
IN CAMPO CONTRO L'ALZHEIMER
In campo contro l’Alzheimer
Il sistema nervoso, di cui il cervello costituisce la centrale operativa fondamentale, è l’organo del corpo dotato della massima complessità. Nell’uomo, un centinaio di miliardi di cellule nervose, denominate neuroni, oltre a un numero ancora maggiore di elementi di supporto per la loro crescita e per l’espletamento delle funzioni più importanti, le cellule gliali, formano un intricato, estremamente complesso sistema di collegamento con il resto dell’organismo. Grazie agli studi condotti principalmente nel 20° sec. sono stati compresi, fino al livello dei costituenti molecolari, i meccanismi che sono alla base del funzionamento di questi elementi cellulari e si è così spiegato il codice di comunicazione tramite il quale i neuroni inviano e ricevono i segnali dagli altri elementi cellulari con i quali sono collegati. Il sistema nervoso, tuttavia, è caratterizzato, più di ogni altro organo o tessuto dell’organismo, da un paradosso. Nonostante tutte le conoscenze acquisite e in continuo progresso, il neurologo si trova spesso in una situazione di frustrazione e di senso di impotenza. Grazie a tutte le tecnologie a disposizione, che permettono di analizzare le funzioni e disfunzioni cerebrali a livello microscopico e submicroscopico, è in grado di identificare la lesione che colpisce il cervello del paziente spesso in modo estremamente preciso. In questo compito è aiutato anche dal fatto che la struttura del cervello permette di valutarne eventuali malfunzionamenti come un elettricista o un informatico sono in grado di identificare un circuito alterato, poiché le vie nervose tramite le quali si elaborano le informazioni o si ricevono o inviano messaggi sono perfettamente conosciute. Tuttavia, sebbene il medico sia in grado di raggiungere una diagnosi precisa dell’affezione che colpisce il cervello del paziente, molto spesso non ha a disposizione gli strumenti, soprattutto quelli di natura farmacologica, per curare la malattia della quale ha formulato brillantemente la diagnosi.
Tutte le malattie denominate neurodegenerative, fra le quali si annoverano il morbo di Parkinson, la sclerosi laterale amiotrofica, la sclerosi multipla, il morbo di Huntington, la malattia da prioni detta della mucca pazza e, ovviamente, il morbo di Alzheimer (mdA), sono caratterizzate da questo tremendo paradosso. Inoltre, gli studi condotti negli ultimi decenni hanno portato alla luce diverse analogie molecolari fra queste malattie che, tuttavia, non hanno permesso fino a oggi di trarne nuovi determinanti attacchi terapeutici.
Del mdA, per es., i cui costi umani e sociali raggiungono soglie elevatissime e che è destinato ad aumentare proporzionalmente all’invecchiamento della popolazione, si conoscono ormai le cause che ne sono alla base, ma ancora oggi non esiste una cura efficace che possa almeno contribuire ad arrestare il lento, inesorabile progresso fino alla morte, in totale amenza, di chi ne è affetto.
La degenerazione e la successiva morte di intere popolazioni neuronali preposte alle funzioni cognitive che si verificano in questa affezione sono dovute al malfunzionamento di due proteine, la proteina tau e la proteina APP (Amyloid Precursor Protein, proteina precursore dell’amiloide).
Sintomi clinici
La diagnosi si basa, inizialmente, sulla valutazione di sintomi neuropsicologici e comportamentali legati a disturbi della memoria, del linguaggio e della percezione visivo-spaziale. I primissimi sintomi si manifestano di solito a carico di quel tipo di memoria denominata breve, per contrapporla all’altro tipo, la memoria di lunga durata. La prima riguarda i ricordi collegati con gli avvenimenti accaduti negli ultimi minuti o nelle poche ore precedenti; se questi ricordi rivestono i connotati di particolare importanza o interesse, essi vengono fissati tramite meccanismi biochimici distinti ma integrati con quelli che presiedono alla memoria breve e possono anche permanere per tutta la vita.
Si distinguono per comodità tre fasi nel decorso della malattia. Nel primo stadio il soggetto manifesta perdita della memoria breve, con difficoltà a ricordare anche parole di uso quotidiano come pane, latte, seggiola ecc., e dimostra difficoltà anche a ricordare il significato dei numeri; presenta minore attenzione per il lavoro o gli hobby che un tempo lo interessavano fortemente.
In un secondo stadio il paziente inizia a dimostrare difficoltà nel riconoscere i parenti e gli amici, si aggira senza scopi in casa o nei suoi dintorni e spesso deve essere riaccompagnato alla propria abitazione, tanto che i parenti sono costretti a lasciargli in tasca il proprio indirizzo. Si manifestano i primi cambiamenti comportamentali accompagnati da ansietà, insonnia e mutamenti di personalità.
Nell’ultimo stadio non riconosce più alcuno, non comprende le parole e ha difficoltà a vestirsi, a mangiare e deglutire nonché a controllare la minzione e la defecazione. La morte sopraggiunge per l’ulteriore aggravarsi di questi sintomi.
Epidemiologia e costi sociali
La malattia fa parte della più vasta categoria delle demenze senili che colpiscono il 5,3% degli uomini e il 7,2% delle donne dopo i 65 anni di età. Ciò è attribuito al fatto che queste ultime hanno un’aspettativa di vita maggiore e quindi il rischio, che è proporzionale all’età, aumenta di concerto. Il mdA costituisce il 50-80% di tutte le demenze senili, mentre un altro 11-24% dei casi è di origine vascolare. In Italia il numero di pazienti affetti da mdA assomma a circa 600.000÷700.000 ed è destinato ad aumentare rispetto alla popolazione per l’incremento progressivo dell’età e poiché l’Italia è fra le nazioni con il più alto numero di anziani. È stato calcolato che negli Stati Uniti il costo annuo per singolo paziente, comprese le spese mediche e i costi indiretti come la perdita di reddito, è pari a 150.000 dollari. Nel nostro Paese la famiglia svolge ancora un ruolo cardine nell’assistenza al malato di mdA e tuttavia i costi arrivano a superare i 45.000 euro annui per i pazienti più gravi.
Mentre l’incidenza delle principali cause di morte (fattori circolatori come infarto o danni ischemici e tumori di varia natura) è in lenta ma progressiva diminuzione grazie alle terapie farmacologiche e gli stili di vita e alimentari, per quanto concerne il mdA si ha paradossalmente un incremento dovuto all’allungamento della vita e, forse, ad altre cause ancora sconosciute.
