
La grande scienza. Malattie genetiche, cervello e comportamento
La grande scienza. Malattie genetiche, cervello e comportamento
Malattie genetiche, cervello e comportamento
Le neuroscienze cognitive hanno le proprie radici in campi diversi quali la neurobiologia, la psicologia, le scienze linguistiche e computazionali; esse si avvalgono dei risultati raggiunti in ciascuna di queste discipline per formulare teorie che permettono di costruire una rappresentazione integrata della mente umana e dei suoi fondamenti biologici. Le recenti rivoluzioni nei campi della visualizzazione della struttura cerebrale e della genetica molecolare hanno fornito alle neuroscienze cognitive la possibilità di esaminare il succedersi di eventi evolutivi, a partire dal livello genetico e neurobiologico fino a quello cognitivo.
Alcune ipotesi sui legami fra i geni e il comportamento possono essere chiarite mettendo a confronto lo sviluppo delle funzioni cognitive, della funzione e della struttura cerebrale in bambini normali e in bambini che presentano fenotipi anormali. Recentemente, sono stati condotti studi di questo tipo su soggetti con sindrome di Turner, autismo, neurofibromatosi e due sindromi genetiche con ritardo mentale: le sindromi di Williams e di Down. Tali programmi di ricerca stanno mettendo in luce evidenze sperimentali convergenti per capire le componenti cognitive e, allo stesso tempo, forniscono modelli per collegare geni e comportamento.
Il progresso raggiunto da questo tipo di approccio può essere mostrato attraverso ricerche recenti che mettono a confronto le sindromi di Williams e di Down. Gli individui affetti da tali sindromi manifestano anomalie linguistiche, cognitive e a livello di sviluppo neurologico che, se per alcuni versi simili, presentano tuttavia differenze marcate in relazione all'esatta natura dei deficit cognitivo e comportamentale di ciascuno (Bellugi et al. 1994; Korenberg et al. 2001). Mentre la sindrome di Down è stata ben caratterizzata, fino a poco tempo fa si conosceva poco sulla sindrome di Williams; il motivo risiede, in parte, nella rarità di tale malattia, con un'incidenza di 1 su 25.000 nati vivi tale da essere riconosciuta come sindrome specifica solo dal 1961. Fino ad alcuni anni fa, le diagnosi venivano effettuate dai genetisti clinici principalmente sulla base della tipica morfologia della faccia associata a ritardo mentale, difetti cardiaci, anomalie nel metabolismo del calcio, difficoltà di crescita nell'infanzia e ritardo nello sviluppo. Tuttavia, recentemente, gli studi condotti dai genetisti molecolari sulla patogenesi della sindrome di Williams hanno reso possibile l'identificazione del difetto genetico responsabile: un'emizigosi attorno al locus dell'elastina nel cromosoma 7 (Korenberg et al. 1996; 2001); attualmente è perciò possibile effettuare la diagnosi genetica. Queste nuove conoscenze genetiche hanno, di conseguenza, determinato la nascita di una nuova branca di ricerca volta a scoprire i legami esistenti fra geni e comportamento nella sindrome di Williams.
Gli studi condotti in parallelo su bambini normali e su altri con sindrome di Williams, sindrome di Down, autismo o lesioni focali all'emisfero destro o sinistro hanno evidenziato che, in particolare, i soggetti con sindrome di Down rappresentano un gruppo di controllo relativamente omogeneo e ben definito, rispetto alla più grande popolazione caratterizzata da ritardi mentali, per il confronto con la sindrome di Williams. Inoltre, ricerche recenti dal punto di vista neurobiologico e genetico pongono, a pieno diritto, la sindrome di Down in un'area di interesse per quanto riguarda la relazione fra geni e comportamento.
Sindrome di Williams e sindrome di Down
Dissociazione fra linguaggio e altre funzioni cognitive
La precisa relazione fra la struttura del linguaggio (grammatica) e altri aspetti delle funzioni cognitive è un argomento teorico fortemente dibattuto. I maggiori modelli teorici di acquisizione del linguaggio presentano punti di vista alternativi sulla relazione fra i domini cognitivo e linguistico. Gli studi condotti su popolazioni atipiche, come su individui con sindrome di Williams e di Down, possono essere determinanti per risolvere questo problema, dissociando aree funzionali che normalmente sono profondamente intrecciate, e fornendo informazioni sul substrato neurale sottostante a queste.
Ritardo nelle abilità cognitive generali
Gli individui che vengono colpiti dalle sindromi sopra citate sono normalmente classificati come ritardati mentali, secondo la definizione ufficiale dell'American association on mental deficiency.
Uno studio specifico colloca la media dei quozienti d'intelligenza (QI) intorno a 55, con un grado di variabilità da 40 a 100 (Bellugi et al. 1997). Sebbene alcuni adulti con sindrome di Williams siano autonomi, la maggior parte vive o studia in ambienti protetti. Per poter essere confrontati, i soggetti con sindrome di Williams e di Down sono stati selezionati in modo da essere accoppiati per età, QI globale e percorso educativo.
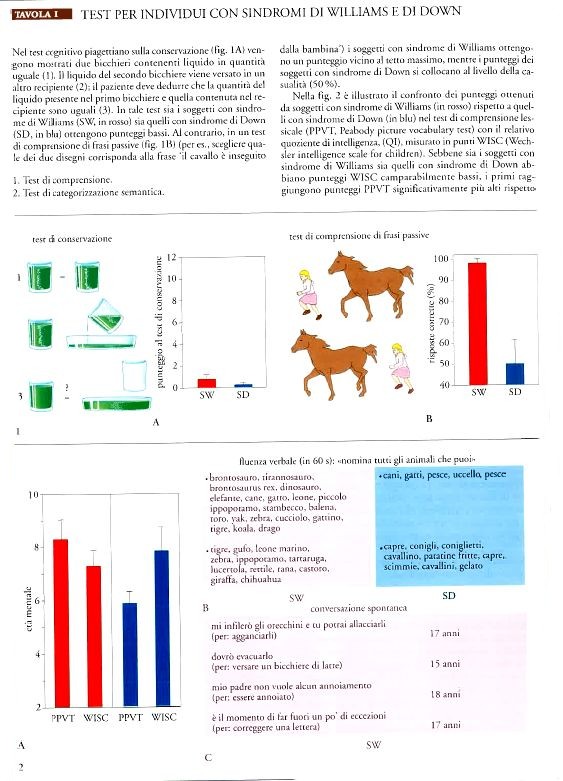
Prove ulteriori di intelligenza generale mettono in evidenza che i soggetti con sindrome di Williams e di Down risultano essere parimenti compromessi. Attraverso una batteria di test concettuali e di soluzione di problemi, entrambi i gruppi mostrano lo stesso deficit (Bellugi e Wang 1998; Bellugi et al. 2001). Un esempio è il test piagettiano che comprende la conservazione del numero, del peso e della materia (Bellugi et al. 1994). Queste prove, in individui normali, sono risolte precocemente nel corso dello sviluppo cognitivo: mentre i bambini piccoli normali sono in grado di superare con facilità le prove cognitive sulla conservazione, al contrario gli adolescenti con sindrome di Williams o con sindrome di Down non ne hanno le capacità (Tav. Ia e Ib).
