
genetica, malattia
genetica, malattia
Malattia causata da una anomalia nel materiale genetico di un individuo. Le malattie g. hanno una frequenza complessiva nella popolazione intorno al 3,8÷5,0%. I difetti genetici sono la causa principale di interruzione della gravidanza nei paesi sviluppati, e almeno metà degli aborti spontanei è dovuta ad anomalie cromosomiche del feto. Circa il 30% della mortalità infantile dopo la nascita, nei paesi sviluppati, è dovuta a malattie g.; inoltre, si stima che difetti genetici, anche se minori, siano presenti in almeno il 10% degli adulti. Sono state identificate diverse migliaia di malattie g. con sintomi clinici definiti; oggi tuttavia è opinione comune che nello sviluppo di ogni malattia abbiano un ruolo sia fattori genetici che ambientali. Le principali banche dati che raccolgono tutte le mutazioni note nell’uomo sono lo Human Mutation Database e lo OMIM (➔) per i caratteri mendeliani.

Classificazione delle malattie genetiche
Le mutazioni svantaggiose che sono alla base delle patologie possono introdurre sia una perdita che un’acquisizione di funzione; esse di solito alterano una sequenza codificante (attraverso sostituzioni non sinonime, introduzione di codoni nonsense, slittamento del modulo di lettura, introduzione di ripetizioni ‘in tandem’), o modificano sequenze intrageniche importanti per l’espressione genica (siti di splicing, elementi conservati di sequenze non tradotte come i promotori e altre regioni di controllo). Anche le mutazioni che alterano gli RNA o i complessi delle ribonucleoproteine (RNPs) sono alla base di molte malattie ereditarie: riguardano lo splicing dei messaggeri, la traduzione, la formazione dei ribosomi. Possiamo classificare le malattie genetiche in quattro tipi principali: a gene singolo; multifattoriali, cromosomali, e mitocondriali.
Malattie a gene singolo o mendeliane
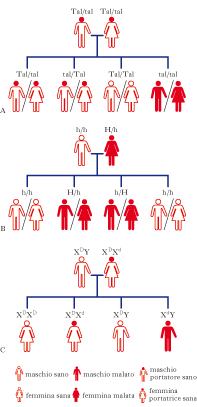
Sono dovute a cambiamenti o mutazioni nella sequenza del DNA di un gene singolo. Sono note di 6.000 malattie genetiche a gene singolo, che si verificano con la frequenza di uno su 200 nei neonati. I genotipi alternativi alla base di queste malattie danno origine a classi di fenotipi discreti e ben distinguibili; pertanto esse sono ereditate secondo gli schemi delle leggi di Mendel. Vengono classificate come dominanti, quando il fenotipo clinico si manifesta sia negli individui eterozigoti che omozigoti per il gene mutato, e recessive, quando la malattia si manifesta solo negli individui omozigoti per la mutazione. In base alla localizzazione del gene sui cromosomi vengono poi suddivise in autosomiche e legate all’X. Molte malattie genetiche mendeliane presentano caratteristiche cliniche variabili anche tra i membri affetti della stessa famiglia (espressività variabile), e in alcuni casi non tutti gli individui che posseggono il gene mutato manifestano la malattia (penetranza incompleta). La variabilità intrafamiliare è dovuta alla combinazione degli effetti di altri geni non associati (geni modificatori) e di influenze ambientali. La trasmissione ereditaria delle malattie monogeniche avviene secondo le seguenti modalità:
• Eredità autosomica dominante. Una persona affetta ha di solito almeno un genitore affetto. Sono colpiti entrambi i sessi ed è trasmessa da entrambi i sessi. Il figlio di genitori uno affetto (eterozigote) e l’altro no ha il 50% di probabilità di essere affetto e le anomalie compaiono di solito in ogni generazione. Esempi di malattie autosomiche dominanti sono la malattia di Huntington e la sindrome di Marfan. Non tutti gli individui che posseggono il gene mutato manifestano la malattia (penetranza incompleta) e il grado di manifestazione fenotipica dei sintomi può presentare ampie variazioni (espressività variabile).
• Eredità autosomica recessiva. Molte di queste malattie sono dovute a mutazioni in enzimi delle vie metaboliche. Gli individui affetti di solito sono figli di individui non affetti, o portatori asintomatici (che possiedono una copia di un gene di una malattia g. recessiva, e non manifestano la malattia). Il figlio affetto riceve una copia del gene difettivo da entrambi i genitori. La probabilità per un figlio, di entrambi i sessi, di contrarre la malattia è di 1 su 4. Il rischio di contrarre una malattia recessiva aumenta se i genitori sono consanguinei: infatti è maggiore la probabilità di ereditare lo stesso gene raro da un antenato comune. Malattie autosomiche recessive sono la fibrosi cistica, la fenilchetonuria, l’anemia falciforme.
• Eredità recessiva legata all’X. Le mutazioni sui geni localizzati sul cromosoma X, che non hanno corrispettivi sull’Y, sono alla base delle malattie genetiche legate al sesso. Sono colpiti quasi esclusivamente i maschi. I maschi affetti di solito nascono da genitori sani; normalmente la madre è un’eterozigote asintomatica e può avere parenti maschi affetti. Le femmine possono essere affette se il padre è affetto e la madre è eterozigote o, occasionalmente, in seguito a inattivazione non casuale dell’X. Esempi di malattie recessive legate all’X sono la distrofia muscolare di Duchenne e l’emofilia A.
• Eredità dominante legata all’X. Sono malattie molto rare (per es., ipofosfatemia e incontinentia pigmenti), che colpiscono entrambi i sessi, ma prevalentemente le femmine. Il figlio di una femmina affetta, a prescindere dal sesso, ha una probabilità del 50% di essere affetto. Un maschio affetto avrà tutte le figlie affette e tutti i figli sani.
Malattie multifattoriali
Queste malattie genetiche (anche dette complesse o poligeniche) sono dovute all’influenza di mutazioni su geni multipli in combinazione con fattori ambientali. Per es., geni diversi che influenzano la comparsa del tumore al seno sono stati identificati su 7 diversi cromosomi. Oggi molte delle malattie più comuni sono considerate multifattoriali, ad es. le malattie cardiovascolari, l’ipertensione, la malattia di Alzheimer, il diabete, i cancro, e l’obesità.
Malattie cromosomiche
Le anomalie cromosomiche che possono avere come risultato una malattia sono dovute alla perdita di intere copie di cromosomi, o alla presenza di copie extra, o a rotture e riunioni di grandi frammenti (traslocazioni), o a grandi e piccole delezioni. Ad es., la sindrome di Down o trisomia 21 si verifica quando una persona ha tre copie del cromosoma 21. Altri esempi sono la sindrome di Klinefelter e la sindrome di Turner (presenza di un solo cromosoma X nella femmina).
Malattie mitocondriali
Questo tipo relativamente raro è causato da mutazioni nel DNA contenuto nei mitocondri. La trasmissione avviene attraverso la linea materna. Negli ultimi anni sono state identificate più di cinquanta malattie genetiche dovute a mutazioni nel DNA mitocondriale, che possono manifestarsi a qualsiasi età. Alcuni esempi sono: ritardo nello sviluppo, problemi gastrointestinali, sordità, aritmie cardiache, disturbi metabolici.