
Geochimica
Geochimica
di Mario Fornaseri
SOMMARIO: 1. Introduzione e cenni storici. □ 2. Metodi di studio. □ 3. Alcuni concetti di base: sfere geochimiche e cicli geochimici. Bilanci geochimici. □ 4. Struttura e composizione chimica della Terra: a) generalità; b) la composizione chimica della Terra. □ 5. Struttura e composizione della crosta terrestre. □ 6. Alcuni problemi di petrogenesi: a) differenziazione magmatica; b) il problema dei basalti; c) il problema dei graniti; d) altri tipi di rocce. □ 7. Alla ricerca dei principi che regolano il comportamento degli elementi nel processo magmatico: a) le dimensioni degli atomi e degli ioni nei cristalli; b) raggi ionici e coordinazione; c) le regole di Goldschmidt e le loro eccezioni; d) nuovi criteri e nuovi principi. □ 8. Problemi relativi alle fasi finali del processo magmatico. □ 9. Lineamenti della geochimica del ciclo esogeno: a) l'alterazione: agenti chimici e azioni chimiche; b) fattori che regolano il comportamento degli elementi nei processi di alterazione, trasporto e sedimentazione; c) aspetti geochimici dei processi diagenetici. □ 10. La misura del tempo geologico: a) generalità; b) i metodi del potassio-argon e del rubidio-stronzio; c) i metodi basati sul decadimento dell'uranio e del tono; d) il metodo del carbonio-14. □ 11. Geochimica degli isotopi stabili: a) generalità; b) geochimica degli isotopi stabili dell'idrogeno; c) geochimica degli isotopi stabili del carbonio; d) geochimica degli isotopi dell'ossigeno; e) geochimica degli isotopi dello zolfo; f) geochimica degli isotopi dello stronzio; g) geochimica degli isotopi del piombo. □ 12. I problemi aperti e il futuro della geochimica. □ Bibliografia.
1. Introduzione e cenni storici.
La geochimica, ramo della scienza che si è sviluppato grandemente nel sec. XX, fino ad assumere una spiccata individualità, ha in realtà le sue origini strettamente legate a quelle della chimica. È sufficiente ricordare che la scoperta della maggior parte degli elementi e delle loro proprietà è iniziata con lo studio delle loro combinazioni naturali, cioè dei minerali, nonché dell'atmosfera e delle acque, da cui il nome di ‛chimica minerale' una volta usato per indicare la chimica inorganica. Il nome di geochimica fu usato per la prima volta da C. F. Schönbein, nel 1838, con la completa visione degli stretti rapporti che intercorrono fra la chimica e la geologia.
Problemi di geologia chimica e di chimica minerale sono già ampiamente trattati da J. J. Berzelius (1779-1848), mentre la prima opera in cui vengono organicamente esposti i problemi della geologia chimica è il Lehrbuch der physikalischen und chemischen Geologie del Bishof (1847), seguito dalla Allgemeine und chemische Geologie (1879) del Roth e dalla Chemische Mineralogie del Brauns (1896).
Più tardi la geochimica si profilerà come ramo ben individuato delle scienze della Terra nella classica opera The data of geochemistry di F. W. Clarke (v., 1908), contenente una raccolta sistematica e ragionata di dati analitici sui materiali della crosta terrestre nonché sui gas naturali e sulle acque. Una ventina d'anni dopo la pubblicazione da parte di H. S. Washington (1903) di un catalogo critico dei dati analitici sulle rocce ignee, Clarke e Washington (v., 1924) potevano giungere alla compilazione di quei dati dell'abbondanza degli elementi nella crosta terrestre che per tanti anni hanno resistito alle ingiurie del tempo.
Un contributo essenziale al progresso della geochimica è dovuto agli studi sulla cristallizzazione delle rocce ignee dei petrografi norvegesi W. C. Brögger e J. H. L. Vogt, al quale ultimo si debbono le prime osservazioni sugli elementi in tracce. Le conoscenze sulla chimico-fisica del processo magmatico sono basate sui contributi teorici di P. Niggli e sull'esteso lavoro sperimentale svolto dal Geophysical Laboratory della Carnegie Institution di Washington, iniziato nel 1905 principalmente per opera di N. L. Bowen (1887-1956) e continuato fino ai giorni nostri.
Contemporaneamente si andava sviluppando in URSS una fiorente scuola di geochimica, facente capo a V. I. Vernadsky (v., 1924), i cui trattati hanno avuto ampia diffusione. Un allievo del Vernadsky, A. E. Fersman (v., 1933-1939), pubblicava fra il 1933 e il 1939 un trattato di geochimica in quattro volumi.
La geochimica moderna deve una gran parte del suo sviluppo e delle sue caratteristiche al gigantesco lavoro svolto da V. M. Goldschmidt e dai suoi collaboratori. L'opera del Ooldschmidt s'inserisce in un periodo di sviluppo particolarmente favorevole delle scienze fisiche, specialmente in seguito alla scoperta della diffrazione dei raggi X da parte di M. von Laue nel 1912, scoperta che ha aperto la via alla conoscenza della disposizione degli atomi nella materia cristallizzata. Partendo dallo studio delle strutture cristalline di molti minerali, il Laue poteva così stabilire i parametri cristallochimici che regolano la distribuzione degli elementi negli stessi. Utilizzando poi i metodi della spettroscopia ottica e i raggi X, poteva stabilire le relazioni di abbondanza degli elementi nei materiali terrestri e nelle meteoriti.
Un impulso altrettanto fondamentale ai progressi della geochimica fu determinato dalla scoperta della radioattività naturale a opera di H. Becquerei (1896) per le conseguenze relative al ruolo degli elementi radioattivi nel bilancio termico della Terra e per le possibilità che ne derivarono ai fini della misura del tempo geologico come, appena nove anni dopo, nel 1905, era dimostrato da E. Rutherford.
Similmente, la moderna geochimica isotopica ha le sue radici nelle ricerche fondamentali di F. Soddy (1910), di J. J. Thomson (1913) e di F. W. Aston (1919), mentre la comprensione dei processi di frazionamento isotopico negli ambienti naturali è largamente impostata sui progressi della chimica quantistica.
La geochimica è parte della cosmochimica e ha come oggetto lo studio chimico della Terra in relazione con gli altri corpi del sistema solare e dell'universo conosciuto. Essa studia la composizione della Terra nel suo insieme e nelle sue parti, la distribuzione degli elementi e dei nuclidi nello spazio e nel tempo e tutti i mutamenti chimici connessi con i fenomeni geologici. Tipica materia interdisciplinare, il suo contenuto è spesso mutevole ed è determinato dal suo naturale sviluppo.
Nella fig. 1 sono indicate le principali suddivisioni della geochimica e la posizione che questa occupa fra le scienze.
Per la sua stessa posizione interdisciplinare è evidente che esistono poi vaste aree di sovrapposizione fra la geochimica, la mineralogia, la petrologia, e molteplici interconnessioni fra la geochimica, la paleontologia, la biologia, le scienze dell'atmosfera e dell'idrosfera.
2. Metodi di studio.
Poiché uno degli obiettivi fondamentali della geochimica è lo studio della distribuzione delle abbondanze degli elementi e dei loro isotopi, è comprensibile che l'attenzione dei geochimici si sia rivolta principalmente ai metodi analitici, opportunamente scelti in relazione ai problemi da risolvere e ai livelli di concentrazione da misurare.
La classica analisi per via umida è tuttora largamente usata, particolarmente per la determinazione dei componenti principali delle rocce e dei minerali, alla scala macro, semimicro e micro, in dipendenza dalla quantità di sostanza disponibile per l'analisi.
I metodi classici sono stati affiancati e gradualmente sostituiti da metodi strumentali. La spettrofotometria di assorbimento in soluzione, la spettrofotometria di fiamma e di assorbimento atomico consentono la determinazione rapida e con alto grado di esattezza e di precisione di un grande numero di elementi a tutti i livelli di concentrazione. Da molto tempo è impiegata nella ricerca geochimica la spettrografia di emissione ottica, mentre all'analisi per emissione diretta di raggi X, che un tempo offriva non poche difficoltà sperimentali, si è sostituita con successo l'analisi per emissione secondaria di fluorescenza, un procedimento analitico non distruttivo divenuto oggi di uso comune, che consente la determinazione quantitativa di molti elementi, in un vasto intervallo di concentrazioni. L'analisi per emissione diretta di raggi X ha avuto un rilancio nell'ultimo ventennio nella versione rappresentata dalle microsonde elettroniche e da speciali adattamenti dei microscopi elettronici a scansione.
Particolari problemi analitici trovano la loro soluzione specifica nell'analisi per attivazione con neutroni lenti o veloci e nei metodi di diluizione con isotopi stabili o radioattivi.
Fra i metodi strumentali deve essere fatta menzione dei metodi elettrici, quali la polarografia e la potenziometria; in quest'ultima tecnica notevoli progressi sono stati recentemente realizzati mediante l'impiego di elettrodi specifici.
Se dall'analisi elementare passiamo all'analisi di fase, è nella diffrazione dei raggi X che il problema trova la soluzione di elezione sia dal punto di vista qualitativo che quantitativo. Trasformazioni di fase possono essere studiate con l'impiego della diffrazione di raggi X variando opportunamente la temperatura del campione o mediante la tecnica dell'analisi termica differenziale (DTA). Quest'ultima può essere impiegata anche nella determinazione quantitativa dei componenti minerali nelle loro associazioni.
La spettrometria nell'infrarosso trova applicazione principalmente nello studio dei minerali argillosi, ma il suo impiego si va rapidamente estendendo anche ad altri settori. La spettrometria di massa è forse una delle tecniche analitiche alle quali è maggiormente legato lo sviluppo della geochimica negli ultimi decenni. Per le sue caratteristiche costituisce il metodo di elezione per le analisi isotopiche ed è a fondamento di molti metodi di geocronometria. Trova impiego anche nell'analisi elementare quantitativa con il metodo della diluizione con isotopi stabili o con l'impiego della sorgente a scintilla. E infine una tecnica largamente impiegata in molti problemi di geochimica organica, particolarmente in connessione con la gascromatografia.
Le tecniche analitiche, per avanzate che siano, non rappresentano d'altra parte l'essenza del pensiero geochimico. I processi che si svolgono durante la consolidazione delle rocce ignee possono in parte essere ricostruiti dall'osservazione diretta dei rapporti tessiturali dei minerali nella roccia consolidata, ma un approccio più razionale richiede lo studio di sistemi pluricomponenti di composizione più o meno simile a quella delle rocce naturali. La costruzione dei diagrammi di stato (o di fase) ditali sistemi, per quanto si riferisce alle alte temperature, si svolge sottoponendo a trattamenti termici miscele a composizione nota e comporta l'individuazione delle fasi che rappresentano i prodotti in equilibrio a determinate temperature. Procedimenti di ‛tempera' (quenching) vengono sovente impiegati per prevenire lo smescolamento e le inversioni durante il raffreddamento. Le tecniche attuali consentono l'impiego di alte pressioni, fino a 200 kilobar, in atmosfere controllate.
Similmente, negli anni recenti, si è cercato di simulare in laboratorio le condizioni chimico-fisiche che hanno determinato il metamorfismo delle rocce, mediante lo studio delle trasformazioni indotte da alte temperature e pressioni su sistemi di composizione simile a quelli naturali, in un complesso di ricerche che si possono comprendere nel termine di ‛metamorfismo sperimentale'.
L'effetto della temperatura, della pressione e delle variazioni di composizione chimica è governato dalle leggi della termodinamica; notevoli sforzi sono stati realizzati per una migliore conoscenza delle grandezze termodinamiche fondamentali che interessano i sistemi naturali.
3. Alcuni concetti di base: sfere geochimiche e cicli geochimici. Bilanci geochimici.
La struttura a gusci della Terra è un fatto accertato in base al comportamento delle onde sismiche. Il ‛nucleo', il ‛mantello' e la ‛crosta', oltre che rappresentare delle entità fisicamente differenti, costituiscono anche delle zone di concentrazione di particolari elementi. A essi compete perciò giustamente il nome di ‛sfere geochimiche'. Vi è un generale accordo nel ritenere che nel nucleo il ferro sia l'elemento dominante, accompagnato da nichel e da qualche altro elemento a numero atomico più basso; al nucleo, considerato come sfera geochimica, è stato perciò attribuito il nome di ‛siderosfera' (NIFE, secondo E. Suess, 1855). Il mantello si ritiene generalmente costituito da silicati di magnesio e, in minor misura, di ferro, ed è stato indicato perciò col nome di SIMA; la crosta è prevalentemente costituita da silicati, ma in essa è l'alluminio che segue il silicio in ordine di abbondanza, da cui la denominazione di SIAL.
Il complesso discontinuo delle acque, con caratteristiche chimiche ben individuabili, che si trovano sulla superficie terrestre e nel sottosuolo costituisce l'‛idrosfera'; così pure ben individuabili sono le caratteristiche chimiche dell'‛atmosfera', l'involucro gassoso che circonda la Terra. Estendendo il concetto di sfera geochimica alle parti della Terra interessate dai fenomeni vitali, si è voluto introdurre il concetto di ‛biosfera' per indicare, secondo alcuni autori, l'insieme delle parti dell'atmosfera, dell'idrosfera e della crosta in cui hanno sede i fenomeni vitali e, secondo altri, l'insieme stesso degli esseri viventi.
Il carattere ciclico dei fenomeni geologici è stato intuito per la prima volta da J. Hutton nel 1785. Con le parole ‟we find no vestige of a beginning, no prospect of an end" egli esprimeva l'innumerevole ripetersi del ciclo nel tempo geologico. I suoi concetti, ripresi più tardi dal Lyell come base logica per interpretare la storia della Terra, mantengono ancora oggi intatta la loro validità, come successivamente è stato ampiamente illustrato da O. Linck nel 1912 e, in epoca più recente, sviluppato da H. Termier e G. Termier (v., 1961 e 1963).
Il verificarsi del ciclo geologico sulla Terra è il risultato di un complesso di circostanze favorevoli: una giusta dose di radiazione solare, la presenza di un'atmosfera e dell'acqua sulla superficie terrestre e di condizioni energetiche nell'interno della Terra. Il ciclo geologico generale si può considerare come costituito da quattro cicli interconnessi: il ciclo idrologico, il ciclo tettonico, il ciclo litologico e il ciclo geochimico. Il ciclo litologico considera il processo magmatico, l'alterazione, l'erosione, la sedimentazione, il metamorfismo e, in una parola, le trasformazioni cicliche che le rocce ignee, sedimentarie e metamorfiche subiscono nella crosta terrestre. Il ciclo geochimico considera le migrazioni di carattere ciclico dei singoli elementi e degli elementi nel loro insieme. Il ciclo litologico e il ciclo geochimico, nel loro aspetto generale, sono strettamente connessi, tanto che è talvolta difficile scinderne lo studio.
Molte rappresentazioni schematiche possono essere utilizzate per illustrare il comportamento ciclico della materia. Nella fig. 2 è dato un diagramma del grande ciclo litologico-geochimico generale.
Il cosiddetto ‛grande ciclo geochimico', o ciclo maggiore, può essere seguito a partire da uno qualsiasi dei suoi episodi: per esempio dalla cristallizzazione di masse fuse o magmi i cui prodotti formano masse cospicue di rocce ignee o ‛quasi ignee', ma vengono anche in parte disciolti nell'idrosfera o riversati nell'atmosfera. Le rocce ignee, come d'altra parte qualsiasi altro tipo di roccia, sedimentaria o metamorfica, non sono stabili a contatto degli agenti atmosferici e in seguito ad azioni meccaniche e chimico-fisiche e alle vicende successive di trasporto e di deposito dei loro prodotti di degradazione danno luogo all'accumulo di materiali sciolti, i sedimenti, alla formazione dei quali concorre d'altronde anche l'azione degli organismi. Un complesso di azioni che si verificano nell'atto stesso della sedimentazione o in epoca successiva, a cui si dà il nome di ‛diagenesi', concorre a trasformare i sedimenti, inizialmente incoerenti, in rocce sedimentarie indurite. Pressioni e temperature quali si realizzano nelle zone profonde, accompagnate da eventuali variazioni di ambiente chimico, possono provocare la completa ricristallizzazione delle rocce con cambiamenti di struttura e di composizione mineralogica. Attraverso diversi gradi di ‛metamorfismo', il cui prodotto sono le rocce metamorfiche, si può anche giungere alla fusione differenziale (‛anatessi') o totale della roccia, e alla rigenerazione (‛palingenesi') di masse fuse con il maggiore o minore contributo di materiale magmatico primario. In seno al grande ciclo geochimico, che implica la partecipazione delle sfere geochimiche esterne e l'eventuale contributo di materiali del mantello, si possono poi individuare numerosi cicli minori, uno dei quali presenta un particolare interesse in quanto comprende in modo essenziale i processi di alterazione e di trasformazione di materiali che si svolgono nell'ambito degli agenti atmosferici: esso è noto col nome di ‛ciclo esogeno' o anche di ‛ciclo minore' in senso stretto.
In senso più strettamente geochimico, qualora si passi a considerare i singoli elementi, potranno essere delineati per ognuno di essi i relativi cicli geochimici: si potrà parlare così del ciclo dell'ossigeno, del silicio, del carbonio, dello zolfo e così via. La tendenza moderna della geochimica è diretta a rilevare per ciascuno di essi, oltre all'aspetto qualitativo, le grandezze quantitative caratterizzanti. A questo fine conviene servirsi di un modello a serbatoi, identificabili in particolari zone di concentrazione di un determinato elemento, come per esempio l'atmosfera, gli oceani, i sedimenti, le rocce ignee. Il trasferimento di un elemento dall'uno all'altro dei serbatoi può avvenire per fattori biologici o per fattori fisici. Per ciascun serbatoio può essere definito il ‛tempo di residenza' dalla relazione

dove A rappresenta il contenuto di un certo elemento nel serbatoio e dA/dt la velocità d'introduzione che, in condizioni di regime stazionario, deve uguagliare la velocità di rimozione. Il trasferimento da un serbatoio all'altro è caratterizzato dalla ‛velocità di flusso'.
Come esempio della rappresentazione del ciclo geochimico di un elemento, in fig. 3 è riportato il ciclo del carbonio, nel quale è da rilevare la parte svolta dalla materia vivente.
Se si ammette che tutti i sedimenti e le rocce sedimentarie siano in ultima analisi derivati dall'alterazione delle rocce ignee e che i materiali presenti negli oceani abbiano la stessa origine, chiamando X la quantità totale di rocce ignee alterate, Y la massa totale delle acque oceaniche, Z la quantità totale di sedimenti, xi, yi e zi le percentuali di ogni elemento i rispettivamente nelle rocce ignee, nelle acque oceaniche e nei sedimenti, si avrà la relazione generale
xiX=yiY+ziZ
che è l'equazione fondamentale del bilancio geochimico di un elemento i. Essa può essere verificata in maggiore o minor misura se e in quanto sono valide le premesse.
Sistemi di equazioni scritte per i diversi elementi, essendo noto il valore di Y e ottenibili sperimentalmente i valori di xi, yi e zi possono essere usati per determinare in alternativa i valori di X e di Z e per verificare il bilancio geochimico dei singoli elementi.
4. Struttura e composizione chimica della Terra.
a) Generalità.
Soltanto una piccola parte della Terra è accessibile all'indagine diretta: questa parte è limitata a una porzione della crosta terrestre, all'atmosfera, all'idrosfera e al complesso dellà sostanza vivente nella biosfera. Ciò nonostante i dati geofisici concorrono a fornire informazioni dirette sulla struttura e la composizione dell'interno della Terra e ogni modello chimico che si voglia proporre per la Terra nel suo insieme dev'essere in accordo con le proprietà fisiche osservate.
I dati fisici fondamentali sono il valore della densità media della Terra e del momento d'inerzia. La densità media, quale si ottiene dalla massa della Terra (M=5,98•1027 g) e dal raggio medio terrestre (R=6,371•108 cm), risulta uguale a 5,517 g/cm3. Questo valore è notevolmente maggiore non solo rispetto a quello della densità delle rocce superficiali (densità media 2,6) e delle rocce dei basamenti sottostanti ai continenti (densità media 2,8), ma anche dei valori massimi registrati delle rocce conosciute (d=3,3). Il momento d'inerzia C intorno all'asse polare, quale si ottiene dalla teoria della forma della Terra e dalla precessione degli equinozi, risulta uguale a 0,3305 Ma2, essendo a il raggio equatoriale. Poiché il momento d'inerzia di una sfera omogenea è uguale a 0,4 Ma2, ne segue che la Terra non può essere omogenea ma la sua densità deve crescere fortemente verso il centro.
Lo studio della variazione delle onde sismiche con la profondità ha dimostrato che a determinati valori della profondità si verificano brusche variazioni di velocità delle onde P (onde longitudinali o di compressione) e delle onde S (onde trasversali o di distorsione). Le discontinuità di primo ordine rilevabili nella curva velocità-profondità sono: a) la discontinuità di Mohorovičić, detta anche ‛Moho' per brevità, a circa 35 km di profondità sotto i continenti e 6 km sotto gli oceani; b) la discontinuità di Wiechert-Gutenberg, a 2.900 km di profondità; c) la discontinuità di Jeffreys-Gutenberg a 5.100 km. Come ben noto, queste discontinuità consentono di individuare nella Terra un nucleo interno, un nucleo esterno, un mantello e una crosta.
Diversi modelli sono stati proposti per la distribuzione delle densità nell'interno della Terra. Qualsiasi distribuzione delle densità si voglia proporre, essa dovrà trovarsi in accordo con il valore della densità media, del momento d'inerzia della Terra, della velocità di propagazione delle onde sismiche, e deve fornire un valore accettabile per la densità, non superiore a 3, 4, alla base della crosta. In fig. 4 sono riportati alcuni fra i più autorevoli e recenti schemi di distribuzione della densità nell'interno della Terra (v. Bullen, 1963; v. Anderson, 1967). Le oscillazioni libere della Terra forniscono un ulteriore controllo della distribuzione della densità e, in generale, l'accordo è buono se non perfetto. Nota la distribuzione della densità, dalle velocità Vs e Vp è possibile calcolare i coefficienti di elasticità K e μ, e inoltre la variazione di g della pressione con la profondità.
Una volta ottenuta la distribuzione della densità, possiamo cercare di ottenere qualche informazione sulla composizione dell'interno, essendo la densità in relazione con la composizione e la struttura delle sostanze. Per poter stabilire gli opportuni confronti è però necessario ridurre la densità ρ, misurata a una pressione P, al valore ρ0 corrispondente alla pressione ordinaria; ciò implica la ricerca di un'equazione di stato ρ=ρ(P, T), cioè di una relazione fra pressione, densità e temperatura, applicabile a un corpo solido quale è la Terra; questa ricerca offre molte difficoltà, sia che si voglia procedere sperimentalmente che per via teorica. Alcune equazioni empiriche possono essere ciò nonostante ricavate. F. Birch (1952) ha proposto l'equazione
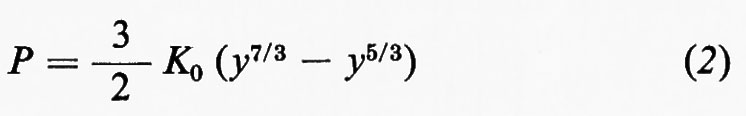
dove K0 è il valore della incompressibilità isoterma KT alla pressione zero e y=ρ/ρ0.
b) La composizione chimica della Terra.
La crosta. - Poiché la crosta terrestre è uno degli oggetti di studio di maggior interesse della geochimica, le questioni relative alla sua composizione verranno trattate in seguito in dettaglio e non verranno qui discusse. Benché non sia facile stabilirne la composizione chimica media, è certo che gli elementi più abbondanti nella crosta sono, in ordine decrescente, O, Si, Al: la parte superiore della crosta continentale ha una composizione da granitica a granodioritica con una densità media di 2,67 g/cm3, nella parte inferiore vi è evidenza di una composizione gabbroide. La sottile crosta oceanica ha prevalentemente una composizione basaltica con una densità media di 2,9 g/cm3.
Il mantello. - Nella ricerca di una ragionevole distribuzione della densità sono emerse chiare indicazioni di una mancanza di omogeneità nel mantello, ascrivibile o a variazioni di composizione chimica o a cambiamenti di fase.
Il ‛mantello superiore', o zona B, dalla discontinuità di Mohorovičić a 200-400 km di profondità, è una zona a densità ridotta costante, pari a 3,2-3,3. Vi sono tre tipi di rocce che hanno densità compresa fra 3,2 e 3,4 e proprietà fisiche simili a quelle che si richiedono per spiegare le proprietà elastiche, e in particolare la velocità di propagazione delle onde sismiche, nel mantello superiore. Queste sono le duniti, le peridotiti e le eclogiti.
Per spiegare la discontinuità di Mohorovičić si possono adottare due ipotesi: secondo la prima si può pensare che si tratti di una discontinuità puramente fisica, dovuta a un cambiamento di fase isochimico da una crosta inferiore gabbroide a un mantello superiore eclogitico. Nella seconda ipotesi la discontinuità, di carattere chimico, porterebbe da una crosta inferiore di media acidità a un mantello pendotitico. Un insieme di argomentazioni di carattere geofisico, geologico e petrologico-geochimico indicano più probabile la seconda ipotesi: in effetti molte peridotiti alpine possono essere interpretate come brandelli del mantello strappati e portati in alto negli stadi iniziali dei ripiegamenti della geosinclinale.
Secondo A. E. Ringwood il mantello superiore si può ritenere costituito da una roccia per cui egli ha proposto il nome di ‛pirolite' (da pirosseno + olivina), corrispondente grosso modo a una miscela di tre parti di dunite e una parte di basalto, contenente da 0,1 a 0,4% di acqua. La fusione frazionata di questa roccia è capace di fornire un fuso basaltico e una dunite residua. La composizione mineralogica della pirolite è variabile secondo la pressione e la temperatura.
Nella ‛zona di transizione', o zona C, da 200-400 a 900- 1.000 km, le condizioni di pressione sono tali per cui sono prevedibili importanti trasformazioni di fase che interessano sia l'olivina sia il pirosseno e la silice. Il Bernal (v., 1936), nei suoi studi sui germanati, la cui cristallochimica presenta strette relazioni con quella dei silicati, aveva osservato che il composto Mg2GeO4 esiste in due modificazioni strutturali, l'una avente la struttura dell'olivina (Mg, Fe)2SiO4, l'altra, stabile ad alta pressione, avente una struttura tipo spinello, a densità più elevata. Analoghe trasformazioni sono prevedibili per l'olivina e sono anche state realizzate in laboratorio. Anche il pirosseno MgSiO3, enstatite, alle alte pressioni subisce una transizione in una struttura tipo ilmenite. Secondo Clark e Ringwood (v., 1964) nella zona di transizione si avrebbe una miscela di olivina e di pirosseno; queste fasi sono stabili fino a una profondità di circa 400 km, a cui corrisponde una pressione di 160 kilobar. A maggiori profondità si verificano principalmente le seguenti reazioni e trasformazioni di fase:
2MgSiO3 (pirosseno)→Mg2SiO4+SiO2 (stishovite)
Mg2SiO4 (olivina)→Mg2SiO4 (con struttura tipo spinello)
Mg2SiO4 (spinello)+SiO2 (stishovite)→2MgSiO3 (tipo ilmenite)
Mg2SiO4 (spinello)→MgSiO3 (tipo ilmenite)+MgO (periclasio).
Queste trasformazioni sarebbero complete a circa 1.000 km di profondità e alla pressione corrispondente di 450 kbar e porterebbero a un incremento di densità, riferita alla pressione ordinaria, da 3,2 a 3,9.
Il ‛mantello inferiore', o zona D, da 1.000 a 2.900 km, verosimilmente omogeneo, consiste probabilmente di una miscela di (Mg, Fe)SiO3 a struttura tipo ilmenite con (Mg, Fe)O avente una struttura tipo periclasio. Si può prevedere che alle alte pressioni che regnano nel mantello inferiore si possa verificare anche la trasformazione della struttura tipo ilmenite in una struttura tipo perovskite e della struttura tipo periclasio in una struttura tipo cloruro di cesio.
Secondo i modelli proposti, l'inomogeneità del mantello è dunque dovuta principalmente a cambiamenti di fase: è verosimile che questi siano anche accompagnati da un incremento del contenuto in ferro, tale da variare di due unità il numero atomico nel passaggio dalla zona B alla zona D.
Circa lo stato fisico del mantello è da rilevare che questo sotto la discontinuità di Mohorovičić presenta una velocità delle onde S di 4,7-4,8 km/s. A partire da una profondità variabile da 50 a 100 km vi è uno strato, che si estende fino alla profondità di circa 350 km, in cui la velocità delle onde S scende a un valore minimo intorno ai 4,3 km/s. Questo strato, detto ‛strato a bassa velocità' (low velocity layer) o ‛astenosfera', è meno rigido rispetto al mantello superiore, probabilmente per la presenza di piccole quantità di liquido interstizialè dovuto a uno stato di fusione incipiente.
L'insieme della crosta e della parte rigida del mantello sovrastante l'astenosfera viene modernamente denominato ‛litosfera'; si tenga presente che quest'ultimo termine era spesso impiegato in passato per indicare la crosta.
Il nucleo. - Il comportamento delle onde sismiche indica che il nucleo terrestre dev'essere fluido nella parte esterna e rigido all'interno. Circa la composizione del nucleo non vi sono dati certi: il componente principale deve essere il ferro, ma non vi sono prove di una particolare abbondanza del nichel, se si fa astrazione dalle argomentazioni sull'alta abbondanza cosmica di questo elemento. Le considerazioni basate sui valori delle densità portano a stimare per i materiali del nucleo un numero atomico medio di circa 23 o forse meno, ciò che induce alcuni autori ad ammettere che nel nucleo esterno vi sia un contenuto dal 14 al 20% di silicio e forse di carbonio e di idrogeno. Il deficit di zolfo riscontrato nelle rocce superficiali e in quelle che presumiamo derivate dal mantello, in contrasto con la sua notevole abbondanza cosmica, rende probabile la presenza di questo elemento nel nucleo esterno. Ciò si adatterebbe bene, fra l'altro, ad alcune teorie recenti sull'evoluzione chimica del sistema solare (v. Lewis, 1974).
