
Immunologia
Immunologia
di Guido Forni e Jack Strominger
Sommario: 1. Il sistema immunitario: a) pressioni evolutive; b) resistenza alle invasioni dei microrganismi; c) controllo delle cellule dell'organismo. 2. Strategie operative: a) sinopsi dei meccanismi; b) significato biologico. 3. L'immunità aspecifica: a) barriere fisiche; b) difese di tipo umorale; c) la fase cellulare; d) lo spegnimento della reazione. 4. Il self: a) necessità di riconoscere il self; b) le glicoproteine HLA; c) segnalazione della propria individualità molecolare. 5. La risposta specifica: a) presentazione dell'antigene; b) attivazione dei linfociti T; c) costimolazione dei linfociti T; d) linfociti T helper; e) linfociti T helper 1 e T helper 2; f) linfociti T killer; g) attivazione dei linfociti B. 6. Strategie della risposta specifica: a) selezione clonale; b) memoria immunitaria; c) generazione delle diversità dei recettori; d) il TCR; e) il BCR. 7. Le immunoglobuline: a) caratteristiche generali; b) commutazione di classe; c) reticolo degli idiotipi. 8. Le cellule natural killer (NK). □ Bibliografia.
1. Il sistema immunitario
a) Pressioni evolutive
La particolare complessità acquisita da alcuni organismi col progredire dell'evoluzione ha permesso loro di adattarsi meglio alle sfide ambientali, in quanto ha reso possibile garantire, all'interno dell'organismo, un ambiente sempre più protetto e scarsamente influenzato dal mondo esterno in cui funzioni complesse possono aver luogo in maniera ottimale. Queste caratteristiche, però, rendono l'ambiente interno degli organismi complessi particolarmente propizio anche per organismi meno evoluti, quali Virus, Batteri, Protozoi e Miceti (microrganismi, in genere), che vi trovano nicchie favorevoli per il loro ciclo vitale e per la loro intensa riproduzione. La comparsa e diffusione di organismi omeotermi, che presentano cioè un ambiente interno a temperatura costante, ha ulteriormente radicalizzato questa situazione: la temperatura costante e l'attivo metabolismo energetico che a essa si accompagna offrono ai microrganismi le condizioni ottimali per una riproduzione pressoché illimitata. Ciò ha favorito la diffusione di quei microrganismi che sono riusciti a elaborare strategie per accedere all'interno degli organismi omeotermi e ad adattare le proprie esigenze di vita in modo da sfruttarne le condizioni particolarmente favorevoli.
b) Resistenza alle invasioni dei microrganismi
I vantaggi a cui i microrganismi vanno incontro penetrando negli organismi superiori sono così grandi da aver dato origine a una grandiosa e mortale guerra biologica tra gli invasori e l'organismo che cerca di difendersi, guerra che ha segnato parte del mondo microbico il quale ha dovuto individuare varie strategie di invasione. Spesso queste strategie costano care al microrganismo che, per realizzarle nel migliore dei modi, si può trovare limitato nella sua mobilità ambientale, o deve investire un numero non piccolo di geni. A loro volta, di fronte al problema vitale costituito dal continuo tentativo di invasione, gli organismi superiori sono stati costretti a mettere a punto strategie di difesa sempre più raffinate per respingere o, comunque, limitare l'attacco microbico.
c) Controllo delle cellule dell'organismo
L'elevata complessità degli organismi superiori non ha soltanto influenzato l'evoluzione di parte del mondo microbico, ma ha anche posto problemi di controllo interno, in quanto la loro difesa è spesso basata sul rapido rinnovamento delle popolazioni cellulari, così da garantire che numerose funzioni vengano esplicate da cellule giovani, in grado di operare efficacemente. La necessità di produrre continuamente numeri elevatissimi di cellule nuove comporta un certo rischio di errore, in particolare per quanto riguarda la copiatura del DNA: questi errori portano in alcuni casi alla morte della cellula, in altri, invece, alla formazione di cellule mutate, che non funzionano o che funzionano male. Le cellule mutate devono essere individuate ed eliminate, in modo da impedire che, proliferando, diano origine a vaste popolazioni di cellule non funzionanti. Molto più raramente, quando più mutazioni si sommano tra loro, possono nascere delle cellule che presentano un comportamento anomalo, non rispondono più ai segnali di controllo dell'organismo e, moltiplicandosi in maniera incontrollata, possono dare origine a un tumore. Di fronte a questi pericoli gli organismi complessi hanno messo a punto dei sistemi di controllo basati sul riconoscimento delle proteine codificate dal complesso maggiore di istocompatibilità (Major Histocompatibility Complex, MHC), chiamato nell'uomo HLA (Human Leukocyte Antigens; v. immunologia: Istocompatibilità).
Le invasioni dei microrganismi e le mutazioni delle cellule dei propri tessuti costituiscono, quindi, un continuo pericolo per la sopravvivenza degli organismi più complessi. Inoltre, la penetrazione di cellule estranee attraverso lesioni dei tessuti di rivestimento potrebbe indurre comportamenti cellulari del tutto diversi da quelli propri dell'organismo invaso; le cellule estranee, trovato un ambiente favorevole, potrebbero moltiplicarsi e dare origine a un individuo chimerico, costituito da cellule di due organismi differenti, l'invaso e l'invasore. È proprio la presenza del sistema immunitario che rende assai improbabili questi eventi, e anzi il controllo immunitario è così efficace nel riconoscere e uccidere le cellule estranee da aver permesso la messa a punto di particolari strategie di riproduzione, per cui l'ovulo viene fecondato all'interno dell'organismo da spermatozoi estranei, che vi penetrano ma che non possono sopravvivervi. Addirittura, nei Mammiferi, non solo gli spermatozoi di un individuo estraneo penetrano all'interno della femmina ma qui, fecondato l'uovo, danno origine a un nuovo organismo che per tutta la durata della gestazione rimane in intimo contatto con i tessuti della madre: il sistema immunitario impedisce la fusione tra i tessuti e le cellule della madre e quelli del feto, che rimangono così individui separati e immunologicamente ben distinti. Il sistema immunitario, in questo caso, funziona così bene che le fusioni dei due tessuti rappresentano un raro evento patologico.
2. Strategie operative
a) Sinopsi dei meccanismi
Il sistema immunitario è costituito da un insieme di molecole e di cellule, sia circolanti nel sangue e nella linfa, sia presenti nei vari tessuti e nelle secrezioni, che cooperano tra loro in maniera integrata per proteggere l'organismo dalle infezioni e per difenderne l'integrità. Nella parte non corpuscolata del sangue (plasma e siero) e nei tessuti sono presenti numerose molecole, o sistemi di molecole, che, in maniera aspecifica, esercitano potenti azioni anti-microbiche: queste molecole costituiscono la fase ‛umorale' dell'immunità aspecifica, così chiamata perché nell'antichità i liquidi dell'organismo venivano definiti ‛umori'. Esse non solo uccidono direttamente i microrganismi invasori, ma attivano le cellule immunitarie e le guidano nella zona dell'invasione. Le cellule immunitarie presenti in circolo nel sangue, i leucociti, prendono tutte origine da un precursore comune situato negli organi emopoietici, da cui si differenziano maturando. Le cellule dell'immunità aspecifica comprendono i monociti, che si differenziano in macrofagi nei vari tessuti ai quali sono destinati, e i granulociti, suddivisi, a seconda del contenuto dei loro granuli citoplasmatici, in neutrofili, eosinofili e basofili: queste cellule uccidono in modo rapido ed efficace i microrganismi, dopo averne riconosciuto alcune proprietà ‛generiche' e caratteristiche. Le cellule dell'immunità specifica sono i linfociti: a differenza di granulociti e monociti, ogni linfocito esprime sulla propria membrana un diverso tipo di recettore attraverso il quale interagisce specificamente con un unico tipo di molecola estranea, che viene denominata antigene (v. immunologia e immunopatologia: Immunologia generale). I linfociti si suddividono in due classi, i linfociti T e i linfociti B. I linfociti T riconoscono l'antigene solo quando questo è parzialmente racchiuso (presentato) in una tasca formata dalle glicoproteine HLA; dopo il riconoscimento, i linfociti T ‛uccisori' (killer) eliminano le cellule che presentano sulla membrana lo stesso antigene associato alle glicoproteine HLA. I linfociti di un'altra sottoclasse, i cosiddetti linfociti T ‛aiutanti' (T helper, TH), invece, aiutano i linfociti B che, riconosciuto l'antigene in forma naturale (cioè non associato alle proteine HLA) e ricevuti i segnali rilasciati dai linfociti T helper, iniziano a produrre gli anticorpi (immunoglobuline, Ig) che con grande specificità si legano all'antigene. Le immunoglobuline, che costituiscono la fase umorale dell'immunità specifica, in presenza del complemento determinano la morte dei microrganismi o delle cellule contro cui sono specificamente dirette; inoltre favoriscono la fagocitosi dei microrganismi a cui si sono legate e guidano, rendendola specifica, l'attività citolitica dei leucociti (Antibody Dependent Cellular Cytotoxicity, ADCC). Granulociti, monociti e linfociti cooperano tra loro utilizzando principalmente, come codice di comunicazione, le citochine, molecole che essi secernono in particolari momenti della risposta immunitaria: le citochine svolgono prevalentemente un'azione locale, sono caratterizzate da una vita media generalmente variabile dai 30 minuti alle due ore e agiscono su tutte le cellule che ne esprimono il recettore. Quest'ultimo solo per alcune citochine è sempre espresso dalle cellule bersaglio, mentre per tutte le altre viene espresso soltanto in alcune fasi del ciclo cellulare. Le citochine che agiscono come mediatori delle cellule immunitarie sono denominate interleuchine (abbreviate con la sigla IL- seguita dal numero caratteristico); alcune citochine, caratterizzate principalmente dalla capacità di attrarre i leucociti così da aumentarne la concentrazione (attrazione chemiotattica o chemiotassi), vengono denominate chemochine.
Oltre alle cellule dell'immunità aspecifica e specifica, svolgono un ruolo importante anche le cellule endoteliali della parete dei vasi sanguigni, in quanto regolano la localizzazione nei tessuti delle cellule immunitarie favorendone l'accumulo là dove è avvenuta un'invasione microbica.
Da questo riassunto schematico dei suoi modi di funzionare, si può notare che il sistema immunitario opera su due registri, quello della reattività aspecifica e quello della reattività specifica. L'immunità aspecifica è, da un punto di vista evolutivo, quella più antica, basata su meccanismi di reazione più primitivi, molto potenti, caratterizzati da un tempo di attivazione di pochi minuti o di qualche ora, che agiscono ad ampio spettro, cioè riconoscendo ciò che è estraneo a grandi categorie molecolari; nella maggior parte dei casi questi meccanismi sono in grado di eliminare rapidamente gli invasori senza lasciarne traccia e senza mantenere alcun ‛ricordo' dell'evento. Al contrario, l'immunità specifica, caratterizzata da vari meccanismi in grado di riconoscere solo lo specifico antigene che ha innescato la reazione e di reagirvi con grande precisione, è lenta, in quanto i meccanismi stessi richiedono alcuni giorni per iniziare a essere operativi: ciò spiega perché l'attivazione delle raffinate risposte specifiche abbia luogo in casi relativamente rari, e solo in seguito e in conseguenza dell'attivazione dei meccanismi dell'immunità aspecifica. L'attivazione dell'immunità specifica determina l'aumento del numero dei linfociti che reagiscono con l'antigene e che per anni, dopo che la reazione è terminata, conserveranno una memoria immunitaria di questa esperienza antigenica e saranno pronti, in seguito a una nuova penetrazione dello stesso antigene, a dare origine a una risposta secondaria più intensa ed efficace.
b) Significato biologico
Quando si cerchi di comprendere il significato biologico dei meccanismi delineati, è necessario considerare che il sistema immunitario è il risultato di una guerra continua e mortale che ha avuto luogo durante l'evoluzione e si ripete ancor oggi più volte al giorno; essa vede coinvolti da un lato l'organismo che tende a mantenere inviolata la propria individualità, dall'altro gli invasori, costituiti da mutazioni di cellule all'interno dell'organismo stesso e da acidi nucleici e da cellule estranei. Di fronte a strategie d'invasione sempre più micidiali e ai sempre più frequenti pericoli di mutazioni - la cui probabilità aumenta mano a mano che l'organismo diventa più complesso - e di contaminazione da parte di agenti estranei, l'organismo ha risposto con la messa a punto di strategie di difesa progressivamente più complesse e raffinate. Spinto dalla drammatica necessità di limitare, in qualsiasi modo e a qualsiasi prezzo, la violenza dell'attacco di elementi estranei, il sistema immunitario si è evoluto senza badare né al numero di geni, né al numero di cellule progressivamente implicati, e senza essere condizionato da problemi di spreco o ridondanza: nel sistema immunitario, anzi, la ridondanza costituisce un'importante strategia per essere efficiente.
3. L'immunità aspecifica
a) Barriere fisiche
Il corpo umano è ricoperto dalla cute che, a livello degli orifizi, si trasforma in mucosa, un tipo di rivestimento più delicato che tappezza gli organi interni in diretta connessione con l'esterno. Oltre a costituire una barriera resistente ed elastica, difficile da penetrare, entrambi questi tipi di rivestimento sono associati a una serie di efficienti meccanismi antimicrobici. Le cellule dello strato corneo della cute, desquamandosi continuamente, allontanano un elevato numero di microrganismi (oltre 107 al giorno), la cui moltiplicazione inoltre è impedita o rallentata dal pH acido (3,5-5,9) della cute, mentre gli acidi grassi insaturi a lunga catena secreti dalle ghiandole sebacee esercitano una potente azione antimicrobica; i batteri saprofiti che si sono adattati a vivere sulla cute, a loro volta, impediscono, tramite un'intensa azione antibiotica, la crescita degli altri microrganismi che tentino di insediarvisi. Le mucose sono protette principalmente dal muco - una sostanza vischiosa che ne ricopre la superficie, secreta dalle ghiandole mucipare - il quale, per il suo contenuto in immunoglobuline, complemento, lisozima e altre sostanze ad azione antibatterica, costituisce una barriera letale per i microrganismi; questi, una volta intrappolati nel muco, difficilmente riescono a liberarsi, e finiscono per venire uccisi ed eliminati col muco stesso.
La resistenza meccanica della cute e delle mucose e l'efficacia dei sistemi difensivi che vi sono associati rendono difficile la penetrazione dei microrganismi all'interno del corpo. Alcuni di essi, però, hanno messo a punto particolari meccanismi per superare queste difese: in genere la penetrazione è resa possibile dalla loro aggressione in numero elevato (elevata carica microbica) o dalla loro capacità di sfruttare situazioni che determinano una diminuzione dei meccanismi di difesa (temperature eccessivamente fredde o eccessivamente calde, climi molto umidi o molto secchi, alterazione della normale flora microbica) o soluzioni di continuo dei rivestimenti (ferite, ustioni, microtraumi delle mucose).
Superate le barriere, il microrganismo si deve confrontare con le difese immunitarie, prima con quelle aspecifiche, poi, nel caso riesca a sopravvivere, con quelle della reattività immunitaria specifica.
b) Difese di tipo umorale
Nei liquidi organici si trovano molecole dotate di azione antibatterica che vengono immediatamente attivate dalla interazione con particolari strutture molecolari o dalla scissione di particolari legami chimici, tipici del mondo microbico e assenti nelle cellule dell'individuo, la cui presenza costituisce il segnale di allarme caratteristico di una invasione microbica.
Un tipico esempio di quest'azione immediata è costituito dal lisozima, enzima di cui sono ricchi vari liquidi organici, che, scindendo il legame 1-4 tra acido N-acetilmurammico e N-acetilglucosammina di alcuni polisaccaridi batterici, è in grado di causare la morte di tutti i batteri la cui parete cellulare sia costituita da quei polisaccaridi.
Il complemento è invece un sistema di molecole che si attivano sequenzialmente ‛a cascata': un componente attivato trasforma da inattive ad attive più molecole di un secondo componente, le quali a loro volta attivano più molecole di un terzo componente, e così via (v. immunologia e immunopatologia: Immunologia generale). In seguito all'interazione con particolari sostanze estranee (spesso componenti delle superfici batteriche), i primi componenti della via di attivazione detta ‛alternativa', anticorpo-indipendente, si uniscono per formare un enzima attivo (la C3-convertasi): dopo essersi depositata sulla superficie del batterio, la C3-convertasi attiva numerose molecole del componente successivo (il C3); la reazione prosegue con il coinvolgimento, sulla superficie del batterio, di un numero sempre maggiore di altri componenti (C5, C6, C7, C8, C9). Risultato finale di questa serie di eventi è la comparsa, sulla parete cellulare, di lesioni irreparabili, dalle quali fuoriescono componenti essenziali, con conseguente morte (lisi) del batterio. La rapida attivazione della via alternativa e l'efficacia della sua azione antibatterica fanno del complemento un importante componente della reattività immunitaria non specifica. Il suo ruolo è, però, più articolato, poiché oltre che direttamente da alcuni componenti batterici, il complemento viene attivato anche dalle immunoglobuline che abbiano legato l'antigene, così che, complementando l'azione di alcune classi di immunoglobuline, agisce come un elemento tipico della reattività specifica. In questo secondo tipo di attivazione, detto ‛della via classica', anticorpo-dipendente, le immunoglobuline unitesi all'antigene (una cellula, un batterio, una struttura molecolare) attivano il complemento con la loro parte costante (il frammento Fc) legando il componente C1; questo a sua volta attiva i componenti C4 e C2 che si uniscono a formare la C3-convertasi, dopodiché la via classica procede allo stesso modo di quella alternativa. Occorre comunque tenere presente che l'attivazione del complemento secondo la via classica ha luogo solo quando siano già stati prodotti gli anticorpi verso quel particolare antigene, il che richiede almeno quattro o cinque giorni dal momento dell'infezione.
Sia nelle tappe della via alternativa, sia in quelle della via classica i vari componenti del sistema, passando dalla forma inattiva a quella attiva, vengono scissi in due frammenti: il frammento di maggiori dimensioni si deposita sulla parete cellulare batterica o sulla membrana cellulare dove ha avuto luogo l'attivazione e, insieme ai componenti successivi, determina la formazione delle lesioni che causano la morte del microrganismo o della cellula estranea. Questo depositarsi dei fattori attivati del complemento, inoltre, rende le cellule e i microrganismi molto più facilmente fagocitabili (opsonizzazione) da parte dei macrofagi e dei granulociti.
I frammenti dei fattori complementari che non vengono direttamente coinvolti nella reazione si diffondono nei tessuti, dove esercitano azioni molto importanti per la progressione del processo infiammatorio: C3a, C5a, C4a attirano chemiotatticamente sia i granulociti neutrofili, sia i macrofagi e i linfociti, e, interagendo con le mastcellule e i basofili, inducono il rilascio di istamina, sostanza che provoca un notevole aumento delle dimensioni dei vasi sanguigni (vasodilatazione). Questi componenti del complemento, inoltre, agiscono direttamente sulle cellule endoteliali determinando ancora vasodilatazione, cioè un ulteriore aumento della portata del vaso e della sua permeabilità: in tal modo, nella zona dov'è avvenuta l'invasione affluisce una maggiore quantità di sangue e dai vasi fuoriescono sia siero, che accumulandosi nel tessuto dà luogo alla formazione dell'edema, sia cellule della reattività aspecifica.
Nei liquidi dell'organismo esistono numerosi altri meccanismi che, attivati rapidamente, uccidono i microrganismi o ne inibiscono la riproduzione: le proteine cationiche β-lisine, rilasciate dai granulociti e forse dalle piastrine; le bacteriocidine, che in presenza di complemento si legano alle cariche negative presenti sulla parete della cellula batterica; la tufsina, un tetrapeptide che deriva dalla digestione enzimatica delle catene pesanti delle immunoglobuline e che, depositandosi sulla parete dei microrganismi, ne favorisce la fagocitosi da parte dei macrofagi; il sistema lattoperossidasi-tiocianato-H2O2, che determina la morte di una grande varietà di microrganismi; gli interferoni IFN-α e IFN-β, che inibiscono la replicazione e l'azione patogena virale; le proteine della fase acuta, che attivano il complemento, favoriscono la fagocitosi dei batteri su cui si sono deposte e impediscono che questi utilizzino i metalli pesanti essenziali al loro metabolismo; il sistema della coagulazione; il sistema della fibrinolisi; il sistema delle chinine. Alcuni di questi sistemi sono simili a quello del complemento e vengono attivati con meccanismi sequenziali ‛a cascata'. Spesso i microrganismi penetrano a causa di lesioni traumatiche della cute che determinano l'immediata attivazione del sistema della coagulazione del sangue; questa può essere innescata, oltre che dalle lesioni delle pareti dei vasi, anche dalla sola presenza di strutture microbiche con cariche negative. Una volta che il danno che ha portato alla coagulazione sia stato riparato, il coagulo viene rimosso tramite l'attivazione di una serie di enzimi che, attaccando la fibrina, ne causano la distruzione. Numerosi enzimi ad attività proteolitica possono scindere le molecole precursori e determinare, al pari della semplice presenza di strutture ricche di cariche elettriche negative, l'attivazione di due chinine (bradichinina e lisilbradichinina) che provoca un'intensa vasodilatazione, l'aumento della permeabilità vasale e la sensazione di dolore.
