggi però si riconosce che l'esatto rapporto per tali substrati è 3, mentre è 2 per i substrati a maggiore potenziale, come il succinato o il glicerolo-1-fosfato. I substrati a basso potenziale, come il 2-chetoglutarato e il piruvato, producono una molecola di ATP in più, la cui sintesi è legata alla riduzione del NAD+ da parte dei chetoacidi (nel caso del piruvato, ciò avviene solo in alcuni microrganismi).
Si ammette generalmente che un rapporto P : O di 3 nel caso di substrati connessi al NAD+ implica la presenza di tre siti di fosforilazione nella catena di reazioni di trasferimento degli elettroni che intervengono nell'ossidazione del NADH da parte dell'ossigeno: tali siti sono indicati rispettivamente come siti I, II e III. L'ulteriore fosforilazione legata all'ossidazione del 2-chetoglutarato appartiene alla categoria delle fosforilazioni ‛a livello del substrato'.
Si è cercato a lungo, al principio degli anni cinquanta, soprattutto da parte di Lehninger e Slater, di localizzare questi siti di fosforilazione nella catena respiratoria (v. bioenergetica). Oggi in genere si ritiene che i siti I, II e III si trovino tra il NADH e l'ubichinone, tra l'ubichinolo e il citocromo c e tra il citocromo c e l'ossigeno, rispettivamente, secondo lo schema
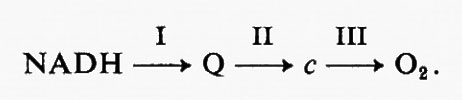
Agli inizi degli anni cinquanta, dopo la scoperta del coenzima A e della partecipazione dei tioesteri come composti intermedi sia nella reazione catalizzata dalla fosfogliceraldeidedeidrogenasi sia nella fosforilazione a livello dei substrati associata all'ossidazione di un a-chetoacido, sembrò ovvio ipotizzare un meccanismo simile per i tre siti di fosforilazione nella catena respiratoria. La proposta formale di questo meccanismo fu avanzata per la prima volta da Slater nel 1953. Il suo meccanismo era cosi indicato:
AH2 + B + C ⇄ A ~ C + BH2
A ~ C + ADP + P1 ⇄ A + C + ATP,
in cui AH2 e B rappresentano due costituenti contigui della catena di trasferimento di elettroni, mentre C è un legante. L'oligomicina inibisce la reazione del composto A ~ C con l'ADP e il fosfato, ma non ha effetto sulla formazione di A ~ C connessa al trasferimento di elettroni. D'altra parte un agente disaccoppiante della fosforilazione ossidativa indurrebbe la scissione di A C e quindi promuoverebbe l'idrolisi dell'ATP aggiunto dall'esterno.
Una caratteristica essenziale di questa ipotesi, che la differenzia da una precedente di Lipmann, per il resto analoga, è che la reazione primaria di conservazione energetica non coinvolge il fosfato inorganico. Ciò fu stabilito subito dopo il 1960, principalmente grazie alle ricerche svolte nei laboratori di L. Ernster, B. Chance ed E. Ch. Slater. In effetti oggi si sa che la sintesi di ATP da ADP e P1 non è che uno dei modi di conservare l'energia liberatasi per il trasferimento di elettroni all'interno dei mitocondri. La conservazione di energia legata al trasferimento di elettroni si trova non solo nella respirazione intracellulare degli animali, delle piante e dei microrganismi, ma anche nella fotosintesi, in cui gli elettroni derivano da donatori formatisi in una reazione fotochimica. Le proprietà dei sistemi trasduttori di energia nella respirazione e nella fotosintesi sono però talmente simili, che è molto probabile che in entrambi i casi operi lo stesso meccanismo, per cui lo sperimentatore può scegliere il sistema più appropriato al disegno sperimentale progettato, con buone probabilità di poter estendere i risultati dal sistema in esame all'altro. In tutti i casi il passaggio di elettroni può essere utilizzato non solo per la sintesi di ATP a partire da ADP e Pi contro un gradiente di potenziale chimico (cioè a concentrazioni di ATP, ADP e Pi molto lontane dall'equilibrio termodinamico), ma può anche essere utilizzato per la riduzione del NADP+ da parte del NADH, anche in questo caso contro un gradiente di potenziale chimico. Può inoltre essere utilizzato per il trasporto di cationi dall'esterno all'interno dei mitocondri contro un gradiente di concentrazione, ma non in direzione inversa: in effetti il movimento di cationi nel senso del gradiente di concentrazione da dentro a fuori può essere utilizzato per formare ATP.
La membrana interna del mitocondrio e le altre membrane trasduttrici di energia catalizzano, in conclusione, quattro diverse reazioni, che possono generare o utilizzare energia, in equilibrio l'una con l'altra. Quindi esse devono avere una connessione, indicata nella fig. 1 col nome di ‛pressione energetica'.
L'aver compreso, nel decennio successivo al 1960, che la sintesi dell'ATP non è il meccanismo primario per la conservazione dell'energia e che i mitocondri e gli altri organelli trasduttori di energia possono accoppiare la respirazione con reazioni endoergoniche a prescindere dalla presenza di fosfato inorganico fece divergere per qualche tempo l'interesse principale dalla sintesi dell'ATP. Oggi si segnala un riflusso verso questo argomento, in quanto è stato chiarito che la funzione fisiologica importante, specie nel mitocondrio, è appunto la sintesi dell'ATP, che viene poi convogliato verso compartimenti extramitocondriali che abbisognano di energia. Ciò fu chiaramente dimostrato da Harary e Slater per cellule contrattili isolate di cuore.
Il tentativo di confermare una delle implicazioni della cosiddetta ipotesi chimica della fosforilazione ossidativa, e cioè la conservazione dell'energia senza sintesi di ATP, ha stimolato un'attiva ricerca dei composti intermedi ad alta energia (A ~ C) supposti in tale ipotesi. Molti ritennero di aver dimostrato la presenza di tali composti intermedi e anche di averli isolati, ma in ogni caso le loro conclusioni si dimostrarono, per una ragione o per l'altra, prive di fondamento.
Implicita nell'ipotesi chimica formulata da Slater è la formazione di un legame covalente tra un componente (A) della catena trasportatrice di elettroni e un legante (C). Nel 1964 P. D. Boyer avanzò la supposizione che l'energia potesse essere convogliata in un cambiamento conformazionale delle proteine trasportatrici di elettroni, senza formazione di legami covalenti; questa variante all'ipotesi chimica, spesso indicata con il nome di ipotesi conformazionale, viene oggi accettata da molti ricercatori, compreso l'autore di questa rassegna.
Tuttavia, mentre molti erano impegnati in una febbrile caccia ai composti intermedi ad alta energia, P. Mitchell affermò che essi, come la pietra filosofale, potevano non esistere; egli formulò l'ipotesi che il processo primario di conservazione dell'energia nella catena trasportatrice di elettroni fosse lo stabilirsi di un gradiente di protoni che può servire a sintetizzare ATP invertendo il flusso di una ATP-asi traslocatrice di protoni. Tale ingegnosa e stimolante ipotesi ha avuto un'influenza dominante sugli sviluppi delle conoscenze nell'ultimo decennio.
Altre due tappe importanti tra la fine degli anni cinquanta e l'inizio degli anni sessanta sono rappresentate dal lungo e difficile tentativo di frazionare la catena trasportatrice di elettroni e il sistema che sintetizza l'ATP, rispettivamente da parte di D. E. Green e di E. Racker. A chi aveva sperimentato quanto la fosforilazione ossidativa fosse sensibile a minime lesioni della membrana mitocondriale, poté sembrare vano il tentativo di scomporre quel sistema complesso come era stato fatto con ottimi risultati per altri sistemi biochimici, per es. nello studio del meccanismo della glicolisi oppure della fosforilazione a livello del substrato. Tuttavia Green è riuscito a suddividere la catena trasportatrice di elettroni in tre frammenti corrispondenti ai tre siti di fosforilazione e cioè la NADH-Q-reduttasi (complesso I), la QH2-citocromo-c-reduttasi (complesso III) e la citocromo-c-ossidasi (complesso IV), oltre a un frammento specificamente associato all'ossidazione del succinato (complesso II). Racker, dal canto suo, ha potuto dividere i mitocondri in particelle trasportatrici di elettroni e fattori solubili, ambedue incapaci di effettuare isolatamente la fosforilazione ossidativa, che invece possono effettuare se ricombinati. Negli ultimi anni egli è stato in grado di ricostituire specificamente i siti I e III della fosforilazione adoperando rispettivamente la NADH-Q-reduttasi e la citocromo-c-ossidasi insieme con i fattori solubili. Uno dei fattori fosforilanti (F1) è la ATP-asi mitocondriale, che nel sistema completo costituisce il passaggio finale per la sintesi dell'ATP.