Preservazione delle abilità linguistiche
Nell'ambito di questo generale ritardo cognitivo, tuttavia, il linguaggio dei soggetti con sindrome di Williams si distingue in maniera marcata da quello dei soggetti di controllo con sindrome di Down, così come dal linguaggio di altri gruppi che presentano un ritardo mentale. Uno dei tratti distintivi dei soggetti con sindrome di Williams, dato l'alto livello di deficit cognitivo, può essere proprio la loro elaborazione linguistica straordinariamente competente.
Grammatica. Nei test di comprensione di frasi passive, negative e condizionali, i punteggi ottenuti dai soggetti con sindrome di Williams sono superiori rispetto a quelli dei soggetti di controllo con sindrome di Down (Bellugi et al. 2001). In un test di comprensione di frasi passive, per esempio, i soggetti adolescenti con sindrome di Williams hanno ottenuto un punteggio vicino al tetto massimo. L'abilità nello scoprire e nel correggere le anomalie della sintassi dipende dalla conoscenza della costruzione sintattica e dalla capacità di riflettere sulle strutture grammaticali di una frase. Queste sono abilità metalinguistiche sofisticate che possono essere padroneggiate facilmente dopo l'acquisizione delle regole grammaticali e che possono non essere mai sviluppate, in maniera completa, in alcune popolazioni a rischio che comprendono bambini con danni cerebrali o con sindromi genetiche. È stato riscontrato che la competenza linguistica dei soggetti con sindrome di Williams si estende anche ai test di abilità metalinguistica (Bellugi e Wang 1998). Inoltre, l'analisi del linguaggio espressivo spontaneo dei soggetti adolescenti con sindrome di Williams mostra che essi, normalmente, sono in grado di formulare frasi ben strutturate e corrette dal punto di vista grammaticale. Generalmente questi soggetti usano marcatori della morfologia in modo appropriato e corretto, inclusi quelli per il tempo e il modo, così come verbi ausiliari e articoli. Inoltre, questi soggetti utilizzano in maniera caratteristica una ricca varietà di forme grammaticali complesse, tra cui frasi passive, proposizioni condizionali e relative, sebbene siano presenti errori occasionali e anche alcuni sistematici (Losh et al. 1997; Bellugi et al. 2001).
Invece, il linguaggio dei soggetti di controllo con sindrome di Down è più semplice e meno vario nella costruzione sintattica e presenta spesso errori e omissioni sia morfologici sia sintattici. Queste differenze nella competenza linguistica, sia nei test di produzione sia in quelli di comprensione, mettono in evidenza come, nel contesto di un deficit cognitivo generale ampiamente diffuso, le abilità linguistiche nei soggetti con sindrome di Williams siano notevolmente preservate.
Semantica insolita: una caratteristica della sindrome di Williams. Attraverso una serie di studi, i soggetti con sindrome di Williams sembrano mostrare una propensione per parole inusuali, atipiche sia rispetto ai soggetti normali sia a quelli con sindrome di Down. Nonostante il loro basso QI, gli adolescenti con sindrome di Williams sono riusciti a indicare il disegno appropriato con parole come 'canino', 'abrasivo' e 'solenne' nel test di comprensione lessicale PPVT (Peabody picture vocabulary test). In un test di categorizzazione semantica, ai soggetti è stato chiesto di nominare tutti gli animali che riuscivano a pensare in un minuto. Gli adolescenti con sindrome di Williams hanno fornito molte più risposte rispetto agli adolescenti con sindrome di Down, tante quante i controlli normali. Il gruppo di soggetti con sindrome di Down ha fornito un numero di risposte inferiore, non sempre comprese nella categoria degli animali (per es., 'gelato'). Degno di nota è il fatto che i soggetti con sindrome di Williams siano stati in grado di produrre non solo parole tipiche della categoria, ma abbiano operato anche scelte lessicali a bassa frequenza, non primitive, quali 'yak' (bue del Tibet), 'chihuahua', 'stambecco', 'condor', 'avvoltoio', 'unicorno', 'tigre dal dente a sciabola', molto più frequentemente rispetto ai controlli normali di pari età mentale. Quindi, sembra che la conoscenza delle parole non usuali, la loro elaborazione e la loro scelta siano una caratteristica dei soggetti con sindrome di Williams. Si noti che questa caratteristica è diversa sia dai disturbi semantici che accompagnano altre patologie cliniche (afasia e demenza) sia dagli errori prodotti occasionalmente da soggetti normali (lapsus); infine, è decisamente diversa dalle limitazioni semantiche caratteristiche di altri gruppi con ritardo mentale (Bellugi et al. 1994; 2001).