Date le numerose incertezze relative che ancora oggi persistono circa la composizione del mantello e del nucleo, si comprende quanto sia insicuro ogni calcolo della composizione media della Terra, il risultato del quale dipende esclusivamente dal modello adottato. Un risultato attendibile è stato ottenuto da B. Mason (v., 1966), che ha fatto seguito ad altri tentativi antecedentemente effettuati da H. S. Washington (1925) e da P. Niggli (1928), partendo dall'ipotesi di un nucleo avente la composizione del ferro-nichel delle condriti con il 5,3% di FeS, e di un mantello e di una crosta aventi una composizione simile a quella della parte silicatica della media delle condriti, con quantità minori di fosfati e di ossidi. I risultati del calcolo sono riportati in tab. I.
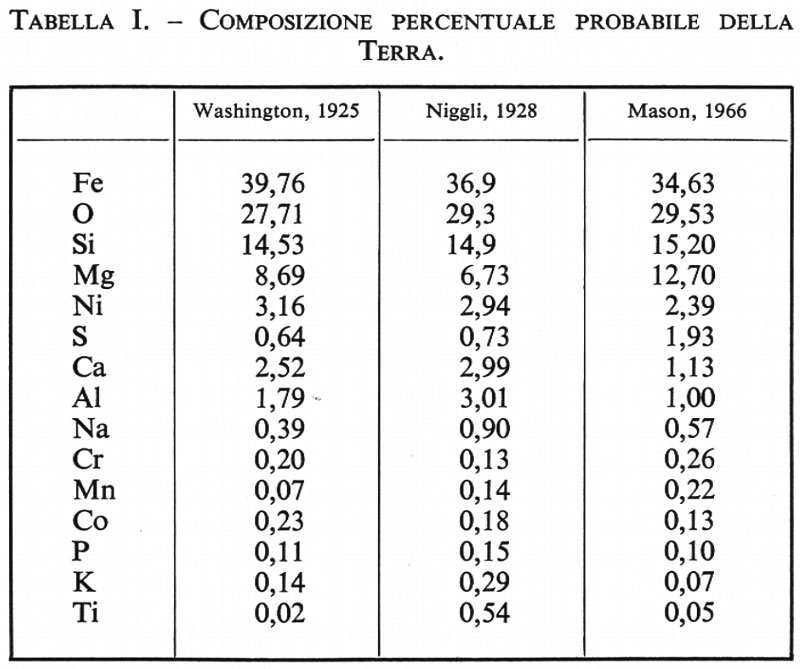
Per quanto insicuri possano risultare questi dati, a causa delle incertezze nelle ipotesi adottate, si può considerare come molto vicino al vero il fatto che Fe, O, Si e Mg siano nella Terra gli elementi più abbondanti. La somma delle loro percentuali ammonta al 90%: Ni, S, Ca, Al, Na, Cr, Mn, Co, P, K, Ti seguono in ordine di abbondanza. Gli altri elementi sono al livello di tracce e la loro percentuale complessiva probabilmente non supera lo 0,1%.
La questione di quale tipo di meteoriti si accosti meglio alla composizione globale del mantello e della crosta è ancora dibattuta: considerazioni sui rapporti Rb/Sr, K/Rb e K/U in materiali terrestri derivanti dal mantello e nei diversi tipi di meteoriti inducono a ritenere che la Terra occupi una posizione intermedia fra le condriti e le acondriti ricche di calcio.
Le osservazioni su materiali terrestri accessibili non sono tra l'altro forse sufficienti per prevedere il comportamento degli elementi nelle condizioni che si verificano nell'interno. Significativa a questo proposito è la scoperta (v. Fuchs, 1966) nelle condriti e acondriti enstatitiche della djerfisherite K3(Na, Cu) (Fe, Ni)12S14, minerale il cui campo di stabilità si estende nella zona delle alte pressioni. Se, come sembra, la troilite (FeS) e la djerfisherite possono mescolarsi completamente alle alte temperature, il potassio e probabilmente anche il rubidio potrebbero esser presenti in quantità non più trascurabili nel mantello e nel nucleo. Con ciò molte idee correnti sul comportamento geochimico di questi elementi e sulla perdita di essi come elementi volatili nella differenziazione primitiva della Terra dovrebbero essere rivedute.
5. Struttura e composizione della crosta terrestre.
È definita come crosta terrestre quella parte della Terra che è limitata inferiormente dalla discontinuità di Mohorovičić, al di sotto della quale la velocità delle onde P sale bruscamente da circa 6,8 a circa 8 km/s. La discontinuità di Mohorovičić ha un andamento piuttosto irregolare, raggiungendo una profondità massima di circa 70 km al di sotto delle aree continentali e di 10 km al di sotto dei fondi oceanici. La crosta manifesta pertanto caratteristiche diverse nelle aree continentali e nelle aree oceaniche, cosicché si può parlare di una ‛crosta continentale', dello spessore medio di 35 km, e di una ‛crosta oceanica', dello spessore medio di circa 6 km. È inoltre individuabile una discontinuità infracrostale sotto i continenti, detta di Conrad, che distingue una crosta superiore, granitica (Vp=6,1 km/s), da una crosta inferiore gabbroide o basaltica (Vp=6,4-7 km/s). Questa discontinuità non è peraltro una caratteristica costante della crosta.
La parte della crosta accessibile all'indagine diretta per mezzo di trivellazioni o a causa dell'affioramento per dislocazione di rocce profonde si stima essere di circa 16 chilometri. Le nostre conoscenze circa la composizione chimica della stessa derivano da diecine di migliaia di analisi di rocce ignee, sedimentarie, metamorfiche.
Si deve a F. W. Clarke e H. S. Washington (v., 1924) la prima stima attendibile della composizione chimica media delle rocce ignee, da loro ottenuta come media aritmetica di 5.129 analisi scelte. Questa media non è esente da critiche perché non tiene conto della proporzione in cui i diversi tipi di rocce sono rappresentati nella crosta, per la non uniforme distribuzione geografica dei campioni analizzati e perché probabilmente dà eccessivo peso a rocce non comuni; tuttavia ha costituito per molti anni un utile riferimento e probabilmente non è molto lontana dal vero. Successivi perfezionamenti si possono considerare i criteri seguiti da Poldervaart (v., 1955), da Vinogradov (v., 1962), da Taylor (v., 1964), da Horn e Adams (v., 1966), da Wedepohl (v., 1969), da Ronov e Yaroshevsky (v., 1969).
Da molti anni era stato osservato che la distribuzione di frequenza della silice nelle rocce ignee è bimodale, con due massimi di frequenza intorno a 73,0 e 52,5% di SiO2 (v. Richardson e Sneebsy, 1922); questo consegue dal fatto che i basalti e i graniti sono, fra le rocce crostali, i tipi più rappresentati. Il problema della composizione media della crosta si riduce allora essenzialmente alla stima del rapporto graniti:basalti. Su questa base Vinogradov (v., 1962) ha calcolato la composizione media di una crosta standard costituita da due parti di rocce ‛felsiche' (graniti e granodioriti) e una parte di rocce ‛mafiche' (gabbri e basalti). S. R. Taylor, in base alla considerazione che vi sono buone ragioni per ritenere che non si sia verificato un frazionamento degli elementi delle terre rare durante il processo sedimentario, partendo dal contenuto di terre rare nelle rocce sedimentarie calcola in 1:1 il rapporto basalti:graniti che le hanno generate e giunge di conseguenza a stabilire la composizione media delle rocce ignee.
A. Poldervaart (v., 1955) ha considerato la crosta come costituita da quattro principali unità: la crosta continentale, le catene ripiegate recenti, la piattaforma e le scarpate continentali. Per ciascuna di esse ha stimato la composizione media tenendo conto della composizione e delle quantità relative delle diverse rocce e da una media ponderata finale ha ottenuto i valori riportati in tab. II.

Ronov e Yaroshevsky (v., 1969) riprendono il modello di Poldervaart introducendo numerosi nuovi dati analitici. Essi considerano la crosta suddivisa nelle unità fondamentali: continentale, subcontinentale e oceanica; ciascuna di queste costituita a sua volta da uno strato sedimentario, uno strato granitico e uno strato basaltico (v. fig. 5); per ciascuna di queste unità i suddetti autori calcolano volumi, massa e composizione chimica, e pervengono così al calcolo della composizione chimica media della crosta al di sopra della Moho per uno spessore medio di 20 km.
La tab. II contiene inoltre i dati di Wedepohl (v., 1969) che si riferiscono alla crosta continentale superiore, ottenuti da una media ponderata dei valori analitici relativi ai diversi tipi di rocce intrusive (S. R. Nockolds, 1964).
Partendo da considerazioni quantitative sui cicli geochimici dei diversi elementi Horn e Adams (v., 1966), utilizzando metodi matematici e con l'impiego di calcolatori elettronici, giungono a stabilire per 65 elementi le relazioni di abbondanza per i singoli tipi litologici e per i quattro domini sedimentari degli scudi continentali, delle cinture mobili (mobile belt shelf) e delle unità pelagiche e semipelagiche.
La tab. II contiene i dati della composizione chimica media della crosta, espressi in ossidi, secondo i vari autori citati, esclusi quelli di Horn e Adams, per i quali si rinvia alla memoria originale. Si può notare come, nonostante la scarsa affidabilità del criterio usato, la media di Clarke e Washington non si discosti in modo sensibile da quella calcolata in epoca recente da Ronov e Yaroshevsky (v., 1969). È in ogni caso evidente come nella crosta terrestre l'ossigeno sia l'elemento più abbondante. L'abbondanza dell'ossigeno, 46,6% in peso, diventa ancora più appariscente qualora si esprima in volume, tenendo conto del raggio ionico di questo elemento. Risulta che circa il 92% in volume della crosta è costituito da ossigeno. Da questo punto di vista la crosta terrestre, come del resto anche il mantello, si può considerare come costituita da un impacchettamento di grossi atomi di ossigeno tenuti assieme, per valenza elettrostatica, da piccoli cationi metallici (‛ossisfera' secondo Goldschmidt). Fanno seguito all'ossigeno, in ordine di abbondanza, Si, Al, Fe, Ca, Na, K, Mg, Ti, P. Questa sequenza di abbondanza distingue chiaramente la composizione della crosta da quella dell'intera Terra e anche del mantello ed è indizio che particolari processi differenziativi hanno presieduto alla sua formazione. Gli elementi menzionati ammontano a oltre il 99% in peso. Rimane meno dell'1% a disposizione di tutti gli altri elementi, che sono rappresentati nella crosta in quantità variabili da un migliaio a meno di 10-8 ppm. In tab. III è riportata la composizione elementare media della crosta secondo Taylor (v., 1964). Gli elementi la cui concentrazione è superiore all'1% sono definiti ‛elementi maggiori', quelli la cui concentrazione è compresa fra 1% e 0,1% sono chiamati ‛elementi minori', quelli la cui concentrazione è inferiore a 0,1% (1.000 ppm) ‛elementi in tracce'. Nonostante la loro scarsa abbondanza, l'importanza geochimica ed economica degli elementi minori e degli elementi in tracce non è per questo da sottovalutare. Basterà ricordare che fra essi troviamo elementi quali il rame, l'argento, il piombo, lo zinco, il mercurio e l'oro.
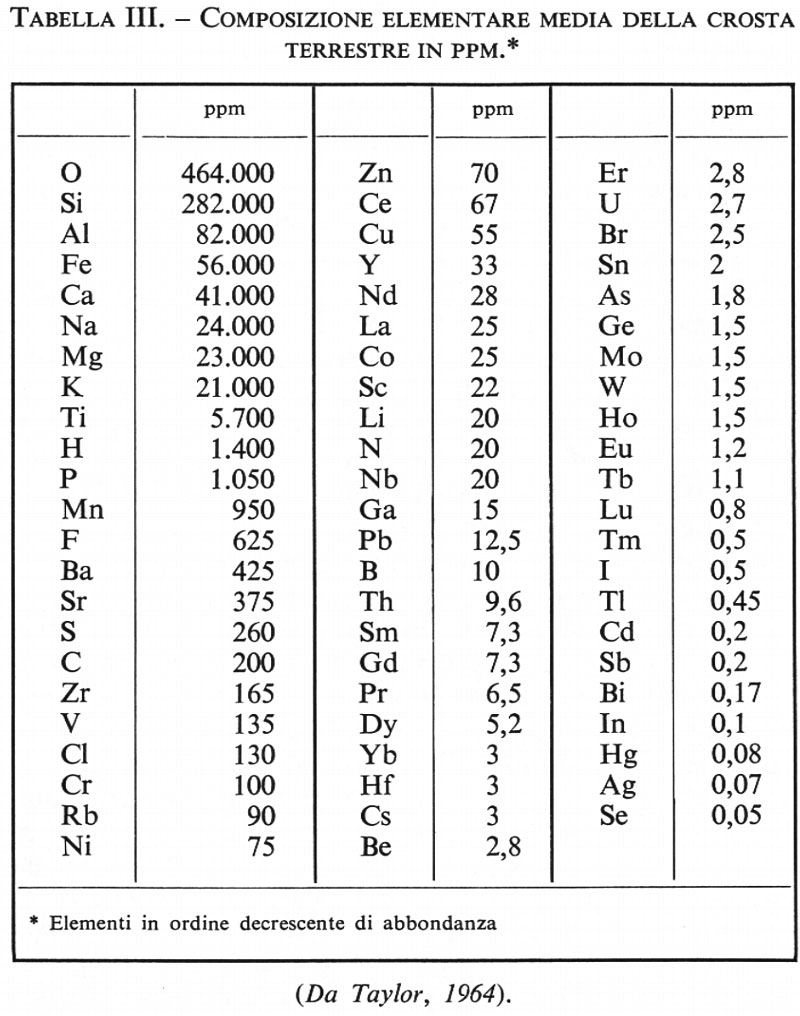
I lineamenti della distribuzione degli elementi nella crosta non si scostano peraltro in modo fondamentale da quelli che regolano l'abbondanza degli elementi nel sistema solare: il decrescere dell'abbondanza col crescere del numero atomico si verifica anche nella crosta terrestre, con l'unica eccezione per l'idrogeno e l'elio, che hanno abbandonato la Terra, mentre la validità della legge di Oddo-Harkins (la quale afferma che gli elementi a numero atomico pari sono più abbondanti di quelli a numero atomico dispari) risulta semplicemente dal fatto che essa fu enunciata per la prima volta per la crosta terrestre. È sufficiente osservare che gli elementi più abbondanti nella crosta (O, Si, Fe, Ca, Mg, Ti) hanno numero atomico pari e ne costituiscono l'87%, mentre quelli che hanno numero atomico dispari (Al, Na, K) non rappresentano che il 12,7%. Le eccezioni alla regola di Oddo-Harkins sono però numerose nella crosta terrestre: 11Na è più abbondante di 12Mg; 13Al è più abbondante di 12Mg; 15P è più abbondante di 16S; 25Mn è più abbondante di 24Cr; ciò indica quanto efficaci debbano essere stati i processi differenziativi che hanno condotto alla formazione della crosta.
È da sottolineare il fatto che molti elementi relativamente abbondanti nella crosta terrestre, come Zr, Rb, Ce, Ga, Eu, sono assai poco disponibili, mentre elementi relativamente meno abbondanti sono viceversa facilmente accessibili a basso costo, come per es. Cu, Pb, B, Sn, As, Sb, Cd, Hg. Questo comportamento apparentemente paradossale può essere messo in relazione con i processi geochimici fondamentali di cui si parlerà in seguito.
6. Alcuni problemi di petrogenesi.
Le rocce ignee e i loro derivati costituiscono una grande parte della crosta terrestre e i processi magmatici sono responsabili della storia geochimica di molti elementi.
L'osservazione dei fenomeni vulcanici ci dimostra l'esistenza di masse fuse, dalla consolidazione delle quali si originano le rocce ignee. Per logica estrapolazione noi pensiamo che queste masse fuse possano trovarsi anche in zone profonde della crosta terrestre e si possano consolidare anche senza giungere in comunicazione con l'esterno. Secondo una definizione di Rittmann (v., 1967) i magmi sono dei fusi silicatici che si trovano entro o al di sotto della crosta terrestre e che contengono in soluzione componenti volatili e in sospensione cristalli di minerali. Questi ultimi possono anche mancare nei magmi surriscaldati. In base al contenuto di gas il magma, secondo T. A. Jaggar (v., 1936), si distingue in: ‛ipomagma', magma sottosaturo di gas che può esistere solo in profondità, dove la pressione idrostatica è maggiore della pressione di vapore dei gas; ‛piromagma', soprassaturo di gas, l'eccesso dei quali forma una fase gassosa libera, in grado di esistere dove la pressione di vapore supera la pressione idrostatica; ‛epimagma', povero di gas avendo già perduto molti dei suoi componenti volatili nell'atmosfera per attività vulcanica. Questa distinzione non è priva d'importanza perché il contenuto in gas di un magma ne condiziona in modo fondamentale le modalità di eruzione.
La consolidazione dei magmi in profondità dà origine alle rocce intrusive, mentre per consolidazione in superficie si formano le rocce effusive o vulcaniche. Per un'esauriente classificazione delle rocce ignee, su base mineralogica quantitativa, dev'essere fatto riferimento ai trattati di petrologia e in particolare alla memoria di Streckeisen (v., 1967).
a) Differenziazione magmatica.
Diversi argomenti geologici e petrografici dimostrano con evidenza che esistono famiglie di rocce diverse fra loro ma collegate da variazioni continue di composizione e costituenti un'unità genetica derivante da un magma originario che è stato interessato nel tempo da un processo di differenziazione. Tali rocce si chiamano consanguinee o comagmatiche.
La differenziazione magmatica è il risultato dei processi chimico-fisici e delle leggi che regolano la cristallizzazione dei sistemi a più componenti, complicate, nei sistemi naturali, da fenomeni di squilibrio e dalla presenza di elementi volatili. I fenomeni dominanti durante il raffreddamento di masse magmatiche sono lo smescolamento allo stato liquido, la formazione di alti gradienti di concentrazione, la cristallizzazione frazionata di componenti che vengono sottratti all'equilibrio chimico, l'arricchimento progressivo dei componenti volatili nel residuo fuso. Quest'ultimo fenomeno può portare a un particolare stadio in cui si può avere la cristallizzazione da una fase in condizioni supercritiche. I gas magmatici, liberantisi nelle fasi finali del processo, sono capaci di trasportare considerevoli quantità di elementi pneumatofili, ossia di quegli elementi capaci di partecipare in maggiore o minor misura alla composizione della fase gassosa.
Uno schema generale degli stadi in cui si può pensare si svolga la cristallizzazione di un magma generico in condizioni intrusive può essere il seguente:
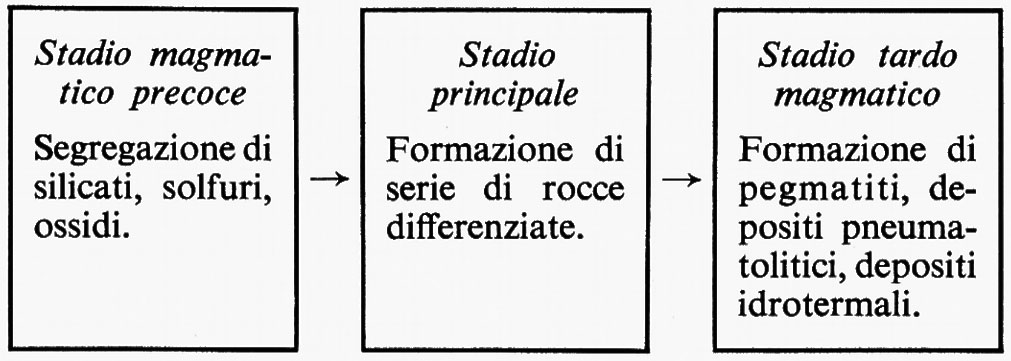
È all'opera di N. L. Bowen, dei suoi collaboratori e dei suoi successori che si deve lo sviluppo delle ricerche che hanno posto su base razionale lo studio della cristallizzazione delle rocce ignee. Le esperienze condotte in laboratorio su masse fuse artificiali e l'osservazione macro- e microscopica delle rocce hanno posto in evidenza che la sequenza di cristallizzazione dei diversi minerali dai fusi silicatici naturali avviene attraverso complesse serie di reazioni, fra le quali se ne possono individuare tre di importanza fondamentale.
1. I minerali femici formano una ‛serie discontinua' di reazioni. Il primo minerale che si separa da un magma di composizione basaltica è l'olivina. I cristalli di olivina (Mg, Fe)2SiO4 di prima segregazione reagiscono con il residuo fuso, più ricco di silice, per formare pirosseno secondo la reazione
(Mg, Fe)2SiO4+SiO2→(Mg, Fe)2Si2O6.
I pirosseni a loro volta reagiscono con il residuo fuso che si è arricchito in acqua per formare anfiboli e questi ultimi infine si trasformano in biotite. Ognuna di queste reazioni si verifica a una temperatura definita o in un ristretto intervallo di temperatura, da cui il carattere di discontinuità. Questo carattere riflette anche il fatto che il passaggio fra l'uno e l'altro termine della serie comporta bruschi cambiamenti di struttura cristallina.
2. I plagioclasi durante il processo di cristallizzazione reagiscono con continuità con il liquido; di conseguenza i primi cristalli separati modificano la loro composizione, da termini ricchi in calcio a termini ricchi in sodio. La cristallizzazione dei plagioclasi avviene in un lungo intervallo di temperatura, senza sostanziali mutamenti strutturali, e dà luogo perciò a una ‛serie continua' di reazioni.
3. I feldspati alcalini seguono parimenti una serie continua di reazioni.
In fig. 6 sono riportate in modo schematico le serie di reazioni discusse.
Il principio di reazione dimostra che, se i cristalli separati dal magma vengono sottratti all'equilibrio per cause meccaniche, per esempio per separazione dovuta alla gravità, il residuo fuso assume una composizione diversa da quella iniziale e pertanto offre una conveniente spiegazione dei fenomeni di differenziazione magmatica.
La fig. 7 mostra, a titolo di esempio, la possibile evoluzione di un magma gabbrico (o basaltico) secondo i principi ora esposti.
Lo schema esposto è però di validità molto limitata, poiché l'andamento della cristallizzazione può essere molto diverso a seconda dei valori della pressione e del contenuto in acqua. Un magma basaltico nelle condizioni di pressione del mantello superiore darebbe luogo alla separazione primaria di omfacite, un pirosseno ferro-alluminifero contenente sodio, e di granato: l'andamento della differenziazione sarebbe molto diverso da quello prevedibile a pressione ordinaria, né si avrebbe la formazione di fusi granitici residuali.
La differenziazione di un magma basaltico può seguire schemi differenti da quello esposto, e sovente da magmi olivin-alcali-basaltici si hanno prodotti finali trachitici o fonolitici. L'andamento della cristallizzazione può infine essere grandemente influenzato dal contenuto in acqua: da magmi molto ricchi in acqua si può avere la cristallizzazione precoce della biotite; ciò implica una separazione precoce del potassio, con la conseguenza che non si avrà più la separazione finale del feldspato potassico.
Che d'altra parte lo schema di Bowen non abbia validità generale era stato riconosciuto dallo stesso autore. Esso è valido, per esempio, per le rocce ignee della regione di Oslo, per le rocce caledoniane della Scozia occidentale, per il batolite della California meridionale e in genere per le rocce ignee delle cinture orogeniche. In altri casi la differenziazione può seguire linee diverse, come per es. nelle intrusioni di Skaergaard, nella Groenlandia meridionale, e del Bushveld in Sudafrica, e nel ‛complesso Stillwater' del Montana. In questi casi la differenziazione, anziché a un arricchimento finale di silice e di potassio, conduce a liquidi residuali ricchi in FeO. La pressione parziale dell'ossigeno durante la cristallizzazione ha indubbiamente un'influenza determinante nel processo di differenziazione.
Gli schemi di differenziazione osservati dipendono poi, ovviamente, dal tipo iniziale di magma; fra le serie più note possiamo ricordare le seguenti:
Tholeiiti→andesiti→daciti→rioliti
Basalti olivinici→basalti sodici→trachiti→fonoliti
Granodioriti→graniti
Andesiti→daciti→rioliti.
Basalti e graniti, incluse le granodioriti, sono riconosciuti fra i costituenti litologici fondamentali della crosta terrestre. Questi due tipi di rocce sono stati e sono oggetto di vivaci discussioni circa la loro origine.
b) Il problema dei basalti.
Un insieme di argomenti geofisici, l'abbondanza dei basalti e la relativa uniformità della loro composizione indicano che il magma basaltico deve provenire da zone piuttosto profonde e si deve essere formato per fusione parziale del mantello solido in seguito a processi limitati nello spazio e nel tempo, probabilmente nelle celle di convezione termicamente non omogenee.
Se si accetta l'ipotesi della provenienza dal mantello dei magmi basaltici, si ripropone necessariamente la questione della composizione del mantello e delle relazioni fra quest'ultima e quella dei basalti. Qualora si ammetta che il mantello sia costituito da rocce eclogitiche, non vi è alcuna difficoltà a porre in relazione queste ultime con i basalti, perché la composizione chimica è la medesima, variando soltanto la composizione mineralogica. Questa ipotesi non è in generale molto accettata e oggi si tende ad attribuire al mantello una composizione peridotitica. La provenienza dei basalti da un mantello peridotitico è comprovata non solo dalla maggiore diffusione delle rocce peridotitiche rispetto alle eclogiti, ma dalla presenza di inclusi peridotitici negli alcali-basalti.
Ricerche recenti hanno dimostrato che nelle peridotiti sono talvolta presenti spinelli alluminiferi nonché pirosseni contenenti molecole diopsidiche e molecole giadeitiche. Ciò è sufficiente per asserire che dalle peridotiti vi è la possibilità di ottenere molecole plagioclasiche, per es. con le reazioni:
Al2O3+CaMgSi2O6+MgSiO3→CaAl2Si2O8+Mg2SiO4
NaAlSi2O6+2MgSiO3→NaAlSi3O8+Mg2SiO4
e perciò di ottenere magmi basaltici.
Nonostante che le rocce basaltiche siano relativamente uniformi, tuttavia è possibile rilevare alcune differenze che portano alla distinzione di due tipi fondamentali: a) basalti tholeiitici; b) olivin-alcali-basalti.
I basalti tholeiitici abissali o basalti abissali, così denominati perché maggiormente rappresentati fra le rocce delle dorsali oceaniche, sono rocce sature o leggermente soprassature di silice: essi mostrano un rapporto SiO2/alcali più alto rispetto agli alcali-basalti (v. fig. 8), non contengono olivina se non in condizioni di disequilibrio e sono caratterizzati dalla presenza di due o tre pirosseni.
I basalti abissali sono caratterizzati da alti valori del rapporto K/Rb (v. fig. 9) e da un basso tenore dei cosiddetti ‛elementi incompatibili'. Con questo termine Ringwood ha voluto indicare un gruppo di elementi, fra i quali K, Ti, P, U, Th, Ba, Rb, Cs, Zr, Hf e gli elementi delle terre rare, che, per ragioni cristallochimiche, non sono facilmente accolti nei minerali principali del mantello, in contrasto con elementi quali Mn, Cr, Ni, Cu, Co, Ga, V, Y, che vi sono più facilmente incorporati e che vengono chiamati ‛elementi compatibili'. Le tholeiiti abissali presentano inoltre una distribuzione di tipo condritico degli elementi delle terre rare (v. fig. 10).
Questi caratteri si riscontrano anche nelle tholeiiti degli archi insulari (tholeiiti di arco). I basalti tholeiitici continentali di altopiano (plateau) presentano delle caratteristiche piuttosto differenti in quanto, nei confronti dei precedenti, mostrano un relativo arricchimento nelle terre rare leggere.
Gli olivin-atcali-basalti sono, in varia misura, sottosaturi di silice (v. fig. 8) e il rapporto silice/alcali è più basso rispetto ai basalti tholeiitici. Sono caratterizzati dalla presenza di olivina in equilibrio, della molecola normativa NaTiAlSiO6 e contengono di norma un solo pirosseno diopsidico. Gli alcali-basalti sono di regola molto arricchiti in elementi incompatibili e presentano un arricchimento negli elementi leggeri delle terre rare (v. fig. 10).
Fra i due tipi descritti vi è poi una serie di termini intermedi di difficile classificazione. In molte regioni le prime estrusioni hanno composizione tholeiitica e sono seguite da emissioni di olivin-alcali-basalti.
Diverse ipotesi sono state prospettate per spiegare l'origine dei due tipi di basalti: secondo alcuni gli alcali-basalti derivano dalle tholeiiti, secondo altri vale il processo opposto. Si è pensato che i due tipi di rocce prendano origine da un magma intermedio, oppure che entrambi i tipi di rocce siano originati damagmi provenienti dal mantello a diversa profondità. Il contenuto in terre rare degli alcali-basalti delle placche oceaniche e continentali è in accordo con una loro derivazione da una fusione parziale di peridotiti a idrogranato alla sommità dell'astenosfera nell'ambito della zona a bassa velocità del mantello. Il problema è ancora oggetto di molte discussioni.
Di notevole importanza petrogenetica sono i prodotti della differenziazione dei magmi basaltici: la differenziazione degli olivin-alcali-basalti conduce alla formazione di residuati fonolitici o fonolit-trachitici. Le tholeiiti originano per differenziazione andesiti basaltiche e riodaciti. Fusi residuali di composizione granitica possono così originarsi da basalti tholeiitici.
c) Il problema dei graniti.
I graniti e le granodioriti, in forma di ammassi o batoliti, costituiscono di prevalenza i nuclei delle catene montuose; la stessa loro forma di giacitura implica una discussione sulla loro origine. Se è infatti accettabile considerarli come rocce magmatiche nel senso definito, è d'altra parte difficile concepire l'esistenza nella crosta terrestre di masse magmatiche di composizione granitica e di dimensioni batolitiche.
Inoltre, se è ammissibile e dimostrabile che fusi residuali ricchi in silice e di composizione granitica si possano formare per differenziazione di magmi gabbroidi, un tale processo non può determinare la formazione di una quantità tale di fusi residuali da giustificare i quindici chilometri di spessore dello strato granitico.
Le stesse osservazioni di campagna si prestano d'altra parte a interpretazioni molteplici e talora contraddittorie. Mentre, infatti, in alcuni casi vi sono chiare indicazioni che i graniti si siano formati per consolidazione di una massa fusa dotata di mobilità e capace di notevoli spostamenti, in molti altri si hanno prove dirette che i graniti si siano formati in posto in seguito a processi di metamorfismo di alto grado di rocce preesistenti: passaggi graduali a rocce metamorfiche o sedimentarie, presenza nel granito di xenoliti di rocce incassanti nei quali è possibile leggere la storia di un progressivo metamorfismo fino all'assimilazione.
