
La grande scienza. Il folding delle proteine all'interno della cellula
La grande scienza. Il folding delle proteine all'interno della cellula
Il folding delle proteine all'interno della cellula
Le proteine, sintetizzate come catene polipeptidiche che si estendono in modo spazialmente non strutturato, devono raggiungere una conformazione tridimensionale stabile per poter svolgere le loro funzioni biologiche. Da decenni è noto che tutte le informazioni necessarie a un polipeptide denaturato per il raggiungimento di una conformazione stabile e funzionale sono contenute all'interno della sua stessa sequenza amminoacidica. In teoria, quindi, la conformazione biologicamente attiva di una proteina dovrebbe essere desumibile una volta che sia nota la sua struttura primaria. Sebbene, in vitro, sia stato dimostrato che numerose proteine sono in grado di assumere spontaneamente una struttura terziaria, in vivo, tuttavia, il processo che porta all'assunzione di una corretta conformazione sterica coinvolge un complesso intreccio di interazioni tra enzimi diversi e chaperon che assistono il polipeptide nel conseguimento della sua conformazione nativa finale. In questo articolo verranno discusse le possibili funzioni delle diverse specie molecolari coinvolte nel folding delle proteine.
La comprensione dei meccanismi utilizzati dai polipeptidi per raggiungere una struttura tridimensionale che li renda in grado di svolgere correttamente le loro funzioni biologiche è stata notevolmente favorita, negli ultimi anni, da numerosi studi sulla chimica fisica e la biologia cellulare di questo processo. Sebbene la conoscenza di tale argomento sia lontana dall'essere completa, è tuttavia opinione generale che il folding proceda attraverso molteplici passaggi distinti. Un gran numero di interazioni sia all'interno della stessa catena in fase di ripiegamento sia, in vivo, con enzimi specifici per il ripiegamento e anche con proteine coadiuvanti (chaperon molecolari) si deve creare e rompere nel corso di questo processo molto complesso.
L'attenzione che il problema del folding delle proteine ha ricevuto nel passato e la sfida che esso sicuramente costituisce per gli scienziati nel futuro sono certamente giustificate dal fatto che questo tema ha un valore fondamentale per la biochimica cellulare. Aggiungendosi alla delucidazione del codice genetico, che è stata ottenuta parecchi decenni fa, la comprensione di come le proteine si ripiegano aiuterà a chiarire il modo in cui la Natura è in grado di tradurre un'informazione genetica monodimensionale in strutture biologiche specifiche. Un eventuale successo nella scoperta di un 'codice di folding' migliorerebbe in maniera specifica il contenuto di informazione degli enormi progetti sui genomi che attualmente producono sequenze di DNA a un ritmo sempre maggiore. Bisogna notare che l'aumentata domanda di predizioni attendibili sulla struttura sterica delle proteine non può essere soddisfatta né dall'analisi cristallografica né da quella spettroscopica mediante risonanza magnetica nucleare (NMR, nuclear magnetic resonance) poiché entrambe queste tecnologie soffrono di limitazioni pratiche. Pertanto la comprensione dei meccanismi e delle vie che conducono al folding potrebbe rivelarsi uno strumento di incalcolabile valore nella traduzione di sequenze amminoacidiche in strutture tridimensionali.
Al di là del fondamentale interesse che questo settore riveste per gli scienziati, errori nel folding delle proteine sono stati recentemente associati a diverse malattie umane. In linea di principio, qualsiasi errore che porti alla perdita di una proteina correttamente funzionante si può tradurre in un fenotipo difettivo. Tra gli errori nel folding si possono annoverare la destabilizzazione dello stato nativo o di uno stato intermedio della proteina, l'eccessivo prolungarsi di un legame con uno chaperon molecolare o con enzimi implicati nel folding, la formazione di vie preferenziali collaterali non funzionali o di stati conformazionali tossici e, infine, il folding in compartimenti intracellulari sbagliati. Le malattie correlate a errori nel folding delle proteine possono attualmente essere classificate in tre categorie a seconda del tipo di difetto sotteso: incapacità delle proteine ad andare incontro a folding, formazione di strutture steriche tossiche e, infine, sbagliata localizzazione intracellulare delle proteine dovuta a folding errato (Thomas et al. 1995).
Un esempio di malattia connessa con folding errato è la fibrosi cistica (CF), causata da mutazioni nel gene che codifica la proteina regolatrice transmembrana della fibrosi cistica (CFTR, cystic fibrosis transmembrane regulator), responsabile della formazione di un canale per il cloro dipendente dall'ATP. La più comune tra queste mutazioni è la delezione di un singolo amminoacido nel sito di legame dell'ATP che porta a un difettoso folding di CFTR. Tale mutazione comporta, nel processo di maturazione di CFTR, un cambiamento nella sensibilità alla temperatura che si traduce non in una destabilizzazione della proteina nativa ma, piuttosto, nella destabilizzazione della proteina nello stato intermedio di folding; ciò suggerisce che il difetto risiede lungo la via che porta al folding stesso. Come conseguenza di questa situazione, alla normale temperatura corporea, la proteina, che non ha raggiunto una conformazione attiva, è incapace di raggiungere la sua localizzazione finale alla superficie cellulare e tende, invece, ad accumularsi nel reticolo endoplasmatico dove si associa con molecole chaperon e infine viene degradata. È significativo che molecole CFTR mutate possano comunque andare incontro a un folding corretto a temperature inferiori a quella corporea e, una volta giustamente conformate, essere perfettamente funzionali anche a 37°C.
Nel caso dell'encefalopatia spongiforme degli ovini e della sindrome della mucca pazza, l'agente patogeno è una proteina, detta anche prione o PrP, che non è andata incontro a un folding corretto. I prioni endogeni sono proteine, presenti nei neuroni, che contengono molte α-eliche. Il prione infettivo responsabile dell'encefalopatia spongiforme degli ovini è invece essenzialmente strutturato a foglietti β. Il processo infettivo si instaura, probabilmente, perché il prione mal conformato è in grado di indurre un cambiamento strutturale nei prioni endogeni, facendo loro assumere la struttura propria dei prioni infettivi. In questo modo si ha un aumento del contenuto in foglietti β delle proteine, tale da rendere i prioni infettivi e causare la malattia. Nell'esempio qui riportato, quindi, un folding sbagliato si traduce in strutture estremamente stabili, che risultano tossiche per la cellula.
Lo scopo di questo saggio è quello di fornire una descrizione generale dei processi che portano al folding delle proteine; si rimanda, pertanto, il lettore interessato a conoscere in dettaglio le malattie da errato folding proteico a una letteratura più specializzata. In ogni caso una comprensione più approfondita delle vie biologiche che presiedono il folding delle proteine potrà offrire, senza dubbio, un contributo positivo all'ideazione di terapie più efficaci per il trattamento di tali malattie. Al di là di questi aspetti medici, la comprensione dei meccanismi di folding proteico riveste notevole importanza nel campo delle biotecnologie, in virtù della crescente domanda di proteine ricombinanti conformate in modo corretto e biologicamente attive.
Il folding e il refolding delle proteine 'in vitro'
Mentre la struttura tridimensionale di numerose proteine è stata risolta nei più piccoli dettagli, meno definite sono le vie che i polipeptidi, ancora lineari, seguono per ottenere la loro conformazione nativa biologicamente attiva. Dai primi esperimenti di Christian B. Anfinsen sul refolding (rinaturazione) in vitro della ribonucleasi denaturata divenne chiaro che tutte le informazioni essenziali richieste per determinare la conformazione finale di una proteina sono contenute all'interno della stessa catena polipeptidica (Anfinsen 1973). Da allora risultati simili sono stati ottenuti non solo con molti altri piccoli polipeptidi costituiti da un singolo dominio ma anche con proteine di dimensioni maggiori e più complesse: se si scelgono condizioni corrette per il refolding, un enzima denaturato raggiungerà la sua conformazione nativa in assenza di ogni altra proteina. Rimane ancora in parte oscuro come l'informazione contenuta nella catena polipeptidica si traduca in una struttura tridimensionale univoca.
Recentemente, attraverso l'analisi comparata di proteine a struttura nota, sono stati identificati numerosi polipeptidi con una struttura terziaria complessivamente simile, oppure con domini simili, anche se privi di un'evidente somiglianza nella struttura primaria. Inoltre mutazioni nella sequenza amminoacidica spesso non hanno alcuna influenza sul raggiungimento di un corretto stato conformazionale finale. Da quanto si è detto in precedenza si evince che, per ottenere un corretto folding, appaiono importanti non soltanto alcuni amminoacidi chiave, ma anche la natura generale della sequenza amminoacidica.
A prima vista le strutture steriche che potrebbero generarsi a partire da una singola catena polipeptidica sono innumerevoli. È stato calcolato che il tempo richiesto da un polipeptide abbastanza lungo per sondare ogni possibile stato conformazionale fino a trovare il suo stato nativo è paragonabile all'età dell'Universo. Di fatto, tuttavia, un polipeptide denaturato impiega una scala temporale fisiologica per riassumere spontaneamente la corretta conformazione sterica, una volta che l'agente denaturante venga rimosso per diluizione in soluzione acquosa. Alla luce di queste osservazioni gli studi sul refolding delle proteine si sono focalizzati sull'esame degli stati intermedi di folding e sulla caratterizzazione delle vie seguite durante la rinaturazione proteica. I biofisici hanno realizzato considerevoli progressi nella definizione delle vie seguite da un polipeptide durante il processo di cambiamento strutturale che lo porta ad assumere uno stato conformazionale biologicamente attivo.
Gli stati conformazionali stabili delle proteine
Durante il refolding in vitro di proteine denaturate la catena polipeptidica potrebbe, in linea teorica, assumere numerosi e diversi stati conformazionali. Molti di questi sono in realtà non sufficientemente stabili da essere individuati fisicamente, e perciò rimangono ignoti. Altri, tuttavia, sono più stabili e possono quindi essere isolati in quantità adeguata tale da permetterne lo studio sperimentale. Il verificarsi di questi stati intermedi di folding ha portato all'ipotesi che il folding avvenga attraverso vie definite e mediante la cooperazione di interazioni multiple che stabilizzano lo stato nativo di una proteina.