L'imponente reazione attivata in pochi minuti da questi sistemi umorali determina la morte degli invasori: solo nel caso di microrganismi particolarmente aggressivi, o che abbiano elaborato particolari sistemi per resistere, diverrà necessario l'intervento delle cellule dell'immunità aspecifica.
c) La fase cellulare
L'attivazione dei sistemi umorali determina il rilascio di molecole che da una parte segnalano l'invasione alle cellule endoteliali dei vasi esistenti nella zona, dall'altra agiscono come richiamo chemiotattico per i leucociti.
Le cellule endoteliali rispondono rapidamente a questi segnali provocando una vasodilatazione, cui fa seguito un aumento del volume e una diminuzione della velocità del flusso ematico (v. tono vasale). Ai segnali infiammatori le cellule endoteliali rispondono anche producendo esse stesse sostanze ad azione vasodilatatrice, le prostacicline, ed esprimendo sulla membrana una serie di molecole d'adesione a cui si legano particolari recettori dei leucociti in circolo nel sangue: inizialmente la selectina P, la selectina E e le molecole ELAM, successivamente le molecole ICAM-1 e VCAM-1. Interagendo con queste molecole, i leucociti rallentano la loro corsa e iniziano a rotolare, sempre più lentamente, sulle pareti del vaso, fino a che, in seguito all'interazione con altre molecole d'adesione, si fermano, dapprima ancora ondeggianti sotto la spinta del flusso ematico, poi adesi fortemente. Le cellule endoteliali, esprimendo le varie molecole d'adesione, favoriscono anche l'inizio della coagulazione del sangue (atteggiamento protrombotico) nei vasi della zona infiammata, limitando così il pericolo che i microrganismi si possano diffondere nel circolo sanguigno dando luogo a una condizione di sepsi. Se questa si realizza, i macrofagi e le altre cellule infiammatorie rispondono rilasciando quantità elevate di vari fattori proinfiammatori, di Tumor Necrosis Factor (TNF-α) e di interferone γ (IFN-γ), attivi sulle cellule endoteliali, che provocano coagulazione intravasale diffusa, fuoriuscita di grande quantità di plasma dai vasi e diffusione di microtrombi in grado di ledere gravemente la funzione del rene, del fegato e del polmone, dando così luogo al grave quadro anatomo-clinico dello shock settico, spesso mortale.
I leucociti adesi alla parete vasale attraversano l'endotelio e, attratti chemiotatticamente dai segnali liberati dai sistemi di difesa umorale, dai segnali prodotti dagli stessi microrganismi invasori e dalle citochine secrete da altri leucociti, migrano verso il sito della reazione. I granulociti neutrofili ed eosinofili sono i primi ad arrivarvi, sia perché le cellule endoteliali esprimono dapprima molecole d'adesione che selettivamente interagiscono con i neutrofili, sia perché sono dotati di movimenti attivi, sia perché proprio grazie alle caratteristiche del loro nucleo plurilobato (diapedesi) riescono a sgusciare attraverso piccoli scollamenti che inizialmente si formano tra le cellule endoteliali. Arrivati a contatto con i microrganismi, i neutrofili fagocitano efficacemente quelli opsonizzati dai fattori del complemento e dalle proteine della fase acuta. I microrganismi fagocitati vengono uccisi nei vacuoli di fagocitosi che si sono fusi con i lisosomi: dapprima, tramite la produzione di radicali reattivi dell'ossigeno, ne vengono denaturate le proteine, poi un'ampia serie di enzimi proteolitici digerisce le proteine denaturate (v. immunologia clinica e immunopatologia).
I granulociti possono anche degranularsi, cioè rilasciare all'esterno il contenuto dei loro granuli (ricchi di enzimi proteolitici e di numerose sostanze ad attività antimicrobica) e uccidere in tal modo sia microrganismi che cellule eucariote senza doverli fagocitare; possono inoltre secernere un gran numero di citochine (IL-1, IL-4, IL-8, IFN-α, TNF-α, fattori che stimolano la proliferazione delle cellule emopoietiche midollari) e di altre molecole biologicamente attive (attivatore del plasminogeno, metaboliti dell'acido arachidonico), con le quali regolano la chemiotassi e l'attivazione di altri leucociti.
Alcuni tipi di invasori, come i parassiti, richiamano invece selettivamente i granulociti eosinofili, che, possedendo un diverso corredo enzimatico, esercitano nei loro confronti azioni più efficaci, utilizzando, comunque, meccanismi reattivi simili a quelli dei neutrofili.
I basofili e le mastcellule agiscono come sentinelle tissutali, disposte nei luoghi più comunemente esposti all'infezione microbica. A seconda del tessuto nel quale sono localizzate, queste cellule secernono una serie differente di mediatori che hanno un ruolo importante nell'induzione dell'infiammazione (istamina, serotonina, fattori chemiotattici per i granulociti, eparina, prostaglandine, leucotrieni, condroitinsolfati).
I macrofagi maturano dai precursori midollari per passare nel sangue come monociti e poi differenziarsi nei tessuti acquisendo morfologie e funzioni assai diverse (macrofagi alveolari, pleurici, peritoneali, macrofagi fissi e liberi nella milza, istiociti nel connettivo, cellule di Kupffer nel fegato, osteoclasti, cellule della microglia, cellule di Langerhans nella cute). Nel processo infiammatorio, i macrofagi, che arrivano nella zona dell'infiammazione uno o più giorni dopo i neutrofili, si comportano come questi ultimi, anche se sono in grado di rispondere a un più ampio repertorio di stimoli chemiotattici e attivanti. Una volta ‛messi in azione' dall'associazione tra segnali che fanno seguito all'attivazione dei sistemi umorali di difesa (metaboliti rilasciati dai Batteri e varie citochine), i macrofagi presentano una forte aderenza ai substrati e una grande mobilità della membrana plasmatica, consumano grandi quantità di glucosio e di O2, producono elevate quantità di radicali reattivi dell'ossigeno e secernono grandi quantità di metaboliti dell'acido arachidonico e di altre sostanze. Il macrofago attivato è una cellula straordinariamente attiva che fagocita efficientemente i microrganismi, uccide cellule alterate e secerne una grande varietà di molecole che esercitano importanti funzioni all'interno del sistema immunitario.
I macrofagi sono anche le cellule che permettono il diretto collegamento tra l'immunità aspecifica e la risposta specifica; esse costituiscono un esempio di sistema di difesa primitivo, che ha sfruttato le caratteristiche specifiche del suo modo di funzionare - la fagocitosi e l'uccisione di microrganismi e cellule estranee - per presentare in maniera ‛professionale' l'antigene ai linfociti T: nel loro citoplasma, infatti, le cellule batteriche e le varie molecole fagocitate vengono digerite in piccoli frammenti di 8-15 amminoacidi, i quali si legano alla tasca delle glicoproteine HLA che verranno espresse sulla membrana del macrofago, dove i frammenti dell'antigene potranno venire riconosciuti dai linfociti T.
L'invasione microbica, dapprima percepita dai meccanismi dell'immunità umorale aspecifica, innesca una reazione cellulare che distrugge l'antigene o almeno lo circoscrive limitandone la diffusione. In questa reazione le cellule endoteliali e i leucociti rilasciano una serie di segnali che coordinano la reazione stessa e che pongono l'organismo in uno stato di allarme. Tra le citochine che vengono rilasciate sono particolarmente importanti le citochine IL-1, TNF-α, IFN-γ, IL-6 e le chemochine (IL-8, MCP-1 e altre), che contribuiscono sia all'iperespressione di molecole d'adesione da parte delle cellule endoteliali, sia alla migrazione per diapedesi e attrazione chemiotattica dei leucociti e alla loro attivazione funzionale. Inoltre l'IL-1, l'IFN-γ, il TNF-α e l'IL-6, agendo come pirogeni interni, inducono la febbre legandosi ai recettori espressi dalle cellule dei nuclei dell'ipotalamo anteriore: l'aumento della temperatura corporea è un meccanismo di difesa basato sul fatto che molti microrganismi vivono stentatamente a temperature più elevate (38-42 gradi), alle quali invece le cellule del sistema immunitario lavorano più efficacemente. Inoltre l'IL-6, principalmente, e altre citochine inducono la liberazione in circolo da parte del fegato delle proteine della fase acuta, tra le quali la proteina C reattiva e la proteina che lega il mannosio, interagendo con alcuni carboidrati della parete cellulare batterica, opsonizzano i Batteri e attivano la cascata del complemento.
Tra le citochine prodotte durante l'infiammazione, particolari associazioni tra chemochine sopprimono la replicazione del virus dell'AIDS, mentre gli IFN svolgono un'intensa azione antivirale, regolano positivamente l'espressione degli antigeni di membrana da parte delle cellule della reattività aspecifica e costituiscono segnali molto importanti per l'attivazione dei macrofagi, delle cellule natural killer e dei linfociti T.
d) Lo spegnimento della reazione
I macrofagi esercitano, oltre a una potente azione di difesa, anche un'importante funzione di pulizia, rimuovendo i frammenti di microrganismi e di cellule morte; la fibrinolisi, d'altra parte, determina la progressiva rimozione dei coaguli sanguigni, così che il tessuto ove ha avuto luogo l'infiammazione ritorna alle condizioni normali. Quando il danno delle cellule dell'organismo è stato particolarmente rilevante e il tessuto non può venir completamente ripristinato, ha luogo una proliferazione di fibroblasti e di cellule epiteliali che dà origine a una cicatrice.
Nel caso in cui la reazione aspecifica e la susseguente reazione specifica non riescano a eliminare i microrganismi o la sostanza estranea, si manifestano un'intensa proliferazione dei fibroblasti e la trasformazione dei macrofagi in cellule epitelioidi e in cellule giganti multinucleate: queste cellule, unitamente ad alcuni linfociti T, formano una specie di vallo cellulare che isola la zona infettata dal resto dell'organismo, dando origine al granuloma.
4. Il self
a) Necessità di riconoscere il self
Il passaggio dalla reattività immunitaria aspecifica a quella specifica ha comportato la messa a punto di strategie operative nuove, caratterizzate da un raffinato riconoscimento delle molecole antigeniche. Ciò ha posto un problema fondamentale per l'organismo: discriminare con precisione le proprie molecole e le proprie cellule da quelle alterate o estranee, cioè riconoscere il self (‛se stesso') che deve essere protetto, e il non-self (‛non se stesso') che deve essere ucciso e distrutto. Il non-self può essere una sostanza estranea, un microrganismo, una cellula di un'altra specie animale, una cellula di un altro individuo della stessa specie, il figlio rispetto a entrambi i genitori o i genitori rispetto al figlio, una cellula propria che è andata incontro a mutazioni o che è alterata da un'infezione virale.
b) Le glicoproteine HLA
Il problema di come segnalare la propria individualità cellulare e molecolare è stato risolto utilizzando le caratteristiche poligeniche e polimorfiche dei geni che codificano le glicoproteine di classe I e II del sistema maggiore di istocompatibilità, espresse sulla membrana cellulare (v. immunologia: Istocompatibilità, vol. XI). Nell'uomo, la regione HLA è stata identificata nel braccio corto del cromosoma 6; essa comprende almeno 6 geni (A, B, C, E, F, G), che codificano diverse glicoproteine HLA di classe I, e almeno 5 geni (DP, DN-DO, DM, DQ, DR) che codificano diverse glicoproteine HLA di classe II espresse simultaneamente dalle stesse cellule. Inoltre, poiché questi geni sono co-dominanti, le cellule di un individuo esprimeranno tutte le glicoproteine codificate dai geni HLA ereditati dalla madre e tutte quelle codificate dai geni ereditati dal padre. Per caratterizzare il self vengono quindi utilizzati molti geni HLA (carattere poligenico del sistema). Ciò però non basterebbe se, per ognuno di questi geni, non esistessero numerosi alleli, tra loro alquanto diversi: i numerosi geni che codificano le glicoproteine HLA sono cioè polimorfici, in quanto gli individui di una popolazione posseggono tipi differenti dello stesso gene. Il carattere poligenico e il polimorfismo genico del sistema HLA fanno sì che le possibili combinazioni siano molto numerose, così che la probabilità che due individui posseggano gli stessi geni HLA è molto bassa. Oltre a quelli del sistema HLA, esistono altri geni polimorfici sparsi nel genoma umano che codificano differenti molecole proteiche polimorfe, gli antigeni minori di istocompatibilità.
Quando i linfociti T e B individuano sulla membrana di una cellula glicoproteine HLA estranee o alterate, si innesca una violenta reazione che porta alla rapida e precisa uccisione della cellula diversa. Nel caso dei trapianti di organo questa reazione determina il rigetto dell'organo estraneo (v. chirurgia dei trapianti).
c) Segnalazione della propria individualità molecolare
Le glicoproteine HLA di classe I e II di ogni individuo esercitano un ruolo ancora più complesso, in quanto segnalano anche i polimorfismi molecolari dell'individuo stesso ai linfociti T, guidandone l'attività. Quando vengono espresse sulla membrana cellulare, esse formano delle strutture globulari piuttosto simili tra loro, che nella parte volta all'esterno presentano una fenditura, una tasca, formata da un ‛pavimento' a nastri β e da due ‛pareti' ad α-elica. Nonostante questa generale somiglianza strutturale, le varie glicoproteine HLA codificate dai diversi geni presentano, nella zona più esterna dove è localizzata la tasca, sequenze di amminoacidi differenti: proprio tali sequenze sono riconosciute dai linfociti T e B, che in tal modo riescono a riconoscere le cellule parte di ‛se stesso' o diverse dal ‛se stesso'. Alla diversità delle sequenze amminoacidiche è anche dovuta la lieve differenza tra ‛pavimenti' e ‛pareti' delle varie tasche, che costituisce una caratteristica di grande importanza. Le glicoproteine HLA di classe I e II, dopo essere state sintetizzate nell'ergastoplasma, vengono espresse sulla membrana cellulare. Durante il loro assemblaggio e la migrazione verso la membrana, nella loro tasca finiscono, grazie a meccanismi particolari, frammenti peptidici di proteine prodotte dalla cellula stessa o fagocitate dall'ambiente esterno, come accennato a proposito dei macrofagi: generalmente, i peptidi contenenti tra i 5 e i 9 residui amminoacidici, che derivano dalle proteine prodotte dalla cellula stessa, vanno a legarsi alla tasca delle glicoproteine HLA di classe I, mentre quelli contenenti 12-20 residui amminoacidici, provenienti dalle proteine fagocitate, vanno a legarsi alle glicoproteine di classe II. Sulla membrana cellulare, le glicoproteine HLA la cui tasca è occupata da peptidi di varia natura segnalano in tal modo all'esterno non solo il proprio polimorfismo, ma anche quali proteine la cellula stia sintetizzando o abbia fagocitato. Poiché esiste un gran numero di glicoproteine diverse appartenenti sia alla classe I che alla classe II, sono molto numerose anche le tasche diverse alle quali, in base alle caratteristiche del loro ‛pavimento' e delle loro ‛pareti', i vari peptidi possono legarsi. Potenzialmente ogni tasca è in grado di alloggiare un migliaio di peptidi diversi, favorendo quello che si adatta meglio.
5. La risposta specifica
a) Presentazione dell'antigene
L'arrivo di un microrganismo viene rapidamente percepito dai meccanismi della reattività aspecifica, la cui attivazione a sua volta favorisce l'innesco della reattività specifica dei linfociti T e B. Infatti il microrganismo, variamente aggredito dai meccanismi della reattività aspecifica, viene fagocitato più efficacemente dai macrofagi o da altre cellule che caratteristicamente presentano l'antigene (genericamente denominate Antigen Presenting Cells, APC), viene digerito nel vacuolo di endocitosi e porzioni di 5-20 amminoacidi vengono presentate sulla membrana associate alle glicoproteine HLA di classe II, di cui le APC sono particolarmente ricche. Le APC fagocitano e presentano anche le proteine estranee, i frammenti delle cellule alterate e altri antigeni che non vengono riconosciuti dall'immunità aspecifica.
Il vacuolo di endocitosi, contenente il microrganismo o altro materiale antigenico, si fonde con un vacuolo post-Golgi che sta veicolando verso la membrana cellulare glicoproteine HLA di classe II, alle cui tasche, in seguito a questa fusione, si legano alcuni frammenti peptidici dell'antigene digerito. Le glicoproteine HLA procedono nel loro viaggio e vanno a inserirsi sulla membrana, dove ‛presentano' verso l'esterno la tasca contenente il peptide.
Se invece il microrganismo penetra in una cellula e vi sopravvive, i peptidi derivanti dalle sue proteine verranno presentati sulla membrana cellulare in associazione alle glicoproteine HLA di classe I. In effetti, le proteine virali e cellulari che vengono prodotte in eccesso o in maniera anomala vengono scisse da un complesso insieme di enzimi proteolitici, il proteasoma, in numerosi peptidi che sono poi attivamente trasportati da particolari molecole trasportatrici (TAP1 e TAP2) dal citosol all'ergastoplasma, dove possono legarsi alle tasche formate dalle glicoproteine HLA di classe I appena sintetizzate (v. immunologia: Istocompatibilità).
b) Attivazione dei linfociti T
I linfociti T circolano continuamente nei tessuti con il sangue e con la linfa, e stazionano per un certo periodo nella regione paracorticale dei linfonodi. Con i loro recettori di membrana essi interagiscono con le molecole HLA espresse dalle cellule con cui vengono a contatto: ciò avviene soprattutto nei linfonodi, dove per via linfatica arrivano dai vari tessuti anche le APC che hanno fagocitato l'antigene. Un recettore dei linfociti T, il CD4 o il CD8, interagisce con una parte costante, cioè identica in tutti gli individui, delle molecole HLA: il CD4 lega la parte costante delle glicoproteine HLA di classe II, mentre il CD8 quelle di classe I. Poiché esprimono solo il CD4 o solo il CD8 (sono o CD4 + o CD8 +), i linfociti T maturi prenderanno contatto solo con glicoproteine HLA di classe I o II. Mentre i recettori CD4 e CD8 sono eguali in tutti i linfociti T, il recettore destinato al riconoscimento dell'antigene (T Cell Receptor, TCR) è differente in ogni linfocito. I TCR sono costituiti da due catene proteiche (α e β oppure γ e δ) simili, ma molto più corte, a quelle delle immunoglobuline, la cui parte più esterna è costituita da una sequenza di amminoacidi che varia da linfocito a linfocito: la variabilità fa sì che ogni linfocito possegga una particolare specificità, cioè leghi molecole differenti. Tuttavia, la conformazione del TCR è tale da consentire di legare solo le glicoproteine HLA, poiché nel timo, durante il differenziamento, vengono lasciati maturare funzionalmente solo dei linfociti T il cui TCR interagisce con un legame a bassa affinità (cioè troppo debole per attivare i linfociti) con le molecole HLA dell'individuo contenenti i peptidi normali dell'individuo stesso. Quando invece le glicoproteine HLA legano un peptide estraneo lineare, il complesso glicoproteina HLA-peptide assume una nuova conformazione in grado di legarsi ad alta affinità con il TCR di qualche linfocito T: è infatti probabile che tra i milioni di linfociti T con TCR diversi ve ne sia uno, o più di uno, capace di stabilire un legame ad alta affinità. Il legame costituisce un importante stimolo per il linfocito T, che viene in tal modo preattivato.
c) Costimolazione dei linfociti T
Perché i linfociti T preattivati dall'interazione del TCR con le molecole HLA che presentano un peptide estraneo raggiungano la completa attivazione è necessaria la concomitanza di altri segnali. Sulla membrana dell'APC devono essere espresse le molecole CD80 o CD86 (B7.1 e B7.2) che, interagendo con i rispettivi recettori sulla membrana dei linfociti T (CD28 e CTLA-4), forniscono uno dei più importanti segnali di costimolazione. L'interazione del CD45 e delle varie molecole d'adesione dei linfociti T con i rispettivi ligandi espressi dalle APC è altresì necessaria per la completa attivazione dei linfociti T, che è anche favorita da alcune citochine secrete dalle APC. In assenza dei segnali costimolatori, il linfocito preattivato va incontro a una forma di suicidio (apoptosi) o rimane funzionalmente bloccato.