Il grande contributo di B. Chance alla comprensione del meccanismo della fosforilazione ossidativa è consistito, durante questo periodo, nel dimostrare l'importanza del controllo della respirazione, cioè il controllo del trasferimento di elettroni, da parte della reazione di fosforilazione.
2. Reversibilità della fosforilazione ossidativa.
Prima di discutere lo stato attuale del problema riguardante il meccanismo della fosforilazione ossidativa bisogna accennarne un aspetto, e precisamente la sua reversibilità globale. Si considera generalmente la riduzione dell' ossigeno ad acqua da parte dei sistemi biologici come irreversibile in assenza di energia luminosa e di un sistema fotochimico, presente nelle piante verdi e nei microrganismi fotosintetici, capace di utilizzare questa energia luminosa per scindere l'acqua in ossigeno ed equivalenti di riduzione.
Nei suoi primi studi Chance stabili che i ‛punti di incrocio', cioè i punti della catena trasportatrice di elettroni rispetto a cui i singoli trasportatori che si trovano più verso il substrato diventano maggiormente ossidati per aggiunta di ADP e fosfato, mentre i trasportatori che si trovano più verso l'ossigeno diventano maggiormente ridotti, sono posti tra i citocromi c ed a, ma non tra il citocromo a e l'ossigeno. Questo fatto sembrava indicare che il segmento compreso tra il citocromo a e l'ossigeno non fosse sotto il controllo della fosforilazione ossidativa o, in altre parole, che la reazione con l'ossigeno fosse essenzialmente reversibile.
Recenti acquisizioni, tuttavia, hanno alquanto modificato questo punto di vista: in primo luogo punti di incrocio sono stati trovati, in altri sistemi, tra il citocromo a e l'ossigeno; inoltre è stato chiaramente dimostrato che mitocondri respiranti possono sintetizzare ATP contro circa 16 kcal/mole di potenziale fosfato definito dall'equazione

(in cui −ΔG0 è l'energia libera standard di idrolisi dell'ATP nelle condizioni sperimentali considerate e i termini in parentesi quadre rappresentano le attività termodinamiche di quelle specie).
Il valore di 16 kcal/mole rappresenta circa il 95% di quello previsto ammettendo che i mitocondri catalizzino l'equilibrio reversibile:
NADH + H+ + ½O2 + 3 ADP + 3 Pi ⇄
⇄ NAD+ +H2O + 3ATP +3H2O.
Infine Bienfait e Slater hanno messo in evidenza l'ossidazione dell'acqua, indotta dall'ATP, da parte dell'acido ossalacetico con liberazione di ossigeno molecolare che veniva intrappolato per l'ossidazione di butanolo, marcato con 14C, ad acido butirrico, catalizzata da particelle di Acetobacter mesoxydans aggiunte al sistema; venivano anche aggiunti fosfoenolpiruvato e piruvatochinasi per mantenere elevato il potenziale fosfato. La formazione di acido butirrico indotta dall'ATP si è dimostrata sensibile all'atractiloside, un inibitore dell'ingresso dell'ATP nei mitocondri, e all'oligomicina o ad agenti disaccoppianti che inibiscono la sua utilizzazione, e anche al cianuro o al solfuro che inibiscono la reazione con l'ossigeno; inoltre la reazione dipende dalla quantità di mitocondri, di piruvatochinasi e di particelle batteriche.
In tal modo è possibile dimostrare sperimentalmente la reversibilità della reazione globale sopra riportata, benché l'equilibrio di essa sia spostato completamente a destra.
3. Stato attuale delle ricerche sul meccanismo della fosforilazione ossidativa.
Le ricerche sul meccanismo della fosforilazione ossidativa stanno procedendo su tre fronti: a) natura della catena trasportatrice di elettroni; b) natura del sistema per la sintesi dell'ATP; c) meccanismo di accoppiamento tra catena trasportatrice di elettroni e sistema di sintesi dell'ATP.
a) Catena trasportatrice di elettroni.
Nel 1950 fu possibile completare la conoscenza delle catene attraverso cui si ossidano il NADH (allora detto CoIH2) e il substrato ad alta energia, il succinato (v. fig. 2); esse comprendono i quattro citocromi di D. Keilin, la diaforasi di H. K. von Euler, l'Atmungsferment di Warburg (identico al citocromo a3 di Keilin) e un fattore non identificato introdotto per spiegare gli effetti di alcuni inibitori. Tuttavia, anche se tale fattore non è stato ancora identificato, a partire dal 1950 è stato possibile aggiungere all'elenco molti altri componenti in grado di accettare elettroni dal NADH o dal succinato. Green e Morton hanno trovato grandi quantità di un chinone liposolubile, l'ubichinone. L'affinarsi delle tecniche spettroscopiche, specialmente ad opera di Chance, ha portato alla scoperta di nuovi citocromi e gli studi di H. Beinert sulle proprietà magnetiche delle membrane trasduttrici di energia hanno rivelato la presenza di rame specificamente legato a enzimi e di altri due tipi di ferro oltre a quello dei citocromi. Sensibili metodi per lo studio della risonanza paramagnetica alla temperatura di pochi gradi Kelvin, introdotti di recente nei laboratori impegnati nello studio della bioenergetica, hanno portato alla scoperta di una serie di centri paramagnetici distinti.

La tab. I mostra i differenti tipi di accettori di elettroni e il numero di ciascuno di essi presente nei mitocondri; molti di essi sono stati trovati anche in altre membrane trasduttrici di energia. Benché sia probabile che ciascuno dei circa 30 centri elencati nella tab. I abbia un ruolo, non sembra ragionevole ipotizzare che tutti accettino elettroni in modo sequenziale in una catena respiratoria lineare o con diramazioni semplici come rappresentato nella fig. 2. Uno dei grandi problemi della bioenergetica è di determinare il significato biologico di questa molteplicità di accettori di elettroni. E possibile che almeno alcuni di essi abbiano una funzione di controllo: è stato ipotizzato, per esempio, che il passaggio di elettroni tra i citocromi b e c1 sia regolato dallo stato ossidoriduttivo di una ferro-solfoproteina.
Green ha dimostrato che i trasportatori di elettroni interessati all'ossidazione del NADH sono presenti in tre componenti ad alto peso molecolare che per aggiunta di due sostanze a basso peso molecolare, l'ubichinone (coenzima Q) e il citocromo c, possono ricostituire un sistema capace di ossidare il NADH per mezzo dell'ossigeno (v. fig. 3); egli ha chiamato ‛complessi' (I, III e IV) tali componenti, che però possono anche essere considerati ‛molecole' proteiche formate da più catene polipeptidiche, alcune delle quali contengono accettori di elettroni come gruppi prostetici. Per es. il complesso IV, detto anche citocromo-e-ossidasi, contiene due gruppi eme a e due atomi di rame per molecola, mentre la elettroforesi su gel in presenza di dodecilsolfato dimostra la presenza di almeno 6 differenti catene polipeptidiche. Il complesso III, detto diidroubichinone-citocromo-c-reduttasi, contiene almeno 2 e forse anche 4 differenti citocromi b, probabilmente 2 gruppi ferro-zolfo, il citocromo c1 e almeno 2 o 3 atomi di ferro ad alto spin che differiscono considerevolmente per potenziale redox.
b) Meccanismo per la sintesi dell'ATP.
Spetta a Racker il merito di aver isolato l'enzima, detto F1 o ATP-asi mitocondriale, responsabile della sintesi dell'ATP. Questo enzima, che ha un peso molecolare dello stesso ordine di quello dei complessi di Green (le aree dei vari componenti disegnati nella fig. 3 sono proporzionali ai pesi molecolari), è unito a un frammento di membrana indicato come CF0: tale unione avviene tramite una proteina a basso peso molecolare detta OSCP (proteina che conferisce la sensibilità all'oligomicina). Il sistema F1-OSCP-CF0 può essere separato dalle proteine trasportatrici di elettroni: questo cosiddetto complesso ATP-asico catalizza non solo l'idrolisi dell'ATP, ma anche l'incorporazione di fosfato inorganico radioattivo nell'ATP. Quindi, benché, come ci si può attendere, questo complesso non sia capace di operare la sintesi ‛netta' di ATP in assenza di una fonte esterna di energia, esso rappresenta una parte del congegno di sintesi dell'ATP. Tale congegno è presente anche nei promitocondri, privi di citocromi.
