glioblastoma
glioblastoma
La forma più frequente di neoplasia primitiva cerebrale; rappresenta anche la forma più maligna di neoplasia astrocitaria. L’incidenza in Europa e in America Settentrionale è di ca. 3÷4 casi per 100.000 abitanti per anno. Può manifestarsi in qualunque età, ma colpisce prevalentemente persone adulte con un picco d’incidenza tra i 45 e 75 anni. Insorge prevalentemente nella sostanza bianca sottocorticale degli emisferi cerebrali e la localizzazione fronto-temporale è fra le più frequenti. L’infiltrazione tumorale spesso si estende nella corteccia adiacente e, attraverso il corpo calloso, nell’emisfero contro-laterale. Rara è l’insorgenza a livello cerebellare o del midollo spinale. Nei bambini colpisce i gangli della base o il tronco cerebrale. Clinicamente si manifesta particolarmente nei pazienti anziani, con una storia clinica breve che non supera i tre mesi di durata (g. primario); nei pazienti più giovani rappresenta un’evoluzione di un astrocitoma di grado più basso (g. secondario).
Sintomatologia
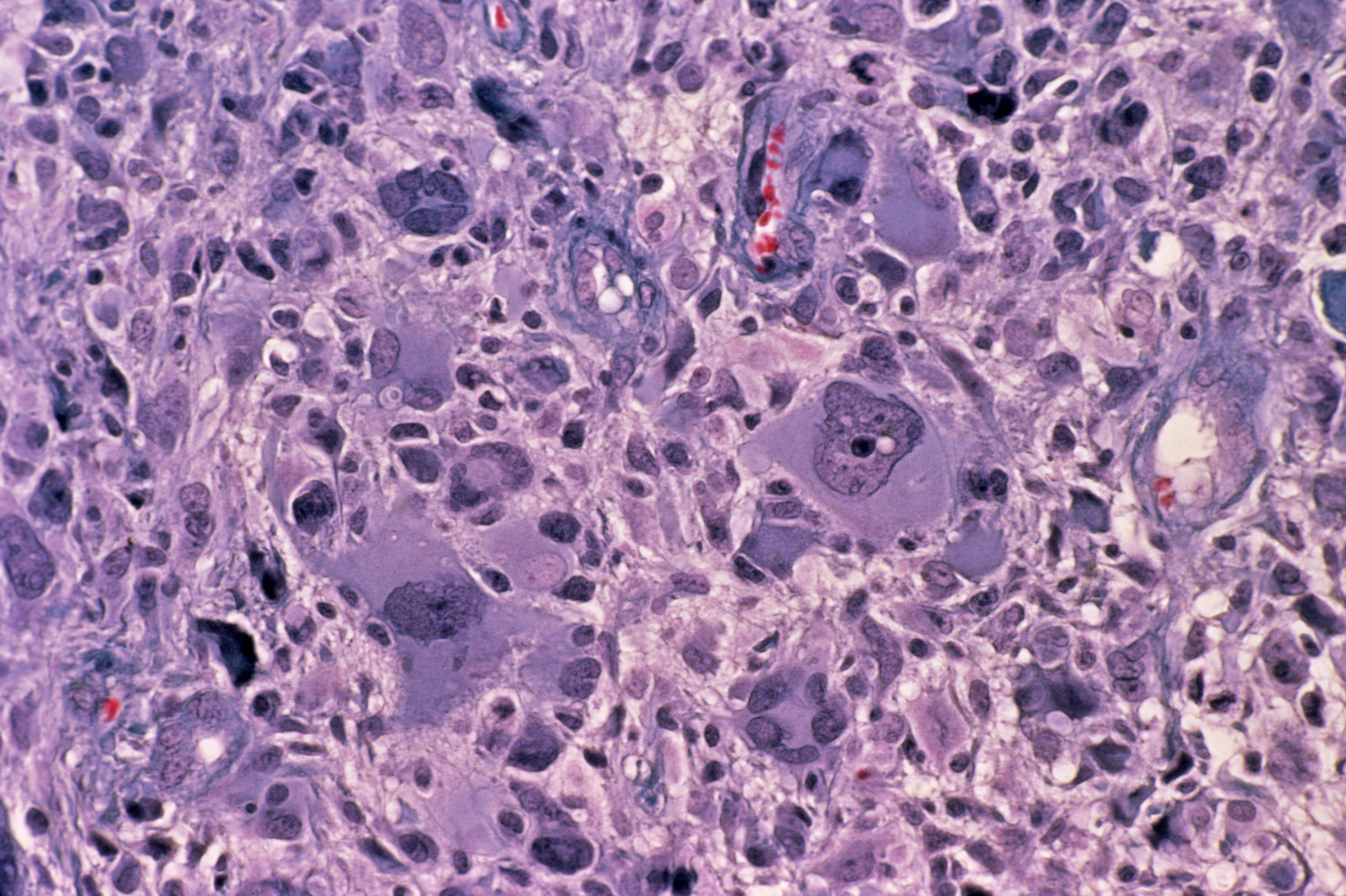
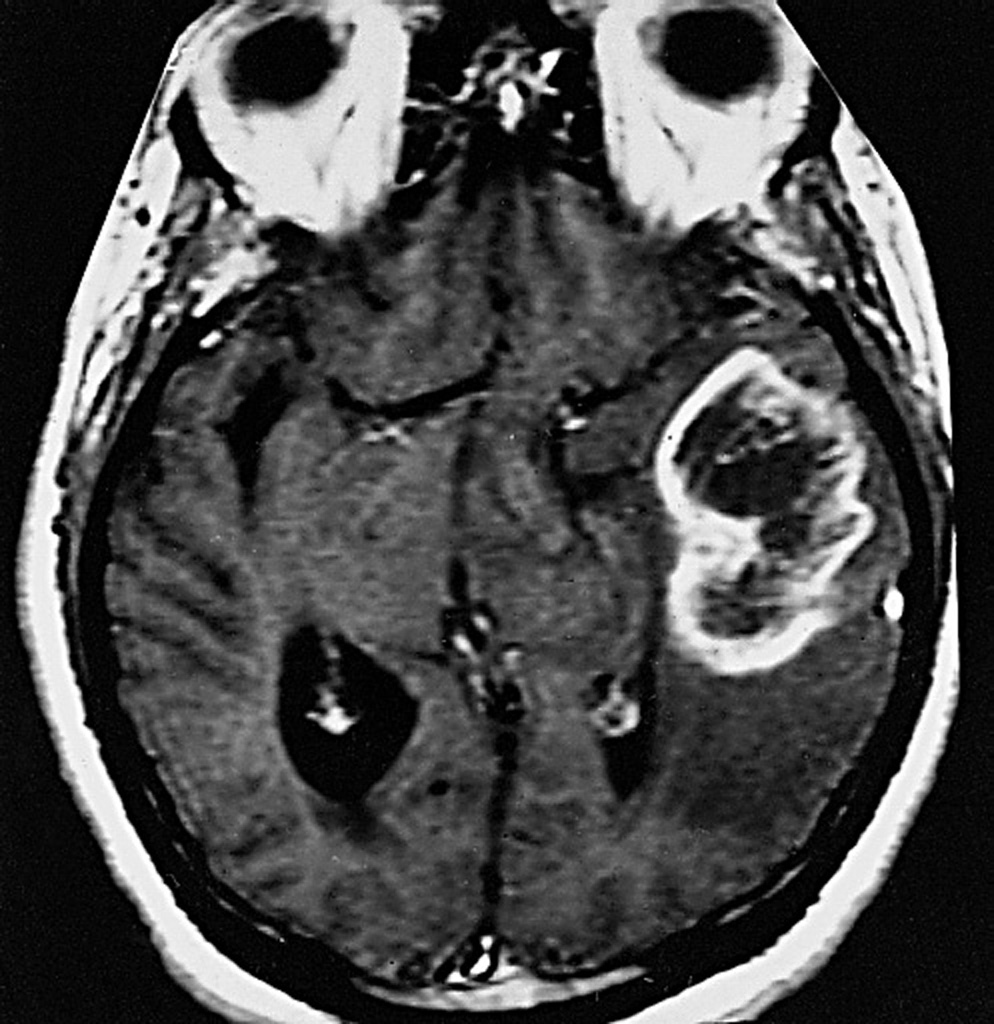
La sintomatologia clinica è legata, nella maggioranza dei casi, all’aumento della pressione intracranica (cefalea, nausea/vomito, edema papillare). In un terzo circa dei pazienti si manifesta con crisi epilettiche. Macroscopicamente il g. si presenta come una lesione relativamente circoscritta che contrasta con la diffusa infiltrazione del tessuto circostante, invariabilmente presente all’esame microscopico. Ha un aspetto variegato con un’area