Si è così andata formando in alcuni autori la convinzione che le rocce in questione si formino nella crosta terrestre attraverso un processo di ‛granitizzazione', che vuole significare un trasferimento di materia attraverso processi di diffusione allo stato solido, o mediante l'intervento di una fase vapore. Questo modo di vedere rappresenta una posizione estrema non più sostenibile, in quanto i coefficienti di diffusione nei silicati solidi si sono dimostrati troppo piccoli per giustificare i fenomeni considerati; altrettanto dicasi per le tensioni di vapore. Anche se si può invocare un trasferimento di materia lungo le superfici limite dei granuli, vi sono buoni argomenti per ritenere che molti graniti si siano piuttosto formati per consolidazione di masse fuse, quali che siano l'origine e la storia geologica di tali masse.
Fondamentale contributo hanno portato a questo proposito le ricerche di Tuttle e Bowen (v., 1958) sul sistema ternario
SiO2−NaAlSi3O8−KAlSi3O8
e di von Platen (v., 1965) sul sistema
SiO2−NaAlSi3O8−CaAl2Si2O8−KAlSi3O8,
in entrambi i casi condotte a elevate pressioni di vapor d'acqua. Il sistema quarzo-albite-ortoclasio in presenza di vapor d'acqua può rappresentare con una buona approssimazione la composizione di molti graniti alcalini poveri in componenti femici. Il diagramma di fig. 11, relativo a una pressione di 2.000 kg•cm-2 di vapor d'acqua, mostra una linea ‛cotettica' fra gli eutettici qz-ab e qz-or con una ‛pozza' di minima temperatura. Le miscele di diversa composizione nel loro processo di cristallizzazione danno liquidi residuali la cui composizione tende verso quella corrispondente al minimo D. Si verifica che molti graniti hanno una composizione che si avvicina a quella corrispondente a detto minimo, ciò che sta a dimostrare che i graniti poveri in femici e ricchi in alcali possono formarsi per cristallizzazione di masse fuse.
È necessario osservare che la miscela di composizione D è l'ultima a cristallizzare da un fuso, ma è anche quella che si forma per prima in un processo di fusione frazionata. Sotto questo aspetto la granitizzazione può assumere un nuovo significato se viene concepita come la formazione di rocce attraverso un processo di fusione parziale e progressiva, con limitato movimento del materiale fuso.
Esperienze recenti di von Platen (v., 1965) hanno dimostrato che l'aggiunta di componenti femici non modifica in modo sensibile la situazione. Lo studio del sistema qz-ab-an-or a una pressione di vapor d'acqua di 2.000 kg•cm-2 ha dimostrato che in presenza di anortite il campo del feldspato si estende a spese del quarzo: in esso compare quale nuova fase il plagioclasio (v. fig. 12). I tre campi di esistenza convergono in un eutettico ternario quarzo-alcalifeldspato-plagioclasio. Con l'aumento del contenuto in calcio si estende il campo del plagioclasio e l'eutettico si sposta verso il lato qz-or, di modo che si ottengono fusi residuali ricchi in questi due componenti.
A differenza dei graniti alcalini, i punti corrispondenti alle composizioni dei graniti ricchi di calcio non si addensano in corrispondenza dell'eutettico, per cui non è più lecito considerare i secondi come rappresentanti di ultimi prodotti di cristallizzazione; le granodioriti e le quarzodioriti vengono piuttosto interpretate come prodotti di stadi antecedenti di differenziazione di magmi femici. Ciò non significa che le esperienze di von Platen portino a una conclusione in contraddizione con quelle di Bowen e Tuttle; se infatti esse possono confermare il modello precedente di Bowen, secondo cui i graniti possono derivare per differenziazione da magmi basaltici, d'altro canto possono indicare che liquidi di composizione compresa fra E ed F si possono formare quale primo prodotto di fusione progressiva di qualsiasi materiale silicatico.
In conclusione, le osservazioni geologiche e gli argomenti chimici indicano non solo come possibile ma come altamente probabile per molti graniti un'origine per metamorfismo di alto grado di materiali crostali, attraverso un processo di fusione graduale; è quindi pienamente giustificato l'uso del termine ‛magmi granitici', anche se tali magmi si sono formati attraverso processi di anatessi. Rimane pur sempre possibile, ed è stata in alcuni casi, sia pure molto rari, verificata, l'ipotesi di un'origine per differenziazione di magmi femici, così che lo schema generale della differenziazione magmatica proposto dal Bowen conserva, sia pure con le dovute limitazioni, la sua validità.
d) Altri tipi di rocce.
Oltre ai basalti e ai graniti, altri tipi di rocce ignee hanno in questi ultimi anni attratto l'attenzione dei petrologi e dei geochimici per il loro significato nella storia dell'evoluzione geochimica della crosta.
Le ‛andesiti', rocce vulcaniche a chimismo intermedio costituite da plagioclasi di media acidità, pirosseni e orneblenda, assieme alle rioliti, sono le più importanti rocce effusive rappresentate nelle zone orogeniche, per esempio nella cordigliera americana; sono anche presenti in alcune provincie medio-atlantiche. Vi sono prove evidenti che i magmi andesitici possano derivare per differenziazione da magmi basaltici secondo lo schema
olivin-tholeiite→tholeiite→andesite (riolite).
Tale è il caso delle andesiti del vulcano terziario Thingmuli nell'Islanda orientale. D'altra parte, poiché alcune considerazioni sulle abbondanze di K, Rb, Th, U, Li e delle terre rare escludono per le andesiti dei territori orogenici una parentela con magmi granitici, si fa strada l'idea che molti magmi andesitici siano primari e che si siano originati per fusione in tre o più stadi di materiali del mantello lungo la zona di Benioff che si affonda sotto i continenti, a una profondità variabile da 80 a 200 km, con contributi insignificanti di materiale crostale (v. fig. 13). Ciò è confermato anche dall'abbondanza degli elementi minori nelle andesiti (v. Taylor e White, 1966).
Una derivazione di liquidi simili per composizione alle andesiti calcio-alcaline per fusione parziale diretta del mantello superiore idrato intorno a 15 kbar è anche comprovata da recenti studi sperimentali sulle relazioni di fase nel sistema forsterite-plagioclasio-silice-acqua a 15 kbar di pressione in condizioni di saturazione di H2O (v. Kushiro, 1974).
Le ‛rocce ultrafemiche', costituite essenzialmente da pirosseno o da olivina o da associazioni di olivina con pirosseno o anfibolo, costituiscono un altro problema ancora non completamente risolto nella petrogenesi.
Alcune peridotiti sono chiaramente da considerare come ‛cumuliti', ossia sono da ritenere derivate da accumulo stratificato di olivina e/o di pirosseno per differenziazione dovuta alla gravità durante la cristallizzazione di magmi gabbrico-basaltici. Tale è il caso delle peridotiti dei citati complessi Stillwater, del Bushveld e del sill di Palisades nel New Jersey.
Per altri è evidente il carattere intrusivo. In questi casi vi sono alcune difficoltà nello spiegarne il meccanismo, perché l'intrusione di magmi di composizione peridotitica richiede alte temperature (circa 1.400 °C) per ammettere le quali non vi sono prove geologiche convincenti, benché l'assenza di fenomeni di metamorfismo termico di contatto possa trovare anche una spiegazione. È stato ipotizzato che l'intrusione di materiale ultrafemico si sia verificata sotto forma di una sorta di emulsione di cristalli di olivina e/o di pirosseno in un mezzo che potrebbe essere un liquido basaltico interstiziale o vapor d'acqua compresso. Un problema strettamente connesso è quello della serpentinizzazione. Due meccanismi si possono immaginare: il primo, rappresentabile con la reazione
3Mg2SiO4+SiO2+4H2O→2 (OH)4Mg3Si2O5.
comporta un aumento di volume del 70%; il secondo, a volume costante, comporta la rimozione di MgO e di SiO2:
5Mg2SiO4+4H2O→2 (OH)4Mg3Si2O5+4MgO+ SiO2.
Non vi è unanimità di giudizio sui due possibili processi, benché molti autori siano in favore del primo.
Poiché è noto che il serpentino non è stabile al di sopra di 500 °C a qualsiasi pressione, è chiaro che la serpentinizzazione dev'essersi verificata al di sotto di questa temperatura.
Estremamente indicativi sono i dati relativi alla composizione isotopica dell'idrogeno e dell'ossigeno nelle serpentiniti; essi dimostrano molto chiaramente la partecipazione di acque meteoriche profonde al processo di serpentinizzazione, mentre le serpentiniti oceaniche della dorsale medio-atlantica rivelano l'influenza dell'acqua marina, come è dimostrato anche dal rilevante contenuto in boro di queste rocce.
Per l'insieme dei loro caratteri le rocce ultrafemiche si possono considerare come, prodotti di differenziazione del mantello e probabilmente costituiscono il residuo refrattario presente localmente nel mantello superiore (v. fig. 13), spinto in alto da scorrimento plastico negli stadi precoci di ripiegamenti di geosinclinali. Ciò spiega anche la presenza di pirosseni alluminiferi, che richiedono per la loro stabilità condizioni di alta pressione.
7. Alla ricerca dei principi che regolano il comportamento degli elementi nel processo magmatico.
Lo studio del comportamento degli elementi nei processi geologici è stato uno degli obiettivi fondamentali della geochimica fin dall'inizio dello sviluppo di questa disciplina su basi razionali. In tale senso il Goldschmidt aveva impostato la sua opera fondamentale Geochemische Verteilungsgesetze der Elemente (v. Goldschmidt e altri, 1923-1937) e invero fin da quell'epoca la conoscenza delle strutture cristalline e delle dimensioni degli atomi e degli ioni nei cristalli ha consentito non solo di gettare le basi della cristallochimica, ma di formulare i principi generali che regolano il comportamento degli elementi.
I prodotti finali del processo magmatico, le rocce ignee, sono per la maggior parte costituiti da aggregati di minerali cristallizzati; si comprende perciò come nelle leggi della cristallochimica siano stati ricercati in prima istanza i principî che regolano la distribuzione degli elementi. Gli aspetti della cristallochimica che più strettamente sono coinvolti nel comportamento geochimico degli elementi sono quelli relativi alla natura del legame chimico, alle dimensioni degli atomi e degli ioni nei cristalli, alla loro coordinazione, alle trasformazioni di fase (polimorfismo), alla miscibilità allo stato solido (isomorfismo), alle azioni stabilizzatrici e perturbatrici del campo cristallino e alla valutazione dell'energia reticolare.
Un'interpretazione più generale del comportamento geochimico degli elementi in un processo magmatico dev'essere basata sullo studio dei coefficienti di distribuzione tra le diverse fasi solide o tra fasi solide e fasi liquide, coefficienti che a loro volta sono legati alle variazioni del potenziale chimico nel passaggio dall'una all'altra fase. Soltanto alcuni di tali aspetti verranno considerati in questa breve trattazione.
a) Le dimensioni degli atomi e degli ioni nei cristalli.
Fra i tipi di legame chimico che maggiormente interessano i composti naturali dobbiamo considerare il legame atomico, omeopolare o covalente, il legame metallico e il legame ionico, un particolare caso del legame elettrostatico. Nei cristalli a legame metallico gli atomi dell'elemento metallico sono a contatto fra loro e la semidistanza minima fra atomo e atomo può essere assunta, come ‛raggio atomico' dell'elemento. Similmente possono venir calcolati i valori dei ‛raggi covalenti' dalle distanze interatomiche in cristalli a legame covalente: questi valori possono anche essere ottenuti con metodi spettroscopici e dallo studio delle molecole gassose per mezzo della diffrazione elettronica.
In molti composti naturali inorganici il tipo di legame ha un'elevata percentuale di carattere ionico; la misura delle distanze interioniche in questi tipi di cristalli fornisce i cosiddetti ‛raggi ionici' degli elementi. Per la grande diffusione in natura dei minerali a legame parzialmente ionico, è la conoscenza dei valori dei raggi ionici che ha maggiormente attratto l'attenzione dei mineralogisti e dei geochimici.
Si possono enumerare molti metodi di determinazione del raggio ionico, ma i valori dei raggi ionici in uso nella geochimica si riferiscono solo ad alcuni di essi, che sono maggiormente noti. I primi valori dei raggi ionici nei cristalli furono ottenuti dal Landé (v., 1920) in base a considerazioni sulle interdistanze anione-catione e anione-anione in serie di composti cristallizzati di tipo AB aventi struttura come NaCl. Una volta stabilito che due anioni sono a contatto, la metà dell'interdistanza può essere assunta come raggio dell'anione. Indipendentemente Wasastjerna (v., 1923) otteneva valori concordi dei raggi ionici da misure della rifrazione molare degli ioni. Partendo dai dati di Landé e di Wasastjerna e utilizzando i valori delle distanze interioniche nei cristalli a legame ionico, Goldschmidt (v., 1926) poteva calcolare i raggi ionici per molti elementi nei diversi stadi di ossidazione. Un metodo semiempirico è stato sviluppato da L. Pauling (v., 1927) basandosi sulla relazione, valida per una serie di ioni isoelettronici, come ad es. Na+, Mg2+, Al3+... .

dove R1 è il raggio ionico, Z il numero atomico, K una costante e S una ‛costante di schermo'. I valori così ottenuti sono quelli degli ioni considerati come univalenti. I raggi degli ioni plurivalenti R2 vengono ottenuti da quelli univalenti R1 mediante la relazione
Rz=R1Z-2/(n-1) (4)
ove Z è la carica (valenza) dello ione e n l'esponente di Born. Quest'ultimo valore deriva dall'espressione del potenziale di repulsione fra ioni ϕ=be2/rn, dove b è un coefficiente di repulsione e r l'interdistanza.
I raggi ionici di Goldschmidt e di Pauling riportati nei testi sono calcolati per numero di coordinazione sei. Se il numero di coordinazione è diverso da sei, dev'essere apportata una correzione secondo la formula
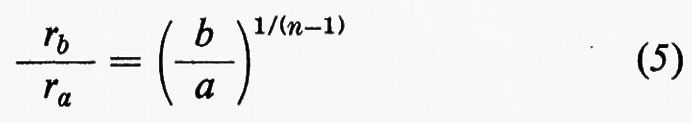
dove rb è il raggio ionico per numero di coordinazione b, ra quello per numero di coordinazione a, b/a il rapporto fra i due numeri di coordinazione ed n ancora l'esponente di Born.
L. Ahrens (v., 1952) ha preso in esame le possibili correlazioni fra raggio ionico e potenziale di ionizzazione e ha potuto stabilire una serie di relazioni empiriche che legano le due grandezze. Così, per esempio, per una serie di ioni di uguale carica (Na+, K+, Rb+, Cs+; Mg2+, Ca2+, Sr2+, Ba2+) si trova un'espressione del tipo

dove R è il raggio ionico, K una costante e V il potenziale di ionizzazione; per gli ioni di uno stesso elemento a carica differente si trova

dove Rx+ è il raggio dell'atomo x volte ionizzato, n una costante che risulta uguale a 0,27 per tutti gli elementi, Vx il potenziale di xma ionizzazione e K una costante caratteristica di ciascun elemento.
Nelle serie isoelettroniche si ha una variazione regolare del raggio ionico col potenziale di ionizzazione e altre regolarità si osservano per effetto della contrazione lantanidica. Si possono infine mettere in evidenza delle correlazioni fra raggio ionico e numero atomico nelle serie a 8 elettroni e a 18 elettroni periferici.
Le interdipendenze così poste in evidenza consentono di determinare o di prevedere il valore del raggio ionico per elementi per i quali non si dispone di dati cristallografici sufficienti. In questo modo sono stati determinati i raggi incogniti di Ag2+, At7+, Au3+, I5+, Np7+, Pa5+, P06+, Re7+ e altri.
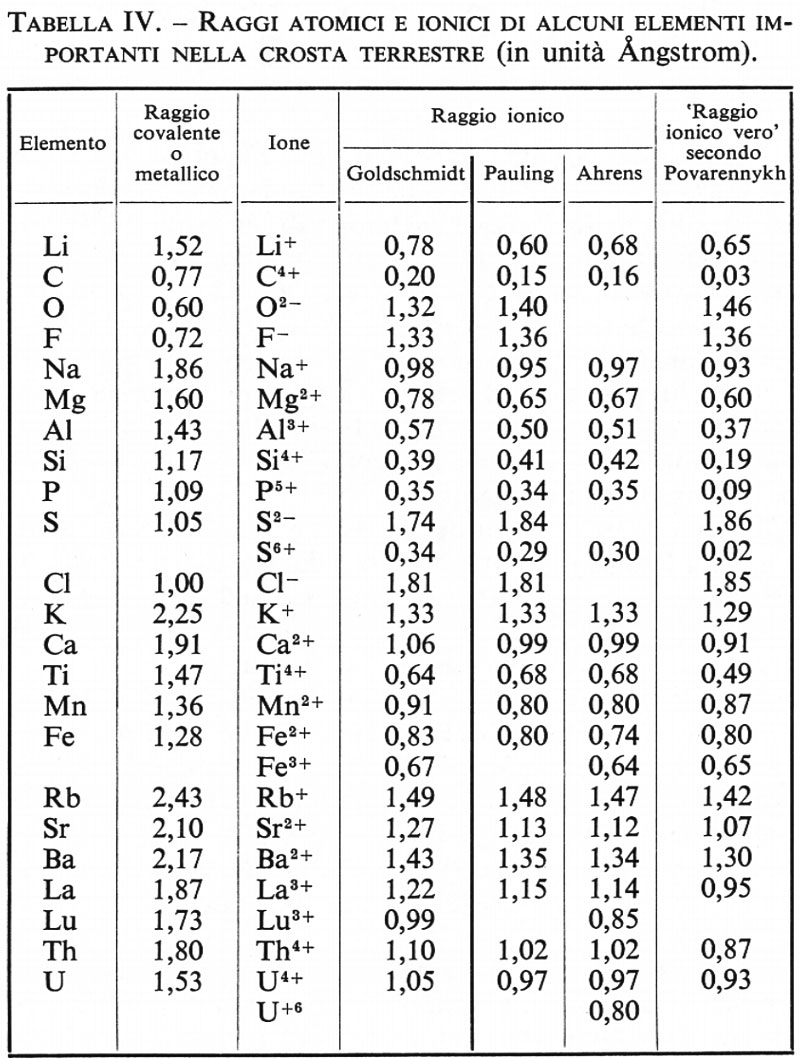
A. S. Povarennykh (v., 1964) ha giustamente rilevato che l'opinione diffusa che le dimensioni degli ioni siano definite e costanti è dovuta all'influenza dominante del pensiero di Goldschmidt e di Fersman sulla concezione sterica delle strutture cristalline. I valori dei ‛raggi ionici' di molti elementi presentano notevoli discrepanze in diversi composti a diversa percentuale di carattere ionico. I metodi correntemente usati per misurare il raggio ionico non ne forniscono in realtà il ‛valore vero', perché molti elementi (B, Si, Ti, P, V) non sono in condizione di formare con l'ossigeno veri composti ionici, ma danno luogo piuttosto a ossidi di carattere semicovalente. Soltanto per i cationi meno elettronegativi i cosiddetti raggi ionici si accostano di più al raggio ionico vero, ma col crescere della elettronegatività, se il calcolo è fatto in relazione all'ossigeno, se ne possono scostare notevolmente. Si può prevedere che il raggio effettivo di un elemento in un composto possa variare in continuità in dipendenza dal tipo di legame, entro limiti che possono esser dati dal raggio dell'elemento nei composti a puro carattere ionico da un lato e dall'altro dai cosiddetti ‛raggi atomici' o ‛raggi metallici'.
‟Ne segue che per determinare il raggio effettivo di un determinato elemento in un composto è necessario cono- scere lo stato di legame chimico (grado di covalenza) in questo composto e i valori dei raggi ionico e covalente di questo elemento" (ibid., p. 151).
L'autore assume come valore del ‛raggio covalente' il raggio metallico e come ‛raggio ionico vero' quello ottenuto per estrapolazione a grado di covalenza zero.
Il grado di covalenza o, se vogliamo, la percentuale di legame ionico è, come è ben noto, legato alla differenza di elettronegatività delle due specie atomiche fra cui si verifica il legame.
La tab. IV contiene i valori dei ‛raggi ionici veri' secondo Povarennykh, dai quali si possono poi calcolare i raggi effettivi negli ossidi, nei solfuri e in qualsiasi altra combinazione.
b) Raggi ionici e coordinazione.
Poiché ogni struttura cristallina rappresenta una condizione di equilibrio, corrispondente a un minimo di energia potenziale, tale condizione imporrà anche il miglior adattamento fra le particelle coordinanti e le particelle coordinate. La struttura di un cristallo ionico è determinata dalla disposizione per cui ciascuno ione è attorniato dal massimo possibile di ioni vicini di carica opposta, compatibilmente con la condizione che la distanza di equilibrio catione-anione sia uguale alla somma dei due raggi ionici. Ne risulta che il numero di particelle coordinate - o numero di coordinazione - attorno a uno ione centrale deve dipendere essenzialmente dal rapporto dei rispettivi raggi ionici, secondo la cosiddetta ‛regola di Magnus'. Così se il rapporto rc/ra fra il raggio del catione e il raggio dell'anione è compreso fra 0,15 e 0,22 si trova che ogni catione si circonderà di tre anioni, cioè avrà numero di coordinazione 3; se rc/ra è compreso fra 0,22 e 0,41 si avrà coordinazione 4 e la figura di coordinazione sarà un tetraedro o più raramente un quadrato. Per valori di rc/ra compresi fra 0,41 e 0,73 si avrà coordinazione ottaedrica (n. c. = 6); per valori compresi fra 0,73 e 1 il numero di coordinazione varia da 8 a 12 con poliedri di coordinazione costituiti da un cubo (n. c. = 8), da un prisma triangolare a facce centrate (n. c. = 9) o da un cubo-ottaedro (n. c. = 12) (v. fig. 14).
La regola di Magnus consente una previsione abbastanza precisa del numero di coordinazione. In realtà l'accordo fra il numero di coordinazione previsto e quello osservato e, di norma, abbastanza buono nei composti ossigenati e nei composti a carattere ionico in genere; in altri casi si verificano frequenti eccezioni: per esempio nei cristalli di AgI, CdS, ZnS, ZnO l'argento, il cadmio e lo zinco presentano numero di coordinazione 4 anziché 6; in BeS, BeSe, BeTe il numero di coordinazione del Be è 4 anziché 3. Ciò può essere spiegato dal notevole grado di covalenza del legame in questi composti per cui i legami ibridi tetraedrici sp3 risultano prevalenti. Nuovamente è necessario porre in guardia contro il preconcetto di considerare gli ioni come sfere rigide e di pretendere di spiegare tutto con semplici considerazioni geometriche, mentre non debbono essere trascurate le configurazioni dei legami di valenza.
La conoscenza dei raggi ionici e dei rapporti di coordinazione nei cristalli è di primaria importanza per comprendere il comportamento geochimico degli elementi nel processo magmatico. In effetti l'uguaglianza o la vicinanza dei valori del raggio ionico di due elementi dà a essi, indipendentemente dalla loro carica e anche, largamente, dal carattere chimico, la possibilità di sostituirsi vicendevolmente nelle strutture cristalline per dar luogo alla formazione - in tutti i rapporti o in rapporti limitati - di soluzioni solide o di cristalli misti. Non solo gli elementi principali possono sostituirsi fra loro nei minerali più comuni delle rocce ignee, ma anche gli elementi in tracce possono così essere accomodati nelle loro strutture in quantità che saranno poi regolate dai rapporti di partizione e dalle condizioni energetiche.
Quando un minerale costituito da una soluzione solida di due o più componenti si separa per cristallizzazione da una massa fusa, possiamo immaginare che si verifichi una competizione fra gli ioni vicarianti presenti nel fuso per raggiungere una posizione nella struttura cristallina. Se prendiamo per esempio la formazione dell'olivina, dal diagramma di stato fayalite-forsterite (v. fig. 15) si deduce che i primi cristalli che si separano dal fuso sono più ricchi di magnesio rispetto al fuso iniziale. In questo caso è lo ione magnesio Mg2+ (raggio ionico = 0,67 Å) che si colloca nella struttura a preferenza dello ione ferro Fe2+ (r. i. = 0,74 Å). Similmente nella formazione dei cristalli misti di plagioclasio i cristalli di prima formazione sono più ricchi di calcio ed è lo ione calcio Ca2+ (r. i. = 0.99 Å) che si colloca nella struttura a preferenza dello ione sodio Na+ (r. i. = 0,97 Å). Ne risulta che il rapporto Fe:Mg nelle olivine e nei pirosseni cresce col procedere della differenziazione e altrettanto si verifica per il rapporto Na:Ca nei plagioclasi.
c) Le regole di Goldschmidt e le loro eccezioni.
Il Goldschmidt aveva espresso nelle seguenti tre regole empiriche il comportamento di distribuzione degli elementi nel corso della consolidazione magmatica: a) se due ioni hanno lo stesso raggio, la stessa carica e costituzione elettronica simile, essi entreranno con la stessa facilità nella struttura cristallina di un minerale (principio di mascheramento); b) se due ioni hanno raggi ionici vicini, ma non uguali, e la stessa carica, lo ione più piccolo entrerà più facilmente nella fase solida, mentre lo ione di dimensioni maggiori rimarrà concentrato nelle frazioni ancora fuse, che cristallizzano a più bassa temperatura (principio di arricchimento); c) se due ioni hanno raggi simili ma carica differente, quello a carica maggiore entrerà più facilmente in una data struttura cristallina.
Queste regole conseguono direttamente dal modello elettrostatico adottato.
Principi analoghi regolano la distribuzione degli elementi in tracce nei minerali principali: quando uno ione di un costituente minore sostituisce uno ione di carica uguale e di raggio ionico simile si parla di ‛mascheramento' (Tarnung): un esempio di questo tipo è dato dalla sostituzione del Ga3+ (r. i. = 0,62) all'Al3+ (r. i. = 0,51 Å). Se uno ione di un costituente minore sostituisce un costituente principale avente carica inferiore si ha la cosiddetta ‛cattura' (Abfangen). Tale è il caso della sostituzione dello Sc3+ (r. i. = 0,81 Å) al magnesio Mg2+ (r. i. = 0,67 Å) e del Pb2+ (r. i. = 1,20 Å) al K+ (r. i. = 1,33 Å). Si può avere cattura anche quando uno ione ne sostituisce un altro a carica uguale ma a raggio maggiore, per es. quando Mn2+ (r. i. = 0,80 Å) sostituisce Ca2+ (r. i. = 0,99 Å). Si osservi come queste sostituzioni si possano verificare, sia pure in misura limitata, indipendentemente non solo dalla carica, ma anche dal carattere chimico dell'elemento. Così il piombo, notoriamente calcofilo, può prendere posto nella struttura dei feldspati e assume in questo caso un comportamento ossifilo.
Un ultimo caso, caratterizzato col termine di ‛ammissione', si verifica quando uno ione di un costituente minore sostituisce quello di un elemento comune avente carica maggiore. Tale è il caso della sostituzione del Li+ (r. i. = 0,68 Å) al Mg2+ (r. i. = 0,67 Å).
Molte ricerche sono state condotte nell'ultimo ventennio sulla distribuzione degli elementi in tracce nel corso della cristallizzazione magmatica; fra esse meritano una menzione particolare quelle di Wager e Mitchell (v., 1951) sulla intrusione di Skaergaard. Per un'ampia rassegna del comportamento dei singoli elementi può esser fatto riferimento al lavoro di S. R. Taylor (v., 1965).
Le tre regole enunciate da Goldschmidt nel 1937 hanno avuto per parecchi anni molta fortuna e sono attraenti per la loro semplicità. Esse sono basate sostanzialmente sul modello elettrostatico e tengono conto del raggio e della carica ionica. Successive ricerche hanno dimostrato che queste regole mantengono una validità generale nel caso di elementi che formano legami altamente ionici, ma hanno posto in evidenza numerose eccezioni, fra le quali vale la pena di ricordare come particolarmente significative le seguenti, enumerate da Shaw (v., 1964).
1. Il Ga è mascherato nei minerali di Al3+ a dispetto del raggio ionico (0,62 Å) che differisce più del 20% da quello dell'alluminio (0,51 Å). I due elementi nei silicati sono geochimicamente coerenti e non vi è alcun indizio di una concentrazione del gallio nelle frazioni tardive.
2. Il Ni (r. i. = 0,69 Å), nonostante il valore del suo raggio ionico, che è maggiore di quello del magnesio (0,66 Å), si accumula nelle frazioni precoci delle olivine.
3. Vi sono numerosi sistemi binari con miscibilità completa allo stato solido per cui il principio di arricchimento non vale; un esempio è dato dalla coppia SrAl2Si2O8 - CaAl2Si2O8 in cui la mancata validità del principio di arricchimento è dimostrata dal confronto dei punti di fusione dei due termini estremi delle miscele isomorfe. Altri casi in cui detto principio non è rispettato sono quelli offerti da sistemi binari con miscibilità completa allo stato solido e con un minimo o un massimo nelle curve di fusione. Tale è il caso che si verifica nel sistema NaAlSi3O8 - KAlSi3O8. In questo caso se il principio di arricchimento è rispettato in una parte del diagramma di stato non lo è, ovviamente, nell'altra.
4. È opportuno poi notare che nei casi in cui si ha sostituzione di uno ione a piccola carica con uno a grande carica si rende necessaria la contemporanea sostituzione di uno ione a grande carica con uno a piccola carica, quest'ultima sostituzione essendo in contrasto con le regole di Goldschmidt.
d) Nuovi criteri e nuovi principî.
Da quanto si è detto emerge chiaramente che la sola considerazione della carica e del raggio ionico non è un criterio sufficiente né universale per giustificare e prevedere il comportamento degli elementi nella cristallizzazione magmatica. Allo scopo di stabilire criteri più validi, numerosi tentativi sono stati effettuati negli anni recenti, in cui altre grandezze sono state prese in considerazione.
Elettronegatività (v. Fyfe, 1951; v. Ringwood, 1955). - Il principio che fa uso di questa grandezza può essere enunciato nel modo seguente: ‟Se consideriamo due ioni aventi uguale carica e raggi non molto differenti, quello dei due che ha più bassa elettronegatività sarà incorporato di preferenza in una struttura cristallina".
