
La grande scienza. Genomica e postgenomica
La grande scienza. Genomica e postgenomica
Genomica e postgenomica
La genetica si è posta, fin dalle sue origini, una serie di domande sui geni: come vengono trasmessi da una generazione all'altra, di che cosa sono fatti, come vengono duplicati e come funzionano. Per rispondere a queste domande la genetica classica utilizza una o più forme varianti di un gene (detto anche allele o gene mutato) che permettono di rilevare l'esistenza del gene stesso e di ipotizzarne la funzione. Gregor Mendel (1822-1884), per esempio, non avrebbe mai potuto affermare che esiste un fattore (gene) per l'altezza dello stelo nel pisello da giardino se non avesse trovato alcune piante nane (mutanti) che in un certo senso soffrono per una mutazione nel gene dell'altezza. La genetica classica ha perciò dedotto l'esistenza dei geni dall'osservazione di come cambia l'aspetto della progenie nata da incroci opportuni fra mutanti e non-mutanti. Per la maggior parte degli organismi viventi tali incroci non possono essere realizzati facilmente o sono improponibili (come nella specie umana). Gli unici organismi animali complessi sui quali è stato possibile utilizzare abbastanza facilmente l'approccio della genetica classica (cioè gli incroci) sono quelli che hanno un ciclo vitale piuttosto rapido e che possono essere allevati numerosi in un laboratorio come, per esempio, il moscerino della frutta (Drosophila melanogaster).
Che i geni fossero costituiti da DNA (acido desossiribonucleico) fu scoperto nel 1944 da Oswald T. Avery (1877-1955) insieme ad alcuni colleghi al Rockefeller Institute di New York. Nel 1953 James D. Watson e Francis H.C. Crick definirono la struttura fisica della molecola di DNA ponendo le basi delle scoperte successive (il ruolo del DNA alla base del codice genetico, la sua capacità di autoduplicarsi e così via). La molecola di DNA ha l'aspetto di una doppia elica nella quale due filamenti paralleli si avvolgono tra loro con un andamento a spirale. Da un punto di vista biochimico il DNA è costituito da quattro tipi di nucleotidi; ogni nucleotide è composto da una molecola di zucchero (il desossiribosio), una di acido fosforico (o fosfato) e da una base azotata. Le basi azotate rappresentano le quattro unità fondamentali del codice della vita e sono: adenina (A), guanina (G), citosina (C) e timina (T). Esse possono appaiarsi secondo la regola della complementarità che consente l'appaiamento di A con T e di G con C; pertanto, le coppie di basi 'legittime' sono soltanto AT, TA, GC e CG. La doppia elica del DNA può essere paragonata a una scala a chiocciola, i cui gradini sono rappresentati dalle coppie di basi e le cui ringhiere sono le molecole di desossiribosio unite fra loro da gruppi di fosfato. Se immaginiamo di separare i due filamenti della doppia elica e di utilizzare ciascuno di essi come calco o stampo per costruire un'altra doppia elica, grazie alla proprietà della complementarità, si producono due doppie eliche identiche a quella originaria. Tale proprietà può essere sfruttata sperimentalmente per portare due frammenti di singole eliche (filamenti) di DNA a riconoscersi tra loro fra milioni di singole eliche 'estranee', cioè non complementari. Il fenomeno del riconoscimento specifico e appaiamento tra molecole complementari viene definito 'ibridazione molecolare'.
Alla metà degli anni Settanta due nuove tecniche hanno cambiato radicalmente il campo della ricerca genetica, consentendo di leggere l'intero patrimonio ereditario (genoma) di ogni organismo vivente. La prima è il 'clonaggio' del DNA, che permette di preparare a volontà 'biblioteche' di DNA (dette appunto genoteche) che rappresentano interi genomi ridotti in piccoli frammenti (si noti che il clonaggio di geni e frammenti di DNA non ha nulla a che fare con la clonazione di individui). La seconda tecnica è rappresentata dall'invenzione dei metodi per sequenziare il DNA, cioè per stabilire con precisione come sono disposte una dopo l'altra le quattro basi azotate del DNA in sequenze lunghe milioni, o miliardi, di basi (per es., il genoma umano è composto di circa 3,1 miliardi di basi, la cui sequenza è stata recentemente determinata dal Progetto genoma umano).
Il Progetto genoma umano

Il Progetto genoma umano rappresenta sostanzialmente l'anello di congiunzione fra ricerca genetica e ricerca genomica. In effetti, l'interesse della comunità scientifica per questo progetto, nonché le notevoli risorse finanziarie investite da diversi governi, per la prima volta nella storia della biologia, in questa operazione di Big science, hanno permesso la transizione dalla ricerca genetica alla ricerca genomica. Sebbene il Progetto genoma umano sia basato e sia progredito grazie all'analisi di organismi più semplici dell'uomo, come vedremo più avanti, è pur vero che l'analisi genetica dell'uomo stesso e delle sue malattie (cioè la genetica medica e, più di recente, la medicina genetica; Tav. I) hanno rappresentato la forza trainante del progetto stesso.
La genetica medica si era dotata di una base anatomica. I genetisti medici ebbero così il loro organo da studiare, il nucleo della cellula e i suoi cromosomi, così come i cardiologi avevano come oggetto di studio il cuore e i neurologi il sistema nervoso. Iniziò allora una rivoluzione, che è ancora in corso e che sta progressivamente portando la genetica al centro della medicina, analogamente a quanto accadde nel XVI sec. quando Andrea Vesalio, nel De humani corporis fabrica libri septem, pose le basi della medicina moderna.
Il secondo sviluppo che ha fornito una base microanatomica alla genetica medica è stata la mappatura dei geni, che ha avuto inizio nel 1968 con la localizzazione di un gruppo sanguigno (il sottogruppo Duffy) sul cromosoma 1. La disposizione lineare dei geni nei nostri cromosomi, spesso paragonata a un filo di perle, è parte della nostra microanatomia. La conoscenza di questa anatomia è risultata molto importante per la medicina, in quanto ha permesso, a partire dal 1986, la scoperta di geni basata sulla loro mappatura e sulle cosiddette tecniche di 'clonaggio posizionale'. Nel caso delle malattie 'mistero' - così chiamate perché la natura della loro anomalia biochimica di base era sconosciuta, come corea di Huntington, fibrosi cistica, distrofia muscolare, ecc. - il procedimento di indagine consiste nel determinare su ciascun cromosoma la posizione esatta del gene da identificare come responsabile della malattia. Ciò si ottiene attraverso lo studio del rapporto di concatenazione, o linkage del gene sconosciuto con marcatori distribuiti sui cromosomi, utilizzando opportunamente i campioni di sangue prelevati dalle famiglie degli individui affetti. È necessario sottolineare che la risorsa più importante per questo tipo di ricerca è rappresentata dalle famiglie dei pazienti, alle quali viene di fatto riconosciuta da tutti i ricercatori una coproprietà dei 'geni-malattia' che vengono scoperti grazie alla loro collaborazione.
È opportuno aggiungere che sebbene non esista ancora nessuna legge che riconosca alle famiglie dei pazienti una coproprietà intellettuale sui 'geni-malattia' clonati attraverso il loro contributo, è anche vero che, finora in un unico caso, la brevettazione di due geni-malattia da parte di una ditta nord- americana ha portato a un regime di monopolio (valido per il momento solo negli Stati Uniti) per quanto riguarda la loro utilizzazione diagnostica. Sull'argomento della brevettazione dei geni umani si è aperto un dibattito molto acceso e diversi paesi europei si sono opposti alla richiesta della ditta americana in questione di estendere tale brevetto anche all'Europa.