Pensare e agire come antidoto fisiologico
Studi condotti in diverse parti del mondo indicano che l’incidenza del mdA è inversamente proporzionale al livello di educazione scolastica e culturale e che, negli Stati Uniti, sarebbe più frequente nella popolazione femminile e nella popolazione di origine africana. Queste conclusioni, tuttavia, non tengono conto della componente alimentare, dello stile di vita e di altre variabili che possono più o meno direttamente favorire l’insorgenza della malattia o accelerarne la velocità a seconda delle capacità economiche della popolazione analizzata. Nel caso della maggiore incidenza nelle donne, per es., negli studi epidemiologici iniziali non si era tenuto conto del fatto che la vita media è più lunga di diversi anni rispetto a quella maschile, aumentando quindi per semplici motivi statistici il rischio di malattia. In uno studio più recente, infatti, nel quale si sono combinati l’età, il genere, gli anni di scolarità delle donne e degli afroamericani si è raggiunta la conclusione che né le une né gli altri presentano fattori di rischio più elevati.
Pertanto, nonostante le analisi epidemiologiche non possano tener conto facilmente di tutti gli effetti collaterali – essendo la forma sporadica della malattia presumibilmente di natura multifattoriale –, una serie di studi condotti sugli animali di laboratorio sottolinea in modo marcato l’importanza di una vita ricca di stimoli raffrontata a una povera. Uno studio pubblicato di recente su una prestigiosa rivista conferma questa conclusione, suffragandola con un insieme impressionante di dati sperimentali. Lo studio è stato necessariamente condotto su animali – topi nel caso specifico – ma induce a concludere che possa essere esteso agli esseri umani.
Ricorrendo alle tecnologie che permettono di manipolare il corredo genetico di un animale, un gruppo di ricercatori che operano in alcune università americane ha creato topi transgenici nel cui corredo genetico sono stati inseriti alcuni geni i quali simulavano, a livello molecolare, e indirettamente comportamentale, una sindrome di mdA. In questi topi, infatti, erano sovraespressi due geni i quali codificano per le proteine (APP e PS1, presinilina 1), che causano la malattia. Un alterato metabolismo di queste proteine, infatti, porta all’accumulo di un peptide denominato β-amiloide (Aβ) che provoca la morte delle cellule nervose circostanti. Questi animali sono stati suddivisi in due gruppi: un gruppo era tenuto nelle gabbie in uso negli stabulari, che non sono certo luoghi confortevoli ma si attengono a norme internazionali severissime per qualità e pulizia. L’altro gruppo era esposto per 3 ore al giorno a un cosiddetto ambiente arricchito, consistente in gabbie di dimensioni più grandi nelle quali gli animali potevano arrampicarsi su ruote rotanti, addentrarsi in piccoli tunnel colorati, giocare a loro piacere. Dopo le 3 ore quotidiane di questo relativo benessere psicofisico e comportamentale il gruppo di animali veniva ricondotto in gabbie analoghe a quelle del gruppo meno fortunato di controllo. Il trattamento in ambiente arricchito veniva nel complesso protratto per sei mesi, al termine dei quali gli animali erano sacrificati e i loro cervelli analizzati con numerose tecniche sofisticate che comprendevano una misura della quantità del peptide neurotossico Aβ, la quantità e l’estensione delle placche (dette senili) alle quali danno luogo i depositi di questo peptide, l’attività della neprilisina, l’enzima cerebrale che normalmente è deputato a distruggere il peptide stesso, e molti altri parametri biomolecolari che caratterizzano nell’uomo e nell’animale il morbo di Alzheimer.
I risultati ottenuti da questo gruppo di ricercatori dimostrano che il mantenimento in un ambiente arricchito (che nell’uomo potrebbe essere assimilato a un ambiente fisicamente e intellettualmente stimolante) esercita un’azione incredibilmente positiva nell’animale, con una marcata riduzione del peptide tossico e delle placche senili, un’aumentata attività dell’enzima che distrugge il peptide e demolisce le placche e una tendenza notevole a una conversione verso un tipo di cervello normale. Lasciamo al lettore le conclusioni di questo studio che costituisce, in ogni caso, per tutti coloro che assistono i pazienti affetti da questa malattia, un invito ad arricchire, per quanto possibile, la vita dei loro assistiti, coscienti che il loro sforzo non è vano o di semplice assistenza passiva, ma può costituire una cura efficace e senza effetti collaterali negativi.
Aspetti genetici e molecolari della malattia
Le cause di questa affezione non sono ancora note e si ritiene che nella maggior parte dei casi essa sia dovuta a una variegata serie di interazioni tra fattori ambientali e componenti genetiche. Appare accertato che queste ultime sono responsabili uniche del 10% circa di ogni mdA e di tutti i casi di mdA che insorgono prima dei 40-50 anni. Nelle forme su base genetica sono coinvolti tre geni. Mutazioni del gene che codifica per la proteina denominata APP, situato sul cromosoma 21, provocano l’1% dei casi di mdA e si manifestano intorno ai 45-55 anni. Mutazioni del gene che codifica per la PS1, situato sul cromosoma 14, causano la malattia, che si manifesta tra i 30 e i 50 anni, nel 4% dei casi. Infine, nell’1% dei casi la malattia è dovuta a mutazioni del gene che codifica per la PS2, situato sul cromosoma 1, e i primi sintomi si manifestano fra i 40 e i 50 anni. Vi sono poi geni che, se in forma mutata, causano una maggiore suscettibilità per l’insorgenza della forma tardiva della malattia, come APOE4, una variante di APOE che svolge un’importante funzione nel metabolismo del colesterolo. È interessante notare che la presenza di APOE4 costituisce anche un fattore di rischio per malattie che colpiscono il cuore. Al contrario di APP e delle preseniline, tuttavia, questo gene rappresenta soltanto un fattore di rischio per l’insorgenza del 60% delle forme tardive. In altre parole, la sua presenza non è di per sé causa di malattia. Sebbene la causa sporadica della malattia (che rappresenta il 90% del mdA) non sia ancora conosciuta, la maggior parte degli specialisti è concorde nel ritenere che, come numerose altre affezioni di tipo degenerativo, le cause siano molteplici. Il fattore di rischio maggiore, come accennato, è legato all’aumento dell’età, a parte le cause strettamente genetiche che analizzeremo in seguito e che rappresentano tuttavia una percentuale minima del mdA. Si calcola che nell’età compresa fra i 65 e i 75 anni l’incidenza sia del 2-3%, che aumenti al 15-20% tra i 75 e gli 85 anni e colpisca il 50% degli individui con età superiore ai 90 anni.