Il criterio della elettronegatività è basato sulla considerazione che il catione a cui spetta il valore più basso della elettronegatività favorisce la formazione di un legame ionico, il quale secondo Ringwood è più stabile. Se tale principio ha avuto successo nello spiegare alcuni casi, fra cui quello citato, in cui le regole di Goldschmidt non sono rispettate, in altri casi anche questo criterio fallisce. Il criterio della elettronegatività è basato, fra l'altro, sul presupposto che il legame ionico sia necessariamente più forte del legame covalente, il che non sembra sia dimostrato.
Funzione del campo. - Ahrens ha introdotto una grandezza F detta ‛funzione del campo', definita come il rapporto V/R, dove V è il potenziale di ionizzazione ed R il raggio ionico, quale misura della capacità di uno ione di vincere la competizione per l'incorporazione preferenziale in una struttura cristallina. Secondo Ahrens ‟quando due ioni della stessa valenza aventi raggi uguali o simili sono in competizione per una data posizione strutturale in un cristallo in via di accrescimento, quello avente la maggiore intensità elettrica giungerà per primo" (v. Ahrens, 1953, p. 14). Questa regola è utile in quanto può differenziare il comportamento di ioni aventi uguale raggio e uguale carica e pertanto non differenziabili in base ai principi cristallochimici convenzionali.
Energie totali di legame. - Nockolds (v., 1966) cerca di ovviare agli inconvenienti menzionati usando una funzione nella quale sono compresi carica ionica, raggio ionico ed elettronegatività, nonché un eventuale contributo dell'energia di stabilizzazione del campo cristallino. Tale funzione, denominata ‛energia totale di legame' (Et), risulta costituita da tre termini, uno dei quali (Ec) rappresenta il contributo del legame covalente, il secondo (Ei) il contributo del legame ionico, entrambi riferiti a legami singoli univalenti, il terzo (Es) è l'eventuale contributo di energia di stabilizzazione del campo cristallino. Si ha pertanto
Et=(Ec+Ei)Zc+Es (8)
essendo Zc la carica formale o valenza del catione.
Valori scelti delle energie totali di legame X−O per diversi ioni sono riportati in tab. V. Il significato dell'energia totale di legame va inteso nel senso che quando due cationi della stessa carica sono capaci di sostituirsi in una struttura cristallina, quello che ha la maggior energia di legame vi sarà incorporato di preferenza, e quando una coppia di cationi, per es. Ca2+−Al3+, sostituisce un'altra coppia di cationi, per es. Na+−Si4+, si verificherà di preferenza quella sostituzione per cui la somma delle energie di legame è maggiore. È da rilevare che l'uso delle energie totali di legame non è ancora sufficiente a giustificare completamente il comportamento degli elementi, particolarmente per quanto si riferisce alle relazioni di fusione di alcuni dei loro composti.
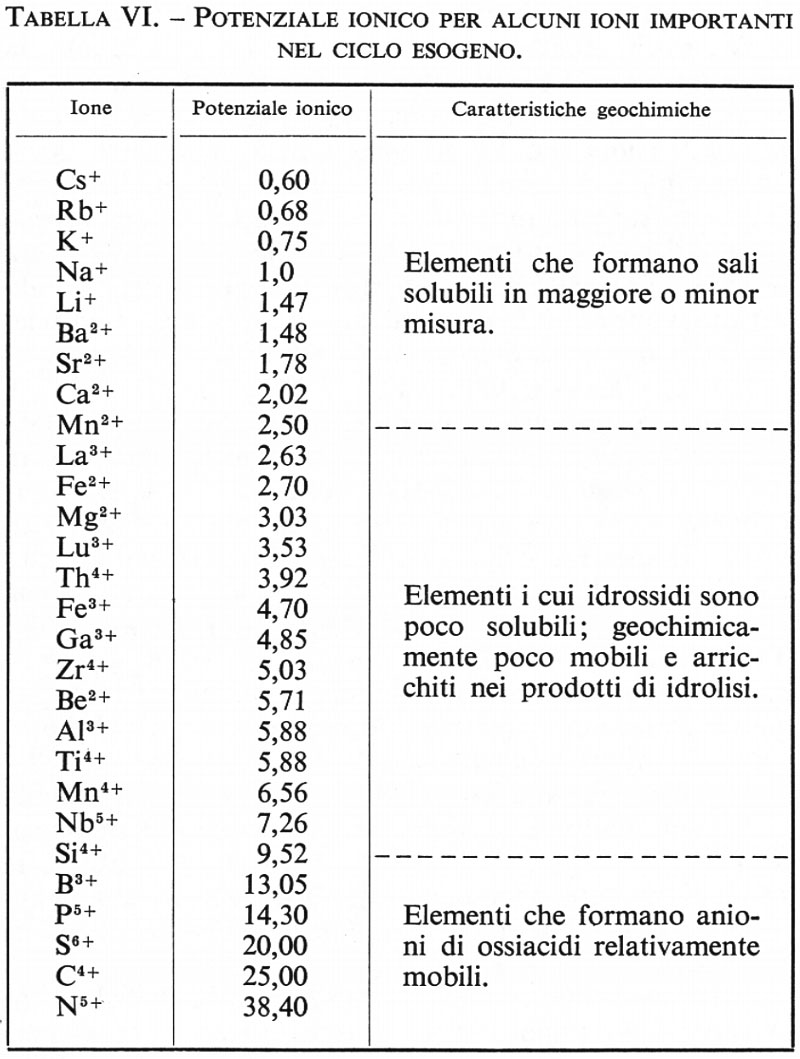
Una delle ragioni delle difficoltà che s'incontrano nella ricerca di principi di validità generale per prevedere il comportamento degli elementi nel corso della cristallizzazione magmatica risiede nel fatto che tale comportamento non è solamente legato alle energie di legame nei composti cristallizzati, ma dipende essenzialmente da tutte le grandezze termodinamiche relative alle reazioni e trasformazioni di fase in cui sono coinvolti i composti degli elementi in questione. Ciò nonostante, l'impiego delle energie totali di legame costituisce un rilevante progresso rispetto ai criteri seguiti in precedenza.
Teoria del campo cristallino. - Il termine Es, nell'equazione precedente, scaturisce dalla teoria del campo cristallino (Crystal Field Theory=CFT). Questa teoria considera le perturbazioni che subiscono gli ioni dei metalli di transizione quando vengono a trovarsi in un ‛campo' ottaedrico, tetraedrico o cubico, quale si viene a determinare nelle strutture cristalline in corrispondenza di spazi o ‛siti' corrispondenti a poliedri di coordinazione rispettivamente ottaedrica, tetraedrica o cubica.
Alla base della teoria è la considerazione che, mentre in un catione isolato di un elemento di transizione (campo sferico) i cinque orbitali d sono degeneri, cioè hanno la stessa energia, la situazione è diversa se s'immagina lo stesso ione collocato in un poliedro di coordinazione (ottaedro, tetraedro, cubo) ai vertici del quale sono collocati gli anioni. In ognuno dei tre casi si verifica una scissione degli orbitali in due sottolivelli aventi differente energia. In campo ottaedrico i cinque orbitali d si scindono nei due gruppi eg e t2g e al gruppo eg spetta l'energia maggiore. In campo tetraedrico la maggiore energia spetta al gruppo t2 e analogamente in campo cubico la maggiore energia spetta al gruppo t2g. Se chiamiamo Δo la differenza di energia fra i due sottolivelli orbitali in campo ottaedrico si può dimostrare che Δt=4/9 Δo e Δc=8/9 Δo (v. fig. 16). Il valore netto dell'energia di stabilizzazione si calcola, nel caso del campo ottaedrico, assegnando un valore +2/5 Δo a ogni elettrone collocato in t2g e un valore −3/5 Δo a ogni elettrone collocato in eg e facendo la somma algebrica; nel caso del campo tetraedrico e del campo cubico, assegnando rispettivamente un valore −2/5 Δt e −2/5 Δt per ogni elettrone collocato in t2 e t2g e un valore di +3/5 Δc e +3/5 Δc per ogni elettrone collocato in e ed eg e sommando algebricamente. La valutazione del parametro Δ può avvenire con procedimenti ottici o termodinamici.
Il risultato netto del calcolo è quello che si chiama energia di stabilizzazione del campo cristallino (Crystal Field Stabilization Energy=CFSE).
È opportuno notare che ioni di elementi di transizione aventi uguale raggio ionico (per es. Cr3+, r. i. = 0,63 Å e Fe3+, r. i. = 0,64 Å) non hanno necessariamente lo stesso valore della CFSE, la quale per il Cr3+ è di 59,6 kcal/mole e per il Fe3+ è uguale a zero. Si comprende quindi come i due ioni, per i quali era prevedibile un analogo comportamento cristallochimico (mascheramento nel senso di Goldschmidt), abbiano viceversa un diverso comportamento nel corso della cristallizzazione magmatica, dovuto all'influenza delle forze del campo.
Per una più ampia trattazione delle applicazioni mineralogiche e geochimiche della CFT il lettore potrà fare riferimento al trattato di Burns (v., 1970).
Processi di frazionamento degli elementi. - Per comprendere il meccanismo del frazionamento degli elementi in tracce nel processo magmatico è necessario riferirsi alla legge di partizione di Nernst applicata a un sistema solido-liquido, secondo la quale il rapporto fra la concentrazione di un elemento nella fase solida e la concentrazione dello stesso elemento nella fase liquida, detto coefficiente di distribuzione K, è costante qualora non varino la pressione, la temperatura e la composizione, ossia
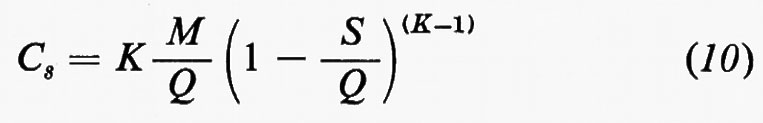
In un processo di cristallizzazione frazionata si può dimostrare (v. Neumann, Mead e Vitaliano, 1954) che la concentrazione di un elemento in tracce nella fase solida in ogni istante è data da
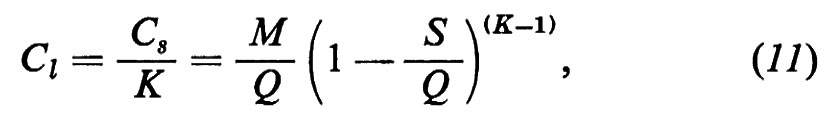
e nella fase liquida restante da
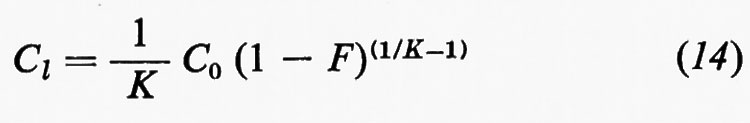
essendo Q la quantità originale del fuso, M la quantità di elemento in tracce nel fuso, S la quantità di fase solida, K il coefficiente di distribuzione.
Le famiglie di curve riportate in fig. 17, A e B, rappresentano la variazione di concentrazione di un elemento in tracce rispettivamente nella fase liquida e nella fase solida in funzione del grado di solidificazione F=S/Q per diversi valori di K. Si può notare come l'effetto di frazionamento sia notevole al crescere di F quando il valore di K è molto diverso dall'unità.
Il caso considerato si riferisce a una sola fase solida cristallina in equilibrio con un fuso e presuppone che K sia indipendente dalla temperatura, dalla pressione e dalla composizione, ciò che in realtà si verifica raramente. Qualora il sistema comprenda due o più fasi solide, l'equazione (10) può essere ancora impiegata usando un coefficiente di distribuzione combinato espresso da
K=r1K1+r2K2+...rnKn, (12)
dove K1, K2 ... Kn sono i fattori di distribuzione per le diverse fasi solide e r1, r2 ... rn i rapporti fra le rispettive quantità e la quantità totale. Nei sistemi in cui si verifica un riassorbimento di una o più fasi solide le equazioni divengono alquanto più complicate.
In un processo di fusione frazionata si può dimostrare (v. Shaw, 1970) che, se ogni frazione di liquido è continuamente rimossa, la concentrazione di un elemento in tracce nel solido è data da
Cs=C0 (1−F)(1/K-1), (13)
la concentrazione in ogni frazione di liquido da
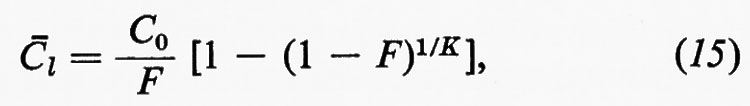
e la concentrazione nel totale del liquido rimosso da
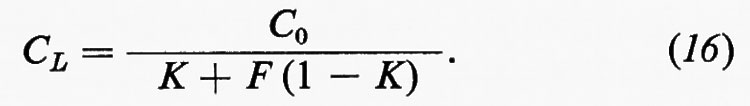
dove C0 è la concentrazione iniziale nel solido, F il grado di solidificazione e K il coefficiente di distribuzione.
Se in luogo di essere continuamente rimosso il liquido rimane per tutto il tempo in equilibrio col solido, la concentrazione dell'elemento in tracce sarà data da
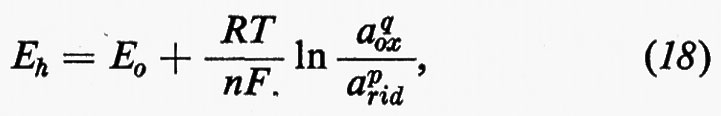
La fig. 18 rappresenta l'andamento di Cl, Ål e CL con il grado di fusione frazionata F nel caso ideale di un solido multifase in cui le quantità relative delle diverse fasi non cambiano durante la fusione parziale per un valore di K uguale a quattro. Se nel processo di fusione frazionata le diverse fasi solide fondono in proporzione differente, le equazioni (13-16) debbono essere convenientemente modificate.
Altri modelli possono essere sviluppati, come quello basato su un processo di raffinazione a zone ispirato ai processi recentemente sviluppati per la purificazione di metalli e di composti organici. In questi processi le impurezze si accumulano in una sezione trasversale fusa di piccolo spessore che scorre lungo un lingotto del materiale solido. Processi analoghi potrebbero verificarsi in scala geologica nei materiali del mantello e potrebbero giustificare l'accumulo dei cosiddetti elementi incompatibili, quali K, Ti, P, Ba, Cs, Rb, Sr, U e Th.
Considerazioni di questo tipo sono oggi alla base di molte speculazioni sull'origine e sull'evoluzione dei basalti. Il ragionamento consiste nel paragonare le distribuzioni osservate con quelle prevedibili in base all'uno o all'altro dei modelli adottati. Si è potuto così vedere, come già accennato al cap. 6, che la grande abbondanza degli elementi a grande raggio ionico nei basalti alcalini non può essere spiegata con un meccanismo di cristallizzazione frazionata, ma piuttosto con l'ipotesi di una fusione parziale delle associazioni mineralogiche che si ha ragione di ritenere siano presenti nel mantello superiore.
8. Problemi relativi alle fasi finali del processo magmatico.
Come indicato schematicamente al cap. 6, le fasi finali del processo magmatico, indicate col termine di ‛stadio tardo magmatico', sono caratterizzate dal progressivo accumularsi dei componenti volatili nel residuo fuso.
I prodotti che si formano in questa fase finale presentano caratteri peculiari, che vanno dalla formazione delle apliti e delle pegmatiti ai depositi pneumatolitici e idrotermali. Tutti questi prodotti presentano un estremo interesse dal punto di vista geochimico sia per l'intenso frazionamento degli elementi che in essi si può constatare, sia per quanto riguarda il meccanismo della loro formazione, sia infine per i problemi relativi al trasporto degli elementi.
Apliti e pegmatiti rappresentano rocce differenziate in senso acido e differiscono sostanzialmente per la tessitura, che è saccaroide nel caso delle apliti, a grana molto più grossa nelle pegmatiti; in queste ultime i cristalli assumono talora proporzioni gigantesche: sono noti cristalli di mica fino a 3 m di diametro, di berillo fino a 6 m, di spodumene fino a 15 m di lunghezza. Le pegmatiti granitiche sono costituite essenzialmente dall'associazione quarzo-feldspato, sovente in tipiche strutture di implicazione; nelle pegmatiti legate a sieniti eleolitiche si ha l'associazione di nefelina con feldspato e in quelle gabbriche di plagioclasio con orneblenda.
Apliti e pegmatiti si considerano entrambe come i prodotti della cristallizzazione di magmi ricchi in acqua. La solubilità dell'acqua in un fuso granitico varia con la pressione, come risulta dalle misure effettuate da Burnham e Jahns (v., 1962) riportate in fig. 19. L'effetto della presenza di acqua in un fuso della composizione indicata si manifesta con un abbassamento della temperatura di inizio di fusione, come è visibile in fig. 20: in presenza del 10% di H2O si possono avere fusi granitici a temperature relativamente basse. La presenza di altri componenti volatili (per es. HF) può abbassare ulteriormente la temperatura di cristallizzazione. Se in un magma saturo di acqua, a una determinata temperatura e pressione, in seguito a raffreddamento ha inizio la separazione di fasi solide, ciò determinerà una sovrassaturazione in acqua e il sistema raggiungerà il cosiddetto ‛secondo punto di ebollizione', in concomitanza con l'aumento retrogrado della tensione di vapore; a questo punto si verificherà nel fuso la formazione di bolle. Questo comportamento, per fusi a composizione quarzoso-feldspatica, è confermato da recenti esperienze di Jahns e Burnham (v., 1969). Raffreddando una massa fusa di questa composizione in presenza di una quantità d'acqua inferiore rispetto alla saturazione, si ottiene dapprima una segregazione abbondante di quarzo e feldspato in forma di aggregato granulare simile per tessitura alle apliti; procedendo nel raffreddamento, la massa diviene satura in acqua e si raggiunge il secondo punto di ebollizione: si separa una fase gassosa in forma di bolle nelle quali si sviluppano grandi cristalli di quarzo e di feldspati, che si alimentano dalla massa silicatica fusa ma si accrescono nelle bolle componendo una tessitura simile a quella delle pegmatiti. Sulla base delle indicazioni fornite da questa esperienza si possono interpretare le apliti come il prodotto di cristallizzazione di fusi in cui non si è ancora verificata la separazione di una fase acquosa, mentre le pegmatiti rappresenterebbero il prodotto della cristallizzazione di un sistema in cui è presente una fase gassosa nella quale i cristalli sono liberi di accrescersi. E necessario tuttavia precisare che, come i fusi a composizione granitica possono rappresentare sia il prodotto estremo di una differenziazione per cristallizzazione frazionata sia il primo prodotto di una fusione frazionata di materiali crostali, anche i fusi pegmatitici possono risultare da un analogo processo. Si conoscono in effetti due tipi di pegmatiti: le pegmatiti complesse e le pegmatiti semplici. Le prime si possono effettivamente considerare come gli ultimi prodotti di consolidazione di magmi granitici; le seconde sono interpretate come il primo prodotto della rifusione parziale di materiali crostali nei processi palingenici.
Da un punto di vista geochimico e mineralogico sono le pegmatiti complesse che presentano il maggior interesse, poiché in esse si ritrovano concentrati tutti quegli elementi che, per le loro carattenstiche cristallochimiche, non possono essere incorporati o lo sono in misura ridotta nelle strutture cristalline dei minerali principali durante lo stadio principale della cristallizzazione magmatica per i seguenti motivi: a) alcuni elementi hanno un raggio ionico troppo piccolo per essere inglobati con i menzionati meccanismi di mascheramento, cattura o ammissione; tale è il caso del litio, per la frazione eccedente quella che può sostituire il magnesio, del berillio e del boro; b) alcuni elementi presentano caratteristiche opposte, vale a dire hanno un raggio ionico eccedente i limiti di tollerabilità nelle sostituzioni: tali sono il rubidio, il cesio e, in parte, le terre rare; c) altri elementi, pur avendo un raggio ionico compatibile con le sostituzioni prevedibili nelle strutture dei minerali più comuni, per ragioni energetiche o di carica ionica possono sostituire solo limitatamente gli elementi principali, come si verifica per Zr, Hf ,Nb, Ta, U, Th.
Gli elementi ricordati raggiungono pertanto nei fusi pegmatici concentrazioni tanto alte da poter dar luogo alla formazione di minerali propri: elementi a scarsa abbondanza cosmica e crostale possono così diventare molto disponibili e sono utilizzabili economicamente.
Le pegmatiti sono costituite prevalentemente da silicati; esse appartengono ancora allo stadio magmatico della cristallizzazione e ne segnano la fine. Graduali passaggi portano, dalla fase pegmatitica, ai successivi stadi pneumatolitico e idrotermale.
Si ritiene correntemente che i depositi pneumatolitici prendano origine da fluidi in condizioni ipercritiche e i depositi idrotermali da soluzioni condensate a temperature inferiori. È molto difficile stabilire se un processo di consolidazione passi o meno per fasi ipercritiche. Attualmente si tende a non annettere eccessiva importanza, ai fini geologici, alla distinzione fra ‛condizioni supercritiche' e ‛condizioni ipocritiche' in quanto, considerazioni di temperatura a parte, i fluidi supercritici a bassa densità non si comportano diversamente, quanto a potere solvente, rispetto ai comuni vapori.
Se si fanno opportune ipotesi sulla composizione di una fase gassosa magmatica, prendendo per esempio come modello la composizione media di gas vulcanici e impostando i calcoli delle costanti di equilibrio sulla base delle energie libere delle possibili reazioni, si possono calcolare le concentrazioni di vari componenti alle diverse temperature (v. Krauskopf, 1967). Ferme restando le ipotesi di partenza, risulta che Cl2 e F2 sono trascurabili in ogni caso; SO2 prevale in condizioni ossidanti; H2S in condizioni riducenti; SO3 è subordinato; la pressione parziale dell'ossigeno è molto bassa. Nelle condizioni indicate Pb, Zn, Cu, Ag, Mo sono stabili come solfuri, Mn come silicato, W e Sn come ossidi. Il piombo presenta la massima volatilità come PbCl2, il molibdeno come ossido, il tungsteno come ossido idrato. A una temperatura di 827 °C, Fe, Mn, Zn, Pb, Cu, Ag, Mo e W hanno una tensione superiore a 10-6 atm e quindi sono presenti in fase gassosa in quantità sufficienti a giustificare l'accumulo di giacimenti metalliferi; a 627 °C, Fe, Mn, Zn, Pb hanno ancora una tensione superiore a 10-6 atm. Possono così essere interpretati molti depositi di magnetite, scheelite e calcopirite che si trovano in prossimità di contatti intrusivi.
Molti depositi di solfuri di metalli pesanti sono da porre in relazione con la fase idrotermale, secondo una scala di temperature decrescenti. La maggiore difficoltà che s'incontra nell'interpretazione della genesi dei depositi idrotermali risiede nel fatto che non sono ancora disponibili dati sufficienti sugli equilibri in cui sono interessati i metalli in soluzione alle alte temperature e pressioni. Questi metalli, d'altra parte, formano adunamenti di dimensioni minerarie in buona parte sotto forma di solfuri. Dai calcoli delle solubilità dei solfuri in diverse condizioni di acidità e di temperatura appare chiaro che le quantità di solfuri che possono essere trasportate dalle soluzioni termali sono in genere troppo piccole per spiegare la formazione, in un tempo e da una quantità di soluzione ragionevoli, di corpi delle dimensioni osservate.
Molte ipotesi sono state formulate per superare queste difficoltà: si è voluto, per esempio, ipotizzare che i solfuri si possano formare da soluzioni separate, contenenti rispettivamente gli ioni S2- e SH- e i cationi metallici, ovvero da sospensioni colloidali contenenti concentrazioni di solfuri largamente superiori a quelle consentite dal prodotto di solubilità. Tali ipotesi non si possono scartare a priori, ma ne mancano prove dirette, in particolare per quanto riguarda la stabilità delle sospensioni colloidali.
Le stesse condizioni di acidità delle soluzioni mineralizzanti sono oggetto di discussione: secondo un possibile modo di vedere, le soluzioni sarebbero, almeno inizialmente, tanto acide da trasportare gli elementi come semplici cationi metallici. In questo caso una diminuzione di acidità o un aumento di alcalinità diventano condizioni favorevoli alla precipitazione dei solfuri o degli ossidi. Dati recenti, ottenuti principalmente dallo studio delle inclusioni fluide nei minerali, indicano che le soluzioni idrotermali non debbono essere lontane dalla neutralità. Si fa strada allora l'opinione che gli elementi metallici, particolarmente quelli che formano solfuri meno solubili, siano trasportati in forma di complessi, come per es. HgS²2-, ZnHS-2, HgHS-2, PbHS-2, AgHS, AuS-, risultanti, almeno formalmente, da reazioni dei solfuri neutri con H2S e con gli ioni S2- e HS-. In questo caso diventano condizioni favorevoli alla precipitazione un aumento di acidità o una diminuzione di alcalinità. Può essere interessante osservare che l'oro metallico è solubile a temperatura ordinaria in soluzioni contenenti HS-, con formazione del complesso molto stabile AuS-. Il lettore potrà trovare un'esauriente trattazione quantitativa degli equilibri che interessano i sistemi acqua-solfuri metallici in relazione ai processi idrotermali in O. Smith (v., 1963), Barnes (v., 1960), Barnes e Kullerud (v., 1961).
Generalmente parlando si può asserire che l'equilibrio interno delle soluzioni idrotermali dipende in larga misura dalla formazione e dalla dissociazione dei complessi in corrispondenza alle variazioni di pressione, temperatura e attività delle specie ioniche. Calcoli eseguiti con l'aiuto di calcolatori sul sistema NaCl-HCl-H2O in equilibrio con galena a temperatura ambiente fino a 350 °C (v. Helgeson, 1964) dimostrano che tali soluzioni possono portare in soluzione da 1 a 600 ppm di piombo, quantità più che sufficienti a spiegare la formazione di depositi idrotermali. A 125 °C si possono riscontrare in soluzione concentrata di NaCl da 20 a 100 ppm di galena a un valore del pH non lontano dalla normalità. I complessi più importanti in questo sistema sono PbCl+, PbCl²4-, H2S, HCl e NaCl. A 300 °C PbCl+ è la specie dominante; a temperature inferiori PbCl+ è dominante nelle soluzioni diluite, mentre PbCl²4- aumenta nelle soluzioni concentrate.
I complessi con i cloruri mostrano di essere probabilmente il fattore più importante nel trasporto e nella deposizione dei minerali di piombo, argento, zinco e di altri elementi di transizione. Carbonati, solfati, disolfuri possono svolgere una parte importante in casi particolari.
9. Lineamenti della geochimica del ciclo esogeno.
a) L'alterazione: agenti chimici e azioni chimiche.
Quella parte del ciclo geochimico che si svolge sotto il controllo degli agenti atmosferici è stata indicata col nome di ‛ciclo esogeno'. In queste condizioni può verificarsi un riciclo degli elementi che ha a sua volta il carattere di un ciclo completo anche se non necessariamente chiuso e che ha perciò ricevuto anche il nome di ‛ciclo minore'. Esso comprende i fenomeni di alterazione, trasporto, sedimentazione, diagenesi e litificazione.
La degradazione meccanica delle rocce produce di per sé ingenti quantità di materiali che possono dar luogo, in seguito a trasporto e deposito, alla formazione di sedimenti clastici, più o meno cementati, dei quali esempi importanti sono il detrito di falda, i depositi glaciali, i depositi fluviali (ciottoli, sabbie) e di litorale. Dal nostro punto di vista una parte decisamente importante spetta alla degradazione meccanica, in quanto essa provvede a suddividere il materiale aumentandone grandemente la superficie specifica e rende così possibile ed efficace l'alterazione chimica.
Gli agenti chimici dell'alterazione superficiale sono notoriamente l'acqua e le sostanze in essa disciolte.
L'acqua stessa, nei confronti dei materiali rocciosi con cui viene a contatto, può comportarsi come solvente, come idratante e come idrolizzante. L'azione puramente solvente dell'acqua, che è in relazione all'elevato valore della sua costante dielettrica, si manifesta, per quanto riguarda l'alterazione delle rocce, nei confronti di quei composti ionici solubili che si possono considerare come sali di acidi forti e di basi forti, in ragione della loro solubilità, nonché delle dimensioni dei granuli. L'azione idratante si esplica nelle trasformazioni di sali anidri in sali idrati, la più vistosa delle quali è quella dell'anidrite in gesso, che implica fra l'altro un aumento di volume del 60% rispetto al volume dell'anidrite originaria. Nelle soluzioni, a causa dell'elevato valore del momento dipolare, le molecole di acqua tendono a disporsi in modo orientato attorno agli ioni, positivi o negativi, causandone l'idratazione, fenomeno che condiziona in modo determinante il comportamento geochimico degli elementi in soluzione.
Se nelle soluzioni sono presenti ioni di acidi deboli o di basi deboli, la loro reazione con l'acqua determina il noto fenomeno dell'idrolisi. Nei processi di alterazione delle rocce l'idrolisi ha importanza soprattutto nei solfati, nei carbonati e nei silicati. Nel caso dei solfati l'esempio più importante è dato dal solfato ferrico: la reazione schematica
2Fe3++3SO²4-+2H2O→2Fe(OH)2++2H++3SO²4-
mostra che si ha liberazione di ioni idrogeno e di conseguenza la soluzione assume reazione acida. In modo analogo si comportano molti solfati metallici che derivano dall'ossidazione dei relativi solfuri, cosicché le acque che circolano in corrispondenza di giacimenti di solfuri assumono di norma reazione acida. Per contro l'idrolisi dei carbonati, rappresentabile genericamente con lo schema
CO²3-+H2O→HCO-3+OH-,
conferisce alla soluzione una reazione alcalina. L'effetto è molto marcato nei suoli di climi aridi, dove sono presenti carbonati alcalini e dove il pH può assumere valori elevati, fino a 10 e oltre, ma è sensibile anche per le acque a contatto con il carbonato di calcio di rocce calcaree. L'acqua pura in contatto con il carbonato di calcio si equilibra a un pH di 9,95. In contatto con l'aria atmosferica, a causa della presenza del CO2 si può calcolare, per una soluzione satura, all'equilibrio un valore del pH di circa 8,4.
Di particolare significato negli ambienti naturali è l'idrolisi dei silicati. Un caso semplice può essere rappresentato dalla forsterite
Mg2SiO4+4H2O→2Mg2++40H-+H4SiO4.