Lo stato di prefolding. Idealmente lo stato privo di folding è il cosiddetto random coil, in cui le conformazioni possibili, anche per una proteina di piccole dimensioni, sono moltissime. In questa fase il polipeptide si dovrebbe trovare in uno stato in cui la sua catena è molto estesa nello spazio e le interazioni non covalenti, che normalmente stabilizzano lo stato nativo, sono inesistenti. Tuttavia, a seconda del tipo di agente denaturante, non si può escludere che si possano creare strutture tridimensionali locali molto instabili. Per esempio, la denaturazione ottenuta mediante agenti caotropici (sostanze che, diminuendo le interazioni idrofobiche interne e, contemporaneamente, riducendo il numero di cluster di acqua che le circondano, destabilizzano l'organizzazione tridimensionale delle proteine), come l'urea o il cloridrato di guanidinio, porta quasi a uno stato di random coil. Al contrario, se la denaturazione è ottenuta in condizioni estreme di pH o di temperatura è possibile osservare la presenza di strutture di interazione localizzate. Questo indica che per mantenere una proteina in uno stato completamente denaturato sono necessari trattamenti fisici o chimici radicali che non si possono certo chiamare fisiologici. È lecito quindi supporre che le proteine, sintetizzate come polipeptidi lineari sui ribosomi, in vivo comincino il folding durante la loro stessa sintesi. Tuttavia evidenze sperimentali suggeriscono che le proteine destinate a essere veicolate a particolari compartimenti cellulari, quali i cloroplasti, i mitocondri e il reticolo endoplasmatico, passino attraverso le membrane intracellulari in una conformazione estesa, o con folding lasso, anche quando il trasporto avviene in modo postraduzionale. Si pensa che a mantenere le proteine neosintetizzate in tale stato di conformazione lassa, necessario per la loro veicolazione verso i vari organelli cellulari, siano gli chaperon molecolari.
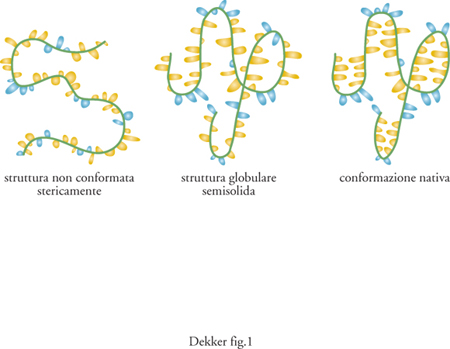
Le strutture globulari semisolide. È stato osservato che molte proteine, in determinate condizioni, si trovano in una conformazione stabile che non è di folding compiuto, ma neanche di completa disorganizzazione sterica (fig. 1). L'esistenza di tale struttura intermedia era già stata proposta su base teorica (Ptitsyn 1995). Il verificarsi di conformazioni globulari semisolide (molten globule) è attualmente ben documentato. Le proprietà caratteristiche di tali strutture sono: la maggior compattezza rispetto allo stato di random coil e la dimensione solo lievemente maggiore rispetto alla proteina nativa; un contenuto in strutture secondarie simile a quello della proteina con folding completo; la presenza di superfici idrofobiche esposte all'esterno, che le rendono suscettibili di aggregazione reciproca; un'entalpia pressoché identica a quella che esse stesse hanno in uno stato privo di folding; un'interconversione, dalla condizione di strutture globulari semisolide allo stato di completa disorganizzazione sterica, rapida e non cooperativa, e invece passaggio allo stato di folding completo lento e cooperativo.
Queste osservazioni suggeriscono che la struttura globulare semisolida sia una molecola collassata, dotata di strutture secondarie simili a quelle della corrispondente proteina nativa, ma priva di strutture terziarie stabili. Sebbene il numero di conformazioni possibili per una struttura globulare semisolida sia ancora smisurato, esso è, tuttavia, molto minore di quello che può essere raggiunto da una proteina completamente disorganizzata. Il significato biologico delle strutture globulari semisolide è oggetto di discussione. In particolare, non è chiaro se esse siano gli intermedi essenziali durante il folding delle proteine in vivo, anche se è stato riportato che il folding di proteine sulla superficie della molecola chaperon GroEL avviene con la formazione di intermedi globulari semisolidi.
Lo stato nativo. Mediante la cristallografia a raggi X e l'analisi spettroscopica NMR è stato possibile esaminare lo stato nativo di molte proteine diverse. Questi studi hanno chiarito alcuni aspetti generali delle proteine globulari il cui folding sia avvenuto compiutamente e che si trovano quindi nello stato nativo. Le caratteristiche che accomunano tutte queste proteine globulari sono la non polarità delle catene laterali che formano la parte interna della struttura e la generale prevalenza di catene laterali idrofiliche esposte alla superficie. Il cuore idrofobico sembra essere il componente più importante per la stabilità della conformazione nativa, mentre la flessibilità è un aspetto comune alla parte superficiale delle molecole. Per la maggior parte delle proteine si conosce soltanto un unico stato di folding completo. Le sole differenze conformazionali sostanziali tra domini di proteine noti possono derivare da proteolisi o, come dimostrato di recente, risiedere nelle alterazioni della struttura secondaria indotte in caso di infezione da prioni. Le interazioni che stabilizzano lo stato nativo sono intrinsecamente deboli, ma la presenza contemporanea di un gran numero di esse e la loro cooperatività hanno come risultato la produzione di strutture stabili (Creighton 1995).
Le vie di folding in vitro
Si ritiene che il refolding in vitro avvenga attraverso diverse vie che coinvolgono uno o più intermedi di folding relativamente stabili. Il processo può avere inizio con un collasso delle regioni idrofobiche all'interno della molecola, o con la formazione di strutture secondarie stabili che forniscono un'intelaiatura per un successivo folding, oppure con la formazione di interazioni covalenti, come i ponti disolfuro, che stabilizzano il polipeptide in specifici stati conformazionali. Ciascuno di questi meccanismi può operare di concerto durante le fasi iniziali del refolding. Come conseguenza di ciò, si formano intermedi di folding che assomigliano alle strutture globulari semisolide sopra descritte. Questi intermedi sembrano trovarsi in un equilibrio rapido con lo stato di denaturazione completa (nell'ordine di millisecondi), mentre sono convertiti allo stato nativo solo molto lentamente. Così, il punto di controllo della velocità di refolding si trova spesso in stadi avanzati del processo, appena prima che la proteina adotti la sua conformazione nativa definitiva. Si ritiene che la conversione degli intermedi di folding nello stato nativo sia un processo cooperativo in cui il verificarsi di una interazione rende stabile la successiva, che a sua volta stabilizzerà la prima interazione (Creighton 1995). Questa 'stabilità combinata' risulterà pertanto più grande di quella ottenibile con la sola somma delle due interazioni. Tale modello implica un'acquisizione della struttura proteica per gradi, in un processo in cui un'interazione cooperativa risulta termodinamicamente favorita rispetto a una non cooperativa.
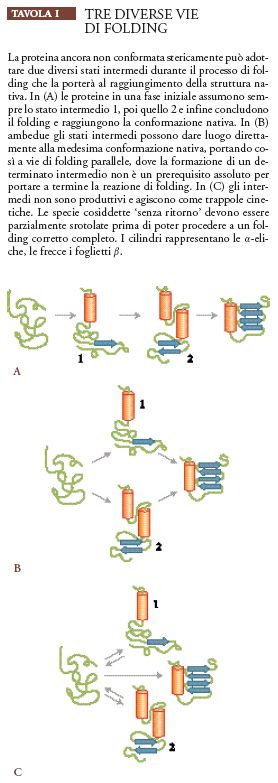
La semplice via lineare di folding che procede attraverso la formazione di una serie di intermedi, uno successivo all'altro (Tav. I), potrebbe non essere applicabile alla maggior parte dei polipeptidi. Esistono varie prove dell'esistenza di vie di folding multiple e indipendenti (Weissman 1995a).
Secondo questa ipotesi il folding di una proteina denaturata potrebbe avvenire attraverso vie distinte e parallele che comportano l'insorgenza di forme intermedie diverse ma che portano a un unico stato nativo ben definito. Durante il folding dell'inibitore pancreatico bovino della tripsina (BPTI, bovine pancreatic trypsin inhibitor), che presenta tre ponti disolfuro, sono stati individuati ponti disolfuro diversi da quelli della proteina nativa. Ovviamente questi legami devono essere rotti durante i successivi stadi del processo di folding; ciò indica che tale processo non deve necessariamente seguire un'unica via e neanche la più diretta.
È sorto il dubbio che gli intermedi osservati non rappresentino stati reali di folding ma che siano invece prodotti di reazioni collaterali non produttive che non conducono allo stato nativo. Questo dubbio è scaturito dalla scoperta che il folding di alcune piccole proteine avviene in modo estremamente veloce (circa 10-50 millisecondi), secondo una modalità 'tutto o nulla' in cui non sono identificabili intermedi di folding. Dal momento che i normali intermedi di folding persistono per centinaia di millisecondi, è possibile che alcuni di essi si comportino da trappole cinetiche, cioè che non abbiano più alcuna possibilità di procedere in nessuna delle direzioni della reazione, forse perché dotati di conformazioni strutturali che sono irrilevanti, o addirittura dannose, per la formazione dello stato nativo finale. A sostegno di questa ipotesi è stato dimostrato che nel folding del BPTI gli intermedi procedono più velocemente verso lo stato nativo quando sono prima denaturati: ciò potrebbe suggerire che essi siano forme intrappolate cineticamente (Weissman 1995a). Il problema dell'importanza cinetica degli intermedi è particolarmente critico quando una reazione di folding procede attraverso fasi multiple, poiché è probabile che gli intermedi che persistono più a lungo siano proprio quelli improduttivi.
Il problema del folding delle proteine 'in vivo'
Gli esperimenti in vitro hanno sicuramente un valore significativo nel definire i tipi di interazioni intramolecolari che guidano il folding delle proteine, tuttavia non riflettono accuratamente i processi di folding delle proteine nascenti all'interno della cellula. Le proteine non completamente strutturate, o in stati intermedi di folding, tendono a esporre superfici idrofobiche all'ambiente acquoso circostante e sono quindi particolarmente inclini all'aggregazione. Dal momento che l'aggregazione è uno dei maggiori problemi in qualsiasi saggio di folding, gli esperimenti in vitro sono spesso eseguiti in soluzioni estremamente diluite e a bassa temperatura. L'aggregazione comporta contatti intermolecolari ed è quindi, al contrario del folding, un processo di secondo ordine. In condizioni di alta concentrazione proteica e di alta temperatura, l'aggregazione aumenterà quindi rispetto al folding in modo esponenziale. In vivo, la temperatura fisiologica e l'alta concentrazione sia delle proteine totali (100-150 mg/ml) sia dei polipeptidi non strutturati favoriranno di gran lunga le interazioni improduttive di aggregazione rispetto alla via di folding corretta. Un'ulteriore differenza tra il folding delle proteine in vitro e quelli in vivo è il livello di complessità di molte proteine all'interno delle cellule. Proteine di membrana altamente idrofobiche, proteine che si assemblano in complessi enzimatici o in microfilamenti e proteine che sono modificate durante o dopo il processo di traduzione mediante legami con lipidi o carboidrati difficilmente seguiranno le vie di folding valide per piccole proteine globulari. Un'ulteriore considerazione è che, nella cellula, le proteine sono sintetizzate per gradi sui ribosomi e, nel caso di proteine dirette all'interno degli organelli, sono traslocate attraverso le membrane in forma estesa; esse dovrebbero quindi, secondo la teoria del folding cooperativo, permanere in forma denaturata almeno fino al compimento della sintesi del primo dominio strutturale.
