
La grande scienza. Apoptosi: un programma speciale di morte cellulare
La grande scienza. Apoptosi: un programma speciale di morte cellulare
Apoptosi: un programma speciale di morte cellulare
L'essere umano è un organismo pluricellulare in cui la divisione cellulare avviene, come in tutte le altre forme di vita eucariotiche, mediante il processo noto come mitosi. Durante la mitosi, le cellule duplicano il proprio DNA e si dividono per formare due cellule figlie, ciascuna delle quali è la replica esatta della cellula genitrice. Nel nostro corpo, la divisione cellulare per mitosi avviene regolarmente: per esempio è responsabile della sostituzione delle cellule che ricoprono il tratto gastrointestinale e le vie respiratorie. Tale tipo di sostituzione avviene con una frequenza di alcuni giorni; inoltre, nonostante in un organismo pluricellulare come quello umano gli eventi mitotici si verifichino costantemente in numero considerevole, le cellule del nostro corpo tuttavia non vanno mai incontro ad alcuna variazione effettiva di numero. Ciò naturalmente implica che per ogni cellula generata con la mitosi, una deve morire per portare in pareggio il bilancio delle cellule nel 'libro dei conti della vita'. La domanda è: come muoiono le cellule? Ora sappiamo che una cellula può morire attraverso due processi diversi, ossia la necrosi e l'apoptosi.
La necrosi è la forma di morte cellulare nella sua definizione classica ed è la conseguenza di un grave trauma subito dalla cellula. Una cellula che muore per necrosi va incontro a un rigonfiamento rapido e incontrollato e alla fine scoppia. Il suo contenuto intracellulare fuoriesce nell'ambiente extracellulare dando luogo, molto spesso, a una risposta infiammatoria, caratterizzata dall'aumento del flusso sanguigno nell'area interessata, dall'afflusso dei leucociti e dal rilascio di varie molecole che mediano la risposta infiammatoria. La morte cellulare per necrosi si osserva raramente in condizioni fisiologiche e le cellule che muoiono a causa di questo processo non hanno alcun controllo sul proprio destino. Tale mancanza di controllo è un aspetto molto importante sul quale torneremo successivamente.
Il secondo tipo di morte cellulare, detto apoptosi, differisce considerevolmente dalla necrosi. In primo luogo, a differenza della necrosi, che è nota da quasi un secolo, l'apoptosi è stata scoperta piuttosto recentemente. In secondo luogo, al contrario della necrosi, l'apoptosi è un processo di morte cellulare controllato geneticamente. Questo capitolo fornirà una rassegna delle attuali conoscenze riguardanti la biologia cellulare e la genetica dell'apoptosi, del ruolo svolto da questo processo nella normale fisiologia e di ciò che accade quando esso non è più soggetto a regolazione, come nell'AIDS e nel cancro.
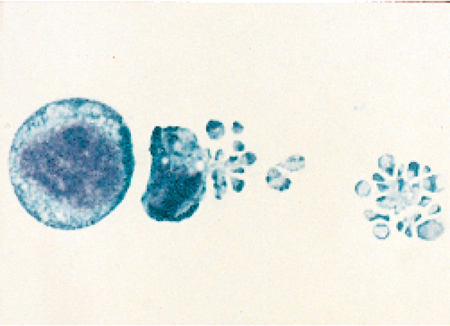
Nel 1972 Andrew H. Wyllie pubblicò insieme ai suoi collaboratori (Kerr et al. 1972) un importante articolo in cui si descriveva il processo di morte cellulare per apoptosi (fig. 1). Questo termine, di derivazione greca, è utilizzato per descrivere la caduta dei petali dei fiori o quella delle foglie dagli alberi. Per quanto apparentemente oscura, l'etimologia del termine apoptosi ha di fatto una certa attinenza con le caratteristiche del processo di morte cellulare in quanto tale. Infatti, le cellule che muoiono per apoptosi si condensano e si staccano letteralmente dalle strutture di supporto tissutali sulle quali stanno crescendo, proprio come le foglie degli alberi che cadono in autunno. Il processo di apoptosi fu identificato nel 1972; tuttavia, prima di allora la letteratura ne aveva riportato molti esempi, sebbene utilizzando una diversa terminologia. Le cellule apoptotiche del timo (tingle bodies), i cheratinociti apoptotici della pelle (sunburn cells) e le cellule apoptotiche del fegato (councilman bodies) sono tutti esempi di apoptosi descritti in anni antecedenti il 1972. Un altro termine utilizzato allo scopo di descrivere il fenomeno di apoptosi prima dell'anno suddetto è stato shrinkage necrosis, necrosi per riduzione di volume (Kerr 1971). Questa definizione costituisce un'accurata descrizione istologica dell'apoptosi dato che la fuoriuscita di acqua è una delle sue caratteristiche chiave; di contro, durante la necrosi, si ha il fenomeno opposto, ovvero il rigonfiamento della cellula dovuto all'afflusso di acqua al suo interno.
Dall'anno della scoperta, passò quasi un decennio prima che l'apoptosi catturasse l'immaginazione degli scienziati. Nella seconda metà degli anni Ottanta gli immunologi e gli embriologi riscoprirono il lavoro di Wyllie, John F. Kerr e Alastair R. Currie del 1972 e iniziarono a esplorare i meccanismi biologici alla base dell'apoptosi sia nel sistema immunitario sia nell'ontogenesi. Ciò ha determinato, verso la fine degli anni Ottanta, un graduale aumento del numero di pubblicazioni in questo campo, fino ad arrivare negli anni Novanta a una vera e propria esplosione di lavori scientifici dedicati a tale argomento. Esistono attualmente circa una dozzina di libri e due riviste specificatamente dedicati allo studio dell'apoptosi. La ragione di questa rapida crescita di interesse è da ascriversi al fatto che gli scienziati hanno capito come la morte cellulare per apoptosi sia un processo biologico fondamentale, di cui conosciamo molto poco. Inoltre l'idea di un processo di morte cellulare che avvenga attraverso 'geni suicidi' ha suscitato un interesse di tipo 'faustiano' sull'argomento. La ricerca tesa a scoprire perché e in che modo una cellula mette in moto il suo programma di morte cellulare ha costituito una delle aree più attive della biologia degli anni Novanta e per una buona ragione: la comprensione del processo mediante il quale una cellula si uccide potrebbe permettere di prolungarne la vita o di accelerarne la morte, un traguardo ambito in diverse patologie umane.