Una volta mappato, il gene-malattia può essere paragonato a un oggetto non identificato, localizzato da un radar, che si trova in mezzo al mare rappresentato dal DNA di un organismo. Di questo oggetto si conoscono le coordinate precise della sua localizzazione (cioè latitudine e longitudine), ma non si conosce la sua natura. Le tappe successive della ricerca consistono nell'avvicinarsi al gene con tutti i mezzi tecnici disponibili, clonarlo, cioè isolarlo e determinarne la sequenza di DNA, e poi studiarne la funzione normale e i difetti (mutazioni) che possono essere causa della malattia. La mappatura dei geni ha quindi contribuito anch'essa a fornire alla medicina una base neovesaliana. Nel corso degli ultimi quindici anni tutte le branche della medicina hanno utilizzato l'identificazione di geni tramite mappatura per fare luce sulle malattie di cui si ignorano le cause.
Come terzo punto di sviluppo, il Progetto genoma umano ha fornito alla medicina genetica la componente anatomica più raffinata: la sequenza completa del genoma. È facilmente intuibile come questa sequenza completa renda oggi relativamente più facile il clonaggio di geni-malattia. Il progetto venne formalmente proposto nel 1988 da Renato Dulbecco, mentre il temine 'genomica' era stato coniato nel 1986 (Tav. I). L'obiettivo di mappare tutti i geni sui cromosomi umani era già sostenuto da 15 anni e di questi negli anni Ottanta ne erano già stati localizzati su specifici cromosomi almeno 700.
Negli Stati Uniti, il Progetto genoma umano ha avuto inizio nel 1990. L'approccio era top-down: in primo luogo si dovevano creare mappe di marcatori con intervalli frequenti lungo i cromosomi, per poi suddividere successivamente ogni cromosoma in segmenti parzialmente sovrapponibili (utilizzabili anche per studi di mappatura di geni-malattia nelle famiglie), e arrivare infine a determinare la sequenza dei nucleotidi AGTC.
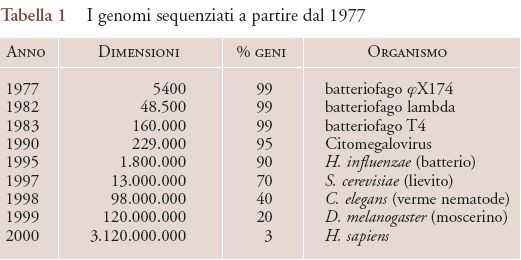
Craig Venter e i suoi colleghi della ditta privata Celera Genomics hanno utilizzato, invece, un approccio bottom-up: l'intero genoma è stato fisicamente spezzato in segmenti sequenziabili singolarmente e, solo alla fine, i pezzi in disordine sono stati assemblati con l'aiuto di computer, per ricomporre il genoma. Questo metodo è stato successivamente denominato shot-gun (letteralmente, colpo di fucile) e la sua efficacia è stata testata per la prima volta nel 1995, producendo la prima sequenza completa di un organismo capace di vita autonoma, il batterio Haemophilus influenzae (tab. 1).
Successivamente il metodo shot-gun è stato utilizzato per completare la sequenza del genoma di altri organismi, tra cui quello preferito dai genetisti, il moscerino della frutta Drosophila e, infine, lo stesso genoma umano. Il completamento di una prima 'bozza' della sequenza del genoma umano è stato annunciato congiuntamente da Francis Collins e Craig Venter il 26 giugno 2000. Per capire quanto è stato rapido lo sviluppo della genetica e della genomica, è sufficiente ricordare che tra la scoperta della struttura del DNA, avvenuta nel 1953, e la versione definitiva della sequenza dell'intero genoma umano, pubblicata nell'aprile del 2003, sono passati soltanto 50 anni.
Genoma umano in numeri, genomica comparata e variabilità umana
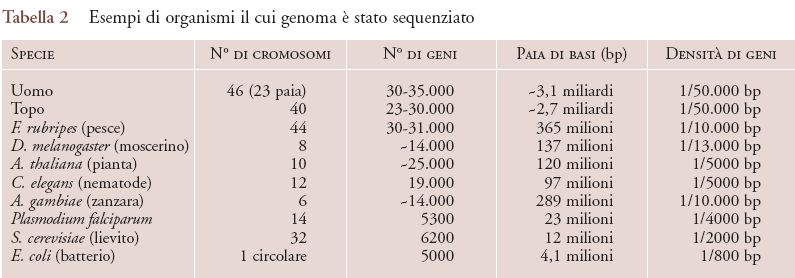
Oltre alla possibilità di clonare geni-malattia con maggiore facilità che in passato, la sequenza completa del genoma umano ci ha fornito una gran quantità di informazioni. Sappiamo oggi che nel nucleo di ogni cellula del nostro organismo (che conta circa 1015 cellule) ci sono 3,1 miliardi di paia di basi. Il gene più lungo nel nostro genoma si chiama distrofina ed è composto di 2,4 milioni di paia di basi. La lunghezza media di un gene è di 100 kb (1 kb=1000 paia di basi). La sequenza del genoma permette di identificare geni usando il computer e algoritmi appositamente sviluppati per questo scopo (il cosiddetto clonaggio in silico, ovvero una sorta di clonaggio virtuale). Questi algoritmi tengono conto della struttura dei geni, degli elementi di regolazione genica e del contesto cromosomico. Le predizioni ottenute al computer possono poi essere verificate sperimentalmente attraverso la corrispondenza con RNA o proteine. Dalla combinazione di predizioni in silico e verifiche sperimentali, si è arrivati alla stima secondo cui i geni presenti nel nostro DNA sarebbero compresi tra 30.000 e 35.000, un numero molto inferiore ai circa 100.000 geni ipotizzati fino a pochi anni fa. La discrepanza tra le due stime è da imputarsi al fatto che le prime regioni dei cromosomi analizzate contenevano un'alta densità di geni, mentre l'intero genoma mostra ampi tratti con pochi o anche nessun gene (i cosiddetti 'deserti genici'). I nostri geni occupano perciò soltanto una piccola porzione, circa il 2%, del genoma umano (tab. 2).
La funzione del resto del genoma e il significato della sua struttura non sono ancora ben chiari, sebbene sia evidente che nelle regioni non occupate da geni (intergeniche) risieda, almeno in parte, l'informazione che determina dove, quando e in che misura i geni devono essere attivati. Ancora più misteriosa è l'evoluzione che ha portato le regioni intergeniche e l'intero genoma a essere occupati da corte sequenze di DNA ripetute migliaia di volte. Le sequenze ripetute nel loro complesso costituiscono circa il 50% del DNA genomico umano.
La genomica è diventata, quindi, la disciplina che studia un intero complesso di sequenze di DNA, sia codificanti sia noncodificanti. L'obiettivo finale della ricerca genomica (o per meglio dire, della ricerca che è alla base del completamento del progetto genoma e cioè della ricerca postgenomica) è quello di capire come sono strutturate e come funzionano le reti (network) in cui i singoli geni sono attivi.
