
Lo sviluppo dei linfociti
Lo sviluppo dei linfociti
I linfociti derivano, durante lo sviluppo embrionale, dalla cellula staminale ematopoietica pluriPotente che ha sede nel midollo osseo. Essi vengono continuamente rigenerati durante tutta la vita. I linfociti T maturano nel timo, mentre i linfociti B si sviluppano all'interno del midollo osseo. Lo sviluppo delle due linee cellulari ha luogo grazie a una selezione di indirizzo funzionale e a una serie di riarrangiamenti dei geni che codificano i recettori specifici degli antigeni. In particolare, lo sviluppo ha luogo a partire da un esiguo numero di progenitori che si espande per proliferazione e differenziazione generando cellule che acquisiscono le strutture recettoriali specifiche per l'antigene, al termine del ciclo maturativo. Sia negli organi linfatici primari (timo e midollo osseo), sia negli organi linfatici secondari (per esempio, i linfonodi), si verifica un fenomeno di selezione negativa per quei recettori linfocitari potenzialmente in grado di dare luogo a processi di autoreattività. Nello stesso tempo le cellule possono essere selezionate per l'ingresso negli organi linfatici periferici preposti alla codificazione e diventare linfociti T e B maturi, capaci di rispondere agli antigeni.
Origine dei linfociti
Una singola cellula, la cellula staminale ematopoietica pluripotente (HSC, Hematopoietic Stem Cell), è in grado di generare tutte le linee cellulari del sangue, quindi anche tutti i linfociti del sistema immunitario (Morrison et al., 1997). Una cellula HSC costituisce il precursore dei 2.10¹² linfociti di un uomo adulto, o dei 2.10⁹ linfociti di un topo adulto.
Questa cellula deve essere in grado di autorinnovarsi e questa capacità, in seguito a divisione, viene mantenuta fino a che, almeno una delle cellule figlie conserva le potenzialità della cellula originale; l'altra cellula figlia può differenziarsi ulteriormente lungo la via delle cellule mieloidi/linfoidi.
Non è ancora del tutto chiaro se il sistema immunitario venga costruito a partire da una singola HSC o da molte. È probabile che le HSC vengano mantenute nel loro stato di sviluppo tramite il contatto con un microambiente di cellule stromali il quale determina anche la dimensione del loro compartimento. Quest'ultimo, una volta che è stato riempito, può entrare in uno stato di riposo e rimanervi, finché sono necessarie cellule più differenziate. Non è ancora stato possibile coltivare le HSC in vitro, quindi non è noto con quale frequenza queste cellule pluripotenti possano dividersi e mantenere il proprio stato di sviluppo né la velocità con la quale possono perdere lo stato di HSC. Nell'uomo e nel topo, le HSC sono state osservate durante tutto l'arco della vita, anche se il loro numero sembra diminuire con l'età. La stima delle frequenze di replicazione delle HSC è stata determinata grazie al fatto che una singola cellula è capace di popolare l'ambiente adeguato di un ospite in via di sviluppo o di un ospite deficitario (per esempio, un animale esposto ad alte dosi di radiazioni) di HSC e dei loro discendenti, in seguito a trasferimento adottivo. Tuttavia, sembra che lo stato preciso di una HSC sia meno stabile quando la cellula viene allontanata dal suo ambiente naturale durante tale trasferimento adottivo, poiché spesso il trasferimento adottivo secondario dall' ospite trapiantato in un nuovo ospite non ha successo (Spangrude et al., 1995).
I compartimenti del sistema immunitario
Circa la metà di tutti i linfociti di un adulto si trova sparsa, come cellule singole, negli epiteli della cute, della lingua e dei tratti respiratorio, riproduttivo e gastrointestinale, dove si pensa che formi la prima linea di difesa e assicuri l'integrità di tali epiteli (Rocha et al., 1992; Boismenu e Havran, 1994; Rocha et al., 1995). Questi linfociti intraepiteliali (lEL, Intra-Epithelial Lymphocyte) sono per la maggior parte linfociti T, e molti di essi esprimono un recettore antigenico delle cellule T (TCR, T Cell Receptor) di tipo γ/δ, sebbene si possano trovare anche lEL che esprimono il TCRα/β. L'altra metà dei linfociti del sistema immunitario è organizzata in strutture extrafollicolari e follicolari all'interno degli organi linfoidi secondari, come la milza o i linfonodi. Di questi, i due terzi sono costituiti da linfociti T, la maggior parte dei quali esprime un TCR di tipo α/β, mentre l'altro terzo è costituito da linfociti B, la maggior parte dei quali esprime un recettore antigenico delle cellule B (BCR, B Cell Receptor) di tipo IgM (immunoglobulina M) (Rolink e Melchers, 1993; Spits, 1994; Kisielow e von Boehmer, 1995; Melchers et al., 1995; Shortman e Wu, 1996; Burrows e Cooper, 1997; Fehling e von Boehmer, 1997; Papavasiliou et al., 1997).
Circa il 10% di tutte le cellule T e B si trova negli organi linfoidi centrali di un adulto, ossia, rispettivamente, nel timo e nel midollo osseo, sotto forma di progenitori, precursori e linfociti immaturi, tutti con vita breve; hanno infatti un'emivita di 2 ÷ 4 giorni.
La maggioranza dei linfociti, ossia il restante 90%, si trova negli organi linfoidi periferici o circola dal sangue alla linfa e viceversa. La maggior parte sopravvive a lungo nell'adulto, con un'emivita di sei settimane e più. llinfociti periferici maturi sono suddivisi in molte sottoclassi che si distinguono dal punto di vista fenotipico e funzionale. Le sotto classi principali di linfociti T sono le cellule T helper (adiuvanti) CD4+ TCRα/β+, le cellule T citolitiche CD8+ TCRα/β+, le cellule T citolitiche TCRγ/δ+, le cellule T lEL con TCR di tipo α/β+ o γ/δ+, le cellule B Bl IgM+ e le cellule B IgM+ convenzionali. Sono note molte altre popolazioni numericamente inferiori di cellule T e B; tra queste vi sono le cellule che hanno già incontrato un antigene estraneo, le quali vengono considerate detentrici della memoria di eventi precedenti (v. il saggio di J. Sprent e D. Tough, nel II volume).
È evidente che ogni giorno muoiono molti linfociti e che questi devono essere rimpiazzati da cellule neo generate e neoselezionate. Con l'aumentare dell'età questa capacità rigenerativa del sistema immunitario diminuisce, mentre aumenta la relativa presenza di cellule che sopravvivono a lungo. Non si conoscono ancora, o sono state analizzate in maniera incompleta, le modalità attraverso le quali, durante la vita, si verifica il tum over delle distinte sottoclassi fenotipiche e funzionali di linfociti del sistema immunitario. Tuttavia, è chiaro che questo processo rigenerativo non si blocca mai completamente. Comunque, non sappiamo se le HSC, o le cellule staminali meno pluripotenti, cioè le cellule linfoidi destinate a generare le linee cellulari T e B, siano effettivamente la fonte di questa continua rigenerazione durante la vita adulta.
Lo sviluppo embrionale

La formazione dei tre strati germinali primari dell'embrione di topo - endoderma, ectoderma e mesoderma - si verifica dopo 6÷7 giorni dall'inizio dell'embriogenesi. Successivamente, il mesoderma si muove in due direzioni, anteriore e posteriore. La parte posteriore dà origine, tra gli altri, ai progenitori delle cellule del muscolo cardiaco, delle cellule endoteliali e di quelle ematopoietiche (fig. 1). I pro genitori putativi comuni delle cellule endoteliali ed ematopoietiche vengono chiamati emangioblasti. Si pensa che durante lo sviluppo embrionale del topo, la prima HSC pluripotente venga generata al decimo giorno di gestazione, quindi in uno stadio cellulare vicino a quello degli emangioblasti. La splancnopleura intraembrionale, nella regione mesonefroaortogonadica, dà inizio alla formazione e all'espansione delle HSC (Cumano et al., 1996; Medvinsky e Dzierzak, 1996). Non si può ancora escludere che le HSC si presentino, in un momento successivo, in più di una sede all'interno dell'embrione poiché, in uno stadio precoce dello sviluppo, il mesenchima e successivamente le HSC, vengono rinvenuti nel sacco vitellino, nel fegato fetale, nella milza e nel midollo osseo. Nell'osso in via di sviluppo le cellule mesenchimali si trovano nella regione subendostea dove si pensa che diano luogo, da una parte a uno strato di osteoblasti e osteoclasti e dall'altra alle HSC.
Le cellule staminali embrionali (ES, Embrionic Stem) possono essere utilizzate per studiare in vitro la predestinazione dell'ectoderma primitivo alla generazione del mesoderma e del mesenchima, e il suo sviluppo successivo di HSC. Esistono infatti numerose linee cellulari ES che sono in grado di crescere in coltura di tessuto. È possibile indurle a sviluppare corpi embrionali che richiamano, almeno in parte, l'effettiva organizzazione morfologica dell'embrione. Ulteriori colture in vitro di corpi embrionali disgregati possono sviluppare cellule eritroidi, mieloidi e linfoidi (Potocnik et al., 1994). Le cellule ES possono venire modificate geneticamente in vitro tramite l'integrazione eterologa casuale del gene transfettato, o l'integrazione omologa della forma mutata di un gene endogeno. Inoltre, lo sviluppo in vitro di cellule ES può essere confrontato con il loro sviluppo in vivo. A questo scopo, le cellule ES vengono iniettate in blastocisti di topo (v. il saggio di M.J. Owen, Topi transgenici e topi knock out come modelli di immunodeficienza). Successivamente, le blastocisti miste vengono reimpiantate in un topo che fa le veci della madre. La prole risultante è chimerica; alcune cellule derivano da blastocisti originali, altre derivano dalle cellule ES iniettate. Ogni volta che vengono generate cellule germinali che derivano dalle cellule ES, il topo chimerico può essere il fondatore di un nuovo ceppo che presenta le mutazioni derivanti dalla cellula ES integrata (Thomas e Capecchi, 1987; Torres e Kiihn, 1997).