Più complessi sono i fenomeni nei silicati di alluminio e di metalli alcalini o alcalino-terrosi. L'esempio più tipico è dato dal K-feldspato. In questo caso raramente la reazione è spinta all'estremo come si potrebbe prevedere dallo schema
KAlSi3O8+8H2O→K++OH-+Al(OH)3+3H4SiO4
ma si arresta a prodotti intermedi molto stabili come la sericite e i minerali argillosi:
3KAlSi3O8+14H2O→
→KAl3Si3O10(OH)2(sericite)+2K++2OH-+6H4SiO4
4KAlSi3O8+22H2O→
→4K++40H-+Al4Si4O10(OH)8(caolinite)+8H4SiO4.
Le reazioni indicate sono molto lente, ma portano all'importante conseguenza che le acque che circolano nei silicati tendono col tempo a perdere ogni eventuale acidità.
Le acque naturali possono contenere numerose sostanze allo stato di soluzione. Primo, in ordine di importanza, il diossido di carbonio. L'acqua, in equilibrio con l'atmosfera, contiene 10-5 moli per litro di acido carbonico. Le acque che circolano nei suoli si arricchiscono ulteriormente di CO2 biogenico. L'acido solforico può essere presente nelle acque nella zona di ossidazione dei solfuri o apportato da esalazioni vulcaniche, ma deve essere ricordata anche l'azione dei solfobatteri che producono H2S e H2SO4 nella decomposizione di sostanze organiche. Un'importanza subordinata spetta all'acido nitrico e ad acidi organici deboli quali gli acidi formico, acetico, propionico e altri che possono essere presenti nelle acque dei suoli.
Dal punto di vista del grado di alterabilità dei diversi minerali, questi possono essere ordinati in una serie ad alterabilità crescente utilizzando criteri diversi. Nella ‛serie di Goldich' si tiene conto delle osservazioni eseguite su profili di alterazione nei suoli; la ‛carta di persistenza' secondo Pettijohn è basata sul criterio della persistenza dei minerali più resistenti nei sedimenti più antichi, mentre la complessità della composizione mineralogica tende ad aumentare con il decrescere dell'età del sedimento. In entrambi i casi si trova che vi sono minerali molto sensibili all'alterazione, come l'olivina e i pirosseni, mentre altri presentano un grado elevato di resistenza, come il quarzo e la muscovite. Ovviamente il grado più elevato di resistenza all'alterazione spetta ai minerali autigeni perché si formano in equilibrio con l'ambiente esterno.
b) Fattori che regolano il comportamento degli elementi nei processi di alterazione, trasporto e sedimentazione.
L'acqua è l'agente più importante nei processi di alterazione; parimenti è in ambiente idrico che si svolgono per la maggior parte i processi di trasporto e di sedimentazione. Si comprende quindi come la sorte degli elementi nel corso del ciclo esogeno sia determinata essenzialmente da quei fattori che ne condizionano il comportamento in soluzione acquosa.
Potenziale ionico e grandezze correlate. - Viene definito come ‛potenziale ionico' (v. Cartledge, 1928) per un dato ione il rapporto fra la carica Z dello ione e il suo raggio ionico R. Il potenziale ionico è una misura dell'effetto polarizzante degli ioni e consente di spiegare in modo semplice alcune proprietà da essi manifestate in soluzione acquosa.
In tab. VI sono riportati i valori del potenziale ionico per i più comuni cationi.
I cationi aventi un basso valore del potenziale ionico (Z/R〈2,5) hanno tendenza a rimanere in soluzione in forma ionica e i loro idrossidi sono solubili in un vasto intervallo di pH. Fanno parte di questo gruppo ioni come Li+, Na+, K+, Rb+, Cs+, Fe2+, Mn2+, Ca2+, Sr2+, Ba2+. Essi hanno tendenza a rimanere in soluzione e perciò sono dotati, sia pure in grado diverso, di elevata mobilità geochimica. Se ne deduce, fra l'altro, che, per un dato elemento, la sua mobilità geochimica dipende dal suo stato di ossidazione: il ferro e il manganese allo stato bivalente sono più mobili rispetto allo stato più ossidato.
I cationi a cui spetta un valore intermedio del potenziale ionico (2,5〈Z/R〈10) hanno tendenza a formare idrossidi insolubili: nelle condizioni di pH prevalenti nelle acque naturali sono geochimicamente poco mobili e tendono ad arricchirsi nei prodotti di idrolisi dove si trovano spesso associati; appartengono a questa categoria Be2+, Al3+, Ga3+, Fe3+, Mn4+, Ti4+, Zr4+, Th4+, U4+, V3+, Nb5+, Ta5+.
I cationi a cui spetta un valore molto elevato del potenziale ionico (Z/R>10) formano viceversa anioni complessi di ossiacidi e come tali rimangono facilmente in soluzione. Così si comportano C4+, N5+, P5+, V5+, B3+, S6+, i quali rappresentano nuovamente un gruppo di elementi geochimicamente mobili. È da avvertire che la capacità di rimanere in soluzione, e pertanto la mobilità geochimica, dev'essere considerata in relazione al prodotto di solubilità delle coppie ioniche interessate. Così, per es., il bario è poco mobile in presenza di solfati e la mobilita del fosforo è grandemente limitata dalla presenza ubiquitaria del calcio.
Il potenziale ionico condiziona il comportamento degli elementi, così che elementi aventi caratteristiche cristallo- chimiche differenti (per carica ionica e per raggio ionico) si possono trovare viceversa associati nei sedimenti, come si verifica per Be, Al, Ga, Ti, Zr, Nb.
È da avvertire che le considerazioni basate sul potenziale ionico sono valide fino a che si confrontano ioni aventi la stessa configurazione elettronica esterna, come nel caso di ioni a 8 e 18 elettroni esterni. Inoltre gli elementi dei sottogruppi b hanno tendenza a formare legami covalenti, perciò la forza del legame metallo-ossigeno non dipende solo dal raggio e dalla carica. Quindi non ci si può aspettare che elementi aventi lo stesso valore del potenziale ionico abbiano necessariamente lo stesso comportamento geochimico se le loro strutture elettroniche e le loro elettronegatività non sono paragonabili.
In conseguenza della struttura polare della molecola d'acqua e del campo elettrico che circonda uno ione, cationi e anioni tendono ad attorniarsi di un numero di molecole d'acqua che è in evidente relazione con il potenziale ionico, nel senso che quanto maggiore è il potenziale ionico tanto maggiore sarà anche l'idratazione. In relazione a questo fenomeno risulta che il raggio dello ione idrato in soluzione sarà in genere diverso dal raggio che lo stesso ione manifesta nei cristalli e sarà tanto maggiore quanto più è grande il numero di molecole d'acqua che lo circondano. È allora prevedibile che se ci si vuole riferire al raggio dello ione idrato Rn, il potenziale ionico effettivo Z/Rh assumerà un valore minore rispetto al valore calcolabile dal raggio ionico R. Così, per es., per il litio, avente numero di idratazione da 10 a 15, il potenziale ionico scende da 1,47 a 0,10-0,27 in corrispondenza ai vari stati d'idratazione, mentre per il cesio, che è meno idrato, il potenziale ionico dello ione idrato può essere stimato fra 0,20 e 0,60. Ciò non è senza conseguenza circa il comportamento geochimico degli elementi negli ambienti acquatici. In effetti, per quanto si è detto in precedenza, in base alla considerazione del solo raggio ionico e della carica, il litio dovrebbe essere arricchito nei sedimenti rispetto agli altri elementi alcalini, mentre i dati sperimentali indicano che l'elemento più arricchito è il cesio. Considerazioni di questo tipo consentono d'interpretare il comportamento dei diversi elementi nei fenomeni di adsorbimento: in genere quanto più gli ioni sono idrati tanto meno risultano adsorbibili; ciò può ancora una volta giustificare l'arricchimento relativo del cesio nei sedimenti argillosi.
Il potenziale ionico è in definitiva una misura dell'effetto polarizzante degli ioni in soluzione. Allo stesso fine si possono prendere in considerazione altre grandezze a esso correlabili, quali i ‛coefficienti di energia di valenza' (v. Fersman, 1936), la ‛funzione del campo' (v. Ahrens, 1953), il ‛potere polarizzante' (v. Goldschmidt, 1926), equivalente alla ‛forza del campo cationico' di Dietzel (v., 1942).
Attività degli ioni idrogeno. - L'attività degli ioni idrogeno, abitualmente espressa in termini di pH=−log aH+, è uno dei fattori più importanti che regolano il comportamento degli elementi in soluzione e quindi le loro caratteristiche geochimiche nel ciclo esogeno. Il pH delle acque naturali può variare nei limiti 0-10, ma i valori estremi sono piuttosto eccezionali: nelle acque crateriche e nelle soluzioni in contatto con pirite in via di ossidazione si registrano i valori più bassi, mentre nelle acque dei suoli alcalini in climi aridi il pH può raggiungere e anche superare il valore 10. L'acqua piovana in equilibrio con il CO2 dell'atmosfera ha un pH di 5,7, soggetto peraltro a variazioni stagionali e geografiche. Nelle acque dei fiumi può variare fra 6 e 8,4 in relazione alle condizioni climatiche e di substrato. Nelle acque oceaniche superficiali si hanno valori fra 8,0 e 8,4: il pH scende a 7,6 negli strati profondi in presenza di sostanza organica in decomposizione.
Il valore del pH condiziona rigorosamente la solubilità degli idrossidi, dei carbonati e dei solfuri. L'influenza del pH sulla solubilità degli idrossidi risulta evidente dal diagramma di fig. 21. Si può osservare come la precipitazione del ferro trivalente inizi già a pH = 2 e sia praticamente completa a pH = 4, mentre la precipitazione del ferro bivalente inizi solo a pH = 7 e sia praticamente completa a pH = 9. Ne risulta che il ferro, in condizioni riducenti, può essere mantenuto in soluzione e quindi trasportato nelle acque naturali più di quanto non si verifichi per il ferro trivalente, il quale può sussistere in soluzione soltanto in condizioni di elevata acidità. La differenza molto piccola di solubilità fra Fe(OH)3 e Al(OH)3 determina il fatto che ferro e alluminio siano sovente associati nei sedimenti idrolizzati, per es. nelle bauxiti, ma in condizioni riducenti il ferro può essere asportato lasciando depositi di puro idrossido di alluminio.
Di particolare interesse è lo studio comparato delle solubilità della silice e dell'idrossido di alluminio in funzione del pH, come è mostrato dalla fig. 22. La solubilità dell'idrossido di alluminio passa per un minimo a un pH compreso fra 5 e 9. La solubilità della silice non varia sensibilmente per valori di pH inferiori a 9 e sale poi piuttosto rapidamente col pH. Per valori di pH inferiori a 4 l'allumina è più solubile della silice: in tali condizioni, peraltro rare negli ambienti naturali, si può avere una rimozione dell'allumina e un arricchimento di silice. Nelle condizioni di acidità corrispondenti alla maggior parte delle acque naturali la solubilità della silice è maggiore di quella dell'allumina. Condizioni di questo tipo presiedono probabilmente alla formazione delle latenti e delle bauxiti.
I fenomeni di precipitazione degli idrossidi sono grandemente complicati dai processi colloidali. Per esempio, il ferro e il manganese, i cui idrossidi manifestano proprietà colloidali, possono rimanere in sospensione ed essere trasportati in quantità superiore a quella consentita dalla solubilità dei loro idrossidi.
Potenziali di ossidoriduzione. - Nei processi di alterazione e di sedimentazione si verificano con frequenza cambiamenti di stato di ossidazione degli elementi che partecipano ai cosiddetti sistemi ossidoriduttori o sistemi OR. Come esempio pertinente possiamo considerare la reazione
4Fe2++O2+10H2O→4Fe(OH)3+8H+.
Come per qualsiasi altra reazione chimica, lo stato di equilibrio per questa reazione può essere ricavato dai valori dell'energia libera di tutte le specie interessate alla reazione. Siccome nelle reazioni di ossidazione e di riduzione si ha a che fare con trasferimenti di elettroni, per stabilire la tendenza di una reazione a procedere in un senso oppure nel senso opposto è opportuno riferirsi al cosiddetto ‛potenziale di ossidoriduzione', ricordando che il potenziale standard di una reazione è legato alla relativa variazione di energia libera dalla relazione
ΔG0=nFE0. (17)
In generale per una reazione di ossidoriduzione
p rid⇄q ox+ne
l'espressione del potenziale di ossidoriduzione sarà
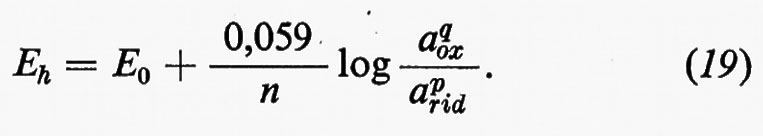
dove R è la costante dei gas, F il faraday, T la temperatura assoluta, aox e arid rispettivamente le attività della forma ossidata e della forma ridotta e n il numero degli elettroni in gioco. Introducendo al posto di R e F i valori numerici e considerando per T il valore di 298 °K = 25 °C, l'espressione diviene
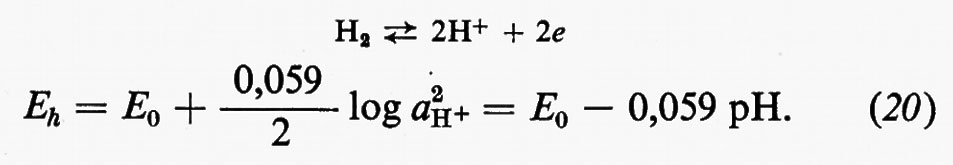
Il termine E0, che è caratteristico di ciascun sistema OR, corrisponde al valore che assume Eh quando l'attività delle specie ioniche è unitaria. I valori del potenziale sono riferiti al potenziale dell'elettrodo a idrogeno caratterizzato dalla reazione
2H++2e⇄H2
preso, per convenzione, come uguale a zero.
Il potenziale normale di ossidoriduzione è una misura della ‛forza' di un ossidante o di un riducente. Ordinando i sistemi OR secondo valori decrescenti di E0 si ha che ciascuno di essi è capace di ridurre tutti quelli che lo precedono e di ossidare quelli che lo seguono.
Nei sistemi OR in cui sono coinvolti ioni idrogeno, Eh dipende ovviamente dal pH in modo facilmente calcolabile. Così, per es., per il sistema
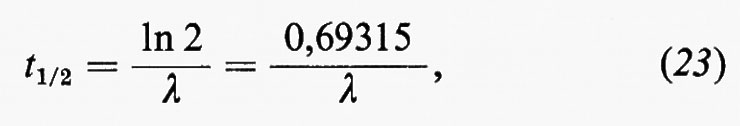
I diagrammi Eh−pH si rivelano particolarmente utili per porre in evidenza le relazioni di ossidazione e riduzione che intercorrono negli ambienti naturali e per definire le condizioni di stabilità delle specie minerali. Le reazioni chimiche in ambiente idrico sono teoricamente limitate a quelle i cui potenziali di ossidazione si trovano fra quelli delle reazioni
H2O⇄1/2O2+2H++2e (B) (Eh=1,22−0,059 pH)
H2⇄2H++2e (A) (Eh=−0,059 pH).
Infatti la forma ossidata di qualsiasi coppia avente un potenziale superiore a quello di (B) decomporrebbe l'acqua con svolgimento di ossigeno e la forma ridotta di qualsiasi coppia avente un potenziale inferiore a quello di (A) decomporrebbe l'acqua con sviluppo di idrogeno. Poiché il pH delle acque naturali raramente è inferiore a 4 e superiore a 9, come è visibile in fig. 23, i potenziali di ossidazione degli ambienti naturali debbono presentare valori compresi fra −0,53 e +0,98 volt. Nella stessa figura è rappresentata la caratterizzazione dei principali ambienti naturali in base ai valori di pH e di Eh.
I diagrammi Eh−pH come rappresentazione dei campi di stabilità di specie mineralogiche. - I diagrammi Eh−pH possono essere impiegati utilmente per la rappresentazione dei campi di stabilità di specie mineralogiche in equilibrio con soluzioni acquose. Come esempi di particolare interesse riportiamo due casi che si riferiscono alle condizioni di stabilità di alcuni comuni minerali di ferro (v. fig. 24) e di manganese (v. fig. 25). Come si vede nel diagramma di fig. 24, è chiaramente definito il campo di stabilità dell'ematite nei confronti della siderite. Con riferimento a questi due minerali si può scrivere un'equazione che pone in relazione le due specie:
2FeCO3+3H2O⇄Fe2O3+2H2CO3+2H++2e.
Dal valore della variazione di energia libera della reazione ΔG0R=+16,87 kcal) si calcola il valore di E0 (=+0,37 volt) e quindi
Eh=E0+0,03 log (a²H2CO3•a²H+)=
=0,37+0,059 log aH2CO3−0,059 pH (21)
Poiché per ogni dato valore della concentrazione totale di CO2 aH2CO3 è funzione del pH, anche Eh sarà funzione del pH, e potrà essere tracciata la corrispondente linea del diagramma di fig. 24 a rappresentare l'equilibrio fra siderite ed ematite. In modo simile possono venire tracciate le linee di demarcazione delle altre specie mineralogiche, facendo opportune ipotesi sulle concentrazioni totali di H2S, HS-, S2-, SO²4- e delle specie carbonatiche H2CO3, HCO-3, CO²3-.
Con analoghi procedimenti può essere tracciato il diagramma Eh−pH per i composti di manganese (v. fig. 25). Come si può osservare, nel caso del manganese il campo di stabilità del carbonato (rodocrosite) è molto più ampio rispetto al corrispondente carbonato di ferro (siderite). Dal confronto dei due diagrammi si può osservare inoltre come il campo di stabilità dell'ossido Fe2O3 sia viceversa molto esteso: ciò sta a indicare che a parità di pH e di Eh il ferro può precipitare come Fe2O3 mentre il manganese, meno ossidabile, rimane in soluzione come ione Mn2+. Il solfuro di manganese non si forma nelle condizioni indicate, ma si può formare se la concentrazione in zolfo totale diviene rilevante rispetto a quella dei carbonati.
Di particolare interesse è infine il diagramma Eh−pH relativo alle specie HSO-4, SO²4-, H2S, HS-, S2-, soprattutto per quanto si riferisce al limite solfati-solfuri (v. fig. 26).
Classificazione dei sedimenti in base ai valori di pH ed Eh. - I valori del pH e del potenziale OR sono grandezze determinanti per quanto riguarda la natura del sedimento.
Nel diagramma di fig. 27 è riportata in modo molto efficace una classificazione dei sedimenti tracciata in base ai valori di pH e di Eh. In esso sono state tracciate delle linee di confine che acquistano il significato di ‛barriera geochimica' (geochemical fence) e indicano i limiti di stabilità di determinate associazioni sedimentologiche. Tali linee di confine sono le seguenti: 1) piano neutro, di significato ovvio, corrispondente a pH=7-8, il più basso valore del pH che ancora consente la formazione di abbondante calcite; 2) limite dei carbonati di Fe e di Mn, definibile in base a quanto è stato detto in precedenza; 3) limite solfati-solfuri, definibile in base a quanto si è detto o più semplicemente in base alla reazione
S2-+4H2O⇄SO²4-+8H++8e
con E0=0,14 volt, il cui Eh è, ovviamente, dipendente dal pH.
Separazione del ferro e del manganese come esempio delle molteplici interazioni dei fattori considerati. - Ferro e manganese, due elementi peraltro simili per molte delle loro caratteristiche cristallochimiche e che per questo si trovano associati e si possono sostituire in larga misura nei silicati delle rocce ignee, si comportano viceversa in modo sostanzialmente differente negli ambienti sedimentari. Come è stato ampiamente illustrato da Krauskopf (v., 1957) il rapporto delle abbondanze del ferro e del manganese nelle rocce ignee è di circa 60. Tale rapporto si osserva anche nelle soluzioni derivanti dall'alterazione delle rocce. Rari sono i casi di soluzioni ricche in manganese, quali si osservano talvolta in regioni vulcaniche. Sono noti depositi sedimentari di ossidi che contengono i due elementi in varie proporzioni, ma sono numerosi i casi di ossidi sedimentari di ferro privi o quasi di manganese e ancora più rilevante è l'esistenza di giacimenti di ossidi di manganese di notevole purezza.
I giacimenti sedimentari di ferro possono formarsi in ambiente lacustre, in torbiere e paludi ove si hanno depositi di idrossidi, siderite, pirite, con eventuali piccole quantità di fosfati e silicati ovvero, in ambiente marino, per sedimentazione in mari bassi. I minerali depositati sono in questo caso prevalentemente gli idrossidi: appartengono a questo tipo anche i giacimenti di ferro ‛oolitico'. Nei mari più profondi il ferro trasportato si deposita per lo più in combinazione con la silice sotto forma di glauconite, chamosite e altri minerali.
Anche il manganese può essere depositato in forma di ‛ocre manganesifere' in ambienti lacustri e palustri e in mari bassi, lungo le linee di spiaggia o in ambienti lagunari. Nel mare profondo si hanno depositi di ossidi di manganese in forma di noduli che possono contenere fino al 50% di Mn.
Di particolare interesse, talvolta anche economico, i giacimenti sedimentari di manganese collegati a rocce vulcaniche, che sono presenti, per esempio, nella regione laziale.
Se immaginiamo di aumentare gradualmente il pH di una soluzione contenente i due elementi, operando in condizioni ossidanti, si verificherà per prima la separazione dell'ossido di ferro e pertanto la soluzione si arricchirà gradualmente in manganese. Il carbonato, i solfuri e gli ossidi di ferro sono tutti, di regola, meno solubili dei corrispondenti composti di manganese; quindi il processo di arricchimento del manganese in soluzione è di carattere generale. Analogamente, se in una soluzione contenente Fe2+ e Mn2+ a un dato pH immaginiamo di aumentare gradualmente il valore di Eh, per esempio sciogliendo ossigeno, il valore di Eh di ossidazione del ferro verrà raggiunto prima che per il manganese; ciò determinerà parimenti una separazione precoce selettiva del ferro. Tutto sembra perciò esprimere una tendenza alla separazione dei due elementi, la quale implica però una separazione precoce del ferro, mentre non si conoscono processi inorganici che possano far prevedere una precipitazione selettiva del manganese nei confronti del ferro. D'altra parte le soluzioni che provocano alterazione delle rocce sono in generale acide per acido carbonico o per altri acidi minerali e tendono ad aumentare il pH col tempo, in seguito a prolungato contatto con le stesse rocce silicatiche o con rocce carbonatiche. Se le cose si svolgessero secondo lo schema descritto, ci potremmo aspettare di trovare in vicinanza del deposito di manganese una deposizione di ferro in quantità notevolmente superiore. Ora, se è vero che le rocce adiacenti ai depositi di manganese sono di norma ricche in ferro, non è però dimostrato che la quantità di ferro in esse contenuta sia sufficiente a comprovare l'ipotesi: non mancano casi in cui gli ossidi di ferro sono praticamente assenti. Non sembra pertanto che l'ipotesi di un isolamento del manganese attraverso una precipitazione precoce dell'ossido di ferro sia applicabile nella generalità dei casi e rappresenti l'unico meccanismo di separazione dei due elementi.
È noto che vi sono batteri capaci di determinare la precipitazione selettiva del manganese e del ferro in forma relativamente pura. Sono stati osservati dei casi in cui ossidi con elevato rapporto Mn/Fe si depositano come rivestimento di alghe incrostanti ciottoli di quarzo in contatto con acque aventi un rapporto Fe/Mn normale; depositi ricchi di manganese si formano in condutture d'acqua; alcuni laghi svedesi presentano depositi ricchi di ferro da una parte e ricchi di manganese da un'altra. L'azione dei batteri non sembra però svolgere una parte essenziale nella formazione dei grandi depositi di manganese. Essa può essere utilmente invocata solo per spiegare la separazione parziale dei due elementi in piccoli giacimenti, quali i depositi di palude, di fiume e di lago.
Si è potuto dimostrare che la precipitazione del manganese è catalizzata dallo stesso MnO2 precipitato. L'attività catalitica di MnO2 ha certamente un posto importante nella geochimica del manganese, ma non è ancora certo che un processo del genere sia sufficiente a giustificare la formazione di grossi depositi di questo elemento.
Sia l'idrossido ferrico sia il diossido di manganese presentano uno spiccato comportamento colloidale, il quale dipende da un grande numero di fattori: in certi casi appare più stabile il colloide di idrossido ferrico, in altri quello del diossido di manganese. Neanche i fenomeni colloidali possono pertanto essere considerati i soli responsabili della separazione dei due elementi.
Come si vede, nessuno dei fattori considerati è in grado di fornire una spiegazione unica della formazione di grandi depositi di manganese. È probabile in molti casi un concorso di diversi fattori. È inoltre necessario tener presente la possibilità che i minerali di manganese, una volta depositati, possano venire ulteriormente concentrati attraverso un processo multistadio di ridissoluzione e riprecipitazione. Molti problemi relativi alla geochimica sedimentaria del ferro e del manganese attendono perciò ancora una soluzione.
c) Aspetti geochimici dei processi diagenetici.
Intendiamo per diagenesi (v. Gümbel, 1868; v. Walther, 1893-1894) quell'insieme di modificazioni subite da un sedimento contemporaneamente o posteriormente alla sua deposizione e che hanno come risultato finale la formazione di una roccia sedimentaria compatta o comunque le modificazioni subite da una roccia sedimentaria in condizioni moderate di temperatura e di pressione. I limiti fra diagenesi e metamorfismo sono talvolta mal definibili: normalmente s'includono nel metamorfismo tutte quelle azioni che in campo di P−T portano a una totale ricristallizzazione della roccia con formazione di facies mineralogiche caratteristiche. La difficoltà di stabilire un limite netto fra diagenesi e metamorfismo risiede principalmente nella diversa attitudine di vari materiali a reagire alle mutate condizioni di temperatura e di pressione. I fenomeni diagenetici si verificano prevalentemente nell'ambito d'azione degli agenti atmosferici o nelle zone in cui vi può essere interazione fra sedimenti e idrosfera e interessano prevalentemente le rocce superficiali e poco profonde. In fig. 28 sono riportate schematicamente le condizioni di temperatura e di pressione nelle quali normalmente si verificano i fenomeni diagenetici.
Il Fairbridge distingue nel processo diagenetico le seguenti fasi successive: la ‛sindiagenesi', che si verifica contemporaneamente alla sedimentazione, l'‛anadiagenesi', che corrisponde a una successiva fase di compattazione e di maturazione, e l'‛epidiagenesi', che si completa durante l'emersione e prima dell'erosione (v. fig. 29).
La diagenesi comprende essenzialmente i fenomeni di compattazione e di cementazione dei sedimenti, di soluzione interstratale, di neoformazione di minerali e di metasomatosi, il risultato dei quali si traduce nel grado di litificazione della roccia.
Diversi materiali possono essere depositati per precipitazione nei pori e costituiscono il cemento della roccia. I materiali più comuni sono il quarzo e la calcite, a cui segue la dolomite; non mancano casi in cui il cemento è costituito da siderite, ossidi di ferro, opale, calcedonio, pirite, baritina, fluorite, fosfati. Nei sedimenti giovani il cemento è sovente costituito da calcite, mentre nei sedimenti più antichi tende a svilupparsi un cemento quarzoso: il tempo favorisce la sostituzione del quarzo alla calcite. Le sostanze cementanti possono provenire dall'esterno per opera delle soluzioni circolanti, ma è anche possibile che esse si originino nel sedimento stesso in seguito a processi di soluzione e rideposizione. Frequenti sono i casi di segregazione irregolare del cemento con formazione di ‛strutture chimiche secondarie'.
I processi di soluzione hanno una parte importante nell'evoluzione dei sedimenti e interessano i diversi minerali in ragione della loro resistenza all'alterazione, con la conseguente persistenza dei minerali più resistenti nei sedimenti più antichi. Vistosi fenomeni di corrosione si possono osservare nei cristalli di staurolite, di granato, di augite. A fenomeni di soluzione interstratale viene ascritta la formazione delle stiloliti, interpretabili come suture di pressione.
Inversamente si possono osservare nei sedimenti fenomeni di accrescimento secondario di minerali e la neoformazione di minerali autigeni come quarzo, feldspati, carbonati, illiti, caolinite, montmorillonite, cloriti, zeoliti, glauconite, tormalina, rutilo, solfati, solfuri, zircone e fosfati.
Gli aspetti geochimici più importanti che interessano la diagenesi sono quelli riguardanti la metasomatosi diagenetica, ossia tutte quelle azioni di carattere chimico in cui si verifica la sostituzione di una specie mineralogica a una preesistente, le più significative delle quali sono i processi di silicizzazione, di dolomitizzazione e di dedolomitizzazione.
I processi di silicizzazione si manifestano con una sostituzione della silice al carbonato di calcio che può essere così schematizzata
CaCO3s+H2SiO3sol+H2CO3→Ca+++2HCO-3+SiO2s+H2O.
I diagrammi di solubilità del carbonato di calcio e della silice amorfa, riportati in fig. 30, mostrano chiaramente come bassi valori del pH siano favorevoli alla soluzione del carbonato di calcio e alla precipitazione della silice, mentre per alti valori del pH la silice tende a passare in soluzione e si ha la precipitazione del carbonato. Risulta pertanto che vi è una reciproca sostituibilità della silice e del carbonato in funzione del pH. È però dubbio che le sole variazioni di pH, quali si possono verificare in condizioni normali negli ambienti naturali, possano essere ritenute responsabili del meccanismo di sostituzione della silice al carbonato perché la solubilità della silice è indipendente dal pH nel campo di valori compresi fra 2 e 9. Oggi si tende ad annettere molta importanza alle reazioni a cui partecipano i minerali argillosi; così, per es., nella trasformazione da montmorillonite a illite e da montmorillonite a caolinite viene messa in libertà della silice; questa e altre reazioni sono poi notevolmente influenzate dal pH. La solubilità della silice amorfa è inoltre condizionata dal suo adsorbimento sulla sostanza organica nonché dall'adsorbimento della stessa sostanza organica su micelle di silice colloidale.