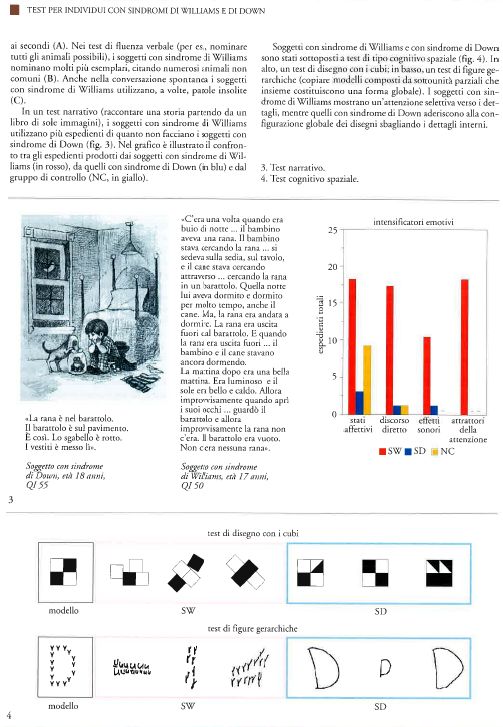
Ricchezza di stati emotivi nel linguaggio della sindrome di Williams. L'interazione fra il linguaggio e gli stati emotivi nei soggetti con sindrome di Williams è stata dimostrata utilizzando una serie di test narrativi; in uno di questi, ai soggetti viene mostrato un libro in cui sono presenti solamente immagini e viene chiesto di raccontare la storia, senza fornire nessun altro aiuto al di là delle figure stesse. Il linguaggio spontaneo prodotto dai soggetti con sindrome di Williams è risultato sofisticato, non solo per quanto riguarda la fonetica e la sintassi, ma anche perché ricco di espedienti grammaticali e affettivi atti a rendere più coerente il racconto e ad attirare l'attenzione dell'ascoltatore. Queste caratteristiche sono in forte contrasto con le prove di linguaggio dei soggetti di controllo con sindrome di Down. I soggetti con sindrome di Williams (Reilly et al. 1990; Jones et al. 2001) sono in grado di produrre, in maniera caratteristica, racconti ben strutturati, stabilendo un chiaro orientamento, introducendo il tempo, i personaggi e, addirittura, i loro stati e comportamenti (C'era una volta, quando era buio di notte), utilizzando clausole subordinate per le informazioni riguardanti avvenimenti passati e previsioni future, stabilendo il problema che è alla base della storia (La mattina dopo la rana non c'era) ed enunciando una sua risoluzione (Guarda e riguarda, lo trovano ...).
Inoltre, i soggetti con sindrome di Williams usavano la prosodia affettiva (cambiamenti di tono, allungamenti vocalici, modificazioni del volume) molto più frequentemente rispetto ai controlli con sindrome di Down e ai coetanei normali. La ricchezza affettiva delle narrazioni dei soggetti con sindrome di Williams si rifletteva anche nelle loro scelte lessicali; i loro racconti includevano commenti frequenti sullo stato emotivo dei personaggi nelle storie (per es., E ah! Si meravigliò, Il cane si arrabbiò e il ragazzo si infuriò), così come espedienti teatrali quali il discorso diretto e gli effetti sonori (E boom, milioni di api uscirono fuori e lo inseguirono per pungerlo). L'utilizzazione di frasi esclamative e di altri espedienti per attirare l'attenzione dell'ascoltatore è evidente nella maggior parte delle storie (All'improvviso splash! L'acqua salì, Guarda e riguarda, lo trovarono con una signora, Santo cielo! Il ragazzo e il cane cominciano ad agitarsi). Questi espedienti erano molto meno frequenti nei soggetti normali ed erano quasi assenti nei soggetti con sindrome di Down.
Nonostante il loro deficit cognitivo, i soggetti con sindrome di Williams non solo sono socievoli ed emotivamente sensibili, ma mostrano anche di essere coscientemente capaci di manipolare gli espedienti linguistici dell'affettività per raccontare storie. Tuttavia, questi soggetti usano lo stesso livello di espressività indipendentemente da quante volte hanno raccontato la storia e da quale sia il loro pubblico, suggerendo in tal modo che la loro espressività estrema possa essere anormale (Reilly et al. 1990; Losh et al. 2000). Questa abbondanza di emotività, sia nella prosodia sia negli espedienti linguistici, è notevolmente diversa rispetto a quella dei soggetti autistici, i quali spesso appaiono privi di risorse sociali e affettive. Infatti, per alcuni aspetti, gli individui con sindrome di Williams e quelli con autismo sembrano essere opposti dal punto di vista sociale, cognitivo e neurale. L'ipersocievolezza, priva della capacità di giudizio sociale sottostante, potrebbe costituire una delle caratteristiche della sindrome di Williams non diversa da quella riscontrata in alcuni pazienti con lesioni frontali descritti da Antonio R. Damasio.
Punti di forza e punti di debolezza nell'area cognitivospaziale
Sia i soggetti con sindrome di Williams sia quelli con sindrome di Down mostrano deficit nel processamento spaziale. Tuttavia, la cognizione dello spazio può essere sezionata, proprio come il linguaggio. Questi due tipi di sindrome sembrano condizionare le abilità cognitivospaziali in modi differenti; inoltre, all'interno delle abilità visuospaziali della sindrome di Williams emergono abilità preservate che potrebbero fornire la chiave di lettura per l'organizzazione sottostante (Bihrle et al. 1989; Bellugi et al. 1999).
Caratteristiche peculiari dei deficit spaziali. Negli individui con sindrome di Williams i disegni spesso mancano di coesione e di organizzazione globale (gestaltica). Il disegno di una casa potrebbe includere finestre, una porta e un tetto, ma le varie parti potrebbero non essere nella giusta relazione reciproca, per esempio, con la finestra disposta dall'altra parte della pagina, fuori dai confini della casa.
Al contrario, un analogo disegno di un soggetto con sindrome di Down tenderebbe a essere molto semplificato, anche se avrebbe contorni e forma chiari, con relazioni appropriate fra gli elementi. Sottoposti al test di 'disegno con cubi' della WISC-R (Wechsler intelligence scale for children - revised), i due gruppi ottengono un punteggio ugualmente scarso. Tuttavia, l'esame del processo mediante il quale hanno ottenuto il proprio punteggio rivela differenze marcate. Sebbene entrambi falliscano nel riprodurre in maniera corretta il disegno, i soggetti con sindrome di Down generalmente aderiscono alla conformazione globale dell'ordinamento dei blocchi, con le configurazioni dei disegni interni sbagliate. I soggetti con sindrome di Williams, al contrario, non riescono a riprodurre la configurazione globale del disegno e sembrano influenzati dai dettagli posizionando i blocchi in un ordinamento apparentemente accidentale, non contiguo. Confrontando il processo di costruzione utilizzato dagli adolescenti con sindrome di Williams con quello dei soggetti affetti da sindrome di Down, risulta che i primi compiono un maggior numero di mosse, ma ciascuna di esse produce come risultato una figura frammentata.