Il sequenziamento completo di molti altri organismi e la creazione di grandi banche dati di sequenze ha aperto, infine, la strada alla genomica comparata. L'identificazione nell'uomo di una sequenza che potrebbe essere un gene, può essere effettuata anche ricercando la stessa sequenza nel genoma di un organismo meno complesso, in cui la sua funzione potrebbe essere già nota o comunque verificabile attraverso esperimenti assolutamente non effettuabili nell'uomo. Per restare nella metafora, si fa uso dell'anatomia comparata.
Gli esseri viventi assumono aspetti e forme diverse ma hanno anche moltissimi caratteri in comune. Tutti conserviamo sequenze di DNA simili o molto simili ereditate dai nostri comuni antenati vissuti milioni di anni fa. In particolare, gli esseri umani condividono molti geni con topi, rane, moscerini, vermi, batteri e lieviti poiché le loro funzioni, essenziali per la vita, sono state trasmesse inalterate di generazione in generazione per lunghissimi periodi di tempo.
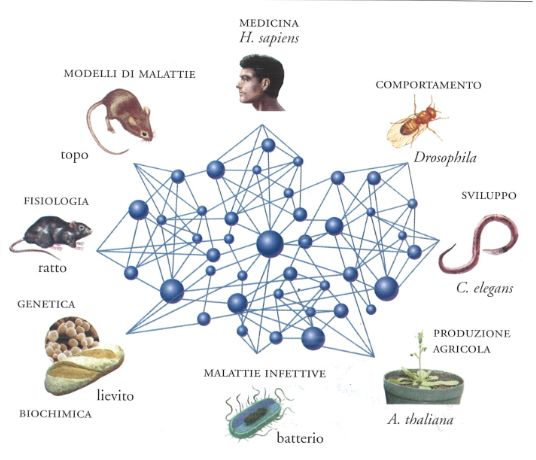
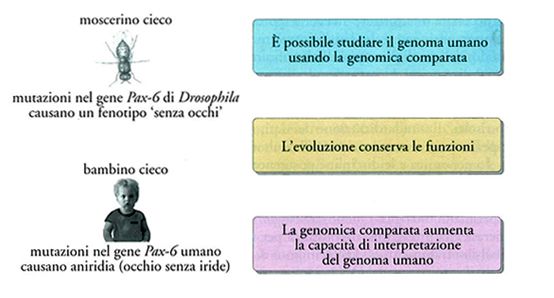
Negli ultimi anni la genomica comparata ha assunto un ruolo molto importante per la scoperta di nuovi geni nell'uomo e per lo studio della loro funzione, come è riassunto schematicamente nella fig. 2 in cui i genomi degli organismi più studiati (topo, Drosophila, Caenorhabditis elegans, lievito) vengono associati alle particolari informazioni che essi forniscono sotto questo specifico profilo (il topo è utilizzato per lo studio di modelli di malattie genetiche, il C. elegans per quello dei geni dello sviluppo e la Drosophila per quello dei geni del comportamento, e così via). Uno dei primi esempi di corrispondenza tra fenotipo umano e di Drosophila causati entrambi da mutazioni in geni omologhi nelle due specie, è riassunto nella fig. 3. Essa mostra come una mutazione del gene Pax-6 di Drosophila causa in questo moscerino un fenotipo 'senza occhi' in modo analogo a quanto avviene nella specie umana in cui la stessa mutazione è associata all'assenza di iride.
In campo medico il principale interesse della ricerca che scaturisce dal completamento del Progetto genoma umano è oggi rappresentato dalla possibilità di identificare geni e mutazioni che causano predisposizione a malattie molto frequenti nella popolazione quali l'ipertensione, il diabete, le malattie cardiovascolari e alcuni tipi di cancro. Gli studi di questo tipo si basano su una strategia alquanto diversa dalla mappatura di geni mediante linkage che ha funzionato egregiamente per l'identificazione di geni e mutazioni che causano malattie rare (quali la fibrosi cistica, la corea di Huntington e la distrofia muscolare). Lo scopo di questo nuovo tipo di ricerca è, infatti, individuare in alcune piccole variazioni della sequenza del nostro DNA le possibili cause di predisposizione a malattie comuni. In ogni individuo della nostra specie la sequenza del DNA è al 99,9% identica alla sequenza di qualsiasi altro essere umano. Le piccole variazioni di cui stiamo parlando riguardano il cambiamento (o la mutazione) di un singolo nucleotide su tutto l'insieme dei 3,1 miliardi di nucleotidi che costituiscono il nostro genoma e vengono perciò chiamate SNPs (single nucleotide polymorphisms). Nel genoma umano questi cambiamenti, o piccole variazioni, fra individui sono stati fino a oggi riscontrati in circa 2,2 milioni di posizioni distinte, ovvero una ogni 1500 basi circa. Anche se molti SNPs non hanno alcun effetto sulla funzione della cellula, alcuni possono predisporre una persona a una malattia o causare una risposta anomala a un farmaco. Così è nato l'interesse della medicina per questo nuovo tipo di studi di associazione fra SNPs e predisposizione alle malattie genetiche complesse e per definire tale approccio innovativo è stato coniato il temine medicina genetica.
La sequenza del genoma umano e quella completa del genoma di altri 99 organismi è stata ottenuta con due anni di anticipo sulle previsioni, un evento quest'ultimo assolutamente straordinario e imprevedibile secondo le aspettative del decennio precedente. I genomi sequenziati sono rappresentativi di quasi tutte le forme di vita, dagli archeobatteri (batteri primitivi, interessanti per comprendere l'evoluzione della vita, ma anche fonte di enzimi con caratteristiche estreme quale, per es., la capacità di funzionare a temperature intorno ai 100°C), ai batteri, agli eucarioti (organismi le cui cellule sono dotate di nucleo). Tra i batteri oltre al classico Escherichia coli, utilizzato come organismo modello e strumento di laboratorio, sono stati sequenziati rappresentanti dei principali batteri patogeni (Salmonelle, Streptococchi, Listerie, Micobatteri) e le loro controparti non patogene; una lista aggiornata dei loro genomi batterici e le informazioni più recenti sulla ricerca in questo settore sono reperibili presso il National human genome research institute (NHGRI). Il confronto tra questi genomi ha già consentito di identificare numerosi geni candidati come responsabili di varie patologie e quindi potenziali bersagli, diretti o indiretti, per il trattamento farmacologico. I batteri sono anche l'oggetto di un importante progetto chiamato Genomes to life (GTL), sponsorizzato dal Dipartimento per l'energia degli Stati Uniti (DOE), che prevede l'integrazione della sequenza del genoma con lo studio del trascrittoma e del proteoma.
Oltre agli eucarioti, già menzionati come organismi modello di rilevante interesse scientifico, sono stati interamente sequenziati, o stanno per essere completati, i genomi di specie di interesse agroindustriale quali il bue, il maiale, la gallina, il riso, il mais e il frumento.
Esistono numerosi siti internet per accedere alle banche dati che contengono la sequenza del genoma dei vari organismi. Tali informazioni sono liberamente accessibili a chiunque perché la maggior parte di questi importanti progetti è stata finanziata da fondi pubblici oppure perché le aziende private (per es., Celera Genomics con il genoma umano o Monsanto con il genoma del riso) hanno deciso di rendere le proprie informazioni parzialmente disponibili. In molti siti specializzati sono disponibili non solo i dati 'grezzi' ovvero la sequenza, base dopo base, del genoma ma anche le banche dati 'annotate' che cioè riportano le informazioni biologiche note associate a ogni sequenza (identità del gene, funzione, omologie con altri geni). Esempi di siti specializzati sono il WormBase, ovvero il progetto di sequenziamento del genoma per C. elegans, e il FlyBase, ovvero il database del genoma per la Drosophila.