L'iniezione di cellule ES in blastocisti può essere utilizzata anche per studiare gli effetti di mutazioni letali a livello embrionale o neonatale nella via ematopoietica della differenziazione. In questa analisi di complementazione blastocistica chiamata RAG- / -, blastocisti di topi RAG-1- / - o RAG-2- / - vengono iniettate con cellule ES che portano una di queste mutazioni letali, introdotta tramite ricombinazione omologa. Poiché la mutazione RAG- / - abolisce lo sviluppo dei linfociti T e B (v. oltre), i linfociti che si sviluppano in questi topi 'complementati' devono derivare dalla cellule ES mutate (Chen, 1996).
Due sono le strategie che vengono seguite per identificare i rari pro genitori precoci delle linee ematopoietiche. Una opera in senso contrario alla direzione dello sviluppo sfruttando una serie di marcatori e di funzioni di un determinato stadio cellulare per ricercare i suoi immediati precursori. Seguendo tale strategia si assume che il precursore esprima già alcuni dei marcatori, o alcuni dei fattori di trascrizione che regolano l'espressione dei marcatori della successiva cellula nota. Inoltre, essa dipende dalla capacità del precursore, non ancora identificato, di svilupparsi in vitro o in vivo, in seguito al trapianto in un ospite che ne è privo, nella successiva cellula nota. La seconda strategia utilizza le tecniche per la produzione di organismi transgenici. È stato osservato che due gruppi di geni, i quali codificano molecole che agiscono alle estremità opposte della cascata di reazioni di trasduzione del segnale, esercitano il controllo principale durante i primi stadi dell'ematopoiesi. Un gruppo è costituito dai geni che codificano una serie di fattori di trascrizione nucleari, come GATA, PU-l, lkaros, Pax5 e altri. L'altro gruppo è formato dai geni che codificano famiglie di tirosinchinasi di membrana e intracellulari, come Flk-2, c-kit e altre. La distruzione specifica di questi geni introduce lesioni genetiche, bloccando lo sviluppo delle cellule ematopoietiche a livello di stadi definiti (Singh, 1996; Georgopoulos, 1997). L'analisi di queste lesioni consente di delineare chiaramente i primi tratti delle 'mappe del destino' dello sviluppo delle cellule eritroidi, mieloidi e linfoidi.
Modalità dello sviluppo linfocitario embrionale e neonatale
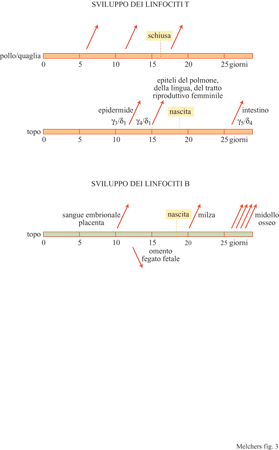
Studi pionieristici (Le Douarin e Jotereau, 1975; Jotereau e Le Douarin, 1982) hanno mostrato che il timo fetale aviario emette tre ondate discrete di cellule T. Tali ondate vengono avviate dall'immigrazione di progenitori linfoidi in un timo ricettivo per la colonizzazione, due volte prima della nascita e una volta dopo (fig. 3). F.V. Jotereau e i suoi collaboratori (1987) hanno dimostrato, nel topo, l'esistenza di ondate di sviluppo di linfociti T paragonabili a quelle aviarie. Anche qui era stata osservata nell'embrione una prima ondata tra il 10° e il 13° giorno, una seconda dopo il 13° giorno, mentre una terza ondata veniva individuata nel topo neonato dal 7° giorno di vita. È probabile che la terza ondata dia inizio a una generazione ininterrotta di cellule T che perdura per tutta la vita. Questa generazione di cellule continua aumenta fino alla pubertà, e poi decresce, anche se non cessa mai completamente (v. figura 3).
Nell'embrione umano i progenitori linfoidi delle cellule T sono stati identificati nel periodo che va dalla 6a alla 8a settimana di gestazione, ben prima della formazione delle componenti preliminari di sviluppo del timo che avviene alla 8a settimana (v. figura 3). Il fegato fetale costituisce una fonte di pro genitori di questi tipi cellulari. Quindi, cellule con marcatori e funzioni dei linfociti T possono svilupparsi al di fuori del timo (Phillips et al., 1992; Poggi et al., 1993; Sanchez et al., 1993). L'embrione umano possiede un sistema immunitario con cellule T e B già alla 10a settimana di gestazione, quindi ben prima della nascita. La stessa cosa si verifica nel feto di agnello. In contrasto, il pollo e il topo nascono praticamente nel momento in cui i primi linfociti maturi reattivi nei confronti dell'antigene compaiono negli organi linfoidi periferici. Questo dimostra che lo sviluppo delle cellule T e B è indipendente dal momento in cui avviene la transizione di un organismo dallo stato embrionaIe a quello perinatale, anche se l'embrione è protetto dalla madre dalla stimolazione da parte di antigeni estranei esclusi gli anticorpi della madre prodotti in risposta a tali antigeni estranei - mentre l'esposizione ad antigeni estranei influenza effettivamente i repertori di linfociti maturi nel momento in cui l'organismo è nato.
Primi stadi di sviluppo delle cellule T
l repertori di cellule T che hanno origine precocemente nell'ontogenesi sono stati analizzati in modo più dettagliato nel topo (Rocha et al., 1992; Kisielow e von Boehmer, 1995). Prima della nascita, vengono generate nel timo due ondate di cellule T TCRγ/δ+ quasi monoclonali (v. figura 3). l topi atimici non sono in grado di produrle, e sembra che non sia neanche possibile rigenerarle tramite il trapianto di HSC in un topo adulto. Le cellule T della prima ondata presentano il segmento V y3 riarrangiato, senza diversità nella regione N, al segmento Jyl che è espresso con il segmento Cyl' La catena o di questo TCRγ/δ è V₈₁, hz C₈. Nella seconda ondata, V y4 è utilizzato nella stessa combinazione, con la stessa catena o, di nuovo senza diversità nella regione N (v. figura 3). Dal momento che gli altri alleli γ e δ dei loci del TCR, che non hanno subito un riarrangiamento produttivo, mostrano eterogeneità nei riarrangiamenti, sembra che le due ondate di cellule T vengano selezionate positivamente da qualche antigene self ancora sconosciuto. La prima ondata popola l'epidermide, la seconda l'epitelio della lingua e del tratto riproduttivo femminile. Non è ancora chiaro fino a che punto queste ondate di popolazioni di cellule T TCRγ/δ+ siano comuni ad altre specie. In stadi successivi della vita, le popolazioni di cellule T TCRγ/δ+ rinvenute in questi epiteli risultano molto più diversificate nei loro repertori di TCR. Le due ondate iniziali sono seguite dalla generazione di cellule T TCRγ/δ+ più eterogenee, che presentano anche regioni N inserite nelle loro unioni V(D)J. Esse popolano la mucosa epiteliale dell'intestino e vengono rinvenute anche negli organi linfoidi secondari e nella circolazione sanguigna. In alcuni individui, gran parte di queste cellule T TCRγ/δ+ circolanti sembra essere oligoclonale. La selezione positiva potrebbe essere la conseguenza dell'esposizione sia ad agenti infettivi, come i micobatteri, sia ad antigeni self. In topi transgenici che esprimono un TCRγ/δ caratteristico di un dato epitelio, è possibile ritrovare cellule T con questo TCR transgenico anche in altri epiteli. Perciò, questi TCR non vengono utilizzati per la migrazione e l' homing (indirizzo) verso una specifica regione del corpo. Poiché è stato osservato che, occasionalmente, le cellule T TCRγ/δ+ esprimono negli epiteli i geni RAG-l e RAG-2 attivi nei processi di riarrangiamento, non è possibile escludere che il primo di tali eventi si verifichi a livello del timo, e che questo sia seguito da un secondo riarrangiamento VJ o da una sostituzione V in un secondo sito, quale l'epitelio. Anche la generazione di cellule T TCRα/β+ inizia nel topo prima della nascita e sembra che porti a un repertorio differente, con TCR privi di regione N prima della nascita, mentre dopo la nascita si osservano inserzioni di regioni N a livello delle giunzioni V(D)J delle catene α e β del TCR (Bogue et al., 1992). Le cellule T TCRα/β+ che popolano le regioni extrafollicolari e follicolari degli organi linfoidi periferici esprimono molecole CDS sotto forma di eterodimeri α/β, ogni volta che si sono differenziate a cellule T citolitiche. Dall'altra parte, le cellule T TCRα/β+ che popolano gli epiteli come lEL, esprimono molecole CDS sotto forma di omodimeri α/α (Rocha et al., 1992; Rocha et al., 1995). Dato ancora più significativo, esse esprimono la subunità y del recettore Fc di tipo l (FcRl-y) come molecola di trasduzione del segnale nel complesso CD3 associato al TCR, anziché la subunità ζ che viene utilizzata nel complesso CD3 delle cellule T TCRα/β+ extrafollicolari e follicolari. Topi atimici immunosoppressi e pazienti umani con la sindrome di DiGeorge (che presentano una displasia simile a quella dei topi atimici immunosoppressi) sono geneticamente privi delle cellule T TCRα/β+ (CDS+ α/β) extrafollicolari e follicolari. Tuttavia, come i topi privi della catena β del TCR (TCRβ- / -), sono in grado di produrre cellule T TCRγ/δ+ e i loro epiteli sono popolati da cellule T lEL presenti in quantità normale. Nel caso dei topi immunosoppressi, ma non di quelli TCRβ-/-, alcune lEL esprimono il TCRα/β. Questo fatto è in eccellente accordo con le modalità extratimiche di sviluppo delle cellule T. Anche se alcune linee cellulari T RAG-l +/RAG-2+ sono state rinvenute negli epiteli, non sono stati ancora localizzati i siti extratimici di sviluppo delle cellule T e le cellule stromali dell'ambiente che inducono tale sviluppo. Riassumendo, è possibile indurre lo sviluppo delle cellule T lEL sia all'interno che all'esterno del timo, mentre sembra che le cellule T extrafollicolari e follicolari abbiano bisogno di trovarsi nel timo per potersi sviluppare. Tale sviluppo nel timo verrà discusso più in dettaglio nei prossimi paragrafi. Topi mutanti privi del gene TCF-l producono la prima ondata embrionale di cellule T TCRα/β+, ma nel topo adulto non è presente la generazione di cellule di questo tipo (Clevers e Grosschedl, 1996). Questo indica che le ondate embrionali di sviluppo delle cellule T e la generazione continua di cellule T nel corso di tutta la vita adulta sembrano seguire, almeno in parte, programmi genetici differenti.