Poiché la concentrazione delle catene proteiche nascenti nella cellula è alta (30-50 μM nel batterio Escherichia coli) queste saranno particolarmente inclini all'aggregazione: deve pertanto esistere all'interno della cellula un meccanismo grazie al quale tali aggregazioni siano impedite. Inoltre si è visto che alcune tappe lente nel folding delle proteine sono catalizzate da enzimi specifici. Negli ultimi anni è divenuto sempre più chiaro il modo in cui le cellule affrontano questi problemi di folding.
Enzimi coinvolti nel folding delle proteine
Due passaggi cruciali per la cinetica del processo di folding in vitro, consistenti nell'isomerizzazione di legami covalenti, possono essere catalizzati da enzimi cellulari purificati. L'enzima proteindisolfuroisomerasi (PDI) catalizza lo scambio tiolo/disolfuro e promuove la formazione, l'isomerizzazione o la riduzione di ponti disolfuro all'interno delle proteine. Gli enzimi peptidilprolilcis-transisomerasi (PPIasi) catalizzano l'altrimenti lenta isomerizzazione dei legami peptidici che precedono i residui amminoacidici di prolina. Ambedue questi enzimi non determinano la via di folding dei polipeptidi, ma piuttosto accelerano il folding di polipeptidi che formano ponti disolfuro o sono ricchi di prolina.
La proteindisolfuroisomerasi
La formazione non catalizzata di legami disolfuro è una tappa lenta nel processo di folding di molte proteine e PDI accelera questa reazione. Poiché solo le proteine della via secretiva contengono legami disolfuro, PDI è localizzato essenzialmente in compartimenti cellulari che sono parte integrante di tale via. In effetti PDI costituisce circa il 2% dell'intero contenuto proteico del reticolo endoplasmatico del fegato di ratto. Grazie alla presenza di questo enzima, in vivo la formazione di legami disolfuro corretti può essere molto rapida. In alcuni casi, come per le catene peptidiche delle immunoglobuline, è addirittura un evento cotraduzionale, cioè avviene prima che sia terminata la sintesi della catena polipeptidica sul ribosoma: appena un intero dominio proteico viene traslocato nel lume del reticolo endoplasmatico si forma contemporaneamente il singolo legame disolfuro interno a quel dominio. È stata dimostrata un'associazione specifica tra PDI e catene immunoglobuliniche neosintetizzate nel reticolo endoplasmatico.
Il ruolo di PDI appare ora abbastanza chiaro. L'enzima facilita la formazione del corretto corredo di legami disolfuro durante il folding ex novo di proteine della via secretiva. Esso non determina la via di folding ma piuttosto catalizza passaggi cineticamente lenti, presumibilmente mediante il rapido ridistribuirsi di legami disolfuro non corretti in presenza di un tiolo con bassa massa molecolare, quale per esempio il glutatione, tanto in forma ridotta quanto ossidata. La direzione in cui avviene il folding e il prodotto finale sono invece determinati dalla proteina stessa, cioè dal corredo nativo di legami disolfuro e da appropriate condizioni di ossidoriduzione.
PDI, nei mammiferi, è un omodimero formato da due subunità con massa molecolare di 57.000 kDa, ciascuna delle quali contiene duplicazioni di quei domini che mostrano una forte omologia con la tioredoxina, una piccola proteina che catalizza molte reazioni di ossidoriduzione e che è presente in tutte le classi di organismi, dai batteri agli eucarioti superiori. Studi di modellistica computerizzata, basati sulla nota struttura tridimensionale della tioredoxina di E. coli, indicano chiaramente che un dimero PDI, funzionalmente attivo, contiene quattro domini simili alla tioredoxina, ciascuno dei quali possiede un sito attivo tiolico posizionato su un'ansa sporgente della superficie della molecola.
Nei batteri la formazione di legami disolfuro nativi richiede la proteina DsbA che assomiglia, come struttura tridimensionale, alla tioredoxina ma che in più contiene un dominio a elica (Freedman 1994). DsbA sembra molto meno efficace di PDI nelle sue qualità di isomerasi, sia su substrati proteici denaturati sia su substrati perfettamente strutturati stericamente. Si può supporre che esso agisca da ossidante trasferendo ai substrati, in modo diretto, il proprio legame disolfuro. Al contrario PDI catalizza in modo efficace, in presenza di un tampone redox a base di glutatione, la conversione di un peptide ridotto alla sua forma disolfuro. L'analisi degli intermedi mostra che il processo avviene perlopiù attraverso la formazione di disolfuri misti tra il glutatione e il polipeptide, il quale poi si isomerizza rilasciando glutatione ridotto e generando il polipeptide con un legame disolfuro intramolecolare. Sia la formazione iniziale dei disolfuri misti sia il loro riarrangiamento successivo sono catalizzati da PDI: si può pertanto concludere che nei processi ossidativi PDI, al contrario di DsbA, agisce essenzialmente come catalizzatore di una successione di scambi tiolo/disolfuro con un ossidante esogeno.
Le peptidilprolilcis-transisomerasi
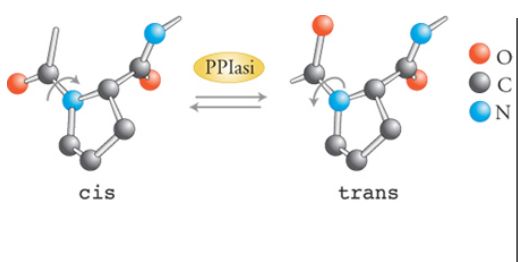
I legami peptidici possono esistere sia in forma cis sia in forma trans (fig. 4). Generalmente, dal punto di vista energetico, la forma trans è mille volte più stabile della forma cis. La situazione è diversa per i legami peptidici tra un amminoacido qualunque e la prolina: in questo caso la forma trans è pur sempre favorita, ma solo di quattro volte. Tuttavia, in modelli cristallografici di strutture proteiche, solo il 6,5% dei residui prolinici si trova nella forma cis, rispetto all'atteso 20% (Creighton 1990). Naturalmente ciascuna prolina presa singolarmente all'interno di una proteina perfettamente strutturata si troverà nella stessa forma isomerica in tutte le molecole. L'isomerizzazione cis-trans spontanea dei legami peptidici prolinici è lenta, con un tempo di dimezzamento di 20 minuti a 0 °C.
L'isomerizzazione di legami peptidici amminoacido-prolina non corretti è, perciò, uno di quei passaggi lenti cruciali nel determinare la cinetica di folding di una proteina nella cellula. La reazione può essere catalizzata da enzimi con attività tipo PPIasica. Le PPIasi sono molto abbondanti e distribuite in modo quasi ubiquitario (si trovano virtualmente in tutti i tessuti e in tutti gli organismi, dai batteri ai mammiferi) e sono presenti in tutti i compartimenti cellulari. Studi in vitro sulle PPIasi hanno mostrato che tali enzimi accelerano il refolding di un ampio spettro di proteine, ma con efficienza variabile. La miglior catalisi di folding operata da questi enzimi è l'aumento della costante cinetica dei folding della ribonucleasi T1 di 100 volte; una simile efficienza di catalisi si osserva anche con le catene leggere delle immunoglobuline. I legami prolinici situati all'interno degli intermedi di folding non sono tuttavia accessibili alle PPIasi.
Le PPIasi caratterizzate sinora possono essere classificate in tre famiglie, strutturalmente non correlate, che hanno preso il nome dai farmaci immunosoppressivi che inibiscono la loro rispettiva attività isomerasica: le ciclofiline legano la ciclosporina A; le proteine leganti FK-506 (FKBP, FK-binding proteins) legano FK-506 e la rapamicina; le parvuline non legano né la ciclosporina A né FK-506. È bene precisare che l'azione immunosoppressiva di questi farmaci non è legata alla loro capacità di inibire le PPIasi, bensì a un ruolo inatteso del complesso farmaco-PPIasi nella regolazione dell'espressione genica.
La reazione di folding della proteina che viene catalizzata da questi enzimi è una rotazione di 180° attorno all'asse C-N del legame peptidico che precede la prolina, che non coinvolge né scissione né formazione di legami covalenti. Perciò le PPIasi possono essere classificate come 'conformasi' (cioè enzimi in grado di modificare la conformazione molecolare) dotate di un'altissima efficienza (Fischer e Schmid 1990).
Chaperon molecolari
Alcune famiglie di proteine, strutturalmente non correlate ma universalmente conservate, aiutano le proteine neosintetizzate ad assumere la loro conformazione nativa. Queste proteine, ora collettivamente denominate chaperon molecolari, si legano ai polipeptidi quando si trovano nello stato totalmente o parzialmente denaturato, impedendone così l'aggregazione. Gli chaperon molecolari sono numerosissimi e l'espressione di molti di loro aumenta notevolmente in diverse condizioni di stress della cellula. Di fatto l'accumulo di proteine che hanno un folding anormale provoca un aumento dell'espressione di questi chaperon. Per ragioni storiche molti di questi chaperon molecolari sono classificati come proteine dello shock da calore (Hsp, heat shock proteins) o proteine da stress. Nel caso di E. coli, a 46 °C, temperatura alla quale la crescita batterica è praticamente nulla, più del 20% di tutti i polipeptidi presenti nella cellula appartiene a questa classe di proteine. Tale enorme accumulo riflette, molto probabilmente, l'aumentato bisogno di chaperon molecolari che contrastino la perdita della corretta conformazione spaziale e la tendenza all'aggregazione delle proteine, fenomeni che si manifestano ad alte temperature. È importante sottolineare che la terminologia 'proteine dello shock da calore' e 'proteine da stress' è in un certo qual modo fuorviante, poiché la maggior parte di queste proteine è importante per la crescita cellulare a tutte le temperature. Nonostante il legame e il rilascio da alcune Hsp siano regolati dal legame e dall'idrolisi dell'ATP, gli chaperon molecolari non possono essere considerati come catalizzatori del folding delle proteine. Più probabilmente essi impediscono l'aggregazione e le interazioni improduttive creando un ambiente regolato e protetto affinché il folding possa avvenire in maniera corretta e con maggiore efficienza.
La maggioranza degli chaperon identificati finora appartiene a famiglie proteiche molto conservate. Nonostante siano state identificate molte classi di chaperon, noi ci occuperemo solo delle famiglie di chaperon Hsp60 (GroEL) e Hsp70 (DnaK) che sono quelle maggiormente studiate (i numeri indicano la massa molecolare approssimativa, in migliaia di dalton, dei membri di ciascuna famiglia). Negli ultimi anni è stato chiarito che questi principali chaperon intracellulari non agiscono da soli. Per assicurare la loro corretta ed efficiente funzione, si sono evolute i cosiddetti 'co-chaperon'. In alcuni casi questi co-chaperon partecipano direttamente all'azione degli chaperon, come DnaJ (Hsp40) di E. coli, che agisce sinergicamente con DnaK. In altri casi hanno un ruolo più indiretto assicurando la corretta funzione e il riciclaggio delle proteine chaperon. Per esempio, sempre in E. coli, le proteine GroES (Hsp10) e GrpE, rispettivamente, lavorano con gli chaperon Hsp60 e Hsp70. Come sarà discusso più avanti, oltre a funzionare individualmente ciascuna con i propri co-chaperon, sembra che Hsp60 e Hsp70, in determinate situazioni, cooperino nel folding delle proteine nei mitocondri, cloroplasti e batteri.
