
Materiali
Materiali
Materiali cementizi di Mario Collepardi
SOMMARIO: 1. Introduzione. □ 2. Gesso. □ 3. Calce. □ 4 Calce idraulica. □ 5. Cemento Portland: a) produzione del cemento Portland; b) idratazione del cemento Portland; c) calore di idratazione del cemento Portland; d) presa del cemento Portland; e) indurimento e resistenza meccanica della pasta di cemento Portland. □ 6. Cementi speciali: a) cemento bianco; b) cementi colorati; c) cemento ferrico; d) cemento pozzolanico; e) cemento d'altoforno; f) cemento soprasolfatato; g) cementi espansivi; h) cemento alluminoso. □ 7. Gli inerti: a) sostanze indesiderabili negli inerti; b) distribuzione granulometrica degli inerti. □ 8. Gli additivi: a) additivi acceleranti; b) additivi ritardanti; c) additivi fluidificanti; d) additivi superfluidificanti; e) additivi aeranti. □ 9. Il calcestruzzo fresco: a) lavorabilità del calcestruzzo fresco; b) segregazione del calcestruzzo fresco. □ 10. Il calcestruzzo indurito: a) resistenza meccanica; b) modulo elastico; c) ritiro e deformazione viscosa; d) durabilità. □ 11. Calcestruzzi speciali. □ Bibliografia.
1. Introduzione.
I materiali cementizi sono prodotti inorganici preparati artificialmente per cottura, a temperature relativamente elevate, di pietre naturali e successiva macinazione. La polvere che così si ottiene, mescolata con acqua, forma una massa di consistenza plastica più o meno lavorabile, che inizialmente perde la sua plasticità (presa) e successivamente diviene rigida (indurimento).
La miscela di acqua e materiale cementizio (‛pasta cementizia') può contenere dispersi anche elementi lapidei, come sabbia, oppure ghiaia o pietrisco; la miscela che ne risulta prende il nome di ‛conglomerato cementizio', e più esattamente di ‛malta' nel primo caso e di ‛calcestruzzo' nel secondo. Sempre più frequentemente vengono aggiunti additivi chimici per migliorare le proprietà delle malte e dei calcestruzzi.
Molto raramente i materiali cementizi sono impiegati per produrre la ‛pasta'; più frequentemente - per motivi economici, ma anche per ragioni tecniche - essi sono utilizzati per preparare la malta e soprattutto il calcestruzzo. Poiché i materiali cementizi, in combinazione con l'acqua, hanno quindi la funzione di legare gli elementi lapidei - detti anche inerti o aggregati - essi sono chiamati anche ‛leganti'.
I leganti sono classificati come ‛aerei' o ‛idraulici' a seconda che l'indurimento della miscela acqua-legante possa avvenire rispettivamente solo all'aria o anche sott'acqua. I leganti aerei comprendono la calce e il gesso, quelli idraulici - molto più importanti - includono il cemento e la calce idraulica. Poiché quest'ultima è attualmente prodotta in quantità trascurabile, i leganti idraulici si identificano di fatto con il cemento. La calce e il gesso sono impiegati per produrre le malte, mentre il cemento è destinato per lo più alla preparazione dei calcestruzzi.
2. Gesso.
Il gesso legante (v. Turco, 1961; v. Collepardi, 1977) è costituito da solfato di calcio semudrato (CaSO4 • 1/2H2O) o da anidrite (CaSO4) o da una miscela dei due solfati ottenuta per cottura a 120-200 °C della pietra di gesso, che si trova in natura sotto forma di CaSO4 • 2H2O. Il processo di fabbricazione del legante consiste in una disidratazione parziale o totale del gesso budrato secondo quanto è indicato rispettivamente nelle reazioni seguenti:
CaSO4 • 2H2O → CaSO4 • ½H2O + ³-2H2O (1)
CaSO4 • 2H2O → CaSO4 + 2H2O. (2)
La polvere che si ottiene per macinazione del gesso cotto ha proprietà leganti, poiché, mescolata con acqua, forma una massa plastica che indurisce progressivamente grazie alla trasformazione del semiidrato e dell'anidrite in gesso budrato. In altre parole, durante l'utilizzazione del gesso legante si verifica un processo di idratazione che è esattamente l'opposto di quello indicato nelle reazioni (1) e (2).
Nel sistema CaSO4-H2O, a temperatura ambiente, il semiidrato e l'anidrite sono metastabili e presentano una maggiore solubilità in acqua rispetto alla fase stabile costituita da solfato di calcio budrato (v. fig. 1). Pertanto, quando si prepara una pasta mescolando gesso legante con acqua, si forma una soluzione satura di semiidrato o di anidrite, ma soprassatura rispetto al gesso budrato. Conseguentemente, dopo un periodo di induzione dovuto alla formazione dei primi nuclei cristallini della fase stabile, cominciano a precipitare i cristalli di CaSO4 • 2H2O. La soluzione tende, quindi, a diventare insatura in semiidrato o in anidrite, cosicché si registra un ulteriore processo di solubilizzazione delle fasi metastabili e di precipitazione di quella stabile. Il processo continua fino alla completa trasformazione del gesso legante nel solfato di calcio budrato, il cui precipitato si presenta sotto forma di cristalli aghiformi. Questi, intrecciandosi tra loro, provocano prima la presa e successivamente l'indurimento della pasta.
I manufatti ottenuti per idratazione del gesso legante non possono essere utilizzati all'aperto, né in ambienti chiusi ma umidi e tanto meno sott'acqua. Infatti, a causa della relativa solubilità del gesso budrato in acqua (circa 2 g/l a 20 °C), il manufatto in gesso può subire un certo dilavamento. Si può in parte ovviare all'azione dilavante esercicitata dall'acqua, trattando il manufatto in superficie con rivestimenti impermeabilizzanti. Tuttavia, a causa delle variazioni termiche o delle sollecitazioni meccaniche che provocano il distacco delle parti superficiali, l'azione protettiva del rivestimento non può essere garantita a lungo. Oltre all'azione dilavante dell'acqua, esiste il problema dell'umidità ambientale, al cui variare i manufatti in gesso subiscono contrazioni e dilatazioni al di sopra dei limiti tollerabili per i materiali da costruzione.
A parte le limitazioni sopra menzionate, il gesso legante può essere ottimamente impiegato come materiale da costruzione per ambienti interni in virtù delle sue particolari caratteristiche: capacità di isolamento termico e acustico, resistenza al fuoco, levigatezza delle superfici finite e leggerezza.
3. Calce.
La calce (v. Boyton, 1966; v. Collepardi, 1977) è il prodotto della cottura del carbonato di calcio, come è indicato nella reazione
CaCO3 → CaO + CO2. (3)
Per poter essere impiegata come legante, la calce viene trasformata in idrossido di calcio secondo lo schema della reazione
CaO + H2O → Ca(OH)2. (4)
L'idrossido di calcio mescolato con acqua dà luogo a una massa plastica capace di indurire per reazione con il diossido di carbonio dell'aria:
Ca(OH)2 + CO2 → CaCO3 + H2O. (5)
Durante la produzione e l'indurimento della calce avviene una reazione, rispettivamente di decarbonatazione e di carbonatazione. Analogamente a quanto si verifica per il gesso, l'indurimento del legante, l'idrossido di calcio, provoca la formazione di un prodotto, il carbonato di calcio, che è anche la materia prima per produrre il legante stesso.
La produzione della calce avviene per decomposizione termica del calcare che nel procedimento industriale alimenta il forno verticale dall'alto. L'ossido di calcio si raccoglie in basso, mentre il diossido di carbonio viene eliminato insieme ai gas della combustione che attraversano dal basso verso l'alto il forno in controcorrente rispetto al materiale in cottura. Alla temperatura di 900-1.100 °C la pressione del diossido di carbonio supera il valore di 1 atm e pertanto a questa temperatura la decomposizione del carbonato può avvenire in un tempo relativamente breve.
Il prodotto ottenuto alla fine del processo di cottura è commercialmente noto con il nome di calce viva o calce in zolle. Esso viene trasformato in calce idrata, detta anche calce spenta, attraverso l'operazione di spegnimento o estinzione, che consiste nel trasformare l'ossido nel corrispondente idrossido, secondo lo schema della reazione (4). A seconda che la quantità d'acqua impiegata nello spegnimento della calce viva sia eguale o di poco superiore a quella stechiometrica, oppure in forte eccesso rispetto a quest'ultima, si ottiene, rispettivamente, la calce idrata in polvere o il cosiddetto grassello. La reazione di trasformazione dell'ossido in idrossido è fortemente esotermica (15,3 kcal per mole) e il calore sviluppato può far raggiungere alla massa la temperatura di 100 °C e quindi far bollire l'acqua aggiunta per l'idratazione; da ciò deriva la denominazione di ‛viva' per la calce costituita da ossido di calcio, e di ‛spenta' per quella trasformata in idrossido.
La calce idratata in polvere viene prodotta in un idratatore dove l'acqua viene spruzzata su un letto di calce precedentemente frantumata e ridotta a una pezzatura di 0,5-1 cm. La quantità di acqua impiegata per l'idratazione è leggermente superiore a quella stechiometrica della reazione (4) per tener conto dell'evaporazione di una parte di essa in conseguenza del forte sviluppo di calore. A causa del riscaldamento, e soprattutto dell'aumento di volume che si verifica nella trasformazione dell'ossido in idrossido di calcio, i granuli di calce si disintegrano producendo una polvere finissima. Affinché l'idratazione possa avvenire in un tempo relativamente breve, è necessario che la calce viva sia porosa, per consentire una rapida penetrazione dell'acqua all'interno dei granuli. La porosità della calce è strettamente legata alla temperatura di cottura, nel senso che quanto più elevata è stata la temperatura nel forno, tanto minore risulta la porosità della calce a causa della sinterizzazione dell'ossido, consistente in un processo di densificazione del materiale. D'altra parte, la trasformazione dell'ossido nell'idrossido deve essere totalmente realizzata durante il processo di estinzione, per evitare l'inconveniente della formazione dei cosiddetti bottaccioli o calcinaroli. Questi sono costituiti da granuli di calce densificata per un eccesso di cottura (calce ‛stracotta' o ‛cotta a morte'), che si trasforma più lentamente in idrossido di calcio proprio a causa della minore porosità dell'ossido. Pertanto, se, durante il processo di estinzione, nei granuli di calce densificata non si completa la trasformazione da ossido in idrossido, la reazione di idratazione continuerà durante l'impiego del legante, e cioè in fase di indurimento. Conseguentemente possono verificarsi il sollevamento e il distacco della malta indurita sovrastante il bottacciolo, a causa dell'aumento di volume che accompagna la trasformazione dell'ossido nell'idrossido di calcio.
Se il calcare impiegato è di tipo dolomitico, e quindi molto ricco in carbonato di magnesio, MgCO3, il prodotto della cottura contiene, accanto all'idrossido di calcio, quantità rilevanti di ossido di magnesio, la cui trasformazione in idrossido è relativamente lenta. In questi casi il processo di estinzione viene accelerato realizzandolo a temperature più alte, preferibilmente in autoclave, per poter arrivare, in presenza di acqua, a 130-200 °C.
Per l'utilizzazione come legante, la calce in zolle (CaO) può anche essere mescolata con un eccesso di acqua, così da ottenere una pasta di consistenza plastica, untuosa al tatto, facilmente modellabile: il grassello. Questo può essere ottenuto anche mescolando acqua con calce idrata, anziché con calce in zolle come avveniva in passato. In questo caso, ovviamente, non si può parlare di estinzione, ma piuttosto di un mescolamento di un solido già idratato, Ca(OH)2, con acqua. Il vantaggio di questo processo deriva dalla maggiore semplicità e sicurezza dell'operazione, anche se il grassello così prodotto è di qualità inferiore (minore plasticità) rispetto a quello ottenuto spegnendo calce viva.
Il grassello, lasciato all'aria in strati sottili, indurisce progressivamente per la combinazione del Ca(OH)2 con il CO2 dell'aria, secondo lo schema della reazione (5). Il carbonato di calcio che si forma precipita nella fase liquida sotto forma di particelle che aderiscono tra loro o ai granuli di sabbia eventualmente presenti nell'impasto.
In genere la calce viene utilizzata sotto forma di malte, cioè di miscele contenenti calce idrata o grassello, sabbia e acqua. La funzione della sabbia è innanzitutto quella di ridurre il ritiro della malta indurita provocato dall'evaporazione dell'acqua e dalla contrazione di volume che si verifica nella reazione di carbonatazione. I granuli di sabbia, infatti, contrastandosi vicendevolmente, rendono più stabile dimensionalmente la malta indurita. Inoltre l'aggiunta di sabbia serve anche a disporre la pasta legante sotto forma di strati sottili avvolgenti i granuli di inerte. In queste condizioni, a causa della maggiore superficie esposta all'azione del CO2, l'indurimento della malta avviene più celermente. In ogni caso le resistenze meccaniche a compressione delle malte di calce sono piuttosto modeste, dell'ordine di una decina di kg/cm2, e quindi nettamente inferiori a quelle raggiungibili, per esempio, con i cementi.
In generale, la calce è impiegata, sotto forma di malta, per il collegamento di pietre, di mattoni, di elementi prefabbricati, o, sotto forma di pasta, per lavori di rasatura e finitura degli intonachi.
4. Calce idraulica.
La calce idraulica (v. Collepardi, 1977) possiede discrete proprietà idrauliche, nel senso che una volta indurita essa può sopportare il contatto con l'acqua. Infatti, accanto all'idrossido di calcio, nella calce idraulica sono presenti anche altri prodotti che sono in grado di indurire e di resistere all'azione dilavante dell'acqua.
Esistono diversi tipi di calci idrauliche che si differenziano per la diversa natura delle materie prime.
Un tempo la calce idraulica veniva prodotta esclusivamente sottoponendo a estinzione il prodotto della cottura dei calcari argillosi naturali (calce idraulica naturale). A causa della presenza di significative quantità di silice e di allumina, contenute nelle argille, si formano, durante la cottura a temperatura non superiore ai 900 °C, il silicato bicalcico, 2CaO • SiO2 (v. cap. 5), e l'alluminato monocalcico, CaO • Al2O3 (v. cap. 6, È h), prodotti capaci di indurire per reazione con l'acqua dando luogo a composti insolubili nell'acqua stessa.
La calce idraulica può anche essere ottenuta sottoponendo a estinzione il prodotto della cottura di una miscela artificiale di argilla e calcare (calce idraulica artificiale). Un altro tipo di calce idraulica viene preparato mescolando semplicemente calce idrata con pozzolana (v. cap. 6, È d) o con loppa d'altoforno, sottoprodotto della produzione della ghisa (v. cap. 6 È e).
5. Cemento Portland.
Il cemento (v. Bogue, 1947; v. Taylor, 1964; v. Lea, 1970; v. Collepardi, 1980) è un legante idraulico, in quanto, mescolato con acqua, dà luogo a una pasta che indurisce e rimane chimicamente stabile anche se conservata sott'acqua. Il cemento Portland, il più importante dei leganti idraulici, si ottiene per macinazione del clinker con piccole aggiunte di gesso budrato o di anidrite (v. cap. 2) ed eventualmente di altri materiali, quali pozzolana, sabbia, calcare, loppa d'altoforno, ecc. La percentuale dei materiali aggiunti al clinker deve essere contenuta entro certi limiti. In particolare, se il tenore di pozzolana o di loppa d'altoforno raggiunge certi valori, il cemento è denominato rispettivamente pozzolanico o d'altoforno.
Il clinker, che è il costituente più importante del cemento Portland (85-95%), è il prodotto della cottura, a circa 1.300-1.500 °C, di una miscela di argilla, calcare, sabbia, ceneri di pirite, ecc. In seguito alle reazioni che avvengono durante la cottura, nel clinker si forma una miscela di silicato tricalcico (3CaO • SiO2), β-silicato bicalcico (β-2CaO • SiO2), alluminato tricalcico (3CaO • Al2O3), e una soluzione solida ternaria di composizione compresa tra gli estremi 6CaO • 2Al2O3 • Fe2O3 e 6CaO • Al2O3 • 2Fe2O3, molto spesso indicata con 4CaO • Fe2O3 • Al2O3. Come componenti minori possono essere presenti il 12CaO • 7Al2O3, il 2CaO • Fe2O3, l'ossido di calcio, l'ossido di magnesio, oltre agli alcali, ai fosfati, ai fioruri e ai solfati, che formano normalmente soluzioni solide con i silicati e gli alluminati di calcio.
Nella chimica del cemento, per brevità, si usa scrivere C, A, F ed S rispettivamente al posto di CaO, Al2O3, Fe2O3, SiO2. In tal modo i costituenti mineralogici del clinker, e cioè 3CaO • SiO2, β-2CaO • SiO2, 3CaO • Al2O3 e 4CaO • Al2O3 • Fe2O3, possono essere indicati rispettivamente con i simboli C3S, β-C2S, C3A e C4AF.
Mescolando separatamente con acqua ciascuno di questi costituenti mineralogici, si ottengono delle paste che induriscono gradualmente secondo l'andamento illustrato nella fig. 2. Si può osservare che, dei quattro composti, solo il C3S e il β-C2S sono in grado di produrre, per reazione con l'acqua, paste dotate di apprezzabile resistenza meccanica. Con il C3S in particolare, a causa di una più alta velocità di idratazione, è possibile raggiungere resistenze meccaniche elevate più rapidamente che con il β-C2S.
a) Produzione del cemento Portland.
La produzione del cemento Portland consiste nel macinare e mescolare le materie prime, nel cuocere la miscela fino a ottenere una fusione parziale (‛clinkerizzare'), cioè fino a ottenere dal 20 al 30% di fase liquida, nel raffreddare piuttosto rapidamente il prodotto della cottura (clinker) e nel macinare il clinker in presenza di gesso biidrato o di anidrite. I processi di fabbricazione si differenziano sostanzialmente nel metodo di mescolamento delle materie prime. Nel cosiddetto processo a secco (v. fig. 3), più diffuso, le materie prime sono frantumate con frantoi rotativi o a mascelle, dosate, mescolate, essiccate, macinate in mulini a sfere e omogeneizzate in sili per insuffiamento di aria; la polvere così ottenuta (denominata ‛farina') viene, infine, inumidita per formare le graniglie che alimentano il forno. Quest'ultima operazione ha lo scopo di evitare che la farina entri nel forno sotto forma di polvere, la quale potrebbe in buona parte essere trascinata fuori dalla controcorrente dei gas di combustione. Negli impianti più moderni la perdita di polvere nella corrente dei gas di scarico viene praticamente annullata con precipitatori elettrostatici, filtri a secco, separatori a cicloni, torri di lavaggio, e la polvere viene rimessa nel ciclo di produzione. Inoltre, al fine di minimizzare il consumo di combustibile, la farina, prima di essere introdotta nel forno, è trattata in un preriscaldatore, dove riceve il calore da parte dei gas ancora caldi (circa 800 °C) provenienti dalla combustione del forno.
Il processo a umido (v. fig. 4) viene, invece, preferibilmente impiegato se le materie prime argillose sono già piuttosto umide (contenuto di acqua superiore al 20-25%); in tal caso le argille sono prima spappolate con altra acqua in apposite vasche e, quindi, mescolate con il calcare e le altre materie prime frantumate a parte, per essere infine macinate a umido in mulini a sfere. La melma così ottenuta, che contiene dal 30 al 40% di acqua, può essere filtrata in filtropresse prima di essere parzialmente essiccata, granulata e inviata al forno per la cottura.
La scelta del processo a secco o a umido dipende fondamentalmente dal costo del combustibile utilizzato nella cottura del clinker e dalla percentuale di umidità presente nell'argilla. Nel processo a umido, per produrre una tonnellata di clinker sono necessarie circa 2,3 tonnellate di melma con 30% di umidità; tenuto conto dell'umidità presente (0,7 tonnellate di acqua per tonnellata di clinker), per la sola evaporazione dell'acqua si richiedono 2.930 MJ (700.000 kcal) per tonnellata di clinker. È evidente che, con l'avvento della crisi energetica degli anni settanta, non solo si è scelto il processo a secco, ma si è anche reso necessario trasformare gli stabilimenti produttivi già esistenti, che utilizzavano il processo a umido, in cementerie con processo a secco. Questa scelta, provocata da ragioni economiche, è stata resa tecnicamente possibile dalla messa a punto di più progrediti sistemi di omogeneizzazione delle materie prime per la produzione della farina con il processo a secco, rispetto al quale la ‛via umida' presentava un tempo il vantaggio di una più efficiente miscelazione e omogeneizzazione delle materie prime per produrre la melma.
La necessità di convertire il processo a umido in quello a secco, unitamente all'esigenza di aumentare la produttività degli impianti, ha comportato anche la modifica dei forni di cottura. Infatti, a causa della maggiore quantità di acqua da evaporare, i forni della via umida presentavano un limite nella capacità produttiva, rispetto a quelli della via secca, per il maggior carico termico - a parità di produzione - al quale veniva sottoposta la zona di cottura. A seguito di questa trasformazione, un impianto con via umida capace di produrre 750 tonnellate di clinker al giorno, con un consumo energetico di 6.700 MJ (1.600.000 kcal) per tonnellata di clinker, può, dopo la conversione nel processo a secco, arrivare a produrre fino a 1.350 tonnellate di clinker al giorno, con un consumo energetico di appena 3.560 MJ (850.000 kcal) per tonnellata di clinker.
I forni sono di due tipi: rotante e verticale. Il primo, più diffuso (v. fig. 5), è costituito da un tubo leggermente inclinato, con pendenza del 3-5%, lungo fino a 200 m e con un diametro fino a 8 m, che ruota lentamente per fare avanzare la miscela da cuocere in controcorrente rispetto ai gas della combustione, a una velocità di qualche decina di metri all'ora. Il consumo di combustibile è tale che occorrono da 3.350 a 6.700 MJ (da 800.000 a 1.600.000 kcal) per tonnellata di clinker, rispettivamente con il processo a secco o a umido, mentre la produzione può variare da diverse centinaia a qualche migliaio di tonnellate al giorno.
I forni verticali, alti da 10 a 20 m e con diametro di 2-3 m, sono alimentati in alto da un granulato, costituito da una miscela di coke e materie prime, che viene preriscaldato dalla corrente ascendente dei gas di combustione. In basso, al di sotto di una griglia, è insufflata l'aria che si preriscalda a contatto con il clinker caldo. In genere il rendimento termico dei forni verticali è migliore (3.350-4.200 MJ, cioè 800.000-1.000.000 kcal per tonnellata di clinker) di quello dei forni rotanti, e tuttavia essi sono stati pressoché abbandonati per la modesta potenzialità (da 10 a 100 tonnellate al giorno) e per la disuniforme distribuzione della temperatura nella sezione del forno, dovuta a possibili cammini preferenziali della corrente gassosa.
Indipendentemente dal tipo di forno, durante la cottura si verificano una serie di reazioni chimiche che portano alla trasformazione delle iniziali materie prime in una miscela di C5S, β-C2S, C3A, C4AF.
Il clinker proveniente dal forno passa in un raffreddatore, che può essere a griglia, a satelliti, o a cilindro, dove viene raffreddato da una corrente d'aria fredda. Dopo il raffreddamento il clinker, unitamente a qualche percento di gesso biidrato o di anidrite, viene macinato in mulini a sfera (v. fig. 6). La polvere viene inviata a un separatore a cicloni, da dove la parte più fine esce per essere insaccata o insilata, mentre quella più grossa ritorna nel mulino per essere ulteriormente macinata. La presenza di additivi coadiuvanti della macinazione generalmente a base di trietanolammina, ligninsolfonato e glicole etilenico - é indispensabile per la produzione di cementi con finezza molto elevata, come quelli a rapido indurimento iniziale.
b) Idratazione del cemento Portland.
Nel clinker di cemento Portand sono presenti, in generale, dal 40 al 60% di C3S, dal 20 al 50% di β-C2S, e dal 20 al 25% di C3A + C4AF. Il C3S è il costituente mineralogico più importante del clinker, sia perché è presente in percentuale maggiore, sia perché più degli altri composti contribuisce alla resistenza meccanica del cemento idratato (v. fig. 2).
La reazione di idratazione del C3S (v. Kondo e Daimon, 1969; v. Collepardi e Massidda, 1971) può essere così schematizzata:
C3S + (3 − y + x) H → CySHx + (3 − y) CH, (6)
dove H sta per H2O, e quindi CH indica Ca(OH)2.
Sebbene il sistema H2O-C3S sia relativamente semplice, soprattutto in confronto a quello molto più complesso acqua-cemento, tuttavia la reazione di idratazione del C3S è una delle più complicate della chimica inorganica. Oltre alle difficoltà di interpretazione del meccanismo e della cinetica di reazione, persistono notevoli incertezze perfino sulla stechiometria e sull'esatta struttura del principale prodotto della reazione (CySHx). La singolarità di questa reazione consiste nel fatto che i suoi coefficienti stechiometrici e, quindi, anche la composizione dell'idrosilicato di calcio CySHx cambiano non solo con le condizioni sperimentali (temperatura, presenza di catalizzatori, ecc.), ma anche durante la reazione stessa. Il valore di x, e cioè il rapporto molare H/S dell'idrosilicato, si porta da 1,5 a 1,0 dall'inizio alla fine della reazione, mentre il valore di y, cioè il rapporto molare C/S,è inizialmente pari a 3, quindi uguale a quello del silicato anidro, e poi diminuisce progressivamente per portarsi a circa 1,5. L'idrosilicato di calcio si presenta sotto forma di particelle fibrose, lunghe qualche μm, che ricoprono i granuli di silicato anidro. All'analisi per diffrazione dei raggi X l'idrosilicato CySHx presenta tre bande diffuse, le cui posizioni corrispondono a quelle dei tre picchi del minerale C5S6H5, noto con il nome di tobermorite (v. fig. 7). Per questa ragione, e per il suo basso grado di cristallinità, il CySHx è spesso indicato con il nome di gel tobermoritico. Più recentemente è stato proposto il nome più vago e meno impegnativo di gel di silicato di calcio idrato.
Oltre alla composizione, anche altre caratteristiche del gel idrosilicatico, quali la morfologia, la porosità, l'area superficiale specifica, variano entro limiti piuttosto ampi, sia durante l'idratazione, sia al variare delle condizioni sperimentali della reazione.
Nell'idratazione del C3S si possono distinguere tre periodi, detti di induzione, di accelerazione e di decadimento. Durante il periodo di induzione, dopo una immediata reazione tra C3S e H2O, l'idratazione praticamente si arresta a causa della formazione di un CySHx (con y ≃ 3 e quindi di composizione molto vicina al silicato anidro) molto poco poroso: questo forma, infatti, una pellicola impermeabile intorno ai granuli di C3S e impedisce il proseguimento dell'idratazione.
Con il periodo di accelerazione ha inizio la trasformazione di questo primo idrosilicato in un altro meno ricco di calcio (y = 2 ÷ 1,5) e più poroso. I primi nuclei del secondo idrosilicato si formano per l'azione dissolvente dell'acqua a contatto con la pellicola esterna e per diffusione degli ioni Ca2+ e OH- dalla pellicola stessa verso la fase liquida, che per questo diviene prima satura e quindi soprassatura in idrossido di calcio. Poiché la trasformazione del ‛primo' nel ‛secondo' idrosilicato è accompagnata da un forte aumento di porosità, la velocità di idratazione aumenta progressivamente, nonostante l'aumento di spessore della pellicola attraverso cui avviene la diffusione delle molecole d'acqua. Quando tutta la pellicola è costituita dal ‛secondo' idrosilicato, la velocità di idratazione è massima. Da questo momento in poi ha inizio il periodo di decadimento e la velocità di reazione diminuisce nel tempo perché, a parità di porosità, lo spessore della pellicola, e quindi la resistenza offerta alla diffusione dell'acqua, va progressivamente aumentando. In effetti, poiché la porosità della pellicola diminuisce per un fenomeno di addensamento del gel idrosilicatico che ‛invecchia', la velocità di idratazione del C3S durante il periodo di decadimento risulta ulteriormente diminuita.
L'idratazione del β-C2S porta praticamente agli stessi prodotti della reazione (6), che si ottengono per idratazione del C3S:
C2S + (2 − y + x) H → CySHx + (2 − y) CH. (7)
La velocità della reazione (7), però, è notevolmente inferiore a quella della (6), com'è illustrato nella fig. 8, dove sono riportate le percentuali dei singoli costituenti idratati in funzione del tempo. La minore velocità di idratazione del β-C2S è probabilmente da ascrivere alla più compatta struttura cristallina del β-C2S rispetto a quella del C3S, cosicché l'ingresso delle molecole d'acqua dentro il reticolo cristallino del β-C2S procede più lentamente.
Le reazioni di idratazione del C3A e del C4AF avvengono con velocità notevolmente superiori a quelle del C3S e del β-C2S (v. fig. 8), ma, poiché i loro prodotti di idratazione sono dotati di scarsa resistenza meccanica (v. fig. 2), gli alluminati contribuiscono relativamente poco allo sviluppo della resistenza meccanica del cemento Portland idratato.
Sia la stechiometria sia la velocità di idratazione del C3A dipendono dalla temperatura e dalla presenza di altri composti, quali il CH e il solfato di calcio, sempre presenti durante l'idratazione del cemento Portland. L'idrossido di calcio si forma, infatti, per idratazione del C3S, mentre il solfato viene aggiunto al clinker durante la macinazione (v. È a).
A temperature inferiori a 15 °C, il C3A forma una miscela di alluminati idrati di calcio esagonali:
2C3A + 27H → C2A • H8 + C4A • H19. (8)
In presenza di CH si forma solo l'alluminato idrato più basico
C3A + 18H + CH → C4A • H19, (9)
con una velocità di reazione più bassa di quella della (8).
A temperature superiori a 15 °C gli alluminati idrati esagonali, C2A • H8 e C4A • H19, sono metastabili e pertanto tendono a trasformarsi nell'idrato cubico, C3A • H6, stabile. A temperature superiori a 50 °C l'idratazione del C3A avviene con formazione diretta della fase stabile del C3A • H6:
C3A + 6H → C3A • H6. (10)
In presenza di gesso biidrato (o di anidrite) il prodotto dell'idratazione del C3A è un sale complesso (C3A • 3CaSO4 • H32), denominato ettringite:
C3A + 3(CaSO4 • 2H2O) + 26H → C3A • 3CaSO4 • H32. (11)
La velocità della reazione (11), che è già inferiore a quella delle (8), (9) e (10), si riduce ulteriormente se la reazione avviene, come si verifica durante l'idratazione del cemento, in presenza di calce. Il rallentamento della velocità di idratazione del C3A, con l'aggiunta di gesso biidrato, è di grande importanza pratica, perché un'idratazione del C3A troppo veloce comporterebbe un troppo rapido aumento della consistenza della pasta di cemento (presa rapida), con la conseguenza che la messa in opera del calcestruzzo risulterebbe impedita. La diminuzione nella velocità di idratazione del C3A in presenza di gesso è generalmente attribuita alla formazione di una pellicola di ettringite che ricopre i granuli anidri di C3A e ne rallenta la velocità di idratazione. L'ulteriore rallentamento della reazione (11) provocato dalla presenza del CH può essere ascritto all'influenza della calce sulla morfologia e sulle dimensioni delle particelle di ettringite che formano la pellicola: in sostanza in presenza di CH si formerebbero cristalli di ettringite più piccoli e la pellicola risulterebbe meno permeabile alle molecole d'acqua.
L'aggiunta di gesso (4-6%) al clinker è stechiometricamente insufficiente a convertire tutto il C3A in ettringite secondo la reazione (11). Pertanto, quando tutto il gesso è consumato, il C3A residuo reagisce con l'ettringite secondo il seguente schema:
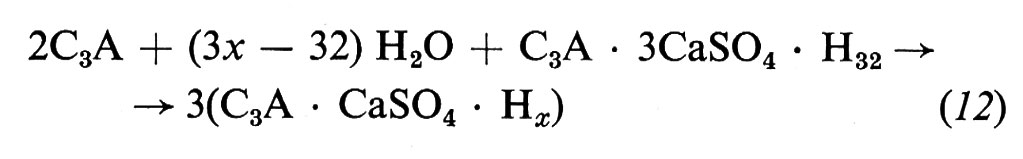
trasformandosi in un altro sale, detto comunemente monosolfoalluminato, di composizione C3A • CaSO4 • Hx, dove x dipende dall'umidità relativa e dalla temperatura. A temperatura ambiente, e con umidità relativa del 50-70%, x è uguale a 12.
Quando anche l'ettringite è tutta consumata per produrre il monosolfato, il C3A residuo reagisce con l'acqua in presenza di calce, secondo la reazione (9), per trasformarsi in C4A • H19. Anche per questo sale, come per il monosolfato, il numero delle molecole d'acqua di cristallizzazione dipende dall'umidità relativa: a temperatura ambiente e con umidità relativa del 20-80% il C4A • H19 perde parte dell'acqua di cristallizzazione e diventa C4A • H13, che può essere anche scritto CaA • Ca(OH)2 • H12, isomorfo al C3A • CaSO4 • H12.
Il monosolfoalluminato, C3A • CaSO4 • H12, e l'alluminato idrato esagonale, C3A • Ca(OH)2 • H12, possono formare soluzioni solide per sostituzione dello ione SO42- con due ioni OH-. Sembra che solo metà del solfato sia sostituibile, cosicché il termine estremo della soluzione solida è C3A • 1/2CaSO4 • 1/2Ca(OH)2 • H12. Ciò significa che, per contenuti di gesso e C3A tali che il rapporto in moli CaSO4/C3A è inferiore a 0,5, il prodotto finale dell'idratazione sarà costituito dal C4A • H13 accanto al C3A • 1/2CaSO4 • 1/2Ca(OH)2 • H12. In altre parole, a seconda del valore del rapporto molare CaSO4/C3A, il prodotto idratato può contenere una o più fasi secondo lo schema riportato nella tab. I.

Rispetto agli altri componenti del cemento, la fase ferrica, spesso indicata con il simbolo C4AF, presenta maggiori difficoltà quando si tratta di esaminarne i prodotti di idratazione. Come si è già detto, il C4AF non è un vero e proprio composto chimico, ma piuttosto una soluzione solida ternaria di composizione variabile entro certi limiti e spesso non ben definita. Inoltre i composti che si formano per idratazione della fase ferrica, C3(A, F) • H6, C4(A, F) • Hx, C3(A, F) • 3CaSO4 • H32, C3(A, F) • CaSO4 • H12 ecc., sono difficilmente distinguibili, con le normali tecniche di indagine (diffrazione dei raggi X, analisi termica, microscopia ottica ed elettronica), da quelli ottenibili per idratazione del C3A.
In generale i composti che si formano per idratazione della fase ferrica sono simili a quelli ottenuti per idratazione del C3A e presentano un rapporto molare A/F più grande di quello del prodotto anidro, cosicché una parte dell'ossido ferrico della soluzione solida ternaria si ritrova, alla fine dell'idratazione, sotto forma di idrossido ferrico.
Sulla base delle informazioni derivanti dallo studio dell'idratazione dei quattro costituenti mineralogici del clinker presi singolarmente, si può tentare di spiegare il più complesso meccanismo dell'idratazione del cemento Portland. Subito dopo aver mescolato l'acqua con il cemento, nella fase acquosa si sciolgono gli ioni Ca2+, SO42-, Na+, K+, oltre a quantità trascurabili di silicati alluminati. In queste condizioni il C3A, e in minor misura il C4AF, si idratano rapidamente formando cristalli esagonali di C2A • H8, C4A • H19 e C4(A, F) • Hx. Dopo alcuni minuti la fase acquosa si satura in solfato di calcio per dissoluzione del gesso, dell'anidrite e del semiidrato (v. È a), cosicché si forma una pellicola di ettringite e di C3(A, F) 3CaSO4 • H32 che, rivestendo il C3A e il C4AF, ne rallenta l'idratazione. Durante questo periodo, detto dormiente o di induzione, anche il C3S risulta ricoperto dalla pellicola del ‛primo' idrosilicato CySHx ricco in calcio e poco poroso, mentre è del tutto trascurabile l'idratazione del β-C2S. A causa della crescente concentrazione di calce nella fase acquosa le idratazioni del C3A e del C4AF risultano sempre più rallentate. Dopo il periodo di induzione, che dura in genere qualche ora, l'idratazione procede con velocità crescente, che raggiunge un massimo intorno alle 5-10 ore, a seconda del tipo di cemento (periodo di accelerazione). L'aumento della velocità di idratazione del cemento è da mettere in relazione con due fenomeni più o meno concomitanti: il primo è quello, già descritto, della graduale trasformazione del ‛primo' nel ‛secondo' idrosilicato, più poroso e più permeabile all'acqua, con conseguente accelerazione dell'idratazione del C3S; il secondo fenomeno consiste nella rottura della pellicola di ettringite (probabilmente causata dalla pressione osmotica o dalla pressione di cristallizzazione) e quindi nella ripresa dell'idratazione del C3A e del C4AF. Nell'ultimo periodo (decadimento), quando i granuli di cemento risultano ricoperti dai prodotti di idratazione, la velocità di reazione è governata dal processo di diffusione dell'acqua attraverso la pellicola dei prodotti idratati. Essa, pertanto, diminuisce progressivamente non solo per l'aumento di spessore della pellicola, ma anche per l'‛invecchiamento' del gel idrosilicatico, che consiste in una diminuzione di porosità della pellicola stessa.
c) Calore di idratazione del cemento Portland.
La reazione tra l'acqua e il cemento Portland è di tipo esotermico. Il processo di idratazione può essere esaminato registrando la velocità di sviluppo del calore di idratazione in funzione del tempo (v. fig. 9).
I quattro costituenti mineralogici del cemento Portland (C3S, β-C2S, C2A e C4AF) si differenziano non solo per la velocità con cui il calore si libera, ma anche per il calore di idratazione complessivamente sviluppato. Questo, misurato in chilojoules per chilogrammo di prodotto completamente idrato, è circa 500 (120 kcal) per il C2S, 250 (60 kcal) per il β-C2S, 880 (210 kcal) per il C3A e 420 (100 kcal) per il C4AF, mentre quello del cemento Portland varia tra 380 e 500 a seconda della percentuale dei vari costituenti mineralogici.
Nelle strutture in calcestruzzo di grandi dimensioni, come per esempio le dighe, dove il rapporto volume/superficie è relativamente alto, e quindi poco favorevole a un rapido smaltimento del calore verso l'ambiente, è consigliabile impiegare cementi con basso calore di idratazione (ricchi quindi in β-C2S e C4AF), per evitare che l'insorgere di gradienti termici provochi variazioni dimensionali differenziali e quindi fessurazioni, talvolta di notevole entità. Da questo punto di vista, oltre al calore di idratazione complessivamente sviluppato, interessa anche la velocità con cui il calore stesso viene liberato. Fortunatamente nei cementi ricchi in β-C2S e C4AF il calore di idratazione, oltre a essere più basso, si sviluppa anche con velocità minore a causa della più bassa velocità di idratazione di questi composti in confronto rispettivamente a quella del C3S e del C3A (v. fig. 8).
d) Presa del cemento Portland.
Subito dopo il mescolamento del cemento con l'acqua, la pasta si presenta come una massa plastica facilmente deformabile. Di solito, dopo qualche ora, essa diviene più consistente e comincia a perdere la sua deformabilità, cioè a far presa.
In modo molto schematico si può pensare di suddividere formalmente l'idratazione del cemento in tre stadi (v. fig. 10): il primo (A) ha inizio subito dopo l'impasto e ha una durata di qualche decina di minuti, il secondo (B) dura qualche ora e corrisponde alla presa, il terzo (C) inizia dopo quasi 24 ore e riguarda l'indurimento.
Subito dopo il mescolamento, durante il primo stadio (v. fig. 10A), la pasta di cemento è costituita da un insieme di granuli di clinker e di gesso, più o meno tondeggianti e di diametro variabile tra qualche μm e qualche decina di μm, dispersi nell'acqua. La mobilità delle particelle solide, e quindi la lavorabilità della pasta, è tanto maggiore quanto più alto è il contenuto d'acqua. A causa dei diversi pesi specifici dei prodotti anidri e di quelli idratati ottenuti dalla reazione di idratazione, per ogni unità di volume di cemento anidro che scompare si formano mediamente circa 2,1 unità di volume di prodotto idratato. Ciò significa che, giorni man mano che l'idratazione procede, una parte dello spazio originariamente occupato dall'acqua viene a essere riempito dai prodotti della reazione tra l'acqua e il cemento. Questo determina durante la presa una minore mobilità delle particelle solide e quindi una maggiore consistenza della pasta. L'effetto è tuttavia quasi irrilevante per il basso grado di idratazione del cemento durante le prime ore. Inoltre i prodotti idratati sono estremamente più fini delle particelle di cemento e si presentano per lo più sotto forma di lamine e di fibre. Sia il maggior sviluppo di area superficiale sia la particolare morfologia dei prodotti idratati contribuiscono a una diminuzione della mobilità delle particelle solide e quindi a un aumento della consistenza della pasta cementizia.
La formazione di alluminati idrati esagonali sotto forma di prodotti laminari, o l'accrescimento dei cristalli di ettringite, gli uni e gli altri capaci di formare dei ponti tra le particelle idratate, possono contribuire in modo determinante all'ulteriore perdita di plasticità dell'impasto e quindi alla presa del cemento.
Tutti questi fenomeni, principalmente dovuti all'idratazione degli alluminati e della fase ferrica, determinano il fenomeno della presa già dopo qualche ora dall'impasto del cemento con l'acqua (v. fig. 10B). Successivamente, soprattutto per l'idratazione dei silicati, si forma un prodotto che va a riempire ulteriormente il volume inizialmente occupato dall'acqua. La natura prevalentemente fibrosa del prodotto idratato determina una struttura di fibre intrecciate che, insieme alle forze di adesione e a quelle di van der Waals, è responsabile dei legami che si stabiliscono tra le particelle solide e quindi della fine della presa della pasta cementizia (v. fig. 10C).
e) Indurimento e resistenza meccanica della pasta di cemento Portland.
Dopo la fine della presa la pasta di cemento Portland diventa sempre più rigida e dura, fino ad assumere l'aspetto di una pietra: questo processo prende il nome di indurimento; la sua misura viene effettuata sollecitando a compressione o a flessione il materiale indurito; il valore della sollecitazione che ne provoca la rottura prende il nome di resistenza meccanica ‛a compressione' o ‛a flessione', rispettivamente. La resistenza meccanica dei manufatti in cemento, e del calcestruzzo in particolare, è indubbiamente la loro proprietà più importante.
Sebbene da un punto di vista pratico si conosca ormai con sufficiente esattezza l'influenza dei vari parametri ‛tecnologici' (lavorabilità, compattazione, rapporto acqua/cemento, tipo e dosaggio di cemento, tipo di inerte, tipo di additivo, durata della stagionatura, temperatura e umidità relativa dell'ambiente) sulla resistenza del calcestruzzo, si è ancora molto lontani dal conoscere, da un punto di vista scientifico, il contributo di tutti i fattori che determinano la struttura della pasta di cemento a livello microscopico e atomico. Ciò dipende dal fatto che questi fattori sono molto numerosi e non tutti possono essere studiati singolarmente. I più importanti tra essi sono: il legame che tiene unite le particelle di cemento idratato, la superficie specifica, la porosità capillare, la forma delle particelle di cemento idratato, l'umidità presente nella pasta cementizia.
Il prodotto della reazione tra l'acqua e il cemento Portland è costituito da una matrice di gel idrosilicatico (CySHx di natura prevalentemente colloidale, nella quale sono dispersi cristalli di idrossido di calcio, di solfoalluminati idrati, ecc., che nell'insieme prende il nome di ‛gel di cemento'.
La pasta cementizia, nella quale si forma il gel di cemento, presenta una struttura microporosa cui si deve un grande sviluppo superficiale. Si distinguono solitamente due categorie di pori (v. Powers, 1960): i pori capillari, situati tra le particelle del gel, sono osservabili con il microscopio elettronico e presentano una dimensione compresa tra qualche centinaio di Å e qualche μm; i pori del gel, presenti dentro il gel di cemento, sono di dimensione variabile da un minimo di qualche Å a un massimo di qualche centinaio di Å (con il valore più frequente situato a 15 Å). Ciò significa che più della metà delle superfici solide sono così vicine da risentire fortemente delle forze attrattive dovute ai legami di van der Waals. Powers (v., 1968) ha calcolato, per esempio, che in un sistema poroso essiccato due superfici solide distanti 6 Å sono attratte da una forza di van der Waals pari a circa 275 MPa; se la distanza è di 8 Å, la forza di attrazione è di circa 120 MPa; a 16 Å è di 15 MPa, a 30 Å è di circa 2 MPa e diviene trascurabile oltre i 100 Å.
Si è molto discusso sulla natura dei legami chimici che tengono unite le particelle di cemento idratato. E stato suggerito (v. Brunauer, 1962; v. Chatterji e Jeffery, 1967; v. Lea, 1970) che la grande differenza tra la resistenza a trazione e quella a compressione, che sono approssimativamente in rapporto di 1: 10, dipenda dalle diverse forze dei legami chimici in gioco. È noto che le forze dei legami di van der Waals, dei legami a idrogeno e dei legami di valenza sono all'incirca in rapporto 1 : 10 : 100. Lea (v., 1970) ha proposto che nella rottura a trazione si debbano vincere le forze di van der Waals, mentre nella rottura a compressione siano coinvolte le forze di legami di valenza; in tal caso, però, il rapporto tra la resistenza a trazione e quella a compressione dovrebbe essere all'incirca 1: 100. Questo disaccordo potrebbe però essere spiegato con il fatto che i legami di valenza, o legami chimici ‛primari', non interesserebbero estensivamente tutta la superficie di contatto tra le particelle.
Una teoria alternativa sulla natura dei legami interparticellari esistenti nella pasta di cemento è stata proposta da Feldmann e Sereda (v., 1968 e 1969). Questi autori postulano che il legame che tiene unite le varie particelle nasca dal contatto solido-solido delle particelle stesse (v. fig. 11). Questo particolare legame differisce dal vero e proprio legame chimico primario, perché nella zona di contatto non esisterebbe una disposizione ordinata di atomi come potrebbe verificarsi, per esempio, durante un processo di ricristallilizzazione. D'altra parte, Feldmann e Sereda escludono che per il legame solido-solido sia essenziale la presenza di molecole di acqua adsorbita tra le particelle, come si verificherebbe, invece, se il legame fosse di tipo ‛secondario', qual è quello dovuto alle forze di van der Waals.
Alcuni ricercatori (v. Wieker, 1974; v. Collepardi e altri, 1971) hanno messo in evidenza la formazione di legami silossanici (O-Si-O) attraverso la policondensazione dei gruppi Si-OH secondo l'equazione seguente:
Si-OH + OH-Si → Si-O-Si + H2O. (13)
Sebbene sia difficile valutare quale sia il contributo apportato alla resistenza meccanica da questo tipo di legame, si può tuttavia ritenere che esso possa contribuire sostanzialmente solo se si ammette che il legame si formi per condensazione di gruppi Si-OH appartenenti a particelle diverse.
Wittmann (v., 1973) ha proposto il cosiddetto ‛modello di Monaco' per la struttura della pasta di cemento. Egli ha trovato che la forza attrattiva di van der Waals tra due superfici solide diminuisce con l'umidità relativa e con la distanza tra le superfici stesse, e ritiene che il legame che tiene unite le particelle di cemento idratato sia all'incirca per il 50% di tipo chimico e per il 50% dovuto alle forze di van der Waals.
Anche l'aspetto morfologico delle particelle di cemento idratato svolge un ruolo importante, sebbene anche in questo caso sia difficile valutare in termini quantitativi il contributo di questo parametro. Molti prodotti che conferiscono caratteristiche leganti si presentano prevalentemente sotto forma di fibre più o meno allungate. L'intreccio di queste fibre può contribuire, anche da un punto di vista semplicemente meccanico, alla resistenza della pasta di cemento.
Tutto ciò dimostra la difficoltà del problema di correlare la resistenza meccanica con la struttura di un sistema molto complesso come quello cementizio. Allo stato attuale delle conoscenze sul cemento idratato, il parametro del quale si può valutare meglio l'influenza sulla resistenza meccanica, a parità di tutte le altre condizioni, è forse la porosità capillare. Powers e Brownyard (v., 1948) hanno trovato che la resistenza meccanica a compressione σc aumenta con il rapporto gel/spazio (x) secondo l'equazione
σc = K xn, (14)
dove n è una costante compresa tra 2,6 e 3, e x è definito come il rapporto tra il volume occupato dal gel di cemento idratato (Vg), inclusi i pori del gel, e il volume totale a disposizione, cioè il volume del gel più il volume dei pori capillari (Vp):