Primi stadi di sviluppo delle cellule B
Anche lo sviluppo delle cellule B durante la vita embrionaIe e neonatale del topo si verifica per ondate, tuttavia in regioni diverse (Melchers, 1979; Marcos et al., 1991). Tra il 10° e il 13° giorno di gestazione (v. figura 3) si osserva una prima ondata di progenitori destinati allo sviluppo delle linee B nel torrente circolatorio embrionale e nelle regioni da esso vascolarizzate come, per esempio, la placenta. In seguito, tra il 13° giorno e la nascita, si sviluppa una seconda ondata di cellule B nel fegato fetale e nell'omento. Dopo la nascita, prima la milza e poi sempre più il midollo osseo cominciano a generare le cellule B. Durante la vita adulta il midollo osseo sembra essere il sito principale per la produzione continua di cellule B; questo sviluppo nel midollo osseo verrà descritto in maggiore dettaglio più avanti.
Anche nell'uomo (intorno alla 6a settimana di gestazione), in altri roditori oltre al topo, e nei ruminanti il fegato fetale è il sito principale dello sviluppo embrionale delle cellule B. Nelle pecore e nei conigli, è stato osservato che le placche di Peyer ileali rappresentano uno dei siti di sviluppo primario dei linfociti. Rimane da stabilire con ulteriori studi da quale stadio di pro genitori venga avviato tale sviluppo (Reynaud et al., 1995; Knight e Winstead, 1997).
Diversamente dai TCR, dove entrambe le catene α e β o quelle y e o presentano, durante la linfopoiesi neonatale e adulta del topo, regioni N inserite nelle giunzioni V(D)J, nel caso delle 19 solo le catene H, e non le catene L, presentano inserzioni di regioni N. Nelle prime ondate di sviluppo embrionale delle cellule B, in modo particolare nel fegato fetale, vengono prodotte catene H in cui non sono presenti inserzioni di regioni N nelle giunzioni V(D)J. In accordo con queste ossevazioni, i precursori dei primi tipi embrionali di cellule B non esprimono l'enzima desossinucleotidiltransferasi terminale (TdT).
Le fasi iniziali dello sviluppo embrionale delle cellule B, come quelle delle cellule T, possono venir distinte dalle successive fasi di sviluppo (neonatale e della vita adulta) nel midollo osseo grazie alla diversa espressione di molecole MHC di classe 11 codificate dai geni del complesso maggiore di istocompatibilità (MHC, Mayor Histocompatibility Complex) e di un gene codificante la catena leggera miosina-simile. Inoltre, la mancanza di espressione del gene ets-l consente ugualmente lo sviluppo embrionale, ma lo abolisce nel topo adulto, mentre la non espressione del gene OBF provoca un effetto opposto (Clevers e Grosschedl, 1996). Anche nel caso dei linfociti B, come per i linfociti T, si può senz'altro concludere che lo sviluppo embrionale segue programmi genetici almeno in parte differenti da quelli dell'adulto.
l primi repertori di cellule della linea B in tutti i siti linfopoietici mostrano una forte inclinazione per la sovrarappresentazione di gran parte dei segmenti genici V H localizzati nella regione 3' all'interno dellocus genico della catena H (Coutinho et al., 1992). Nel topo, questi segmenti appartengono alle famiglie VH7183 e VHQ52, nel coniglio si tratta dei segmenti VHl, nell'uomo dei segmenti appartenenti alla famiglia VH3. Nelle fasi successive dello sviluppo delle cellule B immature e mature, queste famiglie diventano sottorappresentate nei loci delle catene H riarrangiati in modo produttivo, indicando che la selezione delle cellule B sfrutta la specificità di alcune catene Il pesanti H durante lo sviluppo, sia come segnale positivo che negativo. Nel topo, il membro più importante all'interno del repertorio della cellula B, che viene inizialmente sovrarappresentato e successivamente sottorappresentato, è dato dal segmento genico VH81x. Una possibile spiegazione di questa iniziale preferenza e della successiva soppressione della rappresentazione di certi segmenti genici VH verrà data più avanti, quando sarà spiegato più in dettaglio lo sviluppo delle cellule B nel midollo osseo.
Sviluppo linfocitario in vitro
l primi stadi dello sviluppo delle cellule B e T possono anche essere osservati in colture di tessuto. Dopo il 13° giorno, colture di singole cellule in sospensione del timo e del fegato fetali vanno incontro agli stessi stadi di maturazione, da pro genitori iniziali a cellule T e B immature, anche se senza alcuna espansione proliferativa (Melchers et al., 1995; Rolink et al., 1995). Più avanti spiegheremo in dettaglio questi diversi stadi per il midollo osseo.
Nel topo, precursori allo stadio di cellule pro/pre-B I (dal fenotipo c-kit+, CD25-B220+CDI9+ con riarrangiamenti DHJH-) possono essere clonati su cellule stromali preadipociti che in presenza di interleuchina 7 (IL- 7) ricombinante aggiunta dall'esterno. In tale condizione le cellule si espandono tramite divisione cellulare ogni 18 ÷ 24 ore, dando luogo a linee cellulari continuamente in crescita. A uno stadio paragonabile di differenziazione, sia i precursori B sia i precursori T umani o murini non sono in grado di mostrare questa estesa proliferazione senza estesi cambiamenti del loro stato differenziativo.
La rimozione di IL-7 dalle colture di cellule pre-B I in crescita continua induce tali cellule a differenziarsi, senza divisione. La stessa cosa accade in colture di cellule pre-B I isolate ex vivo in assenza di IL- 7. Questa differenziazione comporta una perdita della possibilità di clonare le cellule stromali in presenza di IL- 7, l'induzione dell' apoptosi, un aumento dell'espressione delle proteine attive nel riarrangiamento e riarrangiamenti tra VH e DHJH e tra VL e h, che causano o meno un'interruzione del modulo di lettura del segmento DJ, in modo tale che vengano generate cellule B immature sIgM- e sIgM+.
Lo sviluppo dei primi precursori può essere seguito (Ceredig et al., 1983) anche in colture di timo fetale (FTOC, Fetal Thymus Organ Cultures) e di fegato fetale (FLOC, Fetal Liver Organ Cultures) (Owen et al., 1974). La differenza più rilevante tra queste colture d'organo e le colture di singole cellule in sospensione, sta nella capacità dei precursori non solo di differenziarsi in cellule immature con una modalità regolata normalmente, ma anche di espandersi tramite la proliferazione. Nel caso delle FTOC può essere osservata anche la differenziazione da cellule T immature del fenotipo CD4+ CD8+, a cellule T mature CD4+ o CD8+ 'singole positive', per cui questi sistemi di coltura sono più vicini alle condizioni in vivo di differenziazione, proliferazione e selezione cellulare. Dal momento che, nelle FTOC e nelle FLOC, questi processi possono venire influenzati positivamente e negativamente da ligandi, anticorpi monoclonali, cito chine e altre molecole, questi sistemi sperimentali consentono di studiare molto meglio alcuni dei meccanismi molecolari che caratterizzano la differenziazione in vitro dei linfociti T e B.
Predestinazione alla generazione di linee cellulari T e B
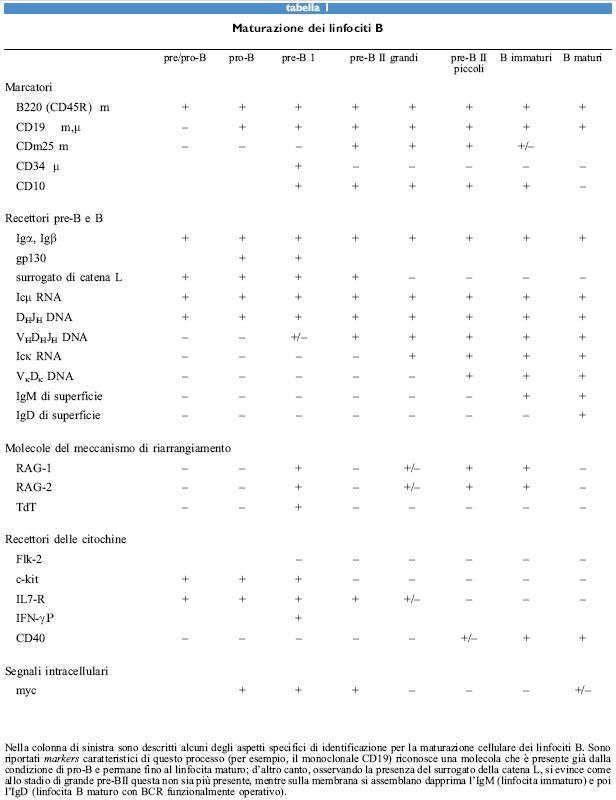
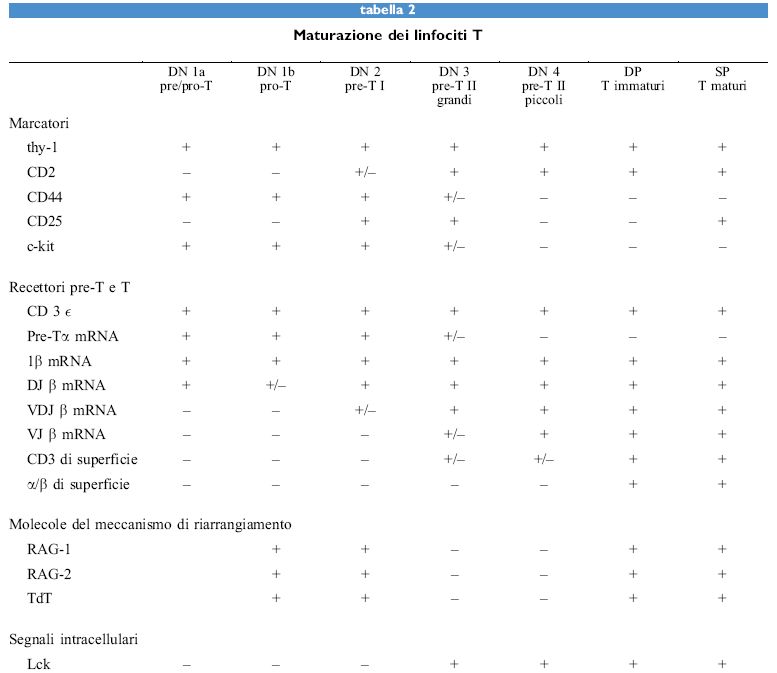
I discendenti di una cellula staminale pluripotente possono essere destinati a diventare eritrociti, granulo citi, mastociti, monociti o macrofagi, megacariociti o piastrine, o linfociti T o B. La predestinazione a tutte le linee cellulari linfoidi, e non solo a quelle che si sviluppano nel timo, deve includere anche l'attivazione dei meccanismi che portano al riarrangiamento dei segmenti genici dei loci del TCR o delle immunoglobuline (tabb. l, 2).