Ipotesi sul funzionamento degli chaperon molecolari
Prima di analizzare le proprietà dei differenti complessi di chaperon ci sembra necessario esporre le nostre idee sul meccanismo molecolare attraverso il quale gli chaperon svolgono la loro funzione. Innanzitutto bisogna distinguere tra facilitazione di un processo e catalisi.
Durante il folding i polipeptidi procedono attraverso stati generalmente separati solo da barriere energetiche molto basse. Proteine piccole, con un unico dominio, spesso assumono molto velocemente il folding corretto senza che nessun intermedio cinetico possa essere fissato. Proteine più grandi iniziano la tappa finale del folding a partire da un intermedio che è separato dallo stato nativo da una barriera energetica molto alta. Il passaggio finale è quello che limita la velocità della reazione ed è fortemente cooperativo. In questo caso quindi l'intermedio finale del folding può essere fissato e analizzato. La velocità con cui appare il prodotto nativo dipenderà dalla concentrazione di questo intermedio e dalla costante di velocità. Questa è correlata alla barriera energetica che c'è tra l'intermedio e lo stato finale (energia di attivazione). La catalisi enzimatica, dovuta agli enzimi descritti prima, aumenta la costante di velocità abbassando questa barriera energetica, spesso stabilizzando lo stato di transizione tra substrato e prodotto. Gli chaperon molecolari, invece, facilitano il folding senza aumentarne la velocità, anzi in molti casi il processo di formazione del prodotto nativo è rallentato. Essi aumentano la quantità di proteina correttamente conformata.
Qualche indizio su come gli chaperon svolgano la loro azione ci viene dagli studi compiuti su SecB (Randall e Hardy 1995). SecB è uno chaperon tetramerico di E. coli che si associa specificamente ai polipeptidi che devono essere secreti. Perché il polipeptide possa essere secreto è essenziale che rimanga in una conformazione estesa fino a che non sia inviato a SecA, che regola il trasporto attraverso la membrana plasmatica. SecB deve quindi competere sia con l'aggregazione sia con il folding delle proteine. Per espletare la sua funzione è necessario che lo chaperon sia molto abbondante e che abbia un'alta velocità di legame e un'alta specificità per i substrati.
Abbondanza. L'aggregazione, poiché comporta contatti tra due o più molecole, è ovviamente un processo almeno di secondo ordine, e quindi la velocità di aggregazione è molto sensibile alla concentrazione degli intermedi di folding. Il folding, invece, è un processo di primo ordine e, quindi, molto meno influenzato dalla concentrazione. Di conseguenza, un incremento della concentrazione degli intermedi di folding liberi porterà a un aumento dell'aggregazione, mentre una diminuzione della loro concentrazione favorirà il folding. Uno chaperon potrebbe agire diminuendo la concentrazione degli intermedi liberi. L'interazione dei polipeptidi con gli chaperon è anch'essa un processo di secondo ordine: questo significa che la concentrazione dello chaperon nella cellula dovrebbe essere uguale o maggiore di quella dei polipeptidi non conformati stericamente. In una cellula in normali condizioni di crescita la concentrazione di SecB (nell'ordine del micromolare) sembra, in effetti, essere sufficientemente alta da favorire il legame con gli intermedi di folding rispetto alla loro aggregazione. Anche la concentrazione di altri chaperon molecolari nella cellula è costitutivamente alta. In condizioni di stress, quando aumenta la concentrazione di polipeptidi che hanno assunto un folding anormale, aumenta anche l'espressione degli chaperon di classe Hsp60 e Hsp70, in modo tale da compensare questo effetto.
Velocità di legame. Se il legame agli chaperon deve competere efficientemente con l'aggregazione, la velocità di legame agli chaperon dovrebbe avvicinarsi al limite di incontro tra chaperon e polipeptide; è stato calcolato, in base a dati ottenuti con SecB, che tutto questo accade (Randall e Hardy 1995). Macromolecole della dimensione di una proteina media collidono con una costante cinetica di circa 109 M−1s−1, mentre la costante che regola l'associazione di SecB con una proteina non conformata stericamente si aggirerebbe intorno a 108 M−1s−1. Lo chaperon potrebbe quindi contrastare efficacemente l'aggregazione.
Oltre che per l'aggregazione, SecB compete per il folding dei polipeptidi. Il collasso di un polipeptide a uno stato compatto e l'acquisizione di una struttura secondaria può aver luogo su una scala temporale di millisecondi. Ciononostante, poiché gli intermedi lungo la via di folding hanno un'energia di stabilizzazione molto bassa e sono in rapido equilibrio, non è possibile trarre conclusioni su quale sia la 'struttura' preferita da uno chaperon. È chiaro tuttavia che i polipeptidi non hanno più caratteristiche leganti nei confronti degli chaperon non appena oltrepassano lo stadio cineticamente limitante del folding, che per molte proteine è uno stadio tardo che coinvolge la formazione di una struttura nativa. Per le proteine precursori che interagiscono con SecB, la costante cinetica di questo stadio viene diminuita dalla presenza del peptide leader. Nel caso di SecB questo favorirebbe fortemente la formazione del legame piuttosto che il folding (Randall e Hardy 1995). Altri chaperon assistono il folding di polipeptidi non ripiegati, e pertanto il folding non è il processo competitivo, ma piuttosto quello favorito.
Specificità. Caratteristica comune degli chaperon è riconoscere polipeptidi solo allo stato non nativo. Questo riconoscimento deve portare a un forte e rapido legame del ligando e al sequestro delle sue superfici idrofobiche così da prevenirne l'aggregazione. Il riconoscimento diretto delle proteine non native potrebbe avvenire in virtù del loro carattere idrofobico solo se anche gli chaperon esponessero alla superficie, permanentemente, un'area idrofobica, ma ciò porterebbe all'aggregazione degli chaperon stessi. Nel caso di SecB è stato proposto, quindi, che il legame al polipeptide non sia dovuto tanto alla sua idrofobicità, quanto piuttosto al riconoscimento di regioni flessibili. Dopo il legame iniziale di SecB con svariate regioni flessibili di una proteina non nativa, avverrebbe un cambio conformazionale all'interno dello stesso chaperon. Solo in questo momento si avrebbe l'esposizione di un sito idrofobico sulla superficie dello chaperon, evento che porta all'identificazione della proteina legata come non nativa. Questo garantirebbe il legame del substrato e allo stesso tempo eviterebbe i problemi di aggregazione di chaperon non complessate.
Le interazioni coordinate su molti siti all'interno di uno stesso ligando portano a un legame ad alta affinità. Poiché ciascuno dei singoli contatti iniziali è a bassa specificità, la velocità del legame si avvicina molto al limite di incontro, permettendo una partizione cinetica dei polipeptidi nella via secretiva piuttosto che nei destini alternativi di aggregazione o di folding. Come altri chaperon risolvano il problema dell'aggregazione è al momento meno chiaro. Gli chaperon appartenenti alla famiglia delle Hsp60 hanno una struttura ad anello e i polipeptidi non conformati strutturalmente si legano all'interno o all'ingresso dell'anello. Gli chaperon di questa famiglia, quindi, hanno anche un sito di legame idrofobico mascherato, che potrebbe prevenire l'aggregazione.
Sebbene il meccanismo molecolare dell'attività degli chaperon non sia ancora chiaro, il caso di SecB fornisce un esempio di come uno chaperon possa risolvere il problema della specificità, della velocità nonché dell'affinità di legame a polipeptidi non nativi.
La famiglia delle Hsp70
Membri della famiglia delle Hsp70 si trovano in tutte le cellule procariotiche ed eucariotiche, nel citoplasma, nel nucleo, nel reticolo endoplasmatico, nei mitocondri e nei cloroplasti. Alcune proteine di questa grande famiglia sono essenziali per la crescita cellulare, altre sono copiosamente espresse in seguito a shock da calore, altre ancora, chiamate Hsc (heat shock cognate, membro della famiglia delle Hsc) sono espresse, invece, in modo costitutivo. L'alto livello di conservazione delle sequenze amminoacidiche nella famiglia delle Hsp70 ha portato alla proposta di un modello generale per la struttura e la funzione delle Hsp70.
Tutti i membri della famiglia delle Hsp70 recano all'estremità amminoterminale un dominio ATPasico altamente conservato seguito da una porzione carbossiterminale, meno conservata, che contiene il sito di legame dei peptidi. Alcuni membri della famiglia possiedono, inoltre, una sequenza segnale amminoterminale o una sequenza carbossiterminale di ritenzione nel reticolo endoplasmatico, a seconda della destinazione finale della proteina Hsp70 in questione.
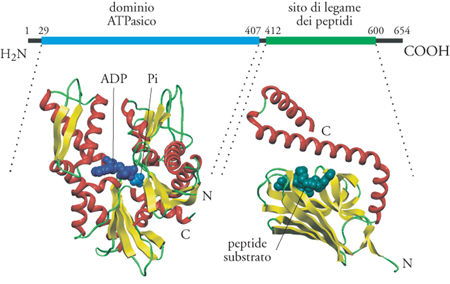
La struttura cristallina del dominio ATPasico amminoterminale della proteina Hsp70 bovina è stata determinata con una risoluzione di 2,2 Å (Flaherty et al. 1990). Essa è dotata di quattro domini strutturali che formano due lobi con in mezzo una fenditura (fig. 6). La tasca di legame per il nucleotide ATP e uno ione magnesio è situata alla base della fenditura. È interessante che la struttura terziaria complessiva del dominio ATPasico assomigli in modo straordinario a quella dell'actina G, nonostante la somiglianza tra le sequenze amminoacidiche di queste due proteine sia irrilevante. Inoltre la tasca di legame del nucleotide è simile a quella della esochinasi, tanto che queste proteine possono essere considerate parte di una superfamiglia strutturale. I residui amminoacidici che interagiscono con il complesso nucleotide-Mg2+ nel sito attivo sono rigorosamente conservati attraverso tutta la famiglia delle Hsp e, pertanto, si può suggerire un loro ruolo nel controllo dell'attività ATPasica delle proteine.
Un frammento di 18 kDa, situato immediatamente dopo il dominio ATIPasico, è sufficiente per il legame ad alta affinità del substrato. Quale sia la specificità di substrato delle Hsp70 è ancora oggetto di discussione. L'analisi di peptidi che si legano alla proteina legante (BiP, binding protein), l'Hsp70 dei mammiferi localizzata nel reticolo endoplasmatico, suggeriva una preferenza per i residui idrofobici. La struttura cristallina del peptide che lega il dominio di DnaK batterico ha dimostrato l'esistenza di un β-sandwich che porta il sito a cui si legano i peptidi e un coperchio ad α-elica che può aprire e chiudere il sito d'ingresso per i peptidi, in un modo che è probabilmente regolato dal dominio ATPasico (Zhu et al. 1996).