Sebbene di quando in quando appaiano nella letteratura articoli che descrivono la sintesi di ATP legata a trasferimento di elettroni da parte di trasportatori di elettroni purificati in assenza di ATP-asi, la partecipazione di questo enzima, almeno nel sistema intatto, è estremamente probabile; basta infatti osservare che: 1) è possibile ricostituire la fosforilazione ossidativa aggiungendo ATP-asi purificata a particelle submitocondriali private di tale enzima; 2) la fosforilazione ossidativa viene inibita per azione di un anticorpo anti-ATP-asi; 3) sono stati ottenuti mutanti di Escherichia coli che mancano di ATP-asi oppure hanno una ATP-asi modificata o un diverso sito di legame per l'ATPasi. Questi mutanti privi di ATP-asi sono incapaci di catalizzare la transidrogenazione ATP-dipendente o la fosforilazione ossidativa; la prima viene ripristinata aggiungendo alle particelle mutanti ATP-asi purificata da ceppi che la contengono.
Sembra del tutto evidente, quindi, che l'ATP-asi, la quale una volta isolata catalizza l'idrolisi dell'ATP ad ADP e fosfato, sia in grado, quando è connessa con la membrana mitocondriale, di sintetizzare ATP da ADP e fosfato. Il problema del meccanismo di fosforilazione ossidativa può allora essere riformulato in questi termini: come può il passaggio di elettroni attraverso i siti accettori di elettroni, per lo più costituiti da metalli, indurre l'ATP-asi a produrre ATP?
c) Meccanismo di accoppiamento tra catena trasportatrice di elettroni e apparato per la sintesi di ATP.
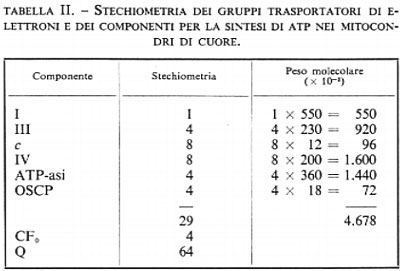
Prima di esaminare il possibile meccanismo di accoppiamento è importante prendere in considerazione le concentrazioni relative dei trasportatori di elettroni e dei componenti legati alla sintesi dell'ATP. Ciò è reso possibile dalla presenza di specifici e forti siti di legame per leganti su ciascuna delle macromolecole proteiche coinvolte nella fosforilazione ossidativa: l'aurovertina si lega all'ATP-asi, l'oligomicina al sito di legame per l'ATP-asi sulla membrana, l'antimicina alla diidroubichinone-citocromo-c-reduttasi, il rotenone alla NADH-ubichinone-reduttasi e il cianuro alla citocromo-c-ossidasi. Si è visto che nei mitoconari di fegato di ratto e in quelli di cuore di ratto vi è un sito di legame per l'oligomicina (sul CF0) e uno tenace per l'aurovertina (sull'ATP-asi) per ogni sito di legame dell'antimicina (sulla diidroubichinone-citocromo-c-reduttasi, complesso III). Dati spettrofotometrici dimostrano esservi due molecole di citocromo c e due di citocromo-c-ossidasi (complesso IV) per ogni molecola di QH2-citocromo-c-reduttasi (complesso III). Esperimenti di combinazione con il rotenone suggeriscono la presenza di una sola molecola di NADH-Q-reduttasi (complesso I) per 4 molecole di QH2-citocromo-c-reduttasi (complesso III). Quindi l'unità minima per la fosforilazione ossidativa contiene circa 30 molecole proteiche per un peso molecolare totale di circa 5 milioni, escludendovi il peso del CF0 (v. tab. II). Lo schema della membrana interna del mitocondrio appare simile a un mosaico, in cui l'unità minima che si ripete deve estendersi su una porzione considerevole di superficie della membrana stessa. Molte delle proteine elencate nella tab. II sono costituite da diverse subunità, spesso differenti fra loro, così che il numero di catene polipeptidiche presenti è di gran lunga maggiore delle sei proteine elencate. Anche senza ulteriori informazioni circa la struttura delle diverse proteine, noi possiamo già trarre una conclusione: è del tutto evidente che per ogni coppia di elettroni che attraversa una proteina trasportatrice di elettroni (I, III o IV) viene sintetizzata, da parte dell'ATP-asi, una molecola di ATP: in qualsiasi modo sia trasmessa all'ATP-asi l'energia liberatasi per il passaggio di elettroni attraverso le proteine trasportatrici, è evidente che ciascuna molecola di ATP-asi deve ricevere un messaggio da più di una proteina trasportatrice di elettroni.
In che modo questa molecola di ATP-asi avverte il passaggio di elettroni attraverso le proteine che li trasportano e come risponde sintetizzando ATP? Possono essere considerate due ipotesi: quella chemiosmotica e quella conformazionale.
4. Ipotesi chemiosmotica.
Secondo questa ipotesi, proposta da Mitchell fin dal 1961 ed estesa nel 1966 e nel 1968, il primo passo per la conservazione di energia nella catena di trasferimento di elettroni è la traslocazione di protoni attraverso la membrana interna dei mitocondri, non compensata dal movimento di cationi in direzione opposta o dal movimento di anioni nella stessa direzione. Questo movimento non bilanciato di particelle positive attraverso la membrana produce un potenziale di membrana; questo potenziale e insieme il gradiente di pH costituiscono una forza motrice nei confronti dei protoni, che può indurre la sintesi di ATP invertendo l'azione di una ATP-asi capace anch'essa di trasferire protoni (v. fig. 4).
In assenza di ADP o Pi il passaggio netto di particelle cariche positivamente verso il polo positivo della membrana si arresterà quando si sarà costituito un potenziale di membrana sufficientemente alto da opporsi a questo passaggio. Teoricamente si può calcolare che in effetti già un indeterminabile, piccolo passaggio netto di cariche positive è sufficiente a costituire un potenziale di membrana così alto che nessuna membrana biologica può resistervi. Ciò nondimeno P. Mitchell e J. Moyle poterono dimostrare la formazione di rimarchevoli quantità di protoni nel mezzo di sospensione quando aggiungevano piccole quantità di ossigeno a una sospensione anaerobia di mitocondri; con quantità limitanti di ossigeno la concentrazione dei protoni formatisi era stechiometrica rispetto alla quantità di ossigeno aggiunta, con un rapporto H+: O di circa 6 se i mitocondri erano stati pretrattati con un substrato connesso col NAD (3-idrossibutirrato), di circa 4 se erano stati pretrattati con succinato.
Questa comparsa di protoni indotta dalla respirazione viene oggi spiegata con il movimento di cationi, presenti nel mezzo di sospensione, verso l'interno dei mitocondri, per influenza del potenziale di membrana: essa comporta la scomparsa del potenziale e quindi la rimozione della forza elettromotrice che impedisce la traslocazione di protoni. In effetti oggi si usa condurre l'esperimento di Mitchell, basato su piccole aggiunte di ossigeno, in presenza di K+ e valinomicina, in quanto il complesso (K-valinomicina)+ passa rapidamente attraverso le membrane biologiche.
In assenza di anioni permeabili, ADP Pi l'energia generata dalla catena di trasferimento di elettroni è fornita dalla forza motrice dei protoni (Δp), il cui valore (in mV) è dato da
Δp = Δϕ − 59 ΔpH.
dove Δψ è il potenziale di membrana proposto da Mitchell e Δ si riferisce alla differenza tra esterno e interno dei mitocondri. Come detto in precedenza, le dimensioni del mitocondrio sono tali per cui, in queste condizioni, ΔpH è molto piccolo e perciò
Δp ≈ Δp
In presenza di K+ e valinomicina, si stabiliscono gradienti di K+ e H+ tali che il termine energetico risulta costituito, in linea di principio, da tre componenti e cioè
Δψ residuo + 59 (ΔpK+ − pH).
in cui Δψ residuo è quella parte del primitivo potenziale di membrana (formatosi per traslocazione non compensata di protoni) non eliminata in presenza di valinomicina.