periferica grigiastra e aree giallastre di necrosi centrale a volte associate a zone rossastre di emorragia. Istologicamente è simile all’astrocitoma (➔) anaplastico con l’aggiunta della presenza di necrosi e proliferazione vascolare o endoteliale. Quest’ultima consiste in piccoli vasi neoformati con cellule endoteliali iperplastiche disposte in più strati. La presenza di questi vasi neoformati privi di barriera emato-encefalica è alla base della visualizzazione radiologica della neoplasia dopo iniezione di mezzo di contrasto. La neoproliferazione vascolare è determinata dalla produzione da parte degli astrociti neoplastici del fattore di crescita vascolare endoteliale (VEGF, Vascular Endothelial Growth Factor) in risposta all’ipossia. La necrosi nel g. assume un aspetto festonato perché le cellule neoplastiche si addensano lungo i bordi con un caratteristico aspetto a ‘pseudopalizzata’. Si riconoscono due varianti istologiche del g.: il g. a cellule giganti, costituito prevalentemente da cellule giganti bizzarre e multinucleate, e il gliosarcoma, in cui alla componente maligna gliale se ne associa una di tipo mesenchimale, sarcomatoso.
Eziologia
L’eziologia del g. è sconosciuta. Non esistono a tutt’oggi (2010) studi che indichino l’azione di fattori ambientali. Il g. origina molto probabilmente da un’instabilità genetica di cellule staminali neurali presenti nel cervello postnatale. Numerosi studi hanno dimostrato la presenza di cellule nei glioblastomi (o in linee cellulari di esso) con le proprietà delle cellule staminali. Tali cellule staminali