Un cervello adulto sano contiene, come si è accennato, circa 100 miliardi di neuroni collegati fra loro tramite terminazioni denominate sinapsi che si calcola assommino a circa 100.000 miliardi. Le cellule nervose comunicano fra loro e con il resto dell’organismo tramite un codice costituito da due tipi di simboli: correnti elettriche (i potenziali d’azione) che viaggiano lungo le fibre nervose e, giunte a livello sinaptico, inducono la liberazione di sostanze chimiche (i mediatori chimici o neurotrasmettitori). Di queste sostanze chimiche ne sono state isolate più di un centinaio, ma la quantità liberata per ogni impulso rimane sostanzialmente eguale; le correnti elettriche, al contrario, possono variare di frequenza ma la loro intensità è costante. Tramite questo sistema di comunicazione binario i neuroni di tutte le specie viventi si scambiano segnali e informazioni che viaggiano attraverso e mediante le strutture sinaptiche che costituiscono, pertanto, la struttura tramite la quale i neuroni comunicano con tutto l’organismo e fra loro. Un potenziale d’azione, generato nel corpo del neurone (il pericario), viaggia lungo l’assone fino a raggiungere le sinapsi ove induce la liberazione del neurotrasmettitore; questo diffonde nello spazio sinaptico e raggiunge una porzione postsinaptica ove genera un nuovo potenziale d’azione che si propaga ad altri neuroni. Si calcola che se tutte le sinapsi fossero attive in un determinato momento il numero di impulsi che vi circolano assommerebbe all’astronomica cifra di 1017.
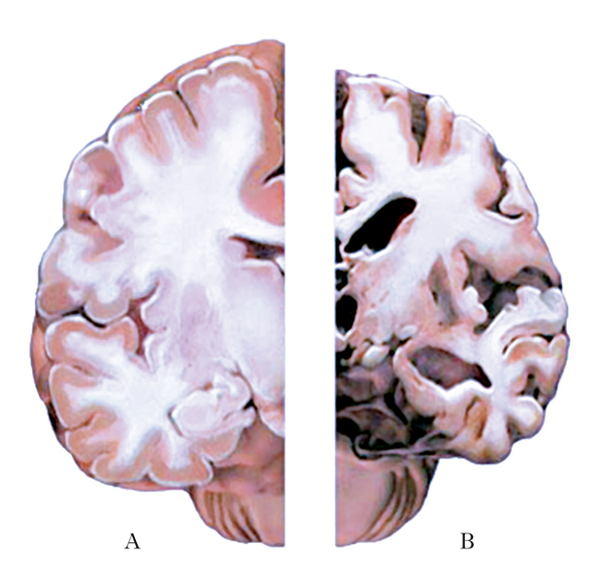
Nel mdA il trasferimento di informazioni fra i neuroni inizia a declinare progressivamente poiché diminuiscono in modo altrettanto progressivo il numero di sinapsi, la loro efficienza e lo stesso numero di neuroni. I sintomi clinici che caratterizzano il mdA, tuttavia, sono da attribuirsi principalmente alla perdita di sinapsi più che alla morte dei neuroni. Nelle fasi terminali della malattia le dimensioni del cervello e il numero di elementi nervosi diminuiscono in maniera drammatica (fig. 1).
Questa progressiva perdita di sinapsi e una loro inefficienza funzionale sono state attribuite all’azione alterata della proteina tau e alla produzione eccessiva di peptidi derivati dall’APP.
Placche e tangles come segni del mdA

Quando il neuropatologo Alois Alzheimer (1864-1915) per primo descrisse un insieme di dati comportamentali e anatomopatologici che in seguito avrebbero connotato con il suo nome la malattia, egli notò due alterazioni che in seguito furono considerate come patognomoniche: all’interno dei neuroni ormai in fase di avanzata degenerazione o già morti egli rilevò l’esistenza di strutture fibrillari denominate neurofibrillary tangles (NFT), che studi successivi rivelarono essere costituite principalmente dalla proteina tau. Inoltre, all’esterno degli stessi neuroni in via di degenerazione egli notò l’esistenza di aggregati di varie dimensioni evidenziabili con particolari colorazioni già allora disponibili, che denominò placche senili (PS) e che in seguito si sarebbe scoperto essere costituite principalmente da un peptide di piccole dimensioni denominato Aβ. Come già detto, questo peptide origina da APP, una proteina situata nella membrana citoplasmatica delle cellule nervose la cui funzione non è ancora stata definita in modo inequivocabile (fig. 2).
APP, il precursore dei peptidi tossici
APP è una proteina di dimensioni relativamente cospicue la cui principale forma, espressa nel sistema nervoso, è costituita da 770 amminoacidi. La maggior parte della molecola si affaccia con l’estremità N-terminale sulla superficie esterna della membrana cellulare e presenta diversi domini di interazione con altre proteine e con metalli come il rame e lo zinco. Questa complessità strutturale rende conto della difficoltà di comprenderne la funzione come si verifica per le proteine che svolgono una singola attività nell’organismo. La porzione di APP che è a contatto con le proteine citoplasmatiche all’interno del neurone (C-terminale) è molto più corta ed è anch’essa in grado di interagire con diverse proteine intracellulari.
Studi in animali e in colture di cellule in vitro hanno evidenziato che questa proteina fa parte di quel tipo di polipeptidi dotati di numerose attività le quali, inoltre, mutano a seconda del periodo di sviluppo del sistema nervoso. Per es., APP svolge un ruolo importante nella fase in cui i neuroni migrano nei vari distretti neuronali muovendosi in stretto contatto con cellule gliali sottostanti che fungono da substrati di adesione. Da questo punto di vista APP assomiglia a quella classe di proteine denominate cell adhesion molecules (molecole di adesione cellulare) le quali svolgono un ruolo molto importante nello sviluppo del sistema nervoso e, in generale, nella costruzione della forma tridimensionale di organi e tessuti. Strettamente collegata con questa funzione sarebbe quella di modulare l’attività dei coni di crescita, le propaggini estreme degli assoni che esplorano l’ambiente circostante per raggiungere il bersaglio cellulare definitivo con il quale stabilire i contatti sinaptici funzionali. Quando poi i neuroni raggiungono le loro sedi di attività definitiva, la parte di APP presente nelle porzioni terminali di assoni e dendriti svolgerebbe un’attività di modulazione sinaptica strettamente collegata con i fenomeni di plasticità cerebrale. Questa funzione ben si accorda con quella di partecipazione ai processi di memorizzazione, come suggerito anche dalla dimostrazione che la quantità di APP espressa sulla membrana risulta notevolmente aumentata negli animali allevati in ambienti arricchiti i quali, come accennato, facilitano funzioni mnemoniche e cognitive.
Il fatto che APP svolga comunque un ruolo o più attività funzionali essenziali per i neuroni è dimostrato dal fatto che topi transgenici ingegnerizzati in modo da non esprimere questa proteina vanno incontro a morte nel corso dello sviluppo.