Nella fig. 12 è schematicamente mostrato il bilanci o di volume per un impasto cementizio preparato mescolando 100 kg di cemento con 42 litri di acqua, cioè con un rapporto acqua/cemento, in peso, di 0,42, considerato mediamente sufficiente a idratare tutto il cemento e definito ‛corretto'.
Se ci si riferisce a un impasto ottenuto da 100 kg di cemento, Vg (in litri) è eguale a 67,90 α, essendo α la frazione di cemento idratato e 67,90 il volume in litri del gel ottenuto per l'idratazione di tutto il cemento.
Il volume (Vp) dei pori capillari, in litri per 100 kg di cemento, è la somma di tre contributi dati rispettivamente: a) dalla contrazione dovuta all'idratazione, 5,85 α, dove 5,85 è la contrazione in litri per 100 kg di cemento completamente idratato (α = 1); b) dall'acqua non combinata, 42 (1 − α), in un impasto con un rapporto acqua/cemento (a/c) ‛corretto' pari a 0,42; c) dall'acqua in eccesso (a/c − 0,42)100 rispetto al valore ‛corretto'. Pertanto si ottiene
Vp = 5,85α + 42(1 − α) + (a/c − 0,42)100. (16)
Sostituendo Vg = 67,90 α e l'equazione (16) nella (15) si ottiene

Inserendo la (17) nella (14) si ricava

La costante K rappresenta la resistenza meccanica, che è massima quando x = 1, quando cioè Vp = 0. Ciò si verifica quando la pasta di cemento è costituita da cemento idratato o cemento anidro e gli unici pori presenti sono quelli del gel. L'esatto valore di K, che secondo Powers si aggira sui 250 MPa, dipende dal tipo di cemento, o più precisamente dal tipo di gel che si può formare con un dato cemento. In altre parole, quando la porosità capillare è assente, la resistenza meccanica del gel dipende da tutti gli altri parametri, quali l'aspetto morfologico, la superficie specifica, il tipo di legame, ecc., che sono stati precedentemente discussi.
L'equazione (18) mette in evidenza che la resistenza meccanica (σc) aumenta all'aumentare del grado di idratazione (α) e al diminuire del rapporto acqua/cemento (a/c). Al fine di ottenere la massima resistenza meccanica il rapporto a/c deve essere il più basso possibile: anche se esso diviene così basso (per es. 0,30) che l'idratazione del cemento non può essere completata, si può egualmente raggiungere un'elevatissima resistenza meccanica. D'altra parte, la completa idratazione del cemento (α = 1) non può essere in pratica realizzata, anche con rapporti a/c molto maggiori di 0,42, a causa dell'impedimento frapposto dalla pellicola dei prodotti idratati che avvolge i granuli di cemento. Si calcola che, anche con rapporti a/c molto elevati - dell'ordine di 0,5-1, quali sovente si impiegano nella pratica - la percentuale di cemento idratato al massimo difficilmente supera l'80% anche dopo stagionature di diversi anni.
Il motivo per cui nella pratica calcestruzzi con rapporti a/c di 0,30 non vengono impiegati, se non in casi eccezionali, è legato al fatto che l'impasto fresco risulterebbe così asciutto da renderne impossibile la messa in opera. Se però esistono sistemi di vibrazione molto efficienti, o se la lavorabilità del calcestruzzo - grazie all'impiego di additivi fluidificanti o superfluidificanti (v. cap. 8, ÈÈ c e d) - è molto elevata e comunque tale da rendere possibile una completa compattazione anche con sistemi di vibrazione ordinari, nessuna preclusione dovrebbe esistere all'impiego di calcestruzzi con rapporti a/c molto bassi: in queste condizioni, infatti, l'incompleta idratazione del cemento - che d'altra parte si verificherà comunque, anche con rapporti a/c più elevati - non influenza negativamente nè la resistenza meccanica nè le altre proprietà del calcestruzzo.
6. Cementi speciali.
Sono cementi speciali (v. Taylor, 1964; v. Lea, 1970; v. Collepardi, 1980) tutti i cementi escluso il cemento Portland ordinario: essi comprendono, oltre ad alcuni cementi Portland di composizione particolare (bianco, colorato e ferrico), il cemento pozzolanico, quello d'altoforno, il cemento alluminoso, quello soprasolfatato e i cementi espansivi. Tutti questi cementi contengono quantità più o meno rilevanti di cemento Portland ordinario, a eccezione del cemento soprasolfatato, nel quale il cemento Portland è presente solo come componente minore, e del cemento alluminoso.
a) Cemento bianco.
Il cemento bianco non presenta la tipica colorazione grigia del cemento Portland, dovuta essenzialmente alla presenza di ossido ferrico e di tracce di altri ossidi, quali quelli di manganese, di cromo, ecc.
Il cemento bianco è impiegato, unitamente a inerti di colorazione chiara, per la confezione di calcestruzzi a vista, quando si desideri realizzare costruzioni di particolare interesse architettonico. La caratteristica essenziale di un cemento bianco è il cosiddetto indice di bianchezza, che rappresenta il rapporto tra il coefficiente di riflessione del cemento bianco e quello del solfato di bario, BaSO4, assunto come riferimento. L'indice di bianchezza di un cemento bianco, normalmente compreso tra 70 e 90, dipende dalla concentrazione dei cosiddetti ioni cromofori, tra i quali gli ioni dei metalli di transizione della prima serie, quali il ferro, il manganese, il cromo, il nichel e il cobalto.
b) Cementi colorati.
I cementi colorati sono ottenuti per colorazione del cemento bianco o del Portland ordinario. Nel primo caso è possibile ottenere tonalità più chiare e una gamma più vasta di colori; nel secondo caso la colorazione grigia del cemento Portland ordinario consente di ottenere solo colori scuri e limitati al rosso e al marrone. L'aggiunta di pigmenti colorati è molto più efficace se fatta in fase di macinazione del cemento, piuttosto che durante la miscelazione del calcestruzzo.
Sia il cemento bianco sia quello grigio possono essere colorati macinando il clinker e il gesso in presenza di pigmenti colorati. Il cemento bianco, inoltre, può essere colorato anche introducendo piccole quantità di ioni cromofori nella materia prima della cottura.
c) Cemento ferrico.
Il cemento ferrico ha un contenuto nullo o molto basso di C3A. Tutta l'allumina e l'ossido ferrico sono contenuti nella fase ferrica C4AF. Ne consegue un basso calore di idratazione (v. cap. 5, È c) e una buona resistenza all'attacco solfatico (v. cap. 10, È d) proprio in conseguenza del basso contenuto di alluminati.
d) Cemento pozzolanico.
Il cemento pozzolanico è ottenuto per macinazione di clinker di cemento Portland, di gesso e di pozzolana. Esso è quindi, in pratica, una miscela di cemento Portland e pozzolana.
La parziale sostituzione del clinker con pozzolana consente di ridurre proporzionalmente il consumo di combustibile necessario alla cottura del clinker. Questo fatto, unitamente alle ottime proprietà del cemento pozzolanico, suscita un notevole interesse per questo cemento in tutti quei paesi dove il problema energetico è molto sentito.
Il calore di idratazione del cemento pozzolanico è più basso di quello sviluppato dal cemento Portland. Pertanto il cemento pozzolanico è da preferirsi al Portland nei lavori effettuati in climi caldi, soprattutto se si tratta di strutture in calcestruzzo di grande mole. Esso fornisce, però, resistenze meccaniche leggermente inferiori a quelle del cemento Portland alle brevi stagionature, soprattutto nei climi più freddi, per il minor contenuto di clinker. Alle lunghe stagionature, invece, le resistenze meccaniche dei manufatti preparati con cemento Portland o pozzolanico sono all'incirca eguali, per la benefica azione della pozzolana che si combina con la calce di idrolisi liberata per idratazione del C3S e del β-C2S (v. reazioni 6 e 7). La pozzolana, infatti, è un materiale, naturale o artificiale, di composizione prevalentemente silicea (v. tab. II), capace di combinarsi a temperatura ambiente con l'idrossido di calcio per produrre composti insolubili in acqua molto simili a quelli ottenuti per idratazione dei silicati del cemento Portland. La combinazione consiste fondamentalmente nella reazione tra la silice tipicamente reattiva della pozzolana e la calce di idrolisi, con formazione di idrosilicati di calcio del tipo CySHx. Pertanto, mentre di per sé la pozzolana non ha caratteristiche leganti, miscelata con calce idrata diventa un legante idraulico (v. cap. 4), che è in grado di produrre malte idrauliche; queste si differenziano dalle malte aeree, a base di calce, sabbia e acqua (v. cap. 3), per il fatto di indurire sott'acqua e per la maggiore resistenza meccanica. A ciò si deve il grande successo di questo materiale fin dall'epoca dei Romani, che con le malte idrauliche a base di pozzolana realizzarono le costruzioni più prestigiose, alcune delle quali sono ancora a testimoniare la durevolezza dei conglomerati a base di pozzolana: i ponti Milvio ed Emilio a Roma, gli archi di Claudio ad Anzio e alcune opere marittime dei porti di Ancona e Civitavecchia sono solo alcuni esempi delle opere realizzate con malte idrauliche a base di calce, pozzolana e acqua.
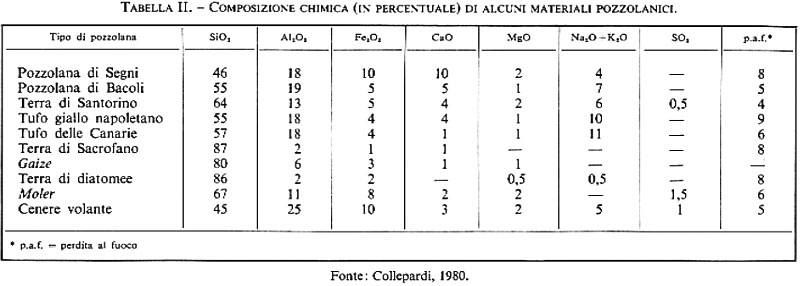
Le pozzolane si dividono in naturali e artificiali. Quelle naturali, a loro volta, possono essere di origine vulcanica, come le pozzolane incoerenti laziali e campane, o come i tufi compatti del napoletano e della Germania (Trass renano), oppure di origine clastica, come le terre di diatomee che si trovano negli Stati Uniti, in Canada e in Danimarca (Moler), oppure di origine mista, come la pozzolana di Sacrofano in Italia e il gaize in Francia.
Le pozzolane artificiali sono sostanzialmente costituite dalle ceneri volanti che si ottengono come residuo della combustione del carbone nelle centrali termoelettriche.
Una caratteristica fondamentale del cemento pozzolanico è la sua elevata resistenza all'azione dilavante dell'acqua in generale, e a quella delle acque contenenti diossido di carbonio in particolare, oltre che una buona resistenza all'attacco solfatico. Inoltre, una pasta di cemento pozzolanico offre una maggiore resistenza alla penetrazione dei cloruri, che potrebbero provocare la corrosione delle armature. Tutte queste proprietà rendono il cemento pozzolanico un legante particolarmente idoneo alle costruzioni idrauliche in genere, e a quelle marittime in particolare.
Sempre dal punto di vista della durabilità, il cemento pozzolanico si comporta meglio del cemento Portland nei confronti di inerti reattivi capaci di provocare indesiderati fenomeni espansivi per la reazione alcali-aggregato (v. cap. 7, È a).
e) Cemento d'altoforno.
Il cemento d'altoforno è un prodotto della macinazione del clinker di cemento Portland, del gesso e della loppa d'altoforno. Esso è quindi una miscela di cemento Portland e loppa. Il cemento d'altoforno è simile, sotto certi aspetti, al cemento pozzolanico, trattandosi in entrambi i casi di un cemento Portland al quale viene aggiunta una certa quantità di pozzolana o di loppa, con lo scopo di produrre un cemento più economico, ma con prestazioni paragonabili a quelle del cemento Portland o addirittura superiori, per alcune applicazioni.
Mentre nel cemento pozzolanico l'aggiunta di pozzolana difficilmente supera il 35%, nei cementi d'altoforno l'aggiunta di loppa può essere compresa entro un ampio intervallo che va da un minimo del 30% (cemento Portland d'altoforno) a un massimo dell'85% (cemento di loppa al clinker). Ciò dipende dal fatto che, a differenza della pozzolana, la loppa può idratarsi per proprio conto purché la fase acquosa contenga altre sostanze (attivatori) capaci di innescare la reazione tra loppa e acqua.
Gli impieghi per i quali il cemento d'altoforno può essere preferito al cemento Portland sono sostanzialmente gli stessi per i quali si raccomanda l'impiego del cemento pozzolanico: lavori in climi caldi, per il minor calore di idratazione, e lavori marittimi, per la buona resistenza ai solfati.
Come il cemento pozzolanico, anche quello d'altoforno presenta resistenze meccaniche più basse del cemento Portland alle brevi stagionature, soprattutto in climi freddi, mentre si possono ottenere risultati sostanzialmente identici alle lunghe stagionature.
La loppa d'altoforno è la scoria eliminata, allo stato liquido, dall'altoforno durante la produzione della ghisa. Esistono diverse utilizzazioni della loppa d'altoforno, ma quella più interessante è proprio la produzione del cemento d'altoforno, per la quale si richiede, però, che la loppa fusa sia raffreddata il più rapidamente possibile, per esempio facendola cadere in grandi masse d'acqua. Lo scopo di questo trattamento è quello di prevenire il processo di cristallizzazione del liquido e di favorire, invece, la solidificazione sotto forma di vetro. Il materiale così temprato si presenta sotto forma di granuli vetrosi misti a frammenti schiumosi e per questo prende il nome di ‛loppa granulata d'altoforno'.
La loppa comprende, quali maggiori costituenti, CaO, SiO2, Al2O3 ed MgO, oltre a quantità minori di MnO, Fe2O2 ed S.
La loppa granulata d'altoforno finemente macinata e mescolata con acqua non è in grado di indurire significativamente. Tuttavia, se la loppa è impastata con acqua in presenza di piccole quantità di calce e di gesso, o di cemento Portland, o di altre sostanze capaci di rendere alcalina l'acqua di impasto, la miscela indurisce comportandosi come un vero e proprio legante idraulico. La loppa granulata è, in sostanza, capace di idratarsi, e quindi di indurire, in presenza di opportuni attivatori. Per questo la loppa si differenzia dalla pozzolana, che è in grado, invece, di indurire solo in presenza di quantità rilevanti di calce idrata con la quale si combina, senza però idratarsi per suo conto come la loppa granulata in presenza di attivatori.
L'effetto degli attivatori sull'idratazione della loppa può essere spiegato ammettendo che la loppa da sola si idrati liberando un po' di Ca(OH)2 nella fase acquosa e formando dei gel impermeabili, prevalentemente costituiti di silice e allumina, che, ricoprendo la loppa non ancora idratata, ne impedirebbero l'ulteriore idratazione. La funzione degli attivatori sarebbe quella di reagire con i gel favorendo la formazione di composti più permeabili.
L'attività idraulica della loppa dipende fondamentalmente dallo stato vetroso del materiale e aumenta con il contenuto di vetro e con la temperatura raggiunta dalla loppa fusa prima della granulazione. L'attività idraulica dipende anche dalla composizione chimica, anche se in modo molto complesso: essa aumenta con il rapporto CaO/SiO2, sino a un valore critico che rende poi praticamente impossibile la granulazione della loppa, in quanto maggiore è il contenuto di CaO, maggiore è la tendenza della loppa fusa a cristallizzare. A parità di rapporto CaO/SiO2, l'attività idraulica della loppa aumenta con il contenuto di Al2O3; a un'eventuale deficienza di CaO - per favorire la granulazione - si può sopperire con una grande quantità di Al2O3.
f) Cemento soprasolfatato.
Il cemento soprasolfatato, prodotto soprattutto in Belgio, ma anche in Germania, Francia e Gran Bretagna, è costituito per l'80-85% da loppa granulata, per il 10-15% da anidrite o da gesso cotto e per il 5% circa da cemento Portland.
I prodotti dell'idratazione sono costituiti fondamentalmente da ettringite, C3A • 3CaSO4 • 32H2O, e da idrosilicato di calcio, CySHx (v. cap. 5, È b). Il primo composto si forma soprattutto durante i primi giorni di idratazione, il secondo alle stagionature più lunghe. I cristalli fibrosi di ettringite sono molto più lunghi (oltre 120 μm) di quelli che si formano nelle paste di cemento Portland (qualche μm). Secondo Mehta (v., 1973) ciò è da ascrivere all'influenza della calce (presente nelle paste di cemento Portland, ma non in quelle di cemento soprasolfatato), che favorisce una cristallizzazione più minuta e quindi la formazione di fibre molto più corte. Secondo Mehta l'adsorbimento di acqua da parte dell'ettringite sotto forma di cristalli piccoli (ettringite colloidale) è la principale causa di espansione delle paste di cemento Portland per attacco solfatico (v. cap. 10, È d); il fatto che i cementi soprasolfatati, contenenti cristalli più grossi di ettringite, non espandano, nonostante le quantità rilevanti di ettringite, sembra essere un'ottima conferma della teoria di Mehta.
Il cemento soprasolfatato presenta, rispetto al cemento Portland, un minor calore di idratazione e una maggiore resistenza agli attacchi chimici. Oltre a resistere all'azione dei solfati e dell'acqua di mare, il cemento soprasolfatato è particolarmente adatto alle strutture in calcestruzzo che debbano venire in contatto con soluzioni acide, purché con pH superiori a 3,5. Inoltre, esso resiste bene anche all'attacco di molte sostanze organiche, quali il fenolo, gli acidi acetico, citrico, tartarico, ecc., purché la concentrazione di queste sostanze non superi lo 0,5%.
g) Cementi espansivi.
Il cemento espansivo è costituito da cemento Portland al quale è aggiunto un agente espansivo (generalmente CaO, MgO o 4CaO • 3Al2O3 • SO3), la cui funzione è quella di provocare un'espansione che, se utilmente impiegata, può ridurre, o addirittura eliminare, gli inconvenienti provocati dal ritiro (v. cap. 10, È c), e in particolare le fessurazioni.
I due eventi, espansione e ritiro, non sono simultanei, ma cronologicamente successivi, come è schematicamente illustrato nella fig. 13. Perché l'espansione possa verificarsi è necessario stagionare con cura l'impasto cementizio, mantenendolo in ambiente umido o sott'acqua. Terminata la stagionatura, se l'acqua evapora dalla pasta di cemento ha inizio il ritiro.
Se si impiega un cemento espansivo, e contemporaneamente si contrasta l'espansione, per esempio mediante l'impiego di un ferro d'armatura, si genera una sollecitazione di compressione nel conglomerato cementizio e uno sforzo di trazione nel ferro. Il successivo ritiro, a seconda che sia inferiore o uguale alla precedente espansione, può rispettivamente ridurre o annullare queste sollecitazioni. Se invece il ritiro è maggiore dell'espansione, il calcestruzzo è sollecitato a trazione, ma lo sforzo di trazione residuo è notevolmente inferiore a quello che si sarebbe generato in assenza di cemento espansivo, e se lo sforzo residuo è inferiore alla resistenza a trazione del materiale, il materiale non si fessura.
Per l'espansione dei cementi espansivi si utilizzano sostanzialmente due tipi di reazione. La prima reazione si basa sulla produzione di ettringite, C3A • 3CaSO4 • 32H2O, per idratazione di miscele contenenti alluminati, o solfoalluminati, con solfato di calcio ed eventualmente con calce. La seconda si basa sulla conversione dell'ossido di calcio nel corrispondente idrossido, con possibilità di sostituire parte del calcio con magnesio. Come si vede, quale che sia il tipo di agente espansivo impiegato, è assolutamente necessario che il conglomerato cementizio sia stagionato in ambiente umido poiché il fenomeno espansivo è sempre connesso con una reazione di idratazione.
h) Cemento alluminoso.
Il cemento alluminoso è ottenuto per cottura di miscele di calcare e bauxite. La calce, ottenuta per decomposizione termica del calcare, si combina con l'allumina della bauxite per formare l'alluminato monocalcico (CaO • Al2O3 = CA):
CaCO3 + Al2O3 → CA + CO2. (19)
Oltre all'alluminato monocalcico, che è il costituente principale del cemento alluminoso, sono presenti altri composti minori (C3S, C4AF, C2F, CA2, C19A7, C2AS, ecc.), che si formano per reazione delle impurezze (silice, ossido ferrico, ecc.) che accompagnano il CaCO3 e l'Al2O3 nel calcare e nella bauxite.
L'idratazione del cemento alluminoso consiste sostanzialmente nell'idratazione dell'alluminato monocalcico con formazione di alluminati di calcio idrati esagonali (CAH10 e C2AH8) e di gel di allumina (AHx):
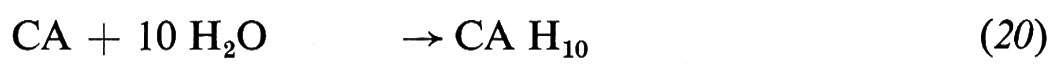
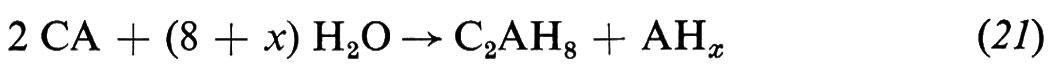
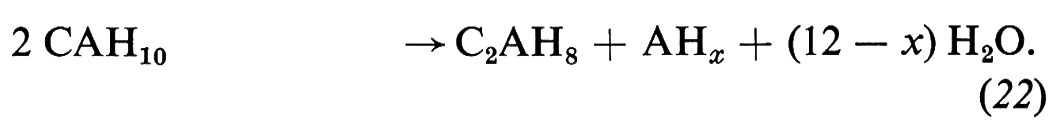
La principale caratteristica del processo di idratazione del cemento alluminoso consiste nella trasformazione degli alluminati idrati esagonali (CAH10 e C2AH8) nell'idrato cubico (C3AH6), favorita dalle alte temperature, dall'ambiente umido e dall'alto rapporto acqua/cemento dell'impasto. La trasformazione degli idrati esagonali in quello cubico,
3CAH10 → C3AH6 + 2AHx + (24 − 2x)H2O (23)
3C2AH8 → 2C3AH6 + AHx + (12 − x)H2O, (24)
è accompagnata da una significativa diminuzione della resistenza meccanica, che costituisce senza dubbio il maggior limite all'impiego di questo cemento.
A temperatura ambiente la trasformazione è molto lenta e può anche non avvenire, se il calcestruzzo è mantenuto asciutto. All'aumentare della temperatura la trasformazione diviene progressivamente più veloce.
La diminuzione di resistenza meccanica è da mettere in relazione con la trasformazione degli alluminati idrati esagonali, con densità di circa 2 g/cm3, nell'idrato cubico più denso (densità di circa 2,5 g/cm3). In sostanza la trasformazione da idrati esagonali in idrato cubico provoca un notevole aumento di porosità, che è la causa fondamentale della diminuzione della resistenza meccanica. Questo decadimento nel tempo delle resistenze meccaniche, non collegato ad alcun fattore patologico, è indubbiamente il maggior limite all'impiego del cemento alluminoso, soprattutto per costruzioni di notevole impegno strutturale.
7. Gli inerti.
In pratica quasi mai il cemento è impiegato da solo con acqua: generalmente la pasta cementizia è mescolata con materiali lapidei che, per il fatto di non partecipare al processo di idratazione, sono chiamati ‛inerti' (v. Orchard, 1976). Questi materiali sono anche comunemente denominati ‛aggregati', termine con il quale si pone l'accento sulla natura particellare degli elementi che compongono l'inerte.
Esistono diverse ragioni per giustificare l'aggiunta dell'inerte alla pasta di cemento. Innanzitutto c'è un motivo di carattere economico, in quanto l'inerte costa molto meno della pasta cementizia, e risulta quindi economicamente vantaggioso disperdere un certo volume di aggregati in una matrice costituita dalla pasta di cemento. Esistono altre ragioni, di carattere tecnico (v. cap. 10), che giustificano l'impiego degli inerti in un conglomerato cementizio. La più importante riguarda il ritiro della pasta di cemento che, per aggiunta dell'inerte, viene diminuito proporzionalmente. Una seconda importante ragione tecnica riguarda la durabilità del calcestruzzo, che può essere migliorata sostituendo una parte della pasta cementizia, materiale relativamente aggredibile dall'ambiente, con un materiale più durevole qual è l'inerte, purché questo possegga determinati requisiti (v. È a). Molte altre proprietà del calcestruzzo indurito (resistenza meccanica, modulo elastico, scorrimento viscoso) dipendono dalla percentuale dell'inerte e possono migliorare significativamente introducendo nell'impasto cementizio un'adeguata quantità di aggregato.
L'unica proprietà del calcestruzzo che, a causa dell'aggiunta dell'inerte, risulta peggiorata rispetto alla pasta di cemento è la lavorabilità dell'impasto fresco (v. cap. 9, È a): se, per esempio, a una pasta cementizia fluida si aggiunge progressivamente un inerte, le proprietà reologiche del conglomerato, e in particolare la fluidità, risultano proporzionalmente più scadenti.
a) Sostanze indesiderabili negli inerti.
Negli inerti, accanto ai minerali principali che compongono le rocce (calcare, quarzo, ecc.), possono essere presenti un certo numero di sostanze, alcune delle quali possono provocare inconvenienti o danni molto gravi nel conglomerato cementizio, e per questo sono definite indesiderabili. Tra esse si possono distinguere quelle che, come le sostanze organiche, interferendo con il processo di idratazione del cemento, provocano un rallentamento nel processo di indurimento del calcestruzzo, senza però provocare danni irreparabili, soprattutto alle lunghe stagionature.
Una seconda categoria di sostanze indesiderabili comprende quelle sostanze che, come la silice reattiva, possono provocare, soprattutto con cementi ricchi in alcali, fessurazioni di tali entità da mettere fuori servizio la struttura in calcestruzzo dopo alcuni anni dalla messa in opera, senza che ci sia stata interferenza con il processo di idratazione del cemento e senza quindi che alle stagionature iniziali si sia manifestato alcun inconveniente.
Anche alcune sostanze a base di solfati e di cloruri e alcune sostanze argillose possono essere classificate fra le sostanze indesiderabili. I solfati possono reagire con gli alluminati del cemento e provocare eccessivi fenomeni espansivi localizzati, con conseguenti fessurazioni nel conglomerato. I cloruri, invece, potrebbero innescare fenomeni di corrosione nei ferri d'armatura. Le sostanze argillose, infine, potrebbero pregiudicare l'adesione tra la pasta di cemento e gli inerti stessi.
Anche se non dovuta a una particolare sostanza, resta da segnalare un'altra caratteristica indesiderabile degli inerti: la ‛gelività', e cioè la tendenza a deteriorarsi in conseguenza dei cicli di gelo-disgelo legati alle variazioni di temperatura. Infatti, quando questa scende al di sotto di 0 °C, l'acqua si trasforma in ghiaccio con aumento di volume. Se gli inerti, a causa della loro porosità, assorbono acqua, questa può diventare fonte di gravi fenomeni espansivi e quindi di deterioramento di tutto il calcestruzzo.
b) Distribuzione granulometrica degli inerti.
Per ottenere un calcestruzzo è preferibile disporre di un aggregato ben assortito, con granuli di diverse dimensioni. Si dice che un inerte di questo tipo presenta una buona distribuzione granulometrica.
La resistenza meccanica di un calcestruzzo prodotto con inerte ben assortito, a parità di lavorabilità e di dosaggio di cemento, è più alta di quella di un conglomerato prodotto con un inerte scarsamente assortito, e cioè formato da granuli di dimensioni pressoché eguali.
Poiché è difficile reperire un solo inerte sufficientemente assortito, e poiché, d'altra parte, la distribuzione granulometrica di un inerte, sia esso naturale o artificiale, è soggetta a variazioni talvolta piuttosto considerevoli, per mantenerne il più possibile costante l'assortimento granulometrico è preferibile combinare due o più inerti al fine di riprodurre una determinata e soddisfacente distribuzione granulometrica.
Siccome nel calcestruzzo la pasta di cemento va a riempire gli interstizi tra i granuli dell'inerte, per realizzare la massima compattezza del conglomerato con il minimo dosaggio di cemento occorre scegliere un aggregato con il minimo contenuto di vuoti interstiziali tra i granuli dell'inerte.
Fuller e Thompson (v., 1907), basandosi su considerazioni di carattere teorico, proposero una curva granulometrica, descritta dall'equazione

e nota con il nome di ‛curva di Fuller', per rappresentare la distribuzione granulometrica con il minor contenuto di vuoti. Nell'equazione (25) p è la percentuale di solido (inerte più cemento) avente dimensione inferiore a d, e D è la dimensione massima dell'inerte (comunemente chiamata ‛diametro massimo').
Bolomey (v., 1947) ha modificato l'equazione (25) partendo dalla considerazione che i calcestruzzi confezionati con inerti secondo la curva di Fuller si presentavano scarsamente lavorabili e aspri. Sulla base di prove sperimentali Bolomey ha proposto l'equazione (26) per descrivere la distribuzione granulometrica dell'inerte ottimale dal punto di vista della lavorabilità, oltre che della compattezza del conglomerato:

Nell'equazione (26) A è una costante che viene scelta in base al tipo di inerte (tondeggiante o angolare) e alla lavorabilità che si richiede, e può variare da 6 a 16. Così, con l'equazione di Bolomey è possibile scegliere la distribuzione granulometrica ottimale in relazione al tipo di inerte disponibile e alla lavorabilità richiesta, fattori ignorati nelle distribuzioni granulometriche (come quella di Fuller) che hanno come unico obiettivo quello di ottenere un inerte con un minimo contenuto di vuoti.
8. Gli additivi.
Gli additivi (v. Collepardi, 1980; v. Ramachandran, 1985) sono dei prodotti chimici che vengono aggiunti in piccole quantità per migliorare le proprietà del calcestruzzo. Essi presentano un notevole interesse dal punto di vista scientifico e tecnologico. Un'analisi della letteratura scientifica e brevettuale nel campo del cemento e del calcestruzzo mostra che il progresso conseguito in questo settore negli ultimi anni è principalmente da attribuirsi allo sviluppo delle conoscenze sugli additivi.
A seconda della loro funzione principale, gli additivi possono essere classificati in acceleranti, ritardanti, fluidificanti, superfluidificanti e aeranti.
In generale, il miglioramento conseguito con l'additivo può essere realizzato, almeno in teoria, variando la composizione del calcestruzzo o la sua tecnologia di applicazione. Tuttavia l'aggiunta degli additivi trova la sua giustificazione nel fatto che il miglioramento di una determinata caratteristica del calcestruzzo, realizzato attraverso il loro impiego, deve risultare più vantaggioso, dal punto di vista tecnico-economico, di qualsiasi altra soluzione alternativa.
L'aspetto economico andrà valutato in relazione al particolare tipo di problema tecnologico che, di volta in volta, deve essere risolto. Il costo del volume unitario di materiale con e senza additivo dovrà essere riferito al calcestruzzo appena confezionato, oppure messo in opera, oppure in servizio dopo un certo numero di anni, a seconda della particolare caratteristica tecnologica che ci si prefigge di migliorare. Per esempio, nel valutare il vantaggio derivante dall'impiego di un additivo accelerante, si possono esaminare i costi derivanti dalle possibili soluzioni alternative, quali l'impiego di un cemento più rapido o il riscaldamento di materiali, facendo riferimento al costo unitario del calcestruzzo nel momento in cui esso ha raggiunto una determinata resistenza meccanica prefissata. Nel caso, invece, che si desideri migliorare la durabilità del calcestruzzo, il maggior costo derivante dall'impiego di un additivo superfluidificante andrà confrontato con le maggiori spese di manutenzione alle quali il calcestruzzo non additivato sarà sicuramente soggetto dopo un certo periodo di servizio.
a) Additivi acceleranti.
Gli additivi acceleranti fanno aumentare la velocità di idratazione del cemento nel periodo di presa o di indurimento. Gli acceleranti di presa accorciano il tempo di presa, mentre quelli di indurimento fanno aumentare la resistenza meccanica iniziale. A eccezione di qualche applicazione, solo gli acceleranti di indurimento presentano un notevole interesse pratico. Essi consentono di ovviare agli inconvenienti provocati dalle basse temperature: per questo motivo essi sono spesso denominati, sia pure impropriamente, additivi antigelo.
L'additivo accelerante di indurimento piu impiegato è il cloruro di calcio. Tuttavia la presenza di rilevanti quantità di cloruro (più dello 0,1% rispetto al peso del cemento) nel calcestruzzo armato, e soprattutto in quello precompresso, può favorire l'innesco di fenomeni corrosivi nei ferri di armatura e pertanto, in queste circostanze, è preferibile impiegare un additivo accelerante privo di cloruri.
Molti altri prodotti sono stati provati al fine di trovare un accelerante che non presentasse i rischi di corrosione del cloruro di calcio nei confronti dei ferri delle strutture armate precompresse. Alcuni di essi sono in effetti impiegati per formulare additivi acceleranti, ma nessuno presenta le caratteristiche del cloruro di calcio e soprattutto il suo basso costo. Tra i prodotti normalmente impiegati i più diffusi sono la trietanolammina, N(CH2−CH2−OH)3, e il formiato di calcio, Ca(HCOO)2, ma molti altri sali quali, per esempio, il nitrato di calcio, il floruro di sodio, il tiosolfato e il tiocianato di potassio possono essere impiegati. L'azione accelerante della maggior parte di questi sali si esplica sull'idratazione dei silicati, e del C3S in particolare.
b) Additivi ritardanti.
L'aggiunta di un additivo ritardante ha lo scopo di rallentare la presa del cemento, per conservare più a lungo la lavorabilità e consentire quindi un trasporto del calcestruzzo a lunga distanza, soprattutto quando la temperatura è molto elevata (v. cap. 9, È a). Naturalmente il ritardo nell'idratazione del cemento, necessario per allungare i tempi di presa, comporta una bassa resistenza meccanica del calcestruzzo alle brevi stagionature.
c) Additivi fluidificanti.
Gli additivi fluidificanti, aggiunti a un impasto cementizio, ne aumentano la lavorabilità a parità di rapporto acqua/cemento, oppure consentono di ridurre l'acqua di impasto a parità di lavorabilità. Pertanto i fluidificanti possono essere anche chiamati riduttori d'acqua.
L'incremento di lavorabilità, espresso come aumento di slump (v. cap. 9, È a), è dell'ordine di 5-10 cm, mentre la riduzione del rapporto acqua/cemento è del 5-10%. Poichè non esiste proprietà del calcestruzzo indurito che non venga migliorata dalla riduzione del rapporto acqua/cemento, i fluidificanti, e ancor più i superfluidificanti, sono gli additivi più interessanti per la tecnologia del calcestruzzo.
Chimicamente gli additivi fluidificanti possono essere classificati in tre gruppi, in base al loro componente principale: il ligninsolfonato, gli acidi idrossicarbossilici e i polimeri idrossilati. Ciascuno di essi può anche contenere, come componenti secondari, altri prodotti, che conferiscono all'additivo fluidificante anche un'azione secondaria accelerante o ritardante.
d) Additivi superfluidificanti.
Gli additivi superfluidificanti si distinguono dai normali fluidificanti solo da un punto di vista quantitativo: la riduzione del rapporto acqua/cemento, che è del 5-10% per un fluidificante, diviene del 20-40% per un superfluidificante. D'altra parte l'aggiunta di un superfluidiflcante, a parità di rapporto acqua/cemento, può trasformare un calcestruzzo asciutto con slump di 1-2 cm in un calcestruzzo fluido con slump superiore a 20 cm (v. cap. 9, È a).
I principali prodotti chimici sui quali si basa la maggior parte dei supeifluidificanti sono dei polimeri solfonati di sintesi, tra i quali i più impiegati in pratica sono il naftalensolfonato condensato con formaldeide (27) e la trimetibìmelammina solfonata condensata con formaldeide (28):
Con l'impiego degli additivi superfluidiflcanti diventa possibile la produzione di calcestruzzi molto fluidi (slump di 20 cm) e quindi molto facili da mettere in opera, senza dover ricorrere a incrementi nel rapporto acqua/cemento, che comporterebbero un peggioramento della qualità del calcestruzzo indurito. Ciò rende conto del grande successo di questi additivi soprattutto nelle costruzioni di difficile esecuzione (alta densità dei ferri di armatura e sezioni sottili) e che richiedano alti valori di resistenza meccanica, di modulo elastico, di durabilità, o bassi valori di ritiro e di deformazione viscosa (v. fig. 14).
Per quanto concerne il meccanismo d'azione degli additivi superfluidiflcanti, come anche di quelli fluidificanti, esso è basato su un'azione disperdente dei granuli di cemento (v. fig. 15). In assenza di questi additivi, nell'impasto cementizio fresco i granuli di cemento - grazie alla presenza di cariche elettrostatiche di segno opposto causate dalla frattura delle particelle di clinker durante la macinazione - si attirano e tendono a formare degli agglomerati (costituiti da numerose particelle) molto ingombranti e pertanto poco mobili, a meno che non si aggiunga molta acqua all'impasto fresco (v. fig. 15A). In presenza degli additivi superfluidificanti, invece, le particelle di cemento che formano gli agglomerati tendono a respingersi l'un l'altra, cioè a disperdersi, formando una struttura reologicamente più faforevole, e quindi più mobile, anche con quantitativi d'acqua relativamente modesti (v. fig. 15B). L'azione disperdente è da mettere in relazione con la repulsione elettrostatica tra le particelle di cemento, tutte caricate negativamente in seguito all'adsorbimento del polimero con i suoi gruppi solfonici.
e) Additivi aeranti.
Gli additivi aeranti sono aggiunti al calcestruzzo per migliorare la resistenza ai cicli di gelo-disgelo attraverso la formazione di un sistema di microbolle d'aria (diametro: 200-300 μm) omogeneamente disperse nel materiale.
L'aumento di volume che si verifica nella solidificazione dell'acqua contenuta nei pori capillari della pasta cementizia provoca un aumento di pressione nell'acqua non ancora congelata, che può arrivare a degradare il calcestruzzo indurito. La presenza di microbolle d'aria, in prossimità di pori capillari pieni d'acqua dove si sta formando del ghiaccio, consente di scaricare la pressione idraulica grazie al trasporto dell'acqua dai pori capillari verso le microbolle d'aria.
Poiché la formazione delle microbolle d'aria avviene solamente nella pasta di cemento, il volume di aria inglobata in un calcestruzzo resistente al gelo dovrà essere tanto maggiore quanto minore è il volume occupato dall'inerte. Ciò significa che il volume d'aria inglobata (solitamente compreso tra il 3 e l'8%) deve crescere al diminuire della dimensione massima dell'inerte.
9. Il calcestruzzo fresco.
Dopo il mescolamento dei suoi ingredienti - acqua, cemento, inerti e additivi - il calcestruzzo deve essere trasportato nel cantiere, gettato dentro le casseforme e compattato mediante vibrazione meccanica o costipazione manuale. Lo stato in cui si trova il calcestruzzo durante queste operazioni è definito ‛fresco' (v. Powers, 1968) ed è caratterizzato da due proprietà del materiale. La prima - detta ‛lavorabilità' - riguarda la capacità del calcestruzzo di muoversi a seguito dell'applicazione di una forza. La seconda proprietà, che prende il nome di ‛segregazione', concerne l'attitudine del calcestruzzo fresco a separarsi in frazioni tra loro diverse - per esempio inerte grosso e malta - con la conseguenza di provocare la formazione di un materiale eterogeneo. Un calcestruzzo ideale dovrebbe essere molto lavorabile e poco segregabile. Sfortunatamente queste due proprietà sono spesso antitetiche e occorre quindi raggiungere un ragionevole compromesso per conferire al calcestruzzo fresco qualità soddisfacenti da entrambi i punti di vista.
a) Lavorabilità del calcestruzzo fresco.
Dopo la messa in opera, all'interno del calcestruzzo possono rimanere dei vuoti, il cui numero dipende fondamentalmente dalla composizione del conglomerato (in particolare: quantità di acqua, dosaggio di cemento e tipo di inerte), dall'efficacia del sistema di compattazione e dalla lavorabilità dell'impasto fresco. A parità di tutte le altre condizioni, più alta è la lavorabilità, minore è il lavoro richiesto per costipare il materiale e più denso risulta il materiale al termine della compattazione: da ciò consegue che un calcestruzzo lavorabile è anche più economico per l'impresa, più affidabile per il progettista e più sicuro per il proprietario dell'opera.
Il sistema più semplice ed economico per migliorare la lavorabilità è quello di introdurre più acqua al momento di preparare l'impasto; se, però, l'operazione è fatta mantenendo costante il dosaggio di cemento, l'aumento dell'acqua si tramuta in un incremento del rapporto acqua/cemento, con tutte le conseguenze negative per la qualità del materiale una volta che sarà indurito (v. cap. 5, È e; v. cap. 10). Pertanto, se si vuole incrementare la lavorabilità del calcestruzzo fresco, come si richiede spesso per getti difficili (sezioni sottili e strutture densamente armate), senza pregiudicare la qualità del materiale indurito, è necessario aumentare sia la quantità d'acqua sia quella di cemento, in modo da mantenere costante il rapporto in peso tra i due componenti. Esistono, però, due limiti a questo tipo di intervento: uno tecnico e l'altro economico. Una quantità eccessiva di cemento comporta un elevato sviluppo di calore di idratazione con possibili fessurazioni nella struttura - soprattutto se di grandi dimensioni - per i gradienti termici che si instaurano tra la parte centrale e quella periferica (v. cap. 5, È c). Inoltre, all'aumentare del dosaggio di cemento, aumentano le deformazioni della struttura in calcestruzzo, per effetto di un ambiente secco o dell'applicazione di un carico costante (v. cap. 10, È c).
D'altra parte, l'aumento del contenuto di cemento comporta un incremento del costo del calcestruzzo. Pertanto si pone il problema di conseguire il miglioramento della lavorabilità del calcestruzzo fresco con sistemi alternativi più economici e che non presentino i limiti tecnici sopra menzionati. L'alternativa più frequente è quella di impiegare additivi fluidificanti, o meglio ancora superfluidificanti, che modificano la reologia del calcestruzzo fresco rendendolo più scorrevole e più docile alla compattazione, senza dover ricorrere a ulteriori aggiunte d'acqua e quindi di cemento (v. cap. 8, ÈÈ c e d). L'altra possibilità, per aumentare la lavorabilità del calcestruzzo fresco, è quella di impiegare inerti con buona curva granulometrica e di grande dimensione (v. cap. 7, È b): maggiore è la dimensione dell'aggregato, minore è la sua superficie specifica (rapporto superficie/volume). Pertanto, inerti con un elevato diametro massimo richiedono meno acqua (e quindi anche meno cemento a parità di rapporto acqua/cemento) per essere rivestiti dalla pasta di cemento. La tab. III mostra l'influenza del diametro massimo dell'inerte sulla quantità d'acqua richiesta per ottenere un determinato livello di lavorabilità. Per esempio, impiegando sempre 200 litri di acqua per m3 di calcestruzzo, la lavorabilità aumenta da 4 a 8 e a 16 cm di slump, impiegando inerti rispettivamente con diametro massimo di 10, 20 e 40 mm.
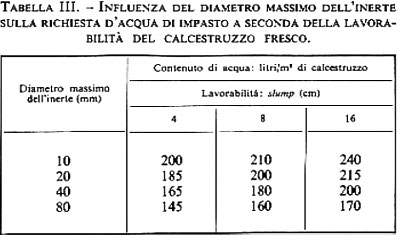
Esistono diversi metodi di misura della lavorabilità, ciascuno dei quali mette in maggior evidenza uno o alcuni aspetti di questa proprietà. L'abbassamento al cono di Abrams, noto anche come slump test, è indubbiamente il metodo più diffuso per la semplicità e rapidità della misura. Il metodo consiste nel determinare l'abbassamento di un calcestruzzo per azione del proprio peso. L'apparecchio è costituito da un tronco di cono aperto alle due estremità (altezza 30 cm, diametro inferiore 20 cm, diametro superiore 10 cm), che viene appoggiato su una base metallica non assorbente e riempito dall'alto con il calcestruzzo fresco. Dopo aver sollevato il cono per mezzo di due maniglie, si misura l'abbassamento (slump) del calcestruzzo rispetto all'altezza originale.
b) Segregazione del calcestruzzo fresco.
La segregazione del calcestruzzo consiste nella separazione dei vari componenti della miscela, a causa delle differenze di dimensioni e di peso specifico delle particelle. Gli elementi più grossi e più pesanti tendono a sedimentare sul fondo della struttura, mentre quelli più piccoli e leggeri, e l'acqua in particolare, tendono a raccogliersi sulla superficie. La raccolta d'acqua in superficie provoca, qualora ne sia impedita l'evaporazione, l'essudazione (bleeding) del calcestruzzo. Di conseguenza si ha la formazione di materiale eterogeneo e soprattutto di una superficie meccanicamente più debole, con conseguenze particolarmente gravi per le pavimentazioni.
Per eliminare o ridurre al minimo la segregazione e l'essudazione è necessario disporre di inerti ben assortiti granulometricamente, impiegando additivi fluidificanti e superfiuidificanti per ridurre l'eccesso di acqua, ed evitare dosaggi di cemento troppo bassi quando l'impasto risulti scarsamente coesivo.
10. Il calcestruzzo indurito.
Il calcestruzzo indurito (v. Collepardi, 1980; v. Neville, 1981) è costituito da un insieme di materiali lapidei inerti, legati tra loro dalla pasta di cemento indurita. Le proprietà del conglomerato dipendono quindi dalla qualità della pasta cementizia (v. cap. 5, È e), dalle caratteristiche dell'aggregato (v. cap. 7) e dal legame che si stabilisce all'interfaccia pasta-aggregato.
Per un dato inerte e una determinata pasta di cemento, cambiando il rapporto in peso aggregato/pasta, è possibile migliorare alcune proprietà: per esempio, all'aumentare del suddetto rapporto diminuisce il ritiro e anche il costo del calcestruzzo.
a) Resistenza meccanica.
Tra tutte le proprietà del calcestruzzo indurito la resistenza meccanica è indubbiamente la più importante. Ciò dipende da molteplici ragioni: innanzitutto la resistenza meccanica del materiale è fondamentale nel calcolo strutturale; in secondo luogo essa è misurabile in modo relativamente semplice e rapido; in terzo luogo si assume - anche se l'assunsunzione non è sempre corretta - che le altre qualità del calcestruzzo migliorino all'aumentare della resistenza meccanica. La pasta aderisce meglio alla superficie ruvida degli aggregati frantumati (pietrisco) che non a quella liscia degli inerti naturali (ghiaia). Tuttavia, anche la composizione chimica e mineralogica degli inerti gioca un ruolo significativo - anche se non ancora del tutto chiarito - nell'influenzare l'adesione della pasta cementizia alla superficie degli aggregati.
A seconda della sollecitazione cui il materiale è sottoposto si può determinare la resistenza meccanica a compressione, a flessione o a trazione. Per il calcestruzzo solo la prima è veramente importante, in quanto le altre sono relativamente modeste, e si affida ai ferri d'armatura l'onere di sopportare i carichi di flessione e di trazione nelle opere in calcestruzzo armato. Inoltre le misure della resistenza meccanica a flessione (σf) e di quella a trazione presentano maggiori difficoltà e incertezze di quella della resistenza a compressione (σc) e si preferisce calcolare le prime mediante equazioni empiriche del tipo della seguente:
σf = 9,5 σc1/2. (29)
I principali parametri che influenzano la resistenza meccanica sono: il rapporto acqua/cemento, il tempo di stagionatura e il tipo di cemento (v. fig. 16). Un rapporto acqua/cemento più basso e un maggior grado di idratazione portano entrambi a una microstruttura più densa della pasta cementizia v. eqq. (15)-(18) - e quindi a un calcestruzzo meccanicamente più resistente.
Gli altri parametri che esercitano un'influenza positiva sulla resistenza meccanica sono: il tipo di inerte (inerti duri, bene assortiti e di grande dimensione); la compattazione del calcestruzzo fresco per l'eliminazione dell'aria intrappolata (vibrazione efficace e alta lavorabilità dell'impasto); la temperatura (alta per le brevi stagionature e bassa per le lunghe stagionature); l'umidità dell'ambiente, per evitare l'evaporazione dell'acqua dal calcestruzzo e quindi l'arresto dell'idratazione del cemento.
b) Modulo elastico.
La deformazione (ε) che subisce un materiale sottoposto a una sollecitazione (σ) dipende dal suo modulo elastico (E) secondo la nota equazione di Hooke
σ = Eε. (30)
Nella fig. 17 sono mostrate, in funzione della sollecitazione di compressione, le deformazioni di una pasta di cemento, di un inerte e di un calcestruzzo. La curva del conglomerato si trova - come avviene per ogni materiale composito - tra quella della matrice legante, che è la pasta di cemento, e quella dei granuli dispersi, che costituiscono l'inerte. Il modulo elastico si identifica con la tangente trigonometrica dell'angolo formato dalla curva nel tratto lineare con l'asse delle ascisse. Poiché l'inerte ha un modulo elastico molto maggiore di quello della pasta di cemento, il modulo elastico del calcestruzzo aumenta con il rapporto inerte/pasta di cemento, purché la quantità di pasta sia sufficiente a riempire tutti gli interstizi tra i granuli dell'inerte. D'altra parte, un aggregato più rigido e una pasta di cemento più compatta, per il minor rapporto acqua/cemento, provocano un aumento del modulo elastico del calcestruzzo a parità di rapporto inerte/pasta.
Nel caso di inerti artificiali (come quelli leggeri), che hanno un modulo elastico paragonabile a quello della pasta cementizia, il modulo elastico del calcestruzzo non dipende sostanzialmente dal rapporto inerte/cemento, come si verifica appunto per i calcestruzzi leggeri (v., a questo riguardo, il prossimo capitolo).
Nella fig. 17 si osserva che per il calcestruzzo la curva sforzo-deformazione è meno lineare di quelle riguardanti i materiali che lo compongono. Ciò è probabilmente da attribuire alla presenza di microfessure che si formano all'interfaccia pasta-aggregato e che si propagano, sotto l'applicazione di un carico, formando angoli diversi rispetto allo sforzo applicato. Conseguentemente, lo sforzo realmente applicato risulta in alcuni punti maggiore di quello nominale riportato nell'ordinata, cosicché la deformazione misurata aumenta più rapidamente di come aumenterebbe in base all'applicazione del carico nominale.
Di notevole interesse pratico sono le equazioni che correlano il modulo elastico con le altre proprietà, per esempio la resistenza meccanica. Occorre, però, tener conto che tali equazioni sono sempre ricavate dall'elaborazione di un numero limitato di dati sperimentali, e che pertanto esse rimangono valide entro le condizioni sperimentali delle prove effettuate. Tutte queste correlazioni sono del tipo
E = K √-σc, (31)
dove K è una costante che dipende dalle unità di misura adottate e soprattutto dalla geometria dei provini (cubici, prismatici, cilindrici) sottoposti a misura.
c) Ritiro e deformazione viscosa.
Oltre a subire deformazioni dimensionali, causate da variazioni di temperatura, ed elastiche, come risposta istantanea a una sollecitazione meccanica (v. fig. 17), il calcestruzzo può deformarsi lentamente e ulteriormente per l'applicazione di un carico costante (deformazione viscosa) o per una variazione dell'umidità relativa dell'ambiente (ritiro igrometrico). Quest'ultima deformazione, a differenza delle altre, è tipica dei solidi microporosi, come il calcestruzzo, ed è invece assente nei materiali, come le leghe metalliche, privi di porosità.
Il ritiro è sostanzialmente connesso con l'evaporazione dell'acqua libera, presente nei pori della pasta di cemento, che si verifica in ambienti non saturi di umidità. Esso è tanto maggiore quanto più è porosa la pasta di cemento e quanto maggiore è il volume di pasta all'interno di un conglomerato. La fig. 18 mostra schematicamente come l'andamento del ritiro nel tempo aumenti con il volume della pasta cementizia (passando da un calcestruzzo a una malta e a una pura pasta di cemento) e con il rapporto acqua/cemento del calcestruzzo (cioè con la porosità del cemento idratato). A parità di rapporto acqua/cemento, il ritiro del calcestruzzo diminuisce aumentando il rapporto inerte/cemento (cioè diminuendo il volume di pasta); questo aumento si realizza scegliendo inerti di maggiori dimensioni.
La deformazione viscosa (v. Neville, 1970) - detta anche scorrimento viscoso, o fluage, o creep - consiste nell'aumento di contrazione nel tempo, subito dopo la deformazione elastica (ε), per applicazione al calcestruzzo di una sollecitazione di compressione costante nel tempo (v. fig. 19). La deformazione viscosa è connessa con la microstruttura della pasta di cemento, che si deforma lentamente sotto l'applicazione di un carico costante (σ). Quanto più densa e compatta (cioè più resistente meccanicamente) è la pasta, tanto minore è la deformazione viscosa (C) del calcestruzzo, come risulta dall'equazione