La morte cellulare durante lo sviluppo
Prima di approfondire la biologia cellulare dell'apoptosi è probabilmente utile chiedersi innanzi tutto il motivo per il quale le cellule abbiano sviluppato un meccanismo sofisticato per morire e in quali circostanze venga attivato il programma di morte cellulare. Negli organismi pluricellulari sembrano molte le situazioni in cui l'apoptosi è di fondamentale importanza per lo sviluppo e la sopravvivenza, una delle quali è la morte cellulare che si verifica nei normali processi fisiologici. Durante lo sviluppo fetale, per esempio, tra le dita è presente una rete di cellule che conferisce a queste strutture anatomiche l'aspetto di una pinna. Con il progredire dello sviluppo da questa pinna si modellano le dita e le cellule interdigitali muoiono per apoptosi. Questo è un chiaro esempio di come il corpo utilizzi il processo apoptotico di morte geneticamente programmata per formare nuove strutture anatomiche. Un altro esempio del ruolo svolto dall'apoptosi si ritrova nell'ontogenesi del sistema nervoso. Durante lo sviluppo il corpo genera neuroni in eccesso che nel sistema nervoso in via di formazione competono tra loro per stabilire le connessioni con altre cellule. I neuroni che non riescono a stabilire queste connessioni muoiono per apoptosi. Di fatto circa il 90% di tutti i neuroni formatisi durante la vita muore per apoptosi; ciò avviene perché essi non riescono a creare con i neuroni vicini connessioni che possano mantenerli in vita. Tale tipo di processo in cui o si attua la connessione oppure si verifica la morte neuronale è di vitale importanza in quanto assicura che il sistema nervoso, il quale dipende strettamente dalla formazione corretta delle connessioni tra le cellule, sia assemblato con estrema cura per formare una rete neurale funzionante. Un ultimo esempio, tratto dal mondo della biologia dello sviluppo e familiare alla maggior parte dei lettori, è il riassorbimento della coda di un girino durante la sua trasformazione in rana; le cellule della coda vengono distrutte per apoptosi e riassorbite (Lockshin e Zakeri 1991).
Altri esempi provengono dall'immunologia nella quale le cellule autoreattive del sistema immunitario vengono eliminate prima che possano nuocere all'organismo, determinando una risposta immunitaria indesiderata a danno dei tessuti normali dell'individuo. Tuttavia, esistono diverse situazioni in cui la regolazione dell'eliminazione delle cellule del sistema immunitario è compromessa provocando l'insorgenza di una serie di malattie autoimmuni molto debilitanti. Di tali malattie discuteremo in seguito.
'Caenorhabditis elegans' e apoptosi
L'organismo utilizzato preferenzialmente come modello per studiare una varietà di processi ontogenetici, in particolare l'apoptosi, è il verme nematode Caenorhabditis elegans. Questo organismo è composto di 1090 cellule di cui 131 muoiono in modo programmato per apoptosi, e ciò lo rende particolarmente interessante. La morte cellulare colpisce particolarmente il sistema nervoso (105 cellule), sebbene muoiano anche altri tipi di cellule, comprese quelle muscolari. Il ruolo centrale nella morte di queste 131 cellule è svolto da due geni chiamati ced 3 e ced 4, i quali vengono attivati selettivamente quando viene avviato il programma di morte cellulare nelle 131 cellule destinate a morire. Se questi geni mutano in modo tale da non poter più compiere la normale funzione, tutte le 131 cellule destinate a morire sopravvivono. Per quanto ne sappiamo, stranamente, i vermi che mantengono queste 131 cellule soprannumerarie appaiono normali. Il riassorbimento e la rimozione delle cellule morte per apoptosi vengono controllati da sette geni le cui mutazioni non influiscono sulla morte o sulla sopravvivenza delle cellule. Ciò sta a indicare l'assenza di un legame diretto tra i geni ced 3 e ced 4 che inducono la morte cellulare e quelli implicati nella rimozione delle cellule morte.
Lo scopo principale della morte cellulare in C. elegans è l'eliminazione delle cellule indesiderate e ciò può avere diverse conseguenze. Per esempio, può essere utile per eliminare le cellule che hanno eseguito un compito specifico durante i primi stadi dello sviluppo, ma che non hanno altre funzioni da svolgere nell'organismo adulto. In secondo luogo, può servire a generare il dimorfismo sessuale. Molti dei geni identificati in questo organismo e implicati nella regolazione dell'apoptosi hanno un corrispettivo negli organismi superiori quali i mammiferi; ciò che rende lo studio di C. elegans così interessante è che questo organismo può essere facilmente manipolato per studi genetici e ontogenetici particolarmente difficili in organismi più complessi. Nel 2002 il premio Nobel per la fisiologia e la medicina è stato attribuito, in compartecipazione, a Robert Horvitz per il raffinato lavoro volto a chiarire alcuni aspetti della genetica dell'apoptosi in C. elegans.
Stress cellulare e apoptosi
In Natura la lotta per la sopravvivenza è costante e gli organismi, nel corso della loro vita, sono soggetti a una varietà di stress naturali. Negli organismi pluricellulari questa lotta sussiste anche a livello cellulare e stranamente la morte cellulare per apoptosi può svolgere un'importante funzione nella risposta di sopravvivenza di un organismo che si trovi ad affrontare uno stress. Una cellula esposta a stress causato da un insulto ambientale o biologico può morire o sopravvivere e questo è un dato di fatto nella vita di ogni cellula. Se la cellula è soggetta a moderati livelli di stress, la sintesi di un gruppo di proteine da stress o da shock termico, o heat-shock proteins, può fornire un certo grado di protezione alla cellula, sempre che l'agente stressante venga eliminato dopo un breve periodo di tempo. Le proteine da stress si rinvengono in quasi tutti i tipi di cellule, dai batteri agli esseri umani, ed è probabile che costituiscano un sistema primitivo di autodifesa. Tuttavia, se la cellula è sottoposta a un insulto, insufficiente a ucciderla subito, ma dal quale le heat-shock proteins non possono proteggerla, allora viene attivato il programma di apoptosi ed essa muore in modo controllato (Samali e Cotter 1996). Ciò è un grande vantaggio per gli organismi pluricellulari, perché, quando una cellula muore per apoptosi, il suo contenuto non è riversato all'esterno, come avviene invece nel processo di necrosi. La fuoriuscita del contenuto cellulare può causare lesioni alle cellule vicine o anche provocarne la morte, con effetti nocivi per la sopravvivenza di altre cellule o dell'organismo stesso. In altre parole, con la morte per apoptosi, una cellula compie l'atto 'altruistico' di salvare le cellule adiacenti. Infine, se la cellula subisce un insulto così grave da non avere il tempo di attivare il programma di apoptosi, la morte avviene allora per necrosi con danni e lesioni ben distinte alle cellule adiacenti causati dal rilascio del contenuto intracellulare da parte della cellula morente.