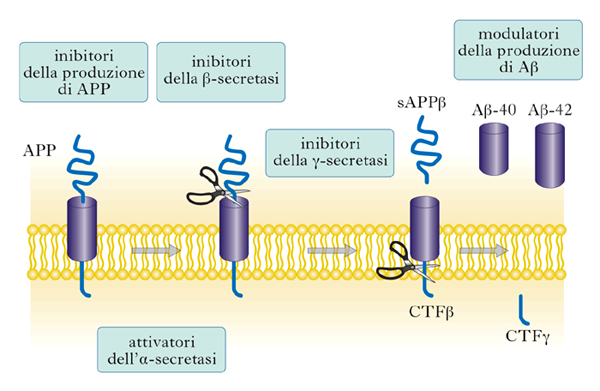
La porzione di APP che è stata oggetto di un crescente numero di studi è la parte transmembrana. In tale contesto, infatti, si verifica il processamento fisiologico o patologico di questo polipeptide a opera delle α-, β- e γ-secretasi. L’azione concertata di β- e γ-secretasi dà origine al rilascio di Aβ-42, il peptide anomalo costituito da 42 amminoacidi, mentre l’α-secretasi rappresenta l’attore principale del processo metabolico fisiologico di APP. Aβ-42 tende ad aggregarsi in fibrille che si accumulano tra una cellula e l’altra dove esercitano un’azione tossica sulle cellule sane circostanti (fig. 3). Anche se non è ancora del tutto chiaro in quale forma, se monomerica, dimerica o polimerica, Aβ-42 eserciti la sua azione tossica, la comunità scientifica riconosce nella produzione eccessiva di questo peptide la causa scatenante della complessa serie di eventi che conducono alla morte neuronale.

In sostanza, il processamento di APP può condurre alla produzione e alla liberazione nel mezzo extracellulare di una proteina ‘buona’, l’α-APP, e di alcuni derivati peptidici non tossici la cui funzione è ancora in larga parte sconosciuta, o può generare la proteina ‘cattiva’, il β-APP, accompagnato in rapida sequenza proteolitica dalla formazione dei peptidi tossici di cui Aβ è il componente principale. La tabella riassume tutte le condizioni che possono provocare la cosiddetta ipotesi della cascata di amiloide.
Il neurologo, e in generale ogni uomo di scienza, può essere portato a esprimere qualche perplessità quando una crescente evidenza sperimentale indica in un evento molecolare unico e relativamente semplice, come quello che riguarda il processamento di APP, la causa scatenante del mdA. Tuttavia, con il progredire delle conoscenze biologiche, un numero crescente di anomalie risulta essere la conseguenza di un’alterata funzione di una singola proteina che conduce a eventi al termine dei quali vi sono esiti catastrofici per l’intero organismo. Uno dei primi casi di questo genere descritti nel campo della biomedicina è riconducibile a diversi decenni fa, quando si dimostrò che un tipo particolare di anemia – che colpisce prevalentemente la popolazione mediterranea – era dovuta alla sostituzione di un singolo amminoacido delle molte centinaia che costituivano una sola proteina, l’emoglobina.
Nel caso delle malattie neurodegenerative, poi, il caso del mdA non è unico: anche il morbo di Huntington o quello da prioni – per citarne solo due dei numerosi conosciuti – sono dovuti a un’anomalia strutturale e funzionale di una singola componente proteica. È da considerare, inoltre, che di solito alla produzione o formazione di un determinato componente tossico si accompagna anche la perdita di funzione del suo omologo sano, e tanto più quella funzione è importante tanto più si genera un duplice danno.
La proteina tau
Più o meno nelle stesso tempo in cui si inizia la demolizione anomala di APP, all’interno degli stessi neuroni la proteina tau subisce una serie di modificazioni postraduzionali e di attacchi da parte di enzimi proteolitici che terminano con la sua parziale demolizione e costituzione di spirali proteiche facilmente identificabili al microscopio elettronico denominate, come accennato, aggregati neurofibrillari o neurofibrillary tangles (NFT). La funzione di tau nell’ambito del sistema nervoso, al contrario di APP, è ben conosciuta e consiste essenzialmente nel permettere l’assemblaggio, l’organizzazione sopramolecolare e la funzione dei microtubuli, strutture cilindriche che svolgono un ruolo essenziale in tutte le cellule eucariotiche partecipando in modo decisivo al movimento di ciglia e flagelli, alla divisione cellulare e in genere a tutte le funzioni che richiedono una struttura in grado di generare lavoro a spese di energia. Nel sistema nervoso, i microtubuli, modulati dalla proteina tau, svolgono due funzioni essenziali: nella fase di crescita delle fibre nervose provvedono, insieme ad altre strutture citoscheletriche come actina e filamenti intermedi, alla crescita delle fibre nervose; essi sono paragonabili alle impalcature create temporaneamente nella fase di costruzione di un edificio. Nell’adulto, quando le fibre nervose hanno stabilito contatti sinaptici con le cellule bersaglio, provvedono al trasporto, all’interno delle fibre nervose stesse, di materiale nutritizio o di supporto funzionale per le terminazioni sinaptiche. Si tratta di un sistema di veicolazione di materiale in entrambi i sensi, anche se con velocità diverse.
Le fibre nervose hanno lunghezze molto differenti: quelle che provvedono alla comunicazione fra neuroni presenti all’interno del cervello hanno, ovviamente, estensioni assai minori di quelle che inviano o ricevono messaggi dall’organismo. Queste ultime possono anche raggiungere la lunghezza di 1 m o più, com’è il caso delle fibre motorie che si dipartono dal midollo spinale per raggiungere la parti estreme di un arto.
Per provvedere alla comunicazione a distanza, pertanto, il sistema nervoso è munito di due sistemi: uno veloce, basato su un preciso codice di comunicazione, al quale si è accennato, che si propaga alla velocità di 100-150 m/s tramite correnti elettriche che si alternano con la liberazione di neurotrasmettitori; un secondo sistema molto più lento ma egualmente indispensabile, che opera all’interno di assoni e dendriti, deputato al trasporto di sostanze e materiali di vario genere. Il trasferimento di materiale, infatti, non potrebbe avvenire per semplice diffusione passiva e necessita, pertanto, di un sistema di trasporto assonale che impiega e consuma energia.
Un ruolo che è emerso più recentemente nell’ambito del sistema di trasporto assonale consiste nel trasportare non solo materiale nutritizio dal pericario alle sinapsi e viceversa, ma anche nel provvedere al trasporto di molecole essenziali per la stessa vita del neurone appar-tenenti alla famiglia delle neurotrofine. Fra queste neurotrofine, come si avrà modo di analizzare in seguito, un ruolo di particolare rilevanza rispetto alle cause del mdA e delle possibili terapie viene svolto dall’NGF (Nerve Growth Factor, fattore di crescita nervoso) e dal BDNF (Brain-Derived Neurotrophic Factor, fattore di crescita derivato dal cervello).