Il ciclo di reazione delle proteine Hsp70
Il modello per l'azione delle Hsp70 è costituito dalla proteina DnaK di E. coli che agisce contemporaneamente con altre due Hsp, DnaJ e GrpE (Georgopoulos 1992). Come ci si potrebbe aspettare, fenotipi simili risultano da mutazioni in ciascuna delle tre componenti del gruppo. Sia DnaJ sia DnaK sono in grado di legare polipeptidi, ma la loro affinità per i substrati è diversa. Sembra che, mentre DnaK preferisce interagire con proteine non strutturate stericamente, DnaJ tenda a legare substrati proteici che contengono strutture secondarie e terziarie (Cyr et al. 1994). Apparentemente queste differenze in specificità di substrato permettono a proteine tipo DnaJ di facilitare l'interazione di proteine nascenti con membri della famiglia Hsp70, che altrimenti non verrebbero legate. La DnaJ di E. coli riconosce una caratteristica strutturale tipica degli intermedi di folding, li stabilizza in una conformazione in cui sono esposti i siti di legame per le Hsp70, e facilita in questo modo il legame del substrato con DnaK. DnaJ agisce anche sulla stessa DnaK stimolando il passaggio chimico limitante nel ciclo di reazione ATPasico di DnaK. La regione amminoterminale di DnaJ, che contiene il dominio conservato J e una regione adiacente ricca di glicine e fenilalanine, è sufficiente per l'interazione con DnaK, poiché tale segmento proteico è in grado di stimolare l'idrolisi dell'ATP della DnaK. A differenza di DnaJ, GrpE promuove lo scambio nucleotidico agendo come un fattore di rilascio dei nucleotidi. GrpE interagisce con un'ansa strutturalmente conservata vicina al sito di legame per l'ATP di DnaK e presumibilmente tramite questa interazione influenza le condizioni di legame dei nucleotidi dello chaperon.
Studi recenti hanno chiarito alcuni aspetti importanti del ciclo ATPasico di DnaK. Da questi risultati è emerso un modello generale per il ciclo di reazioni delle proteine Hsp70. Il legame dell'ATP alle Hsp70 accelera sia la velocità di legame sia il rilascio del substrato peptidico. Le Hsp70, nella loro conformazione con ATP legato, possono essere quindi definite come la forma a 'pronto legame-pronto rilascio' (McCarty et al. 1995). Le Hsp70, nella loro conformazione con ADP legato, rilasciano i polipeptidi solo lentamente e, quindi, tale conformazione è stata all'inizio definita con il termine di 'stato di alta affinità', ma in realtà è molto lenta anche la formazione del legame con i peptidi. La conformazione con ADP legato può essere quindi definita come la forma a 'lento legame-lento rilascio'. Poiché il contenuto di ATP delle cellule in crescita è relativamente alto ci si aspetta che una frazione considerevole delle molecole Hsp70 si trovi nella conformazione legata all'ATP. Poiché la velocità di legame per i substrati non strutturati stericamente è più alta nella conformazione con ATP legato rispetto a quella con ADP legato (McCarty et al. 1995), le Hsp70 interagiranno con i peptidi non strutturati stericamente quando si trovano legate all'ATP. L'idrolisi dell'ATP è stimolata sia dal legame dei peptidi sia da DnaJ. La conformazione di Hsp70 legata all'ADP tenderà ad avere un'interazione stabile con il polipeptide, ed è quindi la principale forma legata ai polipeptidi che si trova negli esperimenti in vitro. La presenza di GrpE induce il rilascio dell'ADP generando una conformazione priva di nucleotidi. Il significato biologico di tale conformazione non è ancora chiaro, anche se ricorda lo stato conformazionale con ADP legato. Probabilmente il fosfato libero viene rilasciato prima dell'ADP, ma ancora non si conosce il significato di tale reazione. Il legame dell'ATP completa il ciclo e porta al rilascio della catena polipeptidica. La conformazione di Hsp70 risultante, cioè quella legata all'ATP, sarà di nuovo pronta per un altro ciclo di legame al rilascio di un nuovo polipeptide.
Da questo ciclo di reazioni si deduce che, all'interno della proteina Hsp70, ci deve essere un qualche tipo di comunicazione tra il dominio ATPasico e il dominio di legame del peptide. Il legame dell'ATP deve portare il dominio responsabile del legame dei peptidi a una conformazione più aperta, mentre il legame dell'ADP lo porta a una più chiusa. In accordo con questa ipotesi, alcuni dati sperimentali mostrano una differente mappa di digestione con proteasi e una differente fluorescenza dell'amminoacido triptofano a seconda che la molecola sia legata all'ATP o all'ADP. Questi dati indicano che il legame dell'ATP comporta un cambiamento conformazionale della molecola. Bisogna sottolineare che questo modello non è necessariamente applicabile a tutte le proteine Hsp70 perché è basato essenzialmente su studi riguardanti la proteina Hsp70 DnaK di E. coli. Per esempio, Ssalp, una Hsp70 citoplasmatica del lievito, sembra possedere interazioni stabili con proteine ancora non conformate stericamente proprio in presenza di ATP, mentre l'idrolisi porta al rilascio dei peptidi.
L'apparente non produttività del ciclo dell'ATP potrebbe sembrare un problema di questo modello. La conformazione di Hsp70 legata all'ATP rappresenta, infatti, lo stato in cui avvengono sia il legame sia il rilascio dei polipeptidi, pertanto dovrebbe essere già sufficiente a svolgere la funzione di chaperon durante il folding delle proteine. Un processo ciclico di idrolisi dell'ATP potrebbe quindi solo ritardare questo processo. Tuttavia un ritardo nel folding potrebbe non essere sconveniente. La produzione di intermedi di folding legati stabilmente porta, infatti, alla riduzione della concentrazione effettiva di proteine non ancora conformate ma inclini all'aggregazione, permettendo un processo di folding più efficiente, seppure più lento. Grazie al ciclo dell'ATP una proteina Hsp70 ha un'alta affinità per i peptidi non ancora conformati stericamente (quando è complessata con ATP), ma li lega anche con grande stabilità (quando è complessata con ADP) ed è inoltre capace di rilasciare la proteina velocemente (quando è complessata con ATP). Questo ciclo regolato di legame-rilascio fornisce allo chaperon il vantaggio di poter svolgere molteplici compiti nella biogenesi delle proteine. Molecole chaperon della classe Hsp70 sono coinvolte in numerosi e diversi processi all'interno della cellula. Dal momento che per gli obiettivi di tale rassegna una discussione di tutte queste funzioni risulterebbe troppo approfondita, ci concentreremo su un unico esempio nel quale diverse Hsp70 svolgono funzioni differenti.
Gli chaperon Hsp70 nella biogenesi delle proteine mitocondriali

Un elegante esempio delle differenti funzioni che le Hsp70 svolgono durante la sintesi e la maturazione delle proteine è dato dalla biogenesi delle proteine mitocondriali nel loro passaggio dal sito di sintesi nel citosol alla localizzazione funzionale nei mitocondri. Le proteine mitocondriali vengono sintetizzate su ribosomi citosolici come precursori, contenenti una sequenza segnale all'estremità amminoterminale. Esse vengono poi trasportate ai mitocondri, traslocate in una conformazione estesa attraverso le due membrane mitocondriali, per assumere infine la conformazione nativa nella matrice mitocondriale. Gli chaperon Hsp70 influenzano tutti gli stadi di questo processo e sono direttamente coinvolti nella traslocazione del polipeptide attraverso le membrane mitocondriali (Tav. II).
Sintesi proteica. Il genoma del lievito Saccharomyces cerevisiae codifica quattordici diverse proteine Hsp70. Come accade in altri eucarioti, queste Hsp70 sono localizzate nel citosol, nei mitocondri e nel reticolo endoplasmatico. Ci sono tre sottofamiglie citosoliche: SSA, con quattro membri, SSB con due, SSE con due e diverse Hsp70 sia mitocondriali sia localizzate nel reticolo endoplasmtico. Le sottofamiglie citosoliche hanno ruoli differenti nella cellula. I membri codificati da SSA sono implicati nel trasporto proteico e hanno un ruolo nella regolazione della risposta al calore e della degradazione proteica. Si pensa, invece, che i membri della famiglia SSB siano coinvolti nella sintesi proteica. Le proteine Ssb si associano ai ribosomi attivamente coinvolti nella sintesi proteica dai quali vengono rilasciate dopo trattamento con puromicina, un antibiotico che causa il rilascio delle catene polipeptidiche nascenti. Inoltre, mutanti nei geni SSB sono ipersensibili ad antibiotici che inibiscono l'allungamento della catena polipeptidica. Queste osservazioni suggeriscono che le proteine Ssb si associano con le catene polipeptidiche nascenti. Si potrebbe immaginare che una proteina Ssb si leghi a una catena nascente appena questa emerge dalla subunità ribosomale 60S, impedendo le interazioni intramolecolari all'interno della catena stessa o tra la catena nascente e la superficie del ribosoma e facilitando, quindi, la fuoriuscita dal ribosoma del resto della catena nel corso della sua sintesi. È da notare che l'espressione dei geni SSB non è indotta da shock da calore; al contrario, questi geni sono abbondantemente espressi in condizione di crescita ottimale, quando la velocità di sintesi proteica è alta.
Trasporto all'organo bersaglio. Le proteine per attraversare le membrane biologiche devono avere una conformazione lassa. Dati sperimentali emersi dallo studio della traslocazione delle proteine nei mitocondri hanno evidenziato la necessità che, durante questi processi, i precursori delle proteine per essere competenti per la traslocazione siano in uno stato particolare e non nativo. Il folding in una struttura terziaria stabile, indotta, per esempio, dal legame di analoghi substrati o cofattori, impedisce la traslocazione delle proteine all'interno del mitocondrio. È stato anche dimostrato che circa 50 residui amminoacidici sono sufficienti a coprire la distanza tra le due membrane mitocondriali. Ciò esclude che le proteine precursori siano in grado di attraversare le membrane nel loro stato nativo, e quindi queste molecole dovrebbero rimanere in uno stato non conformato oppure avere un folding lasso.