Quindi, se si potesse ammettere una completa soppressione del potenziale di membrana in presenza di sufficiente quantità di valinomicina, si potrebbe calcolare il termine energetico misurando le concentrazioni di K+ e H+ dentro e fuori i mitocondri. Mitchell e Moyle, fatta questa ammissione, hanno effettuato tali misure e calcolato che Δp = 227 mV; se, come si è accennato, si potesse ulteriormente ipotizzare che, in assenza di movimento di cationi, il ΔpH tende a zero, anche il potenziale di membrana ipotizzato da Mitchell sarebbe eguale a 227 mV.
Questa è la logica che si trova alla base della determinazione da parte di Mitchell e Moyle del potenziale di membrana nell'ambito dell'ipotesi chemiosmotica.
Se si conosce la stechiometria dei protoni traslocati per ciascun sito di fosforilazione, si può calcolare il potenziale di membrana proposto da Mitchell a partire dal potenziale del fosfato (v. sopra) secondo l'espressione
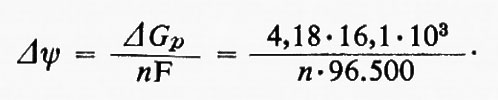
Dagli esperimenti con piccole aggiunte di ossigeno effettuati da Mitchell e Moyle è risultato che n = 2, il che porterebbe a un valore di Δψ = 350 mV, molto maggiore di quello calcolato da Mitchell e Moyle. La causa di questa discrepanza è sconosciuta, ma vi sono tre possibili spiegazioni: a) al contrario di quanto ammesso da Mitchell e Moyle, nelle loro condizioni sperimentali vi è un residuo potenziale di membrana; b) le ‛reazioni di perdita', che dissipano energia, sono più accentuate nelle condizioni sperimentali di Mitchell e Moyle che in quelle usate per misurare il potenziale del fosfato; c) come proposto da G. F. Azzone per i mitocondri e da H. T. Witt per i cloroplasti, n > 2.
Bisogna tener presente che tutti questi calcoli sono fondati sull'ipotesi dell'esistenza del potenziale di membrana proposto da Mitchell e Moyle. Benché si sia stabilito che, in presenza di cationi permeabili, possano instaurarsi un gradiente di protoni e un equilibrio Donnan di cationi, che causano un potenziale di diffusione, rimane un postulato l'esistenza del potenziale così come proposto da Mitchell, risultante cioè da un movimento scompensato di protoni attraverso la membrana. J. T. Tupper e II. Tedeschi hanno trovato mediante misura diretta con microelettrodi un potenziale di lievissima entità; ma poiché non è sicuro che venisse in realtà misurato il potenziale attraverso l'interno della membrana, tale esperimento non può essere considerato conclusivo.
Secondo Mitchell, protoni sono traslocati attraverso l'interno della membrana mitocondriale a livello di tre siti o punti di raccordo, corrispondenti ai tre siti di fosforilazione (v. fig. 5; c'è da aggiungere che Mitchell considera la nicotinammidenucleotidetransidrogenasi come sito addizionale). In ognuno di questi punti due atomi di idrogeno vengono prelevati dalla parte interna della membrana e trasferiti attraverso di essa verso l'esterno, dove mettono in libertà due protoni. Oli elettroni così formatisi vengono ritrasportati attraverso la membrana verso l'interno, dove possono combinarsi con altri due protoni ed essere disponibili, come atomi di idrogeno, per il successivo passaggio. Quindi un postulato essenziale dell'ipotesi di Mitchell è che i vari centri trasportatori di elettroni non sono situati a caso, ma in modo specifico all'interno della membrana.
Una notevole quantità di prove sperimentali è a favore dell'ipotesi di Mitchell.
1. Come previsto dall'ipotesi in questione, sono state chiaramente dimostrate sperimentalmente la liberazione di protoni e l'assunzione di cationi da parte della membrana mitocondriale.
2. È stato pienamente provato l'orientamento all'interno della membrana mitocondriale di alcuni trasportatori di elettroni: il citocromo c è orientato verso l'esterno del mitocondrio, da cui può essere facilmente rimosso per azione di sali e dove è, accessibile all'anticorpo specifico e al ferricianuro; esso invece si trova all'interno delle particelle submitocondriali (che si raggomitolano su se stesse, ma rivoltate, in seguito alla rottura dei mitocondri), dove è non estraibile e inaccessibile all'anticorpo e al ferricianuro. Molte deidrogenasi, situate all'interno dei mitocondri di Mammifero, sono inaccessibili ai loro substrati aggiunti dall'esterno. Come previsto dall'ipotesi, l'ATP-asi si trova all'interno dei mitocondri intatti e all'esterno delle particelle submitocondriali.
3. P. C. Hinkle ha costruito vescicole artificiali contenenti citocromo-c-ossidasi e fosfolipidi che traslocano protoni nell'ossidare il citocromo c per mezzo dell'ossigeno. Introducendo in queste vescicole citocromo e e il complesso ATP-asico, E. Racker è riuscito ad accoppiare l'ossidazione dell'ascorbato con la sintesi di ATP: ha quindi ricostituito il sito 3 della fosforilazione ossidativa. Similmente C. I. Ragan e Racker hanno ricostituito anche il sito 1 usando la NADH-deidrogenasi anziché la citocromo-e-ossidasi.
4. Racker e W. Stoeckenius hanno preparato vescicole contenenti una rodopsina batterica che catalizza, per azione della luce, la fuoruscita di protoni; se si introduce nel sistema. il complesso ATP-asico, ha luogo la sintesi di ATP.
Non pare esservi dubbio che, come ipotizzato da Mitcheli, le proteine trasportatrici di elettroni siano orientate, nelle membrane trasduttrici di energia, in modo che si liberino specificamente protoni da una parte della membrana in coincidenza col trasferimento di elettroni dal substrato all'ossigeno lungo i vari sistemi ossidoriduttivi; inoltre, in presenza di opportuni vettori di cationi (endogeni o esogeni), questi possono essere traslocati attraverso la membrana. E quasi una questione di semantica il chiedersi se considerare la forza motrice di questi spostamenti come un potenziale di membrana; in ogni caso si deve ritenere provata una considerevole parte dell'ipotesi di Mitchell.
Non provati rimangono l'ipotizzata fonte di protoni e l'ipotizzato meccanismo di sintesi dell'ATP.
Secondo Mitchell i protoni deriverebbero per riduzione, da parte di atomi di idrogeno, di nuclei ferrici nelle ferrosolfo-proteine e nelle emoproteine della catena di trasferimento di elettroni:
H + FeIII → H+ + FeII.
Tale ipotesi richiede la presenza di tre trasportatori di idrogeno capaci di trasferirlo attraverso la membrana dall'interno all'esterno; tuttavia, come è stato più volte sottolineato, finora è stato individuato un solo trasportatore con queste caratteristiche, l'ubichinone.
Un secondo postulato dell'ipotesi di Mitchell è che la ATP-asi traslocatrice di protoni, responsabile della sintesi di ATP in presenza di un opportuno gradiente elettrico e di protoni, conterrebbe gruppi permeabili (X- e O-) che migrano nel campo elettrico e si combinano in presenza di protoni a formare un legame covalente X − I (v. fig. 4). Non vi è conferma sperimentale della presenza ditali gruppi.
Sembra più probabile che i protoni traslocati attraverso la membrana mitocondriale derivino dalla parte proteica, più che dal gruppo prostetico. Ad esempio, nell'ossigenazione dell'emoglobina si ha la liberazione di 2,8 grammoioni H+ per mole di emoglobina:
Fe4II + 4 O2 ⇄ (FeII•O2)4 ↔ [FeIII•O2-l4 + 2,8 H+.
Questi protoni, tuttavia, non derivano dall'atomo di ferro ma da catene laterali di amminoacidi della catena polipeptidica, molto distanti spazialmente dall'atomo di ferro. Se fosse possibile orientare l'emoglobina in una membrana in modo che i suoi amminoacidi, responsabili della liberazione di protoni, fossero rivolti verso l'esterno, l'ossigenazione delle vescicole simulanti le membrane porterebbe alla comparsa di protoni nel mezzo di sospensione.
Questi cosiddetti protoni di Bohr derivano da gruppi acidi i cui valori di pK cambiano in conseguenza di un cambiamento di conformazione nella catena polipeptidica scatenato dalla reazione dell'ossigeno con l'atomo di ferro.