I prodotti dei geni RA G-l e RA G- 2 sono indispensabili per questi riarrangiamenti; infatti, la distruzione selettiva di entrambi i geni porta alla totale incapacità di tutti i progenitori delle cellule linfoidi di riarrangiare qualunque segmento genico, sia esso del TCR o dell'Ig. Tuttavia, i loci RAG mutati sono ancora in grado di consentire lo sviluppo dei primi progenitori di entrambe le linee T e B, in cui avviene la trascrizione sterile del locus della catena β del TCR, nel timo, o dellocus della catena pesante (H) dell'Ig, nel midollo osseo.
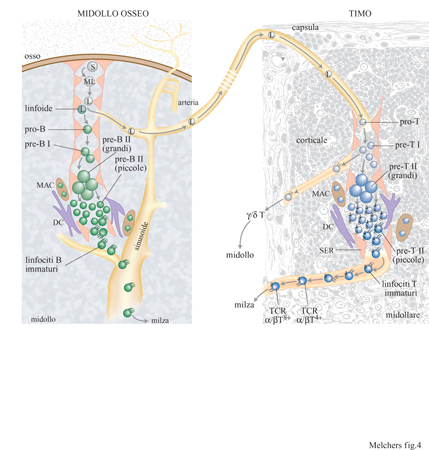
I primi linfociti pro/pre- e pre- I di entrambe le linee T e B, derivanti da topi wild-type e mutanti RAG, esprimono altri componenti coinvolti nel meccanismo di riarrangiamento. Sembra che in questo meccanismo siano coinvolte sia molecole utilizzate in modo ubiquitario sia molecole che sono utilizzate specificamente nei tessuti linfoidi. RAG-l, RAG-2 e TdT sembrano essere specifici per i tessuti linfoidi. È possibile che per la generazione delle varie linee, le cellule debbano migrare in ambienti specializzati dove viene avviato uno speciale programma di espressione genica come primo passo verso la generazione di una determinata linea (fig. 4).
Il timo, l'organo linfoide primario per lo sviluppo delle cellule T
Il caso più impressionante di ambiente specifico in cui viene indotto lo sviluppo linfocitario è costituito dal timo (v. figura 4). Il fatto che alcuni pro genitori presenti in altre parti dell'organismo vengano indotti a migrare nel timo può essere visto come il primo segnale della loro predestinazione a diventare cellule T. Già prima di arrivare alla periferia del timo, questi pro genitori delle cellule T esprimono particolari molecole sulla loro superficie; si ritiene che queste siano coinvolte nel trasferimento delle cellule progenitrici nel microambiente del timo, attraverso gli strati di cellule endoteliali.
Si pensa, inoltre, che lo stretto contatto con lo stroma di cellule epiteliali specializzate nel timo sia essenziale all'ulteriore specializzazione dei progenitori, portando alla maturazione di cellule T che esprimono il TCRα/β o il TCRγ/δ. È probabile che tale contatto induca l'apertura delle regioni cromosomiche dove risiedono i geni delle catene β, γ e δ del TCR, in modo che questi Ioci possano essere trascritti. Gli RNA trascritti sono inizialmente 'sterili', ossia non traducibili in proteine. Essi non contengono, infatti l'informazione per le regioni variabili poiché i geni per la regione V non sono ancora stati riarrangiati. È probabile che i progenitori precoci delle linee T siano in grado di entrare nella via di differenziazione sia della linea con TCRy/8 sia di quella con TCRα/β. Anche se non sono state analizzate in dettaglio singole cellule a uno stadio così precoce di sviluppo, è probabile che esse attivino e trascrivano sterilmente i loci genici sia per il TCRβ sia per il TCR8. A questo punto, l'entrata nella via di differenziazione delle linee T con TCRy/8 o α/β potrebbe dipendere da quale dei due loci attivati subisce inizialmente il primo riarrangiamento V(D)J produttivo, ossia dal fatto che essi producano prima la catena 8 o quella β del TCR (v. oltre).
Il midollo osseo, organo linfoide primario dell'adulto per lo sviluppo dei linfociti B Nell'uomo o nel topo adulti, le cellule staminali pluripotenti non devono migrare lontano per diventare linfociti B. Durante la vita adulta, il midollo osseo è il sito principale per la linfopoiesi delle cellule B, inoltre esso contiene anche le cellule staminali pluripotenti (v. figura 4). Si pensa che le cellule debbano migrare da un ambiente che le mantiene nello stato pluripotente a un altro dove esse vengono indotte a diventare cellule B. Come nel caso delle cellule destinate a diventare linfociti T nel timo, è possibile che il coinvolgimento delle cellule a diventare linfociti B abbia inizio con l'espressione di molecole di adesione che, legandosi alle cellule stromali fanno progredire il differenziamento. L'espressione di queste molecole precede la migrazione. Ci si aspetta che lo stretto contatto dei pro genitori migranti con le cellule stromali che inducono le linee B provochi l'apertura della regione cromosomica che porta il gene per la catena H. In seguito ha inizio la trascrizione sterile del gene della catena H: i diversi segmenti genici destinati a costituire la regione variabile completa non sono ancora stati ricombinati. La macchina per il riarrangiamento viene poi attivata ed è pronta per riarrangiare i segmenti genici corrispondenti. La popolazione di cellule della linea B del midollo osseo non esprime ancora il CD19, ma la tirosinchinasi Flk-2 (v. il saggio di J. Cambier e I. Tamir, Trasduzione del segnale nei linfociti B). Alcune cellule esprimono il surrogato della catena L, mentre altre l'Nk1.l; queste ultime sono i progenitori delle cellule NK (Natural Killer). Quando vengono coltivate in un ambiente appropriato di cellule stromali e citochine, queste cellule si sviluppano ulteriormente e si fermano in un determinato stato di maturazione; il contatto ulteriore con lo stroma e l'esposizione a lL-7 induce la loro proliferazione continua. Queste cellule, chiamate pro/pre- B l, in uno stadio successivo di maturazione diventano CD19+, in esse diminuisce l'espressione di Flk-2, mentre continua a essere espresso c-kit. Nel midollo di un topo giovane, le cellule di pro/pre-Bl sono presenti in una quantità di circa 5 ∙ 10⁶. In presenza di lL- 7 esse sono in grado di proliferare a lungo termine sulle cellule stromali. Il timo di un topo giovane contiene circa 5 ∙ 10⁶ cellule della linea T in due stadi precoci di sviluppo confrontabili. Queste cellule, chiamate cellule pro/pre-T e pre-T l per analogia con le cellule della stirpe B, sono Thyl + CD4CDS- CDT. Le cellule pro/pre- T sono inoltre CDr CD25- CD44+, mentre le pre-T l sono anche CDr/¹ow CD25+ CD44- (v. tabella l).
Riarrangiamenti ordinati degli alleli delle Ig e del TCR
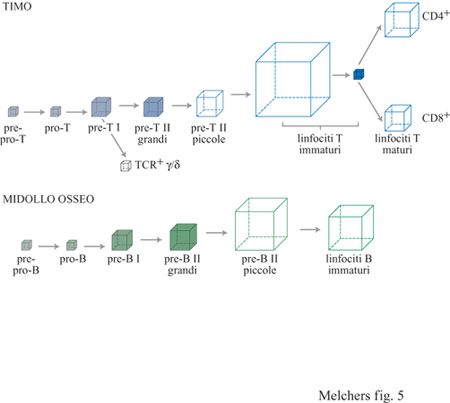
Una volta che le cellule T e B sono state predestinate alla generazione di una linea di linfociti, esse cominciano a riarrangiare i loci genici del TCR e dell'lg rispettivamente e a seguire un programma di sviluppo cellulare in modo che, alla fine, nella periferia emergano cellule TCR + o Ig + in stato di riposo e pronte a rispondere agli antigeni estranei. Gli stadi dello sviluppo, attraverso i quali passano le cellule delle linee T intratimiche con TCRα/β e quelle delle linee B del midollo osseo, e le corrispondenti regole, mostrano somiglianze impressionanti tra le due linee (v. tabella l e figura 4). Per questo motivo le esamineremo insieme, allo scopo di rendere le analogie - e le differenze - più visibili. Sia le cellule destinate a diventare linee T nel timo che quelle destinate a diventare linee B nel midollo osseo, riarrangiano i segmenti genici che codificano il proprio recettore per l'antigene in modo ordinato. In entrambi i casi si inizia con un riarrangiamento del segmento D con il segmento J su entrambi gli alleli, nel caso delle linee T a livello del locus per il TCRβ, e probabilmente anche a livello del locus per il TCRδ; nel caso delle linee B a livello del locus per la catena H. Successivamente al riarrangiamento di D con J sono possibili ulteriori riarrangiamenti ai loci DJ già riarrangiati, in particolare nelle cellule della linea B, dove circa 15 segmenti D e 4 segmenti J sono posizionati nello stesso orientamento all'interno del locus della catena H. Le cellule pre-B I nel midollo osseo che hanno subito il riarrangiamento di DH con JH, esprimono il B220, il CDI9, la tirosinchinasi c-kit, il surrogato della catena L, insieme a due glicoproteine, come recettore pro-B e sono re attive nei confronti delle cellule stromali e della citochina IL-7 (v. tabella l). Nel midollo osseo umano è stata riscontrata una popolazione confrontabile; essa costituisce il 10% di tutte le cellule della linea B. Questa popolazione esprime il CD19, il CD10, il CD34, il VpreB umano e il TdT. In vitro, le cellule stromali e IL-7 mantengono le cellule pre-B I del topo nel loro stato di differenziazione con i segmenti DHJH riarrangiati. Infatti, IL-7 inibisce l'ulteriore differenziazione, mentre la sua rimozione la induce. Queste scoperte hanno suggerito che la stessa cosa potrebbe accadere anche in vivo. Nel midollo osseo, ci si aspetta che le cellule pre-B I riempiano un compartimento che è limitato dalla presenza di una certa quantità di cellule stromali specializzate. Una volta riempito, qualunque ulteriore divisione cellulare sarà asimmetrica, lasciando una cellula figlia in contatto con lo stroma e con IL-7 sotto forma di cellula pre-B I, mentre l'altra cellula figlia perderà i contatti con lo stroma e IL-7, differenziandosi in una cellula pre-B II. È noto che le cellule si dividono due volte al giorno, per cui 5 ∙ 10⁶ cellule pre- B I generano l· 10⁷ cellule pre- B II al giorno attraverso due divisioni asimmetriche (fig. 5).