Gli chaperon molecolari della famiglia Hsp70 aiutano a mantenere nello stato competente per la traslocazione le proteine destinate ai mitocondri, al reticolo endoplasmatico, ai cloroplasti e al nucleo. Nel lievito questa funzione è espletata dalle proteine Ssa. L'eliminazione del gene che codifica Ssa1p porta all'accumulo nel citosol dei precursori della proteina F1β, (subunità β della ATP-sintasi), destinata alla membrana mitocondriale interna, e della proteina di secrezione denominata fattore α: questo suggerisce la presenza di uno stadio comune nel trasporto postraduzionale delle proteine attraverso le membrane mitocondriali e quelle del reticolo endoplasmatico. Essenziale in questo stadio è che gli chaperon Hsp70 siano legati al precursore della proteina da traslocare, in modo da prevenire il folding nello stato nativo. In questo caso è evidente il significato dell'assunzione di una conformazione a rilascio lento da parte delle Hsp citoplasmatiche. Nel trasporto ai mitocondri le preproteine sono inizialmente trasferite a recettori specifici posti sulla superficie della membrana mitocondriale esterna, i quali, a loro volta, presentano la proteina al canale attraverso cui avverrà la traslocazione vera e propria.
L'attività delle Hsp70 citoplasmatiche di lievito è regolata dagli omologhi di DnaJ, cioè Ydj1p e Sis1p. Ydj1p interagisce con le proteine Ssa, mentre Sis1p con le proteine Ssb. Dal momento che Sis1p è associata con piccoli polisomi e con le subunità ribosomali minori (40S), è possibile che sia coinvolta nell'indirizzare le proteine Ssb ai polisomi. Mutanti Ydj1 sono difettivi nella traslocazione delle proteine nel reticolo endoplasmatico e nei mitocondri. Ydj1p stimola l'attività ATPasica di Ssa1p ed è ancorato alle membrane mediante un gruppo lipidico (farnesilico) carbossiterminale, ed è quindi presente in quei siti dove potrebbe interagire con le proteine Ssa durante il processo di traslocazione. È sorprendente che, finora, non siano stati trovati omologhi di GrpE nel citoplasma eucariotico, mentre sono stati identificati numerosi omologhi di DnaK e DnaJ.
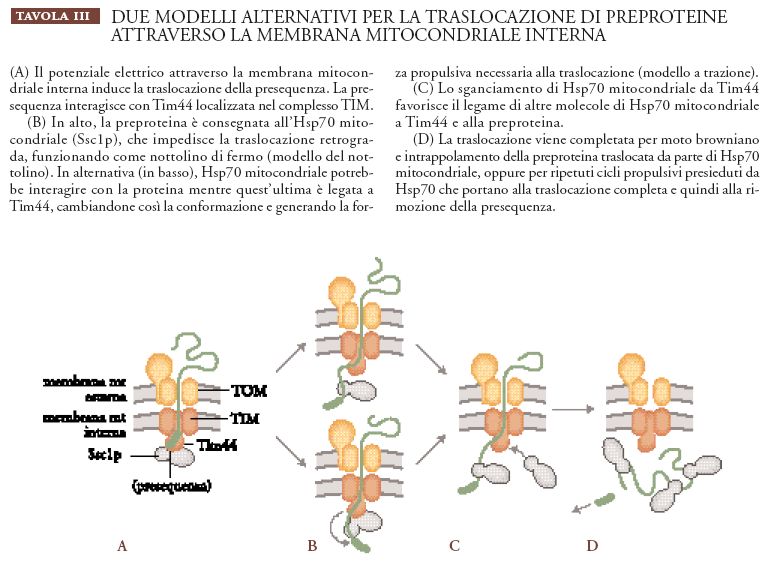
Traslocazione delle proteine. La traslocazione dei precursori proteici attraverso le due membrane mitocondriali richiede sia ATP sia un potenziale elettrico a cavallo della membrana mitocondriale interna. Si pensa che il potenziale di membrana, il cui interno è negativo, fornisca la forza trainante per la traslocazione della sequenza segnale, carica positivamente, della proteina mitocondriale, mentre la richiesta di ATP è attribuita prevalentemente al funzionamento della Hsp70 mitocondriale (Ssc1p nel lievito). Esperimenti su diversi mutanti Ssc1 in cui veniva tolto ATP dalla matrice mitocondriale hanno permesso di stabilire che per completare la reazione di traslocazione è necessario che Ssc1p si leghi ai precursori delle proteine mitocondriali (preproteine) che stanno entrando nel mitocondrio. Al momento sono diffusi due modelli (Pfanner e Meijer 1995) che descrivono l'azione di Ssc1p (Tav. III).
Nel primo modello, chiamato Brownian ratchet (o modello del nottolino browniano), Ssc1p ha un ruolo più passivo durante il processo di traslocazione. Per moto browniano il precursore proteico non strutturato scivola avanti e indietro attraverso il canale ed è intrappolato da Ssc1p dal lato della matrice. Secondo questo modello, Ssc1p avrebbe quindi un'attività 'di bloccaggio'. Alla fine l'intera catena polipeptidica può essere traslocata attraverso le due membrane mitocondriali mediante ulteriori oscillazioni della catena e il legame ad altre molecole Ssc1p. Per il riciclo di proteine Ssc1 legate, è quindi richiesto ATP. Questo modello prevede che, per essere traslocata, la preproteina debba essere in uno stato conformazionale esteso o lasso e spiega così l'importazione nei mitocondri di precursori proteici privi di folding compatto.
Il modello del nottolino browniano non può, però, spiegare facilmente la scoperta che alcuni precursori, come quello dei citocromo b2, vengono attivamente estesi da Ssc1p durante la traslocazione nei mitocondri. È stato perciò proposto un secondo modello in cui Ssc1p ha un ruolo più attivo. Il precursore che sta entrando è legato da Ssc1p dal lato della matrice ed è poi trascinato all'interno del mitocondrio mediante un cambio conformazionale di Ssc1p che rimane legata sia al precursore sia alla membrana. Questo cambio conformazionale genererebbe una forza che potrebbe permettere lo srotolamento (unfolding) del precursore e la sua traslocazione all'interno del mitocondrio. È stata identificata una proteina, Tim44, localizzata sulla membrana interna, che interagisce con Ssc1p e che potrebbe servire da ancoraggio. È interessante il fatto che l'interazione tra Ssc1p e Tim44 è distrutta dal legame dell'ATP allo chaperon. Pertanto, Ssc1p dovrebbe funzionare in equilibrio dinamico tra stato legato alla membrana e stato solubile. Secondo questo modello (modello a trazione) Sscp1 avrebbe quindi un'attività 'traente'. Il cambiamento conformazionale, cui va incontro Ssc1p quando lega l'ATP, costituisce forse il meccanismo molecolare alla base di questa attività traente.
Nei mitocondri di lievito sono stati identificati anche gli omologhi dei co-chaperon di E. coli, DnaJ e GrpE. La proteina GrpE mitocondriale (Mge1p) è essenziale per la crescita cellulare ed è coinvolta nel processo di importazione dei precursori mitocondriali, suggerendo una sua funzione, nel ciclo ATPasico di Hsp70, simile a quella dell'omologo in E. coli. Sorprendentemente, l'omologo di DnaJ (Mdj1p) non è essenziale e non è stato dimostrato che esso abbia un ruolo nella traslocazione dei precursori proteici mitocondriali. Comunque Mdj1p potrebbe avere una funzione nel folding delle proteine che si legano a Ssc1p.
Folding delle proteine. In seguito alla traslocazione nel mitocondrio e alla rimozione della presequenza, le proteine legate a Ssc1p devono assumere la conformazione nativa. Quindi, mentre le molecole Hsp70 sul lato citoplasmatico devono impedire il folding dei precursori proteici, sul lato della matrice proteine molto simili alle Hsp70 devono facilitare questo processo. Molte proteine mitocondriali importate devono essere assemblate nei grossi complessi localizzati nella membrana interna. Molti prodotti genici potrebbero agire come chaperon specializzati nell'assemblaggio di questi complessi nella membrana interna. È stato ben delineato il ruolo di Hsp60 mitocondriale nel folding delle proteine mitocondriali. In primo luogo consideriamo il funzionamento di Hsp60, successivamente descriveremo come Hsp60 e Hsp70 cooperino nel folding di proteine appena importate.
Le chaperonine
Le chaperonine sono una famiglia di molecole chaperon formate da subunità di circa 60 kDa che si assemblano in oligomeri di 14 subunità dalla caratteristica forma a doppio anello. Le chaperonine sono estremamente conservate e si trovano negli eubatteri (GroEL), nei mitocondri (Hsp60) nonché nei cloroplasti (RuBisCO binding protein). Le chaperonine costituiscono così un sottogruppo delle proteine chaperon molecolari (per es., le Hsp70 non sono chaperonine). Il citosol contiene complessi proteici ad anello detti TCP, anch'essi dotati di una struttura a doppio anello, che si pensa siano funzionalmente simili alle GroEL e alle Hsp60 ma che sono costituiti da subunità soltanto debolmente correlate a quelle delle chaperonine. Le proteine TCP mostrano invece una fortissima analogia con le chaperonine degli archeobatteri.
La struttura cristallina della chaperonina maggiormente studiata, GroEL di E. coli, è stata determinata con una risoluzione di 2,8 Å (Braig et al. 1994). GroEL è composta da due anelli formati da sette subunità di 57 kDa impaccate schiena a schiena. Studi strutturali e di mutagenesi hanno portato all'identificazione di un'ampia porzione dell'area funzionale che è posta sulla superficie del canale interno e delle sue invaginazioni. La funzione di GroEL è correlata a quella della chaperonina GroES, la cui struttura cristallina è stata chiarita soltanto di recente. GroES è una proteina essenziale, composta di un singolo anello formato da sette subunità di 10 kDa. Questa proteina forma un complesso asimmetrico con GroEL, in un rapporto stechiometrico di 1:1, legandosi a un'estremità della struttura cilindrica di GroEL. GroES contiene, al centro, un piccolo foro che potrebbe permettere la diffusione di metaboliti verso l'interno oppure l'esterno del cilindro di GroEL. Substrati polipeptidici non conformati stericamente si legano a GroEL nello stesso dominio riconosciuto da GroES, mentre il sito di legame dell'ATP è situato sulla superficie più interna del dominio equatoriale, prossimo al centro del cilindro. Un cambiamento conformazionale potrebbe quindi accoppiare la presenza del nucleotide appropriato con il processo, assistito da GroES, di legame e rilascio dei polipeptidi.
Il meccanismo di azione di GroEL
GroEL può formare un complesso molto forte con varie proteine che si trovano in uno stato strutturale non conformato, mentre ha poca affinità per le proteine allo stato nativo. Studi di microscopia elettronica e di mutagenesi mostrano come polipeptidi che non si trovano allo stato nativo vengano trattenuti all'interno della cavità centrale formata dagli anelli di GroEL. GroEL e GroES, a concentrazioni fisiologiche di ATP e ADP, formano un complesso stabile ma dinamico. Il complesso asimmetrico GroEL-GroES, quando è legato all'ADP agisce, probabilmente, come accettore di polipeptidi poiché si tratta di una specie conformazionale dotata di un'esistenza relativamente lunga e di un'alta affinità per i polipeptidi (Weissman et al. 1995b).