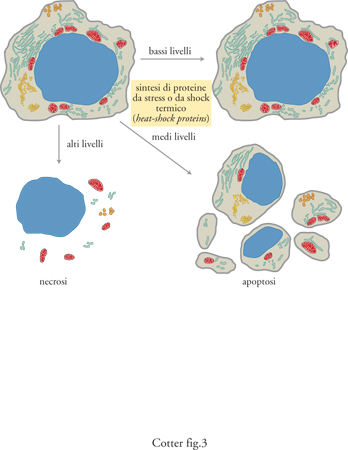
La cellula ha pertanto due livelli di difesa per proteggere sé stessa e l'organismo di cui fa parte. In prima istanza può sopravvivere con l'ausilio delle proteine da stress e quindi, in un secondo momento, tramite l'apoptosi, può morire senza danneggiare le cellule vicine o l'intero organismo (fig. 3).
Caratteristiche biologiche dell'apoptosi
Una delle peculiarità biochimiche più tipiche dell'apoptosi è la rottura del DNA cellulare in frammenti delle dimensioni di un nucleosoma che, dopo elettroforesi su gel di agarosio, appaiono come i pioli di una 'scala' (DNA ladder) (Wyllie et al. 1980). Ciò si verifica a causa dell'attivazione di un'endonucleasi che resta inattiva nella cellula finché non viene avviato il programma di apoptosi. La frammentazione si compie con un processo in due stadi: inizialmente il DNA è suddiviso in grandi pezzi, i quali vengono ridotti in frammenti delle dimensioni di un nucleosoma nel passaggio successivo. È probabile che nel processo descritto intervengano due distinti sistemi enzimatici, in quanto alcune cellule sono capaci di produrre soltanto i frammenti grandi, mentre altre possono produrne entrambi i tipi.
Durante l'apoptosi la morfologia della cellula morente va incontro a una serie di cambiamenti consistenti, uno dei quali è la riduzione di volume dovuta all'espulsione di acqua. Nel corso di questo processo il volume cellulare può ridursi fino al 50% di quello originario, ma non sappiamo quasi niente dei motivi per cui la cellula vada soggetta a questo restringimento o dei modi in cui esso si verifichi. Oltre a questo fenomeno, nella cellula si verifica anche una marcata condensazione della cromatina che origina i nuclei picnotici. In alcune cellule, il nucleo va poi incontro a frammentazione all'interno della cellula. Il successivo cambiamento morfologico evidente è la rottura della cellula in piccole vescicole chiuse chiamate corpi apoptotici, come si vede chiaramente nella fig. 1. Le vescicole vengono poi inglobate e distrutte dai fagociti circostanti. Durante l'intero processo, il contenuto cellulare non fuoriesce nell'ambiente extracellulare, in quanto si conserva l'integrità della membrana plasmatica.
Uno dei motivi per cui gli scienziati hanno impiegato tanto tempo a scoprire l'apoptosi non è da imputarsi al fatto che essa si verifica raramente, ma piuttosto alla rapidità con la quale in condizioni fisiologiche le cellule apoptotiche vengono rimosse. Il riconoscimento e la rimozione delle cellule apoptotiche sembrano correlati all'espressione di nuove molecole sulla loro superficie, che ne consente l'identificazione da parte dei fagociti. Una di queste molecole è il lipide di membrana fosfatidilserina, normalmente localizzato sulla superficie interna della membrana plasmatica, il quale durante l'apoptosi migra verso la superficie esterna agendo come segnale di riconoscimento per la fagocitosi (Fadok e Chimini 2001). Al momento non è chiaro il meccanismo di riconoscimento della fosfatidilserina da parte dei fagociti (fig. 4).
Geni e apoptosi
Grazie al lavoro pubblicato negli anni Ottanta da J. John Cohen e Rick C. Duke (1984) sappiamo che l'apoptosi è un processo controllato geneticamente. Cohen e Duke dimostrarono che gli inibitori della sintesi delle macromolecole ritardano l'apoptosi e che tale processo è probabilmente sottoposto a qualche forma di controllo genetico. Esperimenti successivi eseguiti per diversi anni da vari gruppi di ricerca, incluso il nostro, hanno condotto all'identificazione di due classi di geni coinvolti nella regolazione dell'apoptosi. In una classe si trovano geni quali c-myc e p53 che dirigono il processo apoptotico, nell'altra vi sono geni come bcl-2 e bcr-abl che possono inibire la morte cellulare.
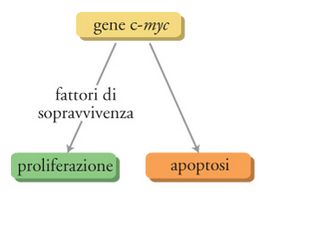
Il gene c-myc è considerato una sorta di 'Giano bifronte' in quanto può guidare sia la proliferazione sia la morte delle cellule (Pelengaris et al. 2002). La direzione che una cellula prende sembra dipendere dalla situazione in cui viene a trovarsi. Per esempio, in presenza dei fattori di sopravvivenza cellulare come il fattore di crescita insulinosimile-1 (IGF-1), l'espressione del gene c-myc induce la proliferazione; invece, in assenza di tali fattori il gene c-myc causa la morte cellulare per apoptosi. Ciò potrebbe essere parte di un meccanismo antierrore per prevenire l'insorgenza di tumori. C-myc è un gene chiave nella regolazione della proliferazione cellulare e perciò ogni mutazione che ne amplifichi l'espressione ha la potenzialità di portare a una proliferazione cellulare incontrollata e al possibile sviluppo di tumori. Tuttavia c-myc non controlla da solo la proliferazione cellulare: deve essere stata prodotta un'adeguata quantità di fattori di sopravvivenza cellulare, la cui sintesi è regolata da altri geni che vengono attivati soltanto quando è necessario. Perciò anche in presenza di un aumento incontrollato dell'espressione di c-myc, il tumore non si sviluppa a meno che non si verifichi un altrettanto incontrollato aumento dell'espressione dei fattori di sopravvivenza. In altre parole, perché si sviluppi una cellula tumorale devono essere mutati almeno due geni chiave, e ciò probabilmente spiega, in parte, perché la conversione di una cellula normale in una tumorale sia un evento relativamente raro, considerata l'elevata frequenza delle divisioni cellulari normali che si verificano nel nostro corpo (fig. 5).

