
leucemia
leucemia
Tumore delle cellule del sangue, caratterizzato da una proliferazione anomala della cellula staminale, non ancora differenziata e con molte potenzialità. Ci sono diversi tipi di l. che si classificano e si riconoscono in base al tipo di cellula affetta e in base al tempo di replicazione di questa. Nel caso di una l. acuta, la cellula che si trasforma e diventa neoplastica perde i meccanismi complessi che ne regolano la proliferazione e la differenziazione, inoltre acquisisce alterazioni cromosomiche e molecolari: queste cellule, che hanno un alto tasso di replicazione, si chiamano blasti. Nel corso del processo leucemico si assiste a un grave sovvertimento degli organi emopoietici, con depressione dell’emopoiesi normale e invasione di organi e tessuti non ematopoietici da parte delle cellule neoplastiche. Questo porta ai sintomi del processo leucemico: la mancata produzione dei globuli rossi, delle piastrine e dei globuli bianchi normali, produce rispettivamente anemia, emorragie e possibili gravi infezioni. Da un punto di vista clinico si distinguono le forme acute dalle forme croniche: queste ultime si caratterizzano per un processo evolutivo lento e graduale, con stabilità clinica.
Eziopatogenesi
Le cause delle l. non sono ancora del tutto chiare: diversi sono i fattori di rischio che possono concorrere a un processo leucemico, ma nessuno di essi è veramente certo. Tra i fattori identificati come predisponenti sono presenti le radiazioni ionizzanti, l’esposizione a sostanze tossiche (come i prodotti derivati dal benzene), alcune sindromi genetiche come la sindrome di Down. Recentemente è stato preso in considerazione l’effetto dell’esposizione ai campi elettromagnetici, altra possibile causa non ancora del tutto certa. Per l’assenza di cause specifiche, non è possibile attuare un piano di prevenzione di queste malattie e non ci sono indagini di screening da effettuare come nel caso di alcuni tumori solidi: è importante quindi saper riconoscere precocemente i sintomi. Possono essere colpiti entrambi i sessi e tutte le età, con alcune fasce colpite in maniera particolare. Le l. si possono distinguere in due tipi, in base alla cellula da cui evolve il clone leucemico: linfoide o mieloide. La prima è più frequente in età pediatrica, mentre la forma mieloide è più frequente nel soggetto adulto.
Diagnosi
L’approfondimento delle conoscenze ha consentito di inquadrare le diverse forme di l. utilizzando metodiche istologiche e citologiche, citogenetiche e molecolari. La diagnosi si pone quindi sulla base di analisi cliniche (emocromo e analisi morfologica del sangue venoso periferico e del midollo osseo), immunofenotipiche (identificazione di antigeni espressi sulla superficie dei blasti mediante citometria a flusso), citogenetiche e molecolari (studio dei cromosomi e dei possibili geni di fusione). L’identificazione di alcune aberrazioni genetiche ha permesso di cambiare l’approccio terapeutico in alcuni casi di l. acuta e cronica (per es., l. acuta promielocitica, il cui trattamento prevede l’uso di retinoidi, e l. mieloide cronica, attualmente trattata con successo con inibitori tirosin-chinasici).
Terapia
Attualmente le strategie terapeutiche impiegate prevedono l’uso di protocolli chemioterapici contenenti diversi farmaci attivi contro i blasti leucemici in associazione; l’avvento delle moderne procedure di trapianto e l’uso di farmaci innovativi, quali gli anticorpi monoclonali o gli inibitori delle tirosinchinasi, hanno modificato la prognosi e la sopravvivenza di alcune forme di leucemia. Nel caso di una forma acuta, lo scopo della terapia applicata è quello di indurre una remissione completa, ovvero il ritorno a una emopoiesi normale con una percentuale di cellule indifferenziate inferiore al 5%. In alcuni tipi di l., in cui è noto il meccanismo molecolare coinvolto nel processo di leucemogenesi, si parla anche di remissione molecolare, intendendo l’assenza di malattia evidenziabile con metodiche di biologia molecolare che coinvolgono un meccanismo complesso di amplificazione (PCR), in grado di determinare la presenza di malattia con una soglia di 10−4 cellule.
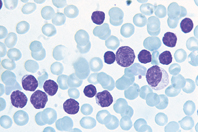
Vengono qui considerati tre tipi di leucemia: le leucemie acute mieloidi (LAM), la leucemia mieloide cronica (LMCr), la leucemia acuta linfoide (LAL), la leucemia linfatica cronica (LLC).