La complessa sequenza di segnali rende l'attivazione dei linfociti T un evento sottoposto a un rigido controllo, reso necessario in quanto una loro anomala attivazione verso componenti normali dell'organismo metterebbe in moto meccanismi distruttivi di grande efficacia.
d) Linfociti T helper
I linfociti T attivati proliferano e danno origine a un clone di cellule figlie identiche, che presentano tutte lo stesso TCR del linfocito progenitore. Quelli CD4 +, quando arrivano in presenza di cellule che esprimano lo stesso peptide estraneo legato alla stessa glicoproteina HLA di classe II che ha innescato l'attivazione, secernono varie citochine. Alcune di queste, come ad esempio l'IL-2, vengono utilizzate sia dallo stesso linfocito T che le ha prodotte (utilizzazione autocrina), sia dai linfociti T CD8 + che, nelle adiacenze, si trovino in uno stato di preattivazione, per iniziare a proliferare. I linfociti T CD4 +, che secernendo l'IL-2 e altre linfochine guidano anche la reattività dei macrofagi, dei linfociti B e delle cellule natural killer (NK), sono detti linfociti T helper.
e) Linfociti T helper 1 e T helper 2
Il repertorio di citochine secrete dai linfociti T CD4 + è quindi importante in quanto da ciò dipende l'attivazione di vie diverse della risposta immunitaria. In genere i linfociti T CD4 + (detti T helper zero, TH0) secernono una grande varietà di citochine. In alcuni casi si differenziano in linfociti CD4 + TH1 o TH2, caratterizzati dall'attitudine a secernere un repertorio di citochine più limitato e più caratteristico: sia la presenza di un'alta densità di complessi HLA-peptide estraneo sulla membrana della APC che ha indotto l'attivazione, sia quella di IL-12 o IFN-γ nel microambiente, inducono la differenziazione dei linfociti TH0 in linfociti TH1 che secernono, in maniera caratteristica, l'IFN-γ, l'IL-12 e favoriscono tanto l'attivazione dei macrofagi e delle cellule NK quanto la produzione di alcune sottoclassi di immunoglobuline; una bassa densità di complessi HLA-peptide estraneo o la presenza nel microambiente di IL-4 (rilasciata, per esempio, dai mastociti) favorisce, invece, la differenziazione dei linfociti TH2 che secernendo l'IL-4 e l'IL-5 promuovono la risposta anticorpale in genere e, in particolare, la secrezione di IgA e IgE. Nei confronti della maggior parte degli antigeni l'attivazione dei linfociti T CD4 + TH1 e TH2 risulta bilanciata o perché la differenziazione dei linfociti da TH0 a TH1 o TH2 non ha luogo, o perché i differenti complessi HLA-peptide, derivati dalla scissione di un antigene, generano indipendentemente i due tipi di cellule helper.
f) Linfociti T killer
I linfociti CD8 +, attivati meno di frequente, secernono citochine e nella maggior parte dei casi divengono linfociti T killer. Questi uccidono le cellule che presentano lo stesso complesso HLA di classe I-peptide della cellula che ne ha innescato l'attivazione tramite la secrezione di perforine, le quali determinano sulla membrana della cellula bersaglio lesioni simili a quelle provocate dal complemento o inducono il loro suicidio tramite la secrezione del Tumor Necrosis Factor. Quest'azione è di fondamentale importanza per eliminare le cellule estranee, le cellule mutate e le cellule infettate da microrganismi: per la loro capacità di uccidere le cellule infettate da virus, i linfociti killer CD8 + costituiscono uno dei meccanismi principali di guarigione dalle malattie virali.
g) Attivazione dei linfociti B
I linfociti B sono cellule essenzialmente statiche, localizzate nei linfonodi a livello dei follicoli linfatici, dove vengono attivate dal contatto con l'antigene che vi giunge tramite il drenaggio linfatico della zona in cui è penetrato. Mentre l'attivazione dei linfociti T è innescata esclusivamente dal riconoscimento delle glicoproteine HLA contenenti peptidi lineari derivati dall'antigene fagocitato, i linfociti B riconoscono l'antigene nella sua forma tridimensionale, cioè naturale. Il riconoscimento inizia con l'interazione tra l'antigene e i recettori di membrana (B Cell Receptor, BCR), costituiti da immunoglobuline di membrana (IgM o IgD monomeriche) associate a molecole in grado di trasdurre il segnale al nucleo (Igα e Igβ). Tuttavia, come nel caso dei linfociti T, questa interazione non attiva completamente i linfociti B ma piuttosto li porta a uno stato di preattivazione. Nel frattempo, l'antigene nativo legato al BCR viene internalizzato e digerito, e i piccoli peptidi che ne derivano vengono riespressi sulla membrana linfocitaria in associazione alle glicoproteine HLA di classe II: in questo modo il linfocito B preattivato si comporta come una APC (è cioè in grado di presentare l'antigene ai linfociti T) pur se molto selettiva, in quanto non fagocitando può presentare solo peptidi dell'antigene che specificamente si è legato alle immunoglobuline del BCR. Se un linfocito T CD4 + attivato riconosce con il suo TCR il peptide associato alle glicoproteine HLA del linfocito B, le due cellule stabiliscono una forte interazione reciproca tramite le varie molecole d'adesione: in quest'interazione il linfocito T riceve e rilascia una serie di segnali di costimolazione e viene stimolato a secernere varie citochine (IL-2, IL-4, IL-5) necessarie per la definitiva attivazione dei linfociti B. I linfociti B attivati proliferano e danno origine a un vasto clone di cellule figlie che si differenziano in plasmacellule e in linfociti B della memoria: le plasmacellule iniziano rapidamente a produrre quantità massicce di immunoglobuline, mentre le cellule della memoria manterranno per anni il ricordo dell'esperienza immunologica trascorsa e, più numerose, saranno pronte a dare origine a una risposta secondaria qualora l'antigene penetrasse una seconda volta nell'organismo.
6. Strategie della risposta specifica
a) Selezione clonale
La reattività immunitaria specifica dipende dalla presenza di linfociti T e B, ciascuno caratterizzato dall'avere un recettore per l'antigene di una sola specificità, diverso da quello di tutti gli altri linfociti della stessa popolazione. L'antigene, in forma naturale per i linfociti B o come peptide lineare associato alle glicoproteine HLA per i linfociti T, si combinerà con i linfociti che presentano il recettore più affine. Una volta attivato, il linfocito prescelto darà origine a un clone di linfociti figli che avranno tutti il recettore identico al linfocito attivato inizialmente. Con un meccanismo tipicamente darwiniano sarà proprio l'antigene che selezionerà il linfocito più adatto a rispondere e che ne stimolerà l'espansione numerica.
b) Memoria immunitaria
La risposta specifica è quindi un fenomeno adattativo: il sistema immunitario si modifica per rispondere più efficacemente all'antigene. L'adattamento consiste in un cambiamento della composizione del sistema immunitario: in conseguenza della selezione operata dall'antigene i linfociti che sono in grado di rispondere divengono significativamente più numerosi e rimangono tali per tutto il tempo in cui si conserverà la memoria dell'esperienza antigenica. Un secondo incontro con lo stesso antigene indurrà l'attivazione dei linfociti di memoria che daranno una risposta immunitaria più efficace (perché sono più pronti a reagire rispetto ai linfociti vergini, che vedono l'antigene per la prima volta) e più intensa (perché sono molto più numerosi). La capacità dei linfociti di mantenere per anni la memoria dell'esperienza antigenica passata e di dare una risposta molto più efficace allorché si trovino nuovamente in presenza dell'antigene costituisce la base della vaccinazione.
c) Generazione delle diversità dei recettori
Poiché la popolazione dei linfociti T e dei linfociti B è composta da milioni di cellule, ognuna con un TCR o un BCR capace di legarsi a un antigene diverso, ci si chiede come possa venir generata questa pressoché infinita varietà di recettori. Ogni TCR e ogni BCR, pur presentando una struttura simile a quella degli altri recettori, ha una specificità propria, poiché le diverse regioni delle catene del recettore sono codificate da un gene diverso: per esempio, una catena del TCR o del BCR può venir codificata da una regione V (variabile) per la parte NH2-terminale (quella più esterna), da una regione D (diversità) per la parte della catena adiacente a quella V, da una regione J (unione) per la successiva porzione della catena, e da una regione C (costante) che codifica la parte COOH-terminale che, attraversata la membrana cellulare, termina nel citoplasma. Per ognuna di queste regioni sono disponibili più geni (per esempio, un centinaio di geni V, una dozzina di geni D, una dozzina di geni J). Questi diversi raggruppamenti di geni sono posti a grande distanza l'uno dall'altro. La variabilità nasce dall'utilizzazione ‛casuale' di uno dei vari geni dei diversi raggruppamenti, così che un numero relativamente basso di geni dà origine a un elevato numero di combinazioni. La ricombinazione genica ha luogo solo durante la differenziazione funzionale del linfocito e comporta un radicale rimaneggiamento di quella regione di DNA. Un gene D, prescelto casualmente, viene contrapposto e poi unito a uno dei vari geni J: per questa unione, i due geni vengono ravvicinati e la lunga sequenza di DNA interposta, con tutti i geni che contiene, viene tagliata via, mentre i due geni vengono ricuciti insieme; con lo stesso meccanismo quei due geni DJ si uniranno a uno dei tanti geni V, tagliando via la lunga regione di DNA interposta.
Teoricamente, già la semplice ricombinazione casuale di questi geni dà origine a circa 1.000-10.000 possibili catene diverse. Poiché la specificità del TCR e del BCR nasce dalla tasca formata da due catene tra loro diverse, la libera associazione delle due catene porta al formarsi di almeno 10 milioni di recettori diversi.
Il processo di ricombinazione genera un numero di recettori ancora più elevato, perché quando due geni vengono uniti (D-J, V-DJ) possono avvenire anche questi fenomeni: 1) l'unione può non essere precisa e vi può essere una perdita o un'aggiunta di nucleotidi; 2) la regione di DNA che codifica la porzione D può venir letta a partire da tre triplette di nucleotidi diversi (tre registri di lettura) e così può dare origine a tre diversi frammenti proteici; 3) nei punti dove il DNA viene tagliato (D-J; V-DJ) un enzima particolare, la terminal-desossinucleotilditransferasi, può aggiungere ai monconi del DNA da 1 a 6 nuovi nucleotidi, dando così origine a nuove sequenze di DNA.
Per ognuna delle due catene del TCR o del BCR questo processo di ricombinazione può aver luogo su entrambi i cromosomi, quello ereditato dalla madre e quello ereditato dal padre. Il processo ha inizio col tentativo di ricombinarne uno, a caso, e se dà origine a un DNA che funziona, il prodotto di questo DNA, cioè la catena del recettore, appena sintetizzata blocca il riarrangiamento dell'altro cromosoma. Spesso, invece, il processo dà origine a un segmento di DNA che non funziona e in questo caso il linfocito tenta la ricombinazione sull'altro cromosoma: se fallisce anche questa volta, il linfocito va incontro ad apoptosi. Quando poi il linfocito maturo, attivato dall'antigene, darà origine a un clone di linfociti, questi erediteranno il recettore nato da questa ricombinazione genica, che rimarrà immutato in tutte le cellule figlie.
d) Il TCR
I linfociti T originati dalla cellula staminale totipotente migrano nel timo, dove si differenziano in linfociti T maturi. Durante la maturazione timica ha luogo il riarrangiamento dei geni del TCR, che è costituito dalle catene α-β o γ-δ. La maggior parte dei linfociti utilizza il recettore formato dalle catene α-β; quelli con recettore γ-δ sono principalmente localizzati nei tessuti delle sottomucose. I geni coinvolti nel riarrangiamento e i principali meccanismi per la generazione delle diversità dei TCR sono riportati nella tab. I.

La varietà dei recettori così generata è molto alta. Molti di questi non interagiscono con le glicoproteine HLA e quindi non funzionano; altri, al contrario, interagiscono con il complesso glicoproteine HLA-peptide dell'individuo con così alta affinità da portare a fenomeni di autoimmunità nel caso che fuoriescano dal timo. Pertanto, durante la maturazione timica, con grande precisione vengono soppressi per apoptosi sia i linfociti che esprimono un TCR che non reagisce con il complesso glicoproteine HLA-peptide del proprio organismo, sia quelli con un TCR in grado di reagirvi con forte affinità; al contrario, i linfociti il cui TCR lega con bassa affinità il complesso glicoproteine HLA-peptide del proprio organismo vengono lasciati maturare, espandere e migrare nel sangue. Di conseguenza, ognuno dei linfociti T maturi in circolo in un individuo presenterà un TCR diverso, ma questi vari TCR saranno tutti caratterizzati dal fatto di interagire male, con bassa affinità, con le glicoproteine HLA di quell'individuo legate a un normale peptide dello stesso organismo.
Il recettore con cui i linfociti T riconoscono il complesso glicoproteine HLA-peptide estraneo è costituito, oltre che dalle due catene del TCR, anche da un insieme di molecole (il CD3), identiche in tutti i linfociti T di tutti gli individui, che trasducono al nucleo il segnale generato dall'interazione tra il TCR e il complesso glicoproteine HLA-peptide estraneo.
e) Il BCR
Sui linfociti B esiste, come si è detto, un recettore analogo a quello dei linfociti T, costituito in parte da una molecola di immunoglobuline della classe delle IgD o IgM, con cui viene riconosciuto l'antigene, e in parte da molecole, identiche in tutti i linfociti B di tutti gli individui, dette Igα e Igβ, che trasducono al nucleo il segnale generato dall'interazione dell'anticorpo di membrana con l'antigene.
Le immunoglobuline, in forma monomerica, sono formate da quattro catene, due pesanti (catene H) e due leggere (catene L): le due catene H e le due catene L sono tra loro eguali. Le catene H delle immunoglobuline che costituiscono il BCR attraversano la membrana cellulare e terminano nel citoplasma. La diversità delle immunoglobuline nasce, con meccanismi molto simili a quelli del TCR, durante la maturazione dei linfociti B nel midollo. La grande differenza esistente tra l'origine del BCR e quella del TCR è costituita dal fatto che i geni delle Ig riarrangiati vanno incontro, con straordinaria frequenza, a mutazioni somatiche: con il progressivo esplicarsi della risposta immunitaria le mutazioni somatiche fanno aumentare l'affinità degli anticorpi e conseguentemente ha luogo la generazione di linfociti B di memoria più affini verso l'antigene di quanto non lo fossero i linfociti B vergini. Le mutazioni somatiche sono anche importanti per la generazione di nuove diversità recettoriali e per lo spegnimento della risposta anticorpale. I linfociti B immaturi il cui BCR incontra un antigene, vanno incontro a morte per apoptosi o, più di frequente, rimangono paralizzati per lungo tempo: in questo modo vengono neutralizzati i linfociti B autoreattivi, che facilmente interagiscono con gli antigeni dell'organismo prima di maturare completamente.
7. Le immunoglobuline
a) Caratteristiche generali
Le immunoglobuline, che costituiscono il prodotto più raffinato del sistema immunitario, sono delle molecole, secrete dalle plasmacellule e presenti in tutti i liquidi organici, in grado di colpire l'antigene a grande distanza dalla cellula che le ha prodotte. La struttura della molecola immunoglobulinica nei liquidi organici è simile a quella delle Ig del BCR: la parte costante delle catene H è un po' più corta, in quanto mancano la regione che attraversa la membrana cellulare e quella che termina nel citoplasma; la parte variabile (il sito combinatorio) è invece costituita da una sequenza amminoacidica eguale a quella del BCR del linfocito B, che si è trasformato in plasmacellula (v. immunologia e immunopatologia: Immunologia generale).
b) Commutazione di classe
Durante il progredire delle risposte anticorpali, segnali dati da varie citochine rilasciate dai linfociti TH fanno sì che i geni che codificano la parte variabile delle catene H (il complesso VDJ) vengano successivamente a trovarsi nelle vicinanze dei diversi geni costanti. Gli stessi geni della parte variabile verranno così associati a parti costanti differenti (commutazione di classe) e si avrà la produzione di IgM, IgD (solo come componenti del BCR), IgG, IgA e IgE. I geni delle varie classi e sottoclassi sono localizzati (nell'uomo sul cromosoma 14) in questo ordine: μ (IgM), δ (IgD), γ3 (IgG3), γ1 (IgG1), α1 (IgA1), γ2 (IgG2), γ4 (IgG4), ε (IgE), α2 (IgA2). Il processo che porta i diversi geni costanti in prossimità della regione VDJ determina la perdita delle sequenze di DNA interposte, per cui può proseguire andando dalle IgM alle IgA2, ma non può tornare indietro.
c) Reticolo degli idiotipi
Durante la risposta immunitaria i cloni di linfociti B che interagiscono con l'antigene si espandono e, differenziandosi in plasmacellule, producono grandi quantità di immunoglobuline con lo stesso sito combinatorio (idiotipo). Durante una massiccia produzione anticorpale, il sito combinatorio può fungere, esso stesso, da antigene inducendo anticorpi specifici (anticorpi anti-idiotipo). A sua volta, il sito combinatorio di questi secondi anticorpi induce la produzione di anticorpi, e così via. Il processo si espande generando un vero e proprio reticolo di anticorpi che si legano l'uno all'idiotipo dell'altro (reticolo degli idiotipi o network di Jerne). Ogni successiva produzione di anticorpi risulta progressivamente di intensità inferiore rispetto a quella che l'ha indotta. Questo reticolo di interazioni regola la produzione di anticorpi e la sua caratterizzazione sta aprendo affascinanti possibilità di manipolazione della risposta immunitaria. Alcuni degli anticorpi di seconda generazione, quelli che interagiscono con il sito combinatorio degli anticorpi che legano l'antigene (e che pertanto vengono considerati come ‛immagine interna dell'antigene'), in determinati casi, come risulta da evidenze sperimentali e cliniche, possono venir utilizzati al posto del vero antigene; potendo interagire con il sito combinatorio del BCR e dare un segnale simile a quello dato dall'antigene, essi sono in grado di indurre la produzione di anticorpi reattivi verso l'antigene.
8. Le cellule natural killer (NK)
Questi linfociti, un po' più grandi della maggior parte dei linfociti e caratterizzati da fini granulazioni citoplasmatiche, esercitano un ruolo importante nell'uccisione ‛naturale', cioè immediata, di alcuni tipi di cellule immature, di quelle infettate da virus e di alcuni tipi di cellule tumorali. Le cellule bersaglio dell'azione delle cellule NK, a causa del particolare stato differenziativo o in seguito all'infezione, presentano una diminuita espressione di alcune glicoproteine HLA di classe I ed è proprio la rarefazione di queste glicoproteine (la perdita del self) che le rende suscettibili all'attività delle cellule NK. Tuttavia, dati recenti suggeriscono che l'espressione di alcune glicoproteine HLA possa anche costituire uno stimolo di attivazione della reattività delle cellule NK.
BIBLIOGRAFIA
Alberts, B., Bray, D., Lewis, J., Raff, M., Roberts, K., Watson, J. D., Molecular biology of the cell, New York 19943 (tr. it.: Biologia molecolare della cellula, Bologna 19963).
Alt, F. W., Blackwell, T. K., Yancopoulos, G. D., Development of primary antibody repertoire, in ‟Science", 1987, CCXXXVIII, pp. 1079-1087.
Benoist, C., Mathis, D., Generation of the αβ T-cell repertoire, in ‟Current opinion in immunology", 1992, IV, pp. 156-161.
Bjorkman, P. J., Parham, P., Structure, function and diversity of class I major histocompatibility complex molecules, in ‟Annual review of biochemistry", 1990, XL, pp. 253-288.
Boehmer, H. von, Kisielow, P., How the immune system learns about self, in ‟Scientific American", 1991, CCLXV, pp. 74-81 (tr. it.: Come il sistema immunitario impara a riconoscere il ‟sé", in ‟Le scienze", 1991, XLVII, 280, pp. 64-74).
Chesnut, R. W., Grey, H. M., Antigen presentation by B cells and its significance in T-B interactions, in ‟Advances in immunology", 1986, XXXIX, pp. 51-94.
Janeway, C. A. Jr., Travers, P., Immunobiology, London 1996.
Jerne, N. K., The immune system, in ‟Scientific American", 1973, CCXXIX, pp. 52-60 (tr. it.: Il sistema immunitario, in ‟Le scienze", 1973, XI, 63, pp. 70-79).
Kuby, J., Immunology, New York 1992.
Paul, W. E., Fundamental immunology, New York 1995.
Autoimmunità di Benvenuto Pernis
sommario: 1. La selezione clonale dei linfociti: a) premessa; b) i linfociti T: selezione positiva e selezione negativa; la soppressione; c) i linfociti B: selezione positiva e selezione negativa. 2. L'autoimmunità: a) caratteristiche generali delle malattie autoimmuni; b) meccanismi del danno tissutale. 3. Modelli di malattie autoimmuni negli animali da laboratorio: a) il lupus eritematoso sistemico del topo; b) l'artrite cronica sperimentale nel ratto e nel topo; c) il diabete autoimmune nel topo; d) l'encefalomielite allergica sperimentale. □ Bibliografia.
1. La selezione clonale dei linfociti
a) Premessa
Del vastissimo campo dell'autoimmunità, questo articolo tratterà solamente le basi generali della tolleranza del sistema immunitario verso gli antigeni propri dell'organismo e presenterà un quadro generale delle numerose malattie autoimmuni umane; solo alcune di queste verranno discusse più approfonditamente, a scopo di esempio, esaminandone i modelli animali, perché in questo campo sono stati recentemente compiuti notevoli progressi che facilitano la comprensione dell'autoimmunità in generale.
La funzione del sistema immunitario è quella di individuare le macromolecole estranee all'organismo e di attivare le difese idonee a eliminare sia queste, sia gli ‛invasori' che le producono (Virus, Batteri, Metazoi), come risulta chiaramente dall'osservazione che gli individui nei quali il sistema immunitario non è sufficientemente sviluppato, oppure è stato inattivato da farmaci, radiazioni o Virus (ad esempio, HIV), soccombono a molteplici infezioni dovute ad agenti diversi. Questa funzione di difesa richiede che il sistema immunitario sia in grado di distinguere le macromolecole proprie da quelle estranee. È questo un problema di non facile soluzione, specialmente nel caso delle proteine, perché non esistono proprietà chimiche o di carattere generale in base alle quali il sistema immunitario possa operare questa distinzione. Pertanto, è necessario che il sistema immunitario, durante lo sviluppo e la maturazione delle sue cellule (i linfociti), ‛impari' a riconoscere e a convivere con le macromolecole (gli antigeni) proprie, conservando e ampliando nella sua memoria la capacità di reagire contro gli antigeni estranei. La base di questo processo di apprendimento, che dura per tutta la vita, risiede nella selezione clonale dei linfociti.