Sono stati ottenuti risultati simili quando ai soggetti è stato richiesto di copiare modelli composti da sottounità parziali che insieme costituiscono una forma globale come, per esempio, una D maiuscola fatta di y minuscole. In questi test, la prestazione dei soggetti con sindrome di Williams rispetto a quella dei soggetti con sindrome di Down rispecchia superficialmente le differenze riscontrate fra i soggetti cerebrolesi con lesioni cerebrali destre e sinistre. Infatti, quando viene chiesto di riprodurre i disegni, entrambi i gruppi falliscono, ma secondo due modi diversi: i soggetti con sindrome di Williams riproducono fedelmente le sottounità parziali sparse sulla pagina e non la forma globale, mentre i soggetti con sindrome di Down mostrano un comportamento opposto, tendono cioè a produrre la configurazione globale senza gli elementi costitutivi (fig. I.4). Questi risultati suggeriscono una tipologia di processamento insolita nei soggetti con sindrome di Williams, e cioè una propensione a rivolgere l'attenzione verso i dettagli a discapito dell'insieme (Bihrle et al. 1989; Bellugi et al. 1999, 2001).
Test sul riconoscimento di volti. Nonostante la grave compromissione nelle abilità cognitivospaziali, esistono ambiti nei quali i soggetti con sindrome di Williams mostrano un risparmio selettivo di abilità. Tali soggetti, al contrario di quelli con sindrome di Down, mostrano una spiccata abilità nel riconoscere, discriminare e ricordare volti familiari e non familiari (Rossen et al. 1996). Questa include abilità relative alla percezione dei volti, così come la capacità di riconoscerli in varie condizioni di luce e di orientamento. I soggetti con sindrome di Williams, nonostante il loro marcato deficit visuospaziale, nei test sul riconoscimento di volti mostrano prestazioni sorprendentemente buone, migliori rispetto a quelle dei soggetti con sindrome di Down, dimostrandosi abili quanto i controlli normali della stessa età. Quindi, nonostante i soggetti con sindrome di Williams presentino gravi deficit nelle abilità cognitive generali, nell'area cognitivospaziale mostrano un modello peculiare di picchi e cadute, una enfasi sul processamento del particolare rispetto al globale, un estremo frazionamento nel disegno e, ancora, un'isola di abilità preservate nel processamento, nel riconoscimento e nel ricordo di volti (Bellugi et al. 1999; 2001; Jones et al. 2001).
Le prove neurocognitive suggeriscono che la sindrome di Williams, al contrario di quella di Down, è caratterizzata da un profilo neurocomportamentale altamente eterogeneo di deficit specifici, con anomalie e capacità preservate sia all'interno sia fra i domini delle più alte funzioni cognitive. La sindrome di Williams, dunque, rappresenta un modello raro di dissociazioni, fornendo l'insolita opportunità di stabilire legami fra i substrati neurali e le basi genetiche della sindrome.
Sindrome di Williams
Stadi di sviluppo
Il profilo neurocognitivo riscontrato negli adolescenti e negli adulti con sindrome di Williams e di Down è per alcuni versi sostanzialmente differente da quello mostrato durante le prime fasi di sviluppo. Gli studi sull'acquisizione delle prime parole e della grammatica condotti su ampi gruppi di bambini affetti da questi due tipi di sindrome rivelano un ritardo generale in molti aspetti del linguaggio. Questi risultati sono sorprendenti, date le marcate differenze riscontrate fra gli adolescenti con sindrome di Williams e quelli con sindrome di Down dove i primi, nonostante il loro significativo deficit cognitivo, mostrano un livello linguistico superiore a quello dei controlli con sindrome di Down (Singer Harris et al. 1997). Tuttavia, mentre entrambi i gruppi di bambini sono ugualmente ritardati rispetto ai controlli normali, emergono percorsi differenziati per il linguaggio e per la comunicazione. In particolare, i bambini Down mostrano un precoce vantaggio per i gesti comunicativi, mentre quelli con sindrome di Williams, in una fase più tardiva dello sviluppo, mostrano una superiorità per quanto riguarda la grammatica. Emergono altre differenze confrontando i tre domini nel corso dello sviluppo, cioè il lessico, le abilità visuospaziali e il processamento di volti. I bambini con sindrome di Down mostrano punteggi ugualmente bassi in tutti e tre i domini, mentre il profilo di sviluppo dei bambini con sindrome di Williams è differente fra i diversi domini. Le funzioni visuospaziali sono significativamente inferiori in tutte le età rispetto a quelle dei bambini con sindrome di Down e non superano mai il livello che equivale all'età di 5 anni (Wang e Bellugi 1994). Per quanto riguarda lo sviluppo del linguaggio, si verifica un ritardo iniziale equivalente nelle due sindromi e una crescita continua, distinta ma ritardata, nel processo linguistico, come l'emergere della grammatica. Il processamento di volti è eccellente sin dalle prime fasi dello sviluppo, in quanto i soggetti con sindrome di Williams ottengono punteggi superiori alla loro età mentale indipendentemente dall'età cronologica.
Basi neurobiologiche
Oltre a chiarire l'organizzazione delle abilità cognitive, le recenti ricerche hanno per oggetto lo studio delle basi neurobiologiche del linguaggio e del pensiero. Questo compito è stato stimolato dallo sviluppo di metodi innovativi per l'analisi della struttura e delle funzioni del cervello in soggetti viventi e pensanti, come gli studi neurofisiologici che utilizzano i potenziali evocati (ERP, event related potentia), la tomografia a emissione di positroni (PET, positron emission tomography), le nuove tecniche per la visualizzazione tridimensionale e, infine, le immagini di risonanza magnetica (MRI, magnetic resonance imaging).
Caratterizzazione neurofisiologica
Le tecniche ERP sono state utilizzate per stabilire la tempestività e l'organizzazione dell'attività neurale nelle aree cerebrali implicate nel processamento, sensoriale, cognitivo e linguistico in soggetti con sindrome di Williams (Neville et al. 1994; Mills et al. 2001). È stato così possibile studiare le due caratteristiche più salienti del profilo comportamentale in soggetti con sindrome di Williams. In primo luogo, è stato analizzato un ciclo di ripristino uditivo per fornire indizi sulle origini della sensibilità agli stimoli acustici, manifestata da molti soggetti con sindrome di Williams. In secondo luogo, è stato esaminato il processamento acustico di frasi che includono anomalie semantiche per verificare se nella sindrome di Williams esso è mediato dalle stesse connessioni che risultano attive nei controlli normali aventi la stessa età.