Ci si aspetta che circa 5.10⁶ cellule pro/preT-I e grandi cellule preT -Il riempiano i compartimenti del timo determinati dalle cellule stromali dai quali vengono generate cellule TCRα/β+ e TCRγ/δ+ (v. figura 5). Ci si aspetta inoltre che le cellule pro/preT - I inizino dei riarrangiamenti di D con J dei loci del TCRδ e del TCRβ, in modo simile a quanto si verifica nelle cellule pro/pre B-1 a livello dei loci per le catene H.
Surrogati delle catene L e α
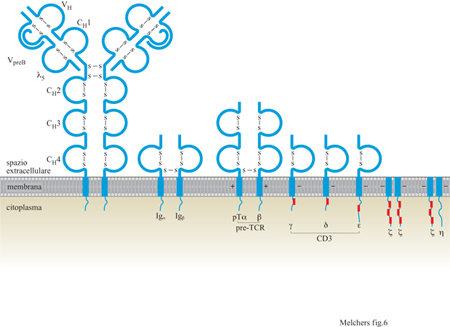
Il surrogato della catena leggera L è una delle prime molecole che indicano la predestinazione per la differenziazione della linea B (Melchers et al., 1993). È codificata da due geni, VpreB e λ₅, che producono due proteine che si associano l'una con l'altra per formare una struttura tipo catena L (fig. 6). Nel topo, i geni per il surrogato della catena L sono localizzati sul cromosoma 22, nell'uomo sul cromosoma 2. Questa catena viene espressa in modo selettivo in tre stati progenitori precoci della linfopoiesi delle cellule B e in nessuna altra cellula del corpo (v. tabella l). Nei primi due stati pro genitori, ossia su cellule che esprimono la molecola specifica della linea B, B220, come del resto la tirosinchinasi c-kit, e che possono esprimere o meno la molecola CDI9, sembra che il surrogato della catena L sia associato a due glicoproteine, gpl30 e gp46. Deve essere ancora chiarita la funzione del complesso Ig precoce di superficie legato alla membrana (il recettore pre-B). In parallelo (v. figura 6), una delle prime molecole che indicano la predestinazione per la differenziazione della linea T è il surrogato della catena a del TCR, chiamata catena pre-Trx (Fehling e von Boehmer, 1997). Le cellule pre-T I esprimono almeno il pTα (v. tabella l). Questo gene, nel topo è localizzato sul cromosoma 17, in posizione telomerica rispetto all'MHC, mentre nell'uomo è localizzato sul braccio corto del cromosoma 6, anche in questo caso vicino all'MHC. Si sospetta che la proteina pTa abbia un partner polipeptidico con il quale si associa allo scopo di formare il surrogato della catena α del TCR e di apparire sulla superficie delle cellule pre- T insieme alla catena TCRβ, ma questo secondo componente non è stato ancora identificato. La proteina pTa potrebbe avere, inoltre, una funzione analoga alla proteina λ5 sul surrogato della catena L, mentre il partner mancante del pTα potrebbe avere proprietà analoghe al VpreB.
Riarrangiamenti di V con DJ: formazione del recettore del pre-linfocita
Il secondo evento di ricombinazione ordinata avviene tra un segmento V e un segmento DJ riarrangiato formato precedentemente sullo stesso cromosoma, ossia tra Vβ e DβJ β o Vδ e DδJδ o VH e DHJH. I riarrangiamenti di V con DJ possono causare o meno un'interruzione del modulo di lettura del segmento DJ, tramite il riarrangiamento DJ a livello dei Ioci del TCRβ, δ o della catena IgH, rispettivamente. Le forme di questi geni riarrangiate in modo produttivo vengono espresse come proteine e trasportate in superficie. La catena δ del TCR appare sulla superficie non appena si realizza un riarrangiamento produttivo tra Vγ e Jγ, e così può essere costruito il TCRγ/δ. La catena β del TCR appare sulla superficie poiché si associa con il surrogato della catena α, il pTα, per formare i recettori pre- T. La catena H dell'lg, analogamente, si associa con il surrogato della catena L per formare i recettori pre-B. Sulla superficie, questi tre tipi di recettori appaiono associati con l'appropriato complesso di trasduzione del segnale, ossia Iga e β per i recettori pre-B e CD3 per i recettori pre-T. L'espressione sulla superficie dei recettori del prelinfocita è cruciale per l'ulteriore sviluppo. Le cellule che presentano sulla propria superficie il recettore pre-B cominciano a proliferare nel midollo osseo; la stessa cosa si verifica per le cellule che presentano il recettore pre-T nel timo. È stato calcolato che queste cellule si dividono da tre a sette volte prima di raggiungere uno stato di riposo, corrispondente allo stadio di sviluppo dei piccoli pre-B II e dei pre-T II rispettivamente (v. figure 4 e 5).
Quando i geni che codificano componenti delle catene surrogato vengono inattivati da mutazioni indirizzate in maniera specifica, come in topi privi della catena λ5 e in topi privi del pre-Ta, viene abolita questa espansione proliferativa nelle cellule delle corrispondenti linee B e T, poiché il recettore del pre-linfocita non può essere formato e depositato sulla superficie. Sembra che la deposizione sulla membrana dei recettori pre-B sia indispensabile affrnché vi sia l'espansione proliferativa, dal momento che l'espansione della linea B non ha luogo nei topi in cui manca la porzione transmembrana della catena μH. In tutti questi mutanti non vengono formati i grossi precursori di tipo 11 circolanti, e tutti i successivi compartimenti immaturi e maturi sono ridotti drasticamente nelle dimensioni (nei mutanti della catena surrogato) o totalmente assenti (nel mutante transmembrana).
La distruzione specifica del gene che codifica la catena pTα non influenza la generazione intratimica di cellule T che esprimono il TCRγ/δ (Fehling e von Boehmer, 1997). Tuttavia, in questi topi privi di timociti con TCRα/β, i precursori delle cellule che esprimono il TCRγ/δ rimangono in piccola quantità e non si espandono nei compartimenti normalmente occupati dai precursori della linea T con TCRα/β. Questo fatto documenta le vie diverse e indipendenti tramite le quali le cellule delle linee T si sviluppano a livello di questi stadi di maturazione nel timo (v. figure 4 e 5).
Possibile esclusione isotipica e allelica da parte dei recettori dei pre-linfociti
Nelle cellule pre-T II sembra che il successo nell'espressione di uno dei due isotipi, γ/δ o α/β, del TRC precluda qualsiasi ulteriore riarrangiamento dell'altro, in modo tale che nel sistema non vengono rinvenute cellule T che esprimono entrambi i TCR, ossia TCRγ/δ+ TCRα/β+. Questa 'esclusione isotipica' nelle cellule pre-T potrebbe seguire le stesse regole dell"esclusione allelica' di riarrangiamenti produttivi V(D)J a livello dei due alleli dei loci del TCRβ e della catena 19H. Non è ancora noto se i riarrangiamenti della catena y del TCR precedano effettivamente quelli della catena o. Se fosse effettivamente così, allora le catene γ potrebbero prendere il posto del surrogato della catena L, e rispettivamente della catena pre-Tα, per l'identificazione e l'amplificazione di cellule con riarrangiamenti V(D)J di successo, cioè produttivi, del/oeus o del TCR. Le cellule che portano loci per la catena β del TCR o per quelle H dell'lg, riarrangiati in modo produttivo, vengono identificate tramite marcatori. Per la linea B, questi marcatori sono c-kiC, CD25+ e le tirosinchinasi syk, lck e fyn espresse selettivamente. Queste cellule vengono chiamate grandi cellule pre-B 11 (perché si trovano nel ciclo cellulare). Per la linea T i marcatori sono CD4- CDS- CD25- e lck. In analogia con lo sviluppo delle cellule B, esse vengono chiamate grandi cellule pre-T II. Nei topi che non esprimono syk lo sviluppo delle cellule B si blocca allo stadio di transizione da cellule pre-B l a grandi cellule pre-B 11. Nei topi che non esprimono lck si verifica lo stesso fenomeno nello sviluppo delle cellule T, a livello della transizione da cellule pre-T l a grandi cellule pre-T II. In questi topi non viene influenzato lo sviluppo di cellule T con TCRγ/δ+. Una delle conseguenze dell'espansione proliferativa è che sia le cellule pre-B II sia quelle pre-T II presentano riarrangiamenti V(D)J produttivi, selezionati positivamente, dei loci per le catene H e per quella β rispettivamente, che predominano sui loci riarrangiati in modo non produttivo, almeno in un allele: infatti, più del 99% di esse esprime, nel proprio citoplasma, rispettivamente le catene β del TCR e H dell'lg. L'amplificazione selettiva mediante PCR (Polymerase Chain Reaetion, reazione a catena della polimerasi) dei riarrangiamenti dei due alle li della catena H in singole cellule pre-B ha evidenziato che metà di esse presenta un allele con riarrangiamento V(D)J produttivo e l'altro allele con riarrangiamento V(D)J non produttivo. L'altra metà delle cellule pre-B presenta un riarrangiamento V(D)J produttivo su un allele, e un riarrangiamento DJ sull' altro allele. Per cui, quando le cellule pre-B che esprimono catene μH con riarrangiamento V(D)J appaiono inizialmente nello sviluppo, esse sono già allelicamente escluse. Questo assicura sin dall'inizio che una cellula B produca solamente una catena μH di un recettore antigene-specifico, un prerequisito essenziale per la validità dell'ipotesi della selezione clonale. Anche se non è stata effettuata un'analisi analoga con la PCR nelle cellule pre-T II per la costituzione degli alle li del TCRβ, ci si può aspettare che siano anch'essi esclusi allelicamente.