Un altro gene chiave che favorisce l'apoptosi e che in determinate circostanze causa la morte cellulare è p53, che risulta mutato nella grande maggioranza dei tumori umani. Per questo motivo esso fa parte di una famiglia di geni, noti come soppressori tumorali, che, quando sono attivi, impediscono lo sviluppo dei tumori. La funzione principale di p53 oggi nota è di bloccare le cellule che hanno subito un danno a livello del DNA nella fase G1, evitando che portino a termine il ciclo cellulare. Tale blocco permette alla cellula di riparare il DNA, e se la riparazione non è possibile, viene avviato il processo di apoptosi (Yonish-Rouach et al. 1991). Se p53 non riesce a svolgere i suoi compiti, per esempio a causa di mutazioni, allora le cellule con il DNA danneggiato possono rientrare nel ciclo cellulare con la possibilità di dare origine al cancro. Il gene p53 quando è mutato non può dare avvio all'apoptosi. Non è chiaro come p53 medi i propri effetti, ma la sua capacità di bloccare le cellule in G1 viene probabilmente realizzata con l'interazione con altri geni, quali waf-1 e gadd 45, noti per il loro coinvolgimento nel controllo del ciclo cellulare (fig. 6). Per il suo ruolo di vigilanza sul ciclo cellulare, il gene p53 è stato soprannominato il custode del genoma.
Proprio come esistono geni che favoriscono l'apoptosi come c-myc e p53, parimenti esiste una classe di geni che inibisce l'apoptosi; in questa categoria rientrano geni come bcl-2 e bcr-abl. Il primo oncogene di cui si è identificato un ruolo di controllo nel processo di apoptosi è stato bcl-2. Il lavoro svolto nel laboratorio di Stanley J. Korsmeyer a St. Louis (USA) ha permesso di dimostrare che le cellule in cui questo gene viene espresso a livelli elevati sono considerevolmente più resistenti all'induzione di apoptosi; anche le cellule in cui questo gene è stato inserito mostrano una resistenza alla morte cellulare (McDonnell e Korsmeyer 1991). È stato dimostrato che l'espressione del gene bcl-2 è associata al linfoma delle cellule follicolari, e ciò ha suggerito che questo tipo di cancro possa essere dovuto più a una mancanza di morte cellulare che a un eccesso di proliferazione. In altre parole, le cellule che alla fine del loro periodo di vita non muoiono, sopravvivono e con la loro immortalità contribuiscono allo sviluppo del tumore. Un'altra conseguenza dell'elevata espressione di bcl-2 è una maggiore resistenza delle cellule ai farmaci; esse non vanno quindi incontro ad apoptosi in seguito a trattamento con agenti normalmente utilizzati per uccidere le cellule tumorali. Lo scenario a nostra disposizione mostra perciò l'esistenza di un gene che, inibendo la morte cellulare, non solo contribuisce allo sviluppo del tumore, ma rende anche più difficile uccidere le cellule tumorali con i convenzionali farmaci contro il cancro. L'espressione di bcl-2 sembra contribuire anche allo sviluppo e alla progressione di altri tipi di cancro come quello della prostata.
Il cancro della prostata è al secondo o al terzo posto tra tutti i tipi di cancro più diffusi nel mondo occidentale; negli Stati Uniti è al secondo posto tra i tipi di cancro più comunemente diagnosticati negli uomini. I pazienti affetti da questa patologia hanno in genere difficoltà a urinare a causa dell'ipertrofia della ghiandola prostatica. La diagnosi è relativamente semplice e il trattamento consiste in un'operazione chirurgica e/o in una chemioterapia. Il cancro della prostata può caratterizzarsi come una malattia bifasica, in cui la fase iniziale risponde al trattamento. Durante questa fase le cellule prostatiche cancerose proliferano in risposta alle proprietà degli androgeni, come il testosterone, che favoriscono la crescita cellulare. Di fatto, per la sopravvivenza, queste cellule sono completamente dipendenti dagli ormoni maschili. Se gli androgeni vengono eliminati, il tumore si rimpicciolisce notevolmente e le condizioni del paziente migliorano sensibilmente. Ciò può essere ottenuto o usando sostanze che antagonizzano gli effetti degli androgeni o con la chirurgia, che nella forma della castrazione può essere utilizzata per rimuovere la fonte degli ormoni. Purtroppo, come altri tipi di cancro, anche quello della prostata inevitabilmente recidiva, con una seconda fase assai più resistente al trattamento e che di solito è fatale. In questa fase, infatti, il cancro recidivante è indipendente dagli androgeni e quando è eliminato l'ormone che ne favorisce la crescita le cellule non muoiono per apoptosi. Prove recenti suggeriscono che un ruolo centrale nel cambiamento del fenotipo tumorale sia probabilmente svolto dall'aumentata espressione di bcl-2. Nelle cellule tumorali che rispondono all'ormone, l'espressione di bcl-2 è minima o assente, mentre è presente quella del gene bax che favorisce la morte cellulare. Quando lo stimolo proliferativo degli androgeni viene rimosso, o chimicamente o con la castrazione, le cellule muoiono in breve tempo. Viceversa, bcl-2 previene questa morte e le cellule tumorali sopravvivono (McDonnell et al. 1993). È probabile che l'espressione di questo gene sia coinvolta nel passaggio del cancro da una fase che inizialmente risponde bene alla terapia a una fase resistente. Tale trasformazione non si osserva soltanto nel cancro della prostata, ma anche in un numero crescente di altri tipi di cancro. Pertanto l'espressione dei geni antiapoptotici, come bcl-2, sembra avere un ruolo determinante nello sviluppo di diversi tumori umani e nel loro trattamento.
La famiglia delle proteine bcl-2 svolge un ruolo fondamentale nel controllare il destino delle cellule. Queste proteine sono regolatori chiave dell'apoptosi, anche se ancora non comprendiamo il meccanismo preciso attraverso cui esse svolgono le loro funzioni. In origine bcl-2 fu scoperto nei linfomi delle cellule B dove la sua espressione veniva amplificata a causa di una traslocazione che metteva il gene sotto l'influenza del regolatore di un forte promotore delle immunoglobuline. Ciò si traduceva nel blocco della normale apoptosi delle cellule B, nell'accumulo di queste cellule 'non morte' e nello sviluppo del tumore. Questa osservazione ha dimostrato che il tumore può insorgere anche in conseguenza del blocco della morte cellulare oltre che per un incremento della proliferazione. Da allora si è aggiunto a questa famiglia un numero sempre crescente di nuove proteine, di cui si può dire che bax sia la più conosciuta. Essa agisce di concerto con bcl-2 per promuovere la morte cellulare. Un'ipotesi di studio molto affascinante sostiene che la principale funzione di bcl-2 sia proprio quella di mantenere bax in uno stato inattivo (Oltvai et al. 1993). A seconda del bilancio tra queste due proteine una cellula può essere indirizzata verso la sopravvivenza o la morte.