Esiste poi una banca dati dove sono depositate tutte le sequenze disponibili di DNA, non solo genomi ma anche singoli geni (completi o parziali) clonati da qualsiasi specie vivente (nonché alcuni esempi di sequenze di DNA ottenute da animali estinti, anche se non si tratta di dinosauri come fantasticamente riportato nella recente filmografia). Questa banca generale è GenBank® negli Stati Uniti oppure l'europea EMBL Nucleotide database e la giapponese DNA database of Japan. Nel 1990, primo anno del Progetto genoma umano, il numero totale di basi di DNA sequenziato e depositato era di 80 milioni; oggi nel 2003 questo numero è aumentato di 500 volte, arrivando, con andamento esponenziale, a 40 miliardi di basi. È opportuno concludere con un riferimento al sito forse più informativo in assoluto per i professionisti ma anche per tutti coloro che sono interessati alle scienze biomediche: si tratta delle pagine e delle banche dati gestite dal National center for biotecnology information (NCBI) che è a sua volta finanziato dai National institutes of health (NIH) degli Stati Uniti. Tale sito consente l'accesso a GenBank®, a OMIM (Online Mendelian inheritance in man) e a PubMed, la quale è la banca dati più completa che riporta le citazioni bibliografiche relative a oltre 4600 riviste scientifiche a carattere biomedico.
Postgenomica
Per assonanza con il termine genoma, l'intero complemento di RNA o di proteine sintetizzate in una cellula sono stati definiti rispettivamente 'trascrittoma' e 'proteoma', il cui studio, a causa della complessità delle analisi e della molteplicità di campi di applicazione, può essere assimilato a un'autentica disciplina scientifica.
Le discipline postgenomiche sono state definite tali perché l'identità dei geni costituisce il loro riferimento primario ed esse sono organizzate in modo simile alla genomica. Molti degli strumenti informatici e di comunicazione (per es., banche dati accessibili tramite internet) utilizzati per i progetti postgenomici sono stati inizialmente sviluppati per rispondere alle necessità dei vari progetti genoma. Le discipline postgenomiche mutuano dalla genomica anche il modello di impresa scientifica su larga scala organizzata sotto forma di consorzi nazionali o internazionali (nuovamente la Big science). L'obiettivo comune di queste discipline è fornire una descrizione globale dei fenomeni, prendendone contemporaneamente in esame ogni aspetto e riservando all'analisi successiva la determinazione dei rapporti funzionali tra gli oggetti osservati. Esse perciò si discostano radicalmente dall'approccio riduzionista, o hypothesis-driven, utilizzato finora in biologia, dove lo studio dei fenomeni biologici viene affrontato analizzando di volta in volta uno o pochi geni (così come proteine o metaboliti).
La descrizione globale dei fenomeni biologici è stata anche soprannominata 'biologia dei sistemi'; uno dei suoi principali vantaggi è che invece di raccogliere soltanto i dati necessari ad accettare o respingere una certa ipotesi, essa riunisce contemporaneamente tutte le informazioni disponibili permettendo, in linea di principio, di identificare fenomeni non ipotizzabili a priori. Un aspetto ulteriore consiste nell'estrema complessità delle informazioni raccolte dalle discipline post-genomiche, che pone problemi inediti, e non ancora del tutto risolti, di standardizzazione dei dati, riproducibilità degli esperimenti e interpretazione dei risultati.
La genomica e le discipline postgenomiche sono state rese possibili da un'evoluzione tecnologica molto rapida che ha portato, tra l'altro, anche all'automazione su larga scala degli esperimenti biologici. Queste tecnologie, tra cui la PCR (polymerase chain reaction, una tecnica per moltiplicare in vitro qualsiasi frammento di DNA milioni di volte), il sequenziamento automatico del DNA, i microarray o DNA chip, e la spettrometria di massa applicata all'analisi di proteine, si sono rivelate cruciali sia per la capacità di produrre grandi quantità di dati sia perché hanno ridotto di diversi ordini di grandezza i tempi e i costi delle analisi. Per esempio, la capacità di sequenziamento del DNA di un laboratorio di medie dimensione è passata, nel giro di un decennio, da mille basi al giorno (al costo di alcuni euro per base) al milione e oltre di basi al costo di pochi centesimi per base. I centri dedicati al sequenziamento del DNA quali il Wellcome trust Sanger center a Cambridge, o il Department of energy joint genome institute in California, producono dati di sequenza al ritmo di 1000 basi al secondo in modalità 24/7, cioè 24 ore al giorno per sette giorni alla settimana.
È evidente che l'approccio riduzionista alla ricerca biologica, prima di essere un'espressione di filosofia della scienza, ha rappresentato fino agli inizi degli anni Novanta una necessità, a causa delle limitazioni tecnologiche di allora.
Il flusso di informazione e le discipline postgenomiche
Alla fine degli anni Cinquanta si arrivò a postulare il cosiddetto 'dogma centrale' della biologia molecolare che afferma che la sequenza di DNA di ogni cellula rappresenta il calco (o lo stampo) da cui l'informazione genetica viene prima 'trascritta' in acido ribonucleico (RNA), che, a sua volta, viene poi tradotto in proteina. In altre parole, il DNA costituisce il supporto fisico dell'informazione biologica, ma questa informazione può essere utilizzata solo se espressa in maniera regolata come RNA e se tradotta, infine, in elementi funzionali, quali per esempio le proteine strutturali. Volendo utilizzare un'analogia informatica, il 'programma della vita' è memorizzato nel DNA come in un hard-disk di computer. Per essere funzionale il programma necessita però che le istruzioni vengano eseguite in sequenze ordinate (la trascrizione) e che le istruzioni si traducano nel prodotto desiderato (le proteine).
Il genoma mantiene invariata nel tempo l'informazione genetica e la trasmette di generazione in generazione nell'ambito della specie e di cellula in cellula durante lo sviluppo embrionale e per tutta la durata della vita individuale. Perciò esso è per sua natura un'entità relativamente stabile la cui sequenza di DNA è costante in tutti gli individui (a eccezione dei rari eventi d'insorgenza di mutazioni), indipendentemente dalle cellule o dai tessuti presi in considerazione, dallo stato metabolico e dai fattori ambientali.
Al contrario l'espressione genica, ovvero i tipi e le quantità degli RNA e delle proteine prodotte seguendo le istruzioni del genoma, dipende profondamente dal tipo di cellula, dal suo stato metabolico e dalla risposta a stimoli esterni. Perciò un neurone e un epatocita differiscono per i geni espressi che svolgono le funzioni specifiche rispettivamente del tessuto nervoso e del fegato, anche se condividono l'espressione dei geni necessari a tutte le cellule per il loro funzionamento (per es., i geni per le proteine strutturali come l'actina, oppure per gli enzimi necessari a produrre energia chimica sotto forma di ATP). Non solo, oggi si è in grado di riconoscere differenze significative nei profili di espressione genica anche a livello della medesima cellula, a seconda del suo stato metabolico o funzionale.
Si può quindi concludere che per ogni genoma esiste un numero estremamente elevato, ancorché finito, di combinazioni di RNA, proteine e metaboliti, ognuna delle quali corrisponde a una modalità di espressione del genoma stesso. L'analisi dei profili di espressione dell'RNA, la proteomica, e gli altri approcci postgenomici ambiscono a catturare la complessità di questi fenomeni nella loro totalità.