L'idea che i protoni evidenziati da Mitchell e Moyle mediante gli esperimenti con piccole aggiunte di ossigeno siano conseguenza di un cambiamento di conformazione di una proteina, più che di una riduzione diretta di gruppi Fe(III) da parte di atomi di idrogeno, porta in successione logica alla cosiddetta ipotesi conformazionale.
5. Ipotesi conformazionale.
L'ipotesi conformazionale è stata preceduta dalla cosiddetta ipotesi chimica (v. sopra), secondo la quale l'energia viene conservata sotto forma di un composto ricco di energia tra uno dei prodotti della reazione di ossidoriduzione e un legante:
AH2 + B + C ⇄ A ~ C + BH2
A ~ C + ADP + Pi ⇄ A + C + ATP.
Secondo tale ipotesi esisterebbero due forme di A, una ad alta energia, ad alto potenziale, combinata (capace di ossidare BH2) e un'altra a bassa energia, a basso potenziale, non combinata (incapace di ossidare apprezzabilmente BH2). Una variante altrettanto possibile prevede la conservazione di energia in una forma altamente energetica, ma a basso potenziale, combinata, di BH2, capace, al contrario della forma non combinata BH2, di ridurre A.
Come per la teoria chemiosmotica, nella teoria chimica si concentra l'attenzione sui gruppi ossidoriduttivi delle proteine trasportatrici di elettroni. L'ipotesi conformazionale, proposta da P. D. Boyer nel 1964, sottolinea l'importanza delle catene polipeptidiche. La forma altamente energetica, ad alto potenziale, di A è costituita ora da una specifica conformazione della proteina A:
AH2 + B ⇄ A* + BH2
A* + ADP + Pi ⇄ A + ATP.
La seconda equazione esprime il concetto che l'energia presente nella forma altamente energetica di A, cioè A*, viene trasmessa all'ATP-asi, che può così catalizzare la sintesi dell'ATP; ciò implica che i trasportatori di elettroni siano in contatto, attraverso CF0 e OSCP, con l'ATP-asi stessa. Ci si aspetterebbe, quindi, di trovare una corrispondenza stechiometrica semplice tra le concentrazioni dei sistemi ossidoriduttivi e l'ATP-asi, come infatti è stato trovato sperimentalmente. L'ipotesi di Mitchell non richiederebbe invece diretto contatto tra essi, benché i trasportatori di elettroni debbano essere orientati specificamente all'interno della membrana, in quanto il potenziale elettrico è a lungo raggio e opera proprio attraverso la membrana. Secondo l'ipotesi conformazionale, cambiamenti strutturali a carico di proteine trasportatrici di elettroni, indotti dall'acquisizione o dalla perdita di un elettrone, si trasmettono dall'ATP-asi per interazioni ‛a corto raggio' che potrebbero anche essere, almeno in parte, di natura elettrostatica.
Possiamo formulare ciò del tutto schematicamente come nella fig. 6, che per semplicità mostra solo una molecola di ATP-asi per molecola di citocromo-c-ossidasi (complesso IV). Si può immaginare, per esempio, che la riduzione del gruppo prostetico nel complesso IV comporti un cambiamento di conformazione della proteina; questo induce, mediante contatti attraverso CF0 e OSCP, un cambiamento di conformazione nell'ATP-asi. La rimozione di elettroni da parte dell'ossigeno ricostituisce il complesso ossidato, ma per influenza della nuova conformazione indotta nell'ATP-asi esso rimane nella conformazione propria del complesso ridotto, almeno per un po'. Dal momento che questa combinazione di conformazioni dell'ATP-asi e del complesso IV è instabile (tende infatti a riconvertirsi nella conformazione originale), la molecola di ATP-asi è energeticamente in grado di reagire con il fosfato e l'ADP (forse in tempi successivi, più probabilmente con meccanismo concertato), dopo di che l'ATP-asi si converte nella conformazione a bassa energia. Questo evento induce il costituirsi della conformazione a bassa energia nel complesso ossidato, completando così il ciclo che catalizza la sintesi di una molecola di ATP per ogni passaggio di due elettroni nel ciclo stesso.
Che la riduzione dell'atomo di ferro della citocromo-c-ossidasi causi un cambiamento di conformazione nella proteina è prevedibile ed è stato già dimostrato direttamente: B. F. Van Gelder e H. Beinert hanno messo in evidenza qualche anno fa la comparsa di un segnale dovuto al ferro ad alto spin nello spettro EPR durante la riduzione della citocromo-c-ossidasi. Studi su composti modello e studi condotti da M. F. Perutz (1970) sull'emoglobina (v. sangue: Emoglobina) hanno dimostrato che l'atomo di ferro che si trova nel piano dell'anello porfirinico nei composti a basso spin, come l'ossiemoglobina, si sposta di 0,4-0,8 Å da questo piano nei composti ad alto spin. Nell'emoglobina l'istidina legata al ferro si sposta solidalmente con questo e tale movimento dà origine a una variazione nella struttura terziaria della catena polipeptidica cui l'eme è connesso.
In una catena di emoglobina isolata, come anche nel caso della mioglobina, non vi è ostacolo a questo cambiamento di struttura terziaria; invece, nell'emoglobina tetramerica, la struttura terziaria della catena polipeptidica è controllata in parte dalle interazioni tra le quattro catene, cioè dalla struttura quaternaria. Queste interazioni sono evidenziate dalla ben nota osservazione che è molto più difficile ossigenare l'emoglobina che una sua catena polipeptidica isolata e che l'ossigenazione di una catena polipeptidica nell'oligomero favorisce l'ossigenazione delle altre catene. Studi di cristallografia a raggi X compiuti da Perutz ne hanno spiegato il motivo: il cambiamento della struttura terziaria nella catena contenente l'atomo di ferro che per primo reagisce con l'ossigeno comporta la rottura dei ponti salini tra la catena stessa e le altre; ne consegue che queste catene adiacenti divengono meno rigide, gli atomi di ferro divengono a spin meno alto, per cui l'ossigeno può reagire più facilmente. Il bilancio del ΔG nella ossigenazione dell'emoglobina può essere indicato cosi:
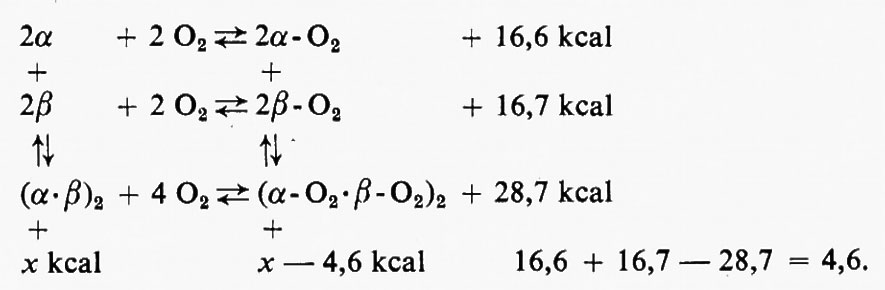
Il − ΔG per la reazione dell'ossigeno con l'emoglobina completamente desossigenata è di 4,6 kcal/mole inferiore alla somma delle reazioni con le catene α e β isolate: questo valore di 4,6 kcal rappresenta l'energia di interazione tra le catene presente nella desossiemoglobina, ma assente nell'emoglobina; in altre parole le catene non ossigenate si associano più facilmente, di 4,6 kcal appunto, rispetto a quelle ossigenate.
Consideriamo questo fatto da un altro punto di vista: bisogna introdurre 4,6 kcal di energia in meno nel sistema quando si stacca l'O2 dagli atomi di ferro nel caso della ossiemoglobina rispetto al caso delle quattro catene isolate. In termini di bioenergetica, là struttura quaternaria dell'ossiemoglobina a basso spin è ad alta energia rispetto alla più stabile, cioè a bassa energia, desossiemoglobina ad alto spin. Parte dell'energia che si libera quando l'O2 reagisce con l'atomo di ferro di una catena polipeptidica nella desossiemoglobina è usata per rompere ponti salmi tra le catene della desossiemoglobina. Le 4,6 kcal di energia sono accumulate nell'energia ‛potenziale' di questi ponti salini che sono scomparsi e l'energia potenziale può sempre essere utilizzata se si dispone di un meccanismo per superare la barriera cinetica.