Oltre a bcl-2 si sono dimostrati potenti inibitori dell'apoptosi anche bcr-abl e v-abl. Per esempio, la leucemia mieloide cronica è caratterizzata dalla traslocazione del gene c-abl dal cromosoma 7 al cromosoma 22. Ciò porta alla produzione della proteina di fusione bcr-abl, in quanto il gene abl viene a trovarsi allineato al gene bcr, inducendo un aumento dell'attività chinasica di Abl. L'aspetto interessante di questo tipo di cancro è che l'incremento massiccio del numero delle cellule mature si verifica senza alcun cambiamento della normale frequenza delle mitosi cellulari (Spiers 1992). Studi ulteriori hanno dimostrato che sia bcr-abl sia il suo corrispettivo virale v-abl sono potenti inibitori dell'apoptosi. Il meccanismo mediante il quale bcr-abl e v-abl mediano i loro effetti non sembra coinvolgere l'azione di bcl-2 o bax, ma un altro sistema effettore ancora del tutto sconosciuto. La leucemia mieloide cronica è una malattia bifasica, la cui fase iniziale è caratterizzata dall'accumulo di un gran numero di cellule mature della serie mieloide. Ciò sembra dovuto a un difetto del processo di morte cellulare per apoptosi, probabilmente causato da un aumento dell'attività chinasica della proteina mutata bcr-abl. La malattia può rimanere in una fase cronica per settimane o anni ma è inevitabile che prima o poi avvenga la transizione a una forma più acuta. Tale stadio acuto, definito talvolta fase accelerata, è particolarmente difficile da curare e di solito è fatale. A livello biochimico si verifica una mutata espressione dei geni che regolano la proliferazione durante la fase acuta della malattia e questo, insieme alla presenza di geni che prevengono la morte cellulare, fornisce alle cellule tumorali tutti gli ingredienti per sopravvivere e proliferare. Una delle questioni biochimiche fondamentali è in che modo l'aumento dell'attività chinasica consenta ad Abl di sopprimere la morte cellulare. Una via di segnalazione chiave è l'attivazione della cascata della chinasi PI3/Akt che impartisce un forte segnale di sopravvivenza alla cellula. L'attivazione di questa via si rivela, dunque, assai importante per la sopravvivenza di molte cellule tumorali (Keeshan et al. 2001).
Enzimi proteolitici e apoptosi
È possibile dedurre il ruolo degli enzimi proteolitici da diverse osservazioni sperimentali, incluso il fatto che gli inibitori delle proteasi sembrano rallentare l'inizio dell'apoptosi. Una seconda osservazione che ha focalizzato le ricerche sul ruolo delle proteasi nell'apoptosi è stata che la sequenza di Ced-3, un gene per la morte cellulare proveniente da C. elegans, è simile a quella dell'enzima convertitore della interleuchina 1β umana (ICE), che nell'uomo è il mediatore dell'infiammazione. Questo enzima è stato inizialmente scoperto nei macrofagi dove sembra tagliare la prointerleuchina I trasformandola nella sua forma attiva; inoltre appartiene a una famiglia di enzimi chiamati cisteinaproteasi (Martin e Green 1995). Partendo dai risultati ottenuti in vari studi sperimentali, nei quali si era stabilita una relazione diretta tra Ced-3 e ICE, diversi laboratori iniziarono a cercare non soltanto altri enzimi simili a ICE, ma anche i loro substrati nell'apoptosi. A mano a mano che la storia di queste ricerche veniva gradualmente svelata vennero scoperti molti enzimi di questo tipo ora denominati caspasi. A tutt'oggi sono stati identificati circa 14 membri di questa famiglia di enzimi caratterizzati dalla capacità di tagliare i loro substrati polipeptidici immediatamente dopo i residui di acido aspartico. Approssimativamente questi enzimi possono essere suddivisi in due gruppi, uno implicato nella fase iniziale dell'apoptosi e l'altro in quella di attuazione. La caspasi-3 è una classica caspasi implicata nella fase di attuazione che può essere attivata dalle caspasi -8 e -9, due enzimi classificabili come iniziatori. Una volta attivata, la caspasi-3 può spezzare una quantità di substrati proteici che portano al collasso e alla morte delle cellule. Tali substrati comprendono l'enzima di riparazione del DNA poliADP-ribosiopolimerasi (PARP). Un secondo bersaglio è costituito dalla proteina ICAD che inibisce l'endonucleasi del DNA chiamata CAD. Una volta rilasciato dal suo inibitore, questo enzima procede al taglio del DNA in frammenti delle dimensioni di un nucleosoma, i quali costituiscono la caratteristica più tipica dell'apoptosi. Dal punto di vista biologico, ha senso la disattivazione degli enzimi di riparazione del DNA come PARP se uno degli obiettivi dell'apoptosi è la frammentazione del DNA.
È stato sorprendente scoprire che il rilascio del citocromo c da parte dei mitocondri di una cellula danneggiata costituisce un passaggio cruciale che porta all'attivazione della caspasi-9, la quale a sua volta attiva la caspasi-3. Ciò si verifica a causa dell'apertura di pori nella membrana mitocondriale che consentono il rilascio del citocromo c. Quest'ultimo, una volta liberato, si combina con numerose molecole sia di procaspasi-9 sia di una proteina chiamata Apaf-1 per formare un complesso proteico (detto apoptosoma), il quale porta all'attivazione della caspasi-9. Una volta attivata, la caspasi-9 attiva a sua volta immediatamente la caspasi-3. La ricerca che ha portato alla scoperta di questa sequenza di eventi ha svelato quindi un ruolo del citocromo c completamente nuovo, rimasto del tutto sconosciuto per cinquant'anni.
Morte cellulare nel sistema immunitario
La sindrome da immunodeficienza acquisita (AIDS) è una malattia caratterizzata dal lento deterioramento del sistema immunitario la cui causa sembra essere una massiccia diminuzione di un gruppo di linfociti T detti cellule CD4 (Cotter 1993). La funzione principale di queste cellule è di controllare e stimolare la risposta immunitaria alle infezioni. Sembra che la morte di tali cellule nei pazienti affetti da HIV, ossia dal virus dell'immunodeficienza umana, avvenga per apoptosi, sebbene gli eventi cellulari che provocano la morte cellulare siano ancora sconosciuti. Ciò che sappiamo con certezza è che, in gran parte, l'apoptosi delle cellule CD4 non è mediata direttamente dal virus dell'AIDS, ma attraverso un meccanismo indiretto. Una serie di prove indica che l'apoptosi ha luogo a causa di un 'corto circuito' cellulare nei segnali che normalmente dovrebbero portare alla proliferazione delle cellule CD4 e alla risposta immunitaria. Ecco ciò che accade. Una volta entrato nella cellula tramite gp120, una delle proteine strutturali del virus che riveste un ruolo fondamentale nel processo che gli consente di prendere contatto con i linfociti CD4, il virus si integra nel genoma cellulare dove rimane finché non è stimolato a replicarsi. La proteina gp120 viene prodotta anche durante il processo replicativo e può essere rilasciata nel siero dove può legarsi ad altre cellule. Se stimolate per dare origine a una risposta immunitaria, queste stesse cellule vanno incontro ad apoptosi invece di proliferare come farebbero in un processo di risposta normale. Perciò, quando una cellula CD4 riceve un segnale di proliferazione, se nello stesso tempo si è legata a una proteina gp120, si comporta esattamente al contrario in risposta ai segnali provenienti dalla proteina virale. Mentre la proteina gp120 e la stimolazione della cellula CD4 attraverso il suo recettore possono essere gli eventi chiave nella stimolazione dell'apoptosi, poco si sa riguardo al modo in cui avviene esattamente l'apoptosi delle cellule CD4. L'effettiva morte cellulare potrebbe essere causata da una molecola della superficie cellulare detta Fas e dal suo ligando (FasL). Negli ultimi anni Fas è diventata una molecola particolarmente importante per la comprensione dei primi passaggi del meccanismo di apoptosi.