I recettori per gli antigeni, sia nei linfociti T che nei linfociti B, sono controllati da geni che durante la maturazione di queste cellule subiscono processi di riarrangiamento nei quali il caso svolge un ruolo fondamentale. Vengono così prodotti cloni diversi di linfociti, ciascuno con un diverso recettore. Il ruolo del caso nella generazione della diversità clonale dei linfociti è essenziale per la produzione di una popolazione di cellule capaci di reagire, in linea di principio, contro antigeni diversi e non prevedibili a priori, quali sono quelli dei potenziali agenti infettanti presenti all'esterno dell'organismo. Tuttavia, proprio l'elemento casuale prospetta due grandi problemi: il primo consiste nella generazione di cellule con recettori completamente inutili; il secondo invece nella produzione di cellule con recettori dannosi, in quanto capaci di reagire efficacemente contro gli antigeni propri dell'organismo. Ovviamente, il sistema immunitario delle cellule mature deve comprendere il minimo numero possibile di cloni inutili e deve eliminare o poter controllare i cloni potenzialmente dannosi. Il rispetto di questa regola fondamentale si ottiene attraverso processi di selezione positiva e di selezione negativa che operano durante la maturazione dei linfociti negli organi linfoidi centrali e che, mediante l'eliminazione della grande maggioranza delle cellule prodotte a quel livello, provvedono a disseminare negli organi linfatici periferici, là dove si svolgerà la funzione protettiva del sistema immunitario, i cloni di cellule potenzialmente utili e non potenzialmente dannose (v. Schwartz e Datta, 19892; v. Sinha e altri, 1990; v. Cohen e Young, 1991).
Consideriamo adesso più da vicino i processi di selezione positiva e negativa dei linfociti T e B, poiché la loro comprensione è necessaria per chiarire sia la funzione normale del sistema immunitario, sia le sue deviazioni patologiche che conducono alle malattie autoimmuni.
b) I linfociti T: selezione positiva e selezione negativa; la soppressione
Come descritto in altri articoli (v. immunologia: Istocompatibilità), i linfociti T sono forniti di recettori clonali che riconoscono frammenti di antigeni proteici inseriti in una ‛tasca' presente nella struttura delle molecole di istocompatibilità. Le strutture molecolari con le quali i recettori dei linfociti T reagiscono sono quindi costituite da un complesso, formato da un peptide e dalla molecola di istocompatibilità (MHC, Major Histocompatibility Complex) che lo presenta. Nel timo, dove avviene la maturazione e la selezione dei linfociti T, normalmente non vi sono antigeni proteici estranei all'organismo; pertanto la selezione dei cloni T, sia quella positiva che quella negativa, è operata dal contatto dei recettori esclusivamente con complessi di peptidi derivati da proteine ‛proprie' e da molecole MHC ‛proprie'.
Siamo dunque di fronte al problema del ‛come' e del ‛perché' le stesse strutture molecolari possano determinare sia la selezione positiva che la selezione negativa dei linfociti T, e di fronte all'apparente paradosso di due risultati opposti conseguenti al contatto dei recettori clonali con gli stessi complessi di peptidi di derivazione endogena, presentati dalle proprie molecole MHC. Su questo argomento, di importanza centrale per tutta l'immunologia e in particolare per il capitolo dell'autoimmunità, si è detto e scritto moltissimo, ma a tutt'oggi non possiamo asserire di essere giunti a un completo chiarimento del problema; tuttavia, particolarmente in seguito a recenti esperimenti condotti in vitro su colture di frammenti di timo, cominciamo a comprenderne gli aspetti fondamentali. Anzitutto sono stati accertati due fatti: 1) i linfociti che durante il processo di maturazione nel timo esprimono sulla loro membrana recettori che non reagiscono contro i complessi MHC-peptidi presenti sulle cellule della matrice linfoepiteliale timica non evolvono ulteriormente, ma sono eliminati mediante l'attivazione di un programma di autodistruzione (v. Surh e Sprent, 1994); 2) al contrario, i linfociti il cui recettore reagisce con il complesso MHC-peptidi presentato loro dalle cellule della matrice timica evolvono fino alla maturazione e continuano a proliferare. Questa è la base della ‛selezione positiva', mediante la quale sopravvivono solo linfociti con recettori capaci di riconoscere peptidi presentati dalle molecole MHC proprie dell'organismo stesso. Il significato funzionale di questo aspetto della selezione positiva è facilmente intuibile, in quanto la funzione ultima dei linfociti T è quella di riconoscere antigeni (peptidi) presentati dalle molecole MHC proprie dell'organismo, e sarebbero inutili quei cloni con recettori non idonei a reagire contro un complesso molecolare del quale le molecole MHC proprie sono parte essenziale. Meno evidente è la funzione, nella selezione positiva, della reattività dei recettori nei confronti di peptidi derivati da proteine endogene, presenti e presentati nel timo. Questo aspetto della selezione positiva sembra, a prima vista, destinato a produrre una popolazione di linfociti T autoreattivi, potenzialmente capaci di generare ogni sorta di fenomeni autoimmunitari. In realtà, la propensione autoreattiva della selezione positiva è limitata dalla selezione negativa, come diremo in seguito. Possiamo tuttavia domandarci, in linea teorica, se non vi sia un profondo motivo biologico per la selezione positiva di cloni linfocitari con recettori reattivi contro molecole MHC proprie che presentano peptidi endogeni. Questo motivo può essere individuato nella somiglianza generale di peptidi potenzialmente derivati dalla frammentazione di antigeni batterici e, soprattutto, virali con i peptidi prodotti dalla frammentazione di proteine endogene. Perciò la selezione positiva nel timo evita la sopravvivenza di tutta una serie di cloni il cui recettore, originato da eventi in parte casuali a livello del DNA cellulare, ha una struttura non idonea a reagire contro i complessi MHC-peptidi che potrebbero essere prodotti nel mondo esterno da potenziali agenti infettanti. In altri termini, la selezione positiva nel suo complesso rinuncia a generare una popolazione di linfociti onnipotente, ma largamente sovradimensionata rispetto allo spazio reale del sistema immunitario, in favore di una popolazione, pur sempre molto ampia, già orientata verso una possibile utilizzazione pratica della propria specificità. A questo punto, è necessario un processo capace di eliminare, già a livello del timo, almeno quell'aliquota di cloni che hanno superato il vaglio della selezione positiva, ma che, essendo provvisti di recettori dotati di un'alta affinità per i complessi MHC-peptidi presenti nel timo stesso, sarebbero sicuramente capaci, una volta maturi e trasferiti nel sistema linfatico periferico, di generare reazioni autoimmunitarie. Questo processo è la ‛selezione negativa', alla quale abbiamo accennato.
Nel loro complesso, dunque, le azioni selettive operanti nel timo eliminano sia i cloni che non reagiscono contro i complessi MHC-peptidi presentati dalle cellule stromali, sia i cloni che reagiscono contro gli stessi complessi con alta affinità. La popolazione di cellule che matura e lascia il timo per colonizzare gli organi linfatici periferici è quindi quella che reagisce con bassa affinità contro i complessi MHC-peptidi ‛propri' ed è incapace di produrre spontaneamente reazioni immunitarie nelle varie sedi dell'organismo. Tuttavia, questa popolazione include cloni in grado di reagire con alta affinità contro complessi formati da molecole MHC proprie e peptidi derivati da antigeni estranei, e quindi capaci di una efficace reazione immunitaria di difesa contro agenti infettanti. Questa capacità è legata alla potenziale omologia, più o meno stretta, fra i peptidi derivati dall'elaborazione (processing) delle proteine proprie e quelli derivati dalle proteine batteriche o virali: in altri termini, un clone selezionato sulla base di recettori che reagiscono in modo approssimativo contro i complessi MHC-peptidi endogeni ha una certa probabilità di possedere un recettore che reagisce bene quando è confrontato con una molecola MHC propria la cui tasca è occupata da un peptide derivato, per esempio, dalla elaborazione di una proteina virale. Un'altra proprietà del sistema, ancorché probabilmente non legata direttamente a motivi funzionali, è la alloreattività, cioè la capacità di una aliquota relativamente alta di linfociti T (attorno al 3%) di reagire energicamente nei confronti di cellule che esprimono molecole MHC allogeniche, cioè codificate da geni alleli presenti nella stessa specie. Questa proprietà - che è stata alla base della scoperta e della definizione degli antigeni di istocompatibilità - si spiega tenendo conto della possibilità che cloni relativamente numerosi di linfociti T, selezionati nel timo sulla base dell'espressione di recettori di bassa affinità per complessi MHC-peptidi propri, siano anche cross-reattivi, con affinità relativamente alta, contro complessi formati da molecole MHC simili (ma non identiche), alle proprie e peptidi uguali a quelli elaborati nel timo dell'individuo nel quale i linfociti sono maturati (v. Blackman e altri, 1990; v. Ashton-Rickardt e Tonegawa, 1994; v. Nossal, 1994; v. von Boehmer, 1994).
Così congegnata, la selezione negativa nel timo può agevolmente controllare la produzione di cloni di linfociti autoreattivi verso le proteine solubili prodotte dall'organismo stesso - sempre che queste possano raggiungere il timo in concentrazione sufficiente per essere internalizzate, frammentate e presentate sulle molecole MHC di classe II delle idonee cellule ivi presenti - e verso tutte le proteine non solubili sintetizzate nel citoplasma delle cellule stromali del timo, i cui peptidi sono presentati in quella sede dalle molecole MHC di classe I. In questo modo, tutte le proteine presenti nell'insieme delle cellule dell'organismo - gli enzimi che partecipano alle funzioni metaboliche generali e le proteine strutturali comuni a tutte le cellule, cioè i prodotti dei cosiddetti ‛geni di mantenimento' (household) - sono protette dalla selezione negativa nel timo (la cosiddetta tolleranza immunitaria ‛centrale'). Di conseguenza, queste proteine raramente divengono autoantigeni, salvo in quelle malattie nelle quali appunto la tolleranza centrale è deficitaria per varie ragioni, come nel lupus eritematoso sistemico e nei suoi modelli sperimentali (v. cap. 3, § a). Un interessante esempio di tolleranza centrale è dato da un esperimento nel corso del quale in un embrione di pollo è stato trapiantato un timo di quaglia; successivamente, nell'embrione è stato innestato l'abbozzo di un'ala di quaglia, che si è normalmente sviluppata.
Tuttavia, la tolleranza centrale difficilmente può operare l'eliminazione di cloni T potenzialmente autoreattivi nei confronti di antigeni di differenziazione espressi da cellule di organi periferici: questi antigeni, specialmente se non solubili ed espressi nelle cellule periferiche in bassa quantità, hanno scarse possibilità di raggiungere il timo e determinare in quella sede la selezione negativa dei cloni T, necessaria per stabilire la tolleranza centrale. In tal caso i cloni potenzialmente autoreattivi lasciano il timo e raggiungono la periferia, dove devono essere controllati dalla tolleranza periferica.
L'esistenza di meccanismi di tolleranza periferica è stata dimostrata nel modo migliore con esperimenti condotti in topi transgenici (v. farmacologia e sperimentazione animale), nei quali è stato introdotto un gene esterno unito a un segmento regolatore capace di determinarne l'espressione in un dato organo: sono stati prodotti, per esempio, topi che esprimevano una molecola MHC di classe II o di classe I allogenica, propria cioè di un altro ceppo di topi, esclusivamente nelle cellule insulari o in quelle acinari del pancreas, a seconda che il gene per la molecola MHC estranea fosse collegato con il segmento regolatore per l'insulina o per l'elastasi. Con lo stesso meccanismo di introduzione di geni estranei nella linea germinale, sono state prodotte linee di topi che esprimevano a livello delle cellule insulari del pancreas una classe di molecole MHC, le molecole I-E, inesistenti nell'organismo di quel particolare ceppo di topi, oppure addirittura una proteina virale (l'antigene T del virus SV40), ovviamente del tutto estranea a quell'organismo. In questi topi, in linea di massima, non si è determinata una reazione immunitaria di cellule T dirette contro il tessuto pancreatico e neppure sono state dimostrate in vitro cellule T capaci di reagire contro gli antigeni transgenici; in altri termini, in questi topi è stata dimostrata una tolleranza ad antigeni che sicuramente non erano espressi nel timo, cioè una tolleranza periferica (v. Burkly e altri, 1990).
La tolleranza periferica differisce tuttavia dalla tolleranza centrale per il meccanismo della sua genesi: i cloni T autoreattivi non sono distrutti dai meccanismi della tolleranza periferica, come accade invece nel caso dei cloni sottoposti alla selezione negativa nel timo, ma sono semplicemente inattivati. Questo meccanismo di inattivazione, detto ‛anergia', permette la sopravvivenza dei cloni di linfociti T interessati, ma li rende incapaci di reagire nei confronti degli antigeni anche se conservano i recettori idonei a tale scopo. Il meccanismo mediante il quale determinati antigeni espressi dai tessuti periferici possono inattivare i cloni T di specificità corrispondente, maturati nel timo e disseminati nel sistema periferico, non è completamente noto; probabilmente è legato al modo col quale questi antigeni sono presentati e alla mancanza, nelle cellule presentanti, di molecole necessarie per la piena attivazione dei linfociti T.
Un'interessante ipotesi chiama in causa, nella tolleranza periferica, la molecola CTLa-4. Questa molecola è omologa del corecettore CD28 e come questo reagisce con alta affinità con le molecole B-7 che si trovano sulle cellule che presentano l'antigene; la sua funzione fondamentale è quella di regolare l'omeostasi dei linfociti T dopo la stimolazione antigenica. Infatti, la molecola CTLa-4 è espressa, a livelli crescenti, nei linfociti attivati e in ultima analisi blocca la continuazione della risposta dei linfociti T, sia interferendo col segnale positivo dato dal CD28, sia provvedendo direttamente a fornire un segnale negativo. In topi nei quali il gene per la molecola CTLa-4 è stato inattivato, le risposte agli antigeni non hanno termine e gli animali muoiono in poche settimane in seguito a un'invasione generalizzata di linfociti proliferanti (v. Waterhouse e altri, 1995). È possibile, peraltro, che la molecola CTLa-4 intervenga nel bloccare sul nascere le risposte dei linfociti T contro autoantigeni, in quanto questi spesso vengono presentati da cellule con limitata espressione di B-7, condizione, questa, che favorisce le molecole CTLa-4 rispetto alle molecole CD28 (v. Kuchroo e altri, 1995).
Poiché i cloni anergici non proliferano, ma neppure scompaiono, si capisce come la tolleranza periferica sia meno stabile della tolleranza centrale. Infatti, nei topi transgenici che esprimono nel pancreas l'antigene virale T SV40, ad esempio, la tolleranza periferica è imperfetta in quegli animali nei quali l'antigene viene espresso più tardivamente durante lo sviluppo. In buona sostanza, la tolleranza periferica è una garanzia solo parziale contro l'autoimmunità, e molte fra le più frequenti malattie autoimmuni, come ad esempio quelle della tiroide, sono probabilmente dovute al venir meno, a un certo punto della vita, di alcuni processi di tolleranza periferica. È interessante notare che alcuni polimorfismi delle molecole CTLa-4 sono stati associati con l'immunità antitiroide della malattia di Graves.
Nella tolleranza periferica sono stati segnalati, in alcuni casi particolari, meccanismi di delezione clonale delle cellule T: ad esempio, in topi transgenici nei quali l'antigene introdotto (la molecola allogenica H-2Kb) era sotto il controllo del promotore della metallotioneina ed era espresso nel fegato. In questi casi, la delezione delle cellule autoreattive avviene probabilmente mediante l'attivazione del sistema Fas/Fas L, come discusso più avanti nel paragrafo dedicato al lupus eritematoso sistemico dei topi MLR-MLR (v. cap. 3, § a; v. Lynch e altri, 1995).
Quando la tolleranza fallisce e dopo che la reazione autoimmunitaria è iniziata, il sistema immunitario ha un'ultima possibilità per evitare o limitare il danno che ne può derivare, attivando l'immunosoppressione. Tuttavia, questa non va considerata come un processo destinato esclusivamente al controllo dell'autoimmunità, ma più in generale come un meccanismo di autoregolazione del sistema immunitario, capace di limitare le reazioni eccessive o troppo prolungate; molti casi di immunosoppressione sono stati infatti osservati non soltanto in malattie autoimmuni, ma anche in malattie infettive croniche, come la lebbra, nelle quali una reazione immunitaria diretta contro gli antigeni del microrganismo può indirettamente danneggiare e provocare mutilazioni nell'individuo infettato. L'immunosoppressione è operata da linfociti T, specialmente della sottopopolazione CD8, ma con partecipazione anche dei CD4, i quali agiscono eliminando o rendendo anergici altri linfociti T impegnati in una determinata reazione immunitaria. L'immunosoppressione è specifica, ma le basi molecolari di questa specificità non sono chiare. Vi sono due possibilità: o le cellule T soppressive reagiscono con l'antigene e liberano linfochine (IL-4, IL-10, TgFB) che sopprimono l'attivazione da parte dello stesso antigene delle altre cellule T; oppure le cellule soppressive riconoscono direttamente il recettore clonale delle cellule T che sono bersaglio della soppressione ed esercitano su queste un'azione citotossica o anergizzante. Perché le cellule T soppressive possano riconoscere il recettore clonale delle cellule T bersaglio, è necessario che queste operino una elaborazione del loro recettore e la presentazione di peptidi corrispondenti sulle loro molecole MHC di classe I, secondo le regole generali per la presentazione di antigeni capaci di essere riconosciuti da parte di altre cellule T: in questo caso, quindi, le cellule T soppressive riconoscono i peptidi derivati dai recettori clonali delle cellule T che sono bersaglio della soppressione. Esperimenti di immunizzazione con peptidi derivati dai recettori T confermano questo meccanismo in alcune condizioni, comprese alcune malattie autoimmuni sperimentali (v. Sercarz e Krzych, 1991).
Concludiamo questo paragrafo sottolineando l'importanza centrale delle cellule T per l'autoimmunità: infatti, anche l'autoimmunità da cellule B, cioè da autoanticorpi, richiede nella maggioranza dei casi la rottura della tolleranza delle cellule T che sono necessarie come coadiuvanti dei linfociti B nella produzione di anticorpi contro gli antigeni proteici, ivi compresi gli autoantigeni.
c) I linfociti B: selezione positiva e selezione negativa
I linfociti B, come i linfociti T, producono i loro recettori clonali per gli antigeni (le immunoglobuline di membrana), sotto il controllo di geni che subiscono processi di rielaborazione somatica secondo regole che includono elementi casuali. Pertanto, come i linfociti T, costituiscono una popolazione che deve essere sottoposta a processi di selezione, sia positiva che negativa. Le modalità e le sedi di questa selezione sono però molto diverse da quelle dei linfociti T, essenzialmente in rapporto alle seguenti fondamentali differenze funzionali tra linfociti B e T: 1) i recettori dei linfociti B reagiscono contro l'antigene direttamente, senza necessità di preparazione dell'antigene e di presentazione dei suoi frammenti da parte delle molecole MHC; 2) i geni per i recettori delle cellule B subiscono processi di rielaborazione non solo nell'organo centrale dove queste cellule maturano (il midollo osseo nei Mammiferi), ma anche in particolari sedi dei tessuti linfatici periferici (i centri germinativi dei follicoli linfatici).
La stimolazione dei linfociti B, come detto precedentemente, non è operata direttamente dagli antigeni proteici, ma richiede l'intervento di particolari linfociti T con specificità per lo stesso antigene; i linfociti B devono preparare frammenti dell'antigene stesso e presentarli alla cellula T cooperante, mediante le molecole MHC di classe II delle quali sono provvisti.
Consideriamo pertanto la selezione dei linfociti B a tre diversi livelli: nel midollo osseo, nei centri germinativi dei follicoli linfatici e nella periferia dei tessuti linfatici. Nel midollo osseo le cellule B non richiedono, come i linfociti T nel timo, una selezione positiva sulla base dei loro recettori completi (una funzione in tal senso di un recettore incompleto, il recettore pre-B, ha tutt'altro significato). Invece, una forma di selezione negativa è possibile, come è stato dimostrato in topi transgenici nei quali era stata indotta la coesistenza di un antigene MHC di classe I e di una maggioranza di linfociti B esprimenti nella loro membrana un recettore immunoglobulinico (prodotto sotto il controllo del transgene) diretto contro lo stesso antigene: in questi esperimenti, la maturazione delle cellule B autoreattive si arresta. Il destino di queste cellule può essere diverso: o la morte per attivazione di un programma di autodistruzione, chiamato ‛apoptosi' (un termine di derivazione greca che indica la caduta di un frutto maturo dall'albero), o il recupero mediante l'espressione di una catena immunoglobulinica leggera diversa da quella impiegata nel primitivo recettore autoreattivo. Il midollo osseo produce così una popolazione di linfociti B largamente purgata dalle cellule autoreattive, ma provvista di recettori capaci di reagire con antigeni estranei all'organismo, generalmente con bassa affinità.
La trasformazione di queste cellule in elementi con recettori ad alta affinità - quindi capaci di produrre in ultima analisi anticorpi efficienti - avviene nei centri germinativi dei follicoli linfatici sotto la selezione positiva degli antigeni estranei all'organismo. In questa sede, per l'attivazione di particolari enzimi, il DNA che controlla la parte variabile delle catene pesanti e leggere delle immunoglobuline subisce rapidi processi di mutazione che modificano la capacità della cellula di reagire con l'antigene esterno concentrato nel centro germinativo. Poiché nei centri germinativi l'antigene è presente sotto forma di complessi antigene-anticorpo depositati sulla superficie delle cellule dendritiche (non linfoidi) del centro stesso, si determinano le condizioni per una rapida selezione positiva delle cellule B con recettori per l'antigene ad affinità superiore a quella della media degli anticorpi sino allora prodotti.