dove K è una costante e σc è la resistenza meccanica a compressione del calcestruzzo.
d) Durabilità.
La durabilità del calcestruzzo (v. Collepardi, 1984) è la sua capacità di durare nel tempo resistendo alle aggressioni dell'ambiente provocate da agenti chimici presenti in natura (per es. solfati e cloruri dell'acqua di mare) o da fenomeni fisici naturali (per es. formazione di ghiaccio). È escluso dal concetto di durabilità che il calcestruzzo possa resistere a lungo all'attacco di particolari agenti chimici artificiali (per es. gli acidi) o di straordinari eventi fisici (per es. un incendio), per quanto sia sempre possibile ridurre i danni di queste aggressioni straordinarie entro limiti accettabili in modo da evitare collassi della struttura.
Si è già detto (v. cap. 8, È e) come l'inglobamento di un certo volume di aria all'interno del calcestruzzo, sotto forma di microbolle, possa allentare le tensioni generate dalla formazione del ghiaccio e proteggere il materiale dal deterioramento.
Per quanto concerne gli attacchi chimici dei solfati e dei cloruri, è necessario innanzitutto produrre un calcestruzzo con il più basso rapporto acqua/cemento (0,35-0,50), al fine di rendere il materiale denso e compatto - v. eqq. (15)-(18) - e quindi impenetrabile agli agenti chimici.
Accanto a queste misure di carattere generale è opportuno adottare alcuni particolari accorgimenti in relazione alle specifiche azioni degli agenti chimici aggressivi. Il solfato, per esempio, esplica la sua azione aggressiva nei confronti del calcestruzzo in quanto reagisce con gli alluminati del cemento producendo un sale (C3A • 3CaSO4 • H32) molto voluminoso, con conseguenti fenomeni espansivi che nel tempo possono provocare fessurazioni e distacchi del calcestruzzo: in questo caso, pertanto, è consigliabile l'impiego di un cemento povero in alluminati, come il cemento ferrico, o resistente ai solfati, come il cemento pozzolanico o quello d'altoforno (v. cap. 6).
Il cloruro, invece, esercita un'azione corrosiva diretta nei confronti dei ferri d'armatura, con conseguente espulsione del calcestruzzo soprastante (copriferro) per il rigonfiamento del ferro che si trasforma in ruggine (idrossido ferrico). Pertanto è necessario frapporre una barriera che ostacoli il cammino del cloruro dall'ambiente verso il ferro: ciò può essere realizzato, oltre che impiegando un calcestruzzo impermeabile con basso rapporto acqua/cemento (0,35-0,40), allungando il cammino del cloruro con un copriferro di adeguato spessore (non minore di 3-4 cm).
11. Calcestruzzi speciali.
Possono essere definiti calcestruzzi speciali tutti quelli che si differenziano sostanzialmente, per una o più proprietà, dai calcestruzzi ordinari. Essi comprendono il calcestruzzo leggero, quello pesante, quello fibro-rinforzato e quello polimero-impregnato.
Un calcestruzzo è definito leggero se la sua densità è compresa tra 300 e 1.800 kg/m3 circa, contro i 2.200÷2.500 kg/m3 del calcestruzzo ordinario.
La produzione del calcestruzzo leggero (v. Short e Kinniburgh, 1978) è resa possibile introducendo dei vuoti nel materiale. In pratica i vuoti possono essere localizzati nell'aggregato (impiegando, per esempio, argilla espansa, polistirene, pomice, ecc.), nella pasta di cemento (utilizzando agenti schiumogeni) o tra la pasta e gli inerti (rinunciando (32) all'impiego di inerti assortiti granulometricamente e provocando la formazione di cavità interstiziali). Ovviamente l'introduzione di questi vuoti rende il calcestruzzo leggero meno resistente meccanicamente, ma consente la soluzione di alcuni problemi tecnologici, quando un'elevata resistenza meccanica non è un requisito determinante per la struttura. La leggerezza del materiale consente, per esempio, di costruire su terreni che per la loro modesta solidità renderebbero difficoltosa la costruzione con un calcestruzzo ordinario. Un'altra caratteristica interessante del calcestruzzo leggero è la sua bassa conducibilità termica, proprietà che è diventata sempre più apprezzabile, in tempi di crisi energetica, per la possibilità di costruire strutture termicamente più isolanti.
Il calcestruzzo pesante (v. Mather, 1965), con densità di 3.000-4.000 kg/m3, può essere ottenuto impiegando inerti pesanti. Esso è per lo più destinato alle strutture che debbano proteggere l'ambiente dalle radiazioni (per esempio nelle opere di ingegneria nucleare) e a tutti quei casi nei quali l'impiego di un calcestruzzo ordinario richiederebbe strutture con sezioni troppo elevate. L'inerte pesante più comunemente impiegato è la barite, BaSO4. I calcestruzzi pesanti possono essere anche confezionati con inerti naturali ricchi in ferro, quali la magnetite, la limonite, l'ematite, la ghetite, ecc. Calcestruzzi ancora più pesanti (5.500 kg/m3) possono essere ottenuti con inerti metallici artificiali (acciaio, piombo, ecc.).
Il calcestruzzo fibro-rinforzato (v. Clifton e Frohnsdorff, 1975) è un materiale composito nel quale, oltre agli ingredienti presenti nel calcestruzzo ordinario, vengono introdotte delle fibre d'acciaio, di vetro, o di materie plastiche. Rispetto al calcestruzzo ordinario, quello fibro-rinforzato è più resistente alla flessione, agli urti, agli scoppi e presenta meno fessurazioni da ritiro. Esso risulta, quindi, particolarmente vantaggioso nella costruzione di piste aeroportuali o di opere militari soggette al rischio di scoppi.
Il calcestruzzo polimero-impregnato (v. Clifton e Frohnsdorff, 1976) viene prodotto introducendo un liquido (‛monomero') in un calcestruzzo precedentemente essiccato e facendo successivamente ‛solidificare' il monomero mediante polimerizzazione. Ne consegue un riempimento parziale o totale delle cavità presenti nel calcestruzzo con un materiale solido quale il polimero. Pertanto il calcestruzzo polimero-impregnato presenta un'elevata resistenza meccanica a compressione, che può arrivare a 100-200 MPa. Anche la resistenza a flessione e a trazione, ma soprattutto la resistenza agli attacchi degli acidi, risultano decisamente più alte di quelle del calcestruzzo ordinario. A parte il maggior costo rispetto al conglomerato ordinario, il calcestruzzo polimero-impregnato presenta interessanti prospettive di impiego per tubazioni, serbatoi, sili, ecc., che debbano stare a contatto con liquidi aggressivi, in particolare gli acidi, capaci di distruggere rapidamente il calcestruzzo ordinario (v. cap. 10, È d).
Bibliografia.
Bogue, R. H., The chemistry of Portland cement, New York 1947.
Bolomey, J., The grading of aggregate and its influence on the characteristics of concrete, in ‟Revue des matériaux de construction et travaux publiques", 1947, pp. 147-149.
Boyton, R.S., Chemistry and technology of lime and limestone, New York 1966.
Brunauer, S., Tobermorite gel. The heart of concrete, in ‟American scientist", 1962, L, pp. 210-229.
Chatterji, S., Jeffery, J. W., Strength development in calcareous cements, in ‟Nature", 1967, CCXIV, pp. 559-561.
Clifton, J., Frohnsdorff, G., Fiber-reinforced cementitious materials, in Cement research progress 1974, American Ceramic Society, Ohio, 1975.
Clifton, J,. Frohnsdorff, G., Polymer-impregnated concretes, in Cement research progress 1975, American Ceramic Society, Ohio, 1976.
Collepardi, M., Materiali inorganici da costruzione, in Enciclopedia della chimica, vol. V, Milano 1977, pp. 34.1-34.47.
Collepardi, M., Scienza e tecnologia del calcestruzzo, Milano 1980.
Collepardi, M., Degradazione e durabilità del calcestruzzo, II parte, in ‟La prefabbricazione", 1984, IX, pp. 567-575.
Collepardi, M., Massidda, L., Hydration of tricalcium silicate, in ‟Journal of the American Ceramic Society", 1971, LIV, pp. 419-422.
Collepardi, M., Massidda, L., Usai, G., La cinetica di invecchiamento del ‛gelo tobermoritico', in ‟Il cemento", 1971, LXVIII, pp. 3-8.
Feldmann, R.F., Sereda, P. J., A model for hydrated Portland cement paste as deduced from sorption length change and mechanical properties, in ‟Matériaux et constructions", 1968, I, pp. 509-520.
Feldmann, R.F., Sereda, P. J., Commento all'articolo ‛Structures and physical properties of cement pastes' di G. J. Verbeck e R. H. Helmuth, in The V international symposium on the chemistry of cements, parte III, Tokyo 1969, pp. 36-44.
Fuller, W.B., Thompson, S.E., The laws of proportioning concrete, in ‟Transaction ASCE", 1907, LIX, pp. 67-143.
Kondo, R., Daimon, M., Early hydratation of tricalcium silicate: solid reaction with induction and acceleration period, in ‟Journal of the American Ceramic Society", 1969, LII, pp. 503-508.
Lea, F.M., The chemistry of cement, Glasgow 1970.
Mather, K., High strenght, high density concrete, in ‟Journal of the American Concrete Institute", 1965, LXII, pp. 951-962.
Mehta, P. K., Mechanism of expansion associated with ettringite formation, in ‟Cement concrete research", 1973, III, pp. 1-6.
Neville, A. M., Creep of concrete: plain, reinforced, and prestressed, Amsterdam 1970.
Neville, A.M., Properties of concrete, London 1981.
Orchard, D.F., Concrete technology, vol. III, Properties and testing of aggregates, London 1976.
Powers, T.C., Physical properties of cement paste, in The IV international symposium on the chemistry of cements, parte III, Washington 1960, pp. 577-609.
Powers, T.C., The properties of fresh concrete, New York 1968.
Powers, T.C., Brownyard, T. L., Studies of the physical properties of hardened Portland cement paste, in Research laboratory, Portland Cement Association Research. department bullettin n. 22, 1948.
Ramachandran, V.S. (a cura di), Concrete admixtures handbook. Properties, science and technology, Park Ridge, N. J., 1985.
Short, A., Kinniburgh, W., Lightweight concrete, London 1978.
Taylor, H.F.W., The chemistry of cements, London 1964.
Turco, T., Il gesso. Trasformazioni, impieghi, Milano 1961.
Wieker, W., New methods for investigations of the hydration processes of Portland cement, in The VI international symposium on the chemistry of cements, Moscow 1974, vol. II, pp. 1-41.
Wittmann, F. M., Interaction of hardened cement paste and water, in ‟Journal of the American Ceramic Society", 1973, LVIII, pp. 409-415.
Materiali ceramici di Antonio Cocco
SOMMARIO: 1. Introduzione. □ 2. Comportamento al calore delle argille. □ 3. Strutture e proprietà meccaniche dei ceramici tradizionali. □ 4. Strutture e proprietà meccaniche dei refrattari. □ 5. Processi di sinterizzazione. □ 6. Teoria di Griffith sulla rottura fragile dei ceramici. □ 7. Strutture e proprietà meccaniche dei ceramici tenaci. □ 8. Conclusioni e prospettive di impiego dei ceramici tenaci. □ Bibliografia.
1. Introduzione.
I manufatti ceramici sono certamente tra i primi realizzati dall'uomo. La produzione di questi manufatti con tutta probabilità e iniziata contemporaneamente in diversi paesi non molto dopo che l'uomo ebbe imparato ad accendere il fuoco. Infatti è presumibile che l'uomo si sia casualmente accorto di come certi tipi di terre (κέραμος = argilla) a contatto con il fuoco diventassero resistenti come la pietra e di come fosse possibile, prima della cottura, dare loro una forma impastandoli con acqua. Da questa osservazione è nata una semplice tecnologia che ha permesso all'uomo, già 17000 anni a.C., la preparazione in Europa delle prime terre cotte (v. Bassani e Giovannoni, 1931). Nei secoli successivi la produzione di manufatti ceramici si è gradualmente sviluppata e 3000-4000 anni a.C. si era in grado di produrre oggetti non solo di uso comune, ma anche artistici e di uso industriale; si conoscevano le più semplici tecnologie per verniciare a fuoco le ceramiche, l'impiego dei torni per formare le paste crude, la preparazione di mattoni cotti. Nel 1000 a.C. si preparava anche un prodotto in qualche modo simile alla porcellana; ciò dimostra che unitamente alle argille s'impiegavano anche altre materie prime.
Con la decadenza dell'Impero romano la tecnologia ceramica non ebbe un significativo sviluppo e solo nel tardo Medioevo, soprattutto in Italia, furono conseguiti notevoli progressi con la messa a punto di coperte e di smalti che facevano assurgere le ceramiche a oggetti di notevole contenuto artistico (v. Cairola, 1981). Il Rinascimento vide fiorire, sia in Italia che nel resto dell'Europa, un notevole interesse artistico e tecnico per i materiali ceramici, stimolato anche dalla conoscenza delle porcellane cinesi preparate con il caolino, un'argilla particolarmente pura e bianca, allora non conosciuta in Europa. Solo però intorno al 1700 in Europa, per merito dell'elettore di Sassonia Augusto il Forte e del chimico Johann Friedrich Böttger, si riuscì a produrre delle porcellane molto simili alle cinesi; questo fatto è importante perché a questi risultati si poté arrivare solo in quanto il problema venne per la prima volta affrontato non più empiricamente, ma utilizzando le conoscenze che allora si avevano della chimica. Questo nuovo modo di procedere certamente dette nuovo impulso alla tecnologia ceramica, anche nella preparazione di materiali sempre più idonei a essere utilizzati nei forni della nuova era industriale.
Nei secoli XIX e XX i fondamenti chimico-fisici delle tecnologie ebbero un rivoluzionario sviluppo. A seguito di ciò, il concetto di materiale ceramico fu allargato: attualmente con questo termine s'intendono quei prodotti risultanti da una cottura a elevate temperature, formati da una o più fasi, cristalline o vetrose, che contengono contemporaneamente elementi metallici e non metallici tra loro uniti da legami misti ionici e covalenti, con prevalenza dei primi (v. Kingery e altri, 1975). Oggi lo studio e la preparazione dei ceramici non sono più, quindi, basati solamente sulle conoscenze chimiche e fisiche delle materie prime e sui risultati dei procedimenti di cottura, ma anche sulle loro strutture elettronica e cristallina e sul loro modo di preparazione. Inoltre nuovi mezzi di indagine hanno permesso di caratterizzare meglio i materiali ceramici, le cui proprietà sono messe in relazione con le microstrutture, con le caratteristiche delle materie prime e con i metodi di fabbricazione impiegati. Questa metodologia di ricerca si è rivelata molto fruttuosa e non solo ha consentito di spiegare in termini scientifici le proprietà e il comportamento in servizio dei materiali ceramici tradizionali, ma ha anche permesso di preparare una vasta gamma di nuovi prodotti utilizzati praticamente in tutti i rami dell'industria (v. Schwartz, 1985).
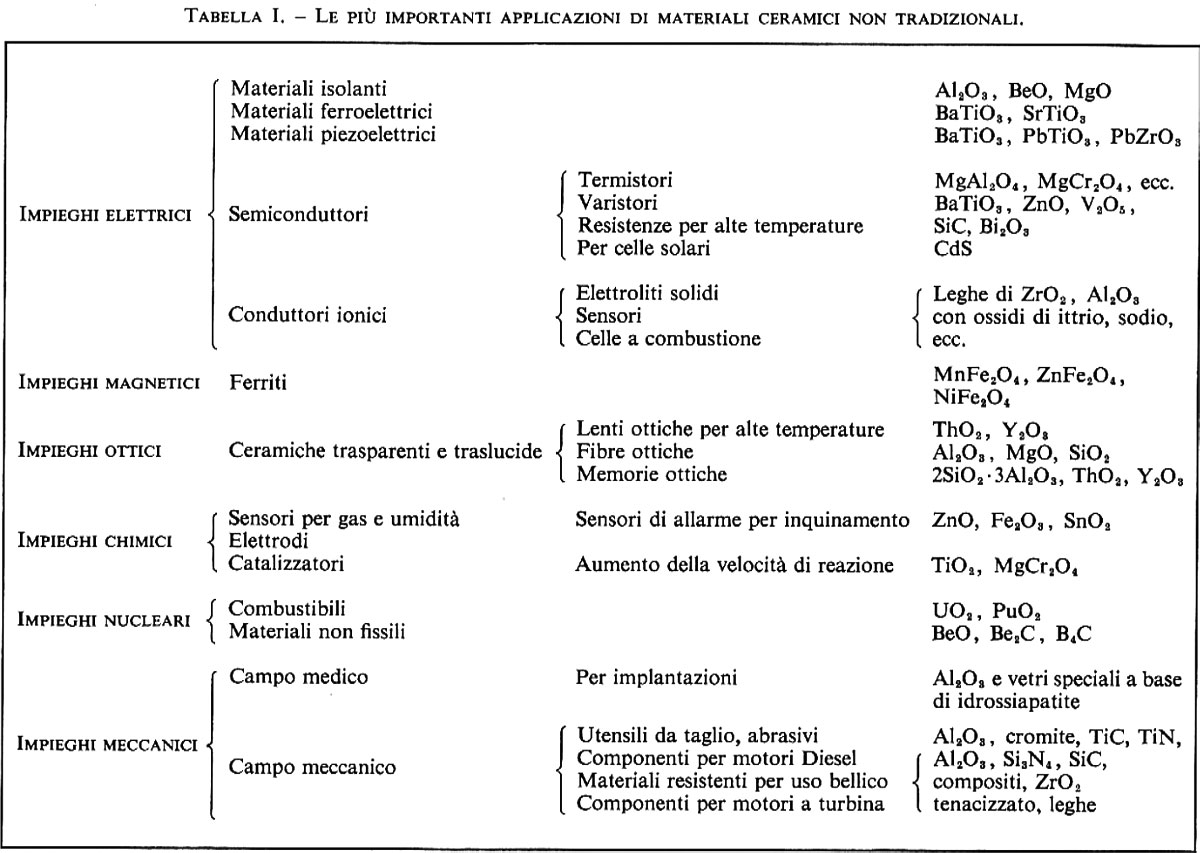
Nella tab. I sono riportate, a titolo di esempio, alcune tra le più importanti applicazioni di questi materiali non tradizionali, messi a punto negli ultimi decenni, che sono conosciuti anche con i nomi di neoceramici, ceramica fine, new ceramics, advanced ceramics, fine ceramies, high technology ceramics e altri ancora. Sono materiali che hanno un vastissimo campo di applicazione e tra i prodotti commerciali sono forse quelli a più elevato valore aggiunto. È però necessario chiarire che questi nuovi materiali non sono, salvo alcune eccezioni, originati da recenti scoperte, ma sono composti ben noti da tempo e alcuni, del resto, anche usati per la preparazione di materiali tradizionali. La novità consiste nei nuovi metodi di preparazione, che, favorendo le reazioni allo stato solido, hanno permesso di raggiungere strutture idonee a utilizzare, alcune volte anche a esaltare, le caratteristiche intrinseche dei composti di base. I nuovi metodi di preparazione consentono di ottenere composti di base molto puri, a granulometria molto fine, e mescole molto ben omogeneizzate; alle polveri così ottenute si applicano nuovi processi di compattazione a caldo e a freddo. Particolare importanza sta anche assumendo la preparazione di miscele per coprecipitazione da soluzioni contenenti i sali dei diversi componenti del materiale ceramico.
Se si procede a un confronto con i materiali metallici, si può subito rilevare che i ceramici hanno: una più elevata resistenza alle alte temperature e alla corrosione, una minore conducibilità termica ed elettrica, un minore peso specifico, una minore resistenza meccanica, una più complessa tecnologia produttiva per ottenere pezzi a geometria precisa. A queste caratteristiche è da aggiungere la larga e diffusa disponibilità in natura delle materie prime necessarie alla loro preparazione, a differenza di quanto avviene per i metalli e in particolare per quelli strategici, idonei alla preparazione delle superleghe. Per chiarire le differenti proprietà tra ceramici e metalli è necessario rifarsi alle caratteristiche di base delle loro strutture (v. Moffat e altri, 1964). I legami chimici nei ceramici sono misti, ionici e covalenti, tra atomi non metallici - elettronegativi - e atomi metallici - elettropositivi. I rapporti percentuali tra i due tipi di legame sono in relazione alla differenza di elettronegatività tra gli elementi che formano il composto. Per quanto gli ossidi abbiano una percentuale di legame ionico in generale ben superiore al 50%, questo non può dirsi per i carburi o gli azoturi, nei quali è di gran lunga prevalente il legame covalente (v. Pauling, 19402). Comunque, il legame che unisce fra loro gli atomi nei ceramici è molto forte; ciò è anche messo in evidenza dall'elevata temperatura di fusione di questi materiali. Diverso è invece il caso dei metalli, in cui gli elettroni di valenza si ‛muovono liberamente', non facendo parte di orbitali molecolari localizzati tra coppie di atomi o attorno ad atomi singoli. Questa disposizione elettronica dà luogo, nei confronti dei ceramici, a una minore energia di legame che, in generale, si manifesta con più bassi punti di fusione. In effetti, mentre le energie di legame nei solidi ionici e covalenti assumono valori che possono anche superare i 1.000 kJ/mole, nei metalli difficilmente arrivano a valori superiori a 800 kJ/mole. Il tipo di legame porta a importanti conseguenze. Nei metalli determina le alte conduttività termiche ed elettriche - per le scarse forze con cui vengono trattenuti gli elettroni di valenza - e le deformazioni plastiche dei reticoli, per il movimento delle dislocazioni quando vengono applicate tensioni di taglio. Nei ceramici il movimento delle dislocazioni è in pratica bloccato e le deformazioni plastiche che si possono generare sono, agli effetti pratici, del tutto trascurabili (v. Hayden e altri, 1965). Conseguenza di ciò è la rottura fragile di questi materiali per la loro scarsa tenacità. Inoltre le strutture elettroniche dei ceramici potrebbero far ritenere che essi non abbiano alcuna possibilità di condurre energia elettrica. Ciò non sempre è vero, in quanto vi sono materiali ceramici semiconduttori elettronici, conduttori ionici e misti. La conduzione elettronica trova la sua spiegazione nella teoria delle bande, mentre la conduzione ionica nei difetti reticolari dei cristalli (v. Rose e altri, 1966).
L'eccezionale sviluppo degli studi e delle realizzazioni industriali fa prevedere una notevole espansione del mercato internazionale di questi nuovi materiali ceramici, nei prossimi 15-20 anni: si presume che il fatturato relativo supererà i 60 miliardi di dollari intorno al 2000. Ciò indipendentemente dai ceramici tradizionali, sia per usi civili che industriali, il cui mercato è quanto mai consistente. Naturalmente questo eccezionale sviluppo è avvenuto a seguito di una intensa attività di ricerca, che è stata soprattutto promossa in Giappone e negli Stati Uniti e, in misura minore, nella Germania Federale, in Inghilterra e in Francia. Altre nazioni, come per esempio l'Italia, pur contribuendo allo sviluppo culturale, si sono tenute al margine di questa rivoluzione tecnologica, con conseguenze che in futuro potranno far sentire il loro peso economico.
Tutto ciò premesso, questo articolo, che non può certamente avere la pretesa di esaurire e approfondire l'argomento, tratterà delle relazioni tra struttura dei materiali ceramici - tradizionali e non tradizionali - e loro proprietà meccaniche. L'articolo seguirà in questo senso l'evolversi delle conoscenze sino agli attuali indirizzi di studio. Partendo quindi dalle strutture dei materiali ceramici più tradizionali, risultanti dall'impiego delle argille, si passerà all'esame strutturale dei refrattari e dei loro meccanismi di compattazione. Si illustreranno quindi i processi di sinterizzazione che, unitamente ai nuovi metodi di preparazione delle materie prime e alle nuove tecnologie di produzione, hanno aperto la strada alla messa a punto di manufatti con strutture meccanicamente più resistenti. Una breve esposizione dei meccanismi di frattura permetterà di orientarsi sulle direttrici di studio che attualmente sono perseguite al fine di realizzare ceramici tenaci, una nuova classe di materiali che unisca i vantaggi dei ceramici alle resistenze meccaniche dei metalli. Nelle conclusioni si farà un accenno alle applicazioni di questi materiali nei motori endotermici.
2. Comportamento al calore delle argille.
Sebbene in questi ultimi anni per la preparazione dei materiali ceramici vengano utilizzate molte materie prime diverse dalle argille, non vi è dubbio che queste rappresentino ancora oggi nella preparazione dei materiali ceramici tradizionali la materia prima di più largo impiego (v. Ravaglioli e Polifrone, 1977). Se quindi si vuole conoscere la struttura di questi materiali ceramici è necessario esaminare il comportamento delle argille alla cottura.
Il termine argilla nell'uso corrente è impiegato in senso molto lato e sta a indicare le terre dotate di plasticità. Questa può essere intesa come la capacità di un materiale di assorbire tenacemente acqua e assumere in conseguenza attitudine a venire plasmato. Una successiva cottura non modifica la forma impartita al pezzo crudo e lo rende meccanicamente resistente. In effetti le argille sono delle rocce, generalmente di carattere sedimentario, provenienti dalla decomposizione di altre rocce. Esse sono formate da componenti plastici, cioè propriamente argillosi, accompagnati da altri minerali non plastici, tra i quali i più frequenti sono il quarzo, il calcare, gli ossidi di ferro, il feldspato e la mica. I minerali argillosi come tali sono dei silico-alluminati idrati in cui il rapporto SiO2/Al2O3 può variare da 1 a 8. I minerali più frequenti hanno rapporti compresi tra 2 e 4 a cui, per esempio, corrispondono rispettivamente la caolinite (Al2O3 • 2SiO2 • 2H2O) e la montmorillonite (Al2O3 • 4SiO2 •H2O) (v. Eitel, 1966, voi. I). Se i minerali argillosi vengono sottoposti a un processo di cottura, a temperatura di 600-700 °C, perdono la loro acqua di combinazione, con conseguente rottura della loro struttura: l'SiO2 e l'Al2O3 possono reagire, se si creano le condizioni opportune, seguendo il diagramma di equilibrio riportato nella fig. 1. In effetti la formazione del composto intermedio, la mullite (3Al2O3 • 2SiO2), avviene con una certa facilità solo a temperature superiori a quelle dell'eutettico, quando la presenza di massa fusa facilita i fenomeni di trasporto. Risulta quindi evidente che, per esempio, una caolinite, che dopo aver perso l'acqua di combinazione ha un tenore di Al2O3 del 46% circa, comincia il suo processo di fusione alla temperatura dell'eutettico e lo termina quando il segmento corrispondente al tenore di Al2O3, perpendicolare all'asse delle ascisse, interseca la curva del liquido. Da ciò si deduce che i minerali argillosi non hanno un punto di fusione proprio, ma fondono progressivamente in un intervallo di temperatura che va sempre più allargandosi quanto più il minerale argilloso è ricco di allumina. Se ora si passa a esaminare le argille, si può vedere che il processo di fusione si complica notevolmente, in quanto, oltre alla presenza di più minerali argillosi, è da considerare la presenza dei minerali accessori che modificano le relazioni di equilibrio precedentemente riportate. Senza entrare nei dettagli, si osserva un notevole abbassamento della temperatura alla quale si forma il primo liquido, mentre la temperatura alla quale si ha la completa fusione della massa, oltre che dalla composizione, dipende dalla granulometria e dalla distribuzione dei minerali accessori (v. Eitel, 1966, voll. III e V). Ne consegue che, da un punto di vista tecnico, le argille si distinguono in base alla loro resistenza piroscopica, che è definita come la temperatura alla quale la punta di una piramide standard a base triangolare, costituita dall'argilla in esame, sfiora la base di appoggio. Più alta è questa temperatura e più l'argilla è resistente al calore (in quanto contiene un più elevato tenore di allumina e una più bassa percentuale di minerali accessori). Le argille che hanno una più bassa resistenza piroscopica sono impiegate per preparare prodotti ceramici che devono essere cotti a bassa temperatura, mentre quelle a più alta resistenza piroscopica sono utilizzate per preparare prodotti che devono essere cotti a temperature più elevate.
3. Strutture e proprietà meccaniche dei ceramici tradizionali.
I prodotti ceramici ottenuti dalle argille possono essere suddivisi secondo lo schema della tab. II, che comprende due distinti gruppi a seconda che la massa cotta risulti porosa o compatta (v. Aliprandi, 1974). Questa caratteristica dipende dalla struttura chimica e dalla tessitura fisica, a loro volta derivanti dalla qualità delle materie prime, dal metodo di fabbricazione, dalle temperature raggiunte. Le strutture chimiche difficilmente sono il risultato di equilibri chimici raggiunti durante la cottura: sono presenti in diverse percentuali, variabili a seconda della qualità dell'argilla e della temperatura di cottura, masse fuse, solidificate, spesso allo stato vetroso, minerali che non hanno reagito, frequentemente corrosi sui bordi, e mullite, non facilmente evidenziabile. Le tessiture fisiche presentano masse fuse, che saldano le masse cristalline, e vuoti, che appaiono come una porosità più o meno fine e diffusa in tutta la massa.
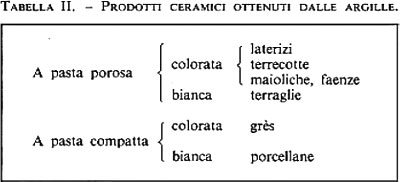
Per i ceramici a pasta porosa colorata si impiegano argille ricche in minerali accessori e conseguentemente a bassa resistenza piroscopica. Le loro temperature di cottura in genere non superano i 1.000 °C e per questo motivo le reazioni tra i minerali presenti nell'argilla di partenza sono poco sviluppate. Di conseguenza nei prodotti cotti si rilevano ancora molti di questi minerali e la percentuale di massa fusa non è mai molto elevata; ciò è in accordo con la loro elevata porosità. Sempre per le basse temperature di cottura è difficile evidenziare granuli di mullite. I prodotti a pasta porosa bianca possono considerarsi un termine di passaggio verso i prodotti a pasta compatta. Le argille di partenza per ottenere un prodotto bianco sono più pure, in particolare meno ricche in ossido di ferro, e spesso sono in tutto o in parte sostituite dal caolino. Unitamente all'argilla è frequente anche l'impiego del feldspato e della mica. Rispetto ai prodotti colorati, sia per le diverse composizioni di partenza sia per le temperature di cottura, in generale più alte (possono anche raggiungere i 1.300 °C), si osservano percentuali di massa fusa più elevate, anche se i prodotti conservano ancora una elevata porosità. Raramente possono essere evidenziati granuli di mullite.
Esaminando i prodotti a pasta compatta è da rilevare in tutti un'elevata percentuale di massa fusa vetrosa, che in questi materiali non ha solamente il compito di cementare i granuli cristallini, ma anche quello di rendere compatto il materiale, riducendone, per quanto possibile, la porosità. Per questo motivo vengono anche chiamati ‛prodotti vetrificati'. Per la preparazione dei grès s'impiegano argille e fondenti con temperature di cottura comprese tra i 1.000 e i 1.300 °C. Nelle porcellane l'argilla viene completamente sostituita dal caolino (v. fig. 2), al quale si aggiungono quarzo e feldspato. Le temperature di cottura oscillano tra i 1.300 e i 1.400 °C. Nelle porcellane la percentuale di massa vetrosa può superare il 60% e in questa si evidenziano granuli di mullite e di quarzo più o meno corroso dalla massa fusa. La porosità si riduce a poche unità per cento.
Questi prodotti ceramici hanno, a freddo, una resistenza meccanica a compressione che va dai 10÷20 MPa, per le terre cotte e i laterizi meno pregiati, ai 50÷100 MPa per le terraglie, ai 200÷300 MPa per i grès, per arrivare infine, nelle porcellane, anche a resistenze dell'ordine dei 500 MPa. Queste resistenze variano a seconda della composizione della miscela di partenza, del metodo di formatura e della temperatura di cottura raggiunta. Si può comunque rilevare che più il materiale cotto è compatto, ricco in masse vetrose e povero di pori, più alta è la sua resistenza meccanica a compressione. Non esiste per questi materiali una stretta relazione tra le resistenze a compressione, a trazione e a flessione. Comunque, le resistenze a trazione per i migliori prodotti vetrificati possono anche superare i 40 MPa, mentre le resistenze a flessione passano da valori di 3 MPa per i materiali porosi a valori anche dell'ordine di 50 MPa per le porcellane. Per questi materiali non possono essere prese in considerazione le resistenze meccaniche a caldo perché i vetri silicei, che legano i granuli, già a temperature di poche centinaia di gradi, a seconda del carico applicato, assumono sotto carico scorrimenti viscosi più o meno rilevanti. Nelle figg. 3 e 4 vengono riportate due microfotografie che evidenziano le strutture delle superfici di frattura di un laterizio e di un grès. È evidente la diversa porosità dei due materiali e la disomogeneità della fase solida del laterizio.
4. Strutture e proprietà meccaniche dei refrattari.
I refrattari sono materiali ceramici impiegati per schermare internamente le apparecchiature in cui si creano elevate temperature. Essi quindi, oltre a un'elevata resistenza piroscopica, o refrattarietà (in generale superiore ai 1.550 °C), devono disperdere il meno possibile il calore generato, devono resistere all'azione corrosiva delle sostanze con le quali vengono a contatto e inoltre devono possedere quelle caratteristiche che permettono il loro impiego per la costruzione di strutture sollecitate sia meccanicamente sia termicamente (v. Norton, 1968).
Sono quindi materiali che rivestono un'importanza determinante in molti processi industriali che altrimenti non potrebbero aver luogo. Peraltro la necessità in molte tecnologie industriali di raggiungere temperature sempre più elevate ha stimolato le ricerche per la messa a punto di materiali sempre più idonei alle richieste. Questi studi hanno portato a una migliore conoscenza dei meccanismi di compattazione e sinterizzazione e hanno costituito il punto di partenza per lo sviluppo di nuove tecnologie per la preparazione di materiali ceramici dotati di nuove e interessanti proprietà. Nella preparazione dei refrattari, oltre alle argille a più elevata resistenza piroscopica, entrano anche molti altri ossidi, quali ad esempio quelli di calcio e magnesio, di alluminio, di silicio, di cromo, di zirconio e altri ancora. A seconda del sistema di fabbricazione i refrattari possono essere suddivisi come è riportato nella tab. III.