Fas appartiene alla famiglia dei TNF (tumour necrosis factor) così chiamati per il loro ruolo nella morte cellulare. Fas è espressa sulla superficie di molte cellule del corpo e, quando si lega al suo ligando naturale, induce l'apoptosi. La maggior parte delle nostre conoscenze su Fas deriva dagli studi effettuati sulle cellule del sistema immunitario, dove questa proteina sembra svolgere un ruolo cruciale nell'apoptosi (Nagata e Golstein 1995). Nel timo, un organo fondamentale del sistema immunitario responsabile dello sviluppo dei linfociti T, l'apoptosi naturale avviene frequentemente. Infatti circa il 99% di tutti i timociti prodotti muore all'interno del timo e sembra che la morte avvenga per apoptosi. I timociti esprimono un elevato livello di Fas e vi sono numerose prove del fatto che la morte cellulare osservata in queste cellule sia mediata da Fas. Le mutazioni nella molecola Fas causano la proliferazione dei linfociti; ciò indica che questa molecola è fondamentale nella regolazione dell'equilibrio tra proliferazione e morte dei linfociti. Fas ha inoltre un ruolo nella distruzione delle cellule tumorali a opera dei linfociti T citotossici. In altri termini, i linfociti T citotossici uccidono le loro cellule bersaglio inducendo l'apoptosi; in questo processo sembra essere coinvolta anche la proteina Fas.
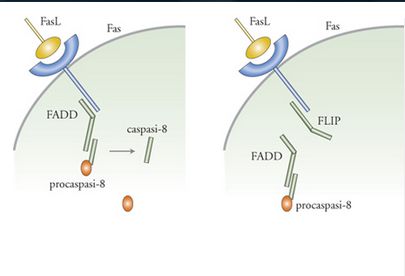
Fas è una proteina recettore transmembrana con una coda citoplasmatica contenente un cosiddetto 'dominio di morte'. Tale denominazione è dovuta al fatto che le mutazioni in questa regione della molecola ne bloccano la capacità di indurre la morte cellulare. Uno dei quesiti fondamentali che i ricercatori si pongono riguarda il modo in cui il recettore Fas determini le principali caratteristiche morfologiche e biochimiche dell'apoptosi. La risposta sembra essere la seguente. Quando il ligando di Fas si lega al suo recettore inizia una cascata di eventi che porta all'attivazione della caspasi. Il primo evento in questa cascata è l'aggregazione del complesso Fas/FasL in modo simile a ciò che si osserva quando molti altri ligandi interagiscono con i rispettivi recettori. Il passaggio successivo prevede la formazione di una struttura composta da più proteine chiamata DISC e situata immediatamente al di sotto della superficie della membrana plasmatica. Componenti chiave di questo passaggio comprendono una proteina chiamata FADD che si lega alla coda citoplasmatica di Fas in risposta all'aggregazione con il recettore. FADD a sua volta interagisce con la procaspasi-8, evento che determina l'attivazione dell'enzima, la quale poi conduce all'attivazione della cascata mediata dalle caspasi e infine all'apoptosi. Al pari di molti altri sistemi biologici, l'attivazione di queste vie implica l'esistenza di un interruttore molecolare che interrompe il processo quando ciò si rivela necessario (fig. 7). Nel caso in questione, l'interruttore che interrompe il processo è una proteina detta FLIP, la quale compete con la caspasi-8 per un sito di legame su FADD; ciò regola la velocità con cui la cellula va incontro ad apoptosi (Irmler et al. 1997).
Morte cellulare nel sistema nervoso
La morte cellulare nel sistema nervoso è stata identificata da quasi un secolo. Nel 1949 Rita Levi-Montalcini dimostrò che nell'embrione di pollo un notevole numero di neuroni andava incontro a morte durante lo sviluppo del sistema nervoso. Si pensò che la morte cellulare osservata fosse parte della complessa sequenza di eventi ontogenetici che porta alla costruzione del sistema nervoso. Questi primi studi aprirono la strada alla comprensione del ruolo svolto dalla morte cellulare nei processi ontogenetici, ma non riuscirono a stimolare l'interesse degli scienziati di allora, forse perché a quei tempi si pensava che la morte cellulare fosse, fondamentalmente, un processo passivo e che, quindi, non meritasse ricerche approfondite. L'interesse per la morte cellulare nel sistema nervoso ebbe nuovo stimolo soltanto negli anni Settanta quando furono presentati i risultati degli importanti lavori sperimentali di Wyllie e dei suoi colleghi. Studi successivi dimostrarono che la morte cellulare, documentata precedentemente dalla Levi-Montalcini, era in realtà l'apoptosi. I neuroni morenti mostravano le classiche caratteristiche morfologiche dell'apoptosi e, inoltre, frammentavano il loro DNA in pezzi delle dimensioni di un nucleosoma. Il fatto che l'apoptosi delle cellule nervose fosse un processo attivo dipendente dalla sintesi di RNA e proteine venne dimostrato coltivando cellule neuronali in assenza di fattori di crescita, come per esempio quello delle fibre nervose (NGF, nerve growth factor). In questo modo le cellule andavano incontro ad apoptosi che poteva essere prevenuta trattandole con un inibitore della sintesi proteica come la cicloesimmide (Martin et al. 1992). Attualmente è noto che durante l'ontogenesi viene prodotta un'enorme quantità di neuroni, i quali sopravvivono se possono formare connessioni con le cellule circostanti, altrimenti muoiono. Tali connessioni non soltanto rendono le cellule vitali, ma sono anche utili nella costruzione del sistema nervoso.