Particolare attenzione merita il meccanismo di formazione delle ‛selci' (chert) che frequentemente si trovano in forma di noduli e di liste in rocce calcaree e danno luogo alla tipica associazione dei ‛calcari con selce'. Vari meccanismi sono stati proposti: può essere invocata una deposizione di silice primaria o singenetica, in forma di masse gelatinose successivamente indurite; un arricchimento secondario o epigenetico per sostituzione secondaria di silice nella roccia ospite o infine una formazione diagenetica precoce per una deposizione penecontemporanea di silice che si può essere verificata dopo la sedimentazione, ma prima della consolidazione. La precipitazione può verificarsi direttamente da acque marine arricchite in silice da emanazioni vulcaniche, o da acque fluviali ricche di silice colloidale, o ancora per dissoluzione, migrazione e susseguente precipitazione di silice di origine biochimica. Rilevanti a questo proposito sono le osservazioni condotte durante la spedizione della Glomar Challenger in esecuzione del Deep Sea Drilling Projéct. Nelle perforazioni sono stati raggiunti orizzonti eocenici a radiolariti spesso fortemente litificate che si estendono dall'Atlantico occidentale a quello orientale e in ogni caso i sedimenti eocenici risultano ricchi di resti di Radiolari, Diatomee, Spugne silicee, non direttamente collegati ad arricchimento in silice dovuto ad attività vulcanica. Questi sedimenti litificati non sono mai emersi e sono associati a sedimenti mobili, ciò che induce a ritenere che la litificazione sia un fenomeno precoce se non istantaneo.
Il problema relativo alla formazione delle dolomie e alla ‛dolomitizzazione' è uno dei più interessanti e discussi. Mentre sembra accertato che in particolari condizioni può verificarsi una precipitazione diretta di dolomite dall'acqua marina, depositi di questo tipo non possono avere estensioni degne di nota se non in formazioni di estremo ambiente evaporitico.
I sedimenti organogeni possono contenere originariamente una percentuale variabile da 0 al 25% di MgCO3 per lo più in forma di una soluzione solida metastabile detta ‛calcite magnesifera'. Il rapporto medio Ca/Mg nei sedimenti marini è di 40:1. Questi valori non sono tali da giustificare una formazione precoce della dolomite. È perciò necessario ammettere che la dolomitizzazione avvenga attraverso un processo di arricchimento secondario, che risulta tra l'altro evidente dai processi di sostituzione con dolomite di fossili e di strutture originariamente calcitiche o aragonitiche.
Meno facile è stabilire se la dolomitizzazione si sia verificata nell'ambiente di deposizione prima del seppellimento o dopo il seppellimento o dopo l'emersione del sedimento.
Si può considerare come provato un processo di dolomitizzazione per diagenesi precoce nell'ambiente stesso di sedimentazione: minuti romboedri di dolomite sono stati talvolta osservati in sedimenti di mare profondo. Significative a questo proposito le osservazioni condotte nell'atollo di Funafuti nelle isole Ellice, ove una perforazione ha attraversato dapprima circa 60 m di sedimenti calcitici con un moderato contenuto di magnesio, mentre i successivi 130 m costituiscono una zona in cui si è verificato un allontanamento del magnesio per lisciviazione e oltre i 190 m il sedimento è completamente dolomitizzato.
Una sostituzione più tardiva può aver luogo per reazione del carbonato di calcio con acque ‛connate' arricchite in magnesio.
La reazione di scambio
2CaCO3+Mg2+→CaMg(CO3)2+Ca2+
comporta una variazione d'i energia libera di −3,59 kcal/mole e può pertanto procedere spontaneamente, confermando così la possibilità di una diagenesi precoce. Può addirittura meravigliare l'assenza quasi sistematica della dolomite nei precipitati marini, poiché l'acqua marina dovrebbe essere sovrassatura rispetto alla dolomite, e la calcite e l'aragonite dovrebbero reagire con l'acqua di mare per formare dolomite. Esperienze di laboratorio hanno dimostrato che la formazione della struttura ordinata della dolomite è un processo termodinamicamente favorito ma estremamente lento, probabilmente per una riluttanza del carbonato di magnesio a precipitare; ciò può essere attribuito da un lato al fatto che lo ione Mg2+ in soluzione è idratato con legami eccezionalmente stabili, dall'altro all'esistenza in soluzione di una ‛coppia di ioni' stabile MgCO3.
Fra i fenomeni metasomatici in fase diagenetica merita particolare attenzione anche il processo inverso di ‛dedolomitizzazione', che implica una sostituzione di calcite o di anidrite alla dolomite, come risulta dalle reazioni
CaMg(CO3)2+CaSO4•2H2O→2CaCO3+MgSO4+2H2O
CaMg(CO3)2+MgSO4→CaSO4+2MgCO3.
La dedolomitizzazione può essere assunta come un esempio dei numerosi fenomeni di riadattamento ambientale che sono descritti col termine di ‛diagenesi retrograda'.
10. La misura del tempo geologico.
a) Generalità.
La valutazione quantitativa del tempo geologico dev'essere un'aspirazione piuttosto antica dell'uomo se, come si legge, già Erodoto (484-424 a.C.) aveva pensato che lo spessore dei sedimenti deposti dal Nilo potesse essere assunto come una misura del tempo preistorico (v. Hamilton, 1965). In effetti la velocità di accumulo dei sedimenti rappresentò, quasi fino ai nostri giorni e in assenza di cognizioni migliori, il mezzo più appropriato per la stima della durata degli eventi geologici e, verso la fine del secolo scorso, il concetto prendeva forma nell'espressione del geocrono di Williams (v., 1893) inteso a significare un intervallo di tempo pari alla durata dell'Eocene, che veniva preso come unità di misura del tempo geologico. Una migliore valutazione della velocità di sedimentazione conduceva qualche anno dopo (v. Goodchild, 1896) a una stima del tempo necessario per il deposito dei sedimenti postcambriani in 704 milioni di anni, valore non del tutto disprezzabile anche alla luce delle acquisizioni più recenti.
Mentre i vari tentativi di stabilire una cronologia geologica assoluta stavano prendendo corpo, nel secolo scorso e anche in quello attuale i geologi e i paleontologi con paziente lavoro analitico potevano stabilire e generalizzare una successione dettagliata di eventi geologici e biologici, universalmente accettata come colonna stratigrafica di riferimento, detta anche scala stratigrafica. Le unità crono-geologiche che figurano in questa scala sono, come è noto, le ere, i periodi, le epoche e i piani o età.
La scala stratigrafica era nota pertanto con grande dettaglio e non attendeva che di essere tradotta in una scala dei tempi.
La grande occasione fu offerta dalla scoperta della radioattività: se la grande diffusione dei metodi radiometrici di misura dell'età di formazioni geologiche è una caratteristica inconfondibile di quest'ultimo ventennio, è doveroso riconoscere che appena otto anni dopo la scoperta della radioattività E. Rutherford, nel 1904, annunciava chiaramente la possibilità di usare il decadimento radioattivo per la misura del tempo geologico e quattro anni dopo poteva assegnare un'età di 500 milioni di anni a un cristallo di fergusonite in base alla misura del contenuto di uranio e di elio. Con i successivi lavori di Boltwood (v., 1907) e Strutt (v., 1905, 1908, 1909, 1910), basati sulla misura dei rapporti U/Pb e U/He, la cronologia geologica era definitivamente orientata in senso quantitativo.
I grandi progressi realizzati in questi ultimi anni sono perciò più il frutto del perfezionamento dei mezzi tecnici a disposizione che di nuove acquisizioni concettuali; tuttavia è innegabile che allo sviluppo di queste tecniche la geologia deve i sostanziali progressi recenti.
Benché i metodi radiometrici non costituiscano l'unica possibilità offerta dalla chimica e dalla fisica alla geologia, essi nondimeno rappresentano, nella misura del tempo geologico, la parte principale. Essi debbono la loro fortuna al fatto che la disintegrazione spontanea dei nuclidi radioattivi decorre con velocità misurabile e con legge definita, costante nel tempo e in ottima approssimazione indipendente dai fattori ambientali: essa pertanto fornisce un metodo assoluto per la determinazione dell'età dei minerali e delle rocce che li contengono, se sono verificate alcune condizioni fondamentali.
Debbono essere infatti noti il processo di disintegrazione, il tipo di decadimento, la natura dei prodotti di decadimento e le costanti di disintegrazione. I prodotti del decadimento debbono essere originariamente assenti nel materiale in esame o la quantità di essi eventualmente presente in origine dev'essere in qualche modo valutabile; infine il sistema deve comportarsi come un sistema chimico chiuso nei riguardi sia dell'elemento radioattivo sia dell'elemento radiogenico, dall'istante in cui l'elemento radioattivo è entrato a far parte della struttura cristallina del minerale ospite.
Le leggi generali del decadimento radioattivo sono troppo note e devono semplicemente essere ricordate: se x0 è il numero di atomi di elemento radioattivo presenti in origine, il numero di atomi xt ancora presenti al tempo t è dato da
xt=x0 e-λt, (22)
dove e è la base dei logaritmi naturali e λ è la costante di decadimento. Il tempo necessario perché gli x0 atomi iniziali si riducanoo a metà, altrimenti detto tempo di dimezzamento, è dato da

mentre la ‛vita media' ‛ di un gruppo di atomi di una specie radioattiva è data da
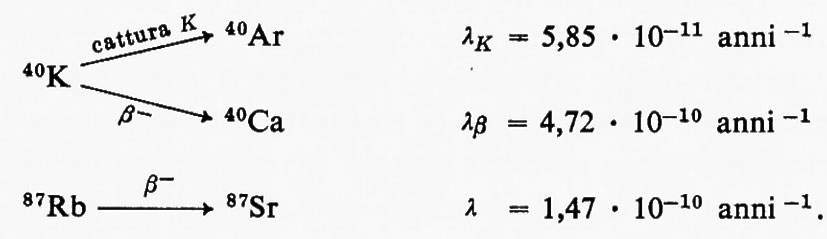
Poiché il numero y di atomi dell'elemento radiogenico che si sono formati è uguale al numero x di atomi dell'elemento radioattivo che sono decaduti, si avrà
y=x0−x=x0 (1−e-λt)=x (eλt−1). (25)
In questo modo il numero y di atomi dell'elemento radiogenico formati al tempo t è dato in funzione del numero x di atomi dell'elemento radioattivo ancora presenti al tempo t, nonché di λ e di t. In questa forma l'equazione generale del decadimento radioattivo è per lo più impiegata ai fini geologici. Se si misura il tempo a partire dal presente contando i tempi positivi verso il passato, t rappresenta l'età del minerale.
Esiste in natura un considerevole numero di nuclidi radioattivi che sono o possono essere usati per determinare l'età dei minerali o delle rocce. Alcuni di questi nuclidi (3H, 14C, 10Be, 22Na, 32Si, 32P, 33P, 39Cl, 39Ar) si formano in continuazione nell'atmosfera per effetto dei componenti della radiazione cosmica; altri (36Cl) per cattura di neutroni nelle rocce superficiali; altri ancora sono sempre esistiti nei minerali che li contengono fin dal momento della formazione di questi ultimi perché fanno parte di quel ‛complesso isotopico' dell'elemento che si è mantenuto più o meno inalterato fin dall'istante della nucleogenesi. Tali sono 40K, 87Rb, 187Re, 232Th, 235U, 238U, per citare i principali.
b) I metodi del potassio-argon e del rubidio-stronzio.
Fra i metodi più largamente in uso, speciale menzione meritano quelli basati sul decadimento del 40K e del 87Rb, che avvengono secondo gli schemi
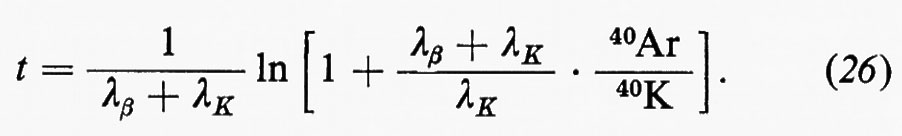
L'interesse di questi due metodi risiede principalmente nel fatto che essi sono basati sul decadimento di due isotopi radioattivi che sono entrambi abbondanti e diffusi nella crosta terrestre e prevedono l'impiego di minerali comuni come i feldspati e le miche, abbondanti sia nelle rocce ignee sia in quelle sedimentarie e metamorfiche; essi inoltre consentono di determinare valori indipendenti dell'età di una stessa roccia servendosi di diversi minerali in essa contenuti; possono fornire valori indipendenti dell'età impiegando i due metodi sullo stesso minerale o sulla roccia in toto e consentono, dato il valore favorevole delle costanti di decadimento, di esplorare praticamente tutto il tempo geologico.
Il metodo di datazione fondato sul decadimento del potassio per cattura K è basato sulla misura del contenuto di argon radiogenico del minerale nonché del contenuto di potassio totale, assumendo che il potassio-40 si trovi in rapporto costante nel complesso isotopico del potassio, e precisamente di 0,0119 atomi per cento. L'età geologica del minerale è data da

Il contenuto in argon-40 viene attualmente misurato per spettrometria di massa con la tecnica della diluizione isotopica facendo uso di uno spike di argon arricchito in argon 38. In questo modo è anche possibile apportare una precisa correzione per la presenza di argon atmosferico, il cui contenuto nel campione può essere stimato dalla quantità di argon-36 presente in esso.
Se la roccia o i minerali in essa contenuti si sono comportati sin dall'origine come ‛sistemi chiusi', l'età K/Ar coinciderà con la vera età della cristallizzazione primaria. Se in tempi successivi si sono verificate perdite di argon, si otterranno in genere età inferiori (età minime) e, in circostanze favorevoli, il rapporto argon/potassio potrà indicare l'età del metamorfismo, di eventi tettonici, del metasomatismo o di mineralizzazioni.
Il metodo di datazione che fa uso del decadimento β del rubidio-87, isotopo che costituisce circa il 28% in atomi del complesso isotopico del rubidio naturale, è basato sulla misura del rapporto 87Sr(radiogenico)/87Rb come indicato dall'equazione
87Sr−87Rb (eλt−1). (27)
Poiché il tempo di dimezzamento del rubidio-87 è molto lungo rispetto all'età della Terra, l'espressione può essere semplificata in
87Sr=87Rb λt (28)
e l'età è data in questo caso semplicemente da
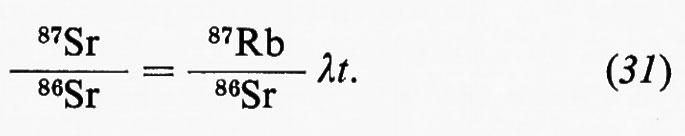
Nel caso ideale di un minerale che non contenga originariamente stronzio comune (non radiogenico) l'età potrebbe quindi essere stabilita facendo uso direttamente della citata espressione e sarebbe sufficiente una determinazione, anche per semplice via chimica o spettrochimica, del contenuto totale di rubidio e di stronzio. Circostanze così favorevoli si verificano molto raramente, poiché la maggior parte dei minerali hanno un contenuto imprevedibile di ‛stronzio comune', la cui composizione isotopica può inoltre variare di diverse unità per cento in dipendenza dall'origine e dalle modalità di formazione del minerale in questione. È possibile conoscere la composizione dello ‛stronzio comune' all'atto della formazione della roccia eseguendo l'analisi isotopica di un minerale presente nella roccia stessa e contenente soltanto stronzio comune, essendo privo di rubidio, come nel caso dell'apatite o del feldspato di calcio. In minerali di questo tipo, infatti, la composizione isotopica dello stronzio non ha subito variazioni dall'epoca della cristallizzazione. Ma anche se la composizione isotopica dello stronzio comune non è direttamente accessibile nel modo indicato, un elegante metodo teorico, sviluppato indipendentemente da Compston e Jeffery (v., 1960 e 1961) e da Nicolaysen (v., 1961) del Bernard Price Institute di Johannesburg e perciò indicato con la sigla di ‛metodo BPI', consente ugualmente di ricavano e di ottenere l'età isotopica della roccia.
L'equazione semplificata
87Sr=87Rb λt (30)
poiché nella pratica si misurano sempre rapporti isotopici riferiti all'isotopo 86Sr può essere scritta
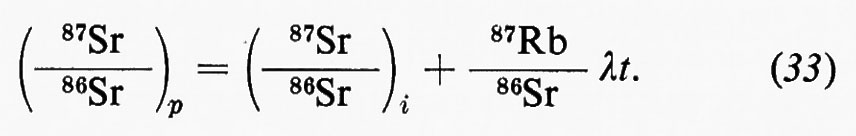
Tenendo conto che 87Sr radiogenico è dato dalla differenza fra 87Sr attualmente presente (87Srp) e 87Sr presente in origine (87Sri) nel minerale e cioè
87Sr=87Srp−87Sri (32)
si avrà

L'equazione (33) è l'equazione di una retta del tipo y=mx+c con m=λt e c=(87Sr/86Sr)i che ci esprime il variare del rapporto 87Sr/86Sr in funzione del rapporto 87Rb/86Sr; questa retta, che ha una pendenza proporzionale all'età t e intercetta sulle ordinate un segmento che è una misura del rapporto (87Sr/86Sr)i all'origine (fig. 31), è detta ‛isocrona': su di essa si allineano i punti corrispondenti ai diversi minerali della roccia, tutti coevi, ai quali corrisponde un diverso valore del rapporto 87Sr/86Sr semplicemente perché ciascuno di essi rappresenta una fase più o meno ricca di rubidio, cioè avente un diverso valore del rapporto 87Rb/86Sr. Sarà pertanto sufficiente proiettare in un diagramma i valori dei rapporti 87Sr/86Sr e 87Rb/86Sr ottenuti dai diversi minerali di una roccia, nonché dalla roccia in toto, per ottenere la retta la cui pendenza fornisce l'età t e la cui intersezione con l'asse delle ordinate dà la composizione iniziale dello ‛stronzio comune'. In conformità ai principi esposti, il significato dell'età così determinata è quello del tempo trascorso dall'istante in cui la roccia è diventata un sistema chiuso: esso può rappresentare perciò l'età della cristallizzazione primaria in assenza di metamorfismo, o dell'ultimo evento metamorfico. In quest'ultimo caso alcune considerazioni sul valore del rapporto (87Sr/86Sr)i possono anche condurre alla stima dell'età del primo evento metamorfico o della prima cristallizzazione.
c) I metodi basati sul decadimento dell'uranio e del tono.
L'uranio-238, l'uranio-235 e il torio-232 decadono secondo uno schema complesso e sono i capostipiti delle tre famiglie radioattive i cui prodotti finali stabili sono rispettivamente 206Pb, 207Pb e 208Pb, oltre all'elio che si forma nel decadimento α di molti termini intermedi. Sul decadimento di questi nuclidi possono venire sviluppati diversi metodi di misura del tempo geologico, di cui il più antico, basato sulla misura del quantitativo d'elio accumulato nei minerali di uranio e di tono, è ormai di interesse storico, così come i metodi basati sull'accumulo totale di piombo. Le determinazioni più accurate di età sono quelle che si basano sull'analisi isotopica del piombo. In un minerale contenente uranio e tono si possono infatti ottenere tre valori indipendenti dell'età dalle relazioni
206Pb=238U (eλ1t−1) (34)
207Pb=235U (eλ2t−1) (35)
208Pb=232Th (eλ3t−1), (36)
dove λ1, λ2 e λ3 e sono le rispettive costanti di decadimento.
Il metodo, detto anche metodo isotopico del piombo, fornisce risultati attendibili in assenza di piombo ordinario. Il piombo comune è infatti costituito dai quattro isotopi 204Pb, 206Pb, 207Pb e 208Pb. La presenza di piombo ordinario nel materiale in questione è pertanto indicata dal piombo- 204, rivelabile dall'analisi isotopica, e richiede l'introduzione di un termine correttivo. La valutazione di quest'ultimo è spesso difficile perché la composizione isotopica del piombo comune presenta una notevole variabilità dato che in quest'ultimo il rapporto fra gli isotopi varia con l'età, come è facilmente prevedibile, e, al di fuori di questo, vi sono dei cosiddetti ‛piombi anomali' nei quali la composizione isotopica non è regolata da alcuna legge apparente (v. cap. 11). Questo inconveniente è però superabile qualora sia possibile stabilire la composizione isotopica del piombo comune analizzando minerali di piombo appartenenti alla stessa area e che la situazione geologica indichi coevi con il minerale o la roccia da datare.
Se ora ci limitiamo a considerare le espressioni (34) e (35) riscritte nella forma

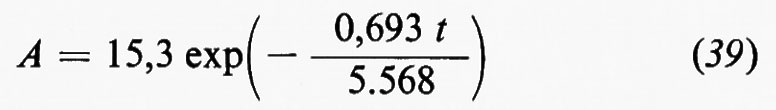
risulta che i rapporti 206Pb/238U e 207Pb/235U variano entrambi in funzione del tempo con la legge espressa dalle menzionate equazioni. Per un certo valore di t avremo pertanto un unico valore di 206Pb/238U e un unico valore di 207Pb/235U. Le coppie di valori 206Pb/238U e 207Pb/235U che corrispondono ai diversi possibili valori di t, riportate in un diagramma avente i due rapporti menzionati rispettivamente in ordinata e in ascissa, danno luogo a una curva detta ‛curva concordia' (v. fig. 32). Questa curva è il luogo dei punti a cui corrispondono età 206Pb/238~U e 207Pb/235U concordanti. Ciò è quanto si verifica nel caso ideale in cui il sistema sia rimasto chiuso, cioè ove non si siano verificati né acquisti né perdite dell'elemento radioattivo (uranio) né dell'elemento radiogenico né dei membri radioattivi intermedi delle famiglie dell'uranio e in cui inoltre sia nota la concentrazione iniziale di 206Pb e 207Pb. Qualora queste condizioni non siano verificate, le due età saranno discordanti e il punto corrispondente cadrà al di fuori della curva menzionata.
I punti che giacciono al di sotto della curva rappresentano casi in cui si è verificata una perdita di piombo, o eventualmente un guadagno di uranio; quelli che giacciono al di sopra della curva rappresentano i casi in cui si è verificata una perdita di uranio o un acquisto di piombo. Wetherill (v., 1956) ha dimostrato che se si verifica al tempo t0 una perdita episodica di piombo senza frazionamento isotopico, in una o più fasi minerali, in un periodo di tempo che sia breve rispetto all'età di quella fase, i punti corrispondenti alla fase in questione giaceranno su una retta, detta anche ‛linea discordia', la cui intersezione inferiore con la curva concordia rappresenterà il tempo t0 in cui si è verificata la perdita episodica e l'intersezione superiore rappresenterà l'età vera della fase (v. fig. 33). Se la perdita episodica si è verificata recentemente, la retta in questione passerà per l'origine. Se poi si è verificata nel tempo una perdita continua di piombo per diffusione, la linea discordia presenterà nel primo tratto un andamento curvilineo.
Le considerazioni esposte rendono conto delle ‛età discordanti' e consentono, in condizioni favorevoli, di risalire da queste ultime alle età vere.
Nelle tre grandi famiglie radioattive dell'uranio-238, dell'uranio-235 e del torio-232 si stabilisce col tempo uno stato di equilibrio radioattivo secolare. A equilibrio raggiunto, mentre il rapporto fra i vari radionuclidi che costituiscono la famiglia rimane costante, varierà viceversa, nel tempo, il rapporto fra l'elemento progenitore (per es., 238U) e l'elemento stabile che rappresenta il prodotto finale del decadimento (per es., 206Pb). In questo modo il rapporto 238U/ 206Pb è funzione dell'età e può essere utilizzato nel modo descritto per la misura del tempo geologico. Avviene talvolta che i diversi radionuclidi derivanti dall'uranio-238, dall'uranio-235 e dal torio-232 risultino, a causa di fenomeni di precipitazione selettiva, inglobati nei sedimenti in quantità relative diverse da quelle previste dall'equilibrio secolare. Così, per esempio, in certi sedimenti marini, si trova un arricchimento di torio-230 (ionio) mentre l'elemento genitore 238U, che è più solubile, si arricchisce nelle acque marine. Se questi materiali sono di età recente, l'equilibrio nella famiglia dell'uranio non è ancora ristabilito e i metodi tradizionali di datazione, che presuppongono appunto tali condizioni di equilibrio, non possono essere impiegati. Al contrario possono essere impiegati i cosiddetti metodi del non equilibrio. Essi sono basati sulla variazione nel tempo del contenuto in radio o in ionio come risultato del disturbo dell'equilibrio nella famiglia dell'uranio al tempo della deposizione del sedimento, nell'ipotesi che il contenuto del genitore radioattivo depositato sia rimasto costante durante il tempo della deposizione a causa dell'uniformità chimica della fase in cui l'elemento genitore è distribuito, come si verifica, per es., nell'acqua di mare. I metodi più noti basati su stati di non equilibrio radioattivo sono quelli che fanno uso delle coppie ionio-torio, protoattinio-ionio, ionio-uranio-234 e protoattinio-uranio-235. Metodi di questo tipo possono essere impiegati con successo nella datazione di sedimenti recenti e di fossili carbonatici marini e lacustri, purché siano verificate alcune condizioni fondamentali.
d) Il metodo del carbonio-14.
Nella misura del tempo geologico un posto del tutto particolare spetta al metodo del carbonio-14 per la possibilità di datare eventi recenti e per l'interesse che ne deriva non solo ai fini della geologia del Quaternario recente, ma anche per le applicazioni nel campo dell'archeologia.
Si conoscono due isotopi stabili del carbonio, il 12C e il 13C, i quali formano rispettivamente il 98,89% e l'1,11% in atomi del carbonio ordinario, e un isotopo radioattivo, il 14C detto anche radiocarbonio, che decade secondo lo schema 14C→14N+β-. Il carbonio-14 si forma in continuazione nell'alta atmosfera per azione di neutroni termici su atomi di azoto, ossigeno e carbonio mediante le reazioni
14N+n→14C+1H
17O+n→14C+4He
15N+n→14C+2H
13C+n→14C+hν
delle quali la prima è la dominante. Gli atomi di 14C così formati entrano in combinazione con l'ossigeno per formare diossido di carbonio radioattivo 14CO2 attraverso un processo non ancora del tutto chiarito; quest'ultimo si mescola con il CO2 inattivo dell'atmosfera e con esso partecipa al ciclo geochimico del carbonio attraverso l'idrosfera e la materia vivente. La quantità di 14C contenuta nel diossido di carbonio atmosferico è molto bassa ed è stimabile a 10-12 g di 14C per ogni g di 12C.
Le piante, attraverso la funzione clorofilliana, e gli animali che di esse si nutrono contengono il 14C in questo stesso rapporto fino a che sono viventi, in quanto durante il periodo di vita il 14C assimilato dalla materia vivente compenserà esattamente quello disintegrato nel tessuto. Quando interviene la morte il processo di assimilazione cessa e rimane attivo solo il processo di disintegrazione. Poiché è conosciuto il tempo di dimezzamento del carbonio-14, che è di 5.568 anni, risulta pure nota la legge esponenziale del decadimento e pertanto la quantità di carbonio-14, espressa dalla sua attività ancora presente in un campione morto, ne rivela l'età, cioè il tempo trascorso dal momento della morte.
L'esperienza indica che nei campioni biologici, al momento della morte, la concentrazione di 14C è tale da fornire un attività media di 15,3 disintegrazioni per minuto per grammo di carbonio. La legge del decadimento sarà allora espressa da
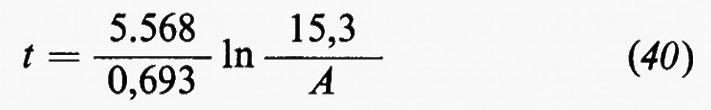
da cui
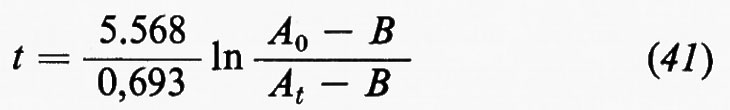
essendo A l'attività del materiale, sempre espressa in disintegrazioni per minuto per grammo di C, e t l'età del campione.
Il dosaggio del radiocarbonio avviene attraverso la misura della sua attività β, il che presuppone la possibilità di misurare attività molto basse. Nella pratica corrente si usa fare riferimento a uno ‛standard di carbonio attuale' ed è necessario tener conto che ogni sistema di rivelazione ha un ‛fondo' (background) B dovuto ai raggi cosmici, alla radio-attività dei materiali di cui è costituito il rivelatore e all'ambiente: si fa perciò uso dell'espressione
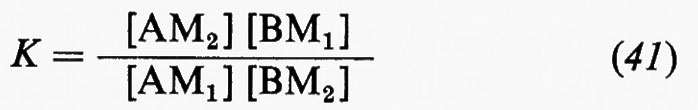
dove A0 e At rappresentano le attività misurate rispettivamente per lo standard e per il campione in esame e B è il ‛fondo'.
W. F. Libby (v., 1952), a cui si deve la scoperta del metodo, determinava l'attività del materiale, ridotto in forma di carbonio elementare, per mezzo di uno speciale contatore di Geiger opportunamente schermato e munito di una corona di contatori in anticoincidenza per eliminare il fondo dovuto alla radiazione cosmica. Successivamente è prevalso l'uso dei contatori proporzionali a gas o di scintillatori liquidi; con le tecniche attuali è possibile misurare età fino a 50.000 anni dal presente e, operando un arricchimento isotopico, si può raggiungere il limite di 70.000 anni.
Il Libby aveva fatto originariamente l'ipotesi che il rapporto 14C/12C fosse rimasto costante negli ultimi 20-30.000 anni; tale costanza è uno dei presupposti del metodo di datazione. Lo stesso Libby aveva però previsto che certe variazioni potevano essersi verificate, benché le misure effettuate con i mezzi di cui disponeva non avessero indicato mutamenti significativi.
Il miglioramento delle tecniche ha consentito successivamente di porre in evidenza delle variazioni che possono essere così raggruppate: a) effetto industriale, o effetto Suess, che consiste in una diminuzione di attività in seguito alla diluizione del diossido di carbonio atmosferico con CO2 inattivo da combustione di carboni e di petroli; l'effetto è rilevabile a partire dal 1860; b) effetto delle bombe atomiche: si stima che negli ultimi venti anni le esplosioni nucleari abbiano pressoché raddoppiato la concentrazione atmosferica del 14C; c) variazioni secolari prima del 1900 d. C.: queste ultime possono essere messe in evidenza dal confronto delle età ottenute col metodo del 14C con quelle di campioni archeologici di età nota o di campioni di legno di Sequoia gigantea o di Pinus aristata di età nota attraverso la dendrocronologia.
Se i primi due effetti sono senza conseguenze sul sistema di datazione qualora si faccia riferimento a opportuni standard, più serio è l'effetto delle variazioni secolari. Oggi si dispone però di precise curve di correlazione fra le ‛età calendario' e le ‛età carbonio-14', per cui è possibile apportare a queste ultime un'opportuna correzione (v. fig. 34).