Si può pensare a due possibili meccanismi che operino per assicurare l'esclusione allelica. Nella metà di tutte le cellule nello stato di transizione da cellule pre-l a cellule pre-ll, il primo riarrangiamento di V con DJ potrebbe essere non produttivo, e il secondo produttivo. Nell'altra metà delle cellule, il primo riarrangiamento potrebbe essere produttivo. In una cellula di questo gruppo, la catena μH o la catena TCRβ prodotta dal primo allele dovrebbe segnalare l'inibizione del riarrangiamento tra V e DJ sul secondo allele. Come potrebbe ottenere questo risultato? Poiché queste cellule esprimono rispettivamente i recettori pre- T e pre-B, una volta prodotte le catene TCRβ o 19H, i pre-recettori potrebbero lanciare un segnale. Quali cambiameni potrebbe provocare il segnale nella cellula? Un'analisi dell'espressione delle molecole coinvolte nel riarrangiamento, in particolare di RAG-l e RAG-2, ha mostrato che l'espressione di questi componenti essenziali diminuisce nelle cellule che esprimono il pre-recettore (v. tabella l). l geni RAG vengono riattivati in uno stadio successivo dello sviluppo quando vengono riarrangiati i geni che codificano la catena L. Un'analisi simile nelle cellule della linea T ha mostrato la stessa diminuzione e lo stesso aumento dell' espressione dei geni RAG. Prima che questi geni vengano riattivati, l'allele con riarrangiamento DJ dovrebbe essere chiuso e reso inaccessibile alle molecole coinvolte nel riarrangiamento. Gli stessi stadi dello sviluppo delle cellule della linea B nell'uomo mostrano una modulazione analoga dell'espressione dei geni RAG.
Riarrangiamenti dei loci genici e generazione di linfociti immaturi
Per poter esprimere l'slgM e il TCRα/β, i precursori dei linfociti immaturi devono entrare in un terzo stadio di riarrangiamenti genici ordinati: i segmenti V della catena α del TCR e i loci κ e λ, della catena L dell'lg devono venire riarrangiati ai segmenti J di questi loci, e anche in questo caso il riarrangiamento può causare o meno un' interruzione del modulo di lettura del segmento DJ (v. figure 2 e 4). Le cellule della linea T vengono trovate con loci riarrangiati nello stadio di transizione da cellule T con TCRα/β, immature grandi circolanti, a cellule T con TCRα/β, immature piccole in stato di riposo CD4+8+. Per quanto riguarda la linea B, si trovano elementi nello stadio di transizione da cellule pre-B 11, grandi circolanti, a cellule piccole pre-B 11 c-kiCCD25+, in stato di riposo, insieme a cellule B immature in stato di riposo che esprimono 19M di superficie (v. tabella l). Nell'uomo, le cellule della linea B paragonabili sono quelle CD19+10+34-VpreB- che esprimono catene μH nel citoplasma o associate con le catene L sotto forma di 19 di superficie. Nel midollo osseo di un topo giovane, 5 ∙10⁶ cellule pre-B l proliferanti generano inizialmente 1 ∙ 10⁷ cellule pre- B II grandi circolanti esprimenti il recettore pre-B; queste si espandono ulteriormente tramite proliferazione formando un pool di 4.10⁷ cellule pre-B 11 in stato di riposo e di 2 ∙ 10⁷ linfociti immaturi in stato di riposo che esprimono 19 di superficie (v. figura 5). Il turn over di questo insieme di cellule in stato di riposo si verifica in conseguenza dell'apoptosi, cioè la morte cellulare programmata. Tali cellule presentano infatti un'emivita di due o quattro giorni. La maggior parte delle cellule generate in questo modo muore senza aver mai lasciato il midollo osseo. Cellule di ogni compartimento sono state rinvenute nel midollo osseo umano, tutte con proprietà sorprendentemente simili, sebbene in numero più abbondante. Nel timo di un topo giovane, 5.10⁶ cellule pre-T l proliferanti generano 1 ∙ 10⁷ ÷ 2 ∙ 10⁷ cellule pre-T II grandi, esprimenti il recettore pre-T, le quali vanno a costituire un insieme di timo citi immaturi doppi positivi CD4+8+ in riposo che, inizialmente, non esprimono il TRC α/β di superficie; tale espressione si verifica però in seguito. Anche in questo caso, la maggior parte di tali cellule va incontro ad apoptosi con un'emivita di due o quattro giorni. Quindi, anche se la maggior parte delle cellule immature del midollo osseo e del timo non lascia mai questi organi ma muore in situ, esse devono essere generate in quantità talmente elevate da assicurare l'accumulo dei pool di linfociti maturi periferici. Nei topi privi di catene surrogato, vengono prodotti normali linfociti B e T periferici rispettivamente, anche se con un ritmo molto ridotto: questi topi impiegano più di sei mesi per accumulare solo metà del loro normale numero di cellule periferiche mature.
Riarrangiamenti secondari nei loci per IgL e per TCRα
Le cellule pre- B II, le piccole cellule pre-T II e i linfociti B e T immaturi continuano a esprimere le molecole coinvolte nel meccanismo di riarragiamento (v. tabella l). In queste cellule possono verificarsi riarrangiamenti secondari, dal momento che i loci per le catene L dell'lg e per quella α del TCR rimangono accessibili. In tal modo, aumentano le possibilità di convertire in produttivi i riarrangiamenti non produttivi di tali loci, rendendo quindi disponibile la cellula per una selezione che si verificherà successivamente. Questi riarrangiamenti possono anche modificare l'espressione del recettore per l'antigene di un linfocita T o B immaturo, sia conferendogli una nuova specificità mediante una catena nuova, sia bloccandone l'espressione a causa della perdita del riarrangiamento produttivo e dell'acquisizione di quello non produttivo (Nemazee, 1993; Radic e Weigert, 1994; Goodnow et al., 1995). Le molecole coinvolte nel meccanismo di riarrangiamento vengono inattivate nel momento in cui le cellule B diventano mature, ma ben poco si sa sui meccanismi molecolari con cui si verifica la progressiva riduzione dell'espressione di tali molecole.
Sviluppo dei linfociti senza espressione di Ig e TCR
Le cellule pro-T e pro-B possono venire indotte a differenziarsi fino ai rispettivi stadi di linfociti T immaturi CD4+8+25- e di linfociti B immaturi (piccole cellule pre-B II c-kiCCD25-), persino quando non sono in grado di esprimere, rispettivamente, il TCR e le catene delle 19, come nel caso dei topi che non esprimono i geni RAG. Questo si verifica naturalmente senza esprimere TCR e BCR. Anticorpi specifici contro il CD3 o l'lgα stimolano questa espansione cellulare in vivo delle rispettive linee T e B; lo stesso fatto viene provocato dall'espressione costitutiva specifica di p 56lck delle cellule T. Ciò dimostra che i primi stadi del differenziamento dei linfociti non richiedono l'espressione del TCR o dell'lg, anche se essa è necessaria per riempire correttamente i successivi compartimenti più maturi con una quantità sufficiente di cellule funzionanti in modo normale, ossia in grado di esprimere il recettore per l'antigene.
Normalmente, la trascrizione dei loci genici per l'lg o il TCR precede il loro riarrangiamento. Nei topi che non esprimono i geni RAG, l'induzione della trascrizione dei loci per la catena α del TCR e le catene L dell'lg viene osservata nelle cellule che si stanno differenziando dagli stadi pre- B l o pre- T l agli stadi pre-B II o pre-T II in vitro. La stimolazione di una cellula pre-B in via di differenziazione con lL-4 e con un anticorpo specifico per il CD40 induce persino lo switehing (commutazione) sμ-sε sul locus della catena H. Questo fenomeno suggerisce che la differenziazione dei linfociti a stadi di cellule completamente mature dipende sia dai contatti tra cellule sia dalle citochine, ma non dall' espressione di recettori antigenici specifici (Rolink et al., 1996; Fehling e von Boehmer, 1997).
Arresto della differenziazione e morte di linfociti immaturi autoreattivi
La diversità dei recettori per l'antigene, generata da un'ampia varietà di ricombinazioni V(D)J e VJ e ulteriormente diversificata dall'inserzione di regioni N all'interno delle loro giunzioni, è talmente elevata che i repertori di linfociti generati originariamente, ognuno con una particolare coppia V(D)JNJ, deve includere cellule che riconoscono autoantigeni. Gli autoantigeni vengono riconosciuti inizialmente nei siti dove vengono prodotti i linfociti, quindi nel timo e nel midollo osseo. Questo riconoscimento porta all'arresto della differenziazione della cellula autore attiva e, di conseguenza, alla sua morte accelerata (Nemazee, 1993; Radic e Weigert, 1994; Goodnow et al., 1995; Kisielow e von Boehmer, 1995).