Lo stesso tipo di interazioni può essere dimostrato nei componenti delle membrane trasduttrici di energia. Sono state dimostrate forti interazioni tra accettori di elettroni nel caso della citocromo-c-ossidasi, sia libera che legata alle membrane: come già detto, Van Gelder e Beinert hanno dimostrato che la riduzione di uno dei due gruppi eme α provoca la comparsa di una configurazione ad alto spin nell'atomo di ferro dell'altro eme a; benché non si possano escludere interazioni dirette tra gli emi o tra un eme e un atomo metallico, sembra probabile che almeno alcune di queste interazioni siano indirette, come nell'emoglobina.
D. Keilin ed E. F. Hartree già nel 1939 avevano dimostrato che solo una delle molecole di eme α si combina con leganti come il cianuro, l'azide e il monossido di carbonio. D. F. Wilson e collaboratori hanno trovato che il legame di un eme α con il monossido di carbonio o con l'azide produce un considerevole aumento del coefficiente di assorbanza dell'altro eme ridotto; in effetti le informazioni in nostro possesso indicano che le due catene polipeptidiche contenenti gli emi α nella citocromo-c-ossidasi sono simili e che il legarsi di una di esse con un legante impedisce che si leghi la seconda; se ciò è vero, dovremmo considerare questo fenomeno come un caso estremo di cooperatività negativa con un'altissima energia di interazione tra le due catene polipeptidiche contenenti gli emi.
Nel presente contesto gli effetti della riduzione di un eme sulle proprietà dell'altro sono di considerevole interesse. Van Gelder e collaboratori hanno dimostrato che, nella ossidasi isolata, ‛in assenza di citocromo c', i gruppi eme vengono titolati con lo stesso potenziale redox (280 mV) con n i nel grafico di Nernst, indicando che entrambi accettano indipendentemente un elettrone; ciò nondimeno nel corso della titolazione sia le bande α che le bande γ si spostano a lunghezza d'onda maggiore. Inoltre l'aumento di ellitticità (misurato con il dicroismo circolare) è maggiore a basso che ad alto potenziale e ciò indica che la riduzione di un eme modifica la simmetria dell'altro. Similmente la riduzione di un eme α aumenta di circa mille volte la reattività verso il cianuro dell'altro ferrieme.
Vi sono anche prove, benché meno dirette, dell'interazione tra differenti molecole di proteine trasportatrici di elettroni: il complesso III, o QH2-citocromo-c-reduttasi, isolato, contiene un singolo sito di legame per l'antimicina, per cui il legame con l'antimicina non mostra cooperatività; invece il legame dell'antimicina con le membrane mitocondriali è inibito e cooperativo in presenza di substrato, proprio come il legame dell'ossigeno con l'emoglobina, il che indica la presenza di interazioni tra le varie molecole di QH2-citocromo-c-reduttasi incorporate nelle membrane.
L'ATP-asi isolata contiene un solo sito di legame tenace per l'aurovertina; il legame dell'aurovertina con i mitocondri di fegato di ratto nello stato 3 è invece inibito e cooperativo, il che dimostra pertanto l'esistenza di interazioni tra le molecole di ATP-asi nella membrana.
Più importante è la dimostrazione di interazioni tra proteine trasportatrici di elettroni e proteine responsabili della sintesi di ATP. Come dimostrato da M. E. Puliman, le membrane contengono un polipeptide che inibisce l'attività di scissione dell'ATP da parte della ATP-asi isolata. Esperimenti più recenti hanno dimostrato che questo inibitore è un piccolo polipeptide che può legarsi stabilmente all'ATP-asi; Van de Stadt ha mostrato che nelle particelle submitocondriali questo inibitore si dissocia dall'ATP-asi quando ha luogo il trasferimento di elettroni accoppiato. R. MS. Bertina e H. S. Penefsky, inoltre, hanno visto che la resa quantica della fluorescenza dell'aurovertina legata all'ATP-asi nei mitocondri aumenta quando ha luogo la fosforilazione ossidativa. A. T. Jagendorf ha trovato un aumento di radioattività nell'ATP-asi dei cloroplasti dovuta a trizio proveniente da acqua triziata quando i cloroplasti venivano energizzati. Tutti questi esperimenti mostrano in modo chiarissimo che la proteina ATP-asi è capace di sentire' gli eventi che si verificano nella catena di trasporto degli elettroni. Vi è anche qualche indicazione del fatto che le proteine che trasferiscono elettroni possono risentire dell'aggiunta di ATP all'ATP-asi: l'aggiunta di ATP a particelle ridotte con NADH in assenza di ossigeno e in presenza di antimicina fa comparire una forma ad alto spin del ferrieme, che non compare in assenza di antimicina; quindi, se si inibisce il flusso retrogrado di elettroni verso il substrato, l'ATP induce una redistribuzione di elettroni all'interno della citocromo-c-ossidasi e si forma un ferrieme ad alto spin. Ciò fa supporre che il ferrieme ad alto spin che si riscontra in queste condizioni sia caratteristico della citocromo-c-ossidasi energizzata. Inoltre si sono visti effetti dell'ATP sullo spettro di assorbimento nel visibile della citocromo-c-ossidasi nei mitocondri, sia nella forma ossidata sia in quella ridotta.
L'aggiunta di ATP a mitocondri ridotti con succinato rimuove l'ostacolo al legarsi di piccole concentrazioni di antimicina a molecole di QH2-citocromo-c-reduttasi nella membrana: ciò suggerisce che l'energizzazione comporti un allentarsi delle interazioni tra le molecole di questa reduttasi all'interno della membrana. Vi sono buone prove, quindi, di un'intima interconnessione tra le conformazioni delle proteine trasportatrici di elettroni e delle proteine che sintetizzano ATP. Benché possa darsi che tali connessioni siano casuali, resta la suggestiva possibilità che esse siano invece fondamentali per il meccanismo di sintesi dell'ATP; la prova definitiva che ancora manca è la dimostrazione che un'ATP-asi in conformazione energizzata possa sintetizzare l'ATP dall'ADP e dal fosfato.
Ogni molecola di ATP-asi mitocondriale, quando viene isolata dai mitocondri di cuore, contiene tre molecole di ATP e due di ADP legate stabilmente con legami non covalenti, che non si possono scambiare a velocità misurabile con nucleotidi adeninici esterni. L'ATP-asi è formata da circa 10 subunità, di 4 tipi diversi: trattando l'enzima con nitrato a O °C si ottiene la dissociazione della molecola nelle sue subunità e il distacco delle cinque molecole di nucleotidi adenosinici; ciò indica che il legame saldo dei nucleotidi è una proprietà della struttura ‛quaternaria'. In aggiunta a questi siti di legame molto tenaci è possibile, con tecniche cinetiche, mettere in evidenza almeno un altro sito più debole coinvolto nell'attività idrolitica dell'enzima isolato.
Slater e Boyer, separatamente, hanno ipotizzato che la funzione dell'energia è di distaccare l'ATP, legato fortemente ma con legami non covalenti, inducendo un cambiamento conformazionale della proteina; in altri termini l'apporto energetico sarebbe necessario dopo che la molecola di ATP si è formata al sito catalitico.
6. Aspetti comuni delle ipotesi proposte circa il meccanismo della fosforilazione ossidativa.
Tutt'e tre le ipotesi (chimica, chemiosmotica e conformazionale) contengono l'aminissione tacita, o implicita, che il flusso di elettroni attraverso le proteine trasportatrici di elettroni causi una variazione del potenziale ossidoriduttivo standard (o normale), sia positiva sia negativa, di alcuni o di tutti i trasportatori di elettroni. Secondo le ipotesi chimica e conformazionale A ~ C o A* sono specie ad alto potenziale del trasportatore A, mentre BH2 ~ C o BH2* sono specie a basso potenziale del trasportatore B. Secondo l'ipotesi chemiosmotica, i trasportatori vicini al polo positivo della membrana accetterebbero elettroni più facilmente che quelli situati nella parte mediana non carica della membrana; in altri termini essi avrebbero un potenziale maggiore di quello abituale. L'energizzazione dei trasportatori di elettroni da parte dell'ATP, tramite l'ATP-asi, porterebbe allo stesso risultato. Quindi l'ATP può invertire l'ordine dei potenziali redox e perciò far fluire gli elettroni all'indietro nella catena, in direzione dei substrati. Per esempio, in termini della teoria chimica o conformazionale, l'ATP abbasserebbe il potenziale redox del citocromo b (o di un trasportatore in equilibrio con esso) così da renderlo capace di ridurre il NAD+. In termini dell'ipotesi chemiosmotica, l'ATP abbasserebbe il potenziale ossidoriduttivo dei componenti prossimi alla superficie interna della membrana e aumenterebbe quello dei componenti prossimi alla superficie esterna, così che il flusso di elettroni sarebbe invertito.