Benché, a causa del piccolo valore del tempo di dimezzamento, il campo di applicabilità del metodo sia piuttosto limitato, tuttavia i risultati che se ne possono ottenere presentano il massimo interesse, in quanto trovano applicazione nella datazione di oggetti storici, preistorici e protostorici, di sedimenti oceanici, di eventi geologici del Quaternario recente, ivi compresi gli eventi glaciali e quelli vulcanici, nel controllo e nella taratura del metodo delle varve, nella datazione dell'epoca di caduta di meteoriti e infine nella da tazione e nello studio del movimento delle acque sotterranee.
I metodi che abbiamo descritto sono certo quelli più largamente in uso nelle ricerche di geocronometria; essi non esauriscono peraltro tutte le possibilità offerte dalla chimica e dalla fisica per la misura del tempo geologico. Ci limiteremo a menzionare, per le sue vaste possibilità d'impiego, la datazione per mezzo delle tracce di fissione nucleare e delle tracce di rinculo a (v. Price e Walker, 1963; v. Huang e Walker, 1967). Questo procedimento può essere classificabile in un gruppo di metodi, fra i quali quello della termoluminescenza, degli aloni pleocroici e degli stati metamittici, che sono basati sui danni provocati dalle disintegrazioni radioattive alle strutture cristalline dei minerali che ospitano nuclidi radioattivi.
È in fase di sviluppo un nuovo procedimento, basato sulla constatazione che gli L-amminoacidi di origine biologica presenti nei sedimenti e nelle ossa subiscono nel tempo un lento processo di epimerizzazione: così, per es., la L-isoleucina si trasforma con il tempo in D-alloisoleucina. Il metodo si rivela molto promettente per la possibilità di datare sedimenti marini nell'intervallo di tempo da 4•104 a 2•106 anni e oltre, superando di molto il campo di applicabilità del metodo del carbonio-14.
Questa rassegna, necessariamente incompleta, dei diversi metodi di valutazione del tempo geologico è tuttavia sufficiente a dimostrare come oggi, con l'impiego dei metodi geochimici, la geologia sia in grado di valutare quantitativamente il parametro tempo e ciò in tutta l'estensione della scala dei tempi, dall'età di formazione della Terra fino agli eventi più recenti. E questa una delle conquiste più significative del nostro secolo.
La scala stratigrafica ha potuto così tradursi in una scala dei tempi, quest'ultima potendo ormai esprimersi in grande dettaglio come appare nella Phanerozoic Time Scale (v. tab. VII) presentata dalla Geological Society di Londra (v. Harland, Smith e Wilcock, 1964). Di grandissimo significato si rivelano poi le datazioni di minerali in rocce precambriane, che in questi ultimi anni sono diventate così numerose da fornire una larga base per la costruzione di una cronologia precambriana e da consentire una correlazione generale dei cicli orogenici precambriani nei diversi continenti (v. Sarkar, 1968).
Tabella 7
Nella fig. 35 sono indicati i limiti pratici di validità dei metodi radiometrici oggi disponibili, unitamente a quelli di altri metodi di cronologia assoluta in relazione alla scala dei tempi.
11. Geochimica degli isotopi stabili.
a) Generalità.
La geochimica classica ha avuto e ha fra i suoi principali obiettivi lo studio del comportamento dell'elemento chimico negli ambienti naturali. Le stesse relazioni di abbondanza degli elementi diventano però più chiare nei loro fondamenti qualora in luogo delle abbondanze degli elementi intesi come specie atomiche si considerino le abbondanze delle specie nucleari, ossia dei nuclidi. È logico considerare il complesso dei nuclidi a cui corrisponde lo stesso valore del numero atomico Z, ossia il ‛complesso isotopico' di ogni elemento: le abbondanze relative ai diversi isotopi di questo, espresse in atomi per cento, vengono definite come ‛abbondanze isotopiche'. Le abbondanze isotopiche riportate nella letteratura scientifica si riferiscono agli elementi così come si rendono disponibili nei processi di estrazione dai materiali naturali e non si debbono considerare come una caratteristica costante di ogni elemento se non in prima approssimazione, ma piuttosto come ‛abbondanze isotopiche convenzionali'.
Benché i diversi isotopi di un elemento, a causa della disposizione dei loro elettroni orbitali, mostrino una stretta analogia di comportamento chimico, tanto che manifestano in generale una strettissima coerenza geochimica, pure si verificano in natura talune condizioni che possono provocare, talora in misura notevole, piccole variazioni delle abbondanze isotopiche.
La geochimica degli isotopi studia le cause delle variazioni di composizione isotopica negli ambienti naturali e i relativi processi, nonché le conseguenze che ne possono scaturire e che trovano applicazione nello studio dei processi geologici.
Le variazioni di composizione isotopica che si possono verificare negli ambienti naturali hanno la loro origine in parte come conseguenza di reazioni nucleari, in parte in seguito a processi di frazionamento di natura chimico-fisica.
Le reazioni nucleari spontanee determinano la formazione di nuove specie nucleari, sia stabili sia radioattive, e conseguentemente sono causa di variazione nella composizione isotopica di certi elementi, per cui si verifica in definitiva una diminuzione, nel tempo, degli isotopi radioattivi e una crescita di quegli isotopi stabili che sono il prodotto finale del decadimento. Si è visto, per esempio, che gli isotopi stabili del piombo 206Pb, 207Pb e 208Pb sono i prodotti finali del decadimento di 238U, 235U, 282Th e pertanto la loro percentuale nel complesso isotopico del piombo tenderà ad aumentare nel tempo. Parimenti, nel tempo, tenderà ad aumentare la percentuale di stronzio-87, di argon-40 e di calcio-40 nel complesso isotopico di questi ultimi elementi. Le menzionate variazioni di composizione isotopica stanno alla base dei metodi di geocronologia isotopica e talune di esse formano oggetto di studi particolari: così, per es., lo studio dell'evoluzione della composizione isotopica del piombo ha determinato lo sviluppo di un particolare settore della geochimica, la ‛plumbologia'.
Anche le reazioni nucleari indotte nell'alta atmosfera dalla radiazione cosmica secondaria provocano la formazione di nuove specie nucleari e perciò anche variazioni nella composizione isotopica: esse sono però in genere trascurabili se si eccettuano quelle relative alla produzione di 14C e di 3H. E da rilevare che il trizio si trasforma per decadimento in elio-3 e il carbonio-14 rigenera per decadimento il nuclide 14N che lo ha prodotto.
I principali processi chimico-fisici che possono determinare effetti di frazionamento isotopico sono le reazioni di scambio isotopico all'equilibrio, le differenze fra le tensioni di vapore di liquidi e di solidi isotopici e gli effetti di natura cinetica.
Le reazioni di scambio fra specie isotopiche si verificano a causa delle piccole differenze fra le proprietà termodinamiche delle diverse specie isotopiche di un determinato composto. Un esempio di reazione di scambio isotopico può essere quella fra diossido di carbonio e acqua:
C18O2+H216O⇄C18O16O+H218O.
Per una generica reazione di scambio in fase gassosa
AM1+BM2⇄AM2+BM1,
dove M1 e M2 sono due isotopi dell'elemento M, la relativa costante di equilibrio

può essere espressa come rapporto delle funzioni di partizione Q delle molecole partecipanti alla reazione
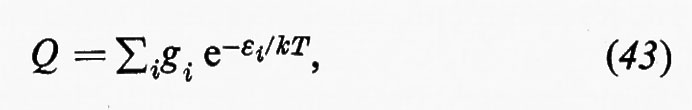
la funzione di partizione essendo definita da

dove εi è l'energia del livello energetico i della molecola, k la costante di Boltzmann, T la temperatura assoluta e gi il peso statistico del livello i, la sommatoria essendo estesa a tutti i livelli energetici della molecola. Questo metodo è generalmente usato essendo noti i rapporti delle funzioni di partizione o quanto meno di grandezze a esse correlabili, che stanno nella stessa relazione con la costante di equilibrio. In genere i valori delle costanti così calcolate non sono molto diversi dall'unità; ciò sta a indicare che, a eccezione di composti di atomi leggeri, gli effetti di frazionamento sono di solito piccoli, ma il frazionamento può diventare rilevante se si verifica attraverso un processo a più stadi. L'ammontare del frazionamento è proporzionale alla differenza di massa relativa (ΔA/A) dei composti isotopici considerati: esso è massimo per la coppia idrogeno-deuterio; gli effetti di frazionamento sono rilevanti fino al numero di massa 40 e sono ancora sensibili fino al numero di massa 80. Per reazioni fra gas ideali a basse temperature vale la relazione in K∝cost/T e a temperature più elevate in K∝cost/T2. Per temperature molto elevate, al crescere della temperatura le costanti di equilibrio tendono all'unità, con la conseguenza che gli effetti di frazionamento isotopico tendono a scomparire. La dipendenza della costante di equilibrio dalla temperatura per alcune reazioni di scambio isotopico in fase gassosa è posta in evidenza in fig. 36. Si può osservare che per alcuni sistemi importanti, come O2−H2O, il valore di K passa da valori maggiori a valori minori dell'unità: ciò significa che il frazionamento isotopico s'inverte per un certo valore della temperatura.
Immaginando che a un sistema costituito da un gas ideale venga fornita l'energia E, se tutte le molecole hanno la stessa massa m, si avrà una distribuzione statisticamente omogenea delle velocità v il cui valore sarà dato da

Se però due molecole differiscono per la composizione isotopica e quindi per le masse m1 e m2, esse si muoveranno con velocità v1 e v2 il cui rapporto sarà dato da
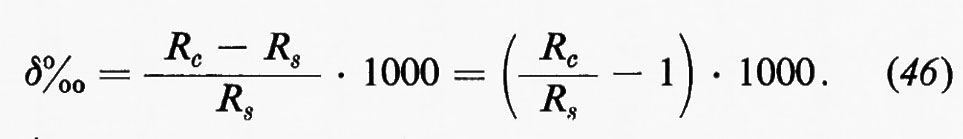
Le molecole più leggere diffonderanno perciò più rapidamente rispetto a quelle più pesanti, determinando in tal modo un effetto di frazionamento per diffusione.
Analoghi effetti si possono prevedere per altri fenomeni fisici quali l'evaporazione, la fusione, la soluzione e l'adsorbimento. Frazionamenti isotopici si verificano così nei processi di evaporazione e di condensazione delle acque terrestri e oceaniche e interessano in particolare il ciclo isotopico dell'idrogeno e dell'ossigeno. In generale la specie isotopica più leggera si concentra, in un equilibrio di due fasi, nella fase meno densa.
Nelle reazioni chimiche unidirezionali, in condizioni di non equilibrio, le molecole più leggere, che si muovono più rapidamente, hanno maggiore probabilità d'interagire con altre molecole rispetto a quelle più pesanti: esse sono dotate, in altre parole, di una maggiore velocità di reazione. Può essere spiegato con questo effetto cinetico il fatto che il carbonio delle piante è isotopicamente più leggero rispetto a quello contenuto nel CO2 atmosferico, in quanto la molecola leggera 12C16O2 viene a contatto della foglia più frequentemente rispetto alla molecola pesante 13C16O2 e di conseguenza viene a partecipare in misura maggiore al ciclo vitale della pianta. Analoga spiegazione può essere data agli effetti di frazionamento degli isotopi dello zolfo osservabili nei processi di riduzione batterica dei solfati.
Vi sono diversi modi di riportare i dati relativi alle composizioni isotopiche; queste possono venire espresse: a) come rapporto atomico assoluto R della coppia di isotopi considerata; b) come differenza Rc−Rs fra il rapporto assoluto Rc del campione e quello Rs di uno standard di riferimento; c) come differenza in parti per mille fra il rapporto Rc nel campione e il rapporto Rs di uno standard, riferita al rapporto dello standard
Formula
Per convenzione R indica il rapporto fra l'isotopo più pesante e quello più leggero.
b) Geochimica degli isotopi stabili dell'idrogeno.
La geochimica degli isotopi stabili dell'idrogeno ha attratto sinora la maggior attenzione per quanto si riferisce al ciclo idrologico, nel quale si manifestano vistose variazioni di composizione isotopica. Per l'idrogeno, come pure per l'ossigeno, si assume come valore di riferimento il rapporto D/H dell'acqua oceanica media (Standard Mean Ocean Water=SMOW). Nelle acque meteoriche il valore δD varia entro limiti molto estesi, da circa −400 a +10‰. In queste acque è stata rilevata una ben definita relazione fra i rapporti D/H e 18O/16O, esprimibile con l'equazione (v. Craig, 1961)
δD=8 δ18O+10 (47)
ricavata da osservazioni eseguite su acque fluviali, lacustri e piovane di tutto il mondo e rappresentata graficamente in fig. 37. Nelle acque superficiali continentali la composizione isotopica dell'idrogeno è determinata da diversi fattori, dei quali il più importante è la temperatura, mentre altri sono da essa dipendenti. Infatti il frazionamento isotopico che si produce durante l'evaporazione è inversamente proporzionale alla temperatura; si comprende pertanto come le precipitazioni tendano a divenire isotopicamente più leggere, con l'aumentare della latitudine e con l'altitudine. I valori più bassi di δD si riscontrano nelle nevi e nei ghiacci polari. Nei processi di evaporazione delle acque oceaniche e delle successive precipitazioni si possono avere effetti multipli di frazionamento. I campioni che nel grafico di fig. 37 si scostano sensibilmente dalla retta provengono da bacini in cui forti fenomeni di evaporazione hanno sensibilmente alterato la composizione isotopica della soluzione residuale.
Nelle falde acquifere la composizione isotopica non subisce ulteriori variazioni, a meno che non intervengano scambi isotopici con l'ossigeno delle rocce; tuttavia questi scambi, alle temperature normalmente osservate, sono molto lenti e si possono ritenere trascurabili. Ne segue che la composizione isotopica delle acque profonde è in relazione a quella delle precipitazioni nella regione di ricarica al tempo di ricarica.
La stretta interdipendenza fra le variazioni di composizione isotopica dell'idrogeno e dell'ossigeno nelle acque meteoriche e superficiali determina il fatto che entrambi questi elementi possono essere impiegati come traccianti isotopici negli studi inerenti alla circolazione delle acque.
Lo studio della composizione isotopica delle acque naturali permette inoltre di porre in evidenza la partecipazione delle acque meteoriche a vari processi geochimici.
Uno dei problemi tuttora aperti è quello riguardante le cosiddette acque iuvenili e le acque magmatiche. Si è definita acqua iuvenile un'acqua che sia derivata da magmi primari e non sia stata mai a contatto con acque superficiali. Non è facile localizzare un'acqua che corrisponda con sicurezza a questi requisiti. La ricerca può essere diretta alle acque delle inclusioni fluide in rocce basaltiche o in minerali come quarzo, calcite, blenda o presenti in minerali idrossilati che si ritengano originati nel mantello o in parti profonde della crosta. Acque di questo tipo mostrano valori di δD variabili da −33 a +83‰: alcuni autori assegnano alle acque iuvenili un valore di δD di circa −48‰.
È evidente che, anche ammesso che si possano accettare i valori esposti come appartenenti ad acque iuvenili, essi non sono purtroppo in alcun modo caratteristici nè diagnostici, poiché qualsiasi acqua può assumere questi valori in seguito a processi di frazionamento e di scambio: infatti valori simili si registrano facilmente anche in acque meteoriche.
Le acque cosiddette magmatiche in generale non sono necessariamente acque iuvenili: si definiscono come tali, infatti, le acque che sono state equilibrate chimicamente e isotopicamente con masse magmatiche o con corpi di rocce ignee. Esse possono derivare da acque iuvenili, come da acque meteoriche o da ‛acque di formazione'. Queste acque presentano valori di δD compresi fra −50 e +90‰ e per esse valgono le considerazioni già esposte per le acque iuvenili.
c) Geochimica degli isotopi stabili del carbonio.
Il carbonio naturale è costituito dai due isotopi stabili, 12C e 13~C, e dall'isotopo radioattivo 14C. Le abbondanze relative convenzionali dei due isotopi stabili sono, in atomi per cento,
12C 98,892
13C 1,108.
Le variazioni della composizione isotopica del carbonio, espresse in unità δ, che si riscontrano nei materiali naturali sono generalmente comprese fra −70 e +60‰, con riferimento a uno standard costituito dal rostro della Belemnitella americana della formazione Pee Dee della Carolina del Sud, contrassegnato perciò con la sigla PDB.
In fig. 38 sono rappresentati i campi di variabilità di δ13C riscontrati in numerosi materiali naturali. I valori positivi più elevati si osservano nella frazione carbonatica di alcune meteoriti dette condriti carbonacee (δ13C=+60‰); il materiale contenente, all'estremo opposto, il carbonio più leggero è il metano, in cui sono stati registrati valori di δ13C fino a −85‰. Particolarmente rilevanti sono gli effetti di frazionamento nel carbonio biogenico. Durante la fotosintesi le piante assimilano di preferenza l'isotopo leggero. Le piante terrestri utilizzano il CO2 atmosferico, in cui δ13C vale in media −7‰, mentre le piante marine assimilano prevalentemente il carbonio proveniente dal bicarbonato dell'acqua, che è alquanto più pesante (δ13C=2‰). La differenza che si riscontra nei campi di variabilità di δ13C dei carbonati marini, di acqua dolce e diagenetici è riferibile alla diversità delle condizioni chimico-fisiche e geologiche che presiedono allà loro formazione e ai processi di equilibrazione che si verificano nella loro evoluzione diagenetica.
d) Geochimica degli isotopi dell'ossigeno.
L'ossigeno è costituito da tre isotopi stabili 16O, 17O e 18O, le cui abbondanze atomiche percentuali relative sono rispettivamente 99,759, 0,0374 e 0,2039. In natura questi rapporti possono variare sensibilmente per i fattori già menzionati. Le variazioni di composizione isotopica dell'ossigeno nei materiali naturali, espresse in δ18O con riferimento allo SMOW, sono comprese fra −50‰ e + 40‰ (v. fig. 39). I valori più bassi si riscontrano nelle nevi e nei ghiacci polari come conseguenza dei meccanismi sopra discussi. Il diossido di carbonio atmosferico ha un valore di δ18O di 41‰, corrispondente al valore di equilibrio con l'acqua oceanica a 25 °C. Le acque magmatiche equilibrate con grandi masse di silicati mostrano valori di δ18O variabili fra meno di 7,0 e 9,5‰. Per quanto si riferisce alle rocce, i più alti valori del rapporto 18O/16O si riscontrano nelle rocce sedimentarie (da 11 a 36‰), i più bassi nelle rocce ignee (da 5 a 14‰), mentre le rocce metamorfiche occupano una posizione intermedia (da 7 a 27‰).
Una delle più interessanti ripercussioni degli studi condotti nell'ultimo ventennio sui frazionamenti isotopici dell'ossigeno si ha nella geotermometria isotopica, basata sul fatto che le costanti di equilibrio delle reazioni di scambio isotopico sono funzione della temperatura. Se tale legge di variazione è nota, è in linea generale possibile, da una misura di K, stabilire la temperatura alla quale il frazionamento isotopico misurato si è prodotto, ammesso che i rapporti isotopici 18O/16O siano rimasti inalterati fino al momento della misura.
Uno dei più noti geotermometri isotopici è quello basato sul frazionamento isotopico dell'ossigeno fra carbonato di calcio e acqua, espresso dalla reazione
1/3CaC16O3+H218O⇄1/3Ca18CO3+H216O
suggerito da H. C. Urey fin dal 1947.
Epstein, Buchsbaum, Lowenstam e Urey (v., 1951 e 1953) in base a misure compiute su conchiglie di molluschi cresciuti a temperatura controllata poterono così elaborare la cosiddetta scala delle paleotemperature espressa dall'equazione
t=16,5−4,3 (δc−δa)+0,14 (δc−δa)2 (48)
in cui δc rappresenta, in unità δ, la composizione isotopica della calcite depositata in equilibrio isotopico alla temperatura t nel guscio del mollusco da un'acqua, l'ossigeno della quale ha la composizione isotopica δa. Con la precisione attualmente raggiungibile nella misura dei rapporti isotopici è possibile mediante questa scala effettuare valutazioni di temperatura con l'approssimazione di ±0,5 °C. Recentemente è stato proposto un secondo geotermometro per misure di paleotemperature basato sul frazionamento isotopico dell'ossigeno fra l'acqua e il fosfato precipitato nel guscio di molluschi (v. Longinelli e Nuti, 1967 e 1973).
La scala delle paleotemperature è soggetta ad alcune limitazioni, la più importante delle quali riguarda l'incertezza della conoscenza della composizione isotopica dell'acqua δa. Essa ha tuttavia consentito importanti deduzioni riguardanti la paleoclimatologia. La scala termometrica è stata recentemente calibrata in laboratorio fino a 750 °C ed è pertanto applicabile a vari problemi geologici. Più in generale il frazionamento isotopico fra qualsiasi coppia di minerali in equilibrio, che è dipendente dalla temperatura, può essere usato come termometro isotopico. Oggi si conoscono le leggi di variazione con la temperatura delle costanti di equilibrio relative a diversi sistemi quarzo-acqua, alcali-feldspato-acqua e muscovite-acqua. Questi geotermometri sono stati utilmente impiegati nello studio di rocce ignee, metamorfiche, sedimentarie e di depositi idrotermali. I metodi isotopici si affiancano così validamente agli altri numerosi metodi chimico-fisici di geotermobarometria.
e) Geochimica degli isotopi dello zolfo.
Dello zolfo sono conosciuti in natura i quattro isotopi stabili 32S, 33S, 34S e 36S, oltre all'isotopo radioattivo 38S, aventi rispettivamente le abbondanze convenzionali relative 91,1, 0,74, 4,2 e 0,016% in atomi. Di particolare interesse sono le variazioni del rapporto 34S/32S. Per lo zolfo lo standard di riferimento è la troilite della meteorite di Cañon Diablo, per la quale il rapporto 34S/32S è di 0,04500. Il campo di variabilità di δ34S nei campioni di zolfo terrestre è di circa il 10%. Rilevanti, nella geochimica isotopica dello zolfo, sono gli effetti isotopici di natura cinetica che si manifestano sia nei processi chimici sia in quelli biologici. I solfuri e lo zolfo derivanti da riduzione batterica tendono ad arricchirsi nell'isotopo leggero. L'effetto è più o meno rilevante a seconda che i sistemi ricevano un maggiore o minore apporto di solfato dall'esterno. Se l'apporto è trascurabile si possono avere effetti isotopici anche del 60‰; se invece è presente un serbatoio di solfato praticamente infinito, come per es. l'acqua oceanica, l'effetto è limitato al 15‰ (v. fig. 40). Si può così interpretare la maggior dispersione dei valori di δ34S rilevabile nei solfuri e solfati sedimentari nei confronti dei solfati marini. I petroli incorporano verosimilmente lo zolfo ridotto accumulato nei sedimenti. Nei solfuri e solfati delle rocce ignee i rapporti isotopici che in essi si osservano sono attribuiti a effetti di frazionamento durante la cristallizzazione del magma.
f) Geochimica degli isotopi dello stronzio.
Le abbondanze isotopiche convenzionali dei quattro isotopi stabili dello stronzio sono le seguenti: 84Sr 0,56; 86Sr 9,86; 87Sr 7,02; 88Sr 82,56 atomi per cento. Come si è visto, il decadimento radioattivo del rubidio-87 genera lo stronzio-87; è pertanto facilmente intuibile che le variazioni di composizione isotopica dello stronzio osservabili in natura siano in ultima analisi da imputare al decadimento del rubidio, essendo gli effetti naturali di frazionamento trascurabili o quanto meno al di sotto dei limiti di rivelabilità.
Nel corso della cristallizzazione magmatica il rubidio e lo stronzio hanno un differente comportamento geochimico poiché il rubidio tende ad arricchirsi col progredire della differenziazione, mentre lo stronzio viene rimosso precocemente dal magma e tende a concentrarsi nei plagioclasi ricchi di calcio della prima cristallizzazione. Le rocce arricchite in rubidio, d'altra parte, tenderanno ad arricchirsi, nel tempo, anche in stronzio-87. Si comprende quindi come la geochimica isotopica dello stronzio sia condizionata anche dal comportamento geochimico dei due elementi.
Abbiamo visto come, attraverso l'equazione (33), sia possibile determinare non solo l'età della roccia, ma anche il rapporto 87Sr/86Sr al momento della cristallizzazione. Questo rapporto dipende dalla storia precedente dello stronzio e dai rapporti Rb/Sr dei sistemi di cui lo stronzio ha fatto parte in precedenza.
Le rocce del mantello superiore, secondo ogni evidenza, sono costituite essenzialmente da silicati di ferro e magnesio: le rocce della crosta sono essenzialmente a base feldspatica e sono, in proporzione, più ricche di silice, di allumina e di alcali; esse sono, di conseguenza, più ricche di rubidio e hanno un rapporto Rb/Sr più alto rispetto al mantello. È perciò prevedibile che le rocce granitiche della crosta abbiano anche un rapporto 87Sr/86Sr più elevato di quello riscontrabile nel mantello.
Se assumiamo come composizione isotopica attuale dello stronzio delle isole oceaniche un valore 87Sr/86Sr pari a 0,7037 e come rapporto 87Sr/86Sr all'origine il valore di 0,699 indicato dalle acondriti basaltiche, introducendo questi valori nell'equazione (33) si ottiene per la zona del mantello superiore, che si può considerare come il luogo d'origine dei basalti oceanici, un rapporto Rb/Sr di 0,025.
Se ora assegnamo alle rocce crostali un'età media di 2,5•109 anni e ammettiamo che lo stronzio in esse contenuto sia derivato, all'epoca della loro formazione, dal mantello, a quell'epoca il rapporto 87Sr/86Sr doveva essere, come nel mantello, di 0,701. Se assumiamo per il rapporto Rb/Sr nella crosta il valore di 0,18 (come indicato dalle rocce degli scudi precambriani) e introduciamo questi valori nell'equazione (33), si ottiene per il rapporto 87Sr/86Sr nella crosta al tempo presente il valore di 0,719, in buon accordo con i dati sperimentali.
Il diagramma di fig. 41, costruito secondo questo modello, mostra l'evoluzione della composizione isotopica dello stronzio nelle regioni d'origine dei basalti e nella crosta continentale.
Secondo questo modo di vedere, le rocce ignee che cristallizzano da un magma derivato dal mantello superiore sono contraddistinte da un rapporto 87Sr/86Sr iniziale che cade nel ‛campo dei basalti', mentre se il magma è generato per parziale fusione, metasomatismo o assimilazione di materiali crostali presenterà valori di tale rapporto significativamente più alti.
Il modello proposto (v. Faure e Hurley, 1963) pecca certamente di eccessiva semplicità: esso ammette fra l'altro che il rapporto Rb/Sr nelle zone del mantello che si assumono come origine dei magmi basaltici sia rimasto costante nel tempo, cioè che il mantello si possa considerare come un ‛serbatoio infinito' di Rb e di Sr. Ciò non è vero in linea generale poiché tale rapporto è probabilmente andato diminuendo nel mantello proprio a causa della formazione di una rilevante crosta ricca di rubidio. In questo caso le linee AB e AC non sarebbero più rette, ma presenterebbero una convessità verso l'alto.
Anche nella sua forma approssimata il modello descritto giova alla comprensione del meccanismo di evoluzione della composizione isotopica dello stronzio nella crosta e nel mantello.
g) Geochimica degli isotopi del piombo.
Il piombo è costituito dai quattro isotopi stabili 204Pb, 206Pb 207Pb e 208Pb, aventi rispettivamente le abbondanze relative convenzionali di 1,4, 25,2, 21,7 e 51,7 atomi per cento. Poiché il decadimento radioattivo dell'uranio e del torio genera come prodotti finali stabili i tre isotopi del piombo 206, 207 e 208, ne segue che durante la storia della Terra si deve essere verificato un aumento nell'abbondanza totale del piombo terrestre. Tale incremento è stato stimato del 20% negli ultimi 3,3 miliardi di anni (v. Wedepohl, 1956) e del 50% negli ultimi 4,5 miliardi di anni (v. Shaw, 1957), il che corrisponde a una variazione del contenuto medio crostale da 10 a 15 parti per milione.
È inoltre prevedibile un'evoluzione nel tempo della composizione isotopica del piombo a partire dalla sua composizione primordiale al momento della formazione della Terra. Quest'ultima si può dedurre da quella del piombo contenuto in certe meteoriti metalliche, che è il piombo meno radiogenico che si conosca.
La composizione isotopica del piombo che noi possiamo immaginare sia stato separato a un certo tempo t dagli elementi radioattivi uranio e tono dipenderà perciò dalla composizione primordiale, dal rapporto (U+Th)/Pb all'origine, dalle costanti di decadimento, dalle abbondanze isotopi che di U e Th e finalmente dal tempo t. Si possono così costruire dei diagrammi o ‛curve di crescenza' che rappresentano l'evoluzione dei rapporti isotopici in funzione del tempo (v. figg. 42 e 43).
I piombi che soddisfano alle condizioni indicate, cioè che hanno subito un solo frazionamento, si chiamano piombi normali o piombi ordinari; per essi la composizione isotopica è in relazione semplice con l'età. Diversamente si comportano i piombi che sono il risultato di due o più processi di separazione, in quanto essi sono costituiti dal mescolamento di piombi provenienti da regioni a differente composizione ovvero provengono dalla rimobilizzazione di minerali preesistenti. Questi ‛piombi anomali' si riconoscono proprio perché i loro rapporti isotopici indicano un'età diversa rispetto a quella delle rocce incassanti.
12. I problemi aperti e il futuro della geochimica.
La tendenza evolutiva di una disciplina si può facilmente tracciare attraverso il suo sviluppo storico. Nella prima parte del nostro secolo, se si esclude il lavoro pionieristico svolto nel settore della chimica fisica dei silicati, la geochimica ha avuto come obiettivo principale l'interpretazione del comportamento degli elementi, e in particolar modo degli elementi in tracce, nei materiali e negli ambienti naturali, quale risultava dalla valutazione del materiale analitico che gradualmente si rendeva disponibile. La ricerca delle leggi generali che regolano la distribuzione degli elementi era impostata prevalentemente sui principi della cristallochimica per la forte influenza che la scoperta della diffrazione dei raggi X e la possibilità di determinare le strutture cristalline esercitarono sulla mineralogia. Apparve sempre più chiaro che tali principi da soli non erano sufficienti; una delle caratteristiche dello sviluppo della geochimica nell'ultimo ventennio è uno sforzo verso la ricerca di principi più generali attraverso una migliore conoscenza delle energie di legame e delle grandezze termodinamiche caratteristiche delle reazioni a cui l'elemento prende parte.