LAM
Sono frequenti nell’adulto con età media superiore ai 60 anni. Si hanno forme primarie o de novo e forme secondarie a patologia mieloproliferativa o a sostanze tossiche. Il gruppo Franco-Americo-Britannico (FAB) ha individuato otto tipi (dalla forma M0 alla M7) in base all’aspetto morfologico e all’origine (monocitaria, eritroide, megacarioblastica, ecc.). La classificazione dell’OMS ha definito 4 sottogruppi: con anomalie citogenetiche ricorrenti; con displasia multilineare; secondarie a chemioterapia e radioterapia; non classificabili. Nel primo gruppo rientrano le LAM con traslocazione (t) dei cromosomi 8 e 21 e con inversione (inv) del cromosoma 16. La forma con anomalia del cromosoma 11q23 (5%) si associa a varie alterazioni citogenetiche; la più frequente è la t (9;11), che coinvolge i cromosomi 9 e 11. La forma con t (15;17), o promielocitica, è caratterizzata dal gene anomalo PML/RARa, da una peculiare sensibilità all’acido retinoico ad alto dosaggio, e ha percentuali di remissione pari al 90%. Per la prognosi, l’età avanzata, le comorbidità e alcune caratteristiche biologiche (precedente mielodisplasia, alcune alterazioni citogenetiche come le monosomie dei cromosomi 7 o 5 e anomalie complesse) sono fattori negativi. Il riconoscimento di lesioni molecolari caratteristiche come le mutazioni che coinvolgono FLT3 (un recettore delle tirosinchinasi) o NPM (proteina nucleare regolatoria) è associato a prognosi rispettivamente infausta o favorevole. L’alterazione di FLT3 si associa a leucocitosi, a forme monoblastiche e a rischio di recidiva precoce molto alto. L’alterazione NPM si associa al cariotipo normale, a buona prognosi con ottima risposta completa alla chemioterapia. Lo scopo della terapia è indurre remissione completa, ossia una percentuale di blasti inferiore al 5%. Poi, si procede con la terapia di consolidamento e con trapianto, dove possibile.
LMCr
Comporta una progressiva proliferazione clonale e accumulo di globuli bianchi maturi, ed è contraddistinta dalla presenza del cromosoma Philadelphia o t (9;22). Ha la massima incidenza tra i 50 e i 60 anni. Può essere: cronica, accelerata (blasti nel midollo inferiori al 30%) e blastica (blasti midollari superiori al 30%). Spesso si rivela da un riscontro occasionale di leucocitosi in analisi di laboratorio. I sintomi, se presenti, in fase cronica sono: senso di ingombro addominale (per splenomegalia) o sintomi legati alla leucocitosi (vertigini, disturbi visivi, trombosi). Durante la progressione possono comparire febbre, sudorazione, dolori ossei, ed emorragie. Il trattamento di prima scelta è l’imatinib, inibitore tirosinchinasico che induce risposte ematologiche nel 95% e citogenetiche in oltre l’80% dei pazienti. Sono inoltre a disposizione inibitori di seconda generazione con potenza superiore, in grado di superare la possibile resistenza farmacologica e gli effetti collaterali. Il trapianto allogenico di cellule staminali emopoietiche consente la guarigione completa anche se è da considerare l’elevata tossicità e mortalità. Con l’impiego degli inibitori tirosinchinasici, il trapianto allogenico è riservato ai pazienti resistenti dopo 2÷3 linee di terapia.
LAL
È la più frequente nel bambino (l’80% di origine B linfoide) con il 70% di possibile guarigione nelle forme pediatriche. Generalmente, l’80% dei pazienti adulti raggiunge la remissione completa e il 30÷40% ha una sopravvivenza a 3 anni con regime di chemioterapia intensivo. Si riconoscono 3 varianti morfologiche (da L1 a L3); immunologicamente si distinguono le forme B (pro-B, common, pre-B, mature) e le T (pro-T, pre-T, corticali, mature). Il 50÷70% mostra un’alterazione citogenetica; nel 20% dei casi la t (9;22) è correlata a una cattiva prognosi. La prognosi è influenzata da diversi fattori negativi: età avanzata, fenotipo B maturo, leucocitosi, mancata risposta iniziale alla terapia steroidea. Il trattamento prevede 3 fasi: induzione, terapia post-remissionale e mantenimento. Il trapianto allogenico di cellule staminali emopoietiche in prima remissione è indicato in pazienti ad alto rischio con donatore compatibile. Per la forma Ph+ ai normali schemi di chemioterapia si associa il trattamento con imatinib.
LLC
Si tratta di una proliferazione di piccoli linfociti (per circa il 98% di tipo B) apparentemente maturi, scarsamente proliferanti, immunologicamente incompetenti e con lunga sopravvivenza. Colpisce i soggetti di età superiore a 60 anni con decorso molto lento a tipo linfocitosi indolente o con segni di compromissione d’organo generalizzata (epatosplenomegalia, linfoadenopatie) insieme a progressiva invasione midollare (pancitopenia). Può inoltre trasformarsi in linfoma non-Hodgkin ad alto grado (sindrome di Richter). I pazienti con cariotipo normale e delezione del cromosoma 13 hanno prognosi favorevole, mentre i pazienti con delezione dei cromosomi 17 e 11 hanno prognosi sfavorevole. I principali fattori prognostici biologici sono: l’espressione mutata o non dei geni per le immunoglobuline, l’espressione della proteina ZAP-70, generalmente espressa dalle cellule linfoidi T normali, l’espressione di alcuni antigeni di superficie (CD38), l’espressione dell’oncogene p53.
L’innovazione terapeutica
Lo scenario terapeutico si è molto modificato con l’introduzione nei primi anni Novanta del secolo scorso degli analoghi delle purine (fludarabina da sola o in associazione), con risposte di lunga durata. In pazienti giovani, in stadio avanzato e con caratteristiche prognostiche sfavorevoli, è giustificata l’associazione di chemioterapici e anticorpi monoclonali e del trapianto di cellule staminali allogeniche e autologhe.