tumorali rappresentano solo una piccola sottopopolazione, ma hanno la capacità di autorinnovarsi e di essere tumorigeniche una volta iniettate in animali. I due tipi di g., primario e secondario, indistinguibili da un punto di vista istologico, sono ben distinti clinicamente e molecolarmente. Il g. primario è quello più frequente e insorge prevalentemente oltre i 50 anni. Il profilo genico è caratterizzato da iperespressione di EGFR, mutazione di PTEN, delezione di p16 e perdita del cromosoma 10. Il g. secondario deriva da progressione maligna di astrocitomi diffusi o anaplastici, insorge in pazienti più giovani ed è caratterizzato da mutazione di p53 e LOH sul cromosoma 10q. Nel 2008 è stata identificata una mutazione del gene della isocitratodeidrogenasi 1 (IDH1) che sembra insorgere esclusivamente nei gliomi diffusi e nei glioblastomi da essi derivati (g. secondari).
Prognosi
Tra i fattori prognostici che influiscono sulla sopravvivenza del g., l’età è uno dei più importanti: i pazienti più giovani (età inferiore ai 50 anni) sopravvivano più a lungo di quelli più anziani. Altri fattori importanti ai fini della sopravvivenza sono la metilazione del geneMGMT, che predice la risposta al trattamento chemioterapico e la mutazione del gene IDH1. La prognosi dei pazienti con g. rimane tuttavia infausta, a causa dell’elevata resistenza alla terapia di questo tumore. Con gli attuali trattamenti di radio- e chemioterapia dopo la resezione chirurgica, la sopravvivenza media globale è di soli 8÷10 mesi e meno del 10% dei pazienti è ancora vivo dopo due anni.