Esaminando ora i refrattari formati, appare chiaro che gli elettrofusi, subendo una fusione vera e propria, risultano compatti, mentre i materiali cotti o non cotti (ma che in effetti subiscono la cottura dopo essere stati messi in opera), non raggiungendo la temperatura di fusione, hanno una porosità che varia dal 10 al 40% circa. In sostanza, nella preparazione dei refrattari formati, non elettrofusi, si parte da un compatto di polveri in cui sono contenute sostanze che a una certa temperatura assumono lo stato liquido, con viscosità più o meno elevata, e hanno il compito di far accrescere e legare i grani cristallini refrattari. In definitiva si ottiene una struttura costituita da cristalli, più o meno sviluppati, saldati da masse fuse, frequentemente allo stato vetroso, con una porosità più o meno accentuata a seconda della compattazione raggiunta. I pori, la massa cristallina e la massa fusa svolgono un ruolo determinante, in quanto da essi dipendono la conducibilità termica, la permeabilità, le resistenze meccaniche a freddo e a caldo e tutte le altre proprietà che caratterizzano il materiale. In particolare la resistenza meccanica a freddo è in larga misura determinata dalla compattezza: meno il refrattario è poroso, più elevata è la resistenza alla compressione e questa può anche essere ottenuta con abbondanza di massa fusa.
Le resistenze a compressione a freddo dei refrattari più comuni sono dell'ordine di 40÷100 MPa, mentre le resistenze a flessione sono valutate tra i 15 e i 40 MPa. Le resistenze a flessione e a compressione di questi refrattari sono quindi confrontabili con quelle dei ceramici tradizionali mediamente compatti. Se ora si passa a valutarne le resistenze a flessione a caldo, queste non possono essere certamente alte, sia per le porosità che per le masse leganti presenti. Queste ultime, che durante la cottura sono allo stato fuso, svolgono un ruolo più o meno determinante a seconda della loro composizione e percentuale e dello stato fisico. In effetti questa fase legante, quando il refrattario è in opera, può non risultare più solidificata e, a seguito di reazioni, la sua composizione, la percentuale e la viscosità possono risultare modificate. Se poi le masse fuse sono allo stato vetroso è necessario considerare le temperature e i carichi che danno inizio agli scorrimenti viscosi. Per fortuna le murature refrattarie scaricano le loro tensioni a compressione e conseguentemente è sufficiente che questi materiali abbiano, per un loro valido impiego a caldo, anche a elevate temperature, modeste resistenze a compressione. A questo proposito vengono effettuate diverse prove; tutte, comunque, devono essere considerate indicative perché nel tempo anche deboli carichi applicati, naturalmente a seconda della temperatura e del tipo di refrattario, possono portare a fenomeni di cedimento delle strutture. È quindi logico che nei refrattari formati per cottura, che del resto rappresentano la stragrande maggioranza, si cerchi di ottenere una buona compattezza con la minor quantità possibile di massa fondente. Questo risultato può essere raggiunto se in particolare la fase liquida reagisce con le parti solide.
Nel caso in cui la fase liquida abbia solo funzione legante, il processo di compattazione si limita, per effetto della tensione di vapore, a un accumulo del liquido ai punti di contatto tra i grani e a un ravvicinamento degli stessi per effetto della tensione superficiale della massa fusa. Nel caso in cui invece la fase liquida reagisca con i componenti solidi, ai fenomeni precedentemente visti si sovrappone un fenomeno di ingrossamento dei grani per i trasporti di massa che la fase liquida è in grado di svolgere con processi di soluzione, diffusione e riprecipitazione della fase solida. Con questo meccanismo ha termine il processo di compattazione. Se in molti punti i grani vengono a diretto contatto tra loro e si verificano le condizioni opportune, la densificazione del materiale può continuare con il processo di sinterizzazione (v. Brophy e altri, 1964). Nella fig. 5 è riportato schematicamente quanto esposto. Nella fig. 6 è riportata una microfotografia di un refrattario magnesiaco in cui si distinguono i tre stadi indicati. La resistenza meccanica a compressione a freddo di questo refrattario è dell'ordine di 65 MPa. Nei refrattari tradizionali lo sviluppo del terzo stadio è abbastanza limitato e per questo, come precedentemente si è detto, la porosità residua ha spesso valori anche elevati. Nella tab. IV sono elencati alcuni dei refrattari di più comune impiego: appare subito evidente che tutti hanno una porosità residua elevata, oltre a contenere massa fusa.
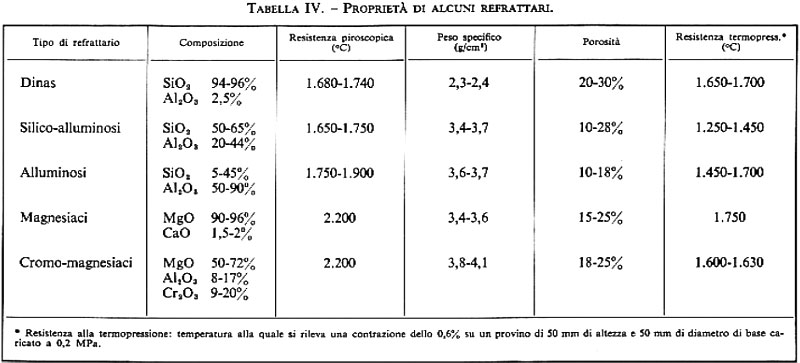
5. Processi di sinterizzazione.
L'attenzione degli studiosi si è quindi rivolta allo studio dei processi con i quali un compatto di particelle fini densifica quando, in assenza di massa fusa, per un certo tempo viene mantenuto a una data temperatura (v. Kingery e altri, 1975; v. Schwartz, 1985). Il processo viene chiamato di ‛sinterizzazione' e la sua ‛forza motrice' (driving force) è data dalla diminuzione dell'energia libera che si estrinseca in una diminuzione dell'area superficiale e quindi della corrispondente tensione superficiale. Per comprendere come avviene il trasporto di massa che porta a una densificazione del prodotto è necessario ricordare che nelle strutture cristalline reali non si ha mai un ordine perfetto. In effetti i reticoli spaziali presentano delle interruzioni, generalmente denominate difetti, localizzate in un punto, lungo una linea o su una superficie (v. Kingery e altri, 1975; v. Moffatt e altri, 1964). I difetti puntiformi sono in sostanza costituiti da nodi reticolari non occupati da atomi (‛vacanze') o da atomi che si localizzano in posizione non dovuta tra altri atomi (‛atomi interstiziali'). I difetti di linea, normalmente denominati ‛dislocazioni', interessano invece più nodi reticolari e tra i più comuni si ricordano quelli a spigolo e a elica. I difetti di superficie si ritrovano sui bordi dei grani dove l'impacchettamento atomico è imperfetto. È opinione comune che i processi diffusionali degli atomi o degli ioni nei reticoli cristallini avvengano per migrazione attraverso i difetti reticolari, la cui concentrazione, a parte quella indotta dalla presenza di impurezze, non dipende solo dalla temperatura ma anche da altri fattori, tra cui la geometria dei cristalli, la tensione superficiale e lo stato di equilibrio meccanico in cui gli stessi cristalli e i loro contorni possono trovarsi. Nella fig. 7 sono schematicamente riportati i meccanismi di diffusione atomica.
Nel caso dei sistemi a un componente, prendendo come modello un compatto di particelle sferiche a contatto tra loro per effetto di una preparazione a pressione, si possono distinguere, durante il processo di sinterizzazione, tre diverse fasi. Nella prima i processi diffusionali per i difetti di superficie dei grani a contatto portano a una coesione delle particelle sferiche. Si ha formazione di ‛colli' (necking stage) sui quali si deposita del materiale sublimato dalla superficie delle particelle. Ciò avviene in quanto la tensione di vapore di un solido varia in funzione della curvatura della sua superficie, e in particolare ove questa è concava la tensione di vapore è minore che non dove è convessa. Il flusso di materia regolato da questo processo è quindi in relazione alla differenza tra le tensioni di vapore delle superfici interessate al processo. Svolge naturalmente anche un ruolo fondamentale la natura chimica della sostanza in esame. Nella fig. 8 viene schematizzato il trasporto per evaporazione-condensazione, che non provoca alcuna contrazione delle due sfere a contatto. Nel secondo stadio ha inizio la contrazione: al processo di evaporazione-condensazione si uniscono la diffusione ai bordi dei grani e la contrazione di volume, che consente un trasporto di materia attraverso la massa. Questo flusso di materia è dovuto alla concentrazione di difetti reticolari più elevata nelle zone concave del collo (che uniscono le particelle) che all'interno della massa e alla superficie dei grani. Si crea quindi un gradiente di concentrazione tra i difetti reticolari che porta a un flusso di materia verso le zone a più alta concentrazione di difetti. Indicando con F il flusso di atomi per cm2 al secondo, si ha F = − D δc/δx, dove c è la concentrazione dei difetti reticolari per unità di volume, x la direzione di diffusione e D il coefficiente di diffusione. Questo dipende dalla temperatura e dall'energia di attivazione E, necessaria al passaggio di un atomo da una posizione reticolare a un'altra vacante, secondo la relazione D = D0 e-E/kT, in cui D0 può considerarsi una costante. In definitiva si arriva, con una diminuzione della tensione superficiale, prima a una struttura policristallina con pori intergranulari tra loro comunicanti, e successivamente a un corpo policristallino in cui i pori rimangono tra loro isolati. Si raggiungono in questo secondo stadio valori di densità dell'ordine del 90% di quella teorica. Solo nel terzo stadio è possibile un'ulteriore densificazione del materiale, se si verifica un flusso di materia verso i pori. Ciò dovrebbe in effetti risultare possibile se la concentrazione dei difetti reticolari è più alta sulla superficie concava del poro che sul contorno e all'interno dei grani. Comunque, con il progredire del processo, si arriva a una rarefazione dei pori e a una loro coalescenza, e nel contempo a un ingrossamento della grana cristallina. Il processo di sinterizzazione tende ad arrestarsi venendo a mancare le condizioni idonee al flusso di materia. Al fine di ottenere la massima densificazione possibile si sono aggiunti all'impasto i cosiddetti ‛attivatori di sinterizzazione', che, ostacolando l'ingrossamento dei grani, dovrebbero favorire il flusso di materia per arrivare alla quasi totale scomparsa dei pori. Nella fig. 9 è schematicamente rappresentato il meccanismo di sinterizzazione. Quanto detto fa ben capire la complessità del processo, che si complica ancor più quando si opera su casi reali e non su modelli. Sono stati studiati anche diversi modelli matematici, che tuttavia sono ben lontani dal rappresentare una trattazione completa (v. Kuczynski, 1979).
Il processo è ovviamente ancora più complesso quando sono in gioco più componenti. Le vacanze presenti sono spesso molto più numerose di quelle che si calcolano allo stato di equilibrio, sia per mancanza di stechiometria dei composti, sia per la presenza di impurezze o sostanze deliberatamente disciolte. L'ossido di zirconio, per esempio, è in grado di sciogliere rilevanti percentuali di ossido di calcio. Ciò comporta che le posizioni cationiche sono completamente occupate, mentre ciò non avviene per le posizioni anioniche, che possono risultare vacanti anche in percentuale di circa il 10%. Per un ossido di zirconio contenente il 14% di CaO sono stati riportati da Kingery e altri (v., 1975), e da chi scrive (v. Cocco e Barbariol, 1963), valori dei coefficienti di diffusione dell'ossigeno, tra 1.200 e 1.300 °C, dell'ordine di 10-6 ÷ 10-7 cm2 s-1. Da misure sperimentali effettuate su una soluzione solida cubica di ZrO2 stabilizzato con CaO sembra che tale diffusione anionica determini la velocità complessiva del processo di sinterizzazione (v. Cocco e Barbariol, 1963).
I processi di sinterizzazione sono certamente influenzati, oltre che dalla natura chimica delle sostanze in gioco, dalla temperatura e dal tempo, dalla tecnologia di preparazione delle polveri, dalla pressatura, dall'ambiente nel quale avviene il processo. Particolare importanza hanno acquistato il sistema di pressatura a caldo (hot pressing) e il sistema di pressatura isostatica con cui si riescono a raggiungere densificazioni elevate. La pressatura a caldo si presenta in pratica insostituibile nella preparazione di nitruri, siliciuri e altri composti che in generale non trovano, per le loro caratteristiche chimiche e fisiche, altri metodi validi di densificazione. Nelle figg. 10 e li sono riportate le microfotografie di due manufatti di allumina. Entrambi sono stati provati a flessione senza averli prima sottoposti ad alcun particolare trattamento. La prima microfotografia (v. fig. 10) evidenzia la struttura di un'allumina pura formata a pressione isostatica e cotta a 1.650 °C; la prova di flessione ha dato valori di 300 MPa. Nella fotografia è indicato con una freccia un difetto. Il processo di densificazione ha raggiunto il 99% del valore teorico. Nella seconda microfotografia (v. fig. 11) è riportata la struttura di un manufatto preparato con allumina con grado di purezza del 96%, ugualmente formato a pressione e cotto alla stessa temperatura. Nella fotografia sono visibili delle cavità, già ripiene di massa fusa formatasi durante la cottura per le impurezze presenti. Il materiale, pur contenendo solo piccole percentuali di massa fusa, ha dato una resistenza a flessione di 150 MPa, la metà di quella precedentemente riportata. Dunque questo esempio mette bene in evidenza l'apporto negativo, ai fini della resistenza meccanica, della massa fusa nella struttura di un materiale densificato.
6. Teoria di Griffith sulla rottura fragile dei ceramici.
Se si considera il tipo di legame, covalente e ionico, e la conseguente impossibilità di deformazione plastica di questi materiali, si ricava che la rottura a trazione di un cristallo ceramico avviene, senza deformazioni plastiche, per stacco dei piani reticolari in corrispondenza di valori teorici di σtraz (carichi specifici a trazione) che dovrebbero risultare dell'ordine di 30.000÷50.000 MPa. Le prove sperimentali, invece, danno valori dell'ordine di 100÷1.000 MPa. In generale si può dire che il σteor si aggira su valori di 10-1E, mentre i σsper su valori di 10-3÷10-4E, dove E è il modulo di Young (v. Jayatilaka, 1979).
A. Griffith riuscì a dare una spiegazione per questo divario di valori. Egli partì dalla teoria di Inglis, secondo la quale ogni intaglio macroscopico costituisce un moltiplicatore di sforzi quando un materiale è messo sotto tensione, e la estese a livello atomico considerando le microfessure superficiali o interne presenti in un pezzo ceramico. Partendo da questi presupposti, Griffith riuscì a preparare sottilissime fibre di vetro, prive di difetti superficiali, del diametro di 2,5 μm con valori di σtraz di 6.500 MPa, mentre i valori medi di resistenza dei comuni vetri sono dell'ordine di 50÷100 MPa. Il valore calcolato per fibre con diametri tendenti a zero è di 14.000 MPa. In effetti, sottoponendo a tensione un monocristallo in cui sia presente una microfessura superficiale si ha una concentrazione di sforzi sul legame all'apice della fessura. Questo legame dovrà quindi ora sopportare uno sforzo molto più elevato del dovuto, in quanto vi si concentrano (v. fig. 12) anche gli sforzi che si sarebbero dovuti scaricare sui legami interrotti. Se tale legame non è in grado di sopportare questo sforzo moltiplicato, si rompe e la microfessura può quindi propagarsi per tutto lo spessore del materiale. Successivamente altri ricercatori inglesi confermarono con studi teorici e sperimentali la teoria di Griffith e lavorando su cristalli di SiO2 e ZnO, privi di difetti superficiali, riuscirono a raggiungere resistenze a trazione dell'ordine di quelle teoriche. Queste strutture, comunemente denominate whiskers, sono le più resistenti mai realizzate.
Partendo da questa teoria si può calcolare la resistenza a flessione di un cristallo mediante l'espressione: σf = Y(2Eγs/C)1/2, dove Y è una costante dipendente dalla geometria del campione e dalle condizioni di prova, E è il modulo di Young, γs l'energia superficiale necessaria a dare inizio alla frattura e C le dimensioni del difetto. Se ora si rappresenta con σfa il carico flettente che innesca la frattura, si ha σfa (1/Y)C1/2 = (2Eγs)1/2. Posto 2Eγs = KIC, chiamato ‛fattore di intensificatore di sforzi' o più comunemente ‛tenacità a frattura', l'espressione precedente può anche essere scritta σfa = Y √-K-I-C-/-C-, evidenziando come il carico di rottura che innesca la frattura fragile aumenti con l'aumentare della tenacità del materiale e con il diminuire delle dimensioni del difetto. Nei materiali ceramici policristallini la propagazione delle fratture viene assicurata dall'energia elastica liberatasi nelle fratture dei grani e viene favorita dai bordi dei grani sui quali la cricca può propagarsi spendendo una quantità minima di energia. D'altra parte lo stesso sistema di preparazione dei manufatti ceramici, basato sulla sinterizzazione di semilavorati costituiti da compatti di polveri cristalline, è tale da comportare nel pezzo la formazione di quei difetti da cui possono partire le cricche. Infatti nei ceramici le tenacità medie a temperatura ambiente sono dell'ordine di 3÷4 MPa √-m, quindi notevolmente più basse di quelle dei metalli, che possono superare anche i 30 MPa √-m. La tab. V riporta alcune proprietà di ceramici a elevata resistenza. Attualmente l'attenzione è particolarmente rivolta, per le loro proprietà intrinseche, al nitruro di silicio (Si3N4), al carburo di silicio (SiC) e alle leghe di ossido di zirconio nelle forme cubica e tetragonale (v. Evans, 1984). Per quanto riguarda il nitruro e il carburo di silicio è da rilevare la natura dei loro legami covalenti e l'influenza che su questi composti ha il sistema di preparazione. A questo proposito la stessa tab. V mette in evidenza l'elevata resistenza meccanica del nitruro di silicio formato per hot pressing, che tuttavia permette solo la preparazione di manufatti in forme semplici. Il nitruro e il carburo di silicio hanno anche la proprietà di resistere bene all'ossidazione a elevate temperature, in quanto si ricoprono di una sottile pellicola di SiO2.

L'ossido di zirconio esiste in tre forme allotropiche (cubica, tetragonale, monoclina) e con aggiunte di CaO, MgO, Y2O3, CeO2 si riescono a ottenere delle leghe in cui l'ossido è in parte in forma cubica e in parte in forma tetragonale. La sua elevata tenacità è dovuta, come si vedrà in seguito, a un'azione di blocco delle cricche per effetto della trasformazione tetragonale-monoclina.
7. Strutture e proprietà meccaniche dei ceramici tenaci.
Da quanto sopra esposto risulta chiaro che per elevare la resistenza a flessione dei ceramici è necessario cercare di ridurre le dimensioni dei difetti e aumentare la tenacità del materiale (v. Creyke e Morrell, 1982). Per ridurre le dimensioni dei difetti si è cercato, migliorando i processi di formatura e di preparazione delle polveri, di preparare materiali con la massima densificazione possibile. Questi sforzi non sono stati ancora coronati da pieno successo, in quanto sarebbe necessario ridurre a 100÷200 nm le dimensioni dei difetti, che, tra l'altro, non risultano individuabili neanche con i più moderni mezzi della tecnica. Poiché allo stato dell'arte si ritiene in pratica impossibile riuscire a preparare manufatti con difetti di dimensioni inferiori ai 10 μm, si cerca di ottenere carichi di rottura più alti tentando di elevare la tenacità del materiale. Dato che la bassa tenacità è una caratteristica intrinseca dei legami chimici che uniscono gli atomi dei ceramici, è necessario aumentarla studiando strutture che possano in qualche modo ostacolare la propagazione delle fessure. I metodi introdotti sono diversi e possono essere divisi in quattro categorie: 1) schermatura delle cricche; 2) interazione delle cricche; 3) realizzazione di compositi; 4) trattamenti superficiali. Vengono qui riportati due esempi.
Mediante il metodo di schermatura delle cricche per trasformazioni di fase si fa in modo che in una matrice meccanicamente resistente (per es. ossido di zirconio cubico, allumina o mullite) sia immersa una seconda fase che, per effetto dello stato tensionale creatosi a seguito della propagazione della fessura in prossimità dell'apice della stessa, subisca una trasformazione di fase, dando origine a una nuova struttura con aumento di volume. Questo genera uno stato di compressione che, opponendosi alla propagazione della cricca, migliora la tenacità del materiale. Nella fig. 13 è riportata una microfotografia di mullite con piccole quantità di massa vetrosa e grani di ZrO2 (circa 10%) in forma tetragonale. Se la cricca nella sua propagazione incontra il grano di ossido di zirconio, questo cambia la sua struttura, che diventa monoclina, con un aumento di volume che dà luogo a uno stato di compressione. Nel caso specifico la tenacità del materiale per la presenza dello ZrO2 tetragonale passa da valori di 2÷3 MPa √-m, propri della matrice, a valori di 5÷6 MPa √-m. Secondo le ultime ricerche sembra che con leghe tetragonali di CeO2 in ZrO2 si siano raggiunti valori anche dell'ordine di 20 MPa √-m. Valori quindi abbastanza vicini a quelli di diverse leghe metalliche.
Il secondo esempio che si riporta è quello di un materiale a matrice ceramica in cui sono immerse fibre unidirezionali o disposte a strati alterni tra loro perpendicolari. Per effetto di simili strutture si ha un aumento della tenacità che è legato a una dissipazione dell'energia elastica, associata alla propagazione della frattura, per deviazione di percorso o per una rotazione del piano di propagazione delle cricche, per scollamento. I compositi si preparano con la tecnica dell'hot pressing. Le matrici sono allo stato vetroso e il materiale deve essere successivamente sottoposto a un trattamento termico per nucleare la fase cristallina. Durante la pressatura a caldo è necessario che non si verifichi un'interazione chimica tra la matrice e le fibre; se ciò avviene le cricche possono propagarsi senza difficoltà. Nella fig. 14 è riportato un esempio di composito con fibre di carburo di silicio incrociate perpendicolarmente a strati, immerse in un materiale vetroso. Nelle figg. 15 e 16 sono riportate due microfotografie relative al diverso comportamento del composito citato, provato a flessione, prima e dopo un ciclo termico. È evidente come in quest'ultimo caso la frattura sia del tutto fragile (v. Sbaizero e altri, 1987). Con tali materiali si è arrivati a ottenere tenacità anche superiori ai 20 MPa √-m, purché naturalmente non vi siano interazioni tra matrice e fibre.
Per aumento di questa tenacità, dunque, i materiali ceramici aumentano la loro resistenza meccanica a trazione e inoltre il diagramma sforzi-deformazioni, che per i ceramici è rappresentabile da un solo tratto lineare relativo al campo delle deformazioni elastiche, assume l'aspetto di quello dei metalli, con evidenti deformazioni di carattere plastico. Nella fig. 17 è appunto riportato il diagramma σ-ε di un composito del tipo suindicato. In esso il σ0 corrisponde alla formazione di cricche nella matrice, il σu al carico di rottura delle fibre seguito dallo sfilamento di queste (pull out), che contribuisce all'aumento della tenacità del materiale. Nella fig. 18 è ancora riportata una microfotografia relativa allo stesso composito, nella quale è ben visibile il benefico effetto delle fibre (v. Sbaizero ed Evans, 1986).
Da quanto detto si può constatare come gli attuali studi sui ceramici stiano avanzando nella direzione giusta. In concreto, mentre nei materiali metallici si ottengono alti valori della tenacità tenendo conto, oltre che del modulo elastico e dell'energia superficiale, anche dell'energia necessaria al lavoro di plasticizzazione 2E(γs + γp), dove γp = 102÷103 γs, nei ceramici si possono ottenere alti valori della tenacità introducendo un fattore strutturale, fs, che moltiplica il valore della tenacità della matrice (2Eγs) × fs.
È infine quanto mai opportuno rilevare la migliore resistenza allo scorrimento a elevate temperature dei ceramici tenaci nei confronti delle superleghe metalliche (v. fig. 19). È questa una importante proprietà che in effetti limita la temperatura di impiego di un materiale.
8. Conclusioni e prospettive di impiego dei ceramici tenaci.
Le strette relazioni che intercorrono tra strutture e resistenze meccaniche dei ceramici risultano ben evidenti da quanto precedentemente esposto. In effetti le resistenze a flessione a freddo dei ceramici tradizionali vanno da 3 a 50 MPa con il diminuire della porosità, anche per effetto della sola massa vetrosa. Peraltro questa fa sì che i ceramici tradizionali, in pratica, non abbiano alcuna resistenza meccanica a caldo. Passando ai refrattari legati da masse fuse, le resistenze a flessione a freddo variano tra 10 e 40 MPa a seconda della loro compattezza. Questi ceramici sono concepiti in modo da potere, a caldo, anche a elevate temperature, resistere a non elevati carichi a compressione. Si è dimostrato quanto sia negativa per questi materiali, ai fini della resistenza meccanica, la presenza di massa fusa. Infatti, anche con materiali tradizionali come l'allumina, quando ben sinterizzati e privi di massa fusa, si possono realizzare resistenze a flessione a freddo dell'ordine dei 300 MPa, mentre bastano piccole percentuali di vetro per abbattere queste resistenze anche di oltre il 50%. Partendo da questa constatazione, per alcuni ceramici si sono potute raggiungere, con i nuovi metodi di preparazione, resistenze a flessione a freddo dell'ordine di 500÷800 MPa, resistenze che questi materiali conservano anche a temperature superiori ai 1.000 °C. I nuovi materiali compositi, a fibre ceramiche in matrice ceramica, con l'incremento dei valori di tenacità hanno consentito un ulteriore progresso, permettendo di superare resistenze a flessione a freddo di 1.000 MPa.
Questi nuovi materiali possono essere impiegati per la costruzione di componenti per motori endotermici, di reattori chimici, di veicoli aerospaziali e, in genere, in tutti quei settori dove vi è necessità di materiali che uniscano un'elevata resistenza meccanica alle caratteristiche intrinseche dei ceramici: elevate resistenze alle alte temperature, all'usura e alla corrosione, accompagnate da basse conduttività termiche e bassi pesi specifici.
Tra le diverse applicazioni è forse opportuno soffermarsi sulla costruzione di componenti per motori endotermici. Già prima del 1970 importanti case automobilistiche avevano promosso programmi di ricerca per verificare se fosse possibile sostituire componenti strutturali metallici con ceramici nei motori termici a ciclo Diesel (v. Cocco e altri, 1985). I vantaggi perseguiti erano quelli di raggiungere temperature più elevate all'interno delle camere di combustione e migliori proprietà isolanti e di resistenza alla corrosione e all'usura. In effetti analizzando il bilancio termico di un motore Diesel, si può subito constatare che simili risultati potrebbero eliminare, o quanto meno ridurre notevolmente, i fluidi di raffreddamento dei motori, ottenendo nel contempo, nei gas di scarico, un elevato contenuto entalpico trasformabile in lavoro utile, con conseguente risparmio energetico. Altro vantaggio non trascurabile sarebbe quello di poter utilizzare, nei Diesel, combustibili meno pregiati degli attuali, anche con elevati poteri corrosivi. In quest'ottica, dopo la guerra del Kippur, gli studi sono stati intensificati in diversi paesi (v. fig. 20).
Certamente i ceramici tenaci richiedono ulteriori studi e sperimentazioni e moltissime sono ancora le difficoltà da superare; tuttavia già si sono ottenuti alcuni risultati concreti: qualche prototipo di motore Diesel ceramizzato è attualmente in fase di sperimentazione sia in Giappone che negli Stati Uniti, mentre in Germania è stato già realizzato dalla Mercedes un motore a turbina parzialmente ceramizzato. Occorreranno ancora diversi anni prima di arrivare a un diffuso impiego dei componenti ceramici nei motori endotermici, ma le prime concrete applicazioni non dovrebbero tardare. Gli esperti hanno previsto un risparmio energetico del 3÷4% sui consumi di petrolio destinato all'autotrazione, se la componentistica ceramica troverà larga applicazione nei motori a ciclo Diesel, consentendo di realizzare l'auspicato motore adiabatico (v. fig. 21).
Questo notevole risparmio energetico è un fatto economico molto importante, ma ancora più importante sarebbe il risultato che potrebbe ottenersi nel campo dell'inquinamento ambientale. Infatti attualmente il motore Diesel, sia per le temperature che si raggiungono al suo interno, sia per le caratteristiche dei combustibili che vi si impiegano, immette nell'atmosfera quantità non trascurabili di particolati organici. Poter elevare le temperature all'interno delle camere di combustione di 200÷300 °C significherebbe rendere meno stabili questi particolati, che potrebbero venire ossidati più facilmente, e ridurre notevolmente la fumosità. Ciò, in definitiva, porterebbe a più basse immissioni, caratteristica che, unitamente al risparmio energetico, potrebbe anche favorire una più larga diffusione dei motori a ciclo Diesel nei confronti di quelli a ciclo Otto.
Un'altra fonte di risparmio energetico è costituita dai nuovi composti superconduttori basati su ossidi ceramici.
È noto che la superconduttività, cioè l'annullamento della resistenza elettrica di certi materiali, ha finora trovato una grossa limitazione nella necessità di impiegare bassissime temperature. Dalle prime osservazioni (1911) su metalli che diventavano superconduttori alla temperatura dell'elio liquido (4,2 K) si è passati (1973) alla preparazione di leghe e composti intermetallici la cui superconduttività si manifesta intorno alla temperatura dell'idrogeno liquido (20,5 K). I progressi maggiori sono stati realizzati nel 1986, quando K. Alex Müller e il suo allievo J. Georg Bednorz, che avevano lavorato con ossidi ceramici per diversi anni nei laboratori dell'IBM a Zurigo, hanno preparato un nuovo ossido di bario, lantanio e rame, la cui resistenza elettrica si annulla intorno a 30 K. Nel corso del 1987 sono stati annunciati, da parte di diversi gruppi di ricercatori, ulteriori miglioramenti: per es., sostituendo il lantanio con l'ittrio, sono state raggiunte temperature di superconduttività fino a 98 K, cioè notevolmente superiori a quella dell'azoto liquido (77,4 K) e quindi facilmente raggiungibili. I superconduttori a temperatura ambiente (circa 290 K) non sono ormai più un miraggio (v. Robinson, 1987).
Le possibili applicazioni ditali superconduttori sono numerosissime: dai calcolatori superveloci ai cavi elettrici con minima dissipazione di energia, ai magneti in grado di generare potentissimi campi, impiegabili negli acceleratori per particelle nucleari, nelle macchine per la fusione nucleare, nei tomografi a RMN e nei treni a levitazione magnetica.
Dell'importanza delle recenti ricerche sui superconduttori ceramici è testimonianza, fra l'altro, il conferimento del premio Nobel per la fisica nel 1987 a Müller e Bednorz.
Bibliografia.
Aliprandi, G., Ceramurgia e tecnologia ceramica, Genova 1974.
Bassani, E., Giovannoni, G., Ceramica, in Enciclopedia Italiana, vol. IX, Roma 1931, pp. 763-777.
Bednorz, J.G., Müller, K.A., Possible high Tc superconductivity in the Ba-La-Cu-O system, in ‟Zeitschrift für Physik, B, condensed matter", 1986, LXIV, pp. 189-193.
Brophy, J. H., Rose, M., Wulff, J., The structure and properties of materials, vol. II, Thermodynamics of structure, New York 1964.
Cairola, A., Ceramica in Italia dalle origini a oggi, Roma 1981.
Cocco, A., Barbariol, I., Meccanismo di sinterizzazione delle soluzioni solide di CaO in ZrO2 con struttura cubica, Istituto di chimica applicata dell'Università di Trieste, pubbl. n. 17, Trieste 1963.
Cocco, A., Lucchini, E., Meriani, S., I neoceramici per lo sviluppo dei motori termici, in Atti delle Giornate di studio su ‛Materiali per tecnologie innovative' (a cura di R. Aiello e A. Nostro), Cosenza 1985.
Creyke, W. E. C., Morrell, R., Design with non-ductile materials, New York 1982.
Eitel, W., Silicate science, vol. I, Silicate structures, New York 1966.
Eitel, W., Silicate science, vol. III, Dry silicate systems, New York 1966.
Eitel, W., Silicate science, vol. V, Ceramics and hydraulic binders, New York 1966.
Evans, A. G., Fracture in ceramic materials, New Jersey 1984.
Hayden, W., Moffatt, W. G., Wulff, J., The structure and properties of materials, vol. III, Mechanical behavior, New York 1965.
Jayatilaka, A.S., Fracture of engineering brittle materials, London 1979.
Kingery, W.D., Bowen, H. K., Uhlmann, D.R., Introduction to ceramics, New York 1975.
Kuczynski, G. C., Sintering processes, New York 1979.
Moffatt, W. G., Pearsall, G. W., Wulff, J., The structure and properties of materials, vol. I, Structure, New York 1964.
Norton, F. H., Refractories, New York 1968.
Pauling, L., The nature of the chemical bond, Ithaca, N. Y., 19402 (tr. it.: La natura del legame chimico, Roma 1947).
Ravaglioli, A., Polifrone, G. G., Materie prime ceramiche, vol. I, I materiali argillosi, Faenza 1977.
Robinson, G. M., La gara nella ricerca sulla superconduttività, in ‟Elettronica oggi", 1987, XLVII, pp. 99-101.
Rose, R., Shepard, L.A., Wulff, J., The structure and properties of materials, vol. IV, Electronic properties, New York 1966.
Sbaizero, O., Evans, A. G., The tensile and shear properties of laminated ceramic matrix composites, in ‟Journal of the American Ceramic Society" (Ohio), 1986, VI, pp. 481-486.
Sbaizero, O., Evans, A. G., Luh, E. Y., Thouless, M. D., Trends in the toughness of fiber reinforced ceramics with the interfacial sliding resistance, in ‟Journal of the American Ceramic Society" (Ohio), 1987.
Schwartz, M. M., Engineering applications of ceramic materials, Ohio 1985.
Materiali metallici di Gernot Kostorz
SOMMARIO: 1. Che cos'è un metallo. □ 2. Proprietà dei metalli. □ 3. Impiego dei metalli. □ 4. Classificazione dei materiali metallici. □ 5. Il processo di fabbricazione dei materiali metallici: a) solidificazione della massa fusa; b) strati sottili; c) metallurgia delle polveri; d) materiali compositi. □ 6. Diffusione nei metalli. □ 7. Trasformazioni di fase: a) diagrammi di fase; b) cinetica delle trasformazioni di fase. □ 8. Plasticità dei materiali metallici: a) misurazione delle proprietà di deformazione e di frattura; b) dislocazioni nei cristalli; c) incrudimento dei metalli puri; d) resistenza meccanica delle leghe; e) deformazione dei metalli amorfi; f) fatica. □ 9. Materiali magnetici e superconduttori. □ 10. Sollecitazione e durata dei metalli. □ 11. Prospettive. □ Bibliografia.
1. Che cos'è un metallo.
I metalli nativi, quali il rame, l'argento e l'oro, sono tra i più antichi materiali usati dall'uomo, ma solamente con l'inizio della produzione di metalli e di leghe a partire dai minerali i materiali metallici raggiunsero una posizione predominante, riflessa nelle denominazioni Età del Bronzo' (in Europa dal 1800 fino al 700 a.C. circa) ed ‛Età del Ferro' (dall 100 a.C. circa). Al giorno d'oggi i metalli e le leghe offrono innumerevoli possibilità applicative. La ricerca attuale si propone, in sostanza, di comprendere e ottimizzare le proprietà dei materiali metallici, come pure di svilupparne nuove combinazioni, anche con materiali non metallici. Metodi e concetti della fisica dello stato solido (v. solidi, fisica dei) vengono utilizzati in misura sempre maggiore.
Accanto allo splendore metallico (alto potere riflettente nel campo della luce visibile), i metalli possiedono un'alta conducibilità sia termica che elettrica, spesso buona duttilità (deformabilità plastica) e, alcuni, particolari proprietà magnetiche. Queste proprietà non sono però sufficienti per una distinzione netta (per es. nei confronti dei semiconduttori, dei polimeri, ecc.). L'interpretazione fisica del legame metallico (v. solidi, fisica dei) fornisce una definizione più precisa: si definiscono ‛metallici' tutti i materiali che possiedono (anche alla temperatura T = 0 K), grazie alla conformazione delle loro bande di energia, cariche libere, ossia bande parzialmente occupate.
I metalli costituiscono oltre l'80% degli elementi. I pochi non-metalli (v. tab. I) diventano metalli a pressioni sufficientemente alte. Miscele (leghe) di metalli tra di loro o con non-metalli forniscono una grande varietà di materiali, le cui proprietà possono venire ulteriormente modificate tramite trattamenti termici, meccanici, termomeccanici e delle superfici.

La scienza dei materiali metallici si prefigge di interpretarne le proprietà macroscopiche a partire dalla relativa struttura microscopica (atomica ed elettronica).
2. Proprietà dei metalli.
L'elevata conducibilità elettrica e termica dei metalli è dovuta alla presenza di portatori di cariche liberi (circa 1023 elettroni/cm3). La fig. 1 illustra la consueta distinzione tra conduttori (elettrici), semiconduttori e isolanti.
La conducibilità elettrica σe è data dal prodotto della densità di portatori di cariche n per la carica q e per la mobilità μ. Mentre nei semiconduttori tipici l'influsso della temperatura T si manifesta primariamente in un aumento di n al crescere di T, la conducibilità tipica dei metalli è caratterizzata da una diminuzione della mobilità al crescere della temperatura (diminuzione del percorso libero medio a causa di un aumento della diffusione di elettroni da parte dei fononi). Quindi, a temperature non troppo basse, la resistenza specifica (ρe = σe-1) dei metalli aumenta in modo lineare con T:
ρe(T) = ρR(1 + xρΔT) (1)
(se ρR = ρe a una temperatura di riferimento, per es. 20 °C), con xρ = coefficiente di temperatura della resistenza elettrica. La tab. II dà alcuni valori di ρR a 20 °C e di xρ.
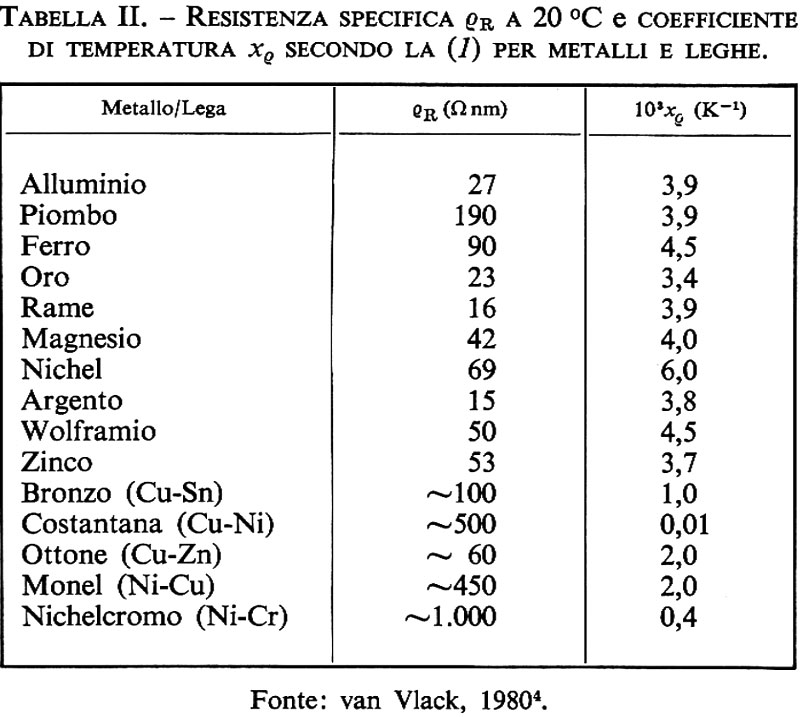
A basse temperature la resistenza diminuisce in modo non lineare con T, fino a raggiungere un valore costante, (normalmente) determinato solo da processi di diffusione (largamente indipendenti dalla temperatura) di impurità e altri difetti (‛resistenza residua', v. fig. 2). La purezza dei metalli viene frequentemente indicata con il ‛rapporto di resistenza residua' cioè il rapporto della resistenza a temperatura ambiente con la resistenza residua, in particolare in presenza di una purezza molto elevata, quando le analisi chimiche delle impurità non sono più attendibili. Le leghe, a seconda della loro composizione, possono possedere un'alta resistenza residua, la quale, per le leghe concentrate, può essere preponderante anche a temperatura ambiente. Di conseguenza il coefficiente di temperatura xρ è piccolo (per es. nel caso di leghe per resistenze elettriche come il nichel-cromo: v. tab. II).
Nei metalli puri la conduzione del calore dipende ugualmente in modo preponderante dagli elettroni di conduzione, sicché il rapporto tra il coefficiente di conduttività termica κ e il prodotto della conducibilità elettrica σe per T risulta essere una costante quasi universale (legge di Wiedemann-Franz, 1853):
κκ/σeT ≃ Le (2)
(Le = costante di Lorentz). La tab. III dà alcuni valori di Le. I particolari della struttura a bande dei metalli puri si riflettono in valori diversi di Le.

Anche la lucentezza dei metalli può essere interpretata in termini di elettroni liberi, poiché la luce visibile può essere fortemente assorbita ed emessa nuovamente da questi elettroni. L'oro (v. fig. 3) e il rame si differenziano dalla maggior parte dei metalli per il colore, poiché il loro potere riflettente varia fortemente già nel campo del visibile, mentre in genere diminuisce solo per lunghezze d'onda minori.
Nei metalli fu osservato per la prima volta il fenomeno della superconduttività (v. criofisica), caratterizzato dalla scomparsa della resistenza elettrica e dall'espulsione completa del flusso magnetico. Occorre tuttavia ricordare che sono stati realizzati recentemente superconduttori non metallici, caratterizzati da una temperatura critica inaspettatamente alta (v. cap. 9).
Analogamente la presenza o meno di proprietà magnetiche non è in generale legata al carattere metallico dei materiali, tuttavia come materiali magnetici vengono di preferenza impiegati metalli e leghe, dato che offrono in aggiunta le tipiche proprietà dei metalli.
Infine la buona deformabilità plastica dei metalli può essere intesa come una conseguenza del legame metallico. A causa del fatto che la direzionalità dei legami è scarsamente pronunciata, le strutture cristalline dei metalli tendono a un riempimento dello spazio quanto più possibile elevato, cioè a tipi di reticolo semplici (v. solidi, fisica dei). Nelle strutture compatte semplici i supporti della deformazione plastica, cioè le dislocazioni (v. solidi, fisica dei), sono facili da produrre e molto mobili, cosicché un impedimento alla deformazione plastica (quindi la resistenza meccanica) si realizza soprattutto attraverso l'impedimento reciproco delle dislocazioni (incrudimento) e attraverso la deviazione dalla struttura del reticolo perfetto (atomi estranei, difetti reticolari, inclusioni non metalliche, fasi estranee precipitate o disperse). Queste inomogeneità controllano molte proprietà macroscopiche, perciò nelle ricerche sui metalli si è sempre interessati a conoscere bene la microstruttura, ossia le proporzioni locali nell'ambito microscopico e submicroscopico, sia sotto l'aspetto strutturale che sotto quello chimico.
3. Impiego dei metalli.
La tab. IV mostra la produzione mondiale di alcuni metalli negli anni 1983 e 1984: l'acciaio (e quindi il ferro) precede di gran lunga gli altri. Ogni anno vengono impiegate molte centinaia di milioni di tonnellate di acciaio in costruzioni della più diversa natura (v. edilizia e costruzioni). Un moderno altoforno oggi può produrre più di 12 kt di ghisa grezza al giorno, circa 500 volte di più che cento anni fa, con un' accresciuta efficacia. Come materiali da costruzione sono importanti, ma più distanziati, le leghe dell'alluminio, del magnesio e del titanio (a causa della loro bassa densità) e le leghe del nichel con l'aggiunta o meno di ferro (a causa della loro resistenza meccanica ad alte temperature). In virtù della loro buona conducibilità elettrica e termica il rame e l'alluminio vengono impiegati in generatori, motori e cavi, in scambiatori di calore (refrigeratori, impianti di condizionamento d'aria), ecc. Il miglior conduttore a temperatura ambiente è l'argento, che però, a causa del suo alto prezzo, viene impiegato come materiale di contatto solo in casi particolari (attrezzi speciali, elettronica). Nella microelettronica viene usato anche l'oro.
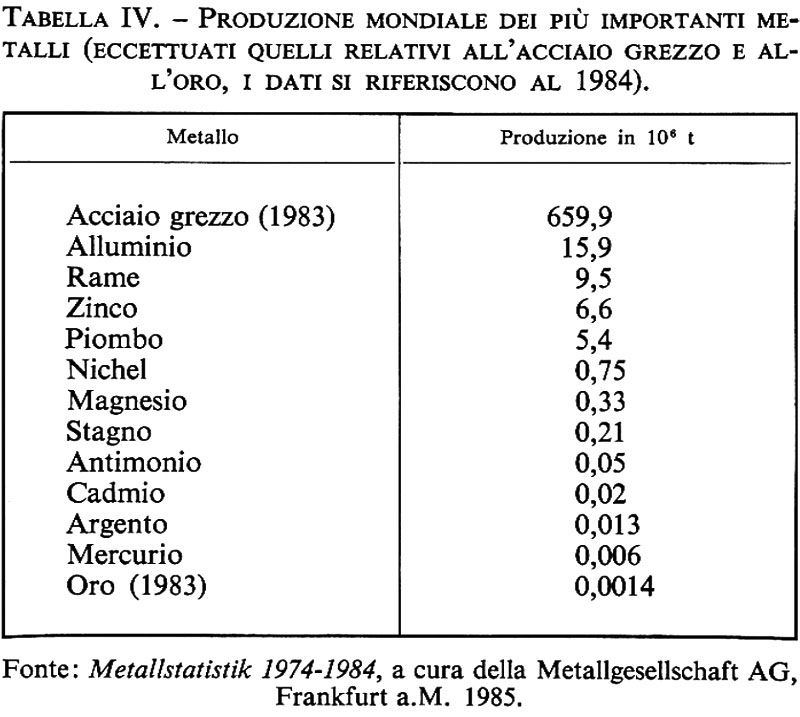
Sono noti molti altri impieghi speciali dei materiali metallici: nell'oreficeria e nella lavorazione artistica sono preferiti i metalli chimicamente stabili (i metalli nobili: argento, oro, platino, rodio); le leghe di ferro, nichel e cobalto forniscono importanti materiali magnetici; le leghe del niobio sono materiali di base della tecnologia della superconduttività; i metalli nobili, i metalli di transizione e le loro leghe trovano impiego nella catalisi eterogenea (v. catalisi eterogenea); lo stagno, lo zinco, il nichel, il cromo e altri metalli vengono impiegati per la protezione dell'acciaio dall'ossidazione e dalla corrosione. Alcune leghe mostrano nelle trasformazioni di fase una ‛memoria' di una forma precedente, che si può utilizzare bene in casi particolari (shape memory alloys; v. meccanica e termomeccanica razionali); le leghe del wolframio vengono impiegate come spirali per lampade a incandescenza e sorgenti di elettroni, a causa del loro alto punto di fusione, mentre il mercurio è notoriamente usato in termometri e barometri per il suo basso punto di fusione e per la sua alta densità.
4. Classificazione dei materiali metallici.
A causa della loro preminente importanza, si distinguono i materiali ‛ferrosi' da tutti gli altri metalli (metalli ‛non ferrosi'). Ai materiali ferrosi appartengono, oltre ai materiali ferrosi ‛puri', quelli che, accanto agli elementi che ad essi si accompagnano in natura (silicio, manganese), contengono, come principale elemento di lega, solo il carbonio. Fra di essi si annovera la ‛ghisa grezza' non malleabile, che può essere ulteriormente elaborata in ‛ghisa da fonderia' (2-6% in peso di carbonio). Per meno del 2% in peso di carbonio si parla di ‛acciaio semplice non legato' (0,03-2% in peso di carbonio), che si lascia forgiare e deformare a freddo. Gli acciai con meno dello 0,6% in peso di carbonio si chiamano anche ‛acciai da costruzione', quelli con contenuto di carbonio più alto ‛acciai per utensili'. Se un acciaio contiene più del 5% circa di altri metalli in lega, per esempio alluminio, silicio, manganese, niobio, cromo, molibdeno, nichel, si chiama ‛acciaio altamente legato', al di sotto del valore indicato ‛acciaio basso-legato'. Altre denominazioni dipendono dalla composizione o dalle proprietà o dall'impiego particolare cui l'acciaio è destinato: si parla, per esempio, di ‛acciaio al cromo-vanadio', di ‛acciaio resistente al calore', di ‛acciaio per valvole', ecc.
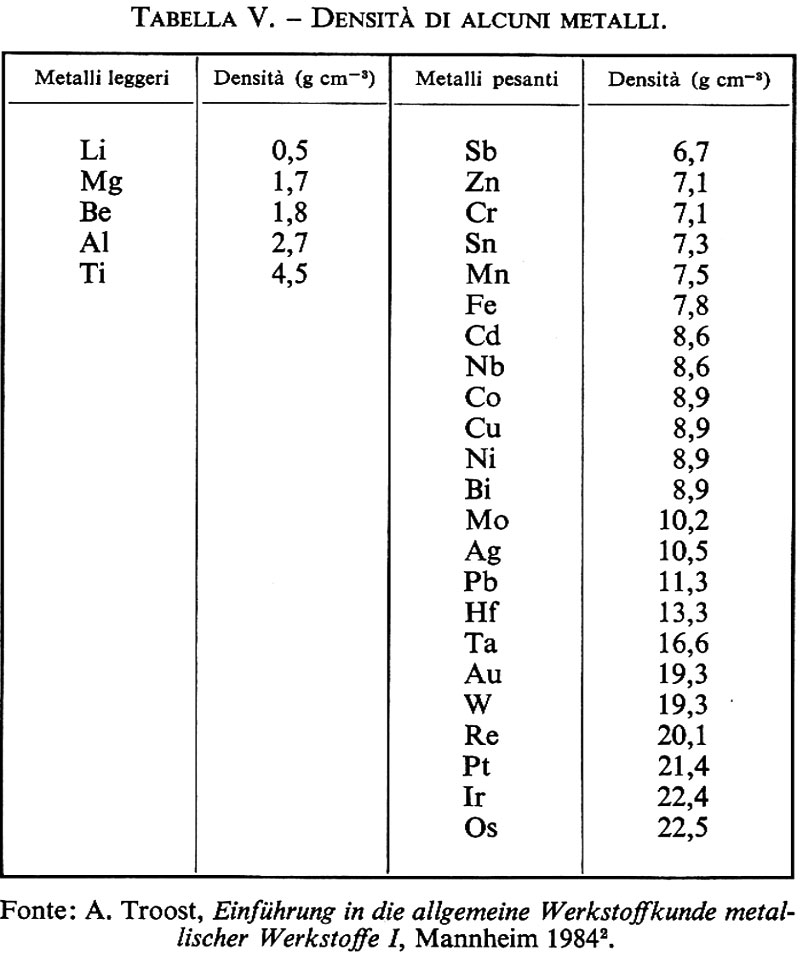