Nei soggetti con sindrome di Williams le risposte ai potenziali evocati uditivi sono risultate normali e ciò indica nessuna alterazione a livello del tronco encefalico. Tuttavia, i dati raccolti con la prova di recupero della funzione uditiva suggeriscono un meccanismo corticale possibile alla base della manifesta sensibilità ai suoni. A otto soggetti con sindrome di Williams e a dieci normali della stessa età sono stati fatti ascoltare toni presentati a livelli di ripetizione differenti (monitorati per turni di frequenza), al fine di determinare il grado di ripristino, o periodo refrattario, delle risposte ai potenziali evocati uditivi. La morfologia generale dei potenziali evocati dagli stimoli tonali (per es., le risposte negative a 100 ms, N100, e positive a 200 ms, P200) è risultata essere molto simile nell'emisfero sinistro e destro in soggetti con sindrome di Williams e in soggetti normali di controllo. Tuttavia, i primi hanno mostrato, rispetto ai secondi, risposte maggiori ai gradi di ripetizione più alti, suggerendo che le loro risposte sono meno refrattarie e quindi più eccitabili rispetto ai soggetti normali. Questo meccanismo risulta evidente unicamente sopra la corteccia temporale ed è specifico allo stimolo uditivo; utilizzando la prova di recupero della funzione visiva i soggetti con sindrome di Williams sono indistinguibili dai soggetti normali di controllo. Nel loro insieme, questi studi suggeriscono che l'iperacusia osservata nella sindrome di Williams potrebbe essere mediata da un'ipereccitabilità specifica entro le aree corticali che vengono utilizzate per il processamento delle informazioni acustiche.
Sono state inoltre registrate con gli ERP le risposte dei soggetti con sindrome di Williams a presentazioni uditive di parole all'interno di frasi. La metà delle frasi era fortemente legata al contesto e terminava con una parola appropriata dal punto di vista semantico, mentre l'altra metà terminava con una parola anomala, per esempio: Prendo il mio caffè con latte e carta. Studi sperimentali precedenti hanno dimostrato che i soggetti normali forniscono un'ampia risposta negativa a 400 ms (N400) in seguito alla presentazione di parole non appropriate per quanto riguarda l'aspetto semantico e questo è considerato un indice del modo in cui si verifica l'organizzazione del lessico mentale. I soggetti con sindrome di Williams mostrano risposte che sono anormali, entro i primi 200-300 ms che seguono l'inizio della presentazione della parola. L'anormalità consiste nella grande positività, non riscontrata nei soggetti di controllo a nessuna età. Questo effetto, che si manifesta solo sopra le regioni cerebrali temporali, può essere collegato con l'iperacusia della sindrome di Williams. L'effetto dell'anomalia semantica è maggiore nei soggetti con sindrome di Williams piuttosto che nei controlli e potrebbe avere relazione con l'insolita facilità dei primi per le parole rare dal punto di vista semantico da loro mostrata in alcuni test (Bellugi et al. 2001). Inoltre, le risposte di questi soggetti non mostrano le asimmetrie attese dell'emisfero sinistro, tipiche dei bambini e degli adulti normali, suggerendo che potrebbe esserci un modello insolito di organizzazione cerebrale sottostante alle abilità linguistiche della sindrome di Williams.
Basi neuroanatomiche
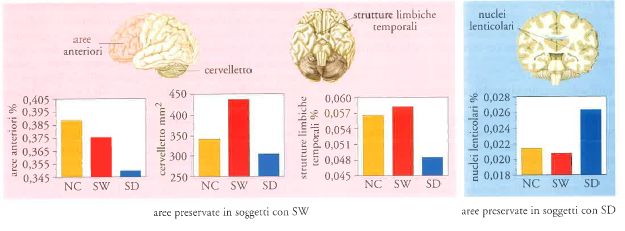
Le nuove tecniche di visualizzazione dell'attività cerebrale attraverso immagini permettono la rappresentazione e l'analisi delle strutture interne del cervello, che non erano possibili in passato. Le tecniche sviluppate nel 1992 da H. Damasio e R. Frank, per esempio, permettono oggi una visualizzazione innovativa e un'analisi tridimensionale del cervello dei soggetti in vita. I nostri studi hanno rivelato che sia la sindrome di Williams sia quella di Down lasciano un'impronta morfologica caratteristica in regioni cerebrali specifiche. Attraverso le immagini di risonanza magnetica sono stati realizzati studi sul volume cerebrale di un gruppo di adolescenti e giovani adulti affetti da sindrome di Williams e di Down (Jernigan e Bellugi 1990; 1994; Hickok et al. 1995a; 1995b). La fig. 1 mostra i risultati della caratterizzazione neuromorfologica dei soggetti con sindrome di Williams rispetto a quelli con sindrome di Down, con il volume cerebrale ridotto in entrambi i gruppi rispetto ai controlli normali della stessa età. I due gruppi presentano differenze considerevoli nel volume delle regioni cerebrali. Innanzitutto, il volume cerebrale anteriore è ridotto in modo sproporzionato nei soggetti con sindrome di Down, mentre è preservato in modo proporzionato in quelli con sindrome di Williams. Inoltre, le strutture limbiche nel lobo temporale mostrano, in proporzione, volumi essenzialmente uguali nei soggetti con sindrome di Williams e nei soggetti di controllo, mentre sono ridotte in modo significativo nei soggetti con sindrome di Down. Invece, il volume del talamo e dei nuclei lenticolari è preservato molto meglio nei soggetti con sindrome di Down rispetto a quelli con sindrome di Williams. È stato riscontrato, inoltre, che le parti anteriori del corpo calloso, così come gli emisferi anteriori, sono preservate nei soggetti con sindrome di Williams, mentre sono ridotte in quelli con sindrome di Down.
Anche l'analisi quantitativa del volume del cervelletto mostra differenze, con il volume ben preservato nei soggetti con sindrome di Williams e ridotto in quelli affetti da sindrome di Down. Analisi strettamente settoriali sono illuminanti: è stato riscontrato che il locus più preservato nella sindrome di Williams è il neocerebello. Delle due porzioni del neocerebello sottoposte ad analisi, sia i vermi sia le tonsille cerebellari mostrano una preservazione volumetrica o addirittura un incremento nei soggetti con sindrome di Williams rispetto ai controlli, mentre entrambe le parti sono volumetricamente ridotte nei soggetti con sindrome di Down. Un risultato di grande importanza ottenuto da questi studi è che le regioni specifiche del neocerebello, di dimensioni maggiori nella sindrome di Williams, si sono rivelate displasiche nell'autismo (Bellugi et al. 1999; Reiss et al. 2001).