La degradazione di tau, pertanto, che si verifica progressivamente dopo l’innesco della produzione di Aβ, porta al collasso dei microtubuli che, a sua volta, si materializza nell’arresto del trasporto assonale e nella morte dell’intero neurone. Tra le macerie cellulari che restano vi sono gli NTF costituiti dai residui di tau e di altre proteine nel frattempo associate a queste strutture.
Baptisti, tauisti e apoptosi
Mentre tutti i neurologi concordano nell’identificare nei tangles e nelle placche senili i due reperti caratteristici del mdA, nell’ultimo decennio sono nate due vere e proprie scuole di pensiero per quanto riguarda la priorità di insorgenza di queste due formazioni. Sono i depositi di Aβ, che formano le placche senili, a iniziare una serie di eventi biochimici (produzione di radicali liberi, aumento nell’entrata di ioni calcio ecc.) che portano all’attacco proteolitico di tau con successivo collasso dei microtubuli e morte della cellula? O, al contrario, i primi eventi sono a carico di tau e il suo alterato funzionamento provoca di conseguenza un alterato metabolismo di APP con conseguente produzione di Aβ e danno tossico alle cellule circostanti in un meccanismo a cascata che si estende a macchia d’olio? I sostenitori della prima ipotesi si definiscono scherzosamente come baptisti, prendendo spunto dal termine che denomina APP. I secondi, per converso, poiché identificano il primum movens dell’evento patogenetico nella proteina tau si definiscono tauisti.
Vi sono argomenti a favore dell’una e dell’altra ipotesi ma la domanda cruciale, che si pone a monte di queste ipotesi è: per quale motivo, e tramite quali meccanismi molecolari iniziali, una di queste due proteine, che svolge un ruolo essenziale nell’economia dei neuroni, va incontro a quei cambiamenti che si materializzano nella formazione di placche e tangles?
Un’ipotesi unificante sulle cause della malattia identifica in processi di apoptosi a carico di vaste popolazioni neuronali l’evento scatenante. Nell’ultimo decennio è emersa una nozione interamente nuova nel panorama della moderna biologia. Ogni cellula non è dotata solo del complesso di geni che presiedono alla sua specializzazione funzionale per divenire cellule nervose, cellule muscolari, cellule epiteliali e così via. Nel nucleo di ogni cellula si trova infatti un gruppo di geni – il cui numero e le cui funzioni si vanno rapidamente identificando – che è devoluto ad avviare con precisione ed efficienza teutonica le cellule verso l’esito finale, cioè la morte. Per questo motivo si parla anche di morte cellulare programmata. Il processo è stato chiamato da coloro che per primi lo hanno descritto (Kerr, Wyllie, Currie 1972) apoptosi, dal termine apóptosis («caduta») che nell’antica lingua greca era usato per descrivere la caduta delle foglie o dei petali dei fiori.
Consideriamo ora due casi, uno in cui il processo di apoptosi svolge un ruolo indispensabile di salvaguardia, l’altro in cui l’attivazione di questo programma genetico può provocare enormi danni all’organismo: il caso dei tumori e quello di malattie neurodegenerative come il morbo di Alzheimer.
Apoptosi ‘benigne’ e apoptosi ‘maligne’
Una nozione popolare di solito errata è che un tumore cresce a dismisura invadendo tutto l’organismo grazie alla sua capacità di moltiplicarsi a ritmo frenetico. Di solito, al contrario, la velocità di replicazione di queste cellule è simile a quella delle cellule normali. Ricordiamo che non vi è tumore che si riproduca così velocemente come un embrione umano che, iniziando da una cellula uovo e da uno spermatozoo, genera nell’arco di nove mesi una massa di 3-4 kg di cellule. Tuttavia, poiché queste cellule sono sane, la loro moltiplicazione si verifica obbedendo a programmi genetici eseguiti secondo una precisa scansione temporale, spaziale e di progressiva specializzazione. Il problema, nel caso dei tumori, non è tanto la loro velocità di replicazione, quanto il fatto che le cellule tumorali muoiono molto meno di quelle normali perché non obbediscono più ai segnali inviati dall’organismo; segnali che hanno la funzione di attivare il programma di morte programmata o apoptosi presente in ogni cellula. Una delle funzioni di questo programma, infatti, è quello di mantenere un equilibrio fra cellule giovani e cellule vecchie, di permettere un continuo rimodellamento funzionale di ogni organo e tessuto eliminando le cellule superflue. In sostanza, mentre in un organismo sano il numero di cellule giovani è sempre strettamente bilanciato da un eguale numero di cellule vecchie che degenerano e muoiono tramite il meccanismo dell’apoptosi, nel caso delle cellule tumorali spesso questo programma è soppresso o alterato e il bilancio favorisce la crescita invece dello stato stazionario.
Un suicidio cellulare programmato male
Consideriamo ora il caso di malattie degenerative del sistema nervoso come il morbo di Alzheimer. Si ritiene che una delle sue principali cause sia dovuta al fatto che i neuroni, di cui siamo dotati fin dalla più tenera età e che non possiamo sostituire con nuove generazioni di cellule come nel caso degli altri organi (salvo nei rari e localizzati casi di coinvolgimento di cellule staminali latenti), attivino il proprio programma endogeno di morte per apoptosi spesso per cause sconosciute, o per mancanza di fattori trofici la cui funzione è tenere bloccato il programma apoptotico.
L’apoptosi svolge un ruolo fondamentale di natura fisiologica nel corso dello sviluppo del cervello e della formazione dei circuiti nervosi eliminando tutti i neuroni che non hanno formato circuiti appropriati e ben funzionanti. In altri termini, tutte le cellule che non hanno formato i circuiti funzionanti seguendo ‘direttive’ in parte di natura genetica e in parte ambientali (tipicamente stimoli sensoriali di varia natura) si autoeliminano e i loro detriti vengono rapidamente digeriti da apposite cellule per evitare un ingombro dannoso e un eccessivo consumo energetico ai neuroni che in questa corsa sono riusciti a stabilire contatti funzionali con altre cellule. Si tratta, come è stato denominato dal neuroscienziato Jean-Pierre Changeux, di una sorta di darwinismo neuronale nel quale sopravvivono i neuroni più adatti al funzionamento dell’organo che essi costituiscono, il cervello.
Ma se questi fenomeni di autoeliminazione venissero ‘accesi’, per cause che considereremo in seguito, nel cervello dell’adulto si potrebbero provocare danni irreversibili. Si verificherebbe, infatti, una progressiva perdita di intere popolazioni neuronali che, nell’arco di qualche anno, condurrebbero l’individuo a quell’insieme di sintomi devastanti descritti in precedenza. Al contrario dei tumori, che non obbediscono all’ordine di attivare i propri geni apoptotici, i neuroni compirebbero l’errore di attivarli perché vengono a mancare i segnali che, di norma, li tengono repressi. In queste cellule, come è comprensibile, il congegno a orologeria che attiva questi geni deve essere bloccato per tutta la vita dell’organismo. Quando questo blocco viene a mancare, le cellule nervose si autoeliminano e, a seconda delle funzioni che esse svolgono nel cervello, vengono a essere gravemente compromesse memoria, intelligenza, movimento.