Naturalmente il processo di ipermutazione può generare, nel centro germinativo, anche cellule B con recettori autoreattivi, ma queste cellule non ricevono il necessario aiuto dalle cellule T presenti in sede e vanno incontro a un processo di autodistruzione per picnosi (apoptosi), del quale vi è ampia evidenza microscopica in tutti i centri germinativi attivi. In questo modo a livello dei centri germinativi ha luogo anche un'estesa selezione negativa delle cellule B autoreattive, per effetto della mancata cooperazione con le cellule T specifiche per l'antigene estraneo. Infine, nei tessuti periferici l'incontro di cellule B mature con un antigene proteico per il quale hanno un'alta affinità può risultare, in assenza di aiuto T-specifico, nella anergia dei cloni di cellule B, con un processo di ‛tolleranza periferica' non molto dissimile dall'analogo esito in anergia al quale possono andare incontro le cellule T.
In definitiva, sia le cellule B che le cellule T sono sottoposte a controlli a diversi livelli, che normalmente riescono a impedire l'autoimmunità. Tali controlli, tuttavia, non possono essere così stringenti da orientare il sistema immunitario verso lo stato opposto, cioè l'immunodeficienza, e alcune maglie deboli della tolleranza immunitaria persistono e possono amplificarsi col processo di invecchiamento del sistema immunitario. Questi punti deboli possono in definitiva aprire la strada a processi autoimmunitari di varia natura, che hanno come bersaglio antigeni diversi dell'organismo, provocando la comparsa di diverse malattie autoimmuni, delle quali tratteremo brevemente.
2. L'autoimmunità
a) Caratteristiche generali delle malattie autoimmuni
Nell'uomo sono state osservate più di 40 diverse malattie autoimmuni; alcune di queste si sviluppano spontaneamente anche negli animali, mentre altre possono esservi indotte mediante procedimenti di immunizzazione.
La frequenza delle malattie autoimmuni nella specie umana è alta, interessando quote dal 5% al 7% della popolazione, con variazioni geografiche per certe malattie. Alcune di queste, come il diabete di tipo I e la sclerosi multipla, sono allo stesso tempo frequenti e seriamente invalidanti, e costituiscono quindi un grave problema di salute pubblica. Nella tab. I è riportato un elenco delle principali malattie autoimmuni umane, suddivise in base ai sistemi che interessano e con l'indicazione del ruolo dei linfociti T autoreattivi e/o dei linfociti B (autoanticorpi) nella determinazione del danno.
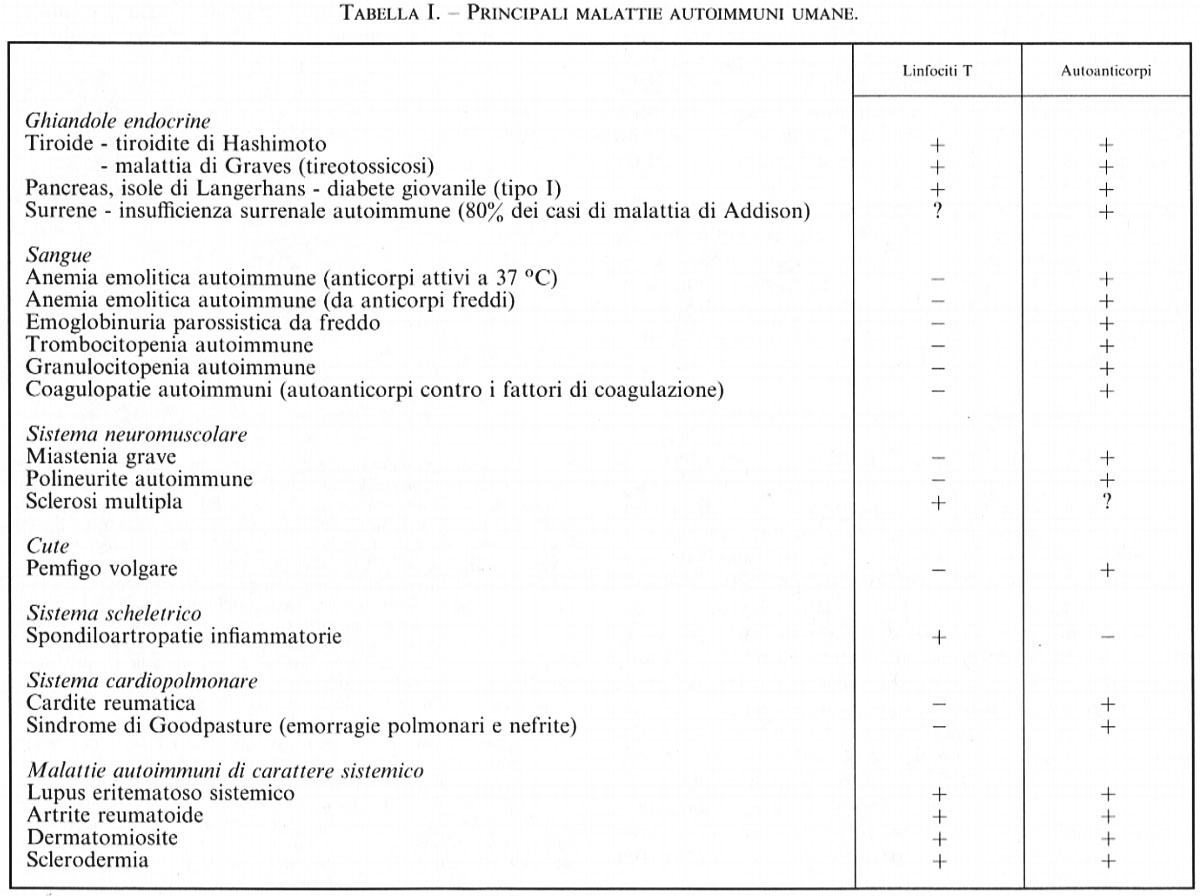
Alcune caratteristiche generali delle malattie autoimmuni meritano attenzione. Anzitutto la loro frequente associazione, più o meno stretta, con determinati alleli dei geni che controllano le molecole di istocompatibilità (MHC). Questa è una caratteristica delle malattie autoimmuni evidentemente legata alla necessità, per i linfociti T autoreattivi, di riconoscere i determinanti degli autoantigeni proteici presentati nella tasca delle molecole MHC: alcuni alleli MHC formano complessi con i peptidi derivati dagli autoantigeni, mentre altri non li formano o formano complessi più labili. Sfortunatamente non sono ancora stati individuati, nella maggior parte dei casi, i determinanti, o epitopi, implicati nelle diverse risposte autoimmuni umane. Esistono probabilmente numerosi epitopi per ogni malattia, e ciò spiega perché singoli alleli MHC siano in prevalenza, ma mai in modo obbligatorio, associati con singole malattie, anche se in misura diversa (il cosiddetto ‛rischio relativo'). Il caso più evidente di associazione MHC-malattia è quello dell'allele HLA-B27 di classe I con le spondiloartropatie infiammatorie; in questo caso il rischio relativo supera il 90%. Evidentemente, nelle spondiloartriti sono in gioco linfociti T autoreattivi della sottopopolazione CD8 +, quelli cioè che reagiscono con peptidi presentati dalla classe I delle molecole MHC. Tuttavia, anche in questo caso gli epitopi (peptidi) degli autoantigeni non sono stati ancora identificati con sicurezza.
Un'altra caratteristica comune a molte delle malattie autoimmuni, specialmente quelle organo-specifiche, è quella di presentarsi dopo un episodio infettivo batterico o virale: possiamo pensare che ciò avvenga per la rottura parziale della tolleranza periferica determinata da una somiglianza (mimicry) fra gli antigeni dell'agente infettante e quelli propri dell'organismo infettato. Un caso specifico è quello relativo alla produzione di autoanticorpi da parte di cellule B che ricevono un aiuto improprio dai linfociti T CD4 + mediante una molecola, o un complesso, che possiede allo stesso tempo sia determinanti capaci di interagire con le immunoglobuline potenzialmente autoreattive dei linfociti B, sia determinanti estranei all'organismo per i quali non vi è tolleranza nella popolazione di linfociti T.
b) Meccanismi del danno tissutale
Infine, un accenno ai meccanismi del danno tissutale causato dalle reazioni autoimmuni, non differenti, in linea di principio, dai meccanismi mediante i quali il sistema immunitario danneggia e quindi elimina gli agenti invasori. I linfociti T CD4 + determinano, laddove incontrano gli autoantigeni, una reazione infiammatoria di ipersensibilità ritardata (DTH, Delayed-Type Hypersensibility) nella quale il danno ai tessuti interessati è legato alla liberazione di una serie di citochine e all'attivazione dei macrofagi. I linfociti citotossici CD8 + uccidono direttamente le cellule bersaglio, esattamente come agirebbero, ad esempio, nei confronti di cellule infettate da un virus.
Gli autoanticorpi provocano danni per vie diverse: possono determinare, ad esempio, la lisi dei globuli rossi mediante l'attivazione del complemento, oppure possono agglutinare i trombociti e determinarne il sequestro nel sistema reticoloendoteliale. Gli autoanticorpi diretti contro recettori cellulari possono agire bloccando la funzione di questi recettori, oppure, al contrario, possono provocare un danno stimolando (funzione agonista) i recettori stessi. Nell'autoimmunità antitiroide, per esempio, autoanticorpi diretti contro il recettore dell'ormone stimolante la tiroide (TSH) possono causare un ipotiroidismo, come nella malattia di Hashimoto; oppure anticorpi un poco diversi, diretti contro lo stesso recettore, possono determinare una stimolazione della funzione tiroidea, come nella malattia di Graves. Inoltre, gli autoanticorpi coesistono solitamente con un'elevata quantità di autoantigeni. Se l'affinità della reazione è sufficiente, si verificano le condizioni ideali per la produzione di abbondanti immunocomplessi i quali, da soli o mediante la fissazione del complemento, sono causa di una rilevante patologia, specialmente a livello dei vasi, dei reni e delle articolazioni.
3. Modelli di malattie autoimmuni negli animali da laboratorio
Malattie autoimmuni che sono insorte spontaneamente in alcuni animali da laboratorio, o che possono essere indotte in questi mediante appropriata immunizzazione, forniscono elementi essenziali per la comprensione delle corrispondenti malattie umane e dell'autoimmunità in generale. Dei numerosi esempi di autoimmunità sperimentale, tratteremo qui di seguito i modelli di lupus eritematoso sistemico, artrite reumatoide, diabete autoimmune ed encefalomielite allergica (modello della sclerosi multipla umana).
a) Il lupus eritematoso sistemico del topo
Un ceppo di topi selezionato alcune decine di anni fa in Nuova Zelanda sulla base del colore del pelo (New Zealand Black, NZB) sviluppa una sindrome autoimmune che ha alcune analogie col lupus eritematoso sistemico umano. Tra queste, in primo luogo vi è un'anemia emolitica dovuta ad autoanticorpi contro un antigene accessibile sulla membrana dei globuli rossi; a questi autoanticorpi si aggiungono successivamente anticorpi anti-DNA, prevalentemente della classe IgM. A questo punto, generalmente nel secondo anno di vita, una parte dei topi NZB sviluppa una glomerulonefrite da immunocomplessi. La nefrite si sviluppa invece più precocemente e più frequentemente (ed è dovuta a immunocomplessi comprendenti anticorpi anti-DNA monoelica della classe IgG) in topi ibridi ottenuti incrociando gli animali del ceppo NZB con topi del ceppo NZW (New Zealand White), che normalmente sono sani. Altri ibridi dei topi NZB con ceppi sani di topi (ad esempio, C57BL/6) non sviluppano alcuna autoimmunità.
Non sono ancora stati identificati i geni responsabili dell'autoimmunità nei topi NZB o nei loro ibridi F1; per conseguenza ignoriamo ancora i difetti molecolari dei linfociti implicati nella sindrome autoimmune.
Un altro ceppo di topi fornisce forse maggiori informazioni: si tratta del ceppo MLR, che sviluppa autoanticorpi con produzione di immunocomplessi e una forma lieve di nefrite nel secondo anno di vita. Infatti, un ceppo congenico, MRL-lpr/lpr, che sviluppa anticorpi anti-DNA e nefrite molto più precocemente e mostra segni di linfoproliferazione diffusa, è caratterizzato dalla mancanza della molecola Fas (altrimenti chiamata APO-1 o CD95). Sempre derivato da MLR è il ceppo MLR-gld/gld, affetto dalla stessa sindrome, caratterizzato però dalla mancanza della molecola che reagisce con la Fas (chiamata perciò Fas-Ligand o Fas-L). Nei ceppi MRL-lpr/lpr e MLR-gld/gld è pertanto carente il sistema Fas/Fas-L. Evidentemente, questo sistema è importante nella regolazione della proliferazione dei linfociti T e, in ultima analisi, nella limitazione dell'autoimmunità, che è presente a bassi livelli nel ceppo originario MRL (v. Lynch e altri, 1995).
È interessante sottolineare che la funzione del recettore Fas è nota: dopo stimolazione, normalmente in seguito all'interazione con la Fas-L, la molecola Fas determina l'inizio di una serie di eventi biochimici intracellulari che culminano con la frammentazione del DNA e la morte cellulare per apoptosi. Sia la molecola Fas che quella Fas-L sono espresse, simultaneamente, anche dai linfociti T dopo ripetuta e prolungata attivazione (è da notare che Fas-L è espressa principalmente dalle cellule T CD8 +), e pertanto la morte per apoptosi dovuta all'interazione Fas/Fas-L colpisce prevalentemente i cloni di linfociti T cronicamente attivati. Evidentemente questa condizione è idonea nella tolleranza periferica e/o nell'immunosoppressione. Tuttavia, è interessante rilevare che la carenza del sistema Fas/Fas-L non interferisce sulla selezione negativa nel timo (v. Surh e Sprent, 1994; v. Lynch e altri, 1995; v. Kotzin, 1996).
b) L'artrite cronica sperimentale nel ratto e nel topo
L'immunizzazione di alcuni ceppi di ratti o topi con collagene di tipo II (il tipo prevalente nelle cartilagini), senza aggiunta di adiuvanti, determina in breve tempo una poliartrite che si accompagna a produzione di anticorpi anti-collagene. La suscettibilità è legata ai geni degli antigeni di istocompatibilità; nel topo, i ceppi di aplotipo H-2q sono particolarmente reattivi, e la reattività è legata in particolare ai geni della regione I-A, che controllano molecole MHC di classe II. Evidentemente, è importante la capacità di determinate molecole MHC di classe II di presentare alle cellule T CD4 + peptidi derivati dal collagene.
Un altro tipo di artrite può essere indotto in ratti di ceppo Lewis mediante una singola iniezione di adiuvante di Freund completo (micobatteri emulsionati in olio e acqua) alla base della coda. La poliartrite che si sviluppa entro un paio di settimane è caratterizzata da granulomi contenenti linfociti e macrofagi, e si risolve spontaneamente entro 30-40 giorni. L'autoimmunità è dovuta esclusivamente a linfociti T, e la malattia può essere trasferita passivamente con queste cellule. Una linea di cellule T, chiamata linea A, ha generato due cloni (A2b e A2c) capaci di trasferire passivamente l'artrite nei ratti Lewis; è stata accertata la specificità dei due cloni, che riconoscono specificamente il peptide comprendente gli amminoacidi 180-188 della proteina HSP65. Questa proteina, del gruppo delle proteine da stress calorico (heat-shock), è molto conservata in natura e probabilmente i linfociti che reagiscono con la HSP65 dei micobatteri presentano una reazione incrociata con un'analoga proteina dei Mammiferi. Per questo motivo è probabile che la proteina HSP65, e altre del gruppo HSP, siano implicate in diversi processi autoimmuni nei quali la rottura della tolleranza è legata alla somiglianza tra proteine batteriche e proteine dei Mammiferi.
È interessante anche rilevare che il clone A2b, se inattivato per radiazione o trattamento con agenti chimici, può essere usato per rendere i ratti Lewis resistenti all'artrite da adiuvante di Freund, se iniettato un mese prima della immunizzazione. Questo processo di ‛vaccinazione' con cellule T che, come vedremo, è possibile anche nell'encefalomielite allergica sperimentale, ha destato molto interesse sia per le sue possibilità pratiche che per il suo significato teorico. Il meccanismo d'azione di questa vaccinazione consiste nell'induzione di cellule T soppressive che riconoscono specificamente il clone usato per la vaccinazione e, per estensione, i cloni artritogenici indotti dal trattamento con adiuvante di Freund (v. Feldmann e altri, 1996).
c) Il diabete autoimmune nel topo
Il diabete autoimmune che si sviluppa spontaneamente nei topi di ceppo NOD (Non-Obesi-Diabetici) verrà discusso più in dettaglio, sia perché costituisce un modello per una grave e diffusa malattia umana, sia perché è stato oggetto di approfonditi studi genetici e immunologici che chiariscono ampiamente, anche se ancora non completamente, le basi sulle quali si fonda e si sviluppa l'autoimmunità.
Il ceppo di topi NOD è stato prodotto casualmente in Giappone, in un allevamento di topi capaci di sviluppare una cataratta spontanea. I topi NOD costituiscono un ceppo di animali isogeni (inbred; v. farmacologia e sperimentazione animale) nei quali si sviluppa, senza che siano necessari procedimenti di immunizzazione sperimentale, una reazione delle cellule T diretta contro le isole di Langerhans del pancreas. Queste strutture mostrano, fra il primo e il terzo mese di vita, una infiltrazione diffusa di linfociti T, sia CD4 che CD8, e anche di linfociti B, cellule dendritiche e macrofagi. Si tratta di un processo autoimmune, definito ‛insulite', che nei mesi successivi evolve con la progressiva distruzione delle cellule insulari β, che producono insulina, alla quale consegue un diabete per cui gli animali possono essere mantenuti in vita solo con la somministrazione di insulina.
Le diverse sottopopolazioni di cellule T hanno un ruolo complesso nella genesi dell'insulite e del successivo diabete, e rivelano capacità differenti, sia nella alterata immunoregolazione, sia nella funzione finale di distruzione delle cellule β produttrici di insulina. Le cellule CD8 appaiono essenziali perché in loro assenza non si manifestano né insulite né diabete, come è possibile osservare nei topi NOD, nei quali il gene per la β-2 microglobulina è stato cancellato e che sono quindi privi sia di molecole MHC di classe I sia di linfociti CD8.
Le cellule CD4 hanno un ruolo ambivalente: infatti, mentre cloni di cellule CD4 con recettori specifici capaci di reagire con antigeni insulari possono indurre insulite e diabete nei topi NOD in età prediabetica, in animali della stessa età esistono anche linfociti CD4 con funzione regolatrice, in grado di prevenire l'effetto patogeno passivo di cellule T prelevate dalla milza di topi NOD con malattia conclamata. È possibile che le cellule CD4 diabetogene appartengano alla sottopopolazione Th1 (che secerne interferone γ e altre linfochine pro-infiammatorie), mentre le cellule CD4 protettive sembrano possedere il fenotipo della sottopopolazione Th2. In conclusione, nei topi del ceppo NOD lo sviluppo dell'autoimmunità contro le isole di Langerhans sembra essere sotto il controllo di cellule T regolatrici, sia CD4 che CD8, che influenzano lo sviluppo e la funzione delle cellule T attive nel determinare le lesioni tissutali; queste ultime, a loro volta, comprendono sia cellule CD4, in grado di determinare il danno tramite le linfochine che producono e l'attivazione dei macrofagi, sia cellule CD8 citotossiche, che uccidono direttamente le cellule insulari β riconoscendo nelle loro membrane peptidi derivati da autoantigeni diversi, presentati dalle molecole MHC di classe I.
La complessità degli eventi autoimmuni che si sviluppano nelle isole di Langerhans dei topi NOD, tramite popolazioni diverse di cellule T, può essere seguita anche analizzando la specificità per gli antigeni dimostrata dai recettori che si trovano sulle membrane di queste cellule, in particolare delle cellule Th1. L'attacco autoimmune inizia invariabilmente con cellule provviste di recettori per due peptidi (il 509-528 e il 524-543) appartenenti a una proteina (la decarbossilasi dell'acido glutammico, GAD) delle cellule β produttrici di insulina; successivamente compaiono cellule Th1 che riconoscono altri peptidi della GAD e poi peptidi di altre proteine insulari. È molto interessante il fatto che nei topi NOD è possibile rinforzare la tolleranza periferica alla GAD mediante iniezioni endovenose di tale proteina effettuate nelle prime 3 settimane di vita; con questo trattamento, anzi, si blocca non solo la risposta contro la GAD, ma l'intero processo autoimmune che conduce al diabete, tanto da far ritenere che nei topi NOD le cellule T anti-GAD costituiscano il ‛tallone di Achille' del sistema immunitario. La stessa condizione si verifica nell'uomo, in cui la GAD costituisce l'antigene cardinale del processo autoimmune che conduce al diabete insulino-dipendente di tipo I. Le ragioni di questa peculiare labilità della tolleranza verso la stessa proteina in due specie separate da più di venti milioni di anni di evoluzione non sono note, ma è comunque evidente che il diabete autoimmune dei topi NOD costituisce un valido modello del diabete umano di tipo I.
Come in altre condizioni (encefalite allergica sperimentale), le cellule Th2, provviste dello stesso recettore per gli antigeni insulari presente nelle cellule Th1, non sono patogene, cioè diabetogene, ma hanno invece un effetto protettivo, probabilmente in conseguenza del diverso spettro delle linfochine che producono nel tessuto dove si localizzano e incontrano il loro antigene, cioè le stesse isole di Langerhans. È interessante notare che le cellule Th2 sono quei linfociti T coinvolti nella cooperazione con i linfociti B per la produzione degli anticorpi; pertanto, la produzione di autoanticorpi anti-GAD, indice della presenza di cellule Th2 anti-GAD, non appare un elemento negativo nel diabete umano di tipo I, anzi è correlata con una prognosi migliore.