Attualmente non sono noti i meccanismi molecolari che portano alla morte cellulare. Dal momento che negli organi linfoidi primari sono stati rinvenuti dei macrofagi, è possibile che la morte, almeno di alcune cellule, sia in realtà ottenuta tramite la fagocitosi delle cellule autoreattive, forse dopo l'incontro con l'antigene e i conseguenti cambiamenti fenotipici. Normalmente queste cellule potrebbero cercare di modificare i propri recettori per l'antigene tramite riarrangiamenti secondari nei loci del TCRα e dell'lgL, rispettivamente, ossia esse potrebbero 'curare' i propri recettori modificandoli geneticamente affinché evitino l' autoriconoscimento. Tuttavia, se il recettore che riconosce l'autoantigene è l'unico presente nel topo, come nel caso di un topo carente nel riarrangiamento che presenta un TCR transgenico o una Ig con specificità per un autoantigene, allora le cellule non sono in grado di evitare l'autoriconoscimento tramite riarrangiamenti genici secondari; in questo caso, si verifica l'arresto dello sviluppo di tutte le cellule. Per quanto riguarda le cellule della linea B a questo stadio di sviluppo, non conosciamo praticamente nulla dei requisiti molecolari e cellulari necessari per tale presentazione dell'autoantigene.
Sembra possibile interferire con l'arresto dello sviluppo. L'iniezione di una forma particolare di un autoantigene può liberare le cellule dall'arresto (Andersson et al., 1995), permettendone lo sviluppo in plasmacellule secernenti autoanticorpi. Questa scoperta suggerisce un possibile scenario per l'induzione di malattie autoimmuni mediate da linfociti B come il Lupus eritematoso. L'arresto della differenziazione, chiamato anche selezione negativa, è il primo segno di induzione della tolleranza, per cui le cellule re attive nei confronti dell'autoantigene, presentate con gli autoantigeni negli organi linfoidi primari, vengono indotte a morire.
Presentazione dell'antigene a cellule T immature nel timo
Gran parte delle informazioni sui geni e sui processi molecolari che controllano la presentazione dell'antigene da parte dei linfociti T deriva da studi condotti sulle cellule che presentano l' antigene del sistema linfoide periferico (Koopman et al., 1997; Pieters, 1997). L'antigene viene captato tramite cellule che presentano l'antigene (APC, Antigen Presenting Cells) specializzate. Esso viene processato, nel caso si tratti di una proteina, tramite la digestione proteolitica fino ai peptidi. Questi ultimi vengono caricati nei siti di legame delle molecole MHC di classe l e II, e i complessi risultanti vengono trasportati sulla superficie cellulare dove sono presentati ai linfociti T. Anche se la captazione dell'antigene, il suo processamento e la sua presentazione non sono stati studiati in maniera molto dettagliata nel timo, ci si aspetta che gli autoantigeni - spesso liberati in seguito alla morte dei precursori della linea T - vengano gestiti e presentati con modalità simili.
Il processamento e la presentazione dei frammenti antigenici da parte delle molecole MHC di classe l e II seguono vie diverse. Dopo la loro sintesi, le molecole di classe II si spostano nella cellula passando, attaverso il reticolo endoplasmatico (ER), prima nei compartimenti del Golgi e poi nei compartimenti post-Golgi e, infine, nei compartimenti endosomali e lisosomali dove vengono caricati i peptidi; dopo di che vengono depositate sulla superficie cellulare. Un elaborato sistema di proteine assiste il montaggio di peptidi all'interno delle molecole MHC di classe II. Alcune, come per esempio la catena invariante (li, lnvariant ehain) mantengono libero il solco fino al trasporto della molecola MHC nel compartimento endo-lisosomale. Qui altre molecole, HLA-DM e HLA-DO, sono incaricate di facilitare il caricamento dei peptidi provenienti dall' esterno. Le molecole MCH di classe I vengono caricate con peptidi processati da proteine prima di lasciare l'ER. La degradazione di proteine in peptidi ha luogo a livello di particolari complessi intracellulari contenenti proteasi chiamati proteasomi, localizzati nel citoplasma; da qui i peptidi vengono trasportati con speciali trasportatori ai siti di caricamento nell'ERo L'accesso ai proteasomi, e di conseguenza il caricamento delle molecole di classe I, avviene attraverso il citoplasma; di conseguenza, tutte le proteine che non vengono sintetizzate non vi arrivano, a meno che utilizzino espedienti particolari.
La differenziazione dei linfociti T immaturi TCRα/β+ che riconoscono i propri autoantigeni con grande avidità viene bloccata portando alla morte cellulare tramite apoptosi, a meno che queste cellule abbiano avuto la possibilità di cambiare la specificità del proprio recettore tramite riarrangiamento secondario dei propri geni che codificano il TCRα; questo permette loro di 'curare' i propri recettori. Se il riarrangiamento provoca una diminuzione dell' avidità per l'autoantigene, vi è la possibilità che le cellule sopravvivano, anche se solo per un breve periodo. I riarrangiamenti secondari a livello dei loci per le catene L dell'Ig o per quella α del TCR portano spesso alla generazione di linfociti immaturi che esprimono - in modo transitorio o stabile - più di una catena L o α. Quindi, a questo stadio dello sviluppo non sempre viene rispettata la regola secondo la quale una cellula produce un solo e unico recettore antigenico specifico. La cellula 'curata' si unisce al pool di linfociti immaturi.
Selezione positiva di linfociti T immaturi nel timo
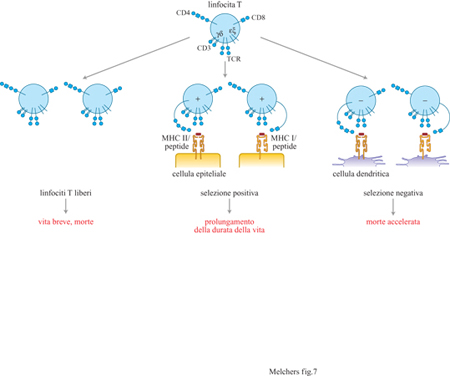
Nel pool timico di linfociti T immaturi, ancora possono trovarsi cellule con TCR con avidità più bassa per i complessi MHC/peptide. Infatti, questi linfociti T immaturi potrebbero riconoscere essenzialmente molecole MHC da sole. Un singolo TCR α/β solubile, che sia specifico per un determinato peptide complessato con una particolare molecola MHC, può legarsi a questa molecola MHC persino quando essa presenta molti peptidi diversi non correlati tra loro. Evidentemente ciò ha lo scopo di stabilizzare semplicemente la conformazione della molecola MHC in modo tale che essa venga riconosciuta dal TCR. Rimane una questione di acceso dibattito se i segmenti V codificati dalla linea germinale dei Ioci α o β, o di entrambi, del TCR vengano selezionati nell'evoluzione per la loro capacità di legare le molecole MHC. Questo non dovrebbe essere il caso di segmenti V di Ioci che codificano recettori per l' antigene, come VH, Vλ, Vδ e Vγ che non devono riconoscere i complessi MHC/peptide per poter diventare funzionali. Questo riconoscimento con minore avidità modifica la vita della cellula T con TCRCα/β. Se il suo TCR si accoppia con l'MHC di classe l, esso utilizza le molecole CD8 coespresse insieme alle molecole CD4 sulla cellula T immatura con TCRCα/β+ per stabilire ulteriori contatti con le molecole di classe l (fig. 7). Questo segnala alla cellula di diminuire l'espressione del CD4, di uscire dal timo, di diventare cellula a vita lunga e di maturare nella periferia. Diversamente, se capita che il TCR si accoppia con molecole MHC di classe II, la cellula utilizza le molecole di CD4 per stabilire ulteriori contatti con le molecole MHC di classe II, diminuisce l'espressione del CD8, esce dal timo, diventa una cellula a vita lunga e matura. Le cellule T mature CD8+, ristrette alle MHC di classe l, sono i precursori delle cellule T killer, mentre le cellule T CD4+, ristrette alle MHC di classe 11, sono i precursori delle cellule T helper. Quindi, le cellule T TCRCi/β+ periferiche che hanno subito i processi di selezione negativa e positiva dovrebbero essere tutte tolleranti nei confronti degli autoantigeni e dovrebbero tutte riconoscere antigeni solo nel contesto delle molecole MHC. La selezione delle cellule T helper viene fatta sulle cellule epiteliali radioresistenti, e non funziona quando le cellule dendritiche (DC, Dendritie Cell) sono le uniche APC capaci di esprimere le molecole MHC di classe II (v. figura 7).
È stato stimato che ogni giorno vengono selezionati solo 2-3 milioni di cellule dal pool di linfociti immaturi, con più di 20 milioni di cellule che lasciano i siti dello sviluppo primario linfoide nella corteccia del timo e nelle zone B del midollo osseo. In seguito i linfociti T immaturi appaiono nella parte midollare del timo e nelle regioni extrafollicolari della milza. Si pensa che questa selezione all'interno del pool di linfociti immaturi non coinvolga la proliferazione cellulare e che possa, o meno, cambiare l'aspettativa di vita delle cellule, che inizialmente è breve.
Selezione positiva delle cellule B immature
Non sono noti fattori genetici in grado di limitare i repertori di cellule B mature. Non è chiaro se le cellule B immature vadano incontro alla selezione positiva tramite i loro recettori antigenici, prima di diventare sensibili e di cominciare a proliferare e a differenziarsi. Tuttavia, topi doppi mutanti CD40-/-/xid (btk-/-) non sono in grado di selezionare un repertorio di cellule B a vita lunga capaci di mediare le risposte cellule T-indipendenti e cellule T-dipendenti. Nonostante tutto, entrambi i ceppi mutanti sviluppano precursori della linea B e cellule B immature nel midollo osseo, in quantità normale e con fenotipi normali. Questo rende almeno plausibile la selezione positiva delle cellule B periferiche a vita lunga; comunque, non esclude la possibilità che la longevità sia indotta solo dall'incontro con un invasore esterno e dopo l'espansione proliferativa di una cellula B matura in stato di riposo.
Riassumendo, una piccola quantità di pro genitori e precursori (circa 5 ∙ 106) si espande negli organi linfoidi primari, tramite proliferazione, dando luogo a 50.10⁶ cellule immature che esprimono il recettore per l' antigene, tutte con vita breve. Solo 2 ∙ 10⁶ ÷ 3 ∙ 10⁶ di queste cellule lasciano il sito di sviluppo linfoide primario nella corteccia del timo e nelle zone B del midollo osseo. Le cellule T immature appaiono poi nella parte midollare del timo da cui escono per spostarsi nelle zone ricche di cellule T delle regioni extrafollicolari della milza. Le cellule B immature escono dal midollo osseo per spostarsi nelle zone ricche di cellule B della milza. Nelle regioni extrafollicolari esse sono presenti come cellule sensibili all'antigene, mature e in stato di riposo, pronte per iniziare una risposta immunitaria contro un antigene estraneo.