Nel 1970 D. F. Wilson e collaboratori fornirono le prove che l'ATP aumenta il potenziale redox di uno dei componenti del citocromo b e abbassa quello del citocromo a3: ciò fu portato da questi autori a sostegno dell'ipotesi chimica o conformazionale. In realtà questi fatti possono essere inquadrati egualmente bene nell'ipotesi chemiosmotica. In ogni caso oggi si ritiene che i cambiamenti di potenziale misurati da Wilson e collaboratori siano solo apparenti, in quanto causati da una maggiore accessibilità dei mediatori di ossidoriduzione impiegati verso alcuni centri trasportatori di elettroni.
Tutt'e tre le ipotesi implicano l'intervento dell'ATP-asi mitocondriale nella sintesi dell'ATP: esse differiscono nella natura dell'accoppiamento fra trasferimento di elettroni e sintesi di ATP.
Se ne può dedurre che tutt'e tre le ipotesi configurano una separazione di cariche alla base del meccanismo di trasmissione energetica fra trasporto di elettroni e sintesi di ATP. Nell'ipotesi chimica, ora abbandonata, l'intermediario sarebbe un composto altamente energetico X ~ I che si puo considerare contenente un legame covalente altamente polarizzato. Nell'ipotesi chemiosmotica la separazione delle cariche avviene attraverso l'intera membrana, il che provoca la scissione X − I in X- e IO- al polo positivo e il riformarsi del legame covalente al polo negativo. Secondo l'ipotesi conformazionale la separazione delle cariche equivale a quella di un ponte salino tra catene polipeptidiche (circa 3 Å). In tal modo si potrebbero distinguere in un certo senso le tre ipotesi in base al grado di separazione delle cariche, che è minimo nel caso del legame covalente polarizzato e massimo nel caso del potenziale di membrana di Mitchell.
Una quarta ipotesi è stata formulata da R. J. P. Williams: similmente a Mitchell, egli ritiene che la forza motrice per la sintesi dell'ATP da parte dell'ATP-asi sia la formazione di protoni per riduzione con atomi di idrogeno di centri carichi positivamente. Nell'ipotesi di Williams, però, i protoni non migrerebbero all'interno della membrana come proposto da Mitchell, ma si muoverebbero verso l'ATP-asi, dove, in un ambiente idrofobico, promuoverebbero l'inverso dell'idrolisi dell'ATP per semplice azione di massa:
H+ + ADP3- + Pi2 ⇄ ATP4- + H2O.
7. Nota finale. Un'ipotesi unitaria sul meccanismo della fosforilazione ossidativa.
Vi sono in tutte le ipotesi avanzate aspetti comuni sufficienti a giustificare il tentativo di proporre un'ipotesi unitaria che contenga elementi di ciascuna di esse.
1. Le proteine trasportatrici di elettroni associate a ciascun sito di fosforilazione sono costituite da diverse catene polipeptidiche (subunità), molte delle quali contengono gruppi prostetici accettori di elettroni.
2. Il passaggio di elettroni attraverso tali proteine (ad esempio la citocromo-c-ossidaài) porta alla formazione di una proteina ossidata con una struttura quaternaria ‛forzata', stabilizzata dalla ATP-asi (in una specifica conformazione, v. fig. 6).
3. Da questa conformazione forzata indotta nelle catene polipeptidiche delle proteine trasportatrici di elettroni deriva un diminuito valore di pK dei gruppi acidi situati nella parte periferica della membrana, in modo che vengono liberati protoni nel mezzo esterno; invece i valori di pK dei gruppi acidi nella parte interna della membrana sono inalterati o addirittura aumentati. Ne risulta la formazione di un gradiente di protoni attraverso la membrana, con la concentrazione maggiore all'esterno, mentre la superficie esterna della membrana contiene cariche negative localizzate e la superficie interna cariche positive; queste cariche localizzate (potenziale zeta) sono responsabili del trasporto di cationi più che di un potenziale di membrana.
4. L'ATP-asi in contatto con le proteine trasportatrici di elettroni in conformazione forzata è in grado di catalizzare la sintesi netta di ATP a partire da ADP e Pi e quindi ritorna alla sua primitiva conformazione. Specificamente, Boyer e Slater hanno proposto che il meccanismo fondamentale di questo processo sia la perduta capacità dell'ATPasi in conformazione sintetizzante di legare l'ATP. Alte concentrazioni locali di protoni potrebbero anche essere implicate nel promuovere la sintesi di ATP tenacemente legato all'ATP-asi, come ipotizzato da Williams.
Bibliografia.
Belitser, V. A., Tsybakova, E. T., The mechanism of phosphorylation as related to respiration, in ‟Biochimija", 1939, IV, pp. 516-535.
Berden, J.A., Slater, E. Ch., The allosteric binding of antimycin to cytochrome b in the mitochondrial membrane, in ‟Biochimica et biophysica acta", 1972, CCLVI, pp. 199-215.
Bertina, R.M., Schrier, P. I., Slater, E. Ch., The binding of aurovertin to mitochondria, and its effect on mitochondrial respiration, in ‟Biochimica et biophysica acta", 1973, CCCV, pp. 503-518.
Boyer, P. D., Carboxyl activation as a possible common reaction in substrate-level and oxidative phosphorylation and in muscle contraction, in Oxidases and related redox systems (a cura di T. E. King, H. S. Mason e M. Morrison), vol. II, New York 1965, pp. 994-1008.
Boyer, P. D., Cross, R. L., Momsen, W., A new concept for energy coupling in oxidative phosphorylation based on molecular explanation of the oxygen exchange reactions, in ‟Proceedings of the National Academy of Sciences", 1973, LXX, pp. 2837-2839.
Chance, B., Williams, G. R., The respiratory chain and oxidative phosphorylation, in Advances in enzymology and related subjects of biochemistry (a cura di F. Nord), vol. XVII, New York 1956, pp. 65-134.
Chang, T., Penefsky, H. S., Aurovertin, a fluorescent probe of conformational change in beef heart mitochondrial adenosine triphosphatase, in ‟Journal of biological chemistry", 1973, CCXLVIII, pp. 2746-2754.
Engelhardt, W. A., Ortho- und Pyrophosphat im aeroben und anaeroben Stoffwechsel der Blutzellen, in ‟Biochemische Zeitschrift", 1930, CCXXVII, pp. 16-38.
Ernster, L., Lee, C. P., Biological oxidoreductions, in ‟Annual review of biochemistry", 1964, XXXIII, pp. 729-788.
Harden, A., Young, W. J., The alcoholic ferment of yeast-juice, in ‟Proceedings of the Royal Society" (Series B), 1906, LXXVII, pp. 405-420.
Harden, A., Young, W. J., The alcoholic ferment of yeast-juice. III. The function of phosphates in the fermentation of glucose by yeast-juice, in ‟Proceedings of the Royal Society", (Series B), 1908, LXXX, pp. 299-311.
Harris, D. A., Rosing, J., Van de Stadt, R. J., Slater, E. Ch., Tight binding of adenine nucleotides and magnesium to beef-heart mitochondrial ATPase, in ‟Biochimica et biophysica acta", 1973, CCCXIV, pp. 149-153.
Hatefi, Y., The functional complexes of the mitochondrial electrontransfer system, in Comprehensive biochemistry (a cura di M. Florkin e E. H. Stotz), vol. XIV, Amsterdam 1966, pp. 199-231.
Hinkle, P. C., Kim, J.J., Racker, E., Ion transport and respiratory control in vesicles formed from cytochrome oxidase and phospholipids, in ‟Journal of biological chemistry", 1972, CCXLVII, pp. 1338-1342.
Hunter, F. E. Jr., Oxidative phosphorylation during electron transport, in Phosphorus metabolism (a cura di W. D. McElroy e B. Glass), Baltimore 1951, pp. 297-330.