Se l’ipotesi di un’accensione anomala di processi apoptotici come causa scatenante del mdA è corretta, dovremmo ipotizzare che questo meccanismo provochi un processamento anomalo di APP e di tau, cioè delle due proteine che tutti gli studi sul mdA indicano quali responsabili primarie. Di fatto, è stato dimostrato che se il processo apoptotico viene attivato in animali appositamente ingegnerizzati, o in colture di cellule nervose provenienti da diverse regioni cerebrali di animali da laboratorio (per es., deprivazione di neurotrofine o interruzione di stimolazioni elettriche), rapidamente il processamento fisiologico di APP viene alterato e si concretizza in una produzione anomala di Aβ e di una sua aggregazione in strutture fibrillari simili a quelle che costituiscono le placche senili. Inoltre, Aβ, diffondendo nel mezzo di coltura neuronale, attacca anche neuroni ancora integri inducendone la morte. Si genera, in sostanza, un circolo vizioso neurotossico che tende a espandersi a tutti i neuroni.
Contemporaneamente a questi eventi, anche la proteina tau viene coinvolta subendo modificazioni, una volta sintetizzata, con fosforilazioni a carico degli stessi amminoacidi che nel mdA sono ritenuti responsabili del distacco di tau dai microtubuli. Questo distacco conduce al loro progressivo disassemblaggio e alla perturbazione del sistema di trasporto assonale seguito dalla degenerazione e morte del neurone.
Nel loro insieme, pertanto, l’induzione di un evento apoptotico tramite manipolazioni analoghe a quelle che si possono anche verificare nel cervello dell’uomo, crea una sindrome molecolare in tutto analoga a quella che nel corso di lunghi periodi s’instaura progressivamente nel cervello umano portando alla progressiva degenerazione neuronale.
Obiezioni all’ipotesi apoptotica
Le due principali obiezioni all’ipotesi che lega causalmente l’instaurarsi del mdA con l’attivazione di apoptosi è rappresentata dal fatto che: a) nel cervello di pazienti deceduti per questa malattia si troverebbero pochi segni di una avvenuta morte per apoptosi dei neuroni colpiti; b) i tempi che accompagnano la sindrome molecolare cui si è accennato sono troppo rapidi nelle condizioni di laboratorio mentre nell’uomo sarebbero molto più lenti.
La morte per apoptosi è caratterizzata da un insieme di eventi intracellulari che permettono facilmente di distinguerla da quella per necrosi, che si verifica in seguito a un trauma o a un’ustione. La ricerca dei marcatori apoptotici da parte di istologi e anatomopatologi in cervelli di persone decedute a causa del mdA ha fornito dati contrastanti e apparentemente non si sarebbero identificate evidenze inequivocabili della presenza di abbondanti quantità di questi marcatori nelle sedi cerebrali maggiormente coinvolte nei sintomi clinici. Questa obiezione, tuttavia, non considera il principio che, ricorrendo a una metafora poliziesca, possiamo definire come il problema del cadavere e del killer. Il cadavere si trova di solito facilmente mentre il killer spesso non è identificabile neppure dopo un’attenta ricerca investigativa. Fuori di metafora, è possibile che nella ricerca si sia facilmente trovato il cadavere (nel caso del mdA, NTF e placche senili), mentre il killer, cioè l’evento apoptotico scatenante, sfugga per la rapidità con la quale gli esecutori della morte per apoptosi, per es. il complesso di enzimi responsabili dell’evento scatenante, vengono rapidamente rimossi dalla zona del ‘delitto apoptotico’. Soltanto studi più approfonditi con tecniche ancor più raffinate potranno risolvere questo problema.
Altre considerazioni di natura analoga valgono anche per la seconda obiezione. Mentre l’instaurarsi in vitro della sindrome molecolare richiede poche ore o al massimo qualche giorno, si calcola che l’instaurarsi della patologia di Alzheimer sia un processo lento, che richiede numerosi anni. Tuttavia, questa obiezione non considera il fatto che il cervello umano è costituito da miliardi di neuroni, mentre una coltura in vitro nella quale si è misurata la cascata di eventi apoptotici solitamente ne contiene poche migliaia. È quindi sufficiente ipotizzare che l’instaurarsi del mdA sia la conseguenza dell’accensione di minuscoli focolai apoptotici, ciascuno formato da poche migliaia di neuroni. Prima che questi microscopici focolai diano luogo a sintomi clinici possono trascorre tempi molto lunghi. Per es., un focolaio di 100.000 neuroni che in vitro va incontro a morte per apoptosi nell’arco di un giorno equivale alla morte di 100 milioni di neuroni in tre anni. In questa fase iniziale il soggetto può ancora non manifestare i sintomi clinici che lo condurranno in seguito dal neurologo. Ricordiamo, infatti, che il repertorio di neuroni che forma il cervello assomma a molte decine di miliardi.
Il ruolo centrale dell’NGF e di altre neurotrofine
È interessante notare che fra i segnali che tengono sotto controllo la morte per apoptosi sia in colture di neuroni sia in animali transgenici figurano l’NGF e altre neurotrofine, la cui mancanza in animali da laboratorio determina una sindrome comportamentale e anatomopatologica simile a quella che caratterizza il mdA. Non è pertanto azzardato ipotizzare che questa affezione neurologica sia dovuta a un diminuito rifornimento di NGF o di altre neurotrofine come IGF-1 (Insuline-like Growth Factor) e BDNF. Questo diminuito rifornimento è provocato da diverse cause, quali una ridotta sintesi da parte delle cellule vicine oppure un arresto o un rallentamento del trasporto assonale retrogrado.
Sebbene nel suo insieme la sindrome molecolare descritta possa dare ampi suggerimenti su quanto potrebbe accadere nell’uomo, saranno comunque necessari molti studi prima di trarre un rapporto di causa ed effetto fra i due fenomeni.