Ricerche affini suggeriscono che l'estesa corteccia prefrontale e il neocerebello, entrambi preservati in modo selettivo nella sindrome di Williams, sono considerati strettamente collegati. Queste due regioni del cervello sono le più sviluppate in Homo sapiens e si ritiene che si siano evolute contemporaneamente. Inoltre, il neocerebello presenta connessioni più estese con la corteccia prefrontale e con altre aree associate che non con le parti più vecchie del cervelletto. La terza area preservata nella sindrome di Williams, il lobo temporale mediale, potrebbe includere porzioni della corteccia associativa uditiva, che si proietta anche sull'area di Broca e su altre aree prefrontali. Il profilo neuroanatomico della sindrome di Williams, che si ottiene dalle nuove tecniche di neuroimaging, sta iniziando così a fornire un contributo alla comprensione dell'organizzazione cerebrale, mostrando un modello morfologico che può essere influenzato da fattori genetici. La scoperta che le regioni anteriore, limbica temporale e neocerebellare sono preservate selettivamente nella sindrome di Williams suggerisce che esse possano essere influenzate da un singolo fattore genetico che regoli il loro sviluppo, oppure che il loro sviluppo sia mutualmente interattivo o, infine, che si verifichino entrambi gli eventi (Bellugi et al. 1994). L'esistenza di una relazione tra cervello e comportamento è di importanza fondamentale per le problematiche affrontate dalle neuroscienze cognitive.
Studi sulla citoarchitettonica cerebrale
Durante gli anni Ottanta sono state condotte numerose indagini sperimentali che hanno portato alla conoscenza delle basi biologiche di molte sindromi di ritardo mentale. Attualmente, è possibile attribuire queste sindromi a fattori che agiscono in epoca prenatale o postnatale e che sono legati a influenze ambientali anomale come tossine, deprivazioni, asfissia, oppure ad anomalie cromosomiche o a livello dei geni mitocondriali. In un ristretto numero di sindromi con ritardo mentale è possibile comprendere i collegamenti tra i fattori esogeni o innati e le malformazioni cerebrali risultanti. A eccezione della trisomia del cromosoma 21 e di altre malformazioni che colpiscono focalmente il cervello, non è possibile comprendere i dati neuropsicologici in relazione ai dati citoarchitettonici del cervello. Studiare la sindrome di Williams, caratterizzata da un deficit cognitivo focale più che generalizzato, offre un'opportunità che non ha precedenti per collegare i risultati sul cervello con specifici profili cognitivi atipici (Bellugi et al. 1997).
I risultati sulla citoarchitettonica cerebrale, ottenuti da studi condotti sul cervello di un individuo maschio di 31 anni con sindrome di Williams, donato alla ricerca scientifica e sottoposto ad autopsia, hanno rivelato che le aree della corteccia frontale posteriore presentavano un volume notevolmente ridotto (Galaburda et al. 1994; Galaburda e Bellugi 2001). Altri risultati hanno evidenziato una esagerata stratificazione orizzontale dei neuroni all'interno degli strati, più marcata nel lobo occipitale, a livello dell'area 17, in cui gli strati superiori assumono un allineamento orizzontale increspato che ricorda una stratificazione neuronale relativa a uno stadio di sviluppo corticale più giovane. Inoltre in tutte le regioni cerebrali si aveva un incremento della densità cellulare e i neuroni erano raggruppati e orientati in maniera insolita. I risultati possono essere messi in relazione con l'insolito processamento visuospaziale presente nella sindrome di Williams e offrono l'opportunità di collegare i dati sul cervello, i deficit cognitivi e le loro basi genetiche.
Studi di genetica molecolare
Questo insolito profilo neurocognitivo rende la sindrome di Williams un modello convincente delle connessioni fra geni e attività cognitive umane. Sta diventando evidente che una particolare organizzazione genomica potrebbe rendere la sindrome di Williams un importante modello sia dell'evoluzione cromosomica umana sia della patologia (Korenberg et al. 1996). La sindrome di Williams è causata da una delezione che include il gene che codifica l'elastina (ELN) e perlomeno altri 10 geni sul cromosoma 7. Nelle ricerche condotte in collaborazione, gli studi sono indirizzati nello sforzo di identificare i geni e di spiegare il meccanismo cromosomico responsabile della sindrome di Williams, al fine di mettere in relazione tali geni con le caratteristiche cognitive e neurali della popolazione (Bellugi et al. 1997). Julie R. Korenberg e i suoi collaboratori stanno costruendo una mappa fisica della regione deleta del cromosoma 7 utilizzando le tecniche di ibridazione in situ a fluorescenza (FISH, fluorescence in situ hybridization) di cromosomi in metafase e in interfase, lo screening di lunghi frammenti di una genoteca, il Southern blot e la sequenza di siti bersaglio (STS, sequence targel siles). Queste mappe sono state clonate in un vettore stabile adatto per l'isolamento di geni e il sequenziamento del DNA e utilizzate per studiare le delezioni in pazienti con sindrome di Williams per i quali sono stati determinati simultaneamente il profilo neurocognitivo, la struttura e la funzione cerebrale. Per la costruzione della mappa fisica sono stati scelti i cromosomi batterici artificiali (BAC, bacterial artificial chromosomes) perché contengono frammenti genomici stabili lunghi oltre 300 kb, con un'attività chimerica bassa, che possono essere facilmente manipolati e sequenziati. In seguito, è stato sviluppato un modello funzionante dell'organizzazione genomica dei cromosomi batterici artificiali che caratterizza la banda 7q11.2 (Korenberg et al. 1996). Questo suggerisce che la regione include duplicazioni cromosomiche altamente omologhe, caratterizzate da un numero di sequenze familiari ripetute, organizzate come una struttura ripetuta e invertita che circonda le regioni occupate dal gene dell'elastina. Si ritiene, perciò, che la sindrome di Williams sia collocata all'interno di una regione copia del cromosoma 7, apparentemente singola, che risulta circondata da una duplicazione genomica relativamente recente - la maggior parte della quale potrebbe essersi duplicata prima nell'evoluzione - localizzata nella banda 7q22. L'erroneo appaiamento meiotico dei sottosettori delle numerose sequenze ripetute in questa regione potrebbe, infine, causare la delezione. Queste ricerche possono fornire gli strumenti per lo studio dell'evoluzione umana, come pure per l'identificazione delle regioni e dei geni responsabili della sindrome di Williams e di altre patologie genetiche (Korenberg et al. 2001; Bellugi et al. 2001).