Anche se ci troviamo ben lontani dalla capacità di descrivere o di capire integralmente il funzionamento di una singola cellula (per non parlare di un intero organismo), il nuovo millennio ha assistito ai primi passi in questa direzione. A tale proposito non vi è dubbio che lo studio della biologia e la nostra comprensione dei fenomeni naturali siano stati rivoluzionati in maniera radicale.
Profili trascrizionali (trascrittoma)
Lo studio dei profili trascrizionali si basa principalmente sulla tecnologia dei microarray (microserie ordinate), noti anche con il nome di 'DNA array' o 'DNA chip', per analogia con i 'chip' della microelettronica.
I microarray rappresentano l'evoluzione, per via moltiplicativa, del concetto di ibridazione specifica tra acidi nucleici, definito da Edwin Southern oltre 25 anni fa e basato sulla proprietà della complementarietà degli acidi nucleici. Southern immaginò che si potesse sfruttare questa proprietà per 'interrogare' molecole legate a un supporto solido tramite molecole di acido nucleico marcate con un isotopo radioattivo del fosforo. Se sul supporto solido vengono trasferite (blotted) le molecole di un gene che si vuole studiare ed esse sono complementari alle molecole marcate, si può formare ('ibridarsi') una doppia elica, in grado di impressionare una pellicola autoradiografica. Il cosiddetto metodo del Southern blot, nella sua versione originale, comportava l'ibridazione di molecole di DNA e poteva analizzare l'ibridazione di una sola sequenza di DNA marcata in ogni esperimento. Analogamente la sua versione per l'analisi dell'RNA (il Northern blot) perlopiù rilevava una sola specie di RNA.
I microarray beneficiano di due innovazioni cruciali rispetto alle precedenti tecnologie di ibridazione. La prima consiste nell'uso di supporti rigidi non porosi al posto delle tradizionali membrane di nylon o di nitrocellulosa. Questo tipo di supporto (per es., vetro trattato chimicamente per legare stabilmente il DNA) ha enormemente facilitato la miniaturizzazione e l'uso di molecole marcate con fluorocromi, invece dei tradizionali isotopi radioattivi. La seconda innovazione tecnologica è rappresentata dalla disponibilità di metodi per la sintesi chimica su larga scala di oligonucleotidi (filamenti di DNA della lunghezza di 20-60 basi) o in alternativa frammenti di cDNA (DNA complementare all'RNA) ottenuti tramite PCR. Con l'uso di queste tecnologie e di vari sistemi (ink-jet, piezoelettrici, fotolitografici) per l'assemblaggio fisico dei DNA chip, si costruiscono serie su scala microscopica (microarray) di migliaia o decine di migliaia di punti indipendenti (attualmente fino a 40.000), ordinati su una superficie di circa 1 cm2. Ogni punto corrisponde a un oligonucleotide o a un cDNA di sequenza diversa. Perciò uno o pochi microarray sono sufficienti per interrogare l'eventuale espressione di ognuno dei 30-35.000 geni del genoma umano. Ovviamente le stesse considerazioni valgono per altri genomi il cui studio ha raggiunto un'evoluzione uguale o maggiore del genoma umano. Per esempio, il lievito della birra Saccharomyces cerevisiae (il primo eucariote il cui genoma è stato completamente sequenziato) ha rappresentato con i suoi 6200 geni ben caratterizzati il principale organismo modello per le applicazioni con i microarray e per gli studi di proteomica.
Un tipico esperimento di microarray per il profilo di espressione comporta i seguenti passi: (1) estrazione e purificazione dell'RNA dalle cellule o dal tessuto di interesse; (2) amplificazione del numero di molecole e marcatura con fluorocromi; (3) ibridazione delle molecole marcate al chip (il microarray con le molecole corrispondenti ai diversi geni); (4) determinazione dei punti che presentano ibridazione agli RNA in esame, esponendo il chip alla lettura da parte di uno scanner ad alta risoluzione; (5) analisi dei risultati al computer.
Attualmente gli ostacoli all'ulteriore evoluzione dell'analisi del trascrittoma risiedono non tanto nella costruzione e manipolazione dei microarray, quanto piuttosto nella riproducibilità, intra e interlaboratorio, degli esperimenti e negli strumenti informatici che consentano di estrarre un significato biologico da questa enorme mole di dati.
In linea di principio i microarray possono essere applicati a tre categorie di esperimenti. La prima riguarda i profili di espressione in sistemi biologici ben caratterizzati per determinare quali geni sono trascritti, o viceversa repressi, nella cellula in risposta a un determinato stimolo esogeno (ambientale, come la presenza o assenza di un nutriente) o endogeno (per es., la divisione cellulare). Ciò permette di identificare la funzione di nuovi geni e confermare la regolazione di quelli noti secondo la logica dell'associazione. Una seconda categoria di esperimenti è rappresentata dall'analisi dei profili trascrizionali di cellule normali e patologiche (per es., tumorali), oppure di cellule normali e infette da un microorganismo. In questi casi, il confronto tra i due profili di espressione può evidenziare i geni coinvolti nei fenomeni patologici. Inoltre l'abilità di distinguere i profili normali da quelli patologici può fornire strumenti per la diagnostica e la prognostica delle malattie. Infine, i microarray vengono utilizzati per caratterizzare sistemi biologici ex novo come nel caso dei batteri termofili oppure degli organismi geneticamente modificati.
Profili proteici (proteoma)
Il termine proteoma fu coniato per indicare tutti i possibili prodotti proteici codificati dal genoma. Lo studio del proteoma, ovvero la proteomica, non è più limitato a tutte le proteine presenti nella cellula, ma si estende alle isoforme proteiche, alle modificazioni postraduzionali subite dalle proteine (glicosilazione, acetilazione, metilazione, fosforilazione), alle loro caratteristiche strutturali e alle interazioni tra proteine, in particolare la formazione di complessi proteici funzionali.
Le più recenti stime del numero di geni presenti nel genoma umano sono comprese tra 30.000 e 40.000. Sulla base dei dati di espressione dell'RNA in diversi tessuti e della descrizione dello alternative splicing (un meccanismo molecolare che determina la sintesi di versioni diverse dell'RNA a partire dallo stesso gene), è stato predetto che le proteine codificate dal genoma umano sarebbero oltre 100.000, senza contare le modifiche postraduzionali menzionate sopra.
Il proteoma è quindi numericamente più complesso sia del genoma sia del trascrittoma. Non solo, mentre gli acidi nucleici (DNA e RNA) possono essere studiati facilmente perché la loro struttura chimica è relativamente semplice e rimane costante nel tempo, le proteine costituiscono oggetti molto più difficili da manipolare. In particolare, l'applicazione al proteoma dei microarray o di tecnologie analoghe è ostacolata dai seguenti fattori: difficoltà a ottenere un gran numero di proteine in forma purificata; grande variabilità nell'abbondanza delle singole proteine (da poche unità a milioni di molecole per cellula) e in generale delle proprietà fisico-chimiche; al contrario degli acidi nucleici, che interagiscono sempre tramite ponti idrogeno tra coppie di basi complementari, le proteine presentano modalità di interazione molto eterogenee, che sono perciò difficili da controllare; non sono disponibili tecniche di amplificazione del materiale biologico di partenza paragonabili alla PCR per gli acidi nucleici.