Per i metalli non ferrosi esistono principî di classificazione diversi a seconda della densità, del punto di fusione, della stabilità chimica, del colore, ecc. In base alla densità e (v. tab. V) i metalli si distinguono in ‛leggeri' (ρ 〈 5 g/cm3) e ‛pesanti'. La tab. VI riporta la suddivisione in metalli a ‛basso, medio e alto punto di fusione'. Ai metalli ‛colorati' (Buntmetalle) appartengono il rame, lo zinco, il cadmio, il bismuto e le loro leghe, specialmente l'ottone (lega rame-zinco). Ferro, cromo e manganese si chiamano metalli ‛neri' (Schwarzmetalle), mentre le leghe di stagno, rame, piombo e antimonio si chiamano metalli ‛bianchi'. Le leghe rame-stagno si chiamano ‛bronzi'.
I materiali metallici vengono anche combinati fra loro e con altri materiali a formare materiali ‛compositi', i cui nomi spesso derivano da quelli dei loro componenti: per esempio i cermets (metalloceramiche) sono formati da materiali ceramici che vengono collocati in diverse forme in una matrice metallica, per esempio carburo di wolframio in cobalto o diossido di uranio in molibdeno.
5. Il processo di fabbricazione dei materiali metallici.
I prodotti di partenza per la produzione controllata dei materiali metallici si ottengono, eventualmente dopo più fasi intermedie, o dal trattamento dei minerali o da una loro lavorazione non convenzionale (per es. già il 4% del Cu si produce per mezzo di biotecnologie) o dal riciclaggio di rottami. Molti metalli possono essere miscelati gli uni con gli altri allo stato liquido senza grosse limitazioni, tanto che la maggior parte dei materiali metallici viene prodotta mediante colata di una opportuna lega allo stato liquido. Normalmente i liquidi metallici solidificano in forma cristallina. A velocità di raffreddamento molto elevate (> 106 K/s), raggiungibili soltanto in strati sottili a contatto con un substrato freddo, si ottengono leghe amorfe. Nel processo di solidificazione vengono decisamente influenzate già molte proprietà del materiale. Il trattamento termico e meccanico (allo stato solido) permette però ancora, particolarmente per le leghe, un'estesa gamma di variazioni delle proprietà. Questi interventi possono riguardare le proprietà massive o anche solamente la superficie dei pezzi finiti. Se un getto riceve già la forma definitiva, che mediante lavorazione successiva deve subire ancora solo dei mutamenti insignificanti, si parla di ‛getto in forma'. Per una lavorazione ulteriore viene prodotto un ‛lingotto', che viene convertito mediante fucinatura, laminazione, stiratura, taglio, ecc. rispettivamente in semilavorato, in pezzi finiti e, alla fine, mediante unione (saldatura, brasatura, ecc.), in struttura finita. Spesso i diversi stadi intermedi sono legati a trattamenti termici energeticamente dispendiosi o a processi di sagomatura dai costi elevati. L'applicazione della ‛metallurgia delle polveri' (v. È c) consente di ottenere in un sol passo, attraverso la pressione e la sinterizzazione di polveri, l'aggregazione delle particelle di polvere in un materiale massiccio e la formatura finale o suscettibile di ulteriore lavorazione. In tal modo si riesce, con le polveri adatte, che si ottengono attraverso la solidificazione in goccioline, tramite la separazione elettrolitica o con la riduzione degli ossidi, ecc., a legare (per es. macinando assieme le polveri: lega meccanica) anche metalli scarsamente miscibili allo stato liquido e a controllarne la porosità. I pori restanti si lasciano ‛impregnare' con altri metalli che fondono a temperature più basse (per es. il wolframio col 12-15% in peso di argento: ‛lega a raffreddamento per evaporazione' negli ugelli dei razzi) oppure vengono impiegati per l'assorbimento di lubrificanti (nei materiali antifrizione). Se infine si tratta di produrre rivestimenti idonei su pezzi massicci in lavorazione, allora si possono usare diversi procedimenti fisici e chimici di stratificazione e impianto.
a) Solidificazione della massa fusa.
Già il processo di solidificazione influisce sulla morfologia delle singole fasi, sulla distribuzione degli elementi nelle diverse fasi solide e quindi sulle proprietà del materiale. Il controllo della struttura risultante dal processo di solidificazione avviene perciò con sempre maggior precisione. Per evidenziare la microstruttura risultante vengono impiegati sia processi di microfotografia ottica ed elettronica (v. microscopia) sia metodi microanalitici, mentre per l'identificazione della struttura su scala atomica servono metodi di diffrazione con raggi X (v. proteine), neutroni (v. neutroni) ed elettroni (v. microscopia).
Il presupposto per la solidificazione di un metallo puro sta nel fatto che, al di sotto di una certa temperatura (la temperatura di fusione), l'entalpia libera (di Gibbs) della fase solida, GS, a parità di pressione e temperatura, è minore di quella della fase liquida GL (v. fig. 4). Per la formazione di una fase cristallina è necessario che venga superata una certa dimensione critica del nucleo di cristallizzazione, la quale diminuisce con l'aumentare del sottoraffreddamento. I nuclei possono formarsi omogeneamente, sulla base di fluttuazioni statistiche, ma in pratica prevalgono i nuclei formati eterogeneamente attorno a impurezze solide e sulle pareti. Partendo da nuclei, capaci di accrescersi, della struttura cristallina stabile, si formano nel liquido piccoli cristalli, per lo più con velocità di crescita dipendente dall'orientazione, fino a quando i diversi cristalliti (‛grani') non vengono a contatto e, uniti lungo i loro bordi (bordi di grano), danno luogo a una struttura policristallina (v. fig. 5). Solo quando si riesce a lasciar crescere nel liquido un singolo nucleo si ottiene un corpo cristallino solido senza bordi di grano, un monocristallo. Dato che i bordi di grano portano già un contributo importante alla resistenza meccanica di un materiale, il controllo della dimensione dei grani durante la solidificazione è un compito tecnicamente importante. Durante la solidificazione la distribuzione della temperatura influisce inoltre sulla distribuzione dell'orientazione dei singoli cristalliti. Si indica con ‛tessitura' ogni deviazione dalla distribuzione statistica casuale dell'orientazione dei cristalliti nell'aggregato policristallino. La fig. 6 mostra l'influenza degli elementi in traccia sulla dimensione dei grani dell'alluminio solidificato, la fig. 7 alcuni monocristalli alimentati dal fuso con ‛cristalli di innesco'.
Nelle leghe il processo di solidificazione è più complicato poiché, anche nel caso di una miscibilità completa allo stato liquido e a quello solido, il diverso andamento dell'entalpia libera, che ora dipende dalla concentrazione, porta a concentrazioni diverse nel liquido e nel solido. Nella fig. 8 sono schematizzate queste relazioni per una lega binaria A-B. Quando a una determinata temperatura T2, tra TA e TB, le curve GL e GS si intersecano (v. fig. 8c), si ottiene la concentrazione di equilibrio in entrambe le fasi attraverso la costruzione della tangente comune (poiché il potenziale chimico di entrambi i componenti in entrambe le fasi è uguale). Perciò si può costruire un grafico temperatura-concentrazione (diagramma di stato binario, diagramma di fase), che mostra i diversi equilibri. Con la solidificazione, che ha luogo ora in un determinato intervallo di temperatura, la massa fusa si impoverisce continuamente di un componente (A nella fig. 8), cosicché con la solidificazione è collegata anche una ridistribuzione delle concentrazioni. Se allo stato solido il raggiungimento dell'equilibrio di concentrazione viene ostacolato (magari a causa di un raffreddamento troppo rapido), si producono delle inomogeneità (‛segregazioni'). Al contrario, facendo passare una zona di fusione lungo una sbarra, nel cosiddetto ‛procedimento di fusione per zone', si possono concentrare le impurezze a un'estremità della sbarra e raggiungere con ciò alti gradi di purificazione.
Se allo stato solido esiste solo una miscibilità limitata, si ha il caso, schematizzato nella fig. 9 per un sistema binario, di un diagramma di fase con eutettico. Nel punto eutettico la massa fusa si scinde, senza un rilevante trasporto di materia, in una miscela bifasica (α e β), la cui morfologia può risultare molto diversa a seconda del volume relativo delle due fasi, della loro velocità di crescita, dell'energia superficiale alle loro interfacce, del sottoraffreddamento e della mobilità atomica (v. fig. 10). Sono di particolare interesse gli eutettici con lamelle e fibre. Con un appropriato pilotaggio della solidificazione (‛solidificazione direzionale') si riesce, per esempio, a disporre le lamelle senza interruzioni lungo una direzione e a ottenere quindi materiali con forti proprietà anisotrope.
I liquidi di composizione approssimativamente eutettica sono anche candidati idonei alla formazione di metalli amorfi (vetri metallici), che ultimamente sono stati studiati intensamente a causa delle loro speciali proprietà. Se la temperatura dell'eutettico è bassa, la formazione e la crescita del nucleo si possono frenare con un raffreddamento molto rapido, cosicché al di sotto della temperatura di transizione vetrosa TG ha origine uno stato vetroso e metastabile, ma con una lunga vita. La fig. 11 mostra diverse possibilità di produzione di ‛metalli amorfi'. Particolarmente diffuso è il processo di trafilatura dal fuso (v. fig. 11 b): in condizioni appropriate si possono ottenere con esso nastri amorfi di spessore inferiore a circa 30 μm e larghi alcuni cm.
b) Strati sottili.
Si possono produrre strati sottili di metalli e di leghe anche galvanicamente oppure chimicamente da soluzioni oppure mediante lo spruzzamento o l'esposizione a vapore di un substrato appropriato (anche in questi casi è possibile ottenere degli strati amorfi). Nei moderni impianti di epitassia a raggi molecolari si dispone di una serie di fonti di evaporazione, le quali (nel vuoto molto spinto) consentono la produzione di strutture artificiali a strati con composizione variabile, monofasiche con modulazioni di concentrazione o polifasiche con superfici limite di fase. Con ciò sono diventate possibili anche combinazioni inconsuete dotate di proprietà di tipo nuovo.
c) Metallurgia delle polveri.
Con metodi (elettro-) chimici o della metallurgia nel vuoto si possono produrre metalli e leghe in forma di polvere. Ancora una volta sono possibili alte velocità di raffreddamento del liquido, in virtù delle quali si ottengono piccole (di circa 0,5 μm di diametro) particelle della composizione più svariata. Per la produzione di semilavorati e di pezzi finiti (anche di forma complicata) le polveri vengono miscelate, pressate e riscaldate, cosicché nei luoghi di contatto si arriva a un legame progressivo (controllabile nei dettagli). Questo ‛processo di sinterizzazione' viene controllato dalla tensione superficiale delle cavità (pori). A seconda dello spessore finale desiderato, si sinterizza anche sotto pressione (sinterizzazione a pressione, HIP = hot isostatic pressing). Processi di metallurgia delle polveri vengono impiegati specialmente per materiali metallici ad alta temperatura di fusione (per es. wolframio, composti intermetallici), per evitare i procedimenti metallurgici di fusione che comportano un alto dispendio di energia, per ottenere pezzi finiti di forma complicata da materiali difficilmente lavorabili, ma in misura crescente anche per la produzione di accoppiamenti insoliti di materiali (fra cui le pseudoleghe, cioè leghe di componenti non miscibili allo stato liquido).
d) Materiali compositi.
Strutture eutettiche, prodotti della metallurgia delle polveri, sistemi di strati eterogenei, ecc. sono annoverati, fin quando siano presenti almeno due fasi con proprietà nettamente distinguibili, fra i compositi. Grazie al contatto intimo, microscopico, di materie diverse si ottengono materiali con nuove proprietà. Altri processi per la produzione di compositi sono la saldatura a pressione, la placcatura per laminazione, la placcatura a spruzzo per compositi a strati, l'applicazione di un componente liquido su di uno solido (immersione nel metallo fuso, infiltrazione di materiali sinterizzati) per compositi a strati e a compenetrazione e l'inclusione di fibre in una matrice per compositi a fibre. La fig. 12 mostra come un composito a fibre si formi da fili metallici rivestiti dal materiale di matrice mediante una deformazione comune. La fig. 13 mostra, come esempio, una sezione di un superconduttore a molti filamenti di niobio, circondati da uno strato di Nb3Sn, in una matrice Cu-Sn.
Con questi processi e altri simili naturalmente si possono anche legare metalli con non-metalli. Di particolare importanza tecnologica è la messa a contatto di semiconduttori, poiché con la crescente miniaturizzazione e integrazione si possono produrre anche giunzioni semiconduttore-metallo sempre più piccole e tuttavia riproducibili. In questi e in tutti gli altri casi si tratta di permettere o di impedire, a seconda del risultato desiderato, l'instaurarsi dell'equilibrio termodinamico. Per molte reazioni allo stato solido la mobilità degli atomi determina la velocità della ‛diffusione' di volume, sulle superfici limite o lungo difetti estesi, e controlla quindi la ‛cinetica' delle trasformazioni.
6. Diffusione nei metalli.
Persino in un metallo cristallino puro vi sono atomi, il cui numero aumenta al crescere della temperatura, in grado di vagare, in modo casuale, attraverso il reticolo. Come nei movimenti browniani, il coefficiente di autodiffusione D è legato all'ampiezza quadratica media a2 dei salti casuali degli atomi dalla relazione

dove ν indica la frequenza di salto. Se assumiamo una ripartizione di Boltzmann delle energie di eccitazione degli atomi (per temperature non troppo basse) e ν0 come frequenza dell'oscillazione dell'atomo che porta a uno spostamento efficace, si ha (k costante di Boltzmann)
ν = ν0 exp(-En/kT), (4)
con ED = energia di attivazione per diffusione (i contributi entropici vengono inclusi in ν0). La fig. 14 conferma la dipendenza esponenziale del coefficiente di autodiffusione dalla temperatura (la misura viene effettuata mediante un tracciante radioattivo deposto in superficie). Le energie di diffusione sono di alcuni elettronvolt (cioè circa 100-600 kJ/Mol) e sono, per ragioni fino a oggi non completamente chiarite, approssimativamente proporzionali al calore di fusione e alla temperatura di fusione (relazione di van Liempt). Per contro si è riusciti recentemente, grazie a misurazioni molto precise, a identificare in modo attendibile il meccanismo principale dell'autodiffusione. La fig. 15 mostra alcuni dei processi di spostamento ipotizzabili. Poiché lo scambio di posto diretto con due partners necessita di un'elevata energia di attivazione, bisogna prendere in considerazione altri processi (scambio ad anello, migrazione di vacanze). Lo scambio di posto con una vacanza risulta il più favorevole per le strutture compatte. Poiché anche il numero delle vacanze - controllato dalla loro energia di formazione EF - dipende in modo esponenziale dalla temperatura, l'ED nell'eguaglianza (4) può essere identificata con
ED = EF + EM , (5)
dove EM indica l'energia di attivazione per la migrazione (quindi per lo scambio di posto). Accurate misurazioni della diffusione, della concentrazione di vacanze e della scomparsa di vacanze in eccesso hanno confermato che il meccanismo per vacanze è il meccanismo di diffusione fondamentale nei metalli cubici a facce centrate, esagonali e anche cubici a corpo centrato. Deviazioni in presenza di temperature più elevate vengono interpretate per lo più come contributo della diffusione su coppie di vacanze, ma occasionalmente, in metalli cubici a corpo centrato, anche su uno scambio quadruplo ad anello. Dieci metalli che possiedono modificazione allotropica cubica a corpo centrato mostrano forti deviazioni (valori bassi di D0 e di ED: per esempio Tiβ, Zrβ), che senza dubbio sono connesse al limitato campo di stabilità della fase cubica a corpo centrato.
Il meccanismo per vacanze viene confermato anche per gli atomi estranei nei posti reticolari (cristalli misti di sostituzione). Se, al contrario, gli atomi estranei sono vincolati agli spazi interstiziali (per es. il carbonio nel ferro, i gas nei metalli), allora, in presenza di una concentrazione non troppo alta, vi sono sempre posti a disposizione, cosicché ED viene determinata solo dall'energia di migrazione EM. ED per la diffusione interstiziale è tipicamente uguale alla metà dell'energia del meccanismo per vacanze.
Nei metalli amorfi, la cui densità solitamente è minore di quella della fase o delle fasi cristalline stabili, questo volume ‛libero', che però non può semplicemente essere considerato equivalente a vacanze isolate, può svolgere il ruolo delle vacanze. Come indicano i più bassi valori di D0 rispetto a quelli dei metalli cristallini, le posizioni di punto di sella (quindi gli stati eccitati per lo scambio di posto) sembra siano raggiunte attraverso un movimento più complesso di parecchi atomi, per cui è possibile anche uno spettro di energie di attivazione. Anche nei vetri metallici gli atomi metalloidici e metallici diffondono con velocità diverse. Le misurazioni della diffusione in questi sistemi devono avvenire a basse temperature (Tmax ≤ TG + 20 K, dove TG = temperatura di cristallizzazione). A causa di ciò i valori di D sono piccoli e queste misure sono molto difficili e affette da errori relativamente grandi.
I bordi di grano e le dislocazioni (v. solidi, fisica dei) accelerano la diffusione poiché anche qui esiste una compattezza minore che nel reticolo perfetto. Corrispondentemente la diffusione si svolge in modo considerevolmente più veloce sulle superfici che nel volume, la qual cosa merita considerazione nella tecnica planare. La fig. 16 illustra questi fenomeni. Da ricerche dettagliate risulta che per la diffusione ai bordi di grano svolge un ruolo importante la ‛natura' del bordo stesso (v. fig. 17). Nelle leghe può essere eventualmente introdotto uno spostamento o un riassestamento dei bordi di grano (migrazione dei bordi di grano indotta da diffusione). Tali processi sono dovuti a differenti coefficienti di diffusione dei componenti per la diffusione ai bordi di grano e hanno luogo quando è trascurabile la diffusione di volume. La spiegazione fino a oggi più convincente (Balluffi e Cahn, 1981) postula un effetto Kirkendall nel bordo di grano con il risultato di flussi di vacanze, che rendono possibile il climbing delle dislocazioni ai bordi di grano.
7. Trasformazioni di fase.
Accanto alla già esaminata trasformazione ‛solido' ('cristallino', ‛amorfo') ⇄ ‛liquido', ha luogo allo stato solido una serie di trasformazioni che sono legate alle variazioni di concentrazione e quindi alla diffusione: ‛trasformazioni di fase controllate dalla diffusione'. Inoltre, si possono avere, nei metalli puri e nelle leghe, transizioni di fase che corrispondono a piccoli, ma correlati cambiamenti di posizione degli atomi, senza che siano necessari ampi processi di diffusione. Poiché la velocità di queste trasformazioni dipende solo debolmente dalla temperatura, esse si chiamano ‛atermiche'.
a) Diagrammi di fase.
Come per la solidificazione, anche per le trasformazioni di fase allo stato solido la diminuzione dell'entalpia libera di Gibbs è la forza motrice per il loro decorso. Nuove interfacce e talvolta tensioni di coerenza elastiche si oppongono alla trasformazione. Le fasi solide stabili a una data temperatura e la loro composizione si trovano, nei sistemi binari (come è stato già schematizzato nella fig. 8 nel caso di due fasi), ricercando la tangente comune alle curve entalpia libera/concentrazione per le diverse fasi possibili. La fig. 18 mostra il diagramma di fase in presenza di un composto intermetallico AB molto stabile. Si hanno due sistemi parziali eutettici. (Per sistemi a più componenti la costruzione avviene in maniera simile e i corrispondenti iperpiani tangenti vengono a toccare le ipersuperfici della entalpia libera. La rappresentazione grafica diventa sempre più difficile al crescere del numero dei componenti, cosicché, a partire da quattro componenti, bisogna accontentarsi di rappresentazioni parziali, cioè su diverse sezioni). Se il composto AB diventa stabile solo a temperature più basse, allora si ha un'altra configurazione, come ad esempio quella della fig. 19, relativa a un sistema con miscibilità quasi continua allo stato solido: se si raffredda un cristallo misto al 50% (fase a, distribuzione casuale di atomi nei posti reticolari) al di sotto della temperatura critica Tc, si forma la fase α′, ordinata, nella quale gli atomi A e B assumono alternativamente le possibili posizioni reticolari, come illustrato nella fig. 20 per un piano atomico di un reticolo cubico. Le trasformazioni ordine/disordine appartengono alle più importanti reazioni allo stato solido. Attraverso la messa a punto di una nuova periodicità del reticolo, una lega con ordine a lunga distanza assomiglia molto di più a un metallo puro per quel che riguarda le proprietà elettroniche. Infatti le onde elettroniche non percepiscono più gli atomi estranei come un disturbo della simmetria di traslazione, bensì, a causa dell'ingrandimento dei vettori della base del reticolo ordinato, sono soltanto influenzate da nuovi confini delle zone di Brillouin, per determinate lunghezze d'onda e direzioni di propagazione.
Le superstrutture che derivano dal reticolo cubico a facce centrate sono per es. L10 (AB, per es. CuAuI) e L12 (A3B, per es. Cu2Au), mentre da quello cubico a corpo centrato si formano frequentemente B2 (AB, per es. CuZn = ottone β′), DO3 (A3B, per es. Fe3Al) e L21 (A2BC, per es. Cu2MnAl, leghe di Heusler). Nella fig. 21 sono rappresehtati alcuni ordinamenti atomici.
Allo stato solido può avvenire anche uno smescolamento, come illustra la serie di diagrammi di fase della fig. 22. Invece di un eutettico si rileva innanzitutto un abbassamento della temperatura di solidificazione e un campo di miscibilità completa allo stato solido, prima che appaia una ‛lacuna di miscibilità' per ulteriore raffreddamento.
Un calcolo dei diagrammi di fase deve partire dalle grandezze termodinamiche per le diverse possibili fasi (anche negli intervalli di temperatura e di concentrazione dove esse non sono stabili). Per mezzo di misurazioni più esatte, di analisi sistematiche delle più disparate reazioni allo stato solido e di valutazioni teoriche, il gruppo internazionale CALPHAD (CALculation of PHAse Diagrams) riesce a prevedere, in misura crescente, la stabilità delle fasi. Calcoli ab initio, che presuppongono una conoscenza su basi microscopiche dei potenziali chimici di tutti gli elementi in tutte le fasi in questione non sono al momento ancora possibili con sufficiente precisione. Il progressivo affinamento dei metodi teorici (per es. della teoria dello pseudopotenziale) lascia sperare però in ulteriori progressi.
Un'interpretazione qualitativa del sorgere di lacune di miscibilità o di fasi ordinate emerge già da uno dei modelli più semplici per le soluzioni solide binarie: quello della ‛soluzione regolare'. Qui, per l'entropia di mescolamento SM, viene assunto il valore della soluzione ideale, che, per un dato numero N di posti reticolari suddivisi tra NA atomi del tipo A e NB atomi del tipo B, è
SM = − NkT(cA lncA + cB lncB), (6)
dove cA = NA/N e cB = NB/N. Per l'energia interna si prendono energie di interazione a coppie εAA, εBB, εAB coll'immediato vicino, per cui εij deve essere tanto più negativa quanto più l'attrazione è forte. Sia Pij la probabilità condizionata di trovare un atomo j nel posto vicino a un atomo i; allora (considerando che PAA + PBA = PAB + PBB = 1 e che PBAcB = PABcA), se Z è il numero di immediati vicini (per es. 12 per la struttura cubica a facce centrate), per l'energia interna E di una lega con N atomi si ha
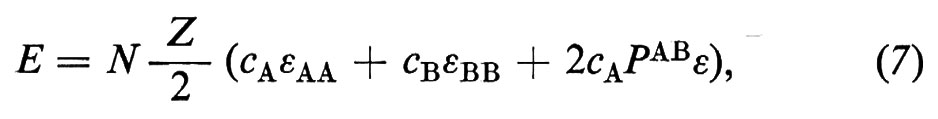
con
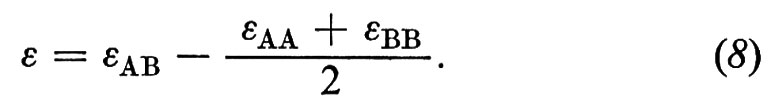
Quando ε = 0, si è di fronte a una soluzione ideale con miscibilità completa. Se ε 〈 0, allora i legami AB sono più stabili dei legami AA e BB ed esiste quindi una tendenza all'ordinamento. Se, al c0ntrario, ε > 0, allora i legami tra vicini uguali sono più stabili: il sistema tende alla separazione dei componenti. Se si pone approssimativamente energia = entalpia, si ottiene da (6) e da (7) per l'entalpia libera della soluzione regolare (riferita a una miscela macroscopica dei due componenti)
ΔGM = NZcAPABε + NkT(cA lncA + cB lncB). (9)
Mentre il secondo termine (contributo entropico) porta sempre a un abbassamento dell'entalpia libera, il contributo positivo del primo termine può prevalere in presenza di temperature sufficientemente basse per ε > 0. Allora una miscela bifasica diventa più stabile della soluzione solida A-B: questo caso è rappresentato dalla fig. 23a, dove è stato posto approssimativamente PAB ≃ cB e dove sono state impiegate temperature ridotte T ′ = kT/Zε. Da ∂(ΔGM)/∂cB = 0 si calcola che al di sotto di una temperatura critica Tc = Zε/2k avviene lo smescolamento. La lacuna di miscibilità data dalla tangente comune ai due minimi locali di ΔGM(cB) diventa sempre più ampia al diminuire della temperatura, cioè la solubilità di A in B e quella di B in A diminuiscono (v. fig. 23b). La seconda curva riportata nella fig. 23b si chiama ‛spinodale' e racchiude la zona in cui la derivata seconda di ΔGM rispetto alla concentrazione diventa negativa. Secondo Hillert, Cahn e Hilliard in quest'ambito si formano continuamente delle piccole fluttuazioni di concentrazione, cosicché è possibile una separazione dei componenti senza formazione di nuclei (‛decomposizione spinodale'). Al contrario nella zona tra la linea di solubilità e la spinodale avviene uno smescolamento con nucleazione e crescita.
Ulteriori trasformazioni allo stato solido traggono origine da instabilità strutturali di una o più fasi interessate. Così, per esempio, il ferro puro solido si presenta in due forme allotropiche: a temperature tra 1.536 °C e 1.392 °C è stabile la fase δ cubica a facce centrate, che a 911 °C si trasforma nella fase γ cubica a facce centrate (austenite), per riapparire a temperatura più bassa (come fase α, ferrite). Con ciò si hanno diagrammi di fase più complicati, a causa delle diverse solubilità degli atomi di lega nelle due fasi. Nella pratica è molto importante il diagramma Fe-C, specialmente al di sotto di circa 6,7% in peso di C (≃ 250% at. di C), composizione del carburo Fe3C, la fase all'equilibrio (metastabile) stechiometricamente più vicina. La fig. 24 mostra l'importante settore del diagramma binario Fe-C, in cui sono disegnate le linee di solubilità (leggermente spostate) dell'equilibrio (metastabile) con grafite.
La solubilità di C in Feγ (austenite) è significativamente più grande della solubilità in Feα (ferrite), poiché la struttura γ cubica a facce centrate mette a disposizione dell'atomo di C siti interstiziali più grandi (v. fig. 25). In entrambe le strutture perciò si arriva a una dilatazione del reticolo, dacché gli atomi di C devono spostare verso l'esterno gli atomi di Fe che li circondano. Nell'austenite tutti gli atomi vicini all'interstizio ottaedrico sono alla stessa distanza, cosicché tutti gli spostamenti sono della stessa grandezza. Nella ferrite l'intorno di un atomo di C nell'interstizio ottaedrico è distorto anisotropicamente, con evidenziazione di un asse. Questa distorsione tetragonale si riflette in effetti anelastici (spostamento degli atomi di C all'applicazione di una tensione meccanica esterna in posizioni per le quali l'asse tetragonale è parallelo a quello cubico sollecitato a trazione). Questi effetti si utilizzano per la determinazione della concentrazione di C e per la misura delle costanti di diffusione di C nella ferrite anche a basse temperature. I campi di distorsione tetragonale forniscono inoltre un contributo importante alla resistenza meccanica della ferrite, poiché ostacolano fortemente il movimento delle dislocazioni (v. cap. 8). Sebbene gli interstizi tetraedrici (v. fig. 25) - assumendo che gli atomi di Fe possano essere visti come sfere in contatto - si presentino all'atomo di C come interstizi grandi quasi il doppio di quelli ottaedrici, tutte le osservazioni fin qui fatte possono essere interpretate senza forzature nel caso di atomi di C su interstizi ottaedrici, in particolare anche la ‛trasformazione martensitica' di Feα-C. Qui gli assi tetragonali degli atomi C si ordinano parallelamente l'uno all'altro, cosicché dalla struttura cubica a corpo centrato si forma una struttura tetragonale a corpo centrato. Per questa trasformazione sono necessari solo spostamenti (coordinati) di dimensioni atomiche, e quindi nessun significativo movimento di diffusione. Se, per mezzo di un rapido raffreddamento, si sopprime l'equilibrio tra ferrite e Fe3C, e quindi la brusca diminuzione del contenuto di C nella fase a al di sotto dell'eutettoide (727 °C), si crea, con alta velocità, al di sotto di una temperatura di soglia MS (temperatura martensite start o temperatura di inizio di formazione della martensite), dipendente dalla composizione, questa fase tetragonale metastabile di grande resistenza meccanica e rigidità. Si osservano trasformazioni martensitiche con caratteristiche atermiche anche nelle leghe non ferrose e molto di frequente anche come fasi di equilibrio a basse temperature. Negli acciai legati possono aversi mescolati insieme anche contributi alla trasformazione martensitica sia atermici che ‛termici', quindi, per una temperatura data, chiaramente dipendenti dal tempo.
b) Cinetica delle trasformazioni di fase.
Le trasformazioni atermiche e le trasformazioni controllate dalla diffusione osservano leggi temporali diverse. Sebbene l'equilibrio stabile, per ogni composizione e per ogni temperatura, sia definito in modo univoco, fino alla sua instaurazione può intercorrere anche una serie di stati metastabili, la cui vita in molti casi dura così a lungo che in pratica essi forniscono dei materiali dalle proprietà utilizzabili. È proprio questa molteplicità delle strutture possibili che permette di impiegare materiali metallici in condizioni così diverse.
Nella formazione di nuove fasi nei solidi, accanto alla differenza nell'entalpia libera che abbiamo introdotto per corpi illimitati, sono di importanza decisiva le superfici limite tra fasi e le deformazioni elastiche (a causa del diverso volume atomico).
Nella teoria classica della nucleazione omogenea, la variazione dell'entalpia libera nella formazione di un nucleo della nuova fase è
ΔG = VΔg + AγG + VΔgS, (10)
con V = volume del nucleo, A = superficie del nucleo, Δg = differenza di entalpia libera specifica, γG = energia specifica di superficie limite e ΔgS = energia specifica di ‛disadattamento' (misfit energy). Per una sfera di raggio R si ottiene (con Δg 〈 0, γG > 0 e ΔgS = 0) l'andamento disegnato nella fig. 26. A causa dei termini positivi della superficie e della deformazione, all'inizio ΔG sale, fin quando, in corrispondenza di un raggio critico
Rc = − 2γG/(Δg + ΔgS), (11)
viene raggiunto un massimo
ΔGm = 16πγ³G/3(Δg + ΔgS)2. (12)
Poiché γG può variare in un ampio ambito, per Rc sono possibili dei valori fortemente differenti per valori confrontabili di Δg. Una superficie limite coerente (v. fig. 27a) con un buon adattamento atomico può richiedere solo alcuni mJ/m2, mentre superfici limite completamente incoerenti hanno bisogno di parecchi dJ/m2. Anche se le costanti reticolari non sono esattamente uguali (caso normale), un'interfaccia coerente è vantaggiosa, almeno fino a quando il termine di deformazione (proporzionale a V!) non prevalga, cioè in presenza di un sottoraffreddamento sufficiente (∣ Δg ∣ ≫ ΔgS) e conseguentemente di raggi corrispondentemente piccoli.
I nuclei il cui raggio è maggiore di Rc possono crescere, secondo la fig. 26, con diminuzione di entalpia libera. La velocità di formazione di tali nuclei in grado di accrescersi dipende in modo esponenziale da −ΔGm/kT. Poiché Δg è in prima approssimazione proporzionale al sottoraffreddamento ΔT = TG − T (dove TG = temperatura di equilibrio, per Δg = 0), ΔGm (eq. 12) diventa proporzionale a (ΔT)-2, cioè la velocità di nucleazione aumenta fortemente al diminuire della temperatura.
Trasformazioni controllate dalla diffusione. - Dato che variazioni di concentrazione sono collegate alla formazione del nucleo, al diminuire della temperatura la frequenza di salto diminuirà secondo la (4). Per ΔGm ≪ ED la velocità di nucleazione è limitata dalla diffusione: sperimentalmente diminuisce quindi con la temperatura. Qualitativamente si ha con ciò la rappresentazione schematizzata nella fig. 28.
Poiché Δg ed ED possono essere fortemente alterate da perturbazioni locali, le superfici, i bordi di grano e le interfacce (altre fasi, pareti del recipiente), come pure le momogeneità nella popolazione dei difetti puntiformi (soprattutto delle vacanze, quando esse influenzano l'energia di diffusione), sono regioni in cui la nucleazione può essere favorita od ostacolata.
La crescita di ‛particelle' di una nuova fase non può essere descritta in termini del tutto generali, anche nel caso di nucleazione omogenea. Per la crescita di particelle isolate (v. fig. 29) è decisivo il rapporto della mobilità delle superfici limite col coefficiente di diffusione (quindi è decisiva la velocità dell'assemblaggio degli atomi diffondenti provenienti dalla superficie limite). In presenza di una elevata mobilità delle superfici limite, la diffusione controlla la velocità e il raggio di una particella cresce secondo un processo di diffusione lineare
R = (2Dαt)1/2, (13)
con t = tempo e α = parametro adimensionale determinato dalla soprasaturazione. Se D al contrario è molto grande, il profilo di concentrazione nella matrice può diventare molto piatto e R diventerebbe proporzionale a t. Per basse soprasaturazioni l'equazione (13) è frequentemente verificata.
Negli stadi più avanzati, quando la matrice è arrivata vicino al valore di equilibrio, rimane come forza motrice per un'ulteriore crescita solo l'energia interfacciale γG Poiché la curvatura di una superficie limite (secondo Gibbs-Thomson) altera la solubilità di un componente in prossimità di un precipitato, ci può sempre essere una corrente di diffusione dalle particelle più piccole a quelle più grandi di un precipitato (v. fig. 30). C. Wagner e Lifschitz Slyosov (WLS) hanno analizzato con precisione questo processo di ridissoluzione e successiva riprecipitazione (maturazione di Ostwald). Per una soprasaturazione trascurabile nella matrice e particelle molto distanti le une dalle altre vale la relazione