Possiamo adesso prendere in esame gli eventi cellulari che si verificano nelle isole pancreatiche dei topi NOD. All'inizio compaiono cellule monoclonali Th1 con specificità anti-GAD e appartenenti alla famiglia di cellule T che esprimono la catena Vβ8 del recettore per gli antigeni; il differenziamento dei membri di questo clone nella direzione Th2 è, almeno inizialmente, limitato. Successivamente, si aggiungono cellule Th con altre specificità e appartenenti ad altre famiglie Vβ, e compaiono anche cellule TCD8 +, cellule B e macrofagi: insomma, un microcosmo immunologico del quale rimane da studiare la specificità e il ruolo di ogni singolo componente nel determinare il risultato finale, cioè la distruzione delle cellule β produttrici di insulina e quindi il diabete.
Una malattia autoimmune interessante come il diabete dei topi NOD ha giustificato un'approfondita analisi genetica. Si è così accertato, grazie ai contributi di vari laboratori, che esistono almeno 14 loci, distribuiti nei vari cromosomi, che contribuiscono in varia misura a determinare la tendenza al diabete spontaneo autoimmune. Un fattore genetico essenziale per la comparsa del diabete è stato individuato nella regione dei geni per la classe II degli antigeni di istocompatibilità, nota nel suo complesso con il nome di Idd-1, la quale presenta almeno due caratteristiche: l'assenza del locus I-E e la presenza nel locus I-A di un allele chiamato I-Ag7. La catena β di questo raro allele contiene un acido aspartico in posizione 57, sucettibile di stabilire un ponte salino con una arginina in posizione 76 della catena α della molecola MHC. È probabile che, nello sviluppo del diabete del topo, questa caratteristica delle molecole di istocompatibilità sia necessaria per legare i peptidi dell'autoantigene dominante (GAD). È molto interessante il fatto che le stesse peculiarità chimiche si ritrovano nell'uomo nella catena β dell'antigene DQ (HLA classe II) che è associato al diabete autoimmune; è questa un'ulteriore conferma della validità del modello dei topi NOD per gli studi sul diabete umano (v. Wucherpfennig e Strominger, 1995; v. Vyse e Todd, 1996).
D'altro canto, i geni diabetogeni inclusi nelle zone da Idd-2 fino a Idd-14 (distribuiti su cromosomi diversi), benché localizzati in queste aree mediante l'analisi di microsatelliti, non sono ancora stati identificati con precisione, per cui non conosciamo ancora le proteine da essi codificate. Quando queste saranno identificate, sapremo indubbiamente molto di più sulla funzione e sulla regolazione dei linfociti T, sia in condizioni normali, sia in presenza di difetti che li predispongono all'autoimmunità. È chiaro che le molecole implicate sono numerose, ed è anche probabile che diverse loro interazioni siano alla base degli errori dei linfociti T che conducono all'autoimmunità. Con questa base genetica non è sorprendente che i topi NOD siano esposti, sia pure con minore frequenza, anche ad altre affezioni autoimmuni interessanti, come per esempio quelle delle ghiandole salivari e lacrimali, della tiroide, delle paratiroidi, della corteccia surrenale e dei testicoli.
In conclusione, i modelli sperimentali nei topi NOD dimostrano che il confine tra il normale funzionamento del sistema immunitario e l'autoimmunità è complesso e, al momento attuale, ancora poco noto. Tuttavia, il progresso delle nostre conoscenze di genetica, biologia molecolare e cellulare, che in questi ultimi anni è divenuto sempre più rapido, fa ben sperare per la soluzione dei problemi legati a una patologia umana così grave e frequente (v. Solimena e altri, 1988; v. Burkly e altri, 1990; v. Solimena e De Camilli, 1991; v. Kaufman e altri, 1993; v. Katz e altri, 1995; v. Wicker e altri, 1995; v. Tisch e McDevitt, 1996; v. Yang e altri, 1996).
d) L'encefalomielite allergica sperimentale
Questa malattia è inducibile sperimentalmente in diverse specie mediante immunizzazione con omogenati di tessuto nervoso o con una proteina della mielina, la proteina basica (BMP). Si tratta di un modello sperimentale di autoimmunità che è stato molto studiato sia perché è facilmente riproducibile, sia perché è considerato un valido paradigma della sclerosi multipla umana (v. anche neuropatologia: Ricerche immunologiche sulla demielinizzazione sperimentale).
Ci occuperemo in modo particolare dell'encefalomielite allergica sperimentale (EAS) inducibile nel topo, perché meglio conosciuta nei suoi parametri immunologici. La sindrome è inducibile con regolarità nei topi adulti (oltre 30 giorni di vita) dei ceppi B10.PL e SJL. L'immunizzazione più semplice è quella che si ottiene iniettando il peptide 1-9 NH2-terminale della proteina basica della mielina, acetilato nel suo gruppo amminico (sequenza Acetyl-Ala-Ser-Gln-Lys-Arg-Pro-Ser-Gln-Arg); questo peptide è legato dalle molecole MHC di classe II dei topi B10.PL (H-2u) ed è presentato alle cellule T CD4. Nei topi del ceppo SJL/J (H-2s) il peptide dominante corrisponde agli amminoacidi 81-104 della regione COOH-terminale della BMP. L'immunizzazione viene effettuata con l'inoculazione di peptidi emulsionati in adiuvante completo di Freund, seguita da due iniezioni di tossina della pertosse, che probabilmente ha il ruolo di permeabilizzare la barriera ematoencefalica. Entro un paio di settimane i topi mostrano segni di paralisi: prima la coda, poi gli arti posteriori, quindi gli arti anteriori. Qualche topo muore per paralisi respiratoria, ma il maggior numero recupera rapidamente in modo pressoché totale. Le ricadute spontanee sono rare nei topi B10.PL, più frequenti invece in quelli del ceppo SJL, nei quali la malattia può assumere un andamento con ricadute ricorrenti che ricorda in qualche modo la sclerosi multipla umana.
L'analisi istologica del sistema nervoso centrale, in particolare del midollo spinale, mostra infiltrazioni perivascolari di linfociti e macrofagi, con aree localizzate di demielinizzazione. I linfociti encefalopatogeni sono cellule CD4 + provviste di recettori (TCR) specifici per i peptidi della BMP presentati dalle molecole MHC di classe II che si trovano sulle membrane dei macrofagi o delle cellule della microglia, particolarmente nelle aree dove l'interferone γ prodotto dai linfociti stimola l'espressione di queste molecole da parte delle cellule suddette.
Le cellule fondamentali nell'induzione della encefalomielite sono dunque le cellule T CD4 + anti-BMP. È interessante notare che i recettori dei linfociti encefalitogeni utilizzano tutti catene β appartenenti al gruppo Vβ8, indipendentemente dalla loro reattività nei confronti di un peptide o di un altro della BMP. La catena α dei recettori è anch'essa ristretta, ma in modo meno rigido, potendo appartenere al gruppo Vα2 oppure, in altri cloni di cellule T, al gruppo Vα4. La restrizione all'uso di catene β della famiglia Vβ8 sembra dunque essere una caratteristica dei linfociti autoimmuni delle EAS, e si ritrova anche in altre malattie autoimmuni del topo. Ripetiamo che questa restrizione è indipendente dalla specificità per gli antigeni; sembra piuttosto che i linfociti T CD4 che hanno recettori la cui catena β appartiene al gruppo Vβ8 siano particolarmente predisposti all'autoimmunità, e cioè alla perdita della tolleranza, probabilmente per motivi inerenti alla loro regolazione.
Oltre ai linfociti T CD4, nelle lesioni si rinvengono anche linfociti T CD8, il cui ruolo è meno chiaro. Alcuni cloni CD8 possono avere un'azione patogena per la loro potenzialità citotossica nei confronti di cellule bersaglio che presentano autoantigeni con le molecole MHC di classe I; altri cloni CD8, al contrario, possono avere un ruolo protettivo per la loro capacità di inibire o sopprimere l'azione dei CD4 encefalopatogeni. Infatti, i topi nei quali il gene per la molecola CD8 è stato escluso (cosiddetto knock-out genetico) sono maggiormente soggetti alle ricadute dell'EAS (v. Jiang e altri, 1992).
Nell'ambito dei cloni specifici per la BMP predominano inizialmente le cellule differenziate nella direzione Th1; queste, trasferite passivamente nei topi singenici, sono capaci di indurre l'EAS. Successivamente compaiono le cellule differenziate nella direzione Th2, che hanno un effetto opposto e inibiscono l'attività encefalitogena delle loro sorelle clonali dotate di fenotipo Th1. Degno di rilievo è il fatto che le cellule Th2, produttrici di IL-4, sono le tipiche cellule che spingono i linfociti B alla produzione di anticorpi; nel corso dell'EAS sono stati evidenziati autoanticorpi diretti contro le BMP e contro altri antigeni del SNC, dei quali però non è stato identificato un ruolo patogeno.
In topi transgenici sono state effettuate interessanti osservazioni sul ruolo patogeno delle cellule T dotate di recettori per il peptide 1-11 della BMP. Linee di topi transgenici per la catena β di tali recettori sono state fatte accoppiare con linee di animali transgenici per la catena α. I transgenici così ottenuti sono stati quindi introdotti per back cross (reincrocio) in topi B10.PL (H-2u), col risultato di ottenere topi le cui cellule potevano presentare il peptide 1-11 delle BMP e il cui sistema linfatico includeva una larga maggioranza di cellule CD4 con recettore per l'autoantigene. Una parte di questi topi sviluppava spontaneamente (cioè senza necessità di immunizzazione) la sindrome EAS, ma solo nel caso che gli animali fossero mantenuti in ambienti convenzionali; i topi mantenuti in ambienti sterili invece non si ammalavano. Questa è una dimostrazione del ruolo dei fattori ambientali (infezioni incidentali) nello sviluppo dell'EAS spontanea in topi geneticamente predisposti, e una possibile indicazione di un analogo ruolo ancillare delle infezioni nell'incidenza dell'analoga malattia umana (sclerosi multipla).
Un passo ulteriore è stato compiuto creando topi transgenici per le catene α e β del recettore encefalitogeno e provvisti di MHC di allotipo H-2u, come quelli sopra descritti, ma nei quali tutta la residua popolazione di cellule CD4 e CD8 dotate di recettori polimorfici, come pure la popolazione di cellule B, era stata eliminata mediante knock-out genetico dell'enzima RAG-1. Questo enzima è necessario per i riarrangiamenti dei geni delle catene TCR (come pure per quelli dei geni per le immunoglobuline) necessari per produrre, durante la maturazione delle cellule, recettori funzionali dotati di diversa specificità. In questi topi, quindi, mancavano le cellule B; inoltre, delle cellule T, che per il 100% esprimono il recettore encefalitogeno, il 98% erano CD4 e solo il 2% CD8. Questi topi progressivamente e spontaneamente venivano colpiti da EAS in una forma sempre letale, senza cioè che in alcuno di essi si verificasse la regressione spontanea che invece si osserva, come già detto, nella maggioranza dei topi normali nei quali la malattia è stata indotta per immunizzazione.
Questo esperimento è importante perché dimostra l'efficienza protettiva, nei confronti di una predominante popolazione di linfociti autoimmuni, di una minoranza di linfociti normali, che ovviamente sono capaci di una azione regolatrice. Questa funzione può essere ascritta alle cellule T CD8, delle quali è stato dimostrato l'effetto nell'inibizione dell'EAS in topi normali, oppure alle cellule CD4 le quali, mediante la produzione di IL-4, possono indurre la differenziazione verso il fenotipo Th2 delle cellule CD4 che esprimono il recettore transgenico.
In ogni caso, questi recenti lavori dimostrano la potenzialità dei nuovi metodi di ingegneria genetica per le analisi dei complessi meccanismi di interazione cellulare che sono in gioco nell'autoimmunità. Tali metodi, applicati allo studio di alcuni modelli fondamentali di malattie autoimmuni sperimentali, sono una promessa di progresso nella conoscenza della immunologia delle malattie umane (v. Sun e altri, 1988; v. Governman e altri, 1993; v. Lafaille e altri, 1994; v. Steinman, 1996).
BIBLIOGRAFIA
Alam, S. M., Travers, P. J., Wung, J. L., Nasholds, W., Redpath, S., Jameson, S. C., Gascoigne, N. R., T-cell-receptor affinity and thymocyte positive selection, in ‟Nature", 1996, CCCLXXXI, pp. 616-620.
Ashton-Rickardt, P. G., Tonegawa, S., A differential-avidity model for T-cell selection, in ‟Immunology today", 1994, XV, pp. 362-366.
Blackman, M., Kappler, J., Marrack, P., The role of the T cell receptor in positive and negative selection of developing T cells, in ‟Science", 1990, CCXLVIII, pp. 1335-1341.
Boehmer, H. von, Positive selection of lymphocytes, in ‟Cell", 1994, LXXVI, pp. 219-228.
Burkly, L. C., Lo, D., Flavell, R. A., Tolerance in transgenic mice expressing major histocompatibility molecules extrathymically on pancreatic cells, in ‟Science", 1990, CCXLVIII, pp. 1364-1368.
Cohen, I. R., Young, D. B., Autoimmunity, microbial immunity and the immunological homunculus, in ‟Immunology today", 1991, XII, 4, pp. 105-110.
Feldmann, M., Brennan, F. M., Maini, R. N., Rheumatoid arthritis, in ‟Cell", 1996, LXXXV, pp. 307-310.
Governman, J., Woods, A., Larson, L., Weiner, L. P., Hood, L., Zaller, D. M., Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity, in ‟Cell", 1993, LXXII, pp. 551-560.
Jiang, H., Zhang, S., Pernis, B., Role of CD8+ T cells in murine experimental allergic encephalomyelitis, in ‟Science", 1992, CCLVI, pp. 1213-1215.
Katz, J. D., Benoist, C., Mathis, D., T helper cells subsets in insulin-dependent diabetes, in ‟Science", 1995, CCLXVIII, pp. 1185-1188.
Kaufman, D. L, Clare-Salzler, M., Tian, J., Forsthuber, T., Ting, G. S. P., Robinson, P., Atkinson, M. A., Sercarz, E. E., Tobin, A. J., Lehmann, P. V., Spontaneous loss of T-cell tolerance to glutamic acid decarboxylase in murine insulin-dependent diabetes, in ‟Nature", 1993, CCCLXVI, pp. 69-72.
Koopman, G., Pals, S. T., Cellular interactions in the germinal center: role of adhesion receptors and significance for the pathogenesis of AIDS and malignant lymphoma, in ‟Immunological reviews", 1992, CXXVI, pp. 21-45.
Kotzin, B. L., Systemic lupus erythematosus, in ‟Cell", 1996, LXXXV, pp. 303-306.
Kuchroo, V. K., Das, M. P., Brown, J. A., Ranger, A. M., Zamvil, S. S., Sobel, R. A., Weiner, H. L., Nabavi, N., Glimcher, L. H., B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy, in ‟Cell", 1995, LXXX, pp. 707-718.
Lafaille, J. J., Nagashima, K., Katsuki, M., Tonegawa, S., High incidence of spontaneous autoimmune encephalomyelitis in immunodeficient anti-myelin basic protein T cell receptor transgenic mice, in ‟Cell", 1994, LXXVIII, pp. 399-408.
Laufer, T. M., DeKoning, J., Markowitz, J. S., Lo, D., Glimcher, L. H., Unopposed positive selection and autoreactivity in mice expressing class II MHC only on thymic cortex, in ‟Nature", 1996, CCCLXXXIII, pp. 81-85.
Lynch, D. H., Ramsdell, F., Alderson, M. R., Fas and FasL in the homeostatic regulation of immune responses, in ‟Immunology today", 1995, XVI, pp. 569-574.
Nossal, G. J. V., Negative selection of lymphocytes, in ‟Cell", 1994, LXXVI, pp. 229-239.
Schwartz, R. S., Datta, S. K., Autoimmunity and autoimmune diseases, in Fundamental immunology (a cura di W. E. Paul), New York 19892, pp. 820-866.
Sercarz, E., Krzych, U., The distinctive specificity of antigen-specific suppressor T cells, in ‟Immunology today", 1991, XII, pp. 111-118.
Sinha, A. A., Lopez, M. T., McDevitt, H. O., Autoimmune diseases: the failure of self tolerance, in ‟Science", 1990, CCXLVIII, pp. 1380-1388.
Solimena, M., De Camilli, P., Autoimmunity to glutamic acid decarboxylase (GAD) in stiff-man syndrome and insulin-dependent diabetes mellitus, in ‟Trends in neurosciences", 1991, XIV, 10, pp. 452-457.
Solimena, M., Folli, F., Denis-Donini, S., Comi, G. C., Pozza, G., De Camilli, P., Vicari, A. M., Autoantibodies to glutamic acid decarboxylase in a patient with stiff-man syndrome, epilepsy and type I diabetes mellitus, in ‟New England journal of medicine", 1988, CCCXVIII, pp. 1012-1020.
Steinman, L., Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system, in ‟Cell", 1996, LXXXV, pp. 299-302.
Sun, D., Ben-Nun, A., Wekerle, H., Regulatory circuits in autoimmunity: recruitment of counter-regulatory CD8+ T cells by encephalitogenic CD4+ T line cells, in ‟European journal of immunology", 1988, XVIII, pp. 1993-1999.
Surh, C. D., Sprent, J., T-cell apoptosis detected in situ during positive and negative selection in the thymus, in ‟Nature", 1994, CCCLXXII, pp. 100-103.
Tisch, R., McDevitt, H., Insulin-dependent diabetes mellitus, in ‟Cell", 1996, LXXXV, pp. 291-297.
Vyse, T. J., Todd, J. A., Genetic analysis of autoimmune disease, in ‟Cell", 1996, LXXXV, pp. 311-318.
Waterhouse, P., Penninger, J. M., Timms, E., Wakeham, A., Shahinian, A., Lee, K. P., Thompson, C. B., Griesser, H., Mak, T. W., Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4, in ‟Science", 1995, CCLXX, pp. 985-988.
Wicker, L. S., Todd, J. A., Peterson, L. B., Genetic control of autoimmune diabetes in the NOD mouse, in ‟Annual review of immunology", 1995, XIII, pp. 179-200.
Wucherpfennig, K. W., Strominger, J. L., Selective binding of self peptides to disease-associated major histocompatibility complex (MHC) molecules: a mechanism for MHC-linked susceptibility to human autoimmune diseases, in ‟Journal of experimental medicine", 1995, CLXXXI, pp. 1597-1601.
Yang, Y., Charlton, B., Shimada, A., DalCanto, R., Fathman, C. G., Monoclonal T cells identified in early NOD islet infiltrates, in ‟Immunity", 1996, IV, pp. 189-194.
Istocompatibilità di Rosa Sorrentino e Roberto Tosi
Sommario: 1. Introduzione. 2. Il complesso maggiore di istocompatibilità nell'uomo o sistema HLA: a) i geni HLA di classe I e II; b) funzione delle molecole HLA di classe I e II; c) polimorfismo dei geni HLA di classe I e II; d) differenze funzionali tra molecole HLA di classe I e II; e) biosintesi delle molecole HLA di classe I; f) biosintesi delle molecole HLA di classe II; g) i geni LMP; h) i geni nella regione HLA di classe III; i) i geni HLA di classe I ‛non classici'. 3. Protezione e autoimmunità: a) self e non-self; b) le malattie autoimmunitarie; c) l'equilibrio tra protezione e autoimmunità; d) il rigetto dei trapianti. 4. Conclusioni. □ Bibliografia.
1. Introduzione
Il rigetto di cellule o tessuti provenienti da altri individui della stessa specie rappresenta un fenomeno precoce nella filogenesi, a partire dalle Spugne e dai Tunicati. Negli animali superiori, le basi scientifiche per lo studio del rigetto dei trapianti sono state poste negli anni trenta da Peter Gorer (v., 1937) in Inghilterra e da George Snell negli Stati Uniti, i quali hanno scoperto che nel topo una singola regione genetica, da loro chiamata H-2, è responsabile del fenomeno del rigetto. Da allora, in tutte le specie analizzate è stata identificata una regione genetica simile, che è stata denominata con termine generale MHC (Major Histocompatibility Complex, complesso maggiore di istocompatibilità) e indicata in ogni singola specie con un nome specifico: H-2 nel topo, HLA nell'uomo, RLA nel coniglio, ecc. L'interesse per l'MHC, che fino agli anni settanta era concentrato sul suo ruolo nel rigetto dei trapianti, ha avuto una svolta in seguito alla scoperta, effettuata negli Stati Uniti da Benacerraf e McDevitt (v., 1972), della sua partecipazione alla regolazione della risposta immune. Da quel momento lo studio dell'istocompatibilità ha portato a un'analisi sempre più approfondita di questa regione genetica, che a sua volta si è rivelata fondamentale per la comprensione non solo del rigetto dei trapianti, ma anche di qualsiasi difesa di tipo immunitario. Pertanto, attraverso l'esame della struttura e della funzione dei geni che si trovano nel complesso maggiore di istocompatibilità umano potremo studiare gli intimi meccanismi che sono alla base di due fenomeni ugualmente importanti: quello del riconoscimento da parte dell'organismo di un aggressore esterno e quello del mancato riconoscimento delle strutture proprie dell'organismo che è in grado di innescare una autoaggressione. Su queste basi, vedremo che anche il rigetto dei trapianti, un fenomeno non ‛naturale' ma indotto da un particolare intervento, comincerà a divenire comprensibile, almeno nelle sue linee essenziali (v. anche chirurgia dei trapianti; v. immunologia e immunopatologia).