Quindi, se non si connettono con i neuroni vicini, i neuroni neoformati muoiono; la formazione di connessioni è pertanto, per i neuroni, un incentivo alla sopravvivenza. L'uso di topi transgenici ha chiarito ulteriormente il ruolo dell'apoptosi nel sistema nervoso. Per esempio, la dimensione del cervello dei topi resi transgenici per il gene antiapoptotico bcl-2 in modo tale che esso sia espresso nel sistema nervoso è una volta e mezza, o due, rispetto a quella dei topi non transgenici. Non sappiamo però se in questi animali esista una reale relazione tra la loro dimensione cerebrale geneticamente modificata e l'intelligenza.
Una delle caratteristiche delle malattie neurodegenerative degli esseri umani è la morte cellulare nel sistema nervoso centrale. Sebbene ancora si discuta sul fatto che la morte cellulare osservata avvenga oppure no per apoptosi, le prove a disposizione sono a favore dell'apoptosi. Con la perdita di cellule si verifica un deterioramento delle funzioni cerebrali che si manifesta in molti modi. Prevenendo la perdita di cellule cerebrali, è probabile che si possa intervenire efficacemente in malattie quali l'Alzheimer e il morbo di Parkinson. Poiché è stato dimostrato che bcl-2 è un potente inibitore della morte cellulare dei neuroni, è probabile che in questo ambito ci sia la possibilità di sviluppare terapie basate sull'utilizzo di bcl-2. Potrebbe anche essere possibile usare tecniche di terapia genica per indirizzare bcl-2 in specifiche cellule cerebrali e prevenirne la morte che si verifica nelle malattie come quelle sopra ricordate. Una strada alternativa potrebbe essere quella di progettare farmaci che provochino un aumento di bcl-2 nelle cellule bersaglio, o anche farmaci che ne simulino gli effetti antiapoptotici.
Prospettive nell'uso di terapie basate sull'apoptosi
Nella sezione precedente si è brevemente accennato alla modificazione dell'espressione dei geni che regolano l'apoptosi, ma potrebbe essere utile approfondire questo argomento considerando che la comprensione del meccanismo che ne è alla base potrebbe essere utilizzata in futuro come forma di trattamento di molte malattie umane. Per esempio, come già ricordato precedentemente, il gene chimerico bcr-abl è la causa scatenante della leucemia mieloide cronica (CML, chronic myeloid leukemia). Il meccanismo sembra essere la soppressione del controllo dei normali livelli di apoptosi. In precedenza si era dimostrato che il trattamento delle cellule con un oligonucleotide anti sense per bloccare la sintesi della proteina bcr-abl inverte la resistenza all'apoptosi (McGahon et al. 1994). Il principio del metodo basato sugli oligonucleotidi anti sense consiste nel sintetizzare un breve oligonucleotide lungo circa 20 paia di basi, il cui bersaglio è la sequenza iniziale dell'mRNA specifico di una particolare proteina. Quando tale oligonucleotide entra nella cellula si lega a quel particolare mRNA bersaglio come se fosse un 'bubble-gum genetico', impedendo la traduzione dell'mRNA in proteina. Così, utilizzando questo approccio, dovrebbe essere possibile bloccare la sintesi di qualsiasi proteina. Nel caso del nostro lavoro sulla proteina bcr-abl, utilizzando questo metodo, abbiamo potuto invertire la resistenza all'apoptosi che si verifica nella CML. Una strategia simile è stata applicata anche al gene bcl-2 nel linfoma delle cellule follicolari. La causa di questo tipo di tumore è l'aumento abnorme dell'espressione di bcl-2; quando tale fenomeno viene invertito utilizzando gli anti sense, le cellule del linfoma vanno incontro ad apoptosi. Inoltre il metodo degli anti sense per diminuire l'espressione di bcl-2 si è dimostrato valido in molti modelli animali utilizzati per studiare lo sviluppo e la progressione dei tumori, al punto che ora viene usato nei trial clinici. Una variante del metodo anti sense descritta recentemente è nota come RNA interference o RNAi. L'RNAi è un processo cellulare che si attiva quando nella cellula entra un RNA a doppio filamento che possiede più di 18 nucleotidi. Ciò causa la degradazione non soltanto dell'RNA invasore ma anche degli RNA a filamento singolo che abbiano identica sequenza. Questa tecnica si è rivelata più facile da usare e più economica di quella basata sugli anti sense.
La tecnica degli anti sense non è l'unica adottabile per cercare di modificare la frequenza dell'apoptosi. Per esempio, il gene p53, che favorisce l'apoptosi, in molti tipi di cancro umano si trova in forma mutata e quindi non funzionale. Di conseguenza molte cellule tumorali non muoiono per apoptosi quando dovrebbero. Tuttavia, se nelle cellule tumorali viene espressa una proteina p53 completamente funzionante, esse vanno rapidamente incontro ad apoptosi. Ciò che sembra accadere in questo caso è che non appena p53 viene espressa nelle cellule tumorali, questa proteina individua le mutazioni del DNA che causano il tumore e immediatamente stimola la morte cellulare per apoptosi. Ciò può essere dimostrato in laboratorio, anche se un problema non indifferente riguarda il modo di inserire una p53 normale nella cellula. A questo scopo possono essere utilizzate diverse tecniche, la più promettente delle quali è forse l'uso di un vettore virale che trasporti il gene p53 nella cellula tumorale bersaglio. Uno di tali vettori di trasporto è l'adenovirus, che con tecniche di ingegneria genetica può essere indotto a trasportare il gene p53 dentro la cellula e, una volta entrato, a esprimere la proteina. Quando ciò accade, nella cellula tumorale il risultato è la morte cellulare. È ovvio che prima che questo approccio possa essere attuato in una valida terapia anticancro bisogna superare alcuni ostacoli scientifici, tra i quali come indirizzare il gene nella cellula bersaglio. Sono quasi nulle le informazioni disponibili riguardanti cosa potrebbe succedere se il gene p53 raggiungesse una cellula normale invece di una tumorale. L'espressione di proteina p53 in tale cellula risulterebbe aumentata e la cellula potrebbe di conseguenza diventare ipersensibile all'apoptosi, cosa che potrebbe rivelarsi non auspicabile. Inoltre, una volta che p53 fosse arrivata alla sua cellula bersaglio, sarebbe effettivamente espressa in quantità sufficientemente elevata da stimolare l'apoptosi? Quando avremo la risposta a tali interrogativi cruciali forse saremo in grado di usare questo tipo di approccio nelle cellule tumorali per far pendere l'ago della bilancia tra mitosi e apoptosi in favore di quest'ultima.