a partire da un raggio iniziale R0. Qui R è il valore medio di una distribuzione teorica di dimensioni, la cui nota caratteristica è una ripida caduta, per il 150% del raggio medio. Sebbene si trovi molto di frequente che R3 è proporzionale a t (anche in presenza di una soprasaturazione ancora significativa), la distribuzione delle dimensioni il più delle volte non è verificata.
Nella maggior parte degli esperimenti i singoli stadi qui registrati non possono certo essere osservati separatamente. Nucleazione, crescita, ridissoluzione per variazione delle dimensioni critiche del nucleo e riprecipitazione sono in molti casi frammiste l'una all'altra. Nella fig. 31 è rappresentato un risultato di una ricerca numerica. Un sistema con una linea di solubilità decrescente (per es. v. fig. 22d) all'istante iniziale t = 0 sia rapidamente raffreddato (temprato) dalla fase omogenea α1 al campo bifasico. La soprasaturazione normalizzata S supera il valore 1 al tempo tS e cresce ulteriormente in modo esponenziale; quindi diminuisce di nuovo dopo l'inizio dello smescolamento (quando comincia la formazione di precipitati α2) e raggiunge il valore i in tempi molto lunghi. Il raggio critico Rc varia corrispondentemente: in presenza di una grossa soprasaturazione Rc è molto piccolo, aumenta però di nuovo al progredire dello smescolamento. Un nucleo formatosi ‛troppo tardi', può così crescere inizialmente, ma anche ridissolversi più tardi. La popolazione in questione si sviluppa così con dipendenza iniziale da S(t) e poi da Δg, ΔgS, D e γG, ma si tratta di una relazione statistica valida per tutta la popolazione nel suo complesso: ogni singolo nucleo può crescere con leggi temporali diverse. Bisogna aspettarsi ulteriori complicazioni in presenza di grossi contributi di ΔgS (distorsioni) e di estese interazioni tra le particelle precipitate. Secondo Nabarro l'energia di deformazione elastica nella formazione di una fase dura in una matrice isotropa elastica più dolce è minore per precipitati a forma di lamine e di aghi che per quelli di forma sferica (v. fig. 32). In presenza di una distorsione molto forte i precipitati coerenti possono essere non sferici già fin dall'inizio, negli altri casi un precipitato coerente inizialmente sferico assumerà un'altra forma crescendo ulteriormente. Con una notevole interazione elastica dei precipitati tra di loro si può in seguito arrivare a ordinamenti quasi periodici e a un allineamento periodico preferenziale di particelle lungo determinate direzioni cristallografiche.
Alcuni aspetti della nucleazione, della crescita e della ridissoluzione sono stati studiati in tempi recenti con diversi metodi ad alta risoluzione. La formazione di precipitati di diversa composizione può ad esempio essere documentata con molta precisione con metodi di diffusione di raggi X o di neutroni, specialmente per mezzo della diffusione a basso angolo (v. fig. 33). La diffusione intorno al raggio primario ha una larghezza caratteristica, inversamente proporzionale alle dimensioni dei precipitati. Con i due tipi di radiazione si possono identificare bene particelle con diametri da circa 1 nm fino a parecchie centinaia di nm. I dettagli delle curve di intensità diffusa contengono anche informazioni sulla forma e la disposizione delle particelle diffondenti. Poiché i metalli assorbono i neutroni molto meno dei raggi X, con misurazioni mediante neutroni è possibile acquisire informazioni su campioni di maggior volume. Volumi tipici interessati dai neutroni sono dell'ordine di 0,1-1 cm3, dai raggi X di circa 1 mm3 e dalla microscopia elettronica in trasmissione di 1 μm3 e anche meno. Attraverso la combinazione di microscopia elettronica, diffusione a basso angolo di neutroni e microscopia ionica a emissione di campo con sonda atomica (AP-FIM) (v. fig. 34) è stata studiata particolarmente a fondo, in tempi molto recenti, la formazione di particelle γ′ coerenti e ordinate (struttura L12) in leghe c.f.c. (cubiche a facce centrate) ricche in Ni (v. fig. 35). La fig. 36 mostra R(t) per una lega di nichel col 14% at. di alluminio. Si vede che all'inizio dello smescolamento R rimane all'incirca costante, per poi crescere aumentando proporzionalmente a t1/3. Da ricerche AP-FIM (v. fig. 37) emerge che i primi nuclei riconoscibili possiedono già la composizione definitiva (23% at. di Al: nucleazione classica). La densità delle particelle, Nv (v. fig. 37a), varia all'incirca proporzionalmente a t-1, come previsto dalla teoria WLS (eq. 14), ma la soprasaturazione (v. fig. 37b) è ancora molto grande. Ricerche numeriche più precise su questo sistema hanno mostrato che la nucleazione, la ridissoluzione e la successiva riprecipitazione si sovrappongono e che il comportamento secondo WLS è solo apparente.
Per trasformazioni ‛discontinue' si intendono reazioni di smescolamento nelle quali il fronte di reazione avanza in un campo monofasico instabile e lascia dietro di sé un campo bifasico. Tali reazioni sorgono in particolare nella solidificazione di liquidi eutettici e nella decomposizione entettoidica, per esempio dell'austenite (Feγ-C) con lo 0,765% in peso di C (v. fig. 24), γ → α + Fe3C. Qui sorgono dietro il fronte di reazione due nuove fasi; il fronte di reazione è costituito da superfici limite di fase (v. fig. 38). In un'altra reazione di precipitazione discontinua ugualmente osservata, il bordo di grano diviene fronte di reazione e la struttura cristallina della matrice rimane invariata, per esempio secondo α → α′ + β; cambia solo la sua composizione. La nucleazione in queste reazioni allo stato solido avviene su un bordo di grano o su un'altra o perturbazione e, in presenza di condizioni appropriate (rapporto delle energie interfacciali delle nuove fasi tra loro e con la fase finale, rapporto reciproco tra le quantità delle fasi, coefficienti di diffusione, ecc.), possono sorgere strutture di smescolamento lamellari. La fig. 39 mostra la decomposizione, tecnicamente importante, della perlite del sistema Feα-C. Come la velocità di nucleazione (v. fig. 28), anche la velocità di decomposizione per le reazioni eutettoidiche è bassa per un piccolo sottoraffreddamento, poi aumenta, per diminuire a un ulteriore sottoraffreddamento a causa della difficoltà della diffusione. La dipendenza dal tempo delle trasformazioni per una data temperatura viene spesso descritta (nonostante le più complicate relazioni esemplificate sopra dal sistema Ni-Al) per mezzo della crescita controllata da diffusione a numero di nuclei costante. Con ciò, a velocità di crescita costante, per la frazione trasformata di un campione (n > 0, dipendente dalle particolarità della trasformazione) si ha in generale
X(t) = 1 − exp(− t/τc)n, (15)
con un tempo caratteristico τc che, attraverso la densità dei nuclei (densità delle lamelle) e la velocità di crescita, dipende dalla temperatura. Questa relazione viene chiamata spesso ‛equazione di Johnson-Mehl' o anche ‛di Avrami'. In un diagramma TTT (tempo-temperatura di trasformazione) si riportano, secondo queste rappresentazioni, curve di trasformazione che indicano l'inizio, gli stadi intermedi e la fine di una trasformazione. La fig. 40 mostra un esempio per un acciaio basso-legato con formazione - di perlite tra circa 690 e 560 °C. Gli elementi di lega sostituzionali (qui Ni, Mn, Cr, Si, Mo) ritardano la formazione della perlite, cosicché si può inquadrare più esattamente la microstruttura programmata. Tra circa 490 e 315 °C si ha un ulteriore campo di trasformazione, chiamato ‛bainite'. Qui si trovano Fe3C finemente suddivisa e dispersa, ma anche rilievi superficiali, che indicano una trasformazione per scorrimento, dunque componenti martensitici. Verosimilmente si separa prima Fe3C, per cui la temperatura MS sale nell'austenite. Se si raffredda abbastanza velocemente, per esempio secondo la curva di raffreddamento n. 575 della fig. 40, si giunge nel campo della formazione atermica della martensite. Qui tra MS (temperatura iniziale della martensite) e MF (temperatura finale della martensite) una certa quantità di austenite si trasforma spontaneamente in martensite: la frazione di austenite trasformata dipende esclusivamente dalla temperatura.
La morfologia dei prodotti di smescolamento ottenuta con un primo trattamento termico può essere ancora modificata con trattamenti successivi, per cui si può giungere a una maggiore molteplicità di prodotti di reazione stabili e metastabili.
Decomposizione spinodale. - Nel campo di instabilità di un sistema con lacuna di miscibilità, cioè dove si ha ∂2G/∂c2 (v. fig. 23), può avvenire, secondo Gibbs, una trasformazione detta ‛continua': il coefficiente chimico di diffusione D di una lega (binaria) è proporzionale a ∂2G/∂c2 e quindi, in quest'ambito, le deviazioni della concentrazione dal valore medio non sono ridotte, bensì aumentate dalla diffusione. La separazione delle fasi comincia dunque senza nucleazione e senza raggiungere preventivamente una concentrazione finale, cioè ‛con continuità'. La fig. 41 illustra la differenza rispetto alla nucleazione classica. Se si sviluppa un profilo unidimensionale di concentrazione come funzione del luogo e del tempo in componenti di Fourier (qui se ne consideri soltanto uno), Q = 2π/λ con λ = lunghezza d'onda dell'onda di concentrazione,
cB(x, t) − c0 = CQeiQx+R(Q)t, (16)
allora questa relazione soddisfa l'equazione di diffusione linearizzata e si ottiene un fattore di amplificazione R(Q) = − DQ2, quindi un valore positivo quando D 〈 0 Secondo questa semplice impostazione, le lunghezze d'onda più piccole (Q grande) crescerebbero alla massima velocità, col che sorgerebbero su scala atomica dei gradienti di concentrazione molto ripidi, che necessitano di un elevato dispendio di energia (poiché coppie disuguali nel caso dello smescolamento sono meno stabili di coppie uguali). Cahn e Hilliard (1958) hanno introdotto una correzione per le leghe non omogenee, che tiene conto, per queste ultime, del contributo di energia dipendente dalla ripidità del gradiente di concentrazione (energia di gradiente). Inoltre, a causa del variare con la concentrazione della distanza interatomica allo stato solido (misurata per es. per mezzo di δ = dln a/dc, con a = costante reticolare per le sostanze cristalline), si ha un termine di energia di deformazione elastica, cosicché infine
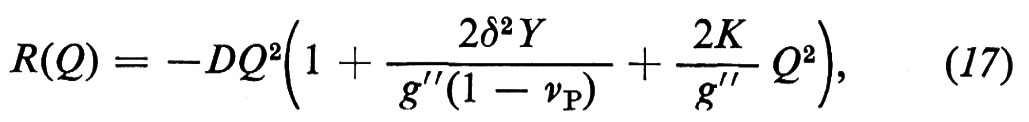
con Y = modulo di Young, νP = coefficiente di Poisson, g′′ = derivata seconda dell'entalpia libera specifica rispetto alla concentrazione e K = costante di proporzionalità per il contributo dell'energia di gradiente, K(dc/dx)2. A causa dell'energia di deformazione, non è la spinodale chimica (g′′ = 0) bensì la spinodale coerente [g′′ + 2δ2Y/(1 − νP) = 0] la curva limite per l'inizio della decomposizione spinodale (esami più accurati forniscono spinodali coerenti dipendenti da Q, giacché bisogna scomporre il campo di deformazione nelle componenti di Fourier). R(Q) però ora diventa positivo solo per piccoli valori di Q, poiché per Q > Qc (Qc si ricava eguagliando a zero l'espressione tra parentesi nell'equazione 17), quando K > 0 e g′′ 〈 0, l'espressione tra parentesi nell'equazione (17) diventa negativa. Qualitativamente R(Q) ha l'andamento mostrato nella fig. 42. Esiste una lunghezza d'onda λm = 2π/Qm, per la quale R(Q) diventa massimo. Da uno spettro delle perturbazioni iniziali si deduce dunque questa modulazione di concentrazione. Nel quadro del modello delineato, che deriva dall'equazione della diffusione linearizzata (quindi D indipendente dalla concentrazione), le misure di diffusione a basso angolo dovrebbero mostrare un'intensità I = I0 exp[2R(Q)t] e quindi, in particolare, un aumento per Q 〈 Qc, una diminuzione per Q > Qc, una posizione di massimo costante nel tempo per Qm = Qc/√-2 e un Qc indipendente dal tempo. Queste caratteristiche si osservano solo molto raramente. È necessario tener conto fin dal principio delle fluttuazioni termiche (Cook 1970) e della tendenza alla ridissoluzione. Attualmente vengono sviluppate diverse estensioni analitiche e numeriche della teoria di Cahn-Hilliard. La fig. 43 mostra un confronto tra curve di diffusione a basso angolo secondo la teoria lineare e secondo un'estensione della teoria e i risultati sperimentali per una lega al centro di una lacuna di miscibilità simmetrica.
Numerose leghe, per es. Cu-Ni-Fe, Cu-Ni-Sn, Cu-Ni-Cr, Fe-Co-Ni-Al, Fe-Cr-Co, ecc., mostrano strutture di decomposizione modulate periodicamente con λ ≈ 10 nm e interessanti proprietà, per esempio elevata rigidità e resistenza meccanica, elevata forza coercitiva magnetica. In questi ultimi anni vengono prodotte anche strutture modulate artificialmente con l'epitassia a raggio molecolare (molecular beam epitaxy = MBE) per mezzo dell'evaporazione alternata di strati diversi. Tali strutture consentono di studiare anche il caso dell'eguagliamento della concentrazione, quindi del mescolamento, a partire da uno stato iniziale ben definito. Nella decomposizione spinodale la caratterizzazione dello stato iniziale è problematica, poiché si tratta di stati di non equilibrio ottenuti mediante un brusco raffreddamento, sui quali hanno un influsso determinante la velocità di raffreddamento e la densità di difetti (dislocazioni e vacanze), senza che sia possibile determinarli bene.
Cinetica di ordinamento. - Per la formazione di uno stato ordinato senza vanazioni di concentrazione, sono necessari spostamenti all'interno della cella elementare (v. fig. 21), cosicché alla fine viene occupato un reticolo parziale su grandi distanze, corrispondentemente al nuovo ‛piano di costruzione' della superstruttura. All'inizio può esserci anche qui un processo di nucleazione, per cui dei ‛domini' ordinati crescono all'interno del cristallo misto disordinato (corrispondentemente alla cinetica di Johnson-Mehl, v. equazione 15) fin quando non si urtano l'uno con l'altro. Poiché l'occupazione di reticoli parziali equivalenti ha luogo in modo non correlato nello stadio di nucleazione, compariranno, ai confini dei domini, dei salti di fase (v. fig. 44). Questi ‛limiti di antifase' poi, analogamente a un processo di maturazione di Ostwald, nel quale l'energia interfacciale provvede all'ingrossamento, vengono eliminati tramite un'ulteriore crescita di dominî appropriati. Le particolarità dell'ordinamento possono presentarsi molto diverse a seconda delle relazioni di simmetria tra la fase ordinata e quella non ordinata e a seconda della mobilità degli atomi. Così si può scarsamente influenzare la trasformazione dell'ottone β, cubico a corpo centrato, nella fase β′ ordinata tra 454 e 468 °C per mezzo di un rapido raffreddamento, mentre per esempio la trasformazione del sistema Cu col 25% at. di Au, cubico a facce centrate, in Cu3Au (a 390 °C) avviene molto più lentamente e può essere completamente bloccata tramite tempra. Nella trasformazione dell'ottone β il parametro di ordinamento η, alla temperatura critica Tc, tende a zero con continuità (trasformazione del secondo ordine), mentre per Cu3Au esiste già a Tc un grado di ordinamento finale (trasformazione del primo ordine), cioè per la formazione del nucleo bisogna superare un'elevata barriera di attivazione (v. fig. 45). Nel modello del reticolo discreto, Cook, de Fontaine e Hilliard (1969) hanno esteso l'idea della decomposizione spinodale anche ai processi di ordinamento. Certamente, nell'eguaglianza (17), g′′ è sempre positivo per il fattore di amplificazione R(Q) nel caso di tendenza all'ordine, quindi è anche D > 0. Ora, poiché le coppie disuguali sono più stabili di quelle uguali, il termine dell'energia di gradiente diventa negativo (K 〈 0) per un Q appropriato, per esempio Q = 2π/a per un reticolo cubico primitivo, cosicché, in presenza di un sottoraffreddamento adatto, R(Q) può assumere anche valori positivi (v. fig. 46). Con ciò si apre una strada per un ordinamento continuo (‛ordinamento spinodale') anche per sistemi per i quali, secondo la teoria classica, il grado di ordine dovrebbe variare a salti.
Trasformazioni atermiche. - Tramite la diminuzione di simmetria di una struttura altamente simmetrica, è possibile un gran numero di trasformazioni di fase atermiche che formano oggetto della ricerca attuale sullo stato solido. Per i materiali metallici, per esempio, appartiene a questa classe la trasformazione ω in leghe di titanio, zirconio e afnio. Invece della trasformazione, controllata dalla diffusione, della fase cubica a corpo centrato ad alte temperature nella fase esagonale compatta stabile a temperature più basse, si forma, dopo un rapido raffreddamento e attraverso un collasso parziale o totale di due piani (111) adiacenti della struttura cubica a corpo centrato, una fase metastabile trigonale o esagonale (fase ω). La causa di questa e di altre trasformazioni è la stabilità precaria della struttura cubica a corpo centrato. In presenza di pure forze centrali questo reticolo è instabile rispetto allo scorrimento su piani (110), ma viene stabilizzato a temperatura sufficientemente alta a causa dei maggiori contributi entropici. Anche la trasformazione atermica più importante, quella dell'austenite rapidamente raffreddata in martensite, ha la sua causa in un'instabilità che, tramite l'impedimento della diffusione degli atomi estranei in soluzione (in particolare C) e la forte soprasaturazione legata a ciò, porta a un ribaltamento coordinato di tutta una porzione del reticolo. Cristallograficamente esistono molte possibilità, tramite una deformazione omogenea, di ottenere da una cella cubica a facce centrate una cella cubica - o tetragonale - a corpo centrato con un rapporto assiale dato. Nella fig. 47 è rappresentata la ‛corrispondenza' che risale a Bain (1924) (per la quale occorrono le minime deformazioni). Quindi ci si aspetterebbe, nell'acciaio parzialmente trasformato,
che per esempio le direzioni fossero [01-1]γ//[11-1]M e [101]γ//[001]M, e parimenti i piani dovrebbero essere (011)γ//(-121)M. Tuttavia si osservano relazioni di orientazione sostanzialmente più complicate che possono anche variare con la composizione. Wechsler, Liebermann e Read (1953) hanno potuto descrivere in modo puramente strutturale tutti i passaggi della trasformazione martensitica tramite una combinazione della deformazione di Bain con una rotazione aggiuntiva (per ottenere le corrette relazioni di orientazione) più uno scorrimento a reticolo invariante (per compensare la variazione di forma). Lo scorrimento a reticolo invariante (v. fig. 48) può aver luogo tramite lo slittamento reciproco di lamelle sottili o attraverso la formazione di regioni con orientazione di gemmazione (un difetto planare con bassa energia specifica) ed è responsabile della microstruttura interna della frazione martensitica della struttura trasformata (v. fig. 49a). Sulle superfici libere si presentano inoltre cambiamenti di forma e deformazioni dell'austenite circostante che portano a effetti di rilievo tipici nei materiali trasformati martensiticamente (v. fig. 49b).
A seconda del tipo di trasformazione inversa si distinguono le martensiti termoelastiche dalle non termoelastiche. Nella fig. 50 è rappresentata la differenza nell'isteresi della trasformazione per questi due tipi. In una lega Fe-30% di Ni, col raffreddamento, crescono spontaneamente nella matrice austenitica delle lamelle di martensite (atermiche), fin quando non hanno raggiunto un certo spessore. Col riscaldamento queste superfici limite non retrocedono, ma la trasformazione inversa comincia attraverso una nuova nucleazione della fase austenitica all'interno di quella martensitica. L'adattamento elastoplastico delle lamelle di martensite nella fase madre austenitica ha portato chiaramente a un ancoraggio delle superfici limite. Nella fig. 50 è visibile un'isteresi molto grande. La situazione è simile negli acciai al carbonio, solo che la trasformazione inversa è influenzata da processi di diffusione. Trattamenti termici diversi (rinvenimenti) portano a diversi prodotti di decomposizione della martensite e a migliorate proprietà meccaniche (bonifica). Nella trasformazione martensitica osservata nelle fasi β cubiche a corpo centrato (ad es. Au-47,5% di Cd) con 1,5 elettroni di valenza per atomo, compare al contrario un'isteresi molto piccola (l'adattamento è sostanzialmente elastico e reversibile): col riscaldamento le superfici limite retrocedono, le lamine di martensite si contraggono e si trasformano di nuovo nella fase madre utilizzando energia elastica immagazzinata. Con ciò vengono mantenute le relazioni di orientazione. Questi aspetti della trasformazione vengono utilizzati nelle ‛leghe con memoria della forma' (v. meccanica e termomeccanica razionali). La trasformazione martensitica per scorrimento può essere indotta anche mediante forze esterne appropriate (per gli acciai si parla di Plasticità Indotta da TRasformazione = TRIP), alle quali è collegato un cambiamento di forma. Nelle martensiti termoelastiche questo cambiamento di forma può, in condizioni appropriate, esser fatto regredire completamente.
La crescita della fase martensitica ha luogo nella maggior parte dei casi a seconda della temperatura (e della sollecitazione meccanica) spontaneamente (la velocità di crescita è limitata superiormente dalla velocità del suono nel materiale in trasformazione). La formazione di una lamella è preferita a causa dell'energia di deformazione favorevole (v. fig. 32); la lamina cresce in larghezza e in spessore. Talvolta si formano anche delle morfologie martensitiche a forma di ago e altre ancora, dipendenti dai processi di adattamento elastici e plastici nella fase madre. Mentre gli aspetti cristallografici e cinetici di crescita della trasformazione martensitica hanno sempre potuto essere compresi compiutamente, la nucleazione rimane ancora non chiarita. Stime dell'energia di nucleazione omogenea (cfr. eguaglianza 10) per trasformazioni martensitiche tipiche nell'acciaio forniscono per ΔGm (v. eguaglianza 12) un valore di circa 105kT, ma con ciò non è possibile raggiungere la consueta concentrazione di 106 nuclei per cm3 in presenza di sottoraffreddamento, attraverso fluttuazioni termiche. Bisogna quindi supporre nuclei preesistenti (speciali ordinamenti appropriati di difetti, per es. gruppi di dislocazioni, microgeminati) o meccanismi di nucleazione discreti, fin qui non osservati, col contributo di dislocazioni e/o di instabilità del reticolo.
8. Plasticità dei materiali metallici.
La deformazione plastica di materiali cristallini ha luogo prevalentemente tramite il movimento delle dislocazioni (v. solidi, fisica dei), talvolta anche tramite formazione meccanica di geminati o trasformazioni indotte da tensione. I metalli amorfi hanno un'elevata resistenza e scarsa deformabilità plastica poiché vengono meno i meccanismi di deformazione legati alla simmetria di traslazione del cristallo.
Nell'uso pratico i materiali metallici sono soggetti a sollecitazioni meccaniche molteplici, complicate e non sempre comprensibili in ogni particolare, combinate con influssi termici ed effetti di superficie. Nello sviluppo e nelle prove dei materiali alcuni metodi standard hanno dato dei buoni risultati.
a) Misurazione delle proprietà di deformazione e di frattura.
Un metodo di prova semplice, per quanto poco chiaro nei dettagli, per determinare la resistenza di un materiale alla deformazione plastica è la ‛prova di durezza': un piccolo penetratore molto duro viene premuto con una forza nota sul materiale da esaminare e le dimensioni dell'impronta sono misurate eventualmente al microscopio. Come numero di durezza viene stabilito il quoziente tra il peso di prova F e la superficie dell'impronta che ne risulta (in MPa, ma spesso anche in kg/mm2). A seconda della forma del penetratore si distingue tra durezza Vickers (HV, penetratore piramidale, angolo di apertura 136°), durezza Rockwell (HRc, penetratore conico, angolo di apertura 120°) e durezza Knoop (penetratore a forma di scalpello). Con una miniaturizzazione adatta si può misurare la durezza con una buona risoluzione locale (≾ 100 μm: microdurezza).
Un'altra prova, che parimenti interessa la massa del materiale e che fornisce una misura del lavoro di deformazione assorbito fino alla frattura è la prova di resilienza. A questo scopo si utilizza un pendolo-maglio, che colpisce un campione intagliato in modo standardizzato. In base all'altezza iniziale h1 e all'altezza di oscillazione del maglio h2 (v. fig. 51a), si determina l'energia ricevuta dal campione, la quale, riferita alla sezione del campione A, viene definita come resilienza specifica (J/m2).
Per i materiali fragili la resilienza è piccola (vengono formate solo due superfici nuove, γK ≃ 2γS, con γS = energia superficiale), mentre γK diventa tanto più grande quanta più energia deve essere impiegata per la deformazione plastica prima della rottura. La fig. 51b mostra risultati tipici per l'acciaio, in particolare la temuta fragilità degli acciai ferritici a temperature intorno allo 0 °C, che dipende sensibilmente dal contenuto di idrogeno.
Per lo studio del comportamento a frattura si è affermata, nell'ambito della meccanica della frattura, una certa standardizzazione. Durante l'allargamento di una cricca vengono rotti dei legami atomici stabili, cosicché si formano delle nuove superfici. In dipendenza dal comportamento plastico viene accumulato nelle vicinanze della cricca anche lavoro di deformazione. L'energia necessaria all'allargamento della cricca viene procurata in parte attraverso l'energia elastica accumulata, in parte attraverso lavoro esterno. Secondo Griffith, in un continuo isotropo elastico, che è soggetto alla tensione σ, per una cricca lenticolare di diametro 2r, si ha come bilancio di energia per una frattura fragile (senza deformazione plastica)
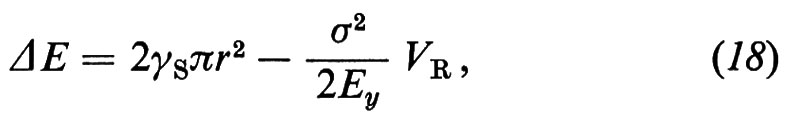
con γS = energia superficiale specifica, Ey = modulo di elasticità e VR = volume dal quale viene liberata, all'apertura della cricca, energia elastica. Se si considera approssimativamente VR = 4πr3/3, si ha come tensione critica per l'allargamento di una cricca su scala atomica
σB = (2γSEy/r)1/2. (19)
I processi di frattura cominciano dunque col massimo della facilità dalle cricche più grosse, preesistenti (flaws). La resistenza alla frattura di un pezzo è quindi limitata dalle dimensioni dei più grossi difetti rimasti (questi possono essere cricche superficiali, ma anche spigoli vivi nelle inclusioni interne). Mentre i termini qualitativi dei modelli di Griffith hanno potuto essere confermati, non è possibile comprendere con essi gli influssi della struttura e della temperatura, e anche i processi plastici all'apice della cricca rimangono non presi in considerazione. Nella meccanica della frattura si definisce tuttavia un parametro caratteristico del materiale (fattore critico di intensità di tensione),
Kc = σB √-π-ac , (20)
che si può determinare sperimentalmente per un intaglio standard di profondità a0, mediante la misurazione del carico di rottura. A seconda del tipo di sforzo (v. fig. 52a), viene attribuito a Kc anche un indice I, II o III; la fig. 52b illustra la misurazione di KIc nella prova di flessione su 4 punti.
Studi più precisi delle proprietà plastiche richiedono la determinazione contemporanea della deformazione e del carico, mentre bisogna accontentarsi di esperimenti semplici a causa della complessità delle possibili modalità di carico. Particolarmente diffusi sono dei procedimenti per la comprensione delle proprietà plastiche sotto un carico (ideale) uniassiale (trazione o compressione anche alternate). Nella prova di trazione uniassiale una provetta di forma allungata con sezione costante (per es. v. fig. 53) viene fissata in appositi afferraggi su una macchina di prova. Mentre un'estremità viene tenuta ferma l'altra é vincolata, mediante una traversa mobile, a un meccanismo motore che dà la velocità di allungamento. Un dispositivo dinamometrico e un estensimetro registrano il carico istantaneo F e la variazione di lunghezza istantanea Δl del campione. Da ciò si possono determinare l'allungamento (nominale) εn = Δl/l0 (l0 = lunghezza iniziale) e la tensione (nominale) σn = F/A0 (A0 = superficie della sezione trasversale). Per una comprensione più esatta della plasticità bisogna riferire allungamento ε e tensione σ alla rispettiva lunghezza l e alla rispettiva sezione A. Poiché il volume di un campione non varia grandemente durante la deformazione plastica, vale Al = A0l0, col che ε = ln(1 + εn), σ = F/A. Per una velocità di allungamento costante ε•n si registrano nella ‛prova di trazione dinamica' F e Δl e si ottengono curve di deformazione di cui nella fig. 54 è riprodotto l'andamento schematico per provette policristalline tipiche. La ripida ascesa iniziale è elasticamente reversibile e segue la legge di Hooke. Quando si raggiunge il pianerottolo di snervamento σ0 comincia il campo plastico, spesso (e in particolare negli acciai ferritici) con un'instabilità (limite di snervamento superiore) in presenza della quale comincia una deformazione plastica locale che si allarga a mo' di bande per tutto il campione (‛bande di Lüders'). Si osserva poi un pianerottolo di snervamento con carico (unitario) al limite di snervamento superiore σh e un carico al limite di snervamento inferiore σu. Un'ulteriore significativa deformazione è possibile solo con un innalzamento della tensione, poiché la provetta diventa più resistente (più dura) a causa della deformazione plastica già subita. Questo incrudimento, misurato attraverso dσ/dε non può però continuare a piacimento. Per dσn/dεn = 0 la deformazione a trazione diventa instabile, il campione subisce una strizione e alla fine si giunge alla rottura.
Per la valutazione tecnica di un materiale sono importanti σ0 e rispettivamente σu, la tensione nominale massima σUTS (UTS, ultimate tensile strength) e l'allungamento totale fino a rottura εB. L'integrale ∫ σde dà il lavoro di deformazione accumulato per unità di volume, che è importante come misura della deformabilità di un materiale (costi di lavorazione, sicurezza in servizio). Nello studio dei materiali si esamina in dettaglio l'influsso di diversi parametri sulla curva di incrudimento. Di importanza del tutto particolare è l'influsso dei bordi di grano nei materiali policristallini. Per eliminarlo vengono deformati anche dei monocristalli che, soprattutto all'inizio della curva di incrudimento, presentano differenze caratteristiche a seconda dell'orientazione, della composizione e della microstruttura (v. fig. 55). Questo comportamento può essere compreso sulla base del tipo di processo plastico elementare cristallograficamente definito (scorrimento delle dislocazioni).
In un altro tipo di sperimentazione lo scorrimento plastico avviene sotto un carico costante (tensione nominale) oppure a tensione reale misurata costante σ (‛prova di scorrimento'). La fig. 56 mostra tipiche curve di scorrimento plastico. Un terzo esperimento standard misura il ‛rilassamento di tensione' dopo l'arresto della macchina di deformazione che ha deformato il campione fino a una tensione σ1. Ora il campione comincia a scorrere, ma contemporaneamente il carico diminuisce, poiché col modulo di elasticità reale di tutto il dispositivo di misura, EM, ogni allungamento aggiuntivo del campione, Δε, produce una variazione di tensione −Δσ = EMΔε. Da σ(t) si può ottenere la sensibilità alla velocità (di applicazione del carico) della tensione di scorrimento. Nella tecnica il rilassamento della tensione spesso è di disturbo, particolarmente nei collegamenti con viti.
Esperimenti simili si possono attuare anche con un carico di compressione lungo un solo asse. Sono possibili altri metodi speciali, accanto alle prove di flessione usuali nella meccanica della frattura; per esempio sollecitazione a torsione di barre e tubi, sollecitazione da pressione di tubi, di corpi cavi, ecc. A causa dell'importanza delle sollecitazioni di segno alterno (v. sotto, È f), vengono condotti esperimenti ciclici (deformazione alterna) di vario tipo. Per l'impiego pratico sarebbe molto utile poter trasformare i risultati di un tipo di esperimento in quelli di un altro: lo si potrebbe fare se esistesse un'equazione di stato meccanica f(ε•, ε, σ) = 0 (con T = cost.). Questo vale, come le più recenti ricerche hanno dimostrato, solo nel caso in cui le microstrutture sono confrontabili. Poiché esse tuttavia possono dipendere chiaramente dal procedimento sperimentale, le equazioni di stato empiriche non possono essere senz'altro estrapolate su grandi intervalli di ε•, ε, σ.
b) Dislocazioni nei cristalli.
Per materiali fragili la resistenza meccanica massima (teorica) si ricava dalla deformazione elastica massima che si può ottenere prima che i legami atomici si rompano. Le tensioni così valutate ammontano circa al 10% del modulo di elasticità Ey. Nei materiali duttili, quindi anche nei metalli, varia la forma dei cristalli, per lo più senza variazione della struttura cristallina (un'eccezione sono le trasformazioni di fase indotte da una sollecitazione: v. cap. 7, È b). Nella deformazione plastica regioni cristalline subiscono uno spostamento reciproco. Nei monocristalli deformati plasticamente si osservano corrispondentemente sezioni spostate tangenzialmente, come rappresentato schematicamente nella fig. 57. La variazione di forma può essere ricondotta perciò a processi di scorrimento microscopici. Se questi processi fossero spostamenti rigidi di parti di cristallo perfetto, l'una rispetto all'altra, allora si otterrebbero per la tensione di scorrimento di materiali duttili dei valori simili a quelli della tensione di rottura (resistenza teorica alla sollecitazione di taglio secondo Frenkel, circa il 10% del modulo di elasticità tangenziale GS). Le ‛dislocazioni' già costituite nella crescita del cristallo e altre prodotte durante la sollecitazione meccanica (v. solidi, fisica dei) sono ora proprio i veicoli adatti alla deformazione plastica. Una dislocazione si caratterizza per il suo vettore di Burgers b e per un elemento lineare ds. La fig. 58 mostra ambedue i tipi di dislocazione chiaramente tra loro distinguibili, a spigolo e a vite, da cui si possono ottenere per combinazione altri casi. Dalla definizione di dislocazione (circuito di Burgers; v. solidi, fisica dei) segue che una dislocazione nel cristallo può cambiare la sua direzione, ma non il suo vettore di Burgers e dunque non può neanche terminare all'interno del cristallo. Essa deve andare fino alle superfici o deve chiudersi su se stessa ad anello. Nelle ramificazioni (nodi) resta costante la somma vettoriale dei vettori di Burgers (per es. b1 = b2 + b3 in un nodo triplo).
Dalla teoria dell'elasticità lineare risulta che, attraverso tensioni sia interne che esterne, sulle dislocazioni vengono esercitate forze che agiscono sempre perpendicolarmente all'elemento lineare ds (Peach e Koehler, 1950):
dF = (b • σ) × ds, (21)
dove σ rappresenta il tensore della sollecitazione effettiva. In una dislocazione a vite b e ds sono paralleli, la dislocazione procederà anche su quel piano che è cristallograficamente ammissibile e in cui opera la maggiore componente della forza. Nelle dislocazioni a spigolo è particolarmente evidente il piano che contiene b e ds. Se si considera infatti (v. fig. 59a) lo spostamento dr di un segmento di dislocazione ds orientato a piacere, allora entrambe le parti del cristallo al di sopra e al di sotto del piano definito da ds e dr vengono spostate l'una rispetto all'altra di b. Se i tre vettori sono linearmente indipendenti, allora si verifica una variazione di volume dV = b • (ds × dr) = dr • (b × ds). dV ≠ 0 indica emissione di vacanze o di atomi interstiziali oppure assorbimento di atomi interstiziali presenti o di vacanze. Un tale movimento provoca un trasporto di materia controllato dalla diffusione. Solo quando (b × ds) è perpendicolare a dr, cioè la dislocazione si muove nel piano che è definito da b e ds, dV risulta uguale a 0 e la dislocazione si può muovere ‛conservativamente'. Questo movimento si chiama ‛scorrimento' (v. fig. 59b), mentre la forma di movimento non conservativa si chiama climbing (o ‛risalita'). Proprio per questa distinzione fondamentale, ha senso scomporre dF secondo la (21) in una componente nel piano di scorrimento (la cui normale è data da n = b × ds) e una componente perpendicolare a questo. La ‛forza di scorrimento' per unità di lunghezza KG operante nel piano di scorrimento (perpendicolarmente alla direzione della dislocazione) ha il valore
KG = τb, (22)
con b = modulo di b e τ = modulo delle componenti della tensione di scorrimento operanti sul piano di scorrimento nella direzione di b. Si mostra inoltre che anche su una dislocazione a vite con lo stesso vettore di Burgers nello stesso piano agisce la stessa forza KG e così anche per tutte le situazioni intermedie. La forza di scorrimento in un dato piano è dunque data, indipendentemente dal carattere della dislocazione (angolo tra ds e b), solo da τb e agisce sempre perpendicolarmente alla linea di dislocazione. Questo fatto ha delle conseguenze importanti. Immaginiamo, per esempio, una dislocazione inizialmente rettilinea ancorata in due punti (per es. su nodi od ostacoli). Senza forze ulteriori la dislocazione che, sulla base della sua energia elastica (≃ GSb2/2 per unità di lunghezza), possiede anche una tensione di linea, andrà in linea retta da un punto all'altro (v. fig. 60a). Se ora sul piano di scorrimento agisce una sollecitazione tangenziale τ in direzione di b, allora KG = τb e la dislocazione si allungherà su un arco di cerchio (ma poiché una dislocazione a spigolo ha una energia maggiore di una dislocazione a vite, la tensione di linea per un segmento di dislocazione a vite sarà maggiore che per un segmento a spigolo e inoltre il suo andamento non sarà esattamente circolare poiché influirà fortemente anche l'anisotropia elastica). Finché non si raggiunge un percorso semicircolare si avrà un equilibrio stabile in corrispondenza del raggio di curvatura Re = dL/dϑ secondo la (23):
τbdL = TL dϑ, (23)
con TL = tensione di linea. Approssimando la TL all'energia per unità di lunghezza GSb2/2, il raggio di curvatura diviene
Re ≃ GSb/2τ. (24)
Quando Re diventa = L/2, cioè con la tensione
τOR = GSb/L, (25)
l'arco di dislocazione diventa instabile, indipendentemente dall'intensità degli ostacoli i bracci della dislocazione girano intorno ai punti di ancoraggio, si muovono verso la parte posteriore degli ostacoli l'uno verso l'altro e si annullano lungo la linea di contatto, cosicché ne deriva un anello di dislocazione completo e un nuovo segmento di dislocazione tra i punti di ancoraggio. Questo meccanismo detto ‛di Orowan', delimita, in linea di principio, la resistenza dei materiali che contengono ostacoli localizzati di qualsiasi tipo al movimento delle dislocazioni. Secondo la formula (25) bisogna cercare di ottenere un piccolo L, quindi un'alta densità di ostacoli, per avere l'innalzamento della ‛tensione di Orowan'. Il meccanismo schematizzato nella fig. 60 mostra anche come, specialmente all'inizio della deformazione, possano essere prodotte delle dislocazioni aggiuntive (‛sorgente di Frank-Read'). Attraverso questi e simili meccanismi di sorgente aumenta la densità delle dislocazioni durante la deformazione plastica. Nella maggior parte delle deformazioni macroscopiche devono essere costantemente fornite ulteriori nuove dislocazioni, poiché solo così diventano comprensibili le grandi deformazioni plastiche e l'incrudimento.
Una singola dislocazione in un cristallo, altrimenti esente da difetti, viene limitata nella sua capacità di scorrimento dal potenziale di Peierls (v. solidi, fisica dei). Nella maggior parte delle sostanze metalliche a struttura compatta, questa intrinseca resistenza alla deformazione è piccola e si osserva solo a basse temperature in esperimenti dinamici. Nei metalli cubici a corpo centrato e nei composti intermetallici, la struttura del nucleo della dislocazione, cioè la disposizione discreta degli atomi nel centro della dislocazione, acquista, tuttavia, un'influenza decisiva sulle caratteristiche macroscopiche della deformazione. Durante la deformazione, inoltre, le dislocazioni avvertono, a causa del proprio campo di deformazione, ogni inomogeneità come un centro locale di interazione attrattiva o repulsiva. Agiscono in tal modo anche altre dislocazioni. Il calcolo di una configurazione di equilibrio per casi complicati è un problema a molti corpi finora irrisolto. Con i grandi calcolatori oggi disponibili è tuttavia possibile compiere estesi calcoli di simulazione e da questi ricavare anche delle connessioni trasferibili alla pratica. Con ciò i problemi della plasticità possono essere limitati, dal lato analitico e dal lato numerico, a calcoli di modello. Qui deve bastare un'esposizione qualitativa dei processi di deformazione.
c) Incrudimento dei metalli puri.
Poiché l'energia delle dislocazioni è proporzionale al quadrato del vettore di Burgers, i più brevi vettori di traslazione della struttura corrispondente si comportano di preferenza come vettori di Burgers. Questi vettori di Burgers giacciono nei piani atomici più compatti e le dislocazioni che giacciono in questi piani sono anche le più mobili, poiché il potenziale di Peierls qui è minimo. Ci si aspetta dunque che siano direzioni di scorrimento le direzioni cristallografiche corrispondenti a piccole distanze interatomiche e che siano piani di scorrimento i piani con indici di Miller semplici. È questo il caso dei metalli cubici a facce centrate (Cu, Ag, Au, Al, Ni, Feγ, ecc.) ed esagonali con grande interdistanza tra i piani di base (Zn, Cd, Mg). Nei metalli cubici a corpo centrato (Feα, V, Nb, Mo, Ta, W, Na, K), i piani {110} e {211} si presentano ambedue come piani di scorrimento (v. tab. VII).

Con l'ausilio della proiezione stereografica (v. fig. 61) si ha una buona visione di insieme dei sistemi di scorrimento ( combinazione della direzione di scorrimento e dei piani di scorrimento). Come esempio la fig. 62 mostra la proiezione di un cristallo cubico: sono messe in rilievo le normali ai piani {111} e le direzioni 〈110>. Per un cristallo cubico a facce centrate sono ora possibili tutti i sistemi di scorrimento che derivano dalla combinazione di un piano {111} con un vettore di Burgers (direzione 〈110>) in esso contenuto; per esempio per (111): [0-11], [-110], [-101] (in ognuno di questi tre casi ci sono segni positivi e negativi dei vettori di Burgers). Ci si aspetta che, a parità di altre condizioni, venga per primo messo in atto quel sistema di slittamento per cui è più alto τ, cioè la componente della sollecitazione tangenziale nel piano di scorrimento e nella direzione di scorrimento. Per una sollecitazione monoassiale σ si ha
τ = σ cosλ cosκ, (26)
dove λ e κ sono rispettivamente gli angoli fra la direzione di scorrimento e l'asse della barretta e fra la normale al piano di scorrimento e lo stesso asse. Il fattore cosλ cosκ si chiama ‛fattore di Schmid' e quando esso solo determina la scelta del sistema di slittamento vale la ‛legge di Schmid della sollecitazione tangenziale'. Come mostrato nella fig. 63, ogni asse della sbarra per un cristallo cubico si può collocare all'interno o al bordo del ‛triangolo standard' tratteggiato e si possono considerare i sistemi di scorrimento più importanti in relazione alla loro posizione rispetto all'asse, ad esempio, per cristalli cubici a facce centrate, il sistema di scorrimento con il più alto fattore di Schmid, (11-1) [101] Hh nella fig. 63 (sistema di scorrimento principale o primario). Dalla rotazione del reticolo nella deformazione a trazione si constata che l'asse A al procedere della deformazione migra effettivamente su [101], almeno finché raggiunge la linea [100]-[111] (‛simmetrale'). Qui i sistemi Hh e Dd (sistema di scorrimento doppio) hanno uguali sollecitazioni tangenziali; tuttavia, poiché il piano H contiene già molte dislocazioni, Dd è attivato solo dopo che l'asse della sbarra ha alquanto sorpassato la sua meta teorica. Le dislocazioni a vite del sistema di scorri mento primario, cioè con vettore di Burgers ½ 〈101 nell'esempio della fig. 63, possono anche deviare sul ‛piano di scorrimento trasversale' Q, come mostrato nella fig. 64 per un anello di dislocazione in espansione. Con ciò, in presenza di un blocco dello scorrimento su un piano primario, le dislocazioni a vite possono cambiare i piani di scorrimento attraverso un duplice processo di scorrimento deviato. Occasionalmente si osserva anche il ‛sistema di scorrimento deviato' Uu (v. fig. 63).
Per la rappresentazione delle curve di deformazione di monocristalli, la componente principale della sollecitazione tangenziale, τ, viene riportata in funzione della ‛deformazione tangenziale o di taglio' γ (slittamento dei piani nella direzione di scorrimento), che si può calcolare dall'allungamento ε e dagli angoli λ, κ. La fig. 65 mostra τ(γ) ricavata da prove di trazione su monocristalli di rame di diversa orientazione iniziale. Si riconosce che l'estensione del tratto indicato con I è degna di nota solo per orientazioni iniziali interne al triangolo standard e che il tratto I è tanto più grande quanto più l'orientazione iniziale è lontana dal simmetrale [100]-[111]. Nel tratto I viene dunque attivato solo il sistema di scorrimento principale. Nel tratto II segue un incrudimento maggiore (approssimativamente lineare), che però al di sopra di una tensione τIII può nuovamente diminuire (riassetto dinamico). In circostanze speciali possono presentarsi nuovamente un maggiore incrudimento (tratto IV) e un ulteriore processo di rilassamento (tratto V), prima che si giunga alla rottura (duttile). Curve di incrudimento simili sono mostrate da tutti i metalli cubici a facce centrate. Per orientazioni al bordo del triangolo scompare il tratto I; a seconda della temperatura e del materiale può scomparire anche il tratto II, quando i processi di riassetto intervengono già fin dall'inizio della deformazione. Nei metalli cubici a corpo centrato si possono parimenti chiaramente distinguere i tre tratti principali; c'è ancora un tratto iniziale ripido: il tratto 0. Nei cristalli esagonali con rapporto assiale sufficientemente grande (Zn, Cd, Mg) si ottiene, per un'orientazione appropriata, come previsto, uno scorrimento facilitato molto esteso (chiamato in questo caso tratto A), poiché è possibile solo lo scorrimento sul piano di base (v. fig. 66).
La sollecitazione tangenziale critica, τ0, cioè la sollecitazione tangenziale necessaria per l'inizio della deformazione plastica, è, nei metalli a struttura compatta e di grande purezza, ampiamente determinata dalla densità delle dislocazioni presenti. Tuttavia non si è ancora riusciti per i metalli a far crescere a grandezze apprezzabili monocristalli completamente esenti da dislocazioni; inoltre col diminuire della densità delle dislocazioni diminuisce parimenti τ0. A causa dei loro campi di tensione, le dislocazioni si ostacolano reciprocamente, come nell'esempio di due dislocazioni a spigolo parallele, mostrato nella fig. 67. A causa del campo di tensione (che decresce con l'inverso della distanza r) della dislocazione che si trova nell'origine, una dislocazione dello stesso segno proveniente da destra viene respinta secondo la (21) nel campo a distanza parimenti proporzionale a 1/r. In un ulteriore avvicinamento è importante l'andamento dettagliato delle componenti della tensione: per la forza di reazione sul piano di scorrimento y = y0, esercitata dalla seconda dislocazione, è decisiva la σxy (v. fig. 68) della prima. Per un intervallo x ≃ 2,4 y0, la forza massima di repulsione per unità di lunghezza è K⊥⊥ ≃ GSb2/[8π(1 − νp)y0], cosicché, secondo la (22), occorre introdurre una sollecitazione tangenziale τf = K⊥⊥/b, detta ‛sollecitazione di scorrimento',
Se si pone quale densità di dislocazioni ρv il numero delle linee di dislocazione che attraversano una superficie unitaria, allora la distanza media tra le dislocazioni è circa 1/√-ρv . Con questo valore per y0 la sollecitazione di scorrimento diventa
τf = αGSb √-ρv . (28)
La costante numerica ‛ può assumere per modelli più realistici altri valori. Questa semplice rappresentazione mostra già la connessione generalmente stabilita tra τf e ρv. In un cristallo reale non tutte le dislocazioni saranno parallele, ma avranno direzioni diverse. Ciò nonostante resta valida la (28). Il piccolo coefficiente di incrudimento nel tratto I si può ora comprendere considerando che la densità di dislocazioni nello scorrimento facilitato si accresce solo di poco: le singole dislocazioni, dunque, possono effettuare grandi percorsi. Nello stesso tempo, tuttavia, vengono prodotte ininterrottamente nuove dislocazioni nelle sorgenti, poiché una semplice stima mostra che, se non se ne producessero di nuove (moltiplicazione), le dislocazioni presenti all'inizio della deformazione dovrebbero abbandonare il cristallo dopo un modesto scorrimento: ogni dislocazione che spazza la superficie dA produce un incremento di deformazione dγ = bdA/V, dove V = L1L2L (con le tre lunghezze degli spigoli di un prisma rettangolare). Con N dislocazioni a spigolo lungo L1 la densità di dislocazione è ρv = N/L2L, così che, con dA = L1dL,
dγ = bρv dL, (29)
con dL = percorso medio delle dislocazioni. Con una densità di dislocazioni iniziale realistica ρv = 106 cm-2 (monocristallo prodotto senza speciali precauzioni), si otterrebbe, per campioni di 1 cm di spessore (dL ≈ 0,5 cm) senza moltiplicazione di dislocazioni, uno scorrimento totale di circa l'1%, mentre si può raggiungere in casi favorevoli più del 100%. Attraverso l'ancoraggio e la formazione di nodi di dislocazioni orientate in modo diverso e attraverso concentrazioni di tensione sulle superfici, possono essere prodotte chiaramente, nei metalli a struttura compatta, sufficienti dislocazioni capaci di scorrere. Il tratto 0 nei metalli cubici a corpo centrato è in rapporto con una difficoltà iniziale della moltiplicazione di dislocazioni. In questi metalli l'influsso del potenziale di Peierls non è ancora cliiaramente separabile da quello degli atomi estranei (O, N, C, H) interstiziali, molto efficaci anche in piccole quantità sulla mobilità delle dislocazioni. Secondo recenti ricerche, al crescere della purezza la netta salita di τ0 al diminuire della temperatura (presentata dai metalli cubici a corpo centrato) decresce progressivamente. Rimane possibile che una parte di questo effetto mostri una dipendenza dalla temperatura che derivi dal potenziale di Peierls. Che la struttura del nucleo di una dislocazione, nella struttura a corpo centrato, influisca fortemente sulla plasticità è dimostrato nell'anisotropia trazione/compressione della sollecitazione critica (v. fig. 69) e nelle altre violazioni della legge di Schmid. La fig. 70a mostra come lo spostamento mediato dal vettore di Burgers ½ [111] di due piani vicini (1-10), invece che in un movimento rigido attraverso elevate barriere di potenziale, possa essere compiuto anche in tre gradi, ¹-8[110] + ¹-4 [112] + ¹-8 [110]. La dislocazione può dissociarsi in ‛dislocazioni parziali' con relativi vettori di Burgers parziali, poiché la somma delle energie delle dislocazioni parziali è minore dell'energia della dislocazione totale. D'altra parte deve esserci tra le dislocazioni parziali un difetto planare (difetto di ‛impilaggio' o stacking fault, SF), poiché proprio attraverso i vettori di Burgers parziali viene interrotta la simmetria di traslazione. La fig. 70b-h mostra scomposizioni schematizzate del vettore di Burgers con partecipazione di difetti di impilaggio coplanari e non coplanari. Anche se le ‛scomposizioni' sono probabilmente molto piccole e non hanno potuto essere rese visibili fino a oggi al microscopio elettronico, l'asimmetria del nucleo delle dislocazioni è confermata continuamente da simulazioni col calcolatore e l'anisotropia trazione/compressione è compresa oggi qualitativamente. Nel reticolo cubico a facce centrate la scomposizione avviene sempre simmetricamente (v. fig. 71), in due dislocazioni parziali di tipo ¹-6 〈211> con un difetto di impilaggio intrinseco, che corrisponde a uno strato di materiale esagonale. Le energie dei difetti di impilaggio sono per Cu, Ag, Au tanto piccole che si può vedere direttamente la dissociazione delle dislocaziolti. Le dislocazioni ne ricevono un carattere planare, in particolare è anche ostacolato lo scorrimento deviato delle dislocazioni a vite dissociate.
Nel tratto II della curva di incrudimento vengono attivate dislocazioni in sistemi di scorrimento secondari. Una teoria quantitativa dell'incrudimento dovrebbe indicare la disposizione e la moltiplicazione delle dislocazioni in ogni momento e da ciò calcolare la sollecitazione di scorrimento, cosa che oggi appare impossibile. Teorie semiempiriche per il tratto II partono da risultati sperimentali ottenuti sia da ricerche sulle bande di scorrimento (dislocazioni che incontrano una superficie e che possiedono una componente del vettore di Burgers perpendicolare a questa superficie vi producono un dislivello osservabile se alcune dislocazioni hanno utilizzato lo stesso piano di scorrimento o piani molto vicini) sia da immagini di dislocazioni prodotte al microscopio elettronico in trasmissione utilizzando sottili campioni ricavati da cristalli deformati (per ottenere risultati realistici le dislocazioni devono essere ancorate sotto carico - ‛inchiodate' -, per esempio attraverso agglomerati di difetti dopo irraggiamento con neutroni veloci o attraverso la produzione di piccoli precipitati che mantengono ferme le dislocazioni). Nel rame si è osservato (Seeger e altri) che nel tratto II vengono attivate sorgenti che di volta in volta emettono un determinato numero n (≃ 20) di dislocazioni singole, il cui percorso L(γ) decresce in modo inversamente proporzionale alla deformazione γ, poiché, attraverso l'attivazione crescente di sistemi di scorrimento secondari, hanno luogo reazioni tra dislocazioni, che conducono a ostacoli immobili. Con γII = deformazione all'inizio del tratto II si ha L = C1(γ − γII)-1 e per un incremento di deformazione dγ si ottiene
dγ = nbC1(γ − γII)- dρS, (30)
con dρS = densità delle sorgenti di nuova attivazione. Se si riconduce l'incrudimento dello stato deformato, secondo Seeger, principalmente alla densità ρS dei gruppi già immobilizzati di n dislocazioni e si considera ogni gruppo come una ‛superdislocazione' con vettore di Burgers nb, allora è (v. eguaglianza 28)
τ = αGSnb √-ρS . (31)
Con (30) e (31) si ha
Con n/C1 = costante, ΘII nel tratto II è ugualmente costante e l'incrudimento è lineare. Secondo la fig. 72 ΘII/GS si trova per molti metalli (e leghe) a facce centrate in un intervallo tra circa 2 • 10-3 e 4 • 10-3. Accanto a questo ‛modello di accumulo' (ogni gruppo viene immobilizzato quando si accumulano n dislocazioni) sono state fatte ancora numerose altre proposte per il tratto Il. Basinski parte dall'esistenza di grovigli tridimensionali di dislocazioni (tangles) con estese regioni relativamente esenti da dislocazioni e dall'esistenza di altrettante dislocazioni sui piani di scorrimento secondari. Queste ultime si chiamano ‛dislocazioni di foresta' perché come alberi penetrano attraverso i piani di scorrimento principali. Le dislocazioni di foresta sono responsabili di ΘII e, attraverso processi di taglio, sono anche responsabili di una parte notevolmente minore di incrudimento dipendente dalla temperatura e dalla velocità. In altri modelli gli ostacoli determinanti per la moltiplicazione o il movimento delle dislocazioni sono i prodotti di reazione tra dislocazioni di foresta e dislocazioni primarie, localizzati in regioni estese e di andamento complesso. Tali regioni costituiscono le strutture a groviglio e a celle che si osservano nei cristalli deformati. I dettagli dipendono anche dall'energia dei difetti di impilaggio γSF, che ha un ruolo nella formazione di prodotti di reazione immobili.
Nel riassetto dinamico, dunque all'inizio del tratto III, si ha a che fare con un processo dipendente fortemente dalla temperatura. Anche senza una tensione esterna, vengono demolite ampiamente, a una temperatura sufficientemente alta, le disposizioni delle dislocazioni prodotte dalla deformazione, mentre con i processi di climbing (risalita), scorrimento deviato e scorrimento semplice i difetti strutturali prodotti dalle dislocazioni vengono risanati e le dislocazioni stesse abbandonano il cristallo o si annullano reciprocamente; complessivamente, dunque, l'energia di deformazione viene diminuita (riassetto e ricristallizzazione). Una tensione esterna produce forze anche per lo scorrimento deviato verticale e il climbing, cosicché entrambi i processi possono mettersi in azione durante la deformazione, se le tensioni sono abbastanza alte. Quanto più piccola è l'energia dei difetti di impilaggio tanto più grande è la dissociazione, e la tensione dev'essere tanto più grande per raggiungere, a una data temperatura, un considerevole scorrimento deviato. La tab. VIII dà i valori dell'energia dei difetti di impilaggio per alcuni metalli cubici a facce centrate. L'alta energia dei difetti di impilaggio dell'alluminio fa si che la deformazione dei monocristalli di alluminio, a temperatura ambiente, incominci già con il tratto III. La fig. 73 mostra le curve di deformazione per monocristalli di nichel a diverse temperature, dalle quali appare evidente la forte dipendenza dalla temperatura di τIII. Per i particolari del meccanismo di scorrimento deviato vengono discussi almeno due modelli (v. fig. 74).
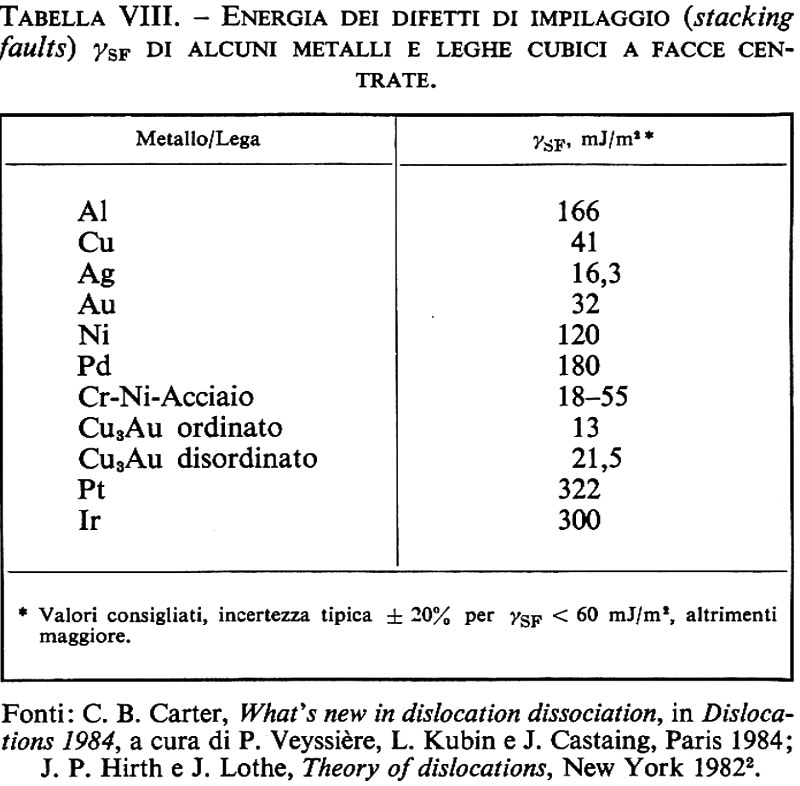
Il riassetto attraverso climbing delle dislocazioni a spigolo produce un ulteriore contributo alla diminuzione dell'incrudimento. In particolare, le dislocazioni con diverso segno, disposte in piani di scorrimento adiacenti e paralleli a formare gruppi di dislocazioni bloccate, possono annullarsi per climbing. Con ciò possono diventare mobili altre dislocazioni. Una distinzione tra processi di scorrimento deviato e di climbing, nella prova di trazione dinamica, non è sempre facile. Quando ha luogo solo lo scorrimento deviato, si trovano sulla superficie tracce dell'attività del sistema di scorrimento deviato, cioè dislivelli nettamente definiti.
I processi tra dislocazioni nella deformazione di monocristalli, rappresentati qui in sintesi, sono fondamentali anche per la plasticità dei policristalli. Tuttavia i bordi di grano fanno si che il movimento delle dislocazioni, iniziato in un cristallite, non possa essere trasmesso senz'altro a un grano adiacente. I bordi di grano sono pertanto ostacoli efficaci al movimento delle dislocazioni. A causa delle orientazioni diverse dei singoli grani, si producono fattori di Schmid diversi nei sistemi di scorrimento orientati diversamente rispetto a una sollecitazione esterna. Per raggiungere una deformazione macroscopicamente omogenea, tutti i grani devono essere deformati insieme, per cui, di volta in volta, è necessario più di un sistema di scorrimento, altrimenti la coesione della struttura policristallina andrebbe perduta (v. fig. 75). Una parte delle dislocazioni è dunque ‛geometricamente necessaria' (Ashby) e fornisce una porzione aggiuntiva di incrudimento, che si sovrappone all'incrudimento (omogeneo) dei singoli grani.
Per la tensione di scorrimento dei policristalli è decisivo il trasferimento dello scorrimento da un grano 1 a un grano 2 adiacente. Se si suppone che lo scorrimento sia stato inizialmente attivato nel grano 1 e una sorgente abbia emesso n dislocazioni, ammassate al bordo del grano (v. fig. 76), da un calcolo di teoria dell'elasticità risulta che esiste, al di là del bordo del grano, una tensione (τ − τ1) √-d-/-LQ, dove d diametro del grano, LQ = distanza della sorgente dal bordo del grano, τ1 = sollecitazione tangenziale critica nel grano 1. Poiché i sistemi di scorrimento nel grano 1 e nel grano 2 sono orientati diversamente, per l'attivazione di una sorgente nel grano 2, questa tensione sarà operante con un fattore di trasformazione m12. La tensione di flusso plastico τ0 sarà raggiunta quando un accumulo di dislocazioni nel grano i riuscirà esattamente ad attivare una sorgente con tensione di sorgente τa nel grano 2, dunque τa = m12(τ0 − τ1) √-d-/-LQ, da cui, per un policristallo (per il quale solo σ può essere indicato macroscopicamente), segue
σ0 = σ1 + ky d-1/2. (33)
Questa equazione, conosciuta come ‛relazione di Hall-Petch', descrive molto bene (con ky = parametro di Hall-Petch) la dipendenza della tensione di flusso plastico dalla grandezza del grano. Come mostra la fig. 77, il parametro di Hall-Petch può tuttavia variare notevolmente. Piccoli aumenti, dunque un debole influsso dei bordi di grano, indicano una rapida attivazione dello scorrimento multiplo nei singoli grani, cosicché non si arriva a forti innalzamenti di tensione ai bordi di grano (specialmente nei metalli puri).
Per la deformabilità e l'incrudimento del policristallo è determinante che una deformazione macroscopica e omogenea, secondo von Mises, di grani comunque orientati e fra loro in contatto richieda per lo meno cinque sistemi di scorrimento indipendenti. Per metalli cubici a facce centrate e cubici a corpo centrato questa condizione è realizzabile; in quelli esagonali si presenta una buona deformabilità plastica per scorrimento solo quando ai due sistemi di scorrimento indipendenti sul piano di base si aggiungono ancora sistemi prismatici o piramidali. A basse temperature la formazione meccanica di geminati può produrre le deformazioni aggiuntive.
Per l'andamento della curva di deformazione di un policristallo sono particolarmente importanti i grani orientati sfavorevolmente, cosicché σ(ε) dovrebbe prima assomigliare alla curva di incrudimento di un monocristallo orientato verso un vertice del triangolo standard (v. fig. 65). Il tratto I, dunque, non compare. La transizione tra il tratto II e il tratto III è attenuata a causa dei vari sistemi di scorrimento e delle tensioni che intervengono; si può tuttavia identificare una parte (debolmente dipendente dalla temperatura e dalla velocità) della curva di deformazione con il comportamento nel tratto II, dove la delimitazione dei percorsi delle dislocazioni avviene sia ad opera dei processi attivi del monocristallo sia, soprattutto, ad opera dei bordi di grano.
La maggior parte dei policristalli ha già all'inizio della deformazione una ‛tessitura': le orientazioni dei grani non sono distribuite in maniera statisticamente isotropa. Durante la deformazione i grani si orientano in ogni caso in modo determinato e dopo una forte deformazione si formano marcate tessiture specifiche di deformazione. La fig. 78 mostra l'analisi dei piani {100} per una lamiera. La tab. IX offre alcune strutture tipiche per fili metallici e lamiere.