Kalckar, H., Phosphorylation in kidney tissue, in ‟Enzymologia", 1937, II, pp. 47-52.
Kalchar, H., The nature of phosphoric esters formed in kidney extracts, in ‟Biochemical journal", 1939, XXXIII, pp. 631-641.
Lardy, H. A., Elvehjem, C. A., Biological oxidations and reductions, in ‟Annual review of biochemistry", 1945, XIV, pp. 1-30.
Lehninger, A. L., Esterification of inorganic phosphate coupled to electron transport between dihydrodiphosphopyridine nucleotide and oxygen, in ‟Journal of biological chemistry", 1949, CLXXVIII, pp. 625-644.
Lehninger, A. L., Oxidative phosphorylation, in ‟Harvey lectures", 1953-1954, IL, pp. 176-215.
Lehninger, A. L., Oxidative phosphorylation in submitochondrial systems, in ‟Federation proceedings", 1960, XIX, pp. 952-962.
Lehninger, A. L., Bioenergetics, New York 1965.
Lehninger, A. L., Wadkins, C. L., Oxidative phosphorylation, in ‟Annual review of biochemistry", 1962, XXXI, pp. 47-78.
Lehninger, A. L., Wadkins, C. L., Cooper, C., Devlin, T. M., Gamble, J. L. Jr., Oxidative phosphorylation, in ‟Science", 1958, CCXVIII, pp. 450-456.
Lipmann, F., Metabolic generation and utilization of phosphate bond energy, in ‟Advances in enzymology and related subjects", 1941, I, pp. 99-162.
Lipmann, F., Metabolic process patterns, in Currents in biochemical research (a cura di D. E. Green), New York 1946, pp. 137-148.
Lohmann, K., Darstellung der Adenylpyrophosphosäure aus Muskulatur, in ‟Biochemische Zeitschrift", 1931, CCXXXIII, pp. 460-469
Meyerhof, O., Carbohydrate and lactic acid exchange in frog muscle, in ‟Pflügers Archiv", 1920, CLXXXV, pp. 11-32.
Mitchell, P., Coupling of phosphorylation to electron and hydrogen transfer by a chemiosmotic type of mechanism, in ‟Nature", 1961, CXCI, pp. 144-148.
Mitchell, P., Chemiosmotic coupling in oxidative phosphorylation, Bodmin 1966.
Mitchell, P., Chemiosmotic coupling and energy transduction, Bodmin 1968.
Mitchell, P., Moyle, J., Stoichiometry of proton translocation through the respiratory chain and adenosine triphosphatase systems of rat liver mitochondria, in ‟Nature", 1965, CCVIII, pp. 147-151.
Ochoa, S., ‛Coupling' of phosphorylation with oxidation of pyruvic acid in brain, in ‟Journal of biological chemistry", 1941, CXXXVIII, pp. 751-753.
Ochoa, S., Efficiency of aerobic phosphorylation in cell-free heart extracts, in ‟Journal of biological chemistry", 1943, CLI, pp. 493-505.
Ogston, A. G., Smithies, O., Some thermodynamic and kinetics aspects of metabolic phosphorylation, in ‟Physiological reviews", 1948, XXVIII, pp. 283-303.
Perutz, M. F., Stereochemistry of cooperative effects in haemoglobin, in ‟Nature", 1970, CCXXVIII, pp. 726-739.
Pullman, M. E., Monroy, G. C., A naturally occuring inhibitor of mitochondrial adenosine triphosphatase, in ‟Journal of biological chemistry", 1963, CCXXXVIII, pp. 3762-3769.
Pullman, M. E, Penefsky, H. S., Datta, A., Racker, E., Partial resolution of the enzymes catalyzing oxidative phosphorylation. I. Purification and properties of soluble, dinitrophenolstimulated adenosine triphosphatase, in ‟Journal of biological chemistry", 1960, CCXXXV, pp. 3322-3329.
Racker, E., Mechanisms of synthesis of adenosine triphosphate, in ‟Advances in enzymology and related subjects", 1961, XXIII, pp. 323-399.
Racker, E., Mechanisms in bioenergetics, New York 1965.
Racker, E., Kandrach, A., Reconstitution of the third site of oxidative phosphorylation, in ‟Journal of biological chemistry", 1971, CCXLVI, pp. 7069-7071.
Racker, E., Stoeckenius, W., Reconstitution of purple membranes vesicles catalyzing light-driven proton uptake and adenosine triphosphate formation, in ‟Journal of biological chemistry", 1974, CCXLIX, pp. 662-663.
Ryrie, I. J., Jagendorf, A. T., Correlation between a conformational change in the coupling factor protein and the high energy state in chloroplasts, in ‟Journal of biological chemistry", 1972, CCXLVII, pp. 4453-4459.
Slater, E. Ch., Mechanism of phosphorylation in the respiratory chain, in ‟Nature", 1953, CLXXII, pp. 975-978.
Slater, E. Ch., Respiratory chain phosphorylation, in Proceedings of the third international congress of biochemistry (a cura di C. Liébecq), New York 1956, pp. 264-277.
Slater, E. Ch., Mechanisms of oxidative phosphorylation, in ‟Review of pure and applied chemistry", 1958, VIII, pp. 221-264.
Slater, E. Ch., Mechanism of uncopuling of oxidative phosphorylation by nitrophenols, in Proceedings of the fifth international congress of biochemistry (a cura di E. Ch. Slater), vol. V, London 1963, pp. 325-364.
Slater, E. Ch., Oxidative phosphorylation, in Comprehensive biochemistry (a cura di M. Florkin e E. H. Stotz), vol. XIV, Amsterdam 1966, pp. 327-396.
Slater, E. Ch., The respiratory chain and oxidative phosphorylation. Some of the unsolved problems, in Biochemistry of mitochondria (a cura di E. Ch. Slater, Z. Kaniuga e L. Wojtczak), London-Warszaza 1967, pp. 1-10.
Slater, E. CH., The coupling between energy-yielding and energy-utilizing reactions in mitochondria, in ‟Quarterly review of biophysics", 1971, IV, pp. 35-71.
Slater, E. Ch., Mechanism of energy conservation, in Mitochondria. Biogenesis and bioenergetics. Biomembranes. Molecular arrangements and transport mechanisms (a cura di S. G. van den Bergh, P. Borst, L. L. M. Van Deenen, J. C. Riemersma, E. Ch. Slater e J. M. Tager), vol. XXVIII, Amsterdam 1972, pp. 133-146.
Slater, E. Ch., The mechanism of energy conservation in the mitochondrial respiratory chain, in ‟Harvey lectures", 1972, LXVI, pp. 19-42.
Slater, E. Ch., The respiratory chain and oxidative phosphorylation, in The molecular basis of electron transport (a cura di J. Schultz e B. F. Cameron), New York-London 1972, pp. 95-114.
Slater, E. Ch., Multiple components of the respiratory chain, in A treatise on electron and coupled energy transfer in biological systems (a cura di T. E. King e M. Klingenberg), New York 1973.
Slater, E. Ch., Electron transfer and energy conservation. Plenary lecture, in Dynamics of energy-transducing membranes (a cura di L. Ernster, R. W. Estabrook e E. Ch. Slater), Amsterdam 1974.
Tager, J. N., Papa, S., Quagliarello, E., Slater, E. Ch. (a cura di), Regulation of metabolic processes in mitochondria, Amsterdam 1966.
Tiesjema, R. H., Muijsers, A. O., Van Gelder, B. F., Biochemical and biophysical studies on cytochrome c oxidase. X. Spectral and potentiometric properties of the hemes and coppers, in ‟Biochimica et biophysica acta", 1973, CCCV, pp. 19-28.
Van Gelder, B. F., Beinert, H., Studies of the heme components of cytochrome c oxidase by EPR spectroscopy, in ‟Biochimica et biophysica acta", 1969, CLXXXIX, pp. 1-24.
Warburg, O., Christian, W., Isolierung und Kristallisation des Proteins des oxydierenden Gärungsferments, in ‟Biochemische Zeitschrift", 1939, CCCIII, pp. 40-68.
Williams, R. J. P., Electron transfer and energy conservation, in ‟Current topics in bioenergetics", 1969, III, pp. 79-156.
Wilson, D. F, Dutton, P. L., Energy dependent changes in the oxidation-reduction potential of cytochrome b, in ‟Biochemical and biophysical research communications", 1970, XXXIX, pp. 59-64.