2. Il complesso maggiore di istocompatibilità nell'uomo o sistema HLA
La presente trattazione sarà limitata al complesso maggiore di istocompatibilità nell'uomo, cioè alla regione HLA. Questa è localizzata sul braccio corto del cromosoma 6, e precisamente nella sottoregione 6p21, nella quale occupa una lunghezza di circa 4 milioni di nucleotidi. Per dare un'idea più concreta, questa lunghezza corrisponde a circa un millesimo della lunghezza totale del genoma umano, cioè, in termini fisici, a circa un millimetro (essendo l'intero genoma lungo circa un metro). Il DNA contenuto nella regione HLA è quantitativamente equivalente a quello di un batterio come Escherichia coli, con la sostanziale differenza che, mentre quest'ultimo contiene circa 10.000 geni che codificano a loro volta 10.000 proteine diverse, la regione HLA contiene solo circa 200 geni: ciò è dovuto al fatto che il genoma degli Eucarioti, a differenza di quello dei Procarioti, contiene in misura preponderante sequenze non codificanti per proteine. La ‛densità' di geni nella regione HLA è di circa 1 ogni 20.000 nucleotidi, che corrisponde grosso modo alla media dell'intero genoma (v. Campbell e Trowsdale, 1993; v. Trowsdale, 1993 e 1995; v. anche genoma).
Una volta definita la fisionomia generale del sistema, passeremo ora all'esame più dettagliato dei geni presenti nella regione HLA e delle loro funzioni.
a) I geni HLA di classe I e II
Per la maggior parte, i geni contenuti nella regione HLA sono coinvolti in un modo o nell'altro nella difesa immunitaria.
I geni HLA ‛classici' sono suddivisi in due classi: I e II. I geni di classe I sono localizzati (cioè ‛mappano') nella parte telomerica della regione, chiamata ‛regione di classe I', quelli di classe II nella parte centromerica, o ‛regione di classe II', mentre la parte compresa fra le due viene chiamata ‛regione di classe III'.
I geni HLA di classe I ‛classici' sono tre: HLA-A, HLA-B e HLA-C. I loro prodotti si associano con una proteina codificata da un gene non-HLA, la β2-microglobulina. Quindi, in ogni individuo sono codificate tre molecole HLA di classe I, chiamate A, B e C. I geni HLA di classe II sono disposti a coppie di geni vicini, in ciascuna delle quali si distinguono un gene A, che controlla una catena proteica α, e un gene B, che controlla una catena β. La catena α e la catena β di ciascuna coppia si associano a formare eterodimeri (cioè proteine composte di due subunità diverse). Ciascun individuo esprime tre tipi di molecole di classe II - DP, DQ e DR - codificate da tre coppie di geni che sono, partendo dal centromero: HLA-DPB1 e DPA1, HLA-DQB1 e DQA1 e HLA-DRB1 e DRA (nella maggior parte degli individui viene inoltre espresso un secondo gene DRB che si associa col prodotto dello stesso gene DRA). Dobbiamo aggiungere che in questa regione, come nel resto del genoma, esistono anche parecchi geni che non sono espressi, in genere a causa di difetti nella loro struttura, e che vengono chiamati ‛pseudogeni'. Per esempio, nella regione di classe II sono pseudogeni DPA2 e DPB2, che non vengono espressi. Ne esistono vari altri nella regione di classe I, sui quali non ci soffermeremo.
Le molecole HLA di classe I e di classe II hanno molte caratteristiche in comune, che le rendono simili sia dal punto di vista strutturale che funzionale. Esse sono glicoproteine, cioè proteine legate covalentemente a catene di carboidrati, inserite nella membrana cellulare, dalla quale sporgono verso l'esterno con l'estremità N-terminale (questa è la configurazione più comune delle proteine integrali di membrana, delle quali solo una minoranza sporge all'esterno con l'estremità C-terminale). Sono composte di quattro ‛dominî' proteici, anche se con diversa distribuzione nei dimeri: nelle catene di classe I tre dominî appartengono alla catena α (α1, α2, α3) e uno è rappresentato dalla β2-microglobulina; nelle catene di classe II due dominî (α1, α2) appartengono alla catena α e due (β1, β2) alla catena β. La più importante caratteristica che esse hanno in comune è comunque quella di possedere, all'estremità esposta verso l'esterno della cellula, una struttura particolare formata da due tratti di sequenze di amminoacidi disposti con conformazione ad ‛α-elica' che delimitano una base formata da otto filamenti con conformazione a ‛nastri β'. L'insieme di questa struttura appare come una fenditura (groove) delimitata da ‛pareti' ad α elica e da un ‛fondo' di nastri β. Lo schema ‛a nastro' della struttura cristallografica mostra una visione frontale della molecola HLA di classe I (la molecola di classe II è sostanzialmnte simile, tranne che in dettagli importanti che prenderemo in considerazione in seguito). Nella parte alta della figura vi sono due parti con struttura ad α-elica; nell'insenatura delimitata dalle due eliche va a inserirsi il peptide antigenico (v. Bjorkman e altri, 1987; v. Brown e altri, 1993; v. Geraghty, 1993).
b) Funzione delle molecole HLA di classe I e II
La funzione della fenditura è quella di accogliere un peptide piuttosto corto, cioè una sequenza di pochi (da 8 a 30) amminoacidi che può derivare da organismi patogeni, come Batteri e Virus. In tal caso, il suo riconoscimento da parte delle molecole HLA è il primo, indispensabile passo perché si instauri una risposta difensiva di tipo immunitario. Si può addirittura affermare che una proteina ‛estranea' può diventare ‛antigene', cioè bersaglio di una risposta immunitaria, solo se darà origine a peptidi (per l'intervento di enzimi proteolitici: questo processo si chiama tecnicamente ‛processazione dell'antigene') e se questi saranno legati da molecole HLA. Vedremo più avanti in dettaglio come, in seguito al riconoscimento HLA-peptidi, si generi, attraverso gradini successivi, una risposta immunitaria specifica di tipo anticorpale o cellulare. Quello che vogliamo considerare per il momento è il seguente problema: in che modo un numero limitato di molecole HLA è in grado di riconoscere la grande varietà di peptidi che possono derivare dal gran numero di microrganismi patogeni che ci circondano? La risposta a questo quesito va ricercata nel grado elevato di flessibilità del legame HLA-peptidi. Nella fenditura della molecola HLA possono trovar posto peptidi con sequenze di amminoacidi diverse e anche, entro certi limiti, di lunghezze diverse, purché rispettino regole abbastanza precise relativamente al tipo di amminoacido presente in due o tre posizioni critiche, chiamate ‛posizioni-ancora': per esempio, per l'attacco di peptidi nonameri a molecole HLA di classe I sono importanti gli amminoacidi nella seconda e nell'ultima posizione del peptide, che vanno a inserirsi in ‛tasche' specifiche (peptide-binding pockets), disposte nella fenditura, nelle quali vengono trattenuti. Quindi, ogni molecola HLA sarà capace di legarsi con alta efficienza a una varietà di peptidi che abbiano nelle posizioni-ancora quei determinati amminoacidi. In un agente patogeno potrebbero però generarsi, per mutazione, varianti in cui questi amminoacidi in posizioni-ancora vengono sostituiti da altri che non consentono il legame alla stessa molecola HLA: ne potrebbero quindi risultare ceppi resistenti, capaci di sfuggire alla difesa immunitaria (v. Falk e altri, 1991; v. Garrett e altri, 1989; v. Stern e altri, 1994).
c) Polimorfismo dei geni HLA di classe I e II
Per rispondere a questa variabilità degli agenti patogeni, l'evoluzione ha selezionato, per ogni gene HLA, non una sola, ma diverse forme che coesistono nella nostra specie. In altre parole, i geni HLA hanno un elevato polimorfismo, anzi costituiscono il sistema più polimorfico nell'uomo. Appare evidente come ci sia un grado altissimo di polimorfismo, fino a circa 100 varianti per i geni HLA-B e -DRB1 e a un numero comunque molto elevato per la maggior parte di questi geni. Per confronto, si consideri che un polimorfismo classico, come quello che determina i gruppi sanguigni AB0, comprende solo quattro alleli. Le varianti alleliche dei geni HLA si diversificano per gruppi, o clusters, di amminoacidi nei dominî della molecola che delimitano la fenditura dove va a inserirsi il peptide, influenzando quindi direttamente la capacità e la specificità di legame dei peptidi con la molecola. Ne consegue che nella specie umana coesistono strutture capaci di riconoscere una grandissima varietà di peptidi; ciascun individuo, però, può possedere al massimo due diverse forme di ciascun gene, cioè una per ciascuno dei suoi due cromosomi 6: quindi, ogni individuo potrà avere al massimo sei diverse molecole di classe I (due al locus A, due al locus B e due al locus C) e sei-otto molecole di classe II (cioè due DP, due DQ e da due a quattro DR). A livello dell'intera specie, questa grande potenzialità di riconoscimento di diversi peptidi potrà esplicarsi al massimo grado: ciò può rappresentare un fattore importante per favorire in ogni caso la sopravvivenza della specie, anche durante epidemie devastanti (v. Hammer e altri, 1993; v. Klein e altri, 1993; v. Kronenberg e altri, 1994).
d) Differenze funzionali tra molecole HLA di classe I e II
Sono ora da prendere in considerazione i meccanismi attraverso i quali si passa dal riconoscimento di un peptide all'avvio di una risposta immunitaria contro di esso. L'incontro tra HLA e peptide avviene all'interno della cellula e il complesso HLA-peptide raggiunge la membrana cellulare, dove viene ‛presentato' per essere riconosciuto da altri componenti del sistema immunitario. Analizzeremo quindi uno per uno gli eventi intracellulari che portano alla ‛presentazione antigenica'. A questo punto i due tipi di molecole HLA di classe I e di classe II, che finora abbiamo considerato insieme perché con struttura simile ed entrambi dotati della capacità di legare peptidi, dovranno essere studiati separatamente nella loro diversa specializzazione.
Una differenza sostanziale tra i due tipi di molecole consiste nel comparto cellulare in cui ha luogo il loro incontro coi peptidi: ciò avviene nel lume del reticolo endoplasmatico nel caso delle molecole di classe I, e in un comparto endosomico specializzato per quelle di classe II; questa differenza ha importanti conseguenze sulla funzione. Infatti, i peptidi che giungono, o piuttosto vengono trasportati, nel reticolo endoplasmatico, sono quelli che si generano nel citoplasma: è questo il caso, per esempio, dei peptidi che derivano dalla ‛processazione' di proteine virali che può avvenire durante il ciclo riproduttivo intracellulare del virus. Invece i peptidi che giungono al comparto endosomiale provengono da proteine ‛internalizzate' dalla cellula per endocitosi e poi ‛processate' da enzimi proteolitici lisosomiali: sono quindi prevalentemente di derivazione batterica, in quanto i Batteri hanno in genere un ciclo riproduttivo che si svolge all'esterno della cellula. Questa specializzazione funzionale, rispettivamente antivirale e antibatterica, delle molecole HLA offre una spiegazione logica, in chiave fisiologica, della diversa distribuzione dei due tipi di molecole nei diversi tipi di cellule dell'organismo: infatti, le molecole di classe I sono presenti in tutte le cellule nucleate (con l'eccezione quindi dei globuli rossi), che sono tutte potenziali bersagli di una infezione virale, mentre le molecole di classe II sono presenti in un numero ristretto di cellule, specializzate nella presentazione di peptidi di proteine esterne e nella induzione di una risposta anticorpale contro di esse. Queste cellule ‛professioniste' della presentazione antigenica sono soprattutto i macrofagi, le cellule dendritiche, i linfociti di tipo B. Cellule di altri tipi possono essere indotte a esprimere le stesse molecole in condizioni di emergenza, per esempio nel corso di infezioni, sotto l'influenza di molecole stimolatorie specifiche, come l'interferone di tipo gamma.
e) Biosintesi delle molecole HLA di classe I
Considerando in dettaglio le molecole di classe I, la catena α nascente viene immessa nel lume del reticolo endoplasmatico e qui si associa con una molecola chaperon chiamata calnexina, e poi con la β2-microglobulina. Questo complesso si lega poi alla proteina di trasporto TAP (Transporter Associated with Antigen Processing, trasportatore associato con la processazione dell'antigene) che è inserita nella membrana endoplasmatica ed è deputata al trasporto di peptidi dal citoplasma all'interno del reticolo endoplasmatico. Questa proteina è composta da due subunità, TAP1 e TAP2, controllate da due geni molto vicini, TAP1 e TAP2, situati nella regione HLA di classe II. Ogni subunità comprende una porzione che attraversa più volte la membrana in modo da formarvi un poro o canale per il passaggio di peptidi. Comprende inoltre un dominio che lega ATP (adenosintrifosfato), e ciò dimostra che il processo richiede energia e quindi la natura attiva e specifica del trasporto dei peptidi. Se il peptide trasportato possiede le caratteristiche strutturali richieste dalla particolare molecola HLA presente, esso verrà immediatamente captato dal complesso catena α-β2-microglobulina all'interno del reticolo endoplasmatico. Ne consegue il passaggio del complesso HLA-peptide nell'apparato di Golgi, dove la proteina verrà glicosilata e potrà così essere trasferita sulla membrana cellulare. Il peptide legato alla molecola HLA è ‛presentato' a una sottopopolazione di linfociti di tipo T funzionalmente differenziati, i linfociti CD8. Questi possiedono innanzitutto il recettore capace di riconoscere il complesso HLA-peptidi, chiamato TCR (T-Cell Receptor; v. anche immunologia); inoltre possiedono una molecola accessoria, CD8, che si lega specificamente a una parte della molecola di classe I diversa da quella che forma la fenditura per il legame col peptide, e che è non polimorfica, cioè è uguale in tutti gli individui. Inoltre, queste cellule possiedono un complesso di molecole effettrici (perforina, granzima A e B) che, in seguito all'interazione cellulare, producono la lisi della cellula presentante il peptide. Perciò questi linfociti vengono chiamati linfociti T citotossici (CTL, Cytotoxic T Lymphocytes). La conseguenza di tutto questo processo è la distruzione della cellula eventualmente infettata dal virus. Il vantaggio di questo ‛sacrificio' per l'individuo è che questa distruzione può avvenire a stadi precoci dell'infezione, prima che il virus abbia avuto il tempo di moltiplicarsi e di invadere molte altre cellule (v. Sette e altri, 1987; v. Powis e altri, 1993).
f) Biosintesi delle molecole HLA di classe II
Le molecole di classe II hanno una maturazione intracellulare molto simile a quella delle molecole di classe I, tranne che per un aspetto molto importante; infatti, subito dopo essere state immesse, appena sintetizzate, nel lume del reticolo endoplasmatico, esse si associano a una proteina chiamata ‛catena invariante' o ‛Ii'. Una parte di questa proteina, legandosi alla regione della molecola deputata all'interazione con il peptide, impedisce di fatto che peptidi di derivazione ‛endogena', cioè provenienti dal citoplasma, possano introdursi nel solco formato dalle molecole di classe II. La catena invariante ha anche una funzione di trasporto, favorendo il progredire delle molecole di classe II nell'apparato di Golgi e successivamente nelle vescicole endosomiali: è qui che le molecole di classe II ‛incontrano' i peptidi provenienti dalla processazione di proteine esogene. Tuttavia, perché i peptidi possano legarsi deve essere prima rimossa la catena invariante, che viene parzialmente degradata dagli enzimi lisosomiali; ne rimane legato alle molecole di classe II un frammento chiamato CLIP (CLass II-associated Invariant chain Peptides, peptidi invarianti associati alla classe II) che occupa la fenditura della molecola e a sua volta viene rimosso, secondo l'ipotesi più accettata, per opera di un'altra molecola, cioè un eterodimero composto da due catene, DMA e DMB, anch'esso controllato da geni nella regione HLA di classe II . Se mancano le molecole DM, ad esempio per una delezione dei geni corrispondenti, le molecole di classe II mantengono il loro legame a CLIP e non possono legarsi ad altri peptidi. La molecola di classe II a cui si legano i peptidi con le caratteristiche strutturali richieste forma un complesso che procederà verso la superficie cellulare. Anche in questo caso, è una sottopopolazione di linfociti T a riconoscere, attraverso il TCR, il complesso HLA di classe II-peptide. Questa sottopopolazione esprime sulla superficie un marcatore chiamato CD4, che si lega specificamente a una parte della molecola di classe II diversa da quella che forma la fenditura per il legame col peptide, e che è non polimorfica. Questa sottopopolazione è definita helper in quanto, in seguito al riconoscimento del complesso specifico, è indotta a produrre fattori solubili, le citochine, che hanno il compito, tra gli altri, di indurre la proliferazione e il differenziamento di un'altra sottopopolazione di linfociti, i linfociti B. Sono questi i linfociti che, differenziandosi ulteriormente, producono e secernono gli anticorpi, cioè molecole capaci di legare e neutralizzare eventuali agenti patogeni. È chiaro che le due risposte - quella che abbiamo visto in precedenza, dipendente dalla presentazione di peptidi da parte di molecole HLA di classe I e mediata dai linfociti citotossici (CTL), e quella anticorpale, mediata da molecole HLA di classe II e linfociti T helper - hanno caratteristiche diverse: la prima, pur portando alla distruzione delle cellule dell'organismo nelle quali un virus si sta replicando, ha il vantaggio di essere rapida, ostacolando così il diffondersi del patogeno; la risposta anticorpale è invece rivolta direttamente contro il microrganismo, ma ha lo svantaggio di esplicarsi in tempi più lunghi, perché richiede stimolazione, proliferazione e differenziamento cellulare, che sono processi relativamente lenti rispetto ai tempi di riproduzione dei microrganismi (v. Lanzavecchia, 1985; v. Fling e altri, 1994).
g) I geni LMP
Nella descrizione del funzionamento delle molecole di classe I e II all'interno della cellula, abbiamo visto come altre molecole siano necessarie perché abbia luogo il processo di presentazione di peptidi. Alcune di queste molecole - per esempio, la catena invariante, la calnexina e anche la β-2-microglobulina, che abbiamo visto associarsi alla catena α per formare le molecole di classe I - sono controllate da geni localizzati in altre parti del genoma; altre, invece, fra le quali abbiamo finora considerato le molecole trasportatrici di peptidi (molecole TAP) e le molecole DM, sono codificate da geni nella regione HLA. Nella regione HLA di classe II si trovano altri due geni, i geni LMP, anch'essi coinvolti in una fase della presentazione non ancora considerata in dettaglio, cioè la processazione dell'antigene. Le proteine intracitoplasmatiche vengono catabolizzate per l'azione di enzimi proteolitici specifici che fanno parte di un complesso specializzato, chiamato proteasoma, molto abbondante nel citoplasma della cellula, dove rappresenta circa l'1% di tutte le proteine solubili; tale complesso è costituito da 14-16 subunità disposte a formare una struttura cilindrica con attività proteolitica, con sequenze amminoacidiche simili tra di loro. Due geni della regione HLA di classe II, LMP2 e LMP7, controllano due di queste subunità, le quali sono in realtà intercambiabili con altre due subunità chiamate in modo diverso da diversi autori (X e Y, oppure subunità 2 e subunità 10), controllate da geni localizzati altrove nel genoma ma con sequenza amminoacidica molto simile. In presenza di γ-interferone che, come abbiamo visto prima, può essere prodotto nel corso di infezioni, l'espressione dei due geni LMP2 e LMP7 aumenta e queste subunità vanno a sostituire le subunità X e Y nel proteasoma . Benché il sistema non sia stato ancora interamente analizzato, è probabile che questo rappresenti un meccanismo con cui la processazione di proteine viene convogliata specificamente verso la produzione di peptidi riconoscibili da parte di molecole HLA. Si stabilirebbe quindi un circuito di stimolazione per il quale a una infezione seguirebbe la produzione di interferone; ciò aumenterebbe sia la quantità di molecole HLA espresse, sia, in modo coordinato, la quantità delle altre molecole necessarie per la processazione, il trasporto e la presentazione antigenica. A questa consegue una maggiore stimolazione di cellule immunocompetenti e una produzione di linfochine che a loro volta amplificano la risposta immunitaria.
h) I geni nella regione HLA di classe III
Passando alla regione di classe III , incontriamo altri geni coinvolti nell'immunità. Essi sono i geni per diversi fattori del complemento - C4, fattore B e C2 - coinvolti quindi sia nella via classica che in quella alternativa dell'attivazione del complemento. Un altro gruppo di geni interessanti è quello dei tre geni omologhi HSP70-1, HSP70-2 e HSP70-HOM, che codificano per le proteine HSP70 (Heat Shock Proteins 70), la cui funzione è quella di fare da chaperon ad altre proteine - o durante la loro sintesi, o quando la loro struttura venga alterata da stress termico o di altra natura (si chiamano infatti, più propriamente, ‛proteine stress') - e quindi apparentemente non è direttamente connessa con la funzione immunitaria. Tuttavia, la loro presenza in questa regione (anche se esistono altri geni della stessa famiglia in altre parti del genoma) potrebbe essere il ‛ricordo' del loro contributo come precursori, nella filogenesi, dei geni HLA classici. È stata infatti avanzata l'ipotesi che i dominî HLA che formano la fenditura per il legame peptidico siano derivati da geni ancestrali HSP: i dati di fatto a favore di questa ipotesi sono che i geni HSP sono molto ‛antichi' e si sono mantenuti con poche variazioni dai Batteri fino all'uomo, che la loro struttura potrebbe essere simile a quella tipica delle molecole HLA (benché immagini cristallografiche non siano disponibili) e che queste molecole sono in grado di legare dei tratti di proteina denaturati, o anche singoli peptidi.