Terapia
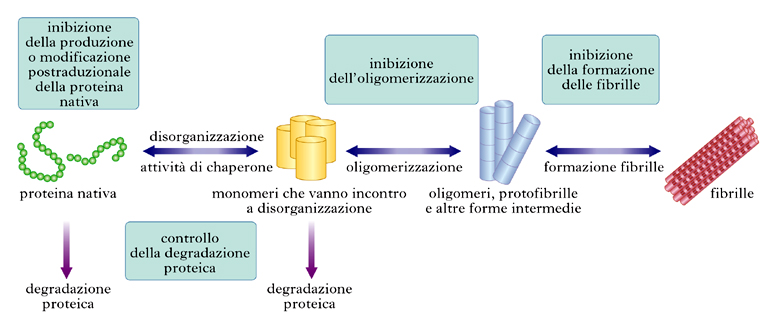
Nello schema della fig. 4 sono riassunte alcune strategie farmacologiche che attualmente sono in opera o sono considerate di possibile utilità terapeutica.
Non vi sono, al momento, cure specifiche così efficaci da poter far regredire la malattia; sostanzialmente vi sono diversi approcci che, in attesa di una terapia risolutoria, permettono al paziente di condurre una vita dignitosa fino alle fasi quasi terminali dell’affezione.
Anticolinesterasici
Gli inibitori della colinesterasi rappresentano attualmente i farmaci di scelta per il mdA. Tali farmaci agiscono inibendo la demolizione da parte di questo enzima del neurotrasmettitore acetilcolina, impiegato dai neuroni colinergici che costituiscono il bersaglio più colpito nelle fasi precoci e successive della malattia. Si tratta di una strategia tesa a limitare il danno e ad attenuare i sintomi più che a migliorare il quadro clinico e anatomopatologico vero e proprio. Tale strategia, tuttavia, è suscettibile di miglioramento anche su quest’ultimo versante poiché gli studi più recenti indicano che un procedimento per rallentare la morte delle popolazioni nervose coinvolte, e possibilmente bloccarla, consiste nel mantenere in un’attività costante tali popolazioni. Come si è accennato nel considerare le cause prime del mdA, un’ipotesi accreditata sostiene che essa è dovuta all’impropria attivazione di eventi apoptotici che vengono scatenati dalla mancanza di neurotrofine o di stimolazioni nervose. La somministrazione di neurotrofine (fra queste, l’NGF appare, da studi in fase ancora sperimentale, la neurotrofina di elezione), ossia di agonisti (cioè attivatori) dei recettori dell’acetilcolina, potrebbe mantenere una stimolazione più costante e in tal modo inibire lo scatenamento del processo apoptotico. L’efficacia di queste terapie costituirebbe, inoltre, una conferma indiretta delle analisi statistiche che attribuiscono un’incidenza maggiore di mdA nella popolazione analfabeta. Questa per la propria condizione culturale e di lavoro, è meno soggetta, rispetto a quella dedita al lavoro intellettuale, all’esercizio continuo delle attività cognitive che coinvolgono, appunto, il sistema colinergico.
Un tipo di farmaci impiegati negli anni Novanta, come si è accennato, è quello basato sull’uso di agonisti dei recettori muscarinici e nicotinici. È interessante notare, a questo proposito, che studi epidemiologici avrebbero dimostrato un ritardo della frequenza di mdA nei fumatori, nei quali l’inalazione di nicotina eserciterebbe un’azione comparabile a quella ottenuta in corso di trattamento terapeutico a base di agonisti degli stessi recettori colinergici di tipo nicotinico. Ricordiamo che il recettore nicotinico è stato così denominato perché fa parte del complesso recettoriale dell’acetilcolina. Come terza generazione di farmaci anti mdA si va impiegando il trattamento congiunto di terapie colinergiche con quelle basate su estrogeni, antinfiammatori o antiossidanti.
Disaggreganti
La quantità di Aβ nel tessuto può essere ridotta stimolandone lo smaltimento. Lo scopo è stato raggiunto usando questo peptide per vaccinare topi trans-genici in grado di produrre grandi quantità di anticorpi anti-Aβ. Rispetto ai controlli, gli animali vaccinati erano capaci di eseguire esercizi più difficili, facendo ipotizzare che, avendo meno amiloide nel cervello, anche la degenerazione neuronale fosse minore. Questo risultato ha indotto a tentare un’analoga sperimentazione sull’uomo: 375 pazienti con demenza di media gravità sono stati selezionati e vaccinati con Aβ. Tuttavia, si è dovuto interrompere la prova prima di poterne giudicare l’efficacia per la comparsa di reazioni avverse nel 5% dei casi. Un’encefalite autoimmune è stata documentata in alcuni dei pazienti morti per cause indipendenti dal vaccino.
Oltre agli anticorpi specifici, anche alcune tetracicline che attraversano la barriera ematoencefalica si sono dimostrate capaci di disaggregare l’amiloide. In una sperimentazione condotta in una vasta popolazione di malati, la rifampicina e la doxiciclina sarebbero state in grado di rallentare l’avanzamento del declino cognitivo per tutta la durata della cura.
Inibitori delle secretasi
Il modo più ovvio per limitare l’accumulo di Aβ nel tessuto è quello di diminuirne la produzione. In teoria questo risultato può essere ottenuto riducendo l’attività delle β- e γ-secretasi. In pratica, lo sviluppo di sostanze anti β-secretasi è resa problematica dalla possibilità che gli inibitori agiscano contemporaneamente su molti substrati. È il caso di eparansolfato ed eparina, inibitori in vitro di β-secretasi, ma usati da tempo come anticoagulanti e perciò poco maneggevoli per una cura di lunga durata.
In condizioni di sperimentazione, la produzione di Aβ-42 viene limitata anche da alcuni antinfiammatori non steroidei, quali ibuprofene, indometacina e sulindac. Questi farmaci favoriscono la trasformazione di Aβ costituito da 42 amminoacidi in un peptide più corto e non neurotossico. Ciò avverrebbe grazie all’azione di queste sostanze sulla γ-secretasi e indipendentemente dal loro effetto antinfiammatorio. L’interesse per queste molecole nacque dall’osservazione che il rischio di demenza sembrava minore in chi ne consumava abitualmente. Tuttavia, l’uso controllato degli antinfiammatori è risultato inefficace nel contenere il deterioramento cognitivo.
Per impedire che Aβ sia prodotto in eccesso e si accumuli, si studia il modo di aumentare l’attività della neprilisina e degli altri enzimi proteolitici che sono in grado di attaccare e demolire Aβ. La ricerca in questo senso viene condotta non soltanto sul piano dell’attivazione farmacologica, ma anche su quello della terapia genica per aumentare la quantità disponibile degli enzimi.
Memantina, statine e antiossidanti
Un altro settore di ricerca farmacologica è offerto dall’azione di Aβ sulle membrane. È verosimile che questo peptide, una volta rilasciato nello spazio extracellulare, interagisca con la membrana cellulare e ne modifichi qualche sua funzione. Uno dei recettori compromessi dall’azione del peptide è il recettore NMDA, N-metil-D-aspartato, per il glutammato che modula l’ingresso del calcio nelle cellule. Una sostanza che riesce a ripristinare il funzionamento normale di questo recettore è la memantina. Sotto la protezione della memantina, i neuroni nonostante la presenza di Aβ non correrebbero più il rischio di morire intossicati dal calcio. Grazie a queste caratteristiche e alla sua tollerabilità, la memantina si propone come un efficace neuroprotettore delle cellule dotate di recettori NMDA.