Un'estremità del cilindro GroEL è 'incappucciata' da GroES lì legata (il lato cis) e ciò dovrebbe impedire l'accesso di una proteina alla regione di legame del peptide, situata nella cavità centrale posta a quella estremità del cilindro. I polipeptidi potrebbero, quindi, legarsi inizialmente all'anello di GroEL non occupato da GroES (il lato trans).
Durante il ciclo di folding, il rilascio e il successivo legame di GroES permetterebbero la formazione di un complesso cis nel quale sia GroES sia il polipeptide sono legati allo stesso anello di GroEL. GroES continuerebbe a svolgere la sua funzione di incappucciatore anche in questo complesso cis, ma questa volta, invece di escludere il polipeptide dalla cavità centrale di GroEL, lo manterrebbe sequestrato al suo interno. La transizione da un complesso trans a uno cis con una sola molecola di GroES legata (modello a pallottola) potrebbe comprendere una fase in cui il complesso è privo di GroES o, in alternativa, il complesso ha un cappuccio GroES ad ambedue i lati del cilindro generando la struttura intermedia rugby. Il legame e l'idrolisi di ATP nell'anello di GroEL, opposto a quello occupato da GroES e dal polipeptide, indurrebbe il rilascio di GroES legato al lato cis e permetterebbe il liberarsi del polipeptide nel citosol.
Dal modello appena descritto del meccanismo d'azione del complesso GroEL-GroES consegue che ci deve essere un limite fisico alle dimensioni dei polipeptidi assistiti dalle chaperonine nel folding. In assenza di co-chaperonine, peptidi di dimensioni fino a circa 35 kDa possono entrare dentro un singolo anello di GroEL. Il legame della co-chaperonina GroES induce, tuttavia, in GroEL un cambiamento conformazionale di vaste proporzioni: approssimativamente il raddoppio del volume della cavità centrale dell'anello. I substrati noti delle chaperonine sono in genere sufficientemente piccoli da essere accolti confortevolmente all'interno, espanso, del complesso. Al contrario, numerose proteine che sono prossime o che superano il limite fisico previsto per essere accolte all'interno della chaperonina non vengono assistite nel folding dal complesso GroEL-GroES.
Le chaperonine potrebbero, quindi, essere attive solo nel folding di quelle proteine cellulari che siano sufficientemente piccole; non è ancora chiaro il meccanismo di folding di proteine di dimensioni maggiori. Inoltre, in normali condizioni di crescita, molte o forse la maggior parte delle proteine più piccole raggiunge il proprio stato nativo di folding senza il coinvolgimento di GroEL. L'esatta percentuale di vie di folding dipendenti o indipendenti da GroEL non è stata ancora determinata.
Le chaperonine e la stimolazione del folding
Una funzione passiva. Sebbene questo modello abbia chiarito che le chaperonine agiscono secondo una modalità regolata legando e rilasciando polipeptidi non conformati stericamente, non è ovvio come questo ciclo di legame e rilascio porti a un aumento dell'efficienza dei folding. Oggi sono argomento di discussione due punti di vista contrastanti riguardo al folding di polipeptidi assistito da GroEL. Alcuni prevedono che il polipeptide sia estruso dalla chaperonina in una conformazione ancora non strutturata stericamente e che il folding finale avvenga all'esterno della chaperonina, nel citosol (Todd et al. 1994; Weissman et al. 1995b). Altri prevedono che il polipeptide vada incontro al folding all'interno del complesso GroEL-GroES e che sia estruso dalla chaperonina solo quando non si possano più verificare eventi di interazione tra il complesso e il polipeptide (Mayhew et al. 1996).
Secondo il primo modello le chaperonine potrebbero agire, come già discusso nel caso di Hsp70 e SecB, attuando un sequestro degli intermedi di folding, riducendo quindi la concentrazione di quei polipeptidi tendenti all'aggregazione: la competizione con l'aggregazione porterebbe, in questo caso, a un aumento netto del folding. Tuttavia, le straordinarie proprietà strutturali delle chaperonine suggeriscono che si possa verificare un meccanismo diverso. Poiché i polipeptidi che devono andare incontro a folding si legano all'interno del cilindro della chaperonina, essi risulterebbero protetti da ogni altra struttura non nativa con la quale si potrebbero aggregare. Le chaperonine sarebbero in grado così di fornire un ambiente protetto nel quale i polipeptidi vanno incontro a folding senza avere alcuna possibilità di aggregarsi. La struttura a più subunità delle chaperonine, con la presenza di molti siti di legame per i polipeptidi e l'ATP, porterebbe a un legame regolato e all'idrolisi di più molecole di ATP; ne consegue che il folding delle proteine potrebbe essere sequenziale. In questo modello alternativo il polipeptide non sarebbe rilasciato dalla chaperonina tutto in una volta, ma sequenzialmente, dominio per dominio. I polipeptidi potrebbero, quindi, incominciare il loro folding mentre sono ancora legati alla chaperonina.
È stato calcolato che il folding completo di una singola molecola polipeptidica coadiuvato da GroEL richiede l'idrolisi di circa 100 molecole di ATP; sono quindi necessari molti cicli di legame e rilascio perché un polipeptide adotti il suo stato nativo finale. Le chaperonine potrebbero legarsi nuovamente a domini che non acquisiscono immediatamente una conformazione nativa e non espongono ancora superfici idrofobiche. I domini che sono andati incontro a folding corretto non hanno più affinità per le chaperonine e rimangono liberi. Il nuovo legame non avviene necessariamente sulla stessa chaperonina ma potrebbe, dopo il rilascio del polipeptide non nativo, verificarsi su una diversa molecola di GroEL (Weissman et al. 1996). Il rilascio di forme non native di una proteina potrebbe anche avere un ruolo fisiologico. Molte proteine che sono substrato di GroEL sono subunità di complessi oligomerici e la oligomerizzazione richiede che le subunità siano in uno stato non perfettamente conformato. Inoltre, il rilascio di forme non native offre alle proteine substrato l'opportunità di interagire con altri sistemi chaperon, per esempio con il macchinario dello chaperon Hsp70, che può essere essenziale per il folding efficiente di alcune proteine.
Una probabile funzione attiva. Come già discusso, il modello più plausibile di azione delle chaperonine è quello che prevede il sequestro dei polipeptidi in un ambiente protetto con un aumento netto del folding. Tuttavia alcune osservazioni sperimentali suggeriscono che il meccanismo non sia esclusivamente passivo ma che, in certe circostanze, le chaperonine possano abbassare in modo attivo la barriera di energia del folding. La prima evidenza è che il folding della malicodeidrogenasi è assistito da GroEL a rapporti stechiometrici molto bassi, pari a una molecola di GroEL per ogni dieci molecole di deidrogenasi. In queste condizioni il 90% del substrato deve essere libero in soluzione, così che l'effetto del sequestro di polipeptidi non conformati stericamente è trascurabile per quanto riguarda la cinetica di aggregazione. La seconda evidenza è che in alcuni casi GroEL attua il recupero, nella via di folding, di intermedi che ne sono usciti e che si trovano stabilmente in una situazione che non evolve né verso il folding né verso l'aggregazione irreversibile. In questo modo, la chaperonina accresce significativamente la velocità di folding (Todd et al. 1994). È probabile che il ruolo attivo delle chaperonine sia legato alla loro funzione di 'srotolamento' di intermedi inclini all'aggregazione o cineticamente intrappolati.
La cooperazione tra diversi chaperon nel folding delle proteine
Proteine appartenenti a differenti classi di chaperon si trovano insieme negli stessi compartimenti cellulari. Esse sono apparentemente non correlate l'una all'altra e hanno un'ampia specificità di substrato e potrebbero, quindi, competere l'una con l'altra per lo stesso substrato e assistere il folding di proteine simili. In effetti, molti chaperon non sono essenziali per la cellula. Per esempio, mutazioni di DnaK non sono letali alle normali temperature; sembra che la perdita della funzione di questo chaperon possa essere compensata dalla espressione di un'altra proteina non correlata con DnaK, ma con funzione sovrapponibile. A temperature elevate DnaK diviene essenziale per la vita, suggerendo quindi che, in condizioni estreme, per prevenire l'aggregazione di proteine, sia necessario l'intero e completo potenziale di tutti gli chaperon della cellula. Altri chaperon sono invece essenziali a tutte le temperature (per es., GroEL E. coli, Hsp60 e Hsp70 della matrice mitocondriale). Ciò suggerisce che questi ultimi chaperon svolgano un ruolo insostituibile, che non può essere assunto da un altro chaperon. Hsp70 mitocondriale è indispensabile poiché presiede all'importazione di tutte le proteine della matrice mitocondriale, anche di quelle essenziali. Le chaperonine sono, comunque, coinvolte nella prevenzione dell'aggregazione o nell'assistenza al processo di folding delle proteine. Sembra che esse abbiano un ruolo in un passaggio essenziale di questo processo che non può essere svolto da un altro tipo di chaperon.
La specificità di substrato di diverse classi di chaperon dovrebbe essere, almeno in parte, differente. Effettivamente, mentre gli chaperon della classe Hsp70 interagiscono principalmente con proteine completamente denaturate, le chaperonine della classe Hsp60 potrebbero interagire con elementi della struttura secondaria e con intermedi della via di folding; esse infatti legano le strutture globulari semisolide. A differenza delle Hsp70, le chaperonine hanno alta affinità per i peptidi corti e i polipeptidi in conformazione estesa. Queste due classi di chaperon potrebbero, quindi, agire in sequenza nel folding di proteine neosintetizzate o appena traslocate.
Il folding delle proteine mitocondriali. Durante la biogenesi delle proteine della matrice mitocondriale i precursori proteici sono traslocati attraverso le membrane mitocondriali, in uno stato conformazionale non strutturato stericamente. Sul lato della matrice la catena polipeptidica interagisce direttamente con la proteina Hsp70 mitocondriale (Ssc1p nel lievito). Questa interazione è transitoria (Manning-Krieg et al. 1991); dopo il rilascio da Hsp70, il folding della proteina, in una conformazione compatta e resistente all'attacco di enzimi proteolitici, avviene con un certo ritardo. È stato anche osservato che il legame alla Hsp60 mitocondriale di una proteina importata nella matrice risulta ritardato rispetto al legame con la Hsp70 mitocondriale. È stato dimostrato che una singola preproteina si lega prima a Hsp70 mitocondriale, poi a Hsp60 e raggiunge lo stato di folding finale solo dopo lo sganciamento dalla chaperonina. Sia il rilascio di polipeptidi da Hsp70 sia il legame a Hsp60 nel mitocondrio sono passaggi che richiedono ATP.