Infine, ancora nella regione di classe III incontriamo due geni estremamente importanti per le reazioni infiammatorie in genere, oltre che per la risposta immunitaria, cioè i geni TNF (Tumor Necrosis Factor) α, β e LTB. Questi geni codificano per citochine che sono state dapprima descritte come fattori citotossici per i tumori, ma la cui funzione si esplica su molti tipi cellulari. Anche queste citochine, come l'interferone gamma, vengono prodotte nel corso di un processo infiammatorio e regolano la risposta immunitaria.
i) I geni HLA di classe I ‛non classici'
Nella regione di classe I troviamo, oltre ai geni ‛classici' di classe I (HLA-B, HLA-C e HLA-A), di cui abbiamo già parlato, altri geni di classe I, come HLA-E, HLA-G, ecc., sulla cui specifica funzione si sta ancora indagando; essi sono strutturalmente simili ai primi, ma sono non polimorfici e hanno una distribuzione tissutale diversa dai geni di classe I classici. Molto recentemente sono stati descritti altri geni, i geni MIC (MHC Class I Chain related), anche questi strutturalmente simili a geni HLA classici, ma con espressione ristretta soltanto ad alcuni tipi cellulari e della cui funzione non si sa ancora nulla.
Risulta sempre più evidente, man mano che si passano in rassegna i geni finora caratterizzati, che la regione HLA ha le caratteristiche di una unità funzionale oltre che fisica, con qualche analogia con gli operoni batterici. Possiamo sostanzialmente considerare questa regione come il ‛ministero degli esteri' della cellula, dove sono concentrati molti dei geni che controllano i meccanismi iniziali della risposta immunitaria e dove vengono prese le decisioni sui tipi di risposta.
Non vogliamo tuttavia dare l'impressione che nella regione siano presenti solamente geni legati alla funzione immunitaria. Vari geni codificano enzimi - per esempio 21-idrossilasi, valil-tRNA-sintetasi - altri codificano componenti del collageno, che probabilmente non hanno alcuna connessione con il sistema immunitario, e molti altri ancora hanno funzioni sconosciute. Inoltre, particolarmente nella regione di classe I, altri geni, probabilmente non molti, potranno essere identificati in futuro, dato che la regione HLA è quella più intensamente analizzata di tutto il genoma umano. Possiamo prevedere che il numero totale di geni della regione sia al massimo di 200, in base a un rapporto di un gene ogni 20.000 nucleotidi, che può considerarsi il limite superiore. Finora ne sono stati identificati almeno la metà. È possibile che lo studio di questa regione, cominciato tanto tempo fa con la scoperta della natura genetica del rigetto dei trapianti e progredito sempre più velocemente grazie alle possibilità aperte dalla rivoluzione tecnologica, riservi ulteriori sorprese nei prossimi anni.
3. Protezione e autoimmunità
a) Self e non-self
Avendo descritto la struttura e la funzione delle molecole HLA, viene spontanea la domanda: come fanno le molecole HLA a discriminare i peptidi che provengono dai microrganismi (proteine non-self) da quelli che provengono dal normale catabolismo delle proteine endogene (proteine self)? Le molecole HLA di per sé non sono in grado di operare questa distinzione. Esse legheranno tutti quei peptidi presenti nel reticolo endoplasmatico (nel caso della classe I) o nel compartimento endosomiale (nel caso della classe II) che soddisfano le regole imposte dalla struttura della fenditura, indipendentemente dal fatto che siano di origine virale o provengano dalle proteine self. La discriminazione del self dal non-self, che è una condizione necessaria perché non ci sia una dannosa reattività verso i propri tessuti, è operata attraverso un processo chiamato ‛tolleranza immunitaria'. Durante la vita fetale, i linfociti T, che originano nel midollo osseo, vengono trasportati nel timo, dove maturano per diventare cellule immunocompetenti, cioè capaci di fornire una risposta immunitaria. La maturazione di queste cellule prevede l'acquisizione di proteine di superficie come CD8 o CD4, che permetteranno loro di riconoscere rispettivamente le molecole HLA di classe I e di classe II, e di altre molecole coinvolte nel riconoscimento delle cellule che presentano l'antigene. Nel timo avviene un doppio processo di selezione: il primo è una ‛selezione positiva', e consiste in uno stimolo alla maturazione delle cellule T che possiedono un recettore (TCR) adatto a riconoscere le proprie molecole di istocompatibilità con una bassa affinità; il secondo processo è una ‛selezione negativa', attraverso la quale tutti i linfociti che possiedono un recettore che riconosce con una affinità più elevata le proprie molecole di istocompatibilità insieme a peptidi endogeni, e quindi che potrebbero dare una risposta anti-self dannosa, vanno incontro a morte spontanea attraverso un processo definito apoptosi. Il processo con cui vengono eliminati i linfociti autoreattivi si chiama appunto ‛tolleranza': le malattie autoimmunitarie sono dovute a una rottura di questa tolleranza.
b) Le malattie autoimmunitarie
Nel circolo ematico degli individui affetti da malattie autoimmunitarie sono presenti anticorpi o linfociti T citotossici diretti contro strutture self, il che comporta un danneggiamento dei tessuti che possiedono gli antigeni self, con conseguente distruzione degli stessi. Le malattie autoimmunitarie rappresentano un enorme problema nel mondo occidentale, dato che interessano circa il 10% della popolazione. Tali malattie possono essere organo-specifiche e non organo-specifiche: appartengono alle prime alcune forme di tiroiditi nelle quali si verifica la distruzione del tessuto tiroideo, il diabete giovanile insulino-dipendente (IDDM, Insuline Dependent Diabetes Mellitus) caratterizzato dalla distruzione delle cellule β del pancreas, l'artrite reumatoide in cui sono interessate le articolazioni, e molte altre; appartiene invece al gruppo delle malattie non organo-specifiche il lupus eritematoso sistemico, in cui l'autoreattività interessa molti tessuti diversi. È interessante notare che le malattie autoimmunitarie sono in buona parte strettamente associate ad alcuni alleli HLA: cioè, nella maggior parte dei pazienti con una data malattia autoimmunitaria sono presenti certi alleli HLA e non altri. Un esempio di forte associazione è quello rappresentato dai pazienti affetti da spondilite anchilosante - una malattia che colpisce le articolazioni della colonna vertebrale e talvolta le periferiche - in cui in oltre il 95% dei casi è stata riscontrata la presenza dell'allele HLA-B27, rinvenibile solo nel 5% circa degli individui sani. Un altro esempio molto interessante è quello rappresentato dall'IDDM: lo studio dell'associazione di questa malattia con il sistema HLA è stato approfondito nel corso degli anni, man mano che venivano scoperti nuovi geni, e ha reso possibile avanzare ipotesi relative all'interpretazione dei meccanismi molecolari alla base di questa associazione. Infatti, la descrizione della prima associazione tra questa malattia e il sistema HLA risale agli anni settanta: si trattava di un'associazione piuttosto debole con alcuni alleli di classe I, gli unici allora conosciuti, in particolare l'allele B8. Successivamente, è stata evidenziata un'associazione con alcuni alleli di classe II, più precisamente DR3 e DR4. Con il progredire delle tecniche per lo studio del polimorfismo, si è visto che in realtà l'associazione più forte era con alcuni alleli di un altro gene di classe II, il gene DQB1, e in particolare con la tripletta che codifica per l'amminoacido nella posizione 57 della catena DQβ. Questa posizione partecipa alla formazione della fenditura dove si lega il peptide, e la presenza di un amminoacido carico negativamente (l'acido aspartico) in questa posizione ostacola lo sviluppo del diabete. Pertanto, l'acido aspartico nella posizione DQβ57 è molto meno frequente nei diabetici che nella popolazione di controllo. Sembra quindi logico ammettere l'esistenza di un peptide proveniente da una proteina specifica delle cellule β del pancreas che soltanto in assenza di una carica negativa in quella posizione del solco può legarsi alle molecole DQ e scatenare in tal modo una risposta contro il self. Rimane tuttavia un problema non risolto: quali siano le proteine self che scatenano questa risposta e perché esistano nell'organismo cellule che riconoscono i peptidi self. Si sta attualmente facendo un grande sforzo per cercare di isolare i peptidi coinvolti nelle malattie autoimmunitarie, dal momento che questo offrirebbe automaticamente la possibilità di approcci terapeutici più specifici di quelli, genericamente immunosoppressori, attualmente utilizzati. Anche se è difficile rispondere a queste domande, è tuttavia possibile prospettare alcune ipotesi. La più accreditata è che alcuni microrganismi patogeni possiedano peptidi, simili nella loro sequenza ad alcuni peptidi self, contro i quali, nel momento in cui infettano un individuo, s'innesca una risposta immunitaria specifica; se, al termine dell'infezione, i peptidi self trovano le molecole di istocompatibilità ‛giuste', che sono presumibilmente le stesse a cui si sono legati i peptidi antigenici, sostituiscono questi ultimi nei solchi delle molecole di istocompatibilità; in tal modo la risposta immunitaria specifica non si spegnerà come avviene normalmente, ma sarà sostenuta da questi nuovi complessi, perpetuando così un meccanismo citolitico che porta alla distruzione del tessuto. Questa interpretazione è ritenuta molto probabile nel caso di alcune malattie autoimmunitarie più strettamente associate al sistema HLA. Molti altri, naturalmente, possono essere i difetti genetici alla base di queste malattie. In alcuni casi, come nel lupus eritematoso sistemico, è possibile che ci siano dei difetti precoci che interessano il processo di selezione timica, impedendo una selezione negativa efficace. Lo studio dei meccanismi molecolari alla base della patogenesi delle malattie autoimmunitarie rappresenta, in conclusione, un campo della medicina tra i più interessanti, al centro del quale si colloca lo studio delle molecole HLA per il ruolo che esse svolgono nella presentazione dell'antigene (v. Adorini e altri, 1988).
c) L'equilibrio tra protezione e autoimmunità
Esistono quindi due effetti della funzione di presentazione dei peptidi ai TCR esercitata dalle molecole HLA: un effetto positivo di protezione dall'attacco di microrganismi e una controparte negativa rappresentata dal rischio di malattie autoimmunitarie. La capacità di riconoscere un maggior numero di peptidi può assicurare una maggiore protezione, ma viene pagata come reattività anti-self; viceversa, la rinuncia a presentare una parte dei potenziali peptidi antigenici diminuisce il rischio di altre malattie. Questi due effetti devono aver trovato un equilibrio nell'evoluzione. Di per sé, la capacità di riconoscere diversi complessi peptide-HLA da parte di specifici TCR sarebbe pressoché illimitata, data l'estrema flessibilità dei meccanismi di riarrangiamento genico che sono alla base della diversificazione dei TCR. Si sono quindi dovuti sviluppare dei meccanismi per limitare il numero di peptidi presentati. Il primo filtro è costituito dai proteasomi, che, come abbiamo visto sopra, comprendono subunità ad attività proteolitica. Questa non taglia indifferentemente in qualsiasi punto la sequenza amminoacidica, ma solo in corrispondenza di determinati amminoacidi. È stato dimostrato che questa specificità viene ulteriormente focalizzata in presenza delle due subunità controllate dai geni di classe II LMP2 e LMP7 della regione HLA. Più precisamente, il proteasoma produrrà solo peptidi che abbiano all'estremità C-terminale o un amminoacido idrofobico - come leucina, isoleucina, valina, fenilalanina, triptofano - oppure un amminoacido basico - come arginina o lisina. Un ulteriore filtro è costituito dal sistema di trasporto dei peptidi dal citoplasma all'interno delle vescicole del reticolo endoplasmico, che, come abbiamo visto, è effettuato dalla proteina TAP, costituita dalle due subunità TAP1 e TAP2, controllate da geni nella regione HLA di classe II. Da parte della molecola TAP vi è un riconoscimento diretto dei peptidi, a seconda della loro struttura, così che vengono preferenzialmente trasportati quelli che hanno una forte affinità di legame per TAP. Non sorprende quindi il dato sperimentale, ottenuto recentemente, secondo il quale i peptidi che hanno un amminoacido C-terminale idrofobico o basico, cioè precisamente quelli generati dall'azione specifica dei proteasomi, possiedono la maggiore affinità per TAP. Inoltre, i peptidi vengono selezionati per il trasporto a seconda della loro lunghezza: solo quelli con un numero minimo di 8 amminoacidi e massimo di 16 potranno essere trasportati. Infine, TAP esercita una ulteriore selezione per la struttura ‛interna' dei peptidi, secondo regole che sono attualmente oggetto di studio. In conclusione, di tutti i peptidi teoricamente possibili, soltanto una frazione molto ridotta è accessibile alle molecole HLA di classe I. Su queste esercitano un'ulteriore selezione le molecole HLA stesse, in quanto ciascun individuo può possedere al massimo 6 diverse molecole HLA (corrispondenti a due alleli per ciascuno dei tre loci HLA-A, -B e -C); conseguentemente vi è una corrispondente riduzione del repertorio di TCR, un minor ‛costo' della selezione negativa di cellule con TCR anti-self e quindi una diminuzione del rischio di reazioni autoimmuni.
d) Il rigetto dei trapianti
All'inizio di questa breve esposizione abbiamo detto come il rigetto dei trapianti sia stato all'origine della scoperta degli antigeni di istocompatibilità. In base a quanto abbiamo appreso sui meccanismi di processazione e di trasporto peptidico possiamo ora meglio interpretare il fenomeno della alloreattività, cioè della reazione immunitaria contro tessuti od organi provenienti da individui della stessa specie. Possiamo ritenere che i diversi individui non differiscano sostanzialmente nella specificità dei meccanismi di processazione (proteasomi) e di trasporto (TAP). In realtà sono state trovate alcune variazioni da individuo a individuo nei geni corrispondenti (LMP2, LMP7, TAP1, TAP2), che tuttavia, almeno nell'uomo, non sembrano essere superiori al livello medio di variazione genica né causare differenze funzionali facilmente rilevabili.
Le differenze fra gli individui sono quindi sostanzialmente riconducibili a due tipi: 1) differenze strutturali nelle proteine citoplasmatiche che vengono processate e da cui si possono quindi originare peptidi diversi, che vengono però ugualmente processati e trasportati; possiamo chiamare questo tipo di differenze ‛antigeni minori di istocompatibilità'; 2) differenze delle molecole di istocompatibilità che, viceversa, sono molto variabili da un individuo all'altro: queste differenze costituiscono gli ‛antigeni maggiori di istocompatibilità'. Nel caso dei trapianti di organo, queste differenze tra donatore e ricevente sono di fondamentale importanza. È ovvio che se due individui sono identici sia per le proteine citoplasmatiche in genere, sia per le molecole HLA, come si verifica nel caso di gemelli monozigoti, il tessuto o l'organo trapiantato verranno visti come self dal ricevente. Nel caso in cui i due individui possiedano le stesse molecole HLA, il che si verifica per esempio tra fratelli nel 25% dei casi, essi saranno compatibili per gli antigeni maggiori di istocompatibilità e, nella maggior parte dei casi, l'organo o il tessuto verrà accettato. Tuttavia, le differenze negli antigeni minori di istocompatibilità potranno essere sufficienti a dare reazioni di incompatibilità: è questo il caso, per esempio, dei trapianti di midollo osseo, in cui sia il ricevente che il tessuto trapiantato sono in grado di innescare una reazione immunitaria che provocherà o mancato attecchimento delle cellule midollari oppure la reazione cosiddetta di GVH (Graft Versus Host, o trapianto contro ospite) in una percentuale di casi piuttosto elevata, anche quando il trapianto avviene tra fratelli identici per HLA (v. anche sangue: Trapianto del midollo).
Naturalmente il caso peggiore è quello di una incompatibilità per antigeni sia maggiori che minori di istocompatibilità. Anche in questo caso, tuttavia, per alcuni organi come cuore, fegato, ecc., la conseguente reazione di rigetto è, in genere, limitata e superabile con l'impiego di farmaci immunosoppressivi.
4. Conclusioni
Negli importanti studi condotti negli anni trenta da Peter Gorer in Inghilterra è stato per la prima volta descritto il fenomeno dell'accettazione o del rigetto dei trapianti. Fu lui a coniare il termine ‛antigeni di trapianto' (o antigeni di istocompatibilità) per le molecole responsabili del rigetto. A più di mezzo secolo di distanza, possiamo affermare che lo studio dell'istocompatibilità ha fornito la chiave per capire il meccanismo del riconoscimento immunitario, cioè il meccanismo attraverso il quale Virus, Batteri e altri patogeni sono ‛rigettati' dai loro ospiti. Nel complesso maggiore di istocompatibilità è stato infatti scoperto un esteso sistema di geni polimorfici che è al centro della risposta immunitaria e che entra in gioco anche nello sviluppo di malattie autoimmunitarie. Il rigetto dei trapianti è quindi interpretabile come l'espressione dei normali meccanismi di difesa, applicata tuttavia non a un aggressore esterno ma a una situazione artificiale come quella del trapianto.
BIBLIOGRAFIA
Adorini, L., Muller, S., Cardinaux, F., Lehmann, P. V., Falcioni, F., Nagy, Z. A., In vivo competition between selfpeptides and foreign antigens in T-cell activation, in ‟Nature", 1988, CCCXXXIV, pp. 623-625.
Benacerraf, B., McDevitt, H. O., Histocompatibility-linked immune response genes, in ‟Science", 1972, CLXXV, pp. 273-279.
Bjorkman, P. J., Saper, M. A., Samraoui, B., Bennett, W. S., Strominger, J. L., Wiley, D. C., Structure of the human class I histocompatibility antigen, HLA-A2, in ‟Nature", 1987, CCCXXIX, pp. 506-518.
Brown, J. H., Jardetzky, T. S., Gorga, J. C., Stern, L. J., Urban, R. G., Strominger, J. L., Wiley, D. C., Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1, in ‟Nature", 1993, CCCLXIV, pp. 33-39.
Campbell, R. D., Trowsdale, J., Map of the human MHC, in ‟Immunology today", 1993, XIV, pp. 349-352.
Falk, K., Rötzschke, O., Stevanovi, S., Jung, G., Rammensee, H.-G., Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules, in ‟Nature", 1991, CCCLI, pp. 290-296.
Fling, S. P., Arp, B., Pious, D., HLA-DMA and -DMB genes are both required for MHC class II/peptide complex formation in antigen-presenting cells, in ‟Nature", 1994, CCCLXVIII, pp. 554-558.
Garrett, T. P. J., Saper, M. A., Bjorkman, P. J., Strominger, J. L., Wiley, D. C., Specificity pockets for the side chains of peptide antigens in HLA-Aw68, in ‟Nature", 1989, CCCXLII, pp. 692-696.
Geraghty, D. E., Structure of the HLA Class I region and expression of its resident genes, in ‟Current opinion in immunology", 1993, V, pp. 3-7.
Gorer, P. A., The genetic and antigenic basis of tumor transplantation, in ‟Journal of pathology and bacteriology", 1937, XLIV, pp. 691-697.
Gorer, P. A., Lyman, S., Snell, G. D., Studies on the genetic and antigenic basis of tumor transplantation. Linkage between a histocompatibility gene and ‛fused' in mice, in ‟Proceedings of the Royal Society" (Series B), 1948, CXXXV, pp. 499-505.
Hammer, J., Valsasnini, P., Tolba, K., Bolin, D., Higelin, J., Takacs, B., Sinigaglia, F., Promiscuous and allele-specific anchors in HLA-DR-binding peptides, in ‟Cell", 1993, LXXIV, pp. 197-203.
Howard, J. C., Restrictions on the use of antigenic peptides by the immune system, in ‟Proceedings of the National Academy of Sciences", 1993, XC, pp. 3777-3779.
Klein, J., Takahata, N., Ayala, F. J., MHC polymorphism and human origins, in ‟Scientific American", 1993, CCLXIX, pp. 78-83 (tr. it.: Polimorfismo MHC e origine dell'uomo, in ‟Le scienze", 1994, LVII, pp. 44-49).
Kronenberg, M., Brines, R., Kaufman, J., MHC evolution: a long term investment in defense, in ‟Immunology today", 1994, XV, pp. 4-6.
Lanzavecchia, A., Antigen-specific interaction between T and B cells, in ‟Nature", 1985, CCCXIV, pp. 537-539.
Madden, D. R., Gorga, J. C., Strominger, J. L., Wiley, D. C., The three dimensional structure of HLA-B27 at 2.1 Å resolution suggests a general mechanism for tight peptide binding to MHC, in ‟Cell", 1992, LXX, pp. 1035-1048.
Powis, S. H., Tonks, S., Mockridge, I., Kelly, A. P., Bodmer, J. G., Trowsdale, J., Alleles and haplotypes of the MHC-encoded ABC transporters TAP1 and TAP2, in ‟Immunogenetics", 1993, XXXVII, pp. 373-380.
Sette, A., Buus, S., Colon, S., Smith, J. A., Miles, C., Grey, H. M., Structural characteristics of an antigen required for its interaction with Ia and recognition by T cells, in ‟Nature", 1987, CCCXXVIII, pp. 395-399.
Stern, L. J., Brown, J. H., Jardetzky, T. S., Gorga, J. C., Urban, R. G., Strominger, J. L., Wiley, D. C., Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide, in ‟Nature", 1994, CCCLXVIII, pp. 215-221.
Trowsdale, J., Genomic structure and function in the MHC, in ‟Trends in genetics", 1993, IX, pp. 117-122.
Trowsdale, J., ‟Both man and bird and beast": comparative organization of MHC genes, in ‟Immunogenetics", 1995, XLI, pp. 1-17.