Anche se esistono microarray costituiti da proteine (o da ligandi delle proteine), come, per esempio, i microarray di anticorpi diretti verso proteine specifiche, la tecnologia più diffusa ed efficace per lo studio del proteoma è oggi una combinazione di tecniche per la separazione delle proteine le une dalle altre (gel elettroforesi bidimensionale, serie di cromatografie in fase liquida su colonne o una combinazione di cromatografia ed elettroforesi) seguita dall'applicazione di metodiche di spettrometria di massa, che determinano le masse dei peptidi o la sequenza amminoacidica propria di ogni proteina. A partire dalla misura esatta della massa dei peptici, o dalle informazioni sulla sequenza amminoacidica, è possibile assegnare ogni prodotto proteico a uno specifico gene, la cui funzione può essere più o meno nota. Di nuovo, come per l'analisi dei profili di espressione, la proteomica acquista significato attraverso l'integrazione con la genomica.
In effetti il processo di integrazione delle informazioni è assolutamente essenziale allo sviluppo odierno delle scienze biomediche e può essere illustrato paragonando la biologia della cellula alla costruzione di un puzzle. Negli anni passati abbiamo trovato i pezzi della cornice, ovvero la sequenza del genoma e l'identità dei geni più facili da studiare. Ora siamo nella condizione di avere accesso a tutti le altre centinaia di migliaia (o forse più realisticamente centinaia di milioni) di pezzi, tramite genomica, microarray e proteomica; se dovessimo posizionarli a uno a uno come nel passato il compito sarebbe immane. Invece, possiamo ragionevolmente immaginare di usare i programmi della bioinformatica per dividere i pezzi prima in gruppi di colore affine (come fa chiunque abbia dimestichezza con il gioco del puzzle) e poi, all'interno dello stesso gruppo, identificare rapidamente i pezzi che si incastrano l'uno con l'altro. In questo senso (funzionale) il risultato degli approcci sistematici può andare ben oltre la semplice somma algebrica dei dati.
Lo studio del proteoma può essere facilitato dall'uso di organismi modello come è ben illustrato dalle ricerche sul lievito: in questo organismo è stato possibile realizzare, a partire dalla sequenza genomica e dall'individuazione sperimentale o 'in silico' dei 6200 geni che codificano per tutte le possibili proteine, tre approcci distinti e in qualche modo complementari. Nel primo si sono presi a uno a uno tutti i 6200 geni e vi sono state introdotte mutazioni sito-specifiche che impedissero la produzione della proteina corrispondente: in questo modo si è cercato di risalire alla funzione di ogni proteina analizzando il fenotipo (ovvero l'aspetto, il comportamento e in generale tutte le caratteristiche misurabili) dei vari mutanti.
Il secondo approccio affronta il problema della funzione tramite la determinazione delle interazioni tra proteine. Uno dei sistemi sperimentali più riusciti utilizza per leggere il risultato dell'interazione un elegante saggio genetico, noto agli specialisti con il nome di two-hybrids system. Nel lievito il saggio è stato applicato a ognuna delle proteine presenti nei confronti di tutte le altre, per un totale di circa 40 milioni (6200×6200) di interazioni. Esperimenti di questa portata sono possibili nel lievito perché esso si presta bene alle manipolazioni genetiche e all'automazione delle stesse con l'uso di robot. Un altro modo per studiare le interazioni tra proteine consiste nell'analizzare la composizione dei complessi proteici. Infatti è noto che quasi tutte le proteine non funzionano come entità isolate; al contrario esse formano complessi multiproteici che sono stati opportunamente paragonati a macchine molecolari. Con metodiche simili alla mutagenesi sito-diretta, viene inserita in ogni proteina una breve sequenza amminoacidica (il cosiddetto 'tag' o 'etichetta' molecolare) che facilita enormemente il riconoscimento e la purificazione non solo della proteina modificata, ma anche dell'intero complesso a cui questa appartiene. Gli specifici componenti del complesso possono poi essere identificati grazie alla spettrometria di massa come già descritto. Con questo sistema sono state identificate nel lievito oltre 200 macchine molecolari e la lista è naturalmente incompleta perché, di nuovo, è stata esaminata soltanto una frazione irrisoria delle possibili condizioni metaboliche e ambientali. Dagli studi sulle interazioni sono già emersi alcuni temi importanti; innanzi tutto la stessa proteina può esercitare la sua attività come membro di più complessi, realizzando l'equivalente cellulare delle economie di scala nelle produzioni industriali. L'inclusione di una proteina in un determinato complesso, inoltre, non è necessariamente automatica, ma può essere soggetta a meccanismi cellulari di regolazione e, infine, se per alcuni dei componenti di un determinato complesso è nota la funzione biochimica, è possibile dedurre sia la funzione degli altri membri sia quella della macchina molecolare nel suo insieme. È interessante notare che l''etichetta molecolare' può essere utilizzata anche per determinare la localizzazione subcellulare di qualsiasi proteina che la incorpori. Addirittura è stato sviluppato un sistema in cui l'etichetta è fluorescente in condizioni fisiologiche: ciò permette di localizzare la proteina (o il complesso di cui fa parte) nelle cellule viventi, ottenendo una rappresentazione dinamica e assai più affidabile della localizzazione intracellulare.
Il terzo approccio, infine, consiste nella produzione e purificazione di tutti i prodotti genici sotto forma di proteine ricombinanti espresse in batteri (o in altri organismi utili a questo scopo). Questo lavoro è necessario per poter determinare la struttura tridimensionale (a livello di risoluzione atomica) di molte delle proteine di interesse.
Il risultato principale di tutti questi approcci è la definizione della funzione delle proteine e delle corrispondenti macchine molecolari: non va dimenticato, infatti, che nonostante gli enormi progressi effettuati in questi anni la funzione di un quarto dei geni (ovvero delle proteine) del lievito e di oltre la metà dei geni dell'uomo è ancora completamente sconosciuta.
Come per il genoma, è possibile reperire in internet informazioni scientifiche aggiornate su ogni aspetto della biologia delle proteine. Per esempio, la Protein data bank (PDB) raccoglie e distribuisce i dati sulla struttura tridimensionale delle proteine e contiene a oggi ben 21.000 strutture.
Il sito ExPASys (Expert protein analysis system) è un server di biologia molecolare che consente l'accesso a Swiss-Prot, con annotazioni sulla funzione, la struttura e le modificazioni di tutte le proteine note (126.000 voci), e alla banca dati EMBL (l'equivalente di Swiss-Prot, ma con predizioni e annotazioni curate automaticamente dal computer invece che da ricercatori). Inoltre, il sito ProSite, fornisce l'accesso ad algoritmi per analizzare e riconoscere le famiglie e i domini funzionali delle proteine.
I progetti di postgenomica

Il contenuto della prossima grande sfida scientifica può essere sintetizzato nel tentativo di conoscere, principalmente tramite la proteomica e l'analisi dell'espressione dell'RNA, la struttura ordinata del programma biologico che il genoma sottende. Questo lavoro è appena iniziato sotto forma di numerosi progetti di ricerca di grandi e medie dimensioni e richiederà molti anni di ricerca per raggiungere i suoi obiettivi, che in un certo senso coincidono con la comprensione scientifica della vita stessa. Una lista parziale dei progetti post-genomici in corso è illustrata nella Tav. II.