I bordi di grano, mentre a temperature moderate aumentano soprattutto la resistenza e l'incrudimento, a temperature superiori a circa 2/3 Tf (Tf temperatura di fusione) possono favorire un ulteriore allungamento plastico. Poiché i bordi di grano non si distribuiscono uniformemente su tutto il campione, lo scorrimento ai bordi di grano è legato alla diffusione sia sui bordi che nel volume intorno a essi. Secondo Ashby si possono raggruppare i diversi prevalenti meccanismi di deformazione in ‛mappe' di deformazione (deformation maps) che presentano qualitativamente tratti simili per tutti i metalli (v. fig. 79).
In circostanze determinate, metalli a grana molto fine, ma specialmente con struttura bifasica, ad alte temperature possono mostrare deformazioni plastiche estreme (fino al 1.ooo%) prima della rottura. Questa ‛superplasticità' ha luogo per una parte considerevole sui bordi di grano, dove speciali configurazioni dei bordi di grano sono particolarmente favorevoli ed è necessario un gioco alterno molto delicato nel dettaglio e implicante procedimenti di climbing per rendere possibile un basso coefficiente di incrudimento. La struttura e la mobilità dei bordi di grano vengono studiate oggi molto intensamente, non solo in relazione alla superplasticità.
d) Resistenza meccanica delle leghe.
La deformazione plastica dei metalli puri viene determinata essenzialmente attraverso l'interazione delle dislocazioni tra di loro e con i bordi di grano. Nelle leghe entrano in gioco, a seconda del tipo e della distribuzione dei diversi atomi, ulteriori meccanismi di interazione. Già il calcolo della sollecitazione tangenziale critica diventa un problema molto complesso, che presenta due aspetti: l'interazione individuale tra la dislocazione mobile e l'ostacolo (incluso il meccanismo di aggiramento o di superamento dell'ostacolo) e la formazione dell'adatto valore medio della sollecitazione tangenziale critica, misurabile macroscopicamente considerando la sovrapposizione dei diversi singoli meccanismi e la flessibilità limitata della dislocazione (tensione di linea).
In tutte le leghe tecniche c'è da attendersi un' aumentata resistenza intrinseca del materiale omogeneo, cioè un contributo all'indurimento degli atomi estranei disciolti omogeneamente. Con ciò si intende un aumento di resistenza attraverso l'interazione reciproca tra singoli atomi disciolti e dislocazioni mobili. (Nei metalli e nelle leghe, con influssi non trascurabili del potenziale di Peierls, gli atomi estranei possono anche diminure la resistenza, mentre, diminuendo localmente il potenziale di Peierls, si può dare inizio al movimento delle dislocazioni). Le interazioni elementari atomi estranei-dislocazioni si comprendono con concetti di teoria dell'elasticità, di termodinamica chimica, di meccanica reticolare e di teoria elettronica. A causa della loro grandezza, che si discosta da quella degli atomi della matrice, gli atomi estranei hanno un campo di deformazione locale, che conduce all'interazione ‛para-elastica' con il campo di tensione della dislocazione. Per un campo di deformazione isotropo (cristallo misto di sostituzione) ci si aspetta, nell'ambito della teoria dell'elasticità isotropa, solo per le dislocazioni a spigolo, una forza di interazione (attrattiva o repulsiva), Fp, proporzionale alla variazione del volume atomico. Il suo ammontare massimo si può valutare pari a circa GSb2 ∣ δ ∣ (δ = a-1 da/dc = variazione delle costanti reticolari con la concentrazione; secondo Hume-Rothery è ∣ δ ∣ ≾ 15% per una buona solubilità in leghe sostituzionali). Spesso è ∣ δ ∣ ≃ 1% e quindi Fp ≃ 10-10 N. Nel caso di grande vicinanza tra atomi estranei e dislocazioni a vite, l'anisotropia elastica e i termini non lineari forniscono forze ancora coafrontabili con il valore precedente. Accanto all'interazione para-elastica si può giungere anche a un'interazione ‛dia-elastica', indotta, poiché l'intorno di un atomo estraneo mostra una diversa polarizzabilità elastica. Se si approssima questa con il ‛difetto di modulo', cioè con la variazione del modulo di elasticità tangenziale rispetto alla concentrazione, η = GS-1dGS/dc, allora la forza di interazione massima diventa Fd ≃ GSb2 ∣ η ∣/3 e, poiché spesso è ∣ η ∣ > ∣ δ ∣, Fd per lo più non è trascurabile rispetto a Fp. Nelle dislocazioni a spigolo, per atomi estranei, Fp ha segni opposti al di sopra e al di sotto del piano di scorrimento; Fd, invece, non cambia di segno. Si ottengono quindi, con un solo tipo di atomi estranei, per dislocazioni a spigolo, due specie di ostacoli con forze massime Fd ± Fp. Nei campi di deformazione anisotropi, la teoria dell'elasticità lineare fornisce, per dislocazioni a spigolo e a vite, quale forza di interazione massima, Ft ≃ GSb2Δε/3. Qui Δε è la differenza degli elementi diagonali del tensore di deformazione relativo all'asse tetragonale, per esempio intorno a un atomo interstiziale nel reticolo cubico a corpo centrato (v. fig. 25). Gli atomi interstiziali che conducono a deformazioni anisotrope sono perciò ostacoli molto più efficaci ai movimenti delle dislocazioni (Δε ≃ 1 è possibile nel reticolo cubico a corpo centrato; per C nel Feα è Δε ≃ 0,41). Un'interazione ‛chimica' (H. Suzuki) è da attendersi tra atomi estranei e dislocazioni dissociate, in particolare nelle leghe cubiche a facce centrate, dove il nastro del difetto di impilaggio rappresenta una striscia di materiale esagonale, che si può arricchire in atomi estranei. Se questo arricchimento è possibile anche durante il movimento delle dislocazioni, l'interazione chimica può produrre non solo un limite di snervamento superiore (fino allo svincolamento dell'ancoraggio statico), ma anche un contributo alla resistenza.
Accanto all' ‛atmosfera di Suzuki' (arricchimento di atomi estranei sostitutivi nei difetti di impilaggio) è possibile naturalmente, con una sufficiente mobilità degli atomi estranei, anche un arricchimento in vicinanza di dislocazioni a spigolo (a seconda della grandezza nel campo di dilatazione o compressione) sulla base dell'interazione elastica (Cottrell). Questa ‛atmosfera di Cottrell' conduce al noto fenomeno dello snervamento negli acciai ferritici (v. fig. 80) e quindi alle corrispondenti instabilità nella curva di trazione. Le dislocazioni, una volta svincolate, possono essere nuovamente catturate durante il loro movimento da atomi estranei che diffondono, come si dimostra con le ripetute instabilità dell'effetto di Portevin-Le Chatelier (jerky flow, serrated yielding, ecc.). Se si interrompe la deformazione dei materiali in cui si possono formare le atmosfere di Cottrell o di Suzuki, si può osservare il fenomeno dell'invecchiamento (ricomparsa di un limite di snervamento dopo parziale diminuzione del carico).
Un ulteriore meccanismo di ancoraggio nei campi di deformazione anisotropa può consistere nel fatto che i dipoli elastici si orientano favorevolmente, di volta in volta, nel campo di deformazione delle dislocazioni (Snoek). Questa ‛atmosfera di Snoek', cioè una nuvola di polarizzazione elastica, può ugualmente spostarsi con la dislocazione.
Per il calcolo della sollecitazione tangenziale critica di un cristallo misto, gli ostacoli per semplicità sono assunti come immobili e si determina la tensione necessaria a spostare una singola dislocazione sul piano di scorrimento (dunque su grandi distanze) (la fig. 81 mostra una successione di posizioni delle dislocazioni in un modello al calcolatore). Si supponga, seguendo Friedel, che una dislocazione si trovi su un piano di scorrimento con ostacoli, la cui sezione d'urto sia molto piccola in confronto alla distanza L (dipendente dalla tensione applicata) degli ostacoli toccati dalla dislocazione (v. fig. 82). La superficie tratteggiata è data, per un raggio di curvatura R, da
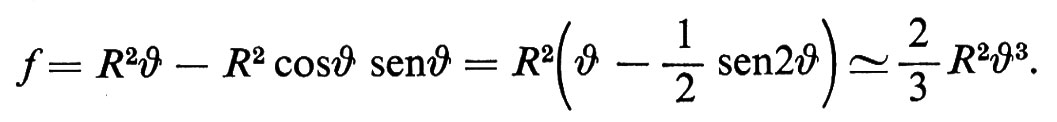
Con ϑ ≃ L/2R e R = TL/τb (v. formula 23) e con la condizione (Friedel, Fleischer) che la sollecitazione tangenziale critica τc sia raggiunta quando la superficie, spazzata fino a toccare l'ostacolo successivo, è esattamente uguale alla metà della superficie media per ostacolo, f = cA/2, con cA = densità di superficie degli ostacoli, si ottiene
L3 = 6TL/(τcbcF). (34)
D'altra parte la sollecitazione tangenziale critica è data da
τcbL(τc) = F0, (35)
con F0 = forza di interazione massima; cioè gli ostacoli o agiscono con la loro forza massima o non agiscono affatto. Da (34) e (35) si ottiene
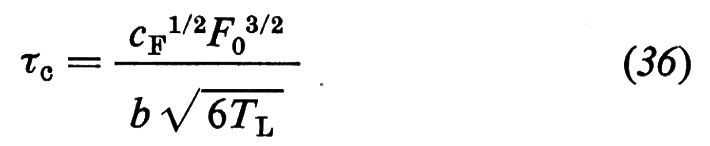
per la sollecitazione tangenziale critica di un cristallo misto. Nonostante le molte limitazioni, questo modello è stato più volte confermato per soluzioni diluite e ostacoli fortemente localizzati, in particolare anche per ferro α con C, N, O e per l'indurimento da carbonio della martensite (v. fig. 83). In concentrazioni un po' più alte (qualche unità per cento nei cristalli misti di sostituzione) i potenziali di interazione dei singoli atomi estranei si sovrappongono, cosicché non si può partire bene da forze di interazione isolate e costanti. La dislocazione è sottoposta all'influsso di molti ostacoli contemporaneamente (Mott). Labusch ha trovato, per questo problema di una dislocazione che si trova sotto tensione in una distribuzione di forze ostacolanti, che la sollecitazione tangenziale critica è proporzionale a cF2/3F04/3TL-1/3, com'è confermato dai risultati di più recenti esperimenti (v. fig. 84). La distinzione sperimentale tra una dipendenza da cF1/2 o da cF2/3 non è sempre possibile.
In queste rappresentazioni della sollecitazione tangenziale critica non si è tenuto conto dell'influenza della temperatura. La si può prendere in considerazione attraverso un' F0 dipendente dalla temperatura. Ostacoli di dimensioni atomiche dovrebbero, a causa dell'aumento delle fiuttuazioni termiche, perdere di efficacia al crescere della temperatura, come si vede dalla fig. 85. Ci si aspetta, tuttavia, un'ulteriore diminuzione al disopra della temperatura ambiente fino al valore del metallo puro. Le cause dell'andamento di τc, (plateau), che in questo campo è quasi indipendente dalla temperatura, sono (nel quadro degli ostacoli atomici fissi) finora non completamente chiarite.
Accanto a una aumentata resistenza intrinseca del materiale omogeneo, è di grande significato, nella tecnica, l'aumento di resistenza attraverso ostacoli più estesi, cioè l'‛indurimento da particelle'. Qui vengono presi in considerazione tutti i prodotti di reazione sia dello smescolamento delle leghe (precipitazione) sia della reazione selettiva con ossigeno o con altri gas (ossidazione interna, formazione di nitruro) o fasi mescolate allo stato fuso o con procedimenti di metallurgia delle polveri (dispersioni). I meccanismi possibili di indurimento e deformazione sono molteplici. Inizialmente si verificano interazioni para-elastica e dia-elastica su precipitati coerenti. Per la forza di interazione para-elastica si trova, in particelle di forma sferica con raggio R,
Fpmax ≃ GSbR ∣ δ ∣, (37)
dove δ = Δa/a, se si assume una dislocazione lineare rigida. Secondo Wiedersich, Fpmax deve essere moltiplicata per un fattore di correzione dipendente dal rapporto β = Fpmax/TL e dalla distanza z del piano di scorrimento dal centro della particella (v. fig. 86). Tale fattore deve essere preso in considerazione soprattutto per ostacoli più forti (angoli all'ostacolo maggiori di 50°).
Quando i contributi para-elastici e dia-elastici sono abbastanza piccoli da rendere possibile una deformazione per scorrimento della particella, prima che si presenti il caso di Orowan (eguaglianza 25; v. fig. 87), allora risultano ulteriori contributi alla resistenza da processi che consumano energia e che diventano efficaci all'ingresso delle dislocazioni nelle particelle. Prima di tutto aumenta sempre nella deformazione per scorrimento l'interfaccia con la matrice, come si vede nella fig. 88. Con l'energia interfacciale specifica γαβ si ha come forza repulsiva massima
Fαβ ≃ 2γαβb. (38)
Questa parte appare tuttavia difficilmente da sola, poiché anche per valori alti di γαβ (10 ... 100 mJ m-2) porta solo a piccole forze (≾ 10-11 N).
Nell'esempio rappresentato nella fig. 88 i precipitati coerenti sono ordinati. Se una singola dislocazione della matrice penetra in una particella ordinata, allora il suo vettore di Burgers non si ‛adatta' più ai vettori di base della sovrastruttura. Secondo la fig. 21a, nella struttura L12, che spesso risulta dal reticolo cubico a facce centrate (composto A3B), ci sono piani (100) occupati alternativamente con soli atomi A o con atomi A e B nel rapporto 50: 50. Nella fig. 89 è schematizzata una sezione, lungo un piano occupato dai due tipi di atomi, di una particella in una matrice cubica a facce centrate. Una dislocazione a spigolo con vettore di Burgers ½ 〈110> nel reticolo cubico a facce centrate è entrata da sinistra nella particella. Questo scorrimento ha disturbato l'ordine sui piani relativi e ha generato una superficie limite di antifase. Con la tensione interfacciale di antifase γAPB (valori tipici si trovano a circa 300 mJ m-2), la forza repulsiva massima diventa, per una particella sferica coerente di raggio R,
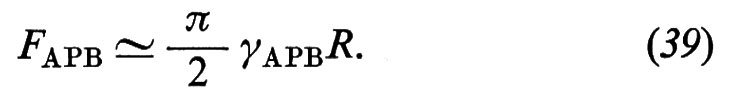
Dunque, per R = 200 Å, per esempio, risulta FAPB ≃ 10-9 N, paragonabile all'alterazione para-elastica quando ∣ δ ∣ è piccolo (eq. 37). Questa forza FAPB deve essere superata dalla prima dislocazione nel piano di scorrimento; la seconda, al contrario, ristabilisce nuovamente l'ordine, viene dunque attirata. In circostanze appropriate si possono vedere coppie di dislocazioni attraversare i precipitati ordinati (‛superdislocazioni' con una banda di bordo di antifase all'interno dei precipitati, analogamente alla dissociazione di dislocazioni semplici nel difetto di impilaggio: v. fig. 90). Questo meccanismo di indurimento è di grande significato per leghe ad alta temperatura a base di nichel (v. fig. 91). Gli elementi della lega Al e Ti formano dei precipitati ordinati di Ni3(Al, Ti) che restano stabili (e ordinati) fino a temperature relativamente alte.
Altri contributi all'indurimento da particelle provengono dalle differenze nell'energia dei difetti di impilaggio, per cui la dissociazione può variare. Si può avere una maggiore sollecitazione tangenziale nella particella stessa (per es. per ordinamento). Nei precipitati incoerenti, le dislocazioni si arrestano davanti alla superficie limite, poiché ora nessun adatto vettore di traslazione è disponibile (v. fig. 27). Fino a quando non si presenta il caso di Orowan e se l'aggiramento tramite risalita o scorrimento deviato non può avvenire, una tale particella può essere sollecitata fino al raggiungimento della sua resistenza teorica allo scorrimento (≃ GSp/5, con GSp = modulo di elasticità tangenziale del precipitato). Con ciò la forza massima che può sostenere una particella diventa Fj ≃ GSp bR/10.
Per il calcolo della sollecitazione tangenziale aggiuntiva Δτc per la deformazione di un cristallo con precipitati, si parte nuovamente dall'eguaglianza (35), dove la lunghezza effettiva L(τc) dipende nuovamente dalla flessibilità della dislocazione e dalla forza degli ostacoli. (La rappresentazione di ostacoli discreti diventa nuovamente incerta quando il volume relativo dei precipitati supera alcune unità per cento. Si deve quindi partire dalla teoria non locale di Labusch). Per ostacoli non troppo forti, si può scrivere la relazione di Friedel-Fleischer (34) come
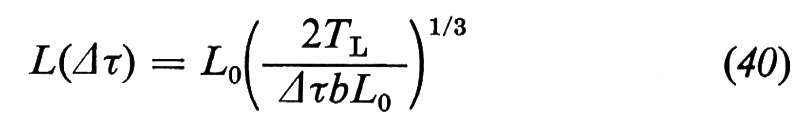
con
L0 ≃ √-2 R/f1/2 (41)
per particelle sferiche con raggio R e frazione volumetrica f (L02 è quindi la superficie media per particella). Per la maggior parte delle forze massime importanti, le equazioni (37), (39), ecc. si possono scrivere
F0 = αGSRb. (42)
Con (40) e (42), segue dalla (35)
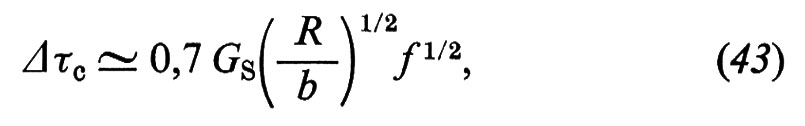
se si pone 2TL ≃ GSb2.
Differentemente vanno le cose per il caso di Orowan (aggiramento delle particelle con formazione di anelli di dislocazione: v. fig. 87): v. eguaglianza (25) con L0 secondo la (41) e un fattore numerico secondo Kocks
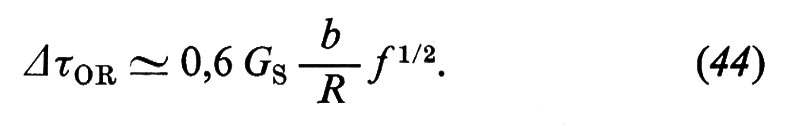
A parità di frazione volumetrica f, anche per piccoli raggi sarà innanzitutto determinante Δτ0 secondo la (43), per un successivo ingrossamento dei precipitati ΔτOR 〈 Δτc, cosicché la resistenza meccanica diminuisce nuovamente. Si può dunque valutare un raggio critico Rc ≃ 0,9b/d, per il quale Δτ diventa massimo. Nelle leghe indurenti si tende a questo stato di indurimento ottimale. Se Δτ diminuisce nuovamente, allora una lega è ‛iperinvecchiata'. Alcuni esempi sono mostrati nella fig. 92. Oltre alla resistenza, viene fortemente influenzato dai precipitati anche l'andamento dell'incrudimento. Coefficienti di incrudimento particolarmente elevati si hanno con l'indurimento mediante particelle incoerenti (indurimento per dispersione), come mostra l'esempio Cu + Al2O3 . Poiché le dislocazioni devono circondare delle particelle, intorno alle particelle stesse si trovano anelli di dislocazioni, che agiscono incrudendo ulteriormente. Oltre al caso degli anelli di Orowan, raffigurato nella fig. 87, si può giungere, secondo Hirsch, all'aggiramento delle particelle anche per mezzo dello scorrimento deviato, come mostrato nella fig. 93.
Le leghe di una composizione appropriata (ad es. 75 : 25, 50 : 50, ecc.) possono anche essere ordinate omogeneamente: così la deformabilità plastica può cambiare in modo decisivo. Un cristallo perfettamente ordinato si comporta in modo simile a un metallo puro, ad esempio τ0 può essere più basso che in un cristallo misto, come già dimostrato da molto tempo per Cu3Au (v. fig. 94). Le dislocazioni singole della struttura disordinata formano con l'ordinamento di tutto il cristallo coppie di dislocazioni (superdislocazioni), come discusso sopra per i precipitati ordinati. L'incrudimento dei cristalli ordinati è in generale maggiore e la loro duttilità minore rispetto a quella dei cristalli disordinati. In tempi recenti, le fasi intermetalliche con la partecipazione di elementi più leggeri, come Al, Si, Ti, V, hanno suscitato un grande interesse, poiché esse offrono una maggiore resistenza meccanica (con minore densità) oppure più alte temperature di impiego (v. fig. 95). La duttilità può essere aumentata attraverso aggiunte appropriate (per facilitare lo scorrimento ai bordi di grano o anche per abbassare il potenziale di Peierls); su questo punto sono in atto intense ricerche.
e) Deformazione dei metalli amorfi.
I vetri metallici mostrano, è vero, una scarsa duttilità, ma nessun incrudimento. Sebbene i concetti della teoria delle dislocazioni siano applicabili anche ai continui non cristallini, sicuramente non sono possibili processi moltiplicativi e reattivi del tipo conosciuto nei cristalli. La deformazione è localizzata in bande di scorrimento nettamente definite e comincia solo con tensioni vicine alla resistenza allo scorrimento teorica. Sotto l'effetto della tensione applicata vengono causati difetti tramite trasposizione di atomi. Tali difetti possono muoversi oppure espandersi e procurare lo scorrimento. Attualmente si discute su diversi modelli. Lo scorrimento viscoso dei vetri metallici avviene al contrario non tramite scorrimenti localizzati, ma attraverso movimenti di flusso viscoso omogenei e controllati dalla diffusione. Entrambi i processi plastici dipendono fortemente dal ‛volume libero', che viene congelato per esempio nella solidificazione, poiché gli atomi non sono ancora ordinati con la densità di impaccamento più alta possibile (amorfa).
f) Fatica.
In seguito a sollecitazione ciclica si riscontra nei metalli e nelle leghe cristalline una serie di particolarità, che hanno la loro origine in speciali reazioni tra dislocazioni. Una conseguenza importante è la limitata durata di pezzi sottoposti a sollecitazione alterna con ampiezze di sollecitazione comprese fra il 50 e il 25% del carico di rottura nel saggio di trazione. Nella tecnica il profilo della sollecitazione è spesso molto complicato, con ampiezze, frequenze e cicli di temperatura molto diversi. Una prova di trazione/compressione simmetrica, con frequenza costante, a temperatura costante e con ampiezza di sollecitazione costante, viene spesso utilizzata per la caratterizzazione della resistenza alle sollecitazioni alterne. Si determina il numero dei cicli di carico N fino alla ‛rottura per fatica' come funzione dell'ampiezza di tensione σé e si ottiene così la ‛curva di Wöhler' (curva S-N, stress νs number; v. fig. 96; N = 1/4 corrisponde al carico di rottura nella prova di trazione). Il campo fino a N = 104 si chiama ‛fatica a basso numero di cicli' o ‛fatica oligociclica' (LCF = low cycle fatigue), al di sopra, fino a circa 106, ‛fatica' (fatigue). Per la maggior parte dei materiali (eccettuati, per es., Cu o Al puri) la curva di Wöhler decorre al di sopra di N ≈ 106 ... 107 quasi orizzontalmente, e si ottiene un limite di fatica (endurance limit) σée. Se si resta con i carichi al di sotto di questo valore, dovrebbe essere sopportato un numero qualsiasi di cicli di carico senza rottura per fatica.
Le reazioni tra dislocazioni, l'inizio e l'allargamento della cricca nella sollecitazione alterna sono processi molto complicati, che vengono indagati da numerosi ricercatori. È fondamentale che una deformazione plastica provocata da una sollecitazione a trazione produca una disposizione delle dislocazioni che non è stabile rispetto all'inversione di tensione (effetto Bauschinger). Una parte delle dislocazioni può effettuare grandi percorsi anche sotto minime tensioni di segno contrario. Nelle numerose alternanze del carico si ottengono infine disposizioni delle dislocazioni completamente nuove, soprattutto dipoli e multipoli. Per una data ampiezza dell'allungamento plastico si raggiunge un valore di saturazione dell'ampiezza della sollecitazione. Lo scorrimento si concentra spesso in regioni nettamente delimitate di alta attività delle dislocazioni, dove i processi di incrudimento e di addolcimento all'incirca si equilibrano (bande di scorrimento persistenti) dando origine a occasionali deformazioni a valanga (strain bursts), che a loro volta conducono a grossolani gradini di scorrimento sulla superficie. Dalla superficie partono la maggior parte delle cricche di fatica. La formazione e l'apertura di una cricca con partecipazione di due sistemi di scorrimento, secondo un modello di P. Neumann, sono rappresentate nella fig. 97. L'apertura delle cricche è, come la fig. 97 lascia vedere, sensibilmente dipendente dallo stato delle superfici neoformate, cosi che mezzi fluidi e gassosi, in cui un materiale metallico viene collocato, possono fortemente influenzare l'espansione della cricca (come pure la sua formazione).
9. Materiali magnetici e superconduttori.
Le proprietà magnetiche e la superconduttività dei sistemi metallici sono di grande importanza nell'elettrotecnica.
Particolare valore tecnico hanno le proprietà ferromagnetiche dei metalli e delle leghe. Il ferromagnetismo è caratterizzato da un forte aumento della densità di magnetizzazione M = (B − B0)/μ0 (con B = induzione magnetica nel materiale, B0 = induzione magnetica nel vuoto, μ0 = permeabilità magnetica del vuoto) in funzione di B0 (= μ0H, misurata in tesla = 104 gauss; H = campo magnetico), nonché da un'isteresi e da una rimanenza (magnetica), cioè una magnetizzazione persistente allo scomparire del campo esterno. La fig. 98 mostra schematicamente l'andamento di M(H) per la curva di prima magnetizzazione di un ferromagnete inizialmente non magnetizzato e la seguente curva di isteresi in confronto con le sostanze diamagnetiche e paramagnetiche. La magnetizzazione di saturazione del ferromagnete è raggiunta quando tutte le regioni del campione hanno allineato i loro vettori di magnetizzazione parallelamente al campo applicato.
Già senza campo, perfino all'inizio della curva di prima magnetizzazione, il campione è magnetizzato quasi ovunque ‛spontaneamente'; le direzioni della magnetizzazione nei ‛domini di Weiss' separati l'uno dall'altro dalle ‛pareti di Bloch' producono macroscopicamente una magnetizzazione totale trascurabile (per es. v. fig. 99). Applicando un campo magnetico si manifestano dunque la rotazione delle magnetizzazioni dei domini elementari e il movimento delle pareti di Bloch che li separano. La ‛magnetizzazione spontanea' si pone nel quadro del magnetismo atomico come mediata da un ‛campo interno'. Premessa per il presentarsi del ferromagnetismo è l'esistenza di momenti magnetici atomici (elettroni non accoppiati) e un'interazione degli atomi magnetici che conduce a un accoppiamento di singoli momenti parallelamente l'uno all'altro. Questo accoppiamento ferromagnetico (campo molecolare) conduce a una magnetizzazione spontanea a cui si oppone tuttavia l'agitazione termica, così che la magnetizzazione di saturazione (identica a essa) decresce all'aumentare della temperatura per scomparire alla temperatura di Curie TC. A temperature più alte si deve osservare il normale comportamento paramagnetico (v. solidi, fisica dei), dove la suscettibilità (per unità di volume) χ, definita come
M = χB0 = χμ0H,
deve ubbidire a una legge di Curie-Weiss

La fig. 100 mostra la magnetizzazione spontanea in funzione della temperatura per il gadolinio, un rappresentante dei metalli delle terre rare, a confronto con la curva teorica calcolata per J = 7/2 (J è il momento angolare totale), corrispondente allo stato fondamentale 8S7/2 di uno ione Gd3+. Il valore assoluto della magnetizzazione spontanea per T → 0 fornisce un momento magnetico di 7,63 μB (μB = magnetone di Bohr) per atomo, vicino al valore aspettato di 7μB per gli stati 4f occupati secondo la regola di Hund. Per i metalli delle terre rare, nei quali gli elettroni spaiati, responsabili del momento magnetico, si trovano al livello 4f, relativamente vicini al nucleo, la teoria della magnetizzazione basata sui momenti magnetici localizzati è confermata. Nei ferromagneti, invece, con ‛elettroni magnetici' nella banda 3d (Fe, Co, Ni con TC = 770, 1.131 e 358 °C) e anche negli altri elementi di transizione delle serie 3d e 4d, il paramagnetismo mostra già forti contributi non localizzati (dove χ non segue più l'eguaglianza 46). Contrariamente al magnetismo atomico, si è qui affermata l'idea del ‛magnetismo a bande'. Le bande d e s vengono considerate unitamente e l'interazione di scambio viene introdotta attraverso un campo molecolare proporzionale alla magnetizzazione media. Elettroni con spins paralleli al campo assumono ora valori di energia diversi da quelli degli elettroni con spins antiparalleli. La fig. 101 mostra schematicamente la sequenza Fe, Co, Ni con le relative bande di second'ordine (bande presupposte rigide). Poiché gli spostamenti interessano principalmente il livello d, il comportamento ferromagnetico scompare nelle leghe di nichel con il 53% di rame. Sebbene molte osservazioni sperimentali confermino la rappresentazione del magnetismo di banda, si conoscono particolareggiatamente anche numerose anomalie, cosicché il modello atomico e il modello a bande spesso vengono utilizzati l'uno accanto all'altro.
Per l'impiego delle leghe e dei metalli magnetici nella tecnica è decisivo il comportamento durante l'inversione magnetica. Si distinguono, a seconda dell'apertura della curva d'isteresi (la cui area dà il consumo di energia per l'inversione magnetica), materiali magnetici ‛duri' e ‛molli'. Di nuovo è, come nella plasticità, la mobilità dei difetti, qui delle pareti di Bloch, quella che controlla un'importante proprietà dei materiali. Nei ferromagneti cristallini l'andamento della magnetizzazione dipende dalla direzione cristallografica; ci sono direzioni ‛facili' e ‛difficili' (anisotropia cristallina: v. fig. 102). Di preferenza i domini di Weiss sono magnetizzati lungo le direzioni facili e la natura della parete di Bloch tra due domini dipende dal cambiamento di orientazione della magnetizzazione. Poiché una variazione brusca dell'orientazione non è favorita, a causa dell'elevata energia di scambio, si presenterà un certo spessore della parete entro il quale la magnetizzazione varia gradualmente (v. fig. 103a). D'altra parte devono essere assunte molte direzioni di magnetizzazione sfavorevoli a causa dell'anisotropia dei cristalli. Da entrambe queste tendenze concorrenti risulta uno spessore stabile (per es. di 100 nm, energia della parete paragonabile alle energie dei bordi di grano). Come il movimento delle dislocazioni attraverso materiali disomogenei, viene influenzato molto fortemente dai costituenti strutturali anche il movimento delle pareti di Bloch, in particolare da precipitati di grandezza appropriata, che possono possedere proprietà magnetiche diverse da quelle della matrice. Il calcolo delle forze di interazione e degli stadi elementari dei movimenti della parete è riuscito solo in pochi casi semplificati. Si distingue tra movimenti di parete reversibili in materiali estesamente omogenei, per i quali diventano rilevanti i processi dissipativi solo a più alte frequenze, e movimenti di parete irreversibili, nei quali devono essere superati gli ostacoli, cosicché una forte isteresi si presenta anche alle frequenze più basse e ad alte forze coercitive (magnete permanente).
Il ferro è l'elemento di partenza più comune anche per la realizzazione di materiali magnetici. Proprietà magnetiche dolci si ottengono con leghe contenenti il 2-3% at. di silicio e decarburate per ricottura in idrogeno, per rendere inefficace il carbonio che causa l'isteresi. In queste leghe possono essere inoltre prodotte, dopo una deformazione appropriata e un trattamento di ricristallizzazione, delle tessiture speciali (per es. la ‛tessitura di Goss' nei lamierini per trasformatori), in cui prevalgono le direzioni di magnetizzazione facile. Le leghe di nichel e ferro (Permalloy, Hypemik, Hymu) hanno suscettibilità magnetiche particolarmente alte con basse isteresi e vengono utilizzate nella tecnica delle comunicazioni (schermatura). Aspettative particolari si pongono nel maggiore impiego dei metalli amorfi (Fe-Ni-B, ecc.) quali materiali magneticamente dolci. Poiché in questo caso viene a mancare l'anisotropia caratteristica dei cristalli, e quindi i bordi di grano, e, se si tratta di materiali di buona qualità, non esistono nemmeno altri ostacoli al movimento delle pareti di Bloch, si ottengono forze coercitive di meno di 1/30 di quelle delle leghe Fe-Si. Accanto all'impiego come nuclei di trasformatori, sono particolarmente utili per la loro flessibilità le lamine di schermatura intrecciate da nastri amorfi sottili.
Per materiali magneticamente duri (in uso per magneti permanenti) vengono ricercati un alto magnetismo residuo e un'elevata forza coercitiva. Per una grande forza coercitiva è favorevole un'elevata anisotropia dei cristalli (vengono resi più difficili i processi di rotazione) e la mobilità delle pareti di Bloch deve essere ridotta attraverso ostacoli appropriati. Nel caso estremo di pareti di Bloch completamente immobili, sono ancora possibili solo processi di rotazione. Ciò vale particolarmente per piccole particelle ferromagnetiche in una matrice: per diametri delle particelle di meno di ~ 100 nm, non ci sono più all'interno pareti di Bloch (particelle a un sol dominio), cosicché a un'alta anisotropia del cristallo segue un'elevata forza coercitiva. Classici materiali magneticamente duri sono - oltre agli acciai al carbonio con Cr e Co - soprattutto leghe di Al-Ni-Co di diversa composizione, che mostrano indurimento per precipitazione. I precipitati fini e coerenti appaiono per smescolamento di una fase cubica a corpo centrato omogenea a elevate temperature. Negli ultimi anni si sono messi in evidenza soprattutto composti intermetallici di cobalto con metalli delle terre rare (per es. samario, ittrio) come materiali magneticamente duri. Essi permettono, a causa dei loro alti valori per il massimo del prodotto BH, che serve come termine di paragone per magneti permanenti, una miniaturizzazione dei componenti attivi negli apparecchi elettromeccanici (per es. stampanti, motori passo-passo, ecc.). La fig. 104 mostra alcune curve di smagnetizzazione. La spiegazione della microstruttura di questi materiali, prodotti spesso attraverso metodi di metallurgia delle polveri, è ancora agli inizi. In considerazione dello scarseggiare mondiale del cobalto, è molto promettente lo sviluppo della polvere microcristallina e di nastri Fe-Nd-B con proprietà ancora più interessanti [(BH)m ≃ 50 MGOe). Come ‛terra rara' il neodimio è relativamente frequente (~ 15% di tutti i minerali dei lantanidi; come elemento è più frequente, per es., del piombo), cosicché possono essere ottenuti anche prezzi più vantaggiosi.
Come esposto nell'articolo criofisica, i ‛superconduttori' di tipo II mostrano raramente curve di magnetizzazione reversibili: il flusso magnetico viene ‛ancorato' ai difetti (flux pinning). La causa risiede nell'interazione tra portatori del quanto di flusso elementare Φ0 (= h/2e = 2 × 10-7 gauss cm2) e fili di flusso (flux lines, vortices) con disomogeneità. Tale interazione conduce a una repulsione o attrazione dei fili di flusso. Solo attraverso l'ancoraggio del flusso è possibile che correnti alte (campi magnetici maggiori di Hc1) possano scorrere senza perdita nei superconduttori di tipo II, perché altrimenti la forza di Lorentz, che opera attraverso la corrente di trasporto sui fili di flusso, causerebbe il movimento dei fili di flusso e con ciò una dissipazione di energia (v. fig. 105). Per ottenere campi magnetici più elevati, questi concetti sono usati nella superconduttività tecnica, al fine di produrre cavi superconduttori con la massima conducibilità. La fig. 106 mostra la corrente critica in funzione del campo magnetico per alcuni superconduttori ‛duri'. I composti intermetallici (per es. Nb3Sn, rappresentante della struttura A15) sono, accanto alle leghe Nb-Ti, particolarmente importanti a causa della loro elevata temperatura critica (Nb3Ge ha, con 23 K, la più alta temperatura di transizione). A causa della loro fragilità, i composti intermetallici si possono a malapena lavorare. Superconduttori a filamento di Nb3Sn si sono potuti realizzare solo attraverso un'abile utilizzazione dei principi della metallurgia. Dapprima si lavorano fili di Nb puro, ben deformabili, insieme con un bronzo Cu-Sn; si ottiene così un filo composito che si riscalda a 700 °C. I fili di Nb hanno uno spessore di pochi μm e durante il riscaldamento si ottiene alla loro superficie, per diffusione preferenziale dello Sn verso l'interno, uno strato Nb3Sn dello spessore ottimale di ~1 μm. Questo strato diviene superconduttore al di sotto della temperatura di transizione (v. fig. 13). Per la stabilizzazione termica del conduttore finito deve essere inserito ancora rame, quale buon conduttore di calore, in buon contatto termico con il materiale superconduttore. La fig. 107 mostra la struttura di un cavo superconduttore pronto. Si spera di realizzare ulteriori progressi nel campo della superconduzione a campo magnetico elevato utilizzando composti del tipo PbMo6S8 (fasi di Chevrel), con i quali sembrano possibili campi magnetici di oltre 40 T.
Le ricerche sulla superconduttività hanno ricevuto recentemente (1986) un nuovo impulso dalla scoperta di una temperatura critica inaspettatamente alta (Tc = 35 K) per un ossido di bario, lantanio e rame (J. G. Bednorz e K. A. Müller, premi Nobel 1987). È inoltre ragionevole aspettarsi ulteriori progressi nel settore della superconduttività ad alto campo dalla successiva scoperta di ossidi metallici superconduttori del tipo YBa2Cu3O9-y (y ≃ 2), con una struttura tipo perowskite distorta ortorombicamente. Attualmente sono state raggiunte temperature critiche fino a 92 K e probabilmente sarà possibile realizzare presto superconduttori tecnicamente utili che basterà raffreddare alla temperatura dell'azoto liquido (anziché alla temperatura, correntemente usata, dell'elio liquido, molto più costoso).
10. Sollecitazione e durata dei metalli.
Accanto alle sollecitazioni meccaniche già trattate, alle quali sono esposti i materiali metallici, hanno una grande importanza, nella valutazione della stabilità e della durata, gli influssi dell'ambiente e del contatto con altri materiali.
Eccetto oro e platino, tutti i metalli sono chimicamente instabili in presenza di ossigeno (o altre sostanze ossidanti). Tuttavia oggetti metallici massivi restano inalterati a lungo, poiché l'ossidazione incomincia alla superficie e un partner di reazione deve diffondere attraverso l'ossido per reagire ulteriormente con il resto del metallo. Sono perciò necessarie energie di attivazione maggiori rispetto ai semplici metalli. Uno strato superficiale continuo di ossido offre dunque, a temperature non troppo alte, una buona protezione contro un'ulteriore ossidazione. La crescita, controllata dalla diffusione, dello strato di ossido segue, all'inizio, sotto l'influenza dei campi elettrici tra le superfici limite metallo-ossido e ossido-superficie esterna, una legge temporale logaritmica e poi parabolica (v. cap. 6). A temperature più elevate, quando la diffusione può risultare accelerata, ci si aspetta che lo spessore dello strato dipenda linearmente dal tempo, quando la reazione di ossidazione, indipendentemente dallo spessore dello strato, determina essa stessa la velocità. Si osserva un analogo comportamento in funzione del tempo quando l'ossidazione comporta un forte cambiamento di volume, cosicché la scaglia di ossido (gli ossidi sono fragili), dopo aver raggiunto un piccolo spessore, si rompe e perciò altro metallo fresco viene esposto all'ambiente ossidante. La grande differenza nel comportamento all'ossidazione dell'alluminio e del ferro si spiega solamente cosi, sebbene in pratica agiscano anche processi elettrochimici.
In presenza di acqua (in ambiente umido o liquido), diventano attive differenze nel potenziale elettrochimico in posti diversi della superficie e si giunge all'ossidazione anodica (‛corrosione elettrochimica'). Già nei metalli puri i bordi di grano sono attivi come anodi locali, poiché gli atomi lì giacenti possiedono un più alto potenziale chimico. Un vantaggio dei metalli amorfi è la loro migliore resistenza alla corrosione a causa dell'assenza di bordi di grano. Altre cause della corrosione, come tensioni interne (variazioni della densità di energia elastica), fluttuazioni di concentrazione e presenza di fasi diverse (precipitati, impurezze), conducono tuttavia, anche indipendentemente dalla struttura dei grani, a processi di corrosione. Riguardo al corso del processo di corrosione in presenza di diversi elementi, decide la posizione dei metalli nella serie delle tensioni (nell'elenco dei potenziali di elettrodo), il cui punto zero è stato fissato arbitrariamente per la reazione H2 → 2H+ + 2e-. La tab. X fornisce alcuni esempi. Si riconosce, tra l'altro, che Sn rispetto a Fe è catodico (più nobile), mentre Zn è anodico. Si comprende da ciò il diverso comportamento del ferro stagnato e di quello zincato (v. fig. 108). Una piccola graffiatura nel sottile strato di stagno (che, fino a quando rimane continuo, offre una buona protezione in molti elettroliti) produce un ‛elemento locale', un posto in cui Fe viene ossidato a Fe2+ e va in soluzione. Uno strato di zinco, anche con interruzioni, perfino un elettrodo di zinco separato ma posto in prossimità della superficie ferrosa, impedisce al contrario la corrosione, poiché Fe agisce da catodo. Lamiere e tubi di acciaio zincati sono perciò materiali adatti a resistere alla corrosione. Particolarità del processo di corrosione dipendono sensibilmente dall'esatta composizione del materiale e dell'ambiente elettroliticamente attivo. Variazioni locali del contenuto di ossigeno nel mezzo liquido possono da sole portare alla corrosione.
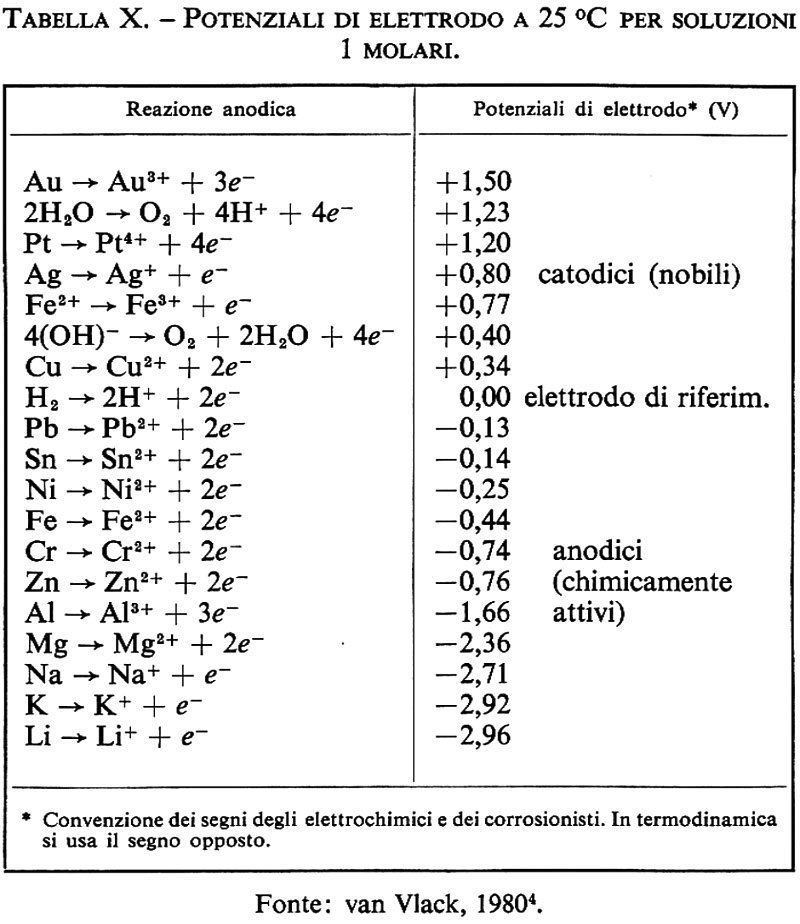
L'acciaio inossidabile viene ottenuto nel modo più facile attraverso aggiunta di cromo. Sebbene il cromo sia anodico rispetto al ferro, viene ridotta fortemente la velocità di corrosione, poiché sulla superficie metallica si formano ioni CrO32- che impediscono l'ulteriore corrosione (‛passivazione'). La passivazione e la galvanizzazione (introduzione di uno strato più nobile attraverso immersione in bagno metallico fuso o attraverso deposizione elettrolitica) proteggono i metalli dalla corrosione. Anche i rivestimenti con materiali chimici inerti (lacche organiche, smalti, ecc.) servono a questo scopo. Un grande numero di processi chimici a umido e a secco (per es. CVD, chemical vapor deposition) e fisici (per es. PVD, physical vapor deposition) vengono oggi applicati per il rivestimento di metalli e sottoposti a ulteriori studi.
Accanto alla rottura e ai processi di corrosione, la cui azione viene accelerata attraverso sollecitazione meccanica alternata (per es. per tensiocorrosione) anche ‛processi di erosione e di usura' sono responsabili della limitazione della durata dei materiali metallici (e dei non metallici). Oltre ad adatti lubrificanti (che ugualmente possono influenzare la corrosione), sono opportuni, per limitare l'usura, o modificazioni speciali delle proprietà superficiali del materiale oppure strati protettivi, che possono essere applicati saldamente (nitruro di titanio, carburo di titanio). La superficie viene usurata dove è presente localmente un'elevata deformazione plastica, spesso accompagnata da innalzamenti di temperatura. I metodi classici di bonifica superficiale (cementazione), particolarmente nella lavorazione del ferro, partono da processi di diffusione e dalla tempra da alte temperature. Nell'acciaio il carbonio e l'azoto sono particolarmente indicati per l'indurimento. Il carbonio viene diffuso in campo austenitico nel quale si scioglie bene (cementazione carburante; 900 °C, ricottura in CO). Dopo il raffreddamento rapido si forma martensite, indurita dai campi di deformazione tetragonali degli atomi di carbonio nei loro siti interstiziali (v. cap. 8, È d). L'azoto viene diffuso in campo ferritico (≾ 500 °C). Lì si formano nitruri, particolarmente AlN, e carbonitruri, che operano come precipitati indurenti. Nitrurazioni in condizioni controllate vengono oggi eseguite in recipienti da vuoto a bassa pressione (~ 10 mbar) che aumenta per una scarica di plasma. Gli ioni di azoto penetrano dappertutto e anche oggetti di forma complicata possono essere ben trattati. La scarica a corona non richiede tensioni particolarmente alte (≤ 1 kV), gli ioni di azoto penetrano ancora più vantaggiosamente nel metallo come azoto neutro (per es. da 2NH3 → N2 + 3H2) e diffondono alle abituali temperature di nitrurazione.
Possibilità completamente nuove si presentano bombardando superfici metalliche con ioni ad alta energia. Mediante ‛impianto ionico' si producono in prossimità della superficie strati di leghe metastabili. Energie tipiche degli ioni sono di 10-500 keV, con le quali si raggiungono profondità di penetrazione di ~100 nm. In questo intervallo si impiantano il 20-50% degli atomi estranei, che, a seconda della temperatura e della cinetica, rimangono in soluzione soprasatura oppure formano precipitati stabili o metastabili. La ripartizione e la profondità degli ioni impiantati possono essere variate variando l'energia. Come esempio la fig. 109 mostra un confronto tra una lega di titanio col 6% in peso di Al e il 4% di V impiantata e non impiantata. Questa lega di titanio fu messa a punto a causa della sua bassa densità e della sua alta resistenza meccanica per l'impiego nella navigazione aerea e spaziale. Oggi viene anche utilizzata sotto forma di giunto sferico nelle articolazioni artificiali dell'anca. Si spera di poter prolungare la durata di queste articolazioni di oltre 400 volte.
Ulteriori processi possibili col contributo di ioni sono rappresentati nella fig. 110a. Attraverso il trasferimento di energia sugli atomi superficiali si giunge a una rimozione superficiale (‛attacco con plasma' e ‛attacco con ioni'). La fig. 110b mostra che anche con laser ad alta potenza diventano possibili processi che modificano la superficie nel modo più diverso. Il ‛trattamento con laser' dopo ricopertura con appropriati strati superficiali permette, attraverso fusione sulla superficie, mescolamento e rapido raffreddamento, la produzione di leghe e di strutture insolite e anche di strati superficiali amorfi (laser glazing) con buone proprietà dal punto di vista della corrosione.
L'irradiazione di elevata energia, naturalmente, non ha solo effetti favorevoli. Come già si vede nella fig. 110b, i metalli, a una densità dell'energia di irradiazione sufficientemente alta, fondono anche nell'intero spessore; ciò rende il laser uno strumento adatto anche a saldare e tagliare. Ma anche al di sotto del punto di fusione tutti i tipi di irradiazione producono dei difetti (‛danno da radiazione'), quando l'energia trasferita ai costituenti atomici del materiale oltrepassa un valore specifico, che nei cristalli dipende anche dalla direzione. In prossimità della superficie si giunge ai menzionati processi di polverizzazione. All'interno del materiale, tuttavia, attraverso urti di spostamento, si producono vacanze e atomi interstiziali che, a seconda della temperatura, possono esistere in configurazioni metastabili e in concentrazioni totali molto al di sopra dell'equilibrio termico e possono influenzare le proprietà del materiale. Nella fisica dello stato solido interessano soprattutto processi semplici, come quelli che predominano, per esempio, nell'irradiazione con elettroni. Con tali processi si possono indagare le relazioni fondamentali. La fig. 111 mostra l'energia massima trasferita su un costituente reticolare per alcune sostanze cristalline in funzione dell'energia degli elettroni (nei moderni microscopi elettronici per metalli vengono utilizzate tensioni di accelerazione comprese tra 100 kV e 1,5 MeV). L'energia di soglia per urti di spostamento è tipicamente di ~ 25 eV. Appare di volta in volta una coppia di Frenkel, consistente in una vacanza e in un interstiziale. Entrambi i difetti singoli possono partecipare, a seconda della distanza e della mobilità, a reazioni correlate o non correlate (ricombinazione, formazione di coppie, agglomerati, ecc.). In quanto difetti atomici, i cosiddetti ‛difetti di punto' influenzano le proprietà macroscopiche sensibili ai difetti: in particolare la conducibilità elettrica e la resistenza meccanica.
Molto meno chiari sono i processi che si presentano nell'irradiazione dei metalli con neutroni veloci (nei reattori nucleari, anche nella fusione nucleare) o con ioni. A causa della maggiore massa del neutrone e degli ioni, l'energia trasferita a un singolo atomo del reticolo è notevolmente e più grande dell'energia di soglia. Il primo atomo espulso dal suo posto assorbe, secondo le leggi della meccanica degli urti, un'energia massima
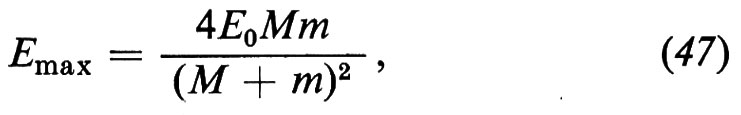
con E0 = energia della particella incidente, m = massa della particella incidente, M = massa del costituente reticolare. Per neutroni con E0 = 1,5 MeV si produce così per il rame un'Emax di ~ 90 keV e in media possono aver luogo per ogni atomo di rinculo primario ~ 500 urti successivi di spostamento. Poiché il cammino libero medio tra due urti decresce al decrescere dell'energia, si giunge, attraverso una ‛cascata di spostamenti', a distanze tra i difetti decrescenti verso la fine del percorso dell'atomo di rinculo. Si formano ‛zone rarefatte' (Seeger), come schematizzato nella fig. 112. Poiché la mobilità delle vacanze è diversa da quella degli atomi interstiziali e la loro disposizione non è correlata a coppie, si giungerà, a seconda della temperatura e della distribuzione dei pozzi di diffusione per entrambi i difetti di punto (superfici, bordi di grano, dislocazioni, precipitati, atomi estranei), al predominio di un solo tipo di difetto, per lo più delle vacanze. Altre reazioni allo stato solido (per es. lo smescolamento) possono essere accelerate o indotte dalle vacanze in eccesso. Ad alte dosi di irradiazione si formano, al di sopra di una temperatura di ~ 1/3 della temperatura di fusione, spazi vuoti (voids e bubbles) attraverso aggregazione di vacanze (in tal caso anche He, che si forma attraverso reazioni nucleari, può partecipare come stabilizzante). Questi agglomerati di vacanze (pori) non solo modificano le proprietà meccaniche, ma conducono anche a variazioni dimensionali macroscopiche di parecchie unità per cento, il che è indesiderato nei ristretti limiti di tolleranza della struttura di un reattore. Sono perciò necessarie intense attività di ricerca nel campo dei materiali per reattori, per capire e controllare meglio anche queste forme di carico estremo.
11. Prospettive.
L'umanità ha dunque un'esperienza lunga e ampia dei materiali metallici; tuttavia la molteplicità delle loro proprietà e delle loro caratteristiche non è stata ancora adeguatamente compresa e utilizzata. La consapevolezza sempre più approfondita dell'ambiente, lo scarseggiare dell'energia e delle materie prime e la crescente concorrenza in campo mondiale stimolano a sviluppare sempre più le possibilità che i materiali offrono in tutti i campi e a ricercare soluzioni nuove. Per questo è necessario il pieno impiego delle conoscenze della ricerca di base e del potenziale insito nei più moderni metodi di ricerca. I metalli saranno sostituiti, non solo occasionalmente, dai non metalli, dei quali subiranno la concorrenza, ma troveranno ulteriori possibilità di applicazione adatte alle loro specifiche proprietà.
Bibliografia.
Barrett, C. S., Massalski, T.B., Structure of metals, Oxford 19803.
Buckel, W., Supraleitung, Weinheim 19843.
Cahn, R. W., Haasen, P. (a cura di), Physical metallurgy, 2 voll., Amsterdam 19833.
Haasen, P., Physikalische Metallkunde, Berlin-New York 19842.
Heck, C., Magnetic materials and their applications, London 1974.
Hornbogen, E., Werkstoffe, Berlin-New York 19874.
Hull, D., Bacon, D. J., Introduction to dislocations, Oxford 19843.
Smallman, R.E., Modern physical metallurgy, London 19854.
Vlack, L. H. van, Elements of materials science and engineering, Reading, Mass., 1980.
Materiali per l'elettronica di Antonio Paoletti e Arnaldo D'Amico
SOMMARIO: 1. Introduzione. □ 2. Materiali semiconduttori: a) silicio policristallino; b) silicio amorfo e silicio amorfo idrogenato; c) struttura cristallina del silicio; d) preparazione del silicio di grado elettronico; e) drogaggio; f) ossido di silicio (SiO2); g) i siliciuri; h) i composti III-V; i) arseniuro di gallio; l) arseniuro di gallio su silicio; m) i composti II-VI e IV-VI; n) nuovi materiali semiconduttori. □ 3. Materiali magnetici: a) registrazione magnetica; b) memorie magnetiche; c) dispositivi magnetoottici; d) dispositivi a microonde. □ Bibliografia.
1. Introduzione.
Lo sviluppo dei dispositivi microelettronici e magnetici di nuova generazione è stato e continua a essere strettamente legato alla sempre migliore conoscenza delle proprietà fisico-chimiche dei materiali che li costituiscono.
Il costante miglioramento delle tecniche diagnostiche tradizionali e di quelle recentemente introdotte, di notevole potenzialità, ha consentito il diffondersi di una considerevole e fruttuosa attività di ricerca nel campo dei materiali, che ha portato a un progresso culturale così sostanziale da sollecitare la revisione della maggior parte delle tecniche di crescita conosciute, con lo scopo di spingerne le prestazioni ai limiti teorici e ottenere così materiali di sempre migliore qualità.
La favorevole circostanza di disporre contemporaneamente di tecniche diagnostiche di notevole accuratezza, di materiali con un sempre minore contenuto di difetti e di adeguate tecnologie di realizzazione di dispositivi ha portato a benefici così evidenti e sorprendenti da far ritenere che il loro impatto scientifico e socioeconomico continuerà proficuamente anche nei prossimi anni.
Lo sviluppo dell'elettronica fino agli anni sessanta insieme a quello successivo, vertiginoso e repentino, della microelettronica fino ai nostri giorni sono conseguenza di uno sforzo globale senza precedenti compiuto nello studio della fisica dello stato solido, nel miglioramento continuo delle tecniche di crescita dei materiali semiconduttori e magnetici, nella loro caratterizzazione strutturale ed elettro-ottica e soprattutto nel miglioramento dei sistemi di controllo dei processi di fabbricazione dei dispositivi, che ha portato a richiedere materiali di sempre più elevata qualità.
L'interesse per i semiconduttori risale a M. Faraday e C. Becquerel. Faraday scoprì che il solfuro d'argento aveva un coefficiente di temperatura negativo e Becquerel nel 1839 iniziò lo studio delle proprietà conduttrici di vari elettroliti. Successivamente, ma solamente nel 1874, fu osservato il fenomeno della rettificazione da parte di F. Braun, che utilizzò sostanze tipo solfuro di piombo (PbS) e pirite, mentre W. Smith nel 1873 aveva osservato per primo la fotoconducibilità nel selenio. In seguito furono intensificate le ricerche per trovare sostanze aventi proprietà analoghe e si ebbero i primi risultati: i solfuri metallici, alcuni ossidi e il silicio furono fra i nuovi materiali quelli più attentamente considerati. Di tutto questo lavoro, che rappresentò la base culturale iniziale per lo sviluppo dei materiali per l'elettronica, K. Lark-Horowitz (v., 1954) ricorda in una sua eccellente pubblicazione le tappe più significative. Un evento importante per le sue conseguenze sullo studio dei materiali per l'elettronica fu la scoperta dell'effetto Hall (1879), cioè la presenza di una tensione trasversa attraverso un conduttore, in cui circola una data corrente, immerso in un campo magnetico.
Sebbene a quel tempo l'elettrone non fosse stato ancora scoperto e l'idea del numero di portatori di corrente non potesse ancora essere utilizzata, tale effetto costituiva tuttavia la chiave per cominciare a distinguere i materiali semiconduttori a differente conducibilità. L'uso sistematico dell'effetto Hall nello studio del CuI è dovuto a K. Baedeker (1909) e, più in generale, nello studio di diversi semiconduttori a J. Königsberger (1907 e 1914). Nel 1915 venne utilizzata per la prima volta la galena (PbS) e nel 1920 si impiegarono il selenio e l'ossido di rame come rivelatori rettificatori. Nel 1933 W. H. Schottky pubblicò la sua teoria sui rettificatori a secco, che inaugurò lo studio della teoria dei semiconduttori su basi quantistiche. Lo sviluppo del radar durante la seconda guerra mondiale fece aumentare l'impiego di diodi a stato solido e ciò dette impulso ulteriore alle ricerche nel campo dei materiali semiconduttori. Successivamente, nel dicembre 1947, W. H. Brattain e J. Bardeen dei laboratori della Bell (Stati Uniti) inventarono il transistore a punta di contatto e pochi mesi dopo W. Shockley fabbricò il primo transistore a giunzione, ancora oggi utilizzato dopo gli ovvi miglioramenti apportati nel corso degli anni. Nel 1958 L. Esaki, sulla spinta del miglioramento del controllo delle tecnologie di processo, progettò e costruì il diodo tunnel, destinato a segnare una tappa significativa nella storia dell'elettronica. All'inizio degli anni sessanta, sulla spinta di una grossa richiesta di calcolatori e di aumenti della produttività a livello industriale, e con la disponibilità di nuove concrete idee a livello di innovazioni circuitali e di dispositivi di nuova generazione (J. Fet, Mosfet, ecc.), prese l'avvio la microelettronica, il cui sviluppo ancora oggi non ha raggiunto i livelli estremi imposti dai limiti fisici di funzionamento dei dispositivi più perfezionati.
Va messo in evidenza che i livelli oggi raggiunti nella microelettronica sono principalmente dovuti ai continui progressi fatti nella conoscenza e nella fabbricazione di materiali per l'elettronica - semiconduttori, dielettrici, metallici - sempre più puri e con un sempre più basso numero di difetti. Nell'ambito dei materiali per l'elettronica è nata una disciplina, chiamata ‛scienza dei materiali'. Essa comprende quella parte della scienza dello stato solido che riguarda le interrelazioni tra struttura cristallina, difetti e microstrutture, composizione chimica e proprietà fisiche, e tiene presenti le potenziali applicazioni tecniche. Nell'ambito di questa disciplina, e con l'aiuto delle nuove tecnologie, si tende al progetto e alla realizzazione (ingegneria molecolare) di materiali nuovi dotati di proprietà fisiche, chimiche e tecniche di particolare interesse scientifico e applicativo. La forte dipendenza delle proprietà dei materiali semiconduttori e dei dispositivi dalla presenza di difetti nei materiali cristallini e nei dispositivi utilizzati - silicio e semiconduttori composti - implica la necessità di impiegare metodi diagnostici estremamente sensibili nel rilevamento e nella caratterizzazione dei difetti. A tale scopo sono sempre più utilizzati, per esempio, i microscopi elettronici tipo TEM, SEM, STEM, ma va detto che esistono numerosissime altre tecniche in campo diagnostico che consentono una descrizione sufficientemente dettagliata dei materiali elettronici. Nel loro insieme, tuttavia, tali tecniche dovranno essere ancora migliorate per spingere le analisi ai più minuti dettagli, al fine di consentire una comprensione migliore dei fenomeni che sono alla base del funzionamento nei dispositivi di nuova generazione (v. Grasserbauer, 1985).
2. Materiali semiconduttori.
I semiconduttori sono quei materiali che hanno valori di conducibilità elettrica intermedi tra quelli dei metalli (104 ÷ 106 ohm-1 cm-1) e degli isolanti (10-22 ÷ 10-10 ohm-1 cm-1). È significativo il fatto che la conducibilità di tali materiali può variare di alcuni ordini di grandezza al variare della temperatura, dell'eccitazione ottica e del contenuto di impurezze, e tale caratteristica li ha resi di notevolissimo interesse scientifico e industriale. I materiali semiconduttori trovano posto nel IV gruppo e in quelli adiacenti della tavola periodica (v. tab. I). Il silicio e il germanio, del IV gruppo, sono chiamati semiconduttori elementari, in quanto composti di una singola specie di atomi. Esistono poi materiali ottenuti dalle combinazioni fra atomi di elementi del III e del V gruppo, del II e del VI, e del IV e del VI, che formano i semiconduttori composti intermetallici.
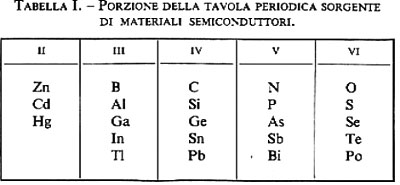