Implicazioni per le neuroscienze cognitive dello sviluppo
Queste linee di ricerca nelle neuroscienze cognitive forniscono indizi per annose questioni teoriche sul linguaggio e sull'organizzazione cerebrale e, inoltre, possono aiutare a stabilire legami fra disturbi genetici specifici, particolari profili neuropsicologici, organizzazioni cerebrali anomale e basi genetiche sottostanti. La sindrome di Williams presenta un profilo cognitivo specifico, caratterizzato da un chiaro deficit; tuttavia il linguaggio è preservato in maniera sorprendente e ciò indica una dissociazione fra esso e le altre funzioni cognitive. I pazienti con sindrome di Williams manifestano anche una ipersocievolezza e stati linguistici affettivi particolarmente evidenti. Essi mostrano, inoltre, una scissione caratteristica all'interno della cognizione spaziale con un'attenzione selettiva verso i dettagli di una configurazione a discapito dell'insieme. Una delle più grandi sfide nella comprensione del cervello e delle attività cognitive consiste nella capacità di operare collegamenti tra discipline diverse nell'ambito delle neuroscienze. Gli studi sui disturbi specifici dello sviluppo neurologico, che presentano un frazionamento delle più alte funzioni cognitive, possono fornire l'opportunità di esplorare alcune questioni centrali delle neuroscienze cognitive che legano tali funzioni all'organizzazione cerebrale e, quindi, al genoma umano.
Bibliografia
Bellugi 1990: Bellugi, Ursula - Bihrle, Amy M. - Jernigan, Terry L. - Trauner, Doris - Doherty, Sneddam, Neuropsychological, neurological and neuroanatomical profile of Williams syndrome, "American journal of medical genetics", Suppl. 6, 1990, pp. 115-125.
Bellugi 1992: Bellugi, Ursula - Bihrle, Amy M. - Neville, Helen J. - Jernigan, Terry L. - Doherty, Sneddam, Language, cognition, and brain organization in a neurodevelopmental disorder, in: Developmental behavioral neuroscience, edited by Megan Gunnar and Charles A. Nelson, Hillsdale, (N.J.), Lawrence Erlbaum Associates, 1992, pp. 201-232.
Bellugi 1994: Bellugi, Ursula - Wang, Paul P. - Jernigan, Terry L., Williams syndrome: an unusual neuropsychological profile, in: Atypical cognitive deficits in developmental disorders: implications for brain function, edited by S. Broman, Jordan Grafman, Hillsdale, (N.J.), Lawrence Erlbaum Associates, 1994, pp. 23-56.
Bellugi, Morris 1995: Bellugi, Ursula - Morris, Colleen A., Williams syndrome: from cognition to gene, Abstracts from the Williams syndrome Association Professional Conference. Special issue, "Genetic counseling", 6, 1995, pp. 131-192.
Bellugi 1996: Bellugi, Ursula - Klima, Edward S. - Wang, Paul P., Cognitive and neural development: clues from genetically based syndrome. The lifespan development of individuals: behavioral, neurobiological, and psychosocial perspective, edited by D. Magnussen (The Nobel symposium, Stockholm, 1994), New York-Cambridge, Cambridge University Press, 1996, pp. 223-243.
Bellugi 1997: Bellugi, Ursula - Mills, Debra L. - Jernigan, Terry L. - Hickok, Gregory - Galaburda, Albert, Linking cognition, brain structure, and brain function in Williams syndrome, in: Neurodevelopmental disorders: contributions to a new framework from the cognitive neurosciences, edited by H. Tager-Flusberg, Cambridge, (Mass.), Massachusetts Institute of Technology Press, 1997.
Bellugi, Wang 1998: Bellugi, Ursula - Wang, Paul P., Williams syndrome: from cognition to brain to gene, in: Encyclopedia of neuroscience, edited by George Edelman and Barry H. Smith, Amsterdam, Elsevier Science Publisher, 1998, 2 v.
Bellugi 1999: Bellugi, Ursula - Lichtenberger, Liz - Mills, Debra, L. - Galaburda, Albert - Korenberg, Julie R., Bridging cognition, brain and molecular genetics. Evidence from Williams syndrome, "Trends in neurosciences", 2, 1999, pp. 197-207.
Bellugi 2001: Bellugi, Ursula - Lichtenberger, Liz - Jones, Wendy - Lai, Zona - St. George, Marie, The neurocognitive characterization of Williams syndrome: a complex pattern of strengths and weaknesses, in: Journey from cognition to brain to gene. Perspectives from Williams syndrome, edited by Ursula Bellugi and Marie St. George, Cambridge (Mass), Massachusetts Institute of Technology Press, 2001, pp. 1-42.
Bihrle 1989: Bihrle, Amy M. - Bellugi, Ursula - Delis, Dean - Marks, S., Seeing either the forest or the trees: dissociation in visuospatial processing, "Brain and cognition", 11, 1989, pp. 37-49.
Bihrle 1991: Bihrle, Amy M., Visuospatial processing in Williams and Down syndromes, "Dissertation abstracts international", 52 (21B), 1991, p. 1048.
Cassady 1997: Cassady, Clare - Bellugi, Ursula - Reilly, Judy S. - Adolphs, Ralph, Hipersociability in Williams syndrome: similarities to bilateral amygdala patients, "International behavioral neuroscience society abstracts", 6, 1997, pp. 2-46.
Damasio, Frank 1992: Damasio, Hanna - Frank, Randall, Three-dimensional in vivo mapping of brain lesions in humans, "Archives of neurology", 49, 1992, pp. 137-143.
Galaburda 1994: Galaburda, Albert - Wang, Paul P. - Bellugi, Ursula - Rossen, Michael L., Cytoarchitectonic anomalies in a genetically based disorder: Williams syndrome, "Neuroreport", 5, 1994, pp. 758-787.