Ringraziamenti
Ringrazio il Dott. Antonio Lanzavecchia per aver letto in modo critico questo articolo. ll Basel Institute for Immunology è stato istituito ed è finanziato da F. Hoffmann-La Roche Ltd., Basilea, Svizzera.
Bibliografia citata
ANDERSSON, J., MELCHERS, F., ROLINK, A. (1995) Stimulation by T cell independent antigens can relieve the arrest of differentiation of immature auto-reactive B cells in the bone marrow. Scand. J. Immunol., 42, 21-33.
BOGUE, M., GILFILLAN, S., BENOIST, C., MATHIS, D. (1992) Regulation of N-region diversity in antigen receptors through thymocyte differentiation and thymus ontogeny. Proc. Natl. Acad. Sci. USA, 89,11.011-11.015.
BOISMENU, R., HAVRAN, W. L. (1994) Modulation of epithelial cell growth by intraepithelial gamma delta T cells. Science, 266, 1253-1255.
BURROWS, P.D., COOPER, M.D. (1997) B cell development and differentiation. Curr. Opin. Immunol., 9, 239-244. CEREDIG, R., McDONALD, H.R., JENKINSON, E.J. (1983) Flow microfluorometric analysis of mouse thymus development in vivo and in vitro. Eur. J. Immunol., 13, 185-190.
CHEN, J. (1996) Analysis of gene function in Iymphocytes by RAG-2-deficient blastocyst complementation. Adv. Immunol., 62, 31-59.
CLEVERS, H.C., GROSSCHEDL, R. (1996) Transcriptional control of Iymphoid development: lessons from gene targeting. Immunol. Today, 17, 336-343.
COUTINHO, A., FREITAS, A.A., HOLMBERG, D., GRANDIEN, A. (1992) Expression and selection of muri ne antibody repertoires. Int. Rev. Immunol., 8, 173-187.
CUMANO, A., DIETERLEN-LIEVRE, F., GODIN, I. (1996) Lymphoid potential, probed before circulation in mouse, is restricted to caudal intraembryonic splanchnopleura. CelI, 86, 907-916.
FEHLING, H.I., VON BOEHMER, H. (1997) Early alphabeta T cell development in the thymus of normal and genetically altered mice. Curr. Opin. Immunol., 9, 263-275.
GEORGOPOULOS, K. (1997) Transcription factors required for Iymphoid lineage commitment. Curr. Opin. Immunol., 9, 222-227.
GOODNOW, C.C., CYSTER, J.G., HARTLEY, S.B., BELL, S.E., COOKE, M.P., HEALY, J.I., AKKARAJU, S., RATHMELL, J.C., POGUE, S.L., SHOKAT, K.P. (1995) Self-tolerance checkpoints in B Iymphocyte development. Adv. Immunol., 59, 279-368.
JOTEREAU, F.V., HEUZE, F., SALOMON-VIE, V., GASCAN, H. (1987) Cell kinetics in the fetal mouse thymus: Precursor cell input, proliferation and emigration. J. Immunol., 138, 1026-1030.
JOTEREAU, F.V., LE DOUARIN, N.M. (1982) Demonstration of a cyclic renewal of the Iymphocyte precursor cells in the quail thymus during embryonic and perinatallife. J. Immunol., 129, 1869-1877.
KISIELOW, P., VON BOEHMER, H. (1995) Development and selection of T cells: facts and puzzles. Adv. Immunol., 58, 87-209.
KNIGHT, K. L., WINSTEAD, C. R. (1997) Generation of antibody diversity in rabbits. Curr. Opin. Immunol., 9, 228-232.
KOOPMAN, J.-O., HAMMERLING, G.I., MOMBURG, F. (1997) Generation, intra-cellular transport and loading of peptides associated with MHC class I molecules. Curr. Opin. Immunol., 9, 80-88.
LE DOUARIN, N.M., JOTEREAU, F.V. (1975) Tracing of cells ofthe avian thymus through embryonic life in interspecific chimeras. J. Exp. Med., 142, 17-40.
MARCOS, M. A., GUTIERREZ, J. C., HUETZ, F., MARTINEZ, C., DIETERLEN-LIEVRE, F. (1991) Waves of B-Iymphopoiesis in the establishment of the mouse B-cell compartment. Scand. J. Immunol., 34, 129-135.
MEDVINSKY, A, DZIERZAK, E. (1996) Definitive hematopoiesis is autonomously initiated by the AGM region. CelI, 86, 897-906.
MELCHERS, F. (1979) Three waves of B Iymphocyte development during embryonic development in the mouse in Cell lineage, stem cells and cell determination. In CelI lineage, stem celIs, and celI determination: proceedings of the International Workshop on celI lineage, stem celIs, and celI determination, a c. di Le Douarin N., Amsterdam, Elsevier Science Publishers, p. 281-289.
MELCHERS, F., KARASUYAMA, H., HAASNER, D., BAUER, S., KUDO, A., SAKAGUCHI, N., JAMESON, B., ROLINK, A. (1993) The surrogate light chain in B-cell development. Immunol. Today, 14, 60-68.
MELCHERS, F., ROLINK, A., GRAWUNDER, D., WINKLER, T.H., KARASUYAMA, H., GHIA, P., ANDERSSON, J. (1995) Positive and negative selection events during B Iymphopoiesis. Curr. Opin. Immunol., 7, 214-227. MORRISON, S.I., WRIGHT, D.E., CHESHIER, S.H., WEISSMAN, I.L. (1997) Hematopoietic stem cells: Challenges to expectations. Curr. Opin. Immunol., 9, 216-221.
NEMAzEE, D. (1993) Promotion and prevention of autoimmunity by B Iymphocytes. Curr. Opin. Immunol., 5, 866-872.
OWEN, U., COOPER, M.D., RAFF, M.C. (1974) In vitro generation of B-Iymphocytes in mouse fetal liver, a mammalian 'bursa equivalent'. Nature, 249, 361-363.
PAPAVASILIOU, F., JANKOVIC, M., GONG, S., NUSSENZWEIG, M.C. (1997) Control of immunoglobulin gene rearrangements in developing B cells. Curr. Opin. Immunol., 9, 233-238.
PHILLIPS, J.H., HORI, T., NAGLER, A., BHAT, N., SPITS, H., LANIER, L.L. (1992) Ontogeny ofhuman natural killer (NK) cells: fetal NK cells mediate cytolytic function and express cytoplasmic CD3 epsilon, delta proteins. J. Exp. Med., 175, 1055-1066.
PIETERS, J. (1997) MHC class II restricted antigen presentation. Curr. Opin. Immunol., 9, 89-96.
POGGI, A., SARGIACOMO, M., BIASSONI, R., PELLA, N., SIVORI, S., REVELLO, V., COSTA, P., VALTIERI, M., RUSSO, G., MINGARI, M. C. (1993) Extrathymic differentiation of T Iymphocytes and natural killer cells from human embryonic liver precursors Proc. Natl. Acad. Sci. USA, 90, 4465-4469.
POTOCNIK, A.J., NIELSEN, P.J., EICHMANN, K. (1994) In vitro generation of Iymphoid precursors from embryonic stem cells EMBO J., 13, 5274-5283.
RADIC, M. Z., WEIGERT, M. (1994) Genetic and structural evidence for antigen selection of anti-DNA antibodies. Annu. Rev. Immunol., 12, 487-520.
REYNAUD, C. A., GARCIA, C., HEIN, W. R., WEILL, J. C. (1995) Hypermutation generating the sheep inununoglobulin repertoire is an antigen-independent processo Cell, 80, 115-125.
ROCHA, B., GUY-GRAND, D., VASSALLI, P. (1995) Extrathymic T cell differentiation. Curr. Opin. Immunol., 7, 235-242.
ROCHA, B., VASSALLI, P., GUY-GRAND, D., Nov, S. (1992) The extrathymic T-cell development pathway. Immunol. Today, 13, 449-454.
ROLINK, A., GHIA, P., GRAWUNDER, D., HAASNER, D., KARASUYAMA, H., KALBERER, C., WINKLER, T., MELCHERS, F. (1995) In vitro analyses of mechanisms of B-cell development. Semin. Immunol., 7, 155-167.
ROLINK, A., MELCHERS, F. (1993) Generation and regeneration of cells of the B-Iymphocyte lineage. Curr. Opin. Immunol., 5, 207-217.
ROLINK, A., MELCHERS, F., ANDERSSON, J. (1996) The SCIO but not the RAG-2 gene product is required for S/l-SE heavy chain class switching. Immunity, 5, 319-330.
SANCHEZ, M.J., GUTIERREZ-RAMOS, J.C., FERNANDEZ, E., LEONARDO, E., LOZANO, J., MARTINEZ, C., TORIBIO, M.L. (1993) Putative prethymic T cell precursors within the early human embryonic liver: a molecular and functional analysis. J. Exp. Med., 177, 19-33.
SHORTMAN, K., WU, L. (1996) Early T Iymphocyte progenitors. Annu. Rev. Immunol., 14, 29-47.
SINGH, H. (1996) Gene targeting reveals a hierarchy of transcription factors regulating specification of Iymphoid cell fates. Curr. Opin. Immunol., 8, 160-165.
SPANGRUDE, G.J., BROOKS, D.M., TUMAS, D.B. (1995) Long-term repopulation of irradiated mice with limiting numbers of purified hematopoietic stem cells: in vivo expansion of stem cell phenotype but not function. Blood, 85, 1006-1016.
SPITS, H. (1994) Early stages in human and mouse T cell development. Curr. Opin. Immunol., 6, 212-22l.
THOMAS, K.R., CAPECCHI, M.R. (1987) Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell, 51, 503-512.
TORRES, R.M., KUHN, R. (1997) Laboratory protocols for conditional gene targeting. Oxford-New York, Oxford University Press.