Il Progetto genoma umano ha rappresentato il primo esempio di Big science in campo biologico, con risorse umane ed economiche dell'ordine di grandezza generalmente utilizzato per alcuni progetti spaziali o di fisica delle particelle. È opportuno notare che il costo elevato del Progetto genoma umano (3 miliardi di dollari) è comunque inferiore a quello di altri progetti internazionali come l'acceleratore di particelle Superconducting super collider (11 miliardi) o la stazione spaziale orbitante (30 miliardi) e, inoltre, si deve sottolineare che il valore d'uso dei risultati del progetto genoma è perpetuo. Per dare un riferimento della scala economica di questi progetti basterà ricordare che nel 2002 l'Italia ha avuto un PIL di circa 1300 miliardi di dollari, una spesa per la sanità di 78 miliardi di dollari e investimenti complessivi per la ricerca pari a circa l'1% del PIL. Altrettanto interessante è il fatto che, se si proiettasse il costo complessivo del Progetto genoma umano utilizzando il costo di sequenziamento del DNA del 1990, quando il progetto ebbe ufficialmente inizio, si otterrebbe una cifra dieci volte superiore (30 miliardi di dollari) al costo effettivamente sostenuto. La differenza favorevole è interamente ascrivibile all'evoluzione tecnologica che ha accompagnato la realizzazione del progetto genoma. Possiamo attenderci dinamiche simili, o anche più impressionanti, per gli sviluppi dei microarray e della proteomica. In virtù del successo del progetto genoma e delle future applicazioni della postgenomica in campo biomedico (ma anche in ecologia, nella generazione di fonti di energia rinnovabili e in ambito agroalimentare), gli investimenti pubblici e privati nella ricerca basata su queste tecnologie appaiono più che giustificati.
A titolo esemplificativo della complessità e delle ambizioni dei progetti di postgenomica, si può descrivere il progetto Genomes to life (GTL) coordinato dal Dipartimento per l'energia degli Stati Uniti. Il programma del GTL prevede i seguenti obiettivi: (1) identificare e caratterizzare sistematicamente tutte le macchine molecolari (i complessi proteici che effettuano le diverse attività enzimatiche della cellula) nei principali microorganismi; (2) descrivere e manipolare i circuiti regolativi che, a partire dalle informazioni genomiche, controllano le macchine molecolari; (3) studiare con microarray e proteomica i microorganismi in diverse condizioni ambientali e, in particolare, la loro capacità di associarsi in vere e proprie comunità microbiche (biofilm); (4) organizzare i dati in un sistema coerente e accessibile alla comunità scientifica.
Il progetto GTL è rilevante sotto molti aspetti; innanzi tutto i microorganismi hanno colonizzato ogni ambiente del pianeta sin dall'inizio dell'evoluzione della vita. Essi costituiscono oltre il 50% della biomassa terrestre e utilizzano ogni sorta di sostanze organiche sia naturali sia sintetiche per il loro metabolismo. Bisogna rammentare che la capacità del pianeta di sostenere la vita è assolutamente dipendente dai microorganismi che partecipano al ciclo dell'acqua, alla produzione di ossigeno e al bilancio dell'anidride carbonica. Infine, è stato stimato che solo una frazione trascurabile (inferiore all'1%) delle specie microbiche è stata studiata almeno una volta in laboratorio e il progetto GTL dovrebbe contribuire a ridurre questo vuoto. La genomica dei microorganismi è facilitata dal fatto che la sequenza del DNA del cromosoma batterico ha in media una lunghezza mille volte inferiore a quella del genoma dei mammiferi. Nel medio e lungo periodo, i risultati del programma GTL contribuiranno alla soluzione di problemi su scala globale quali, per esempio, la riduzione dell'effetto serra, la rimozione dell'inquinamento e la produzione di carburanti da fonti rinnovabili (biofuel).
Infine, parlando di genomica e postgenomica si deve accennare alla bioinformatica che è lo strumento assolutamente cruciale di queste discipline; infatti, essa produce gli algoritmi utilizzati per analizzare i dati, per esempio per confrontare una sequenza di DNA o una proteina di interesse con tutte le sequenze di tutti gli organismi disponibili per trovare sequenze simili o identiche. Allo stesso modo sono stati elaborati, o adattati da altre discipline, algoritmi che individuano specifici profili di espressione dell'RNA associandoli ai geni corrispondenti e rendono possibile identificare pattern di coespressione o coregolazione dei geni. Così definita la bioinformatica rappresenta un settore estremamente specialistico, sofisticato e in rapida evoluzione, nel quale l'aspetto matematico e informatico prevale su quello biologico anche se naturalmente non prescinde mai da esso. In senso più generale, e per la maggior parte degli scienziati, la bioinformatica rappresenta, invece, una raccolta di strumenti per accedere, organizzare e analizzare l'informazione biologica. Si tratta quindi di una definizione che pone l'enfasi sull'utilizzo più che sulla creazione degli algoritmi. La raccolta di questi strumenti si fa sempre più vasta e sofisticata e, grazie a internet, scienziati in qualsiasi parte del mondo possono accedere direttamente alla stessa enorme quantità di informazioni, indipendentemente dalle risorse umane ed economiche disponibili della propria istituzione di appartenenza. Anche questa rappresenta a sua volta una rivoluzione dell'impresa scientifica biomedica le cui conseguenze, certamente positive, non sono ancora state pienamente apprezzate.
Sviluppi e applicazioni della postgenomica
La scoperta delle basi molecolari delle malattie ha compiuto enormi progressi negli ultimi decenni; per esempio, si conoscono molti geni la cui mutazione causa l'insorgenza di tumori, e inoltre sono stati identificati tutti i geni responsabili dei difetti genetici più frequenti di tipo mendeliano e si dispone di strumenti per la diagnostica molecolare degli agenti infettivi patogeni più diffusi.
Allo stesso tempo queste scoperte hanno ancor più evidenziato la complessità dei sistemi biologici e la necessità di applicare le discipline genomiche e postgenomiche alla ricerca biomedica.
In particolare, per alcuni settori della medicina è lecito attendersi ricadute concrete nei prossimi anni grazie agli sviluppi della genomica e della postgenomica. Il primo traguardo consiste, come già descritto, nell'identificazione dei geni che predispongono o contribuiscono all'insorgenza di malattie multigeniche e multifattoriali di elevato impatto sulla popolazione, quali vari tipi di tumori, le malattie cardiovascolari, la demenza e altre neuropatologie, il diabete e così via. La maggior parte di queste scoperte sarà dovuta all'utilizzazione di tecnologie a elevata produttività, come l'analisi degli SNPs che già oggi prevede applicazioni nelle quali vengono analizzati milioni di dati al giorno. Ci si può attendere, in un futuro non troppo lontano, che sia possibile valutare il nostro personale rischio di sviluppare una patologia comune (per es., una malattia cardiovascolare) in base alla particolare combinazione di geni ereditata e quindi poter ricorrere a misure preventive quali una dieta appropriata o farmaci adatti per quel tipo di patologia.
La tipizzazione del DNA genomico per un certo numero di SNPs permette anche, in linea di principio, di studiare come il materiale ereditario individuale influenzi la risposta dell'organismo alla somministrazione dei farmaci, e quindi di trovare per ognuno il trattamento farmacologico più efficace, limitando al tempo stesso gli effetti collaterali. Questo intreccio di genomica e farmacologia (a sua volta un esempio di ricerca postgenomica) è stato chiamato 'farmacogenomica'. Un esempio pratico di come la farmacogenomica rappresenti una forma di 'medicina personalizzata' è il caso del farmaco abacavir nel trattamento delle infezioni da HIV. Il 4% degli individui trattati con questo farmaco sviluppa una specifica reazione di ipersensibilità che determina gravi effetti collaterali. Lo studio del genoma ha identificato i marcatori genetici (SNPs) che comportano per il paziente una probabilità del 97% di sviluppare la reazione negativa. Quindi, è oggi possibile evitare gli eventi non favorevoli con un semplice test sul DNA prima che i pazienti assumano il farmaco.