Una delle caratteristiche interessanti dell'adenovirus è che esso possiede un gene chiamato E1B con funzione antiapoptotica. Ovviamente, qualora il virus dovesse essere usato nella terapia genica come trasportatore di p53, il gene E1B dovrebbe essere eliminato. Fortunatamente ciò può essere effettuato abbastanza facilmente. Ci si potrebbe chiedere in primo luogo come mai un virus come l'adenovirus possegga un gene di quel tipo e se altri virus hanno geni simili. I geni capaci di bloccare l'apoptosi si trovano in molti virus; oltre al gene E1B dell'adenovirus, il virus del vaiolo bovino produce una proteina chiamata crmA che è un inibitore della proteasi che blocca alcuni degli eventi proteolitici chiave nella via del segnale apoptotico. Sui motivi per i quali i virus abbiano simili geni possiamo ipotizzare quanto segue: il principale obiettivo nella vita di qualsiasi virus è quello di replicarsi e, per far ciò, deve infettare una cellula e successivamente deve 'dirottare' l'apparato di replicazione delle cellule verso la produzione di copie di sé stesso. Producendo proteine antiapoptotiche come E1B, crmA o altre, il virus può impedire alla cellula di suicidarsi, facendo in modo che essa continui a produrre copie di sé stesso. La cellula infettata potrebbe voler andare incontro ad apoptosi perché in questo modo impedirebbe al virus di replicarsi e di infettare altre cellule dell'organismo. Purtroppo, molti virus e altri microorganismi che devono replicarsi insieme con le cellule ospiti hanno acquisito la capacità di mantenere in vita le cellule indispensabili al loro processo replicativo.
Bibliografia
Cohen, Duke 1984: Cohen, J. John - Duke, Rick C., Glucocorticoid activation of a calcium-dependent endonuclease in thymocyte nuclei leads to cell death, "The Journal of immunology", 132, 1984, pp. 38-42.
Cotter 1993: Cotter, Thomas G., Cell death in the immune system, "The immunologist", 1, 1993, pp. 181-184.
Fadok, Chimini 2001: Fadok, Valerie A. - Chimini, Giovanna, The phagocytosis of apoptotic cells, "Seminars in immunology", 13, 2001, pp. 365-372.
Irmler 1997: Irmler, Martin e altri, Inhibition of death receptor signals by cellular FLIP, "Nature", 388, 1997, pp. 190-195.
Keeshan 2001: Keeshan, Karen - Mills, Ken I. - Cotter, Thomas G. - McKenna, Sharon L., Elevated Bcr-Abl expression levels are sufficient for a haematopoietic cell line to acquire a drug-resistant phenotype, "Leukemia", 15, 2001, pp. 1823-1833.
Kerr 1971: Kerr, John F.R., Shrinkage necrosis: a distinct mode of cellular death, "Journal of pathology", 105, 1971, pp. 13-23.
Kerr 1972: Kerr, John F.R. - Wyllie, Andrew H. - Currie, Alastair R., Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics, "British journal of cancer", 26, 1972, pp. 239-257.
Lockshin, Zakeri 1991: Lockshin, Richard A. - Zakeri, Zahra, Programmed cell death and apoptosis, in: Apoptosis, the molecular basis of cell death, edited by L. David Tomei and Frederick O. Cope, Cold Spring Harbor, Cold Spring Harbor Laboratory Press, 1991, pp. 47-60.
McDonnell, Korsmeyer 1991: McDonnell, Timothy I. - Korsmeyer, Stanley J., Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14; 18), "Nature", 349, 1991, pp. 254-256.
McDonnell 1993: McDonnell, Timothy I. - Troncoso, Patricia - Brisbay, Shawn - Chung, Leland - Logothetis, Christopher - Hsieh, Jer-Tsong - Cambell, Martin - Tu, Shi-Ming, Bcl-2 expression in the prostate and its association with androgen-independent prostate cancer, in: Programmed cell death, the cellular and molecular biology of apoptosis, edited by Martin Lavin, Dianne Watters, Reading, Harwood Academic, 1993, pp. 179-186.
McGahon 1994: McGahon, Anne - Bissonnette, Reid - Schmitt, Manfred - Cotter, Kate M. - Green, Douglas R. - Cotter, Thomas G., BCR-ABL maintains resistance of chronic myelogenous leukemia cells to apoptotic cell death, "Blood", 83, 1994, pp. 1179-1187.
Martin 1992: Martin, Diane P. - Ito, Akira - Horigome, Kazuhiko - Lampe, Patricia A. - Johnson, Eugene M. jr, Biochemical characterization of programmed cell death in NGF-deprived sympathetic neurons, "Journal of neurobiology", 23, 1992, pp. 1205-1220.
Martin, Green 1995: Martin, Seamus J. - Green, Douglas R., Protease activation during apoptosis: death by a thousand cuts?, "Cell", 82, 1995, pp. 349-352.
Nagata, Golstein 1995: Nagata, Shigekazu - Golstein, Pierre, The Fas death factor, "Science", 267, 1995, pp. 1449-1456.
Oltvai 1993: Oltvai, Zoltan N. - Milliman, Curt L. - Korsmeyer, Stanley J., Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death, "Cell", 74, 1993, pp. 609-619.
Pelengaris 2002: Pelengaris, Stella - Khan, Mike - Evan, Gerard, c-MYC: more than just a matter of life and death, "Nature reviews. Cancer", 2, 2002, pp. 764-776.
Ray 1992: Ray, Carol A. - Black, Roy A. - Kronheim, Shirley R. - Greenstreet, Teresa A. - Sleath, Paul R. - Salvesen, Guy S. - Pickup, David J., Viral inhibition of inflammation: cowpox virus encodes an inhibitor of the interleukin-1 beta converting enzyme, "Cell", 69, 1992, pp. 597-604.
Samali, Cotter 1996: Samali, Afshin - Cotter, Thomas G., Heat shock proteins increase resistance to apoptosis, "Experimental cell research", 223, 1996, pp. 163-170.
Shuey 2002: Shuey, David J. - McCallus, Daniel E. - Giordano, Tony, RNAi: gene-silencing in therapeutic intervention, "Drug discovery today", 20, 2002, pp. 1040-1046.
Spiers 1992: Spiers, Alexander S.D., in: Leukemia, edited by John Alan Whittaker, Oxford, Blackwell Scientific Publications, 1992, pp. 434-467.
Yonish-Rouach 1991: Yonish-Rouach, Elisheva - Resnitzky, Dalia - Lotem, Joseph - Sachs, Leo - Kimchi, Adi - Oren, Moshe, Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6, "Nature", 352, 1991, pp. 345-347.
Wyllie 1980: Wyllie, Andrew H. - Kerr, John F.R. - Currie, Alastair R., Cell death: the significance of apoptosis, "International review cytology", 68, 1980, pp. 251-306.