Galaburda, Bellugi 2001: Galaburda, Albert - Bellugi, Ursula, Cellular and molecular cortical neuroanatomy in Williams syndrome, in: Journey from cognition to brain to gene. Perspectives from Williams syndrome, edited by Ursula Bellugi and Marie St. George, Cambridge (Mass.), Massachusetts Insitute of Technology Press, 2001, pp. 123-146.
Giannotti, Vicari 1994: Il bambino con sindrome di Williams, a cura di Aldo Giannotti e Stefano Vicari, Milano, Franco Angeli, 1994.
Hickok 1995a: Hickok, Gregory - Bellugi, Ursula - Jones, Wendy, Asymmetrical ability, "Science", 270, 1995, pp. 219-220.
Hickok 1995b: Hickok, Gregory - Neville, Helen J. - Mills, Debra L. - Jones, Wendy - Rossen, Michael L. - Bellugi, Ursula, Electrophysiological and quantitative MR analysis of the cortical auditory system in Williams syndrome, "Cognitive neuroscience society abstracts", 2, 1995, pp. 40, 66.
Hirota 1998: Hirota, Hanao - Chen, Xiao Ning - Shi, Z.Y. - Matsuoka, Romiko - Kimura, Michael - Bellugi, Ursula - Lincoln, Alan - Korenberg, Julie R., Williams syndrome (WMS): from cognition to genes, "American journal of human genetics", 63, A138, 1998, p. 775.
Jernigan, Bellugi 1990: Jernigan, Terry L. - Bellugi, Ursula, Anomalous brain morphology on magnetic resonance images in Williams syndrome and Down syndrome, "Archives of neurology", 47, 1990, pp. 529-533.
Jernigan, Bellugi 1994: Jernigan, Terry L. - Bellugi, Ursula, Neuroanatomical distinctions between Williams and Down syndromes, in: Atypical cognitive deficits in developmental disorders: implications for brain function, edited by Sarah Broman and Jordan Grafman, Hillsdale, (N.J.), Lawrence Erlbaum Associates, 1994, pp. 57-66.
Jones 1995: Jones, Wendy - Rossen, Michael L. - Hickok, Gregory - Jernigan, Terry L. - Bellugi, Ursula, Links between behavior and brain: brain morphological correlates of language, face, and auditory processing in Williams syndrome, "Society for neuroscience abstracts", 757, 1995.
Jones 2001: Jones, Wendy e altri, Hypersociability: the social and affective phenotype of William syndrome, in: Journay from Williams syndrome, edited by Ursula Bellugi and Marie St. George, Cambridge (Mass.), Massachusetts Institute of Technology Press, 2001, pp. 43-72.
Korenberg 1996: Korenberg, Julie R. e altri, The genomic organization of Williams syndrome, "American journal of human genetics", Suppl. 59, A306, 1996, p. 1776.
Korenberg 1997: Korenberg, Julie R. e altri, Williams syndrome: the search for the genetic origins of cognition, "American journal of human genetics", 103, 1997, p. 579.
Korenberg 1998: Korenberg, Julie R. e altri, The molecular genetic basis of Williams syndrome. 'Bridging cognition, brain and gene: evidence from Williams syndrome', "Cognitive neuroscience society 1998 annual meeting abstract program", 11, 1998.
Korenberg 2001: Korenberg, Julie R. e altri, Genome structure and cognitive map of Williams syndrome, in: Journey from cognition to brain to gene. Perspectives from Williams syndrome, edited by Ursula Bellugi and Marie St. George, Cambridge (Mass.), Massachusetts Institute of Technology Press, 2001, pp. 147-178.
Losh 1997: Losh, Molly - Reilly, Judy S. - Bellugi, Ursula - Cassady, Clare - Klima, Edward S., Linguistically encoded affect is abnormally high in Williams syndrome children. In poster symposium 'bridging cognition, brain and gene', "International behavioral neuroscience society abstracts", 6, 1997, pp. 53-60.
Losh 2000: Losh, Molly - Bellugi, Ursula - Reilly, Judy S. - Anderson, Diane, Narrative as a social engagement. The excessive use of evaluation in narratives from children with Williams syndrome, "Narrative inquiry", 10, 2000, pp. 1-26.
Mills 2001: Mills, Debra L. e altri, Neurophysiological markets of face processing in Williams syndrome, in: Journey from cognition to brain to gene. Perspectives from Williams syndrome, edited by Ursula Bellugi and Marie St. George, Cambridge (Mass.), Massachusetts Institute of Technology Press, 2001, pp. 73-104.
Neville 1994: Neville, Helen J. - Mills, Debra L. - Bellugi, Ursula, Effects of altered auditory sensitivity and age of language acquisition on the development of language-relevant neural systems: preliminary studies of Williams syndrome, in: Atypical cognitive deficits in developmental disorders: implications for brain function, edited by Sarah Broman and Jordan Grafman, Hillsdal, (N.J.), Lawrence Erlbaum Associates, 1994, pp. 67-83.
Reilly 1990: Reilly, Judy S. - Klima, Edward S. - Bellugi, Ursula, Once more with feeling: affect and language in atypical populations, "Developmental psychopathology", 2, 1990, pp. 367-391.
Reiss 2001: Reiss, Allan L. e altri, Neuroanatomy of Williams syndrome. A high-resolution MRI study, in: Journey from cognition to brain to gene. Perspectives from Williams syndrome, edited by Ursula Bellugi and Marie St. George, Cambridge (Mass.), Massachusetts Institute of Technology Press, 2001, pp. 105-122.
Rossen 1996: Rossen, Michael L. - Klima, Edward S. - Bellugi, Ursula - Bihrle, Amy M. - Jones, Wendy, Interaction between language and cognition: evidence from Williams syndrome, in: Language, learning and behavior disorders: developmental, biological, and clinical perspectives, edited by Joseph H. Beitchman e altri, New York-Cambridge, Cambridge University Press, 1996, pp. 367-392.
Singer Harris 1997: Singer Harris, N.G. - Bellugi, Ursula - Bates, Elisabeth - Jones, Wendy - Rossen, Michael L., Contrasting profiles of language development in children with Williams and Down syndromes, in: Origins of language disorders, edited by Donna J. Thal and Judy S. Reilly, "Developmental neuropsychology", 13, 1997, pp. 345-370.
Wang, Bellugi 1994: Wang, Paul P. - Bellugi, Ursula, Evidence from two genetic syndromes for a dissociation between verbal and visual-spatial short-term memory, "Journal of clinical and experimental neuropsychology", 16, 1994, pp. 317-322.