Un aspetto importante della farmacogenomica, che ha attratto gli investimenti di tutte le multinazionali farmaceutiche, risiede nell'identificazione di nuovi bersagli molecolari per i farmaci; è noto, infatti, che le proteine con cui interagiscono i farmaci attuali sono in totale meno di 500 e che le ricerche di nuovi farmaci con i sistemi di screening tradizionali producono sempre più raramente nuovi bersagli. Le oltre 100.000 proteine identificate dalla proteomica rappresentano invece altrettanti potenziali bersagli per i farmaci del futuro.
Un altro sviluppo della postgenomica di grande interesse medico e commerciale è legato alla diagnostica molecolare: è stato dimostrato che l'identificazione di profili proteici o dell'espressione di RNA caratteristici di specifiche patologie può avere valore diagnostico e prognostico.
Per esempio, è stato riportato che l'analisi del profilo delle proteine del sangue mostra differenze tra il sangue di un individuo sano e quello di pazienti con carcinoma delle ovaie. Sebbene ancora sperimentali (identificano il tumore nel 95% dei casi) questi test sono comunque estremamente incoraggianti per future applicazioni che possiedono il vantaggio sia di non essere invasive sia di rilevare modifiche del profilo proteico già dagli stadi iniziali della malattia.
Diagnostica e predizioni sull'evoluzione delle malattie si possono effettuare anche con i microarray. Un esempio in questo senso è rappresentato dal Lymphochip sviluppato da una collaborazione tra ricercatori del NIH e della Stanford University. Il Lymphochip contiene oltre 18.000 frammenti di DNA corrispondenti a geni associati allo sviluppo normale e patologico dei linfociti umani e permette di caratterizzare in grande dettaglio l'espressione genica dei linfociti umani nelle condizioni più svariate. È stato dimostrato che, utilizzando il Lymphochip, si possono distinguere diversi tipi di un tumore del sangue chiamato DLBCL (diffuse large B cell lymphoma) che rappresenta, dopo il linfoma di Hodgkin, il più frequente tumore delle cellule B. È stato inoltre osservato che i profili trascrizionali permettono di riconoscere due gruppi di pazienti DLBCL la cui prognosi differisce assai nettamente. Tali gruppi, altrimenti non distinguibili, vanno quindi sottoposti a terapie differenziate sin dalla diagnosi con vantaggi per entrambi.
Con la stessa logica del Lymphochip e del tumore DLBCL sono in fase avanzata di ricerca applicazioni simili allestite con appositi microarray per i tumori del seno e il mieloma multiplo.
Conclusioni
Collins, uno dei principali coordinatori del Progetto genoma umano e direttore del National human genome research institute (NHGRI), in un suo editoriale, intitolato Microarrays and macroconsequences, ha analizzato le 'macroconseguenze' della tecnologia genomica e postgenomica sugli aspetti scientifici così come gli effetti che tali studi avranno sulla società. Per questo motivo è stato istituito, sin dall'inizio del Progetto genoma umano, un progetto parallelo denominato ELSI (Ethical, legal and social implications) a sua volta ripartito in quattro aree di attività: (1) privacy ed equità nell'uso e nell'interpretazione delle informazioni genetiche (per es., le eventuali conseguenze della futura disponibilità dell'informazione genetica individuale nei rapporti del cittadino con i datori di lavoro, con le compagnie di assicurazione e con la società in senso lato); (2) adozione a livello clinico delle nuove tecnologie genetiche (quali criteri adottare per l'introduzione delle nuove tecnologie diagnostiche e con quali conseguenze; quando e con quali costi verrà adottata la 'medicina personalizzata' al momento in cui questa sarà disponibile; cercare di capire se l'introduzione della medicina genetica avrà l'effetto di diminuire, oppure al contrario aumentare, il divario nell'accesso alla sanità delle varie popolazioni del pianeta); (3) controversie legate alla ricerca genetica (in particolare esaminare le conseguenze della ricerca genetica in tutti i settori non medici, per es., quello agroindustriale e l'ambiente; come cittadini abbiamo avuto recentemente un'anticipazione del contenuto di questi temi a causa delle controversie suscitate dalla commercializzazione degli organismi geneticamente modificati, OGM); (4) istruzione e informazione dei cittadini e dei professionisti (uno degli obiettivi principali è naturalmente di informare esaurientemente i cittadini in modo che le decisioni sugli usi di queste ricerche possano essere prese democraticamente; altri obiettivi riguardano la valutazione delle conseguenze che le nuove conoscenze scientifiche hanno su specifici aspetti filosofici, legali ed etici; si auspica che i risultati della ricerca genetica potranno modificare i concetti di razza, gruppo etnico, clan, identità familiare e individuale e potranno finalmente definire normali le malattie e le caratteristiche fisiche e comportamentali).
Il progetto ELSI e altri analoghi in Europa hanno probabilmente un'importanza equivalente a quella della ricerca primaria, non ultimo per il fatto che la comunità scientifica non ha alcun interesse a mantenere segreti sulle proprie attività, ma anzi desidera che i risultati della ricerca e le loro applicazioni siano effettivamente compresi, valutati ed eventualmente apprezzati dai cittadini.
È stato scritto che gli storici del futuro indicheranno nel completamento della sequenza del genoma umano l'evento scientifico più significativo del XX sec.; si può sostenere con certezza che quanto fin qui avvenuto non smentisce questa affermazione.
Bibliografia
Collins 2003: Collins, Francis S. - Green, Eric D. - Guttmacher, Alan E. - Guyer, Mark S., A vision for the future of genomics research, "Nature", 422, 2003, pp. 835-847.
Wulfkuhle 2003: Wulfkuhle, Julia D. - Liotta, Lance A. - Petricoin, Emanuel F., Proteomic applications for the early detection of cancer, "Nature reviews cancer", 3, 2003, pp. 267-275.
Siti internet
The institute for genomic research (http://www.tigr.org/).
Genoma programs of the US department of energy office of science (http://DOEgenomes.org).
Genomics & Genetics at the Sanger institute, sequencing genomics projects, Caenorhabditis genome sequencing projects (http://www.sanger.ac.uk/Projects/C_elegans/).
FlyBase. A Database of the Drosophila genome (http://www.flybase.org/).
NCBI GenBank® (http://www.ncbi.nlm.nih.gov/Genbank/index.html).
EMBL Nucleotide sequence Database (http://www.ebi.ac.uk/embl/index.html).
National center for biotechnology information (NCBI) (http://www.ncbi. nim.nih.gov/).
The wellcome trust Sanger institute, (http://www.sanger.ac.uk/).
DOE Joint genome institute, US Department of energy office of science (http://www.jgi.doe.gov/).
PDB The protein data bank (http://www.rcsb.org/pdb/).
ExPASys Molecular biology server - The ExPASy (expert protein analysis system) proteomics server (http://us.expasy.org/sprot/).
ExPASys molecular biology server - PROSITE Database of protein families and domains (http://us.expasy.org/prosite/).
OMIM Online Mendelian inheritance in man (http://www.ncbi.nlm.nih. gov/omim).