L'impostazione razionale dei problemi relativi alla partizione degli elementi tra fasi solide, e tra fasi fluide e fasi solide, si può anch'essa considerare una conquista degli anni più recenti; è facilmente prevedibile un rapido sviluppo di questo e di altri settori della termodinamica geochimica. Attraverso queste considerazioni la geochimica si inserisce profondamente nei grandi problemi della geologia e della geofisica, particolarmente per quanto si riferisce alla composizione del mantello superiore e ai movimenti della crosta in relazione alle possibilità d'interpretazione offerte dal quadro della tettonica globale.
Infatti, se la conoscenza delle modalità di distribuzione degli elementi in tracce, e particolarmente degli elementi incompatibili, ivi inclusi gli elementi delle terre rare, da sola non può univocamente dare una risposta ai grandi quesiti relativi alla genesi dei basalti, alla composizione del mantello superiore e all'evoluzione dei fondi oceanici, essa consente nondimeno d'imporre delle restrizioni alle varie teorie petrogenetiche, siano esse basate su speculazioni teoriche o su modelli derivati dalla petrologia sperimentale.
Similmente la conoscenza dei rapporti isotopici del piombo e dello stronzio impone severe limitazioni alle ipotesi concernenti le relazioni fra i basalti abissali e i basalti insulari e alle zone di provenienza dei rispettivi magmi.
Indubbiamente una delle caratteristiche degli orientamenti della geochimica in questi ultimi anni è l'interpretazione e l'utilizzazione dei dati geochimici nel quadro dell'ambiente tettonico; assai rilevanti sono i risultati ottenuti per quanto riguarda le relazioni fra le caratteristiche geochimiche e il tipo di vulcanesimo le cui relazioni con la situazione tettonica appaiono oggi sempre più chiare. Se d'altra parte oggi si conoscono bene i caratteri geochimici differenziali delle tholeiiti abissali, delle tholeiiti d'arco, delle tholeiiti continentali, dei basalti alcalini delle isole oceaniche e dei loro prodotti di differenziazione, molti problemi relativi alla provenienza e all'interdipendenza dei relativi magmi rimangono ancora oscuri. Similmente ancora aperte rimangono le questioni riguardanti la maggiore o minore omogeneità del mantello e la verifica dell'ipotesi dei diapiri termici o mantle plumes. Molto si attende in proposito dai nuovi più raffinati dati della geochimica degli elementi incompatibili e dai rapporti isotopici dello stronzio e del piombo, che si possono ottenere oggi con un elevato grado di precisione. Il pregevolissimo lavoro che si è compiuto in questi ultimi anni non rappresenta che l'inizio di quella che sarà certamente una delle grandi avventure scientifiche del prossimo ventennio, in cui apparirà più chiara l'origine dei basalti oceanici in relazione alla natura del mantello.
Se, nonostante gli enormi progressi realizzati, a tutt'oggi non si può dare una risposta definitiva ai quesiti riguardanti la composizione e l'evoluzione temporale del mantello superiore, ancora più oscure rimangono, per la mancanza d'informazioni dirette, le conoscenze relative alla geochimica profonda del mantello inferiore e del nucleo. Se i risultati della petrologia sperimentale e della chimica fisica delle alte pressioni consentono di costruire un ragionevole modello per le relazioni di fase del mantello inferiore, molte questioni relative alla distribuzione degli elementi rimangono ancora aperte. Così, per esempio, la valutazione dell'influenza sull'equilibrio chimico del potenziale gravitazionale è ostacolata dall'imperfetta conoscenza della composizione e della densità delle fasi minerali ivi presenti e non è neppure certo che uno stato di equilibrio gravitazionale si debba considerare come raggiunto. Per quanto riguarda il nucleo, pare accertato che il numero atomico medio degli elementi che lo costituiscono debba esser superiore a quello degli elementi del mantello ma inferiore a quello del ferro. Se e in quale misura l'idrogeno, il carbonio, il silicio, lo zolfo e anche MgO e FeO possano trovarsi nel nucleo allo stato di soluzione nel ferro è una domanda alla quale gli sforzi congiunti della geochimica e della geofisica potranno dare forse una risposta nel futuro, anche nel quadro globale dell'origine e dell'evoluzione dei corpi del sistema solare.
Ritornando ai problemi della geochimica di superficie, molto ci si aspetta nel prossimo decennio per quanto si riferisce ai problemi non ancora risolti relativi alle rocce ultrabasiche e al significato di particolari tipi di rocce, quali le rocce alcaline; ancora una volta le prospettive sono aperte nella visione delle interconnessioni fra la geochimica e la tettonica globale.
Ancora più appassionanti si presentano alcuni temi di ricerca che si sono prospettati negli anni recenti. La ricerca di nuovi numeri magici di protoni e di neutroni ha stimolato lo sviluppo di teorie che hanno condotto a prevedere l'esistenza di ‛isole di stabilità' nel campo dei nuclidi più pesanti di tutti quelli sinora conosciuti. Sono in corso tentativi che utilizzano criteri fisici, chimici e geochimici per verificare la possibile esistenza in natura di questi elementi superpesanti. Benché il problema si presenti molto complesso, qualora la ricerca avesse esito positivo, notevoli prospettive sarebbero aperte alla fisica, alla chimica nucleare e alla geochimica, sia per quanto si riferisce all'estensione della legge di periodicità di Mendeleev, sia per la conoscenza dei processi di nucleosintesi nell'interno delle stelle.
Una delle scoperte più sensazionali di questi ultimi anni è senza dubbio quella dei reattori naturali fossili di Oklo nel Gabon. Il punto di partenza è stato un'analisi isotopica dell'uranio proveniente da quel giacimento, che aveva rivelato un tenore singolarmente basso di uranio-235 nei confronti del tenore abitualmente osservato nell'uranio naturale. Il campione aveva infatti rivelato 0,7171%±0,001 in atomi di uranio-235, mentre il tenore nell'uranio naturale è di 0,7202%±0,0006. L'ipotesi di una reazione nucleare a catena verificatasi nei tempi antichi e mantenutasi per lungo tempo, probabilmente per centinaia di migliaia di anni, nel giacimento si è affacciata immediatamente alla mente degli studiosi (v. Bodu e altri, 1972) e ha trovato piena conferma nello studio isotopico dei prodotti di fissione. Le condizioni critiche si sono potute verificare per un insieme di circostanze favorevoli dovute all'età del giacimento, che risale a 1,7 miliardi di anni, alla concentrazione di una grande massa di uranio, a un basso contenuto di elementi capaci di catturare i neutroni e alla presenza di acqua. La scoperta riveste un importanza eccezionale poiché ha posto in evidenza un fenomeno non ancora descritto nella storia della Terra, ma che è possibile si sia verificato più di una volta in circostanze analoghe; è facilmente prevedibile uno sviluppo delle ricerche in questa direzione.
La geochimica organica costituisce uno degli aspetti più recenti e più promettenti delle scienze della Terra. Il suo campo di ricerca è molto vasto. Lo studio delle sostanze organiche presenti nelle meteoriti quali prodotto di reazioni catalitiche fra monossido di carbonio, idrogeno e ammoniaca nella nebula solare ha portato alla recente scoperta, nelle meteoriti, di almeno 17 amminoacidi che si debbono considerare come il prodotto di sintesi abiologiche extraterrestri. E questo un passo molto importante nella conoscenza del meccanismo di formazione delle molecole organiche prebiologiche. Notevoli progressi sono stati realizzati, grazie alle recenti tecniche analitiche, nella conoscenza delle sostanze organiche presenti nei sedimenti e delle modificazioni da esse subite nel tempo. L'obiettivo principale è quello di ottenere quante più informazioni è possibile sulla storia della comparsa della vita sulla Terra (v. biofisica).
Nelle pagine precedenti si è posto principalmente l'accento sull'aspetto fondamentale delle ricerche geochimiche. Dalla conoscenza di principi generali che regolano la circolazione degli elementi in natura sono appunto scaturite le più fertili applicazioni della geochimica, che hanno rappresentato una delle più grosse conquiste dell'ultimo trentennio e che sono destinate a trovare nel prossimo futuro ancora più vaste applicazioni con ampi risvolti di carattere economico e sociale.
La conoscenza dei processi naturali di concentrazione e di dispersione degli elementi, entrambi dipendenti dal tempo, ha consentito da un lato di migliorare le nostre conoscenze e di formulare nuove ipotesi sull'origine di giacimenti di minerali utili, dall'altra di sviluppare un gruppo di metodi di ricerca di giacimenti sepolti, basati sulla rilevazione di quelle anomalie geochimiche che si manifestano in virtù appunto dei processi di dispersione. Questi metodi di prospezione geochimica, particolarmente utili nella ricerca di minerali metalliferi, si affiancano validamente ai metodi geofisici e sono ormai impiegati comunemente. Il crescente interesse che i metodi geochimici presentano nella ricerca di minerali utili è testimoniato dalla recentissima istituzione della International Association of Exploration Geochemists, che conta oltre 500 membri e il cui organo ufficiale è il ‟Journal of geochemical exploration" e che si affianca validamente alla International Association of Geochemistry and Cosmochemistry.
Da un altro punto di vista la conoscenza dei processi di circolazione degli elementi nelle rocce, nelle acque, nei suoli e nell'atmosfera considerati come serbatoi dinamici, dei relativi tempi di residenza e di turnover inserisce profondamente la geochimica nei problemi della gestione ambientale. Il faticoso lavoro svolto dai geochimici nel raccogliere dati analitici relativi alla distribuzione degli elementi nelle rocce, nei suoli e nelle acque ha così reso disponibile un ingente materiale a cui possono attingere oggi gli studiosi dell'ambiente. Lo studio del comportamento delle sostanze inquinanti è reso più facile dalla considerazione che gli elementi inquinanti, una volta immessi nell'ambiente, seguono il ciclo geochimico normale degli elementi e le leggi che lo regolano. È così possibile valutare quantitativamente l'impatto dell'attività umana sull'ambiente, prevederne gli effetti, intervenire con adatti provvedimenti alla regolazione degli effetti dell'inquinamento e, in casi favorevoli, alla rigenerazione ambientale.
Nel quadro della sorveglianza ambientale è infine da segnalare, per i grandi sviluppi che è destinata ad assumere in futuro, la registrazione di opportuni parametri geochimici in vista delle possibilità offerte per la previsione delle eruzioni vulcaniche (v. Tonani, 1971) e dei terremoti (v. Abelson, 1973; v. Hammond, 1973).
La conoscenza sistematica della distribuzione di particolari elementi nelle rocce, nei suoli, nelle acque e nella vegetazione, quale si può acquisire anche a scopo di prospezione mineraria, costituisce, in un altro settore, un materiale di base per la ricerca degli effetti di determinati tipi di rocce o di vegetazione o comunque della presenza di particolari elementi nei materiali naturali sulla diffusione di particolari malattie. Sono state iniziate in questo senso, nei paesi più progrediti, ricerche a vasto raggio ed è prevedibile che il programma ‛geochimica e salute pubblica' si avvierà nel prossimo decennio al massimo sviluppo.
La geochimica è un campo di ricerca i cui limiti, per il suo stesso carattere di interdisciplinarità, sono spesso sfumati e mal definiti. Abbiamo limitato le nostre considerazioni principalmente alla Terra solida, ma è chiaro che vi è stretta interconnessione fra la geochimica e le scienze dell'atmosfera e dell'idrosfera, tanto che si può parlare di una geochimica dell'atmosfera e di una geochimica dell'idrosfera, specialmente per quanto concerne i fenomeni e le azioni che si verificano all'interfaccia atmosfera-oceano, atmosfera-crosta, oceano-fondi oceanici e al ruolo degli organismi nel ciclo terrestre e marino degli elementi. Al concorso delle diverse e numerose discipline interessate spetterà la soluzione dei molti e grandi problemi ancora aperti per quanto riguarda l'origine e l'evoluzione degli oceani e dell'atmosfera terrestre.
Bibliografia.
Abelson, P. H., Observing and predicting earthquakes, in ‟Science", 1973, CLXXX, p. 819.
Ahrens, L., The use of ionization potentials. Part I. Ionic radii of the elements, in ‟Geochimica et cosmochimica acta", 1952, II, pp. 155-169.
Ahrens, L., The use of ionization potentials. Part II. Anion affinity and geochemistry, in ‟Geochimica et cosmochimica acta", 1953, III, pp. 1-29.
Anderson, O. L., Latest informations, from seismic observations, in the earth's mantle, London-New York 1967.
Barberi, F., Innocenti, F., Ferrara, G., Keller, G., Villari, L., Evolution of eolian arc volcanism (southern Thyrrenian sea), in ‟Earth and planetary science letters", 1974, XXI, pp. 269-276.
Barnes, H. L., Mineral zoning and ore transport, in XX Congreso Geológico Internacional-México. Resúmenes de los trabajos presentados, 1956, Ciudad de México 1956, p. 208.
Barnes, H. L., Ore solutions, in Carnegie Institution of Washington yearbook, vol. LVII, Washington 1958, pp. 234-240.
Barnes, H. L., Ore solutions, in Carnegie Institution of Washington yearbook, vol. LIX, Washington 1960, pp. 137-141.
Barnes, H. L. (a cura di), Geochemistry of hydrothermal ore deposits, New York 1967.
Barnes, H. L., Kullerud, G., Relations between composition of ore minerals and ore solutions, in ‟Economic geology", 1957, LII, pp. 825-830.
Barnes, H. L., Kullerud, G., Equilibria in sulfur containing aqueous solutions in the system Fe−S−O and their correlation during ore deposition, in ‟Economic geology", 1961, LVI, pp. 648-688.
Bernal, J. D., Discussion, in ‟Observatory", 1936, LIX, p. 268.
Birch, F., Density and composition of mantle and core, in ‟Journal of geophysical research", 1964, LXIX, pp. 4377-4388.
Bodu, R., Bouzigues, H., Morin, N., Pfiffelmann, J. P., Sur l'existence d'anomalies isotopiques rencontrées dans l'uranium du Gabon, in ‟Comptes rendus hebdomadaires des séances de l'Académie des Sciences (Paris)", 1972, CCLXXV, pp. 1731-1732.
Boltwood, B. B., On the ultimate disintegration products of the radioactive elements. Part II. The disintegration products of uranium, in ‟American journal of science", 1907, XXIII, pp. 77-88.
Bullen, K. E., Introduction to the theory of seismology, London 1963.
Burnham, C. W, Jahns, R. H., A method for determining the solubility of water in silicate melts, in ‟American journal of science", 1962, CCLX, pp. 721-745.
Burns, R. G., Mineralogical applications of crystal field theory, London 1970.
Cartledge, G. H., Studies in the periodic system. I. The ionic potential as a periodic function, in ‟Journal of the American Chemical Society", 1928, L, pp. 2855-2863.
Clark, S. P., Ringwood, A. E., Density, distribution and constitution of the mantle, in ‟Review of geophysics", 1964, II, pp. 35-88.
Clarke, F. W., The data of geochemistry, Washington 1908, 19245.
Clarke, F. W., Washington, H. S., The composition of the earth's crust, in U. S. geological survey, Professional paper n. 127, Washington 1924, pp. 1-117.
Clayton, R. N., Oxygen isotope geochemistry, in Studies in analytical geochemistry (a cura di D. M. Shaw), Toronto 1963, pp. 42-57.
Compston, W., Jeffery, P. M., Anomalous common strontium in granite, in ‟Nature", 1960, CLXXXIV, pp. 1792-1793.
Compston, W., Jeffery, P. M., Metamorphic chronology by the rubidium-strontium method, in ‟Annals of the New York Academy of Sciences", 1961, XCI, pp. 3493-3502.
Craig, H., The geochemistry of the stable carbon isotopes, in ‟Geochimica et cosmochimica acta", 1953, III, pp. 53-92.
Craig, H., Isotopic variations in meteoric waters, in ‟Science", 1961, CXXXIII, pp. 1702-1703.
Curtis, C. D., Applications of the crystal field theory to the inclusions of trace transition elements during magmatic differentiation, in ‟Geochimica et cosmochimica acta", 1964, XXVIII, pp. 389-403.
Damon, P. E., Behaviour of some elements during magmatic crystallisation, in ‟Geochimica et cosmochimica acta", 1968, XXXII, pp. 564-567.
Dietzel, A., The cation field strenghts and their relations to the devitrifying processes, to compound formation, and to the melting points of silicates, in ‟Zeitschrift für Elektrochemie", 1942, XLVIII, pp. 9-23.
Doe, B. R., Lead isotopes, Berlin 1970.
Edwards, A. B., The present state of knowledge and theories of ore genesis, in ‟Proceedings of the Australasian Institute of Mineralogy and Metallurgy", 1956, CLXXVII, pp. 69-116.
Epstein, S., Buchsbaum, R., Lowenstam, H. A., Urey, H. C., Carbonate-water isotopic temperature scale, in ‟Bulletin of the Geological Society of America", 1951, LXII, pp. 417-426.
Epstein, S., Buchsbaum, R., Lowenstam, H. A., Urey, H. C., Revised carbonate-water isotopic temperature scale, in ‟Bulletin of the Geological Society of America", 1953, LXIV, pp. 1315-1326.
Fairbridge, R. W. (a cura di), The encyclopedia of geochemistry and environmental sciences, New York 1972.
Faure, G., Hurley, P. M., The isotopic composition of strontium in oceanic and continental basalt. Application to the origin of igneous rocks, in ‟Journal of petrology", 1963, IV, pp. 31-50.
Faure, G., Powell, J. L., Strontium isotope geology, in Mineral, rocks and inorganic materials, vol. V, Berlin 1972.
Fersman, A. E., Geokhimija, in Enciklopedija Nausk SSSR, 4 voll., Moskva 1933-1939.
Fersman, A. E., Application of VEK's in geochemistry, in ‟Journal of the Academy of Sciences of the USSR", 1936, II, pp. 393-396.
Fleischer, M. (a cura di), Data of geochemistry, Washington 1963.
Fuchs, L. H., Djerfisherite, alkali copper-iron sulphide, a new mineral from the Kota-Kota and St. Mark's enstatite chondrites, in ‟Science", 1966, CLIII, pp. 166-167.
Fyfe, W. S., Isomorphism and bond type, in ‟American mineralogist", 1951, XXXVI, pp. 538-542.
Garrels, R. M., Christ, C. L., Solutions, minerals and equilibria, New York 1965.
Gast, P. W., Trace element fractionation and the origin of tholeiitic and alkaline magma types, in ‟Geochimica et cosmochimica acta", 1968, XXXII, pp. 1057-1086.
Goldschmidt, V. M., Geochemische Verteilungsgesetze der Elemente. VII. Die Gesetze der Kristallchemie, in ‟Skrifter utgitt av det Norske videnskaps-akademi i Oslo", 1926, n. 2, pp. 1-117.
Goldschmidt, V. M., Barth, T., Holmsen, D., Lunde, G., Thomassen, L., Ulrich, F., Zachariasen, W., Geochemische Verteilungsgesetze der Elemente, I-IX, in ‟Skrifter utgitt av det Norske videnskapsakademi i Oslo", 1923-1937.
Goldschmidt, V. M., Barth, T., Holmsen, D., Lunde, G., Thomassen, L., Ulrich, F., Zachariasen, W., The principles of distribution of geochemical elements in minerals and rocks, in ‟Journal of the Chemical Society", 1937, pp. 655-673.
Goldschmidt, V. M., Barth, T., Holmsen, D., Lunde, G., Thomassen, L., Ulrich, F., Zachariasen, W., Geochemistry, Oxford 1954.
Gooldchild, J. C., Some geological evidence regarding the age of the earth, in ‟Proceedings of the Royal Physical Society of Edinburgh", 1896, XIII, pp. 259-308.
Gümbel, C. W. von, Geognostische Beschreibung des ostbayerischen Grenzgebirges oder der bayerischen und oberpfälzer Waldgebirges, Gotha 1868.
Hamilton, E. I., Applied geochronology, New York 1965.
Hammond, A. L., Earthquake predictions: break though in theoretical insight, in ‟Science", 1973, CLXXX, pp. 851-853.
Harland, W. B., Smith, A. G., Wilcock, B., The phanerozoic timescale, London 1964.
Helgeson, H. C., Complexing and hydrothermal ore deposition, Oxford 1964.
Hoefs, J., Applied stable isotope geochemistry, Berlin 1973.
Horn, M. K., Adams, J. A. S., Computer-derived geochemical blances and element abundances, in ‟Geochimica et cosmochimica acta", 1966, XXX, pp. 279-297.
Huang, W. H., Walker, R. M., Fossil alpha particle recoil tracks: a new method of age determination, in ‟Science", 1967, CLV, pp. 1103-1108.
Jaggar, T. A., The mechanism of volcanoes, in ‟Volcanology. Bulletin of the United States Natural Research Council", 1936, LXXVII, pp. 44-71.
Jahns, R. H., Burnham, C. W., Experimental studies of pegmatite genesis. I. A model for the derivation and crystallization of granitic pegmatites, in ‟Economic geology", 1969, LXIV, pp. 843-864.
Kay, R. U., Gast, P. W., The rare earth content and origin of alkali rich basalts, in ‟Journal of geology", 1973, LXXXI, pp. 653-682.
Kern, M. R., Weisbrod, M. A., Thermodynamique de base pour minéralogistes, pétrographes et géologues, Paris 1964.
Krauskopf, K., Separation of manganese from iron in sedimentary processes, in ‟Geochimica et cosmochimica acta", 1957, XII, pp. 61-84.
Krauskopf, K., Introduction to geochemistry, New York 1967.
Krumbein, W. C., Garrels, R. M., Origin and classification of chemical sediments in terms of pH and oxidation-reduction potentials, in ‟Journal of geology", 1952, LX, pp. 1-33.
Kushiro, E., Melting of hydrous upper mantle and possible generation of andesitic magma: an approach from synthetic systems, in ‟Earth and planetary science letters", 1974, XXII, pp. 294-299.
Landé, A., Über die Grösse der Atome, in ‟Zeitschrift für Physik", 1920, I, pp. 191-197.
Larsen, G., Chilingar, G. V. (a cura di), Diagenesis in sediments, Amsterdam 1967.
Lewis, J., L'evoluzione chimica del sistema solare, in ‟Le scienze", 1974, LXX, pp. 35-44.
Libby, W. F., Radiocarbon dating, Chicago 1952.
Longinelli, A., Nuti, S., Composizione isotopica dell'ossigeno nei fosfati. Parte I. Tecnica di misura e scala di temperature isotopiche, in ‟Bollettino della Società Geologica Italiana", 1967, LXXXVI, pp. 115-132.
Longinelli, A., Nuti, S., Revised phosphate-water isotopic temperature scale, in ‟Earth and planetary science letters", 1973, XIX, pp. 373-376.
Mason, B., Principles of geochemistry, New York 1966.
Neuman, H., Mead, J., Vitaliano, C. J., Trace element variation during fractional crystallization as calculated from distribution law, in ‟Geochimica et cosmochimica acta", 1954, VI, pp. 90-100.
Nicolaysen, L. O., Graphic interpretation of discordant age measurements on metamorphic rocks, in ‟Annals of the New York Academy of Sciences", 1961, XCI, pp. 198-206.
Nockolds, S. R., The behaviour of some elements during fractional crystallization of magma, in ‟Geochimica et cosmochimica acta", 1966, XXX, pp. 267-278.
Pauling, L., The sizes of ions and the structure of ionic crystals, in ‟Journal of the American Chemical Society", 1927, XLIX, pp. 765-790.
Platen, H. von, Kristallisation granitischer Schmelzen, in ‟Beiträge zur Mineralogie und Petrographie", 1965, XI, pp. 334-381.
Poldervaart, A., Crust of the earth, New York 1955.
Povarennykh, A. S., Some fundamental problems of crystal chemistry in relation to mineralogy, in Aspects of theoretical mineralogy in the USSR (a cura di M. H. Battey e S. I. Tomkeieff), Oxford 1964.
Price, P. B., Walker, R. M., Fossil tracks of charged particles in mica and the age of minerals, in ‟Journal of geophysical research", 1963, LXVIII, pp. 4847-4862.
Rankama, K., Isotope geology, London 1954.
Rankama, K., Progress in isotope geology, New York 1963.
Rankama, K., Sahama, Th. G., Geochemistry, Chicago 1950.
Richardson, A., Sneebsy, G., The frequency distribution of igneous rocks. I. Frequency distribution of the major oxides in analyses of igneous rocks, in ‟Mineralogical magazine", 1922, XIX, pp. 303-313.
Ringwood, A. E., The principles governing trace element distribution during magmatic differentiation, in ‟Geochimica et cosmochimica acta", 1955, VII, pp. 189-202.
Ringwood, A. E., Phase transformations in the mantle, in ‟Earth and planetary science letters", 1969, V, pp. 401-412.
Ringwood, A. E., The petrological evolution of island arc systems, in ‟Journal of the Geological Society", 1974, CXXX, pp. 183-204.
Rittmann, A., I vulcani e la loro attività, Forlì 1967.
Ronov, A. B., Yaroshevsky, A. A., Chemical composition of the earth's crust, in The earth's crust and upper mantle (a cura di P. J. Hart), Washington 1969, pp. 37-57.
Russell, R. D., Farquar, R. M., Lead isotopes in geology, New York 1960.
Sarkar, S. N., Pre-cambrian stratigraphy and geochronology of peninsular India, Dhanbad 1968.
Saukow, A. A., Geochemie, Berlin 1953.
Schilling, J. G., Sea floor evolution: rare earth evidence, in ‟Philosophical transactions of the Royal Society", 1971, CCLXVIII, pp. 663-706.
Shaw, D. M., Comments on the geochemical implications of lead-isotope dating of galena deposits, in ‟Economic geology", 1957, LII, pp. 570-573.
Shaw, D. M., Interprétation géochimique des éléments en traces dans les roches cristallines, Paris 1964.
Shaw, D. M., Trace element fractionation during anatexis, in ‟Geochimica et cosmochimica acta", 1970, XXXIV, pp. 237-243.
Smales, A. A., Wager, L. R. (a cura di), Methods in geochemistry, New York 1960.
Smith, F. G., Physical geochemistry, Reading 1963.
Streckeisen, A. L., Classification and nomenclature of igneous rocks, Stuttgart 1967.
Strutt, R. J., On the radioactive minerals, in ‟Proceedings of the Royal Society" (Series A), 1905, LXXVI, pp. 88-101.
Strutt, R. J., On the accumulation of helium in geological time, in ‟Proceedings of the Royal Society" (Series A), 1908, LXXXI, pp. 272-277.
Strutt, R. J., The leakage of helium from radioactive minerals, in ‟Proceedings of the Royal Society" (Series A), 1909, LXXXII, pp. 166-169.
Strutt, R. J., On the accumulation of helium in geological time, in ‟Proceedings of the Royal Society" (Series A), 1910, LXXXIV, pp. 194-196.
Taylor, S. R., Abundances of chemical elements in the continental crust, a new table, in ‟Geochimica et cosmochimica acta", 1964, XXVIII, pp. 1273-1285.
Taylor, S. R., The application of trace element data to problems in petrology, in Physics and chemistry of the earth (a cura di L. Ahrens, F. Press, S. K. Runcorn, H. C. Urey), vol. VI, Oxford 1965.
Taylor, S. R., White, A. J. R., Trace element abundances in andesite, in ‟Bulletin volcanologique", 1966, XXIX, pp. 177-194.
Termier, H., Termier, G., L'évolution de la lithosphère. III. Glyptogenèse, Paris 1961.
Termier, H., Termier, G., Erosion and sedimentation, London 1963.
Thode, H. G., Sulphur isotope geochemistry, in Studies in analytical geochemistry (a cura di D. M. Shaw), Toronto 1963.
Tonani, F., Concepts and techniques for the geochemical forecasting of volcanic eruptions, in The surveillance and predictions of volcanic activity, UNESCO, Paris 1971, pp. 145-166.
Tuttle, O. F., Bowen, N. L., Origin of granite in the light of experimental studies in the system NaAlSi3O8−KalSi3O8−SiO2−H2O, in ‟Geological Society of America memoirs", 1958, LXXIV, pp. 1-153.
Urey, H. C., The thermodynamic properties of isotopic substances, in ‟Journal of the Chemical Society", 1947, pp. 562-582.
Verhoogen, J., Possible temperatures in the oceanic upper mantle and the formation of magma, in ‟Geological Society of America bulletin", 1973, LXXXIV, pp. 515-521.
Vernadsky, W., La géochimie, Paris 1924.
Vinogradov, A. P., Average content of chemical elements in the principal types of igneous rocks in the earth's crust, in ‟Geochemistry", 1962, pp. 641-664.
Volborth, A., Elemental analysis in geochemistry, Amsterdam 1969.
Wager, L. R., Mitchell, R. L., The distribution of trace elements during strong fractionating of basic magma. A further study of the Skaerogaard intrusion, East Greenland, in ‟Geochimica et cosmochimica acta", 1951, I, pp. 129-208.
Wainerdi, R. E., Uken, E. A. (a cura di), Modern methods of geochemical analysis, New York 1971.
Walther, J., Einleitung in die Geologie als historische Wissenschaft. Beobachtungen über die Bildung der Gesteine und ihrer organischen Einschlüsse, Jena 1893-1894.
Wasastjerna, J. A., On the radii of ions, in ‟Commentationes physico-mathematicae Societatis Scientiarum Fennicae", 1923, XXXVIII, pp. 1-25.
Wedepohl, K. H., Untersuchungen zur Geochemie des Bleis, in ‟Geochimica et cosmochimica acta", 1956, X, pp. 69-148.
Wedepohl, K. H. (a cura di), Handbook of geochemistry, voll. I, II-1, II-2, II-3, II-4, Berlin 1969-1974.
Wendland, W. M., Donley, D. L., Radiocarbon-calendar age relationship, in ‟Earth and planetary science letters", 1971, XI, pp. 135-139.
Wetherill, G. W., Discordant uranium-lead ages, in ‟Transactions. American Geophysical Union", 1956, XXXVII, pp. 320-326.
Williams, H. S., The elements of the geological time-scale, in ‟Journal of geology", 1893, I, pp. 283-295.
Williams, R. J. P., Deposition of trace elements in a basic magma, in ‟Nature", 1959, CLXXXIV, p. 44.
Wilson, J. T., Mantle plumes and plate motions, in ‟Tectonophysics", 1973, XIX, pp. 149-164.
Yermakov, N. P. e altri, Research on the nature of ore forming solutions, Oxford 1965.