La probabilità che Aβ interagisca con la membrana cellulare depone a favore dell’uso delle statine nella profilassi della malattia, specie negli individui ipercolesterolemici. Le statine diminuiscono la sintesi del colesterolo e sono utilizzate per la cura delle ipercolesterolemie. D’altro canto, la produzione di Aβ diminuisce quando si riduce la disponibilità di colesterolo. Su questa base, si sostiene che il rischio di ammalarsi sia maggiore negli individui ipercolesterolemici e diminuisca in quelli curati con le statine.
In condizioni sperimentali che simulano la malattia, anche i mitocondri soffrono per l’eccesso di Aβ o di APP. Nelle cellule transfettate e nei topi transgenici che producono più APP, questo resta intrappolato nella membrana mitocondriale riducendo sia l’attività citocromo-ossidasica sia la quantità totale di ATP. Aβ, invece, si lega all’alcol-deidrogenasi, modificando il sito di legame del NAD (Nicotinammide Adenine Dinucleotide). L’azione di Aβ e di APP sui mitocondri spiegherebbe la quantità anormale di radicali liberi e lo stress ossidativo, la compromissione dell’attività degli enzimi del metabolismo intermedio e la disfunzione mitocondriale osservati sia nella malattia sia nelle cellule intossicate con il peptide tossico. Si sa, inoltre, che lo stress ossidativo favorisce la produzione di Aβ stimolando l’attività di β-secretasi. Tutti questi dati costituiscono un elemento a favore dell’uso di antiossidanti per la profilassi e la cura della malattia. Mancano tuttavia prove certe della loro efficacia clinica.
Sali di litio
Le chinasi Cdk5 (Cyclin-dependent kinase 5) e Gsk3 (Glycogen synthase kinase 3) controllano la fosforilazione di tau, ma perdono questa loro capacità sotto l’effetto di Aβ. In condizioni sperimentali, i sali di litio sono in grado di evitare ai neuroni le conseguenze del cattivo funzionamento di Gsk3. Come Cdk5, anche questa chinasi promuove la fosforilazione della proteina tau e, nel cervello degli ammalati, si concentra nelle cellule degenerate. L’attività fosforilasica che Gsk3 esercita nei confronti della proteina tau aumenta sotto l’effetto di Aβ-42 e di ApoE4. Ciò fa di questo enzima l’anello che collega i tre elementi più caratteristici della malattia. In questo contesto, i sali di litio hanno una notevole peculiarità, perché non si limitano a inibire l’attività di Gsk3, ma agiscono nello stesso senso anche sulla secretasi, contrastando così la produzione e l’accumulo di Aβ. L’interesse dei sali di litio è giustificato dunque dalla loro doppia azione: sulla causa della malattia, riducendo la disponibilità del peptide tossico, e sul meccanismo principale di degenerazione neuronale, bloccando la fosforilazione della proteina tau. Quindi, somministrati alle dosi ormai ben collaudate dall’uso per la cura della depressione bipolare, i sali di litio potrebbero rivelarsi il mezzo più maneggevole ed efficace per la profilassi della malattia e per la cura del declino cognitivo di lieve entità. Essi hanno fallito, tuttavia, nella prevenzione del deterioramento cognitivo in un gruppo di pazienti che soffrivano di forme depressive cui erano stati a lungo somministrati. Resta da stabilire se il deterioramento di questi ammalati fosse dovuto al mdA.
Cure parentali
Secondo lo stadio nel quale si trova l’individuo affetto da mdA, è stato individuato un insieme di precauzioni.
Per il primo stadio occorre creare nell’abitazione un’area di orientamento spaziale nella quale tenere un insieme di oggetti di uso comune per il paziente quali chiavi di casa, occhiali, orologio, calendario; incoraggiare il paziente a mantenersi in contatto con gli amici, ad ascoltare la musica, a effettuare esercizi fisici; registrare il paziente, se possibile, in un programma di ritrovamento nel caso si perdesse per strada.
Per il secondo stadio è necessario mettere segni sui cassetti e su tutte le strutture di casa più comuni; accertarsi che le luci delle stanze di casa frequentate dal paziente siano sempre accese; mettere tappeti nel bagno e in altri luoghi per evitare scivolamenti; nascondere le chiavi della macchina e installare serrature con chiusura esterna o porre allarmi in caso di apertura dall’interno; effettuare la pulizia dei denti o altre operazioni di uso comune insieme al malato in modo da poter essere imitati dal paziente.
Per il terzo stadio occorre provare a ideare nuovi mezzi di comunicazione con il paziente, per es. tramite vecchie fotografie o con la musica; è necessario, inoltre, ridurre al minimo test medici che implichino il prelievo di sangue o altri test che possano risultare dolorosi per il malato.
Bibliografia
J.F. Kerr, A.H. Wyllie, A.R. Currie, Apoptosis. A basic biological phenomenon with wide-ranging implications in tissue kinetics, «British journal of cancer», 1972, 26, 4, pp. 239-57.
Alzheimer’s disease. Senile dementia and related disorders, ed. R. Katzman, R.D. Terry, K.L. Bick, New York 1978.
J.T. Coyle, D.L. Price, M.R. DeLong, Alzheimer’s disease. A disorder of cortical cholinergic innervation, «Science», 1983, 219, 4589, pp. 1184-90.
P.H. St George-Hyslop, Piecing together Alzheimer’s, «Scientific American», 2000, 283, 6, pp. 76-83.
J. Hardy, D.J. Selkoe, The amyloid hypothesis of Alzheimer’s disease. Progress and problems on the road of therapeutics, «Science», 2002, 297, 5580, pp. 353-56.
R. Adalbert, J. Gilley, M.P. Coleman, A-beta, tau and ApoE4 in Alzheimer’s disease. The axonal connection, «Trends in molecular medicine», 2007, 13, 4, pp.135-42.
D.M. Walsh, D.J. Selkoe, A beta oligomers – a decade of discovery, «Journal of neurochemistry», 2007, 101, 5, pp. 1172-84.
C. Matrone, M.T. Ciotti, D. Mercanti et al., NGF and BDNF signaling control amyloidogenic route and Aβ production in hippocampal neurons, «PNAS», 2008, 105, 35, pp. 13.139-44.
C. Matrone, A. Di Luzio, G. Meli et al., Activation of the amyloidogenic route by NGF deprivation induces apoptotic death in PC12 cells, «Journal of Alzheimer’s disease», 2008, 13, 1, pp. 81-96.