Sulla base di quanto detto si può prefigurare il seguente modello per il folding delle proteine mitocondriali. Durante la traslocazione il polipeptide, che è in configurazione estesa, interagisce inizialmente con la Hsp70 mitocondriale in un modo dipendente dall'ATP. Dopo il rilascio da Hsp70 alcune proteine non raggiungono immediatamente lo stato nativo, ma sono piuttosto indirizzate a Hsp60, che media i successivi passaggi di folding. Hsp70 e Hsp60 perciò svolgono funzioni sequenziali e in parte non sovrapposte nel processo di folding delle proteine mitocondriali. Quale sia l'influenza su questo processo di folding dei differenti co-chaperon presenti nella matrice mitocondriale non è ancora chiaro. Bisogna sottolineare che non tutte le proteine richiedono necessariamente la presenza di Hsp60 per il folding, poiché molte raggiungono il loro stato nativo subito dopo il rilascio da Hsp70.
Il folding delle proteine in E. coli. Una situazione simile si potrebbe configurare per il folding di proteine nel citoplasma di E. coli. In un mutante difettivo per tutte le Hsp, l'aggregazione di proteine non avviene se il mutante è trasformato da un plasmide che causa sovraespressione del complesso chaperon GroEL-GroES o di quello DnaK-DnaJ. Tuttavia, a concentrazioni fisiologiche e a temperature elevate, è richiesta l'espressione di tutte e quattro le proteine; ciò indica che in vivo ad alte temperature sono richiesti entrambi i sistemi chaperon. Questa osservazione suggerisce che anche in E. coli i due sistemi chaperon, in alcuni casi, non hanno specificità o funzioni sovrapposte.
Il ruolo di entrambi questi sistemi chaperon nel folding delle proteine è stato ricostituito in vitro utilizzando componenti isolati da E. coli. È stato dimostrato che il refolding della proteina rodanasi dal suo stato denaturato dipende sia dal complesso GroEL-GroES sia da quello DnaK-DnaJ-GrpE (Langer et al. 1992). In presenza sia di DnaJ sia di ATP, la DnaK impedisce l'aggregazione di rodanasi durante il suo refolding. DnaK, una volta rilasciato ADP, tramite GrpE, si lega all'ATP che permette la liberazione della proteina ancora in uno stato conformazionale non pienamente strutturato, consentendone il trasferimento a GroEL per le fasi di folding finale e per il raggiungimento dello stato nativo. La base per questa azione sequenziale di Hsp70 e di Hsp60 sembra risiedere nella differente specificità che hanno questi chaperon rispetto alle caratteristiche strutturali della catena peptidica.
È stato proposto che la sequenza di eventi sopra descritta sia rappresentativa anche di quanto avviene nel folding di nuove catene polipeptidiche al loro emergere dal ribosoma durante la loro sintesi. Tuttavia, la situazione in presenza di catene nascenti sembra essere più complicata. Le prime proteine con le quali interagiscono le catene nascenti sono, negli eucarioti, le cosiddette 'proteine del complesso associato alle catene nascenti' (NAC, nascent chain associated complex) e, in E. coli, il cosiddetto trigger factor, entrambi associati ai ribosomi e privi di specificità per qualsiasi polipeptide (Rassow e Pfanner 1996). Successivamente i polipeptidi che contengono le sequenze segnale per la secrezione vengono trasferiti, in E. coli, a SecB e, negli eucarioti, al 'complesso riboproteico di riconoscimento del segnale' (SRP, signal recognition particles). I polipeptidi che risultano privi di sequenze segnale, al contrario, potrebbero essere assistiti nel folding dai complessi DnaK-DnaJ e GroEL-GroES. Tuttavia siamo lontani dall'avere un quadro chiaro dei meccanismi di cooperazione tra i differenti chaperon, generici e specifici, sia nel folding sia nel trasporto all'organello bersaglio di proteine neosintetizzate.
Poiché i sistemi chaperon sono strutture che hanno subito poche mutazioni, e pertanto sono estremamente conservate nei compartimenti di molti organismi, le vie di folding descritte per i mitocondri e per E. coli potrebbero rappresentare un meccanismo generale per la prevenzione dei fenomeni di aggregazione e per la stimolazione del folding delle proteine. Persino nel citosol degli eucarioti la sequenza di eventi che porta al folding potrebbe essere la stessa, anche se mancano omologhi diretti di Hsp60 e di GrpE. In questo caso però la funzione di Hsp60 potrebbe essere svolta da TRiC e quella di GrpE da Hip. La funzione specifica di altri chaperon e Hsp è al momento meno chiara. Mentre alcune di queste proteine potrebbero svolgere funzioni specializzate, come, per esempio, SecB nel prevenire il folding delle preproteine secretive in E. coli, altre potrebbero svolgere funzioni vicarianti dei sistemi chaperon Hsp70 o Hsp60. La via Hsp70-Hsp60 non si può, però, applicare indiscriminatamente al folding di tutti i polipeptidi, come indicano alcune proteine che non vanno incontro a folding anche se legate e rilasciate da GroEL. Studi futuri chiariranno il meccanismo d'azione degli chaperon e permetteranno, si spera, di risolvere la questione se gli chaperon siano davvero coinvolti attivamente nel folding delle proteine o se il loro ruolo sia solo quello di prevenire l'aggregazione e, quindi, accrescere la quantità di proteine che abbiano raggiunto una corretta conformazione strutturale.
Bibliografia
Anfinsen 1973: Anfinsen, Christian B., Principles that govern the folding of protein chains, "Science", 181, 1973, pp. 223-230.
Braig 1994: Braig, Kerstin e altri, The crystal structure of the bacterial chaperonin GroEL at 2.8 Å, "Nature" 371, 1994, pp. 578-586.
Creighton 1990: Creighton, Thomas E., Protein folding, "Biochemical journal", 270, 1990, pp. 1-16.
Creighton 1993: Creighton, Thomas E., Proteins: structures and molecular properties, New York, Freeman, 1993.
Creighton 1995: Creighton, Thomas E., Protein folding. An unfolding story, "Current biology", 5, 1995, pp. 353-356.
Cyr 1994: Cyr, Douglas M. - Langer, Thomas - Douglas, M.G., DnaJlike proteins: molecular chaperones and specific regulators of Hsp70, "Trends in biochemical sciences", 19, 1994, pp. 176-181.
Ellis, van der Vies 1991: Ellis, R. John - van der Vies, S.M., Molecular chaperones, "Annual review of biochemistry", 60, 1991, pp. 321-347.
Fischer, Schmid 1990: Fischer, Gunter - Schmid, Franz-Xaver, The mechanism of protein folding. Implications of in vitro refolding models for de novo protein folding and translocation in the cell, "Biochemistry", 29, 1990, pp. 2205-2212.
Flaherty 1990: Flaherty, Kevin M. - De Luca Flaherty, Camille - McKay, David B., Three-dimensional structure of the ATPase fragment of a 70 k heat-shock cognate protein, "Nature", 346, 1990, pp. 623-628.
Freedman 1994: Freedman, Robert B., Protein folding: folding helpers and unhelpful folders, "Current biology", 4, 1994, pp. 933-935.
Georgopoulos 1992: Georgopoulos, Costa, The emergence of the chaperone machine, "Trends in biochemical sciences", 17, 1992, pp. 295-299.
Georgopoulos, Welch 1993: Georgopoulos, Costa - Welch, William, Role of the major heat shock proteins as molecular chaperones, "Annual review in cell biology", 9, 1993, pp. 601-634.
Gething, Sambrook 1992: Gething, Mary-Jane - Sambrook, Joseph, Protein folding in the cell, "Nature", 355, 1992, pp. 33-45.
Hendrick, Hartl 1993: Hendrick, Joseph P. - Hartl, Franz-Ulrich, Molecular chaperone functions of heat-shock proteins, "Annual review of biochemistry", 62, 1993, pp. 349-384.
Langer 1992: Langer, Thomas e altri, Successive action of DnaK, DnaJ and GroEL along the pathway of chaperone-mediated protein folding, "Nature", 356, 1992, pp. 683-689.
Manning-Krieg 1991: Manning-Krieg, Ute C. - Scherer, Philipp E. - Schatz, Gottfried, Sequential action of mitochondrial chaperones in protein import into the matrix, "EMBO Journal", 10, 1991, pp. 3273-3280.
Mayhew 1996: Mayhew, Mark e altri, Protein folding in the central cavity of the GroEL-GroES chaperonin complex, "Nature", 379, 1996, pp. 420-426.
McCarty 1995: McCarty, John S. - Buchberger, A. - Reinstein, J. - Bukau, B., The role of ATP in the functional cycle of the DnaK chaperone system, "Journal of molecular biology", 249, 1995, pp. 126-137.
Morimoto 1994: The biology of heat shock proteins and molecular chaperones, edited by Richard I. Morimoto, A. Tissières and Costa Georgopoulos, New York, Cold Spring Harbor Laboratory Press, 1994.
Parsell, Lindquist 1994: Parsell, Down A. - Lindquist, Susan, Heat shock proteins and stress tolerance, in: The biology of heat shock proteins and molecular chaperones, edited by Richard I. Morimoto, A. Tissières and Costa Georgopoulos, New York, Cold Spring Harbor Laboratory Press, 1994, pp. 457-494.
Pfanner, Meijer 1995: Pfanner, Nikolaus - Meijer, Michiel, Protein sorting. Pulling in the proteins, "Current biology", 5, 1995, pp. 132-135.
Ptitsyn 1995: Ptitsyn, Oleg B., How the molten globule became, "Trends in biochemical sciences", 20, 1995, pp. 376-379.
Randall, Hardy 1995: Randall, Linda L. - Hardy, Simon J.S., High selectivity with low specificity: how Secß has solved the paradox of chaperone binding, "Trends in biochemical sciences", 20, 1995, pp. 65-69.
Rassow, Pfanner 1996: Rassow, Joachim - Pfanner, Nikolaus, Protein biogenesis: chaperones for nascent polypeptides, "Current biology", 6, 1996, pp. 115-118.
Thomas 1995: Thomas, Philip J. - Qu, Bao-He - Pedersen, Peter L., Defective protein folding as a basis of human disease, "Trends in biochemical sciences", 20, 1995, pp. 456-459.
Todd 1994: Todd, Matthew J. - Viitanen, Paul V. - Lorimer, George H., Dynamics of the chaperonin ATPase cycle: implications for facilitated protein folding, "Science", 265, 1994, pp. 659-666.
Weissman 1995a: Weissman, Jonathan S., All roads lead to Rome? The multiple pathways of protein folding, "Chemistry and biology", 2, 1995, pp. 255-260.
Weissman 1995b: Weissman, Jonathan S. e altri, Mechanism of GroEL action: productive release of polypeptide from a sequestered position under GroES, "Cell", 83, 1995, pp. 577-587.
Weissman 1996: Weissman, Jonathan S. - Rye, Hays S. - Fenton, Wayne A. - Beechem, Joseph M. - Horwich, Arthur L., Characterization of the active intermediate of a GroEL-GrOES-mediated protein folding reaction, "Cell", 84, 1996, pp. 481-490.
Zhu 1996: Zhu, Xiaotian e altri, Structural analysis of substrate binding by the molecular chaperone DnaK, "Science", 272, 1996, pp. 1606-1614.