Come è indicato dalla tab. II, numerosi sono i materiali semiconduttori che possono essere realizzati. Fra questi il più utilizzato è il silicio, che costituisce il materiale di base per la maggior parte dei dispositivi a semiconduttore: rettificatori, transistori, circuiti integrati per microprocessori, memorie, ecc. I semiconduttori composti sono largamente utilizzati per quei dispositivi che basano il loro funzionamento sull'emissione o sull'assorbimento di luce. Per esempio, i cosiddetti LED (light-emitting diodes) sono costruiti con materiali tipo GaAs, GaP, GaAsP, GaAlAs. I materiali fluorescenti tipo quelli utilizzati per schermi televisivi utilizzano il materiale semiconduttore ZnS. I sensori di radiazione utilizzano materiali tipo InSb, CdSe, PbTe, PbSe, PbSnTe, HgCdTe. I diodi Gunn e i MESFET (dispositivi a microonde) sono realizzati con GaAs, InP, ecc.
I numerosi materiali semiconduttori disponibili presentano molteplici proprietà utilizzabili per costruire dispositivi e circuiti con elevata flessibilità, capaci di svolgere funzioni elettroniche complesse. Le proprietà elettroottiche dei materiali semiconduttori sono fortemente dipendenti dalle impurezze in essi introdotte. L'inserimento ditali impurezze, che può essere oggi controllato con notevole accuratezza, consente di variare la conducibilità dei semiconduttori su un esteso intervallo di valori e di alterare la natura del processo di conduzione dalla conduzione per cariche negative a quella per cariche positive. Per esempio, l'inserimento di impurezze alla concentrazione di una parte per milione può trasformare il silicio poco conduttore in una varietà a elevata conducibilità elettrica. Lo studio di tali proprietà viene di solito preceduto da quello relativo all'‛arrangiamento' atomico del materiale, cioè al modo in cui atomi uguali o di specie diversa sono impacchettati insieme a costituire il materiale. Si distinguono così materiali semiconduttori monocristallini, policristallini e amorfi.
Un solido monocristallino viene riconosciuto dal fatto che gli atomi sono sistemati con una precisa periodicità, cioè in esso è possibile individuare un insieme di atomi che viene ripetuto isotropicamente per tutto il solido. L'ordine a corto e a lungo raggio è massimo e distribuito in tutto il solido in modo uniforme. Esiste un intervallo di energia proibita - che indicheremo con Eg - che separa il massimo della banda di valenza dal minimo della banda di conduzione. Tutti gli stati elettronici sono di tipo esteso, cioè le funzioni d'onda associate agli elettroni si estendono per tutto il cristallo. Un solido policristallino è composto di zone di materiale monocristallino aventi orientamenti che possono essere anche completamente aleatori. Nella fig. 1 sono rappresentate una fetta di silicio monocristallino e una di silicio policristallino. Nella fig. 2 sono rappresentate sul piano, in modo schematico, la distribuzione ordinata di atomi nel caso di materiale monocristallino (A), la distribuzione disordinata di atomi nel caso di materiale amorfo (B) e la distribuzione aleatoria di aree di materiale monocristallino, tipiche di un materiale policristallino (C). I materiali amorfi non presentano strutture periodiche e sono quindi caratterizzati dall'assenza di ordine a lungo raggio. Per essi si può ancora parlare di bande (sedi di stadi estesi) e di intervalli tra bande. Questi intervalli sono interessati da ‛stati localizzati' la cui funzione d'onda elettronica si estende spazialmente per appena una decina di Å (v. Nagels, 1979).
La distribuzione periodica di atomi in un cristallo è detta anche reticolo (v. Kittel, 19765). In generale ogni tipo di reticolo è formato da un insieme ordinato di celle unitarie. Come esempio si consideri la fig. 3, in cui è rappresentato un reticolo bidimensionale con celle unitarie a forma di parallelogrammi. La cella unitaria OACB ha un atomo in ciascun vertice e ciascun atomo è condiviso con celle uguali adiacenti. Celle unitarie del tipo O′A′C′B′, uguali alla OACB, possono essere individuate tramite una traslazione definita da multipli interi dei vettori a e b. Tali vettori, chiamati ‛vettori della base', determinano una notevole proprietà dei reticoli: punti del reticolo definiti dal vettore traslazione r,
r = na + mb,
con n, m interi, sono indistinguibili.
In tre dimensioni il vettore traslazione diventa
v = na + mb + pc,
con n, m, p interi.
Fra le diverse celle unitarie che possono essere individuate nel reticolo la più piccola viene detta ‛cella primitiva'. Non sempre essa risulta la più conveniente per lo studio dei solidi, ma comunque la sua importanza consiste nel fatto di poter dedurre, dal suo studio, tutte le proprietà fisiche del cristallo. Per esempio, dalla cella primitiva possono essere dedotte le distanze fra gli atomi più vicini e calcolate le forze che tengono il reticolo compatto e la densità e le energie permesse degli elettroni che partecipano al processo di conduzione. Inoltre è di grande importanza nello studio dei cristalli l'individuazione delle eventuali simmetrie: è infatti notevole il fatto che le proprietà fisiche di un materiale semiconduttore devono mostrare le medesime caratteristiche di simmetria della struttura cristallina dello stesso materiale.
a) Silicio policristallino.
Il silicio policristallino presenta proprietà analoghe a quelle del silicio monocristallino, essendo costituito da monocristalli, che sono aleatoriamente orientati nel piano e possono avere dimensioni che vanno da alcuni micron quadrati a millimetri quadrati. Esso può essere ottenuto sotto forma di film sottile tramite deposizione da fase vapore (CVD) o sputtering oppure tramite crescita dal fuso in speciali contenitori. In quest'ultimo caso, dopo taglio meccanico e lappatura, possono essere ottenute fette circolari o quadrate pronte per un eventuale drogaggio. Con tali fette possono essere costruite, per esempio, celle solari a più basso costo, se confrontate con quelle monocristalline, e con rendimenti comparabili. La presenza dei bordi di grano che agiscono come difetti, inficiando le proprietà di trasporto, non consente però l'utilizzazione del materiale policristallino nel contesto della produzione di dispositivi microelettronici.
b) Silicio amorfo e silicio amorfo idrogenato.
Notevole interesse a livello internazionale sta suscitando il silicio amorfo idrogenato, un materiale ottenibile sotto forma di film sottili mediante le seguenti tecniche: sputtering a radio frequenza, glow-discharge, evaporazione, ecc. Il silicio amorfo, per la presenza di stati elettronici nella pseudogap, dovuti ai difetti e alla distorsione dei legami Si-Si, e per i bassissimi valori di mobilità (1 cm2 V-1s-1), non è un materiale per l'elettronica. Esso diventa però interessante per applicazioni elettroniche quando, tramite idrogeno, si passivano i difetti, e ciò accade in quanto atomi di idrogeno si accoppiano a legami liberi del silicio (v. Nagels, 1979). In queste condizioni il silicio amorfo è detto idrogenato (a-Si:H) e presenta interessanti caratteristiche: i suoi tempi di vita sono dell'ordine del microsecondo, è drogabile, presenta elevato assorbimento nel visibile, ha - se intrinseco - elevata resistività (109 ÷ 1010 ohm cm) e può consentire la formazione di giunzioni con profondità di svuotamento di diverse migliaia di Å. A tutt'oggi l'a-Si:H trova impiego nelle memorie ottiche, nelle superstrutture, nei rulli xerografici, come fotoconduttore, nei sistemi di pilotaggio a frequenza televisiva, nella realizzazione di dispositivi a trasferimento di carica, ecc. La velocità di elaborazione dei segnali è però di molto inferiore rispetto a quella consentita dai dispositivi al silicio cristallino. Infatti per il momento non esistono dispositivi, realizzati con il silicio amorfo, funzionanti a frequenze di qualche MHz.
c) Struttura cristallina del silicio.
Il reticolo del silicio (e del germanio) è come quello del diamante (v. Kittel, 19765). Ciascun atomo ha i quattro primi vicini situati ai vertici di un tetraedro regolare che è esattamente contenuto in un cubo (v. fig. 4). Il cristallo non è costruito con una ripetizione periodica di tale cubo, in quanto, se così fosse, ciascun atomo avrebbe otto atomi primi vicini e farebbe parte di una struttura cubica a corpo centrato. Nel reticolo del diamante o del silicio o del germanio ciascuno di tali cubi è separato dal successivo da un cubo uguale, però vuoto. In questo modo un cubo elementare più grande, formato da 4 cubi contenenti il tetraedro e da 4 cubi vuoti, è la struttura periodica (‛cella elementare') che determina l'intero cristallo (v. fig. 5). Il lato del cubo grande, detto parametro reticolare (a), per il silicio vale 5,43072 × 10-8 cm e per il Ge 5,65754 × 10-8 cm. Il reticolo così costruito ha una simmetria cubica rotazionale: come conseguenza la conducibilità elettrica e termica, la mobilità, la costante dielettrica e il coefficiente di diffusione sono isotropi nell'intero cristallo. L'intervallo di energia proibita (Eg) a temperatura ambiente è pari a 0,665 eV per il Ge e a 1,11 eV per il Si, mentre per quanto attiene ai valori di mobilità si ha, nel caso del Ge, μe = 3.900 cm2 V-1s-1 e μl = 1.900 cm2 V-1s-1 per gli elettroni e le lacune rispettivamente, e nel caso del Si μe = 1.350 cm2 V-1s-1 e μl = 480 cm2V-1s-1. Nella tab. III vengono elencate altre proprietà fisiche importanti del germanio e del silicio.

Il germanio può essere utilizzato, se drogato con elementi quali Au, Zn, Pb, Sn, Cu, per la fabbricazione di dispositivi per il medio (λ = 5 ÷ 10 μm) e per il lontano infrarosso (λ > 20 μm). Il suo uso nel campo dei dispositivi per l'elettronica in generale non ha avuto il successo del silicio, di cui parleremo più diffusamente. Va comunque detto che il germanio (e le sue leghe) viene ancora oggi studiato soprattutto grazie alla disponibilità di strumenti diagnostici sempre più sofisticati di caratterizzazione strutturale ed elettroottica.
d) Preparazione del silicio di grado elettronico.
Il silicio utilizzato nei processi di fabbricazione dei dispositivi è monocristallino (nel caso di celle solari può essere usato anche il policristallo di diametro fino a 15 cm), molto puro e ha la forma di fette circolari piatte e lucidate almeno da una parte. La preparazione di tali fette è notevolmente complessa e laboriosa (processo di purificazione). Fortunatamente il materiale di base (SiO2) è abbondante in natura, anche se sotto forma di composto: in molte zone della Terra è possibile trovare il diossido di silicio (SiO2) con contenuto di impurezze minore dell'1%. Questa sabbia viene utilizzata come materiale di partenza per la fabbricazione delle fette. Tre sono le reazioni sequenziali comunemente impiegate per ottenere silicio della purezza necessaria per farne dei dispositivi. Nella prima il diossido di silicio viene miscelato e fatto reagire con il carbonio per formare silicio (Si) puro solamente al 99% e di ossido di carbonio:
SiO2 + C → Si + CO2.
Nella seconda il silicio viene fatto reagire con acido cloridrico per formare triclorosilano secondo la reazione seguente:
Si + 3HCl → SiHCl3 + H2.
A questo punto il triclorosilano può subire alcuni processi di distillazione per aumentare ulteriormente il suo grado di purezza. Nella reazione finale il triclorosilano viene decomposto in camera ad atmosfera controllata tramite l'azione della corrente elettrica per ottenere silicio policristallino ultrapuro. La reazione utilizzata è la seguente:
SiHCl3 + H2 → Si + 3HCl.
Il silicio policristallino è ora pronto per subire il processo di crescita che lo trasformerà in un monocristallo. A tal fine sono due i metodi oggi più utilizzati: Czochralski (CZ) e float zone (FZ). Per la crescita dei cristalli con il metodo CZ viene utilizzato un crogiuolo, contenente pezzi di silicio policristallino, riscaldato tramite resistenza termica o tramite induzione di radiofrequenza (v. Kaldis, 1985). Durante il procedimento di crescita il crogiuolo viene fatto ruotare per evitare o quanto meno ridurre i gradienti termici sul silicio fuso e l'atmosfera ambientale è solitamente controllata con argon. Una volta che la temperatura del silicio fuso è stabilizzata, un pezzo di silicio monocristallino (seme), montato all'estremità di un'asta, viene messo a contatto con la superficie del liquido. Quando il seme è parzialmente fuso inizia la fase del tiraggio, che consiste nel sollevare lentamente con movimenti rototraslatori l'asta. Il silicio aderisce al seme e si solidifica formando via via un lingotto, il cui diametro può essere accuratamente controllato durante tutta la fase di crescita fino a che il contenuto del crogiuolo è esaurito. La crescita FZ utilizza direttamente le barre policristalline preparate elettroliticamente. A un capo della barra viene fissato un seme di silicio monocristallino e il tutto viene messo in una camera ad atmosfera controllata. Il sistema di riscaldamento viene mosso dal basso verso l'alto lungo la barra e viceversa, più volte, in modo da ottenere una zona fusa viaggiante che trasforma il policristallo in monocristallo. Le due tecniche consentono di ottenere lingotti pronti per essere tagliati nelle fette volute, che verranno successivamente lucidate e rese disponibili per i processi di fabbricazione dei dispositivi.
e) Drogaggio.
La conducibilità di un materiale semiconduttore può essere notevolmente influenzata mediante l'inserzione di piccole dosi di elementi detti droganti o impurezze. Essi possono trovare sistemazione nel semiconduttore andando a occupare un posto reticolare (drogaggio per sostituzione) oppure possono inserirsi in posizioni interstiziali, cioè in spazi vuoti fra gli atomi normali. I droganti più tipici per il germanio e il silicio sono dati dagli elementi del III (B, Al, Ga, In, Tl) e V gruppo (P, As, Sb, Bi) del sistema periodico. Nella fig. 6 sono indicati i livelli energetici dei droganti più comuni per il Si e il Ge (v. Sze, 1976). Una volta drogati, a seconda del tipo di drogante, tali semiconduttori possono presentare conducibilità per elettroni o per lacune. La disponibilità di semiconduttori drogati di tipo n (elettroni) e p (lacune) ha consentito la realizzazione di un'enorme varietà di dispositivi elettronici ancora oggi oggetto di intenso studio. L'operazione di drogaggio può essere fatta nei seguenti modi: a) durante l'accrescimento stesso del monocristallo; b) mediante processi di diffusione termica; c) mediante la tecnica della diffusione planare utilizzante sorgenti stabili planari di materiali droganti; d) mediante l'impiantazione ionica, attualmente la tecnica più utilizzata. Il processo di diffusione e quello di impiantazione ionica sono le due tecniche attualmente competitive per l'introduzione di atomi droganti nel silicio. La tecnologia diffusiva è più antica, quella dell'impiantazione è più moderna, ma entrambe trovano ancora posto nel contesto della fabbricazione dei circuiti integrati. Il boro e il fosforo, fra gli elementi droganti del silicio, sono stati e continuano a essere i più importanti e i più utilizzati. Per la tecnologia diffusiva viene richiesta una purezza del materiale drogante più elevata di quella richiesta per l'impiantazione: ciò costituisce talvolta un vantaggio non trascurabile a favore dell'impiantazione.
f) Ossido di Silicio (SiO2).
Il successo del silicio come materiale per l'elettronica è in larga parte dovuto anche alla possibilità piuttosto favorevole di realizzare un isolante (SiO2) basato sull'utilizzazione del silicio stesso, di ottime caratteristiche dielettriche. Infatti i dispositivi MOS-CMOS utilizzano proprio film sottili di diossido di silicio come elementi fondamentali per il loro funzionamento. Il diossido di silicio (SiO2) è il materiale dielettrico standard utilizzato per i transistori a effetto di campo a gate isolato di silicio o di metallo (v. Kasprzak e Gaind, 1980; v. Pantelides, 1978). La sua crescita avviene tramite ossidazione termica del silicio a elevata temperatura (900-1.050 °C), cioè per mezzo di un processo di diffusione limitata in cui, dopo la formazione dei primi strati, l'ossigeno migra attraverso l'ossido di silicio fino al raggiungimento dell'interfaccia Si-SiO2, dove reagisce con il silicio per formare altro SiO2. Sono possibili in pratica almeno due tipi di ossidazioni termiche, ossidazione a secco e ossidazione in presenza di vapore, caratterizzate rispettivamente dalle seguenti reazioni:
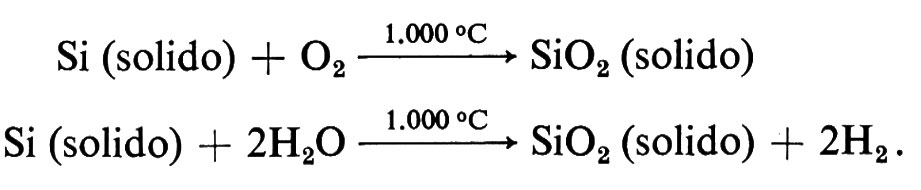
Tali processi causano purtroppo difetti nel silicio e residui di silicio all'interfaccia e, come conseguenza, uno strato non stechiometrico non solamente nell'interfaccia, ma anche nell'ossido di silicio. L'eccesso di silicio nell'ossido e all'interfaccia determina la densità di carica fissa Qss, mentre la mancanza di Si, o di un legame all'interfaccia o in prossimità di questa, determina la densità di stati superficiali veloci Nfs, che sono tra i responsabili nelle limitazioni delle prestazioni dei MOSFET. Il diossido di silicio può anche essere depositato con un altra tecnica detta CVD (chemicai vapor deposition). In questo caso la fetta di silicio viene messa in un reattore a induzione in presenza di un flusso di SiH4 (reagente), CO2 (ossidante), tracce di HCl (per catturare il silicio e formare il composto volatile SiHxCly e di H2, utilizzato come gas di trasporto. Il diossido di silicio che così si forma sulla superficie della fetta ha proprietà simili a quello ottenuto per ossidazione termica. Nella tab. IV sono messe a confronto alcune proprietà dell'ossido termico e di quello ottenuto per CVD. La fig. 7 indica in due dimensioni e in modo schematico che cosa accade al silicio prima (A) e dopo (B) l'inizio dell'ossidazione. Come si vede, la distanza tra gli atomi nell'ossido è diversa da quella nel silicio. Questa è una delle cause della nascita di stati superficiali. Un ossido di silicio di qualità inferiore a quello ottenibile con i metodi sopraddetti può essere ottenuto con la tecnica dello sputtering reattivo a radiofrequenza utilizzando un target di Si e un'atmosfera di scarica ricca di ossigeno, oppure per evaporazione di SiO, sempre in presenza di ossigeno, e per essiccamento di opportune soluzioni contenenti Si e SiO2.
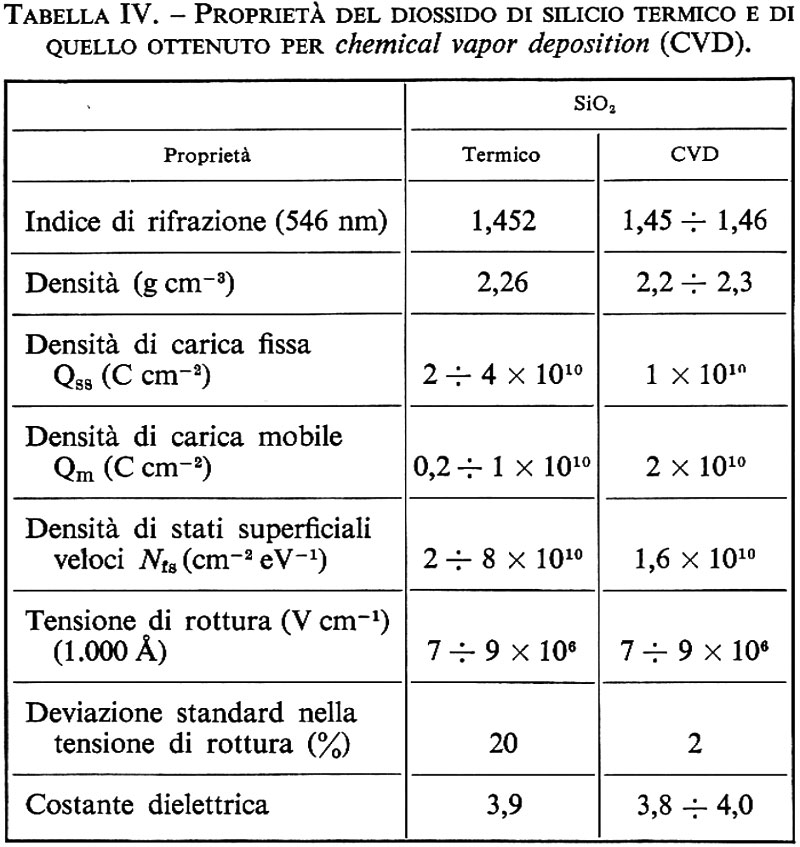
g) I siliciuri.
I siliciuri (v. Ottaviani, 1979; v. Ottaviani e Mayer, 1981; v. Murarka, 1983) sono composti formati dal silicio con alcuni metalli tipo Ta, Ti, W, Mo. Essi possono essere utilizzati a livello di gate per ridurre la resistenza di strato e quindi il tempo di ritardo nella propagazione di segnali sui dispositivi VLSI e UVLSI. Soprattutto a causa della loro elevata compatibilità tecnologica con il silicio e il polisilicio, i siliciuri stanno diventando sempre più utili come materiali di interconnessione per l'elettronica. Tra essi è di rilevante importanza il siliciuro di tantalio TaSi2, che sembra tra l'altro dimostrare caratteristiche interessanti anche quando utilizzato nel primo livello di interconnessione. Analogamente a quanto accade per gli altri siliciuri, il TaSi2 può essere preparato per codeposizione o sputtering del Ta e del Si su polisilicio drogato n in opportuno rapporto - anche se è difficile ottenere le condizioni stechiometriche ideali - e con trattamento termico in atmosfera controllata a 900 °C in fomace o tramite laser o lampade di potenza.
I siliciuri presentano conducibilità vicine a quelle dei metalli e hanno l'utilissima caratteristica di essere stabili a elevata temperatura e soprattutto di essere compatibili con le tecnologie di processo dei circuiti integrati. E probabile che insieme al nitruro di silicio (Si3N4) possano dare un contributo per lo sviluppo dei circuiti a larghissima scala di integrazione nei prossimi anni. Per questo motivo sono tuttora oggetto di intenso studio e lo saranno ancora per molto tempo, in quanto i loro problemi fisico-chimici e la dinamica di crescita non sono ancora stati soddisfacentemente risolti (v. Oppolzer e altri, 1984).
h) I composti III-V.
I composti III-V rappresentano, dopo il silicio, la classe di materiali più importante in campo elettronico ed elettroottico. I più utilizzati sono elencati nella tab. V, dove sono anche mostrate le mobilità degli elettroni e delle lacune. Tali composti sono caratterizzati da un intervallo di energia di tipo diretto, una piccola massa effettiva per gli elettroni e quindi un'elevata mobilità elettronica. I composti della tab. V trovano largo uso in elettronica per elevate frequenze e in particolare i composti 5 e 6 sono impiegati nel campo della rivelazione IR.
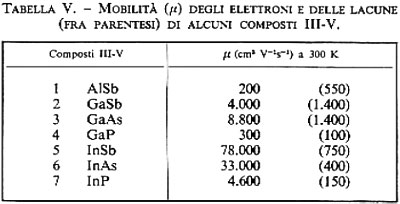
i) Arseniuro di gallio.
Dopo il silicio l'arseniuro di gallio (v. Kaldis, 1985; v. Willardson, 1966-1968; v. Gooch, 1969) è il materiale semiconduttore più importante, oggi largamente usato nel campo delle microonde, dell'ottica integrata e dei laser allo stato solido. Come la maggior parte dei composti III-V, il GaAs cristallizza in accordo col reticolo della blenda, in cui un atomo è al centro di un tetraedro regolare i cui quattro vertici sono occupati da atomi dell'altra specie. Tale struttura è molto simile a quella del diamante, i cui siti sono occupati invece da atomi della stessa specie. La cella cubica del GaAs è mostrata nella fig. 8, dove è visibile un reticolo cubico a facce centrate occupate da atomi di gallio. Gli atomi di arsenico sono posizionati sulle diagonali del cubo e anch'essi formano, a loro volta, un reticolo cubico a facce centrate, spostato però rispetto al reticolo cubico del gallio di 1/4 della diagonale. Nella struttura cristallina del GaAs possono essere trovati numerosi difetti, a causa del processo di crescita non ideale. Difetti di tipo singolo sono le vacanze di gallio e/o di arsenico. Difetti di tipo multiplo sono le bivacanze gallio-arsenico o gallio-gallio.
La crescita del GaAs può essere ottenuta in diversi modi: crescita dal fuso, tiraggio, crescita epitassiale, zone refining, crescita da fase vapore. Il più semplice e antico metodo di crescita è quello da liquido stechiometrico e le tecniche utilizzate differiscono solo per il modo in cui il gradiente di temperatura interessa il fuso. Nel sistema Bridgman il crogiuolo viene spostato in una zona a gradiente termico, nel sistema Stoher il crogiuolo è fisso e il gradiente termico viene programmato nel tempo. Con la tecnica Czochralski, infine, il GaAs viene tirato tramite un seme da un crogiuolo contenente il fuso a una data temperatura. A differenza del diamante e del silicio, che tendono a clivarsi più facilmente lungo il piano (111), dove il numero di legami per unità di area è minimo, il GaAs può essere clivato facilmente lungo il piano (110).
Il GaAs può essere drogato con elementi tipo Zn, Cd, Li, Ge, ecc. per consentire una conduzione per lacune, o con elementi tipo Se, Te, Si, ecc. per una conduzione per elettroni. L'intervallo di energia proibita (Eg) è, come per tutti i semiconduttori, funzione della temperatura e della pressione. Nel caso del GaAs Eg può essere espressa dalla seguente relazione:
Eg = 1,53 + 9,4 × 10-6p − 4,9 × 10-4T,
dove p è la pressione e T è la temperatura. Così, per esempio, alla pressione di 1 atmosfera e a temperatura ambiente (T = 300) Eg è pari a 1,435 eV, mentre a 77 K è pari a 1,52 eV. Il parametro reticolare è pari a 5,6532 Å e la densità è 5,3176 g cm-3, il coefficiente di espansione termica è 6,86 × 10-6K-1, la conducibilità termica è pari a 0,45 W cm-1K-1 a 300 K e 2,5 W cm-1K-1 a 77 K, mentre l'indice di rifrazione è, a 300 K, pari a 3,43 quando l'energia della radiazione utilizzata per la misura è di 1 eV. Nella fig. 9 sono indicati gli elementi droganti possibili per il GaAs. Nel GaAs gli elettroni mostrano una massa effettiva minore di quella esibita dal silicio e dal germanio. Ciò significa che gli elettroni nel GaAs si muovono più velocemente, il che si traduce in dispositivi in grado di funzionare a più elevate frequenze di quelle consentite per il Si e per il Ge.
Per avere un'idea della riduzione della massa effettiva nel GaAs rispetto a quella del silicio, consideriamo la fig. 10, in cui è rappresentata l'energia degli elettroni in funzione della loro quantità di moto. Nel caso del GaAs è possibile passare direttamente dal massimo della banda di valenza al minimo della banda di conduzione dopo aver superato un intervallo di energia di 1,4 eV. Nel caso del silicio è possibile raggiungere il minimo della banda di conduzione (1,1 eV) in modo indiretto, cioè dopo uno spostamento lungo l'asse delle quantità di moto. Tale spostamento è una delle cause della più elevata massa efficace elettronica del silicio: questa è pari a 0,97 volte quella standard, mentre nel GaAs è circa 16 volte inferiore. Un'altra differenza tra il silicio e il GaAs, a favore del GaAs, è messa in risalto dalla fig. 11, dove viene rappresentato l'andamento della velocità degli elettroni in funzione del campo elettrico. Per bassi campi la velocità degli elettroni nel GaAs è molto più elevata di quella nel silicio, ma anche per alti campi la velocità di saturazione elettronica nel GaAs è più del doppio di quella nel silicio. Questa alta velocità elettronica nel GaAs, unita alla dissipazione di potenza che tende a essere minore di quella relativa ai dispositivi al silicio a elevata velocità di elaborazione del segnale, spiega il crescente interesse per questo materiale. Altri vantaggi del materiale GaAs sono legati alla sua elevata resistenza alle radiazioni e alla possibilità di generare dispositivi in grado di funzionare in estesi intervalli di temperatura, per esempio da −100 a 300 °C. Fra i dispositivi al GaAs su cui l'industria elettronica sarà impegnata nel prossimo futuro vanno ricordati i transistori a effetto di campo metallo-semiconduttore di tipo a svuotamento (D-MESFET), di tipo a rafforzamento (E-MESFET) e quelli ad alta mobilità elettronica (HEMT). Infine vanno menzionate le strutture a multistrato tipo GaAs-GaAlAs (superreticoli) come potenziali sistemi di materiali in grado di generare classi di dispositivi ultraveloci di nuova generazione.
l) Arseniuro di gallio su silicio.
L'elevatissima velocità di elaborazione dei segnali, tipica dei dispositivi realizzati con il GaAs, e l'elevatissima densità di componenti funzionanti a ragguardevole velocità, realizzati con il silicio, potranno molto probabilmente essere combinate insieme grazie a una nuova tecnica di crescita di film che consente di ottenere GaAs a bassissimo numero di difetti direttamente su silicio. In questo modo la tecnologia del silicio e quella del GaAs potranno essere utilizzate sulla stessa struttura, con notevoli possibilità di realizzare componenti di nuova generazione che utilizzino le caratteristiche migliori di entrambi i materiali.
Ricercatori dell'Università dell'Illinois sono riusciti per primi a ottenere GaAs su silicio con un numero di difetti pari a circa 103 cm-2, cioè 10 volte inferiore a quello ottenibile con la tradizionale tecnica Czochralski. Essi tagliano il silicio secondo un angolo di 4 gradi rispetto alla direzione (100), creando così scalini sulla superficie alti circa quanto uno strato atomico (2,8 Å) e larghi circa 3,8 Å. Su questa superficie il GaAs può crescere con un numero minore di dislocazioni, in quanto la superficie del silicio così tagliata mostra un parametro reticolare effettivo più vicino a quello del GaAs. Ed è proprio la differenza di parametro reticolare (del 4%) tra Si e GaAs che origina le non volute dislocazioni che dopo la formazione iniziale si propagano fino ad attraversare tutto lo spessore del film di GaAs, inficiando così le proprietà di trasporto. Quelle dislocazioni che malgrado tutto riescono a propagarsi possono essere poi in larga parte bloccate alla superficie del GaAs tramite la crescita di un film correttivo con parametro reticolare di poco superiore a quello del GaAs (per esempio InGaAs). In questo modo il silicio può sostenere crescite di film secondo la sequenza GaAs/InGaAs per consentire la formazione di superstrutture considerate fondamentali per la fabbricazione di nuovi dispositivi elettronici e optoelettronici. La generale soddisfazione che trapela a livello internazionale - derivante dalla notevole prospettiva di disporre di superfici di GaAs a basso numero di difetti ed estese quanto quelle delle fette di silicio oggi commercialmente disponibili, di diametro pari a 15 cm - induce a credere che anche i costi dei dispositivi finali potranno subire consistenti riduzioni.
m) I composti II-VI e IV-VI.
I composti II-VI e IV-VI (v. Abricosov e altri, 1969) sono costruiti con elementi appartenenti ai gruppi II-VI e IV-VI della tavola periodica. Tali composti sono stati intensamente studiati attorno agli anni sessanta e settanta e i loro diagrammi di fase, accuratamente determinati per mezzo di analisi tecniche microstrutturali e a raggi X, sono oggi disponibili nella letteratura specializzata. Questi materiali si possono far crescere dal fuso in forma policristallina (in qualche caso monocristallina), ma possono essere anche prodotti sotto forma di film sottili mediante le seguenti tecniche: sputtering a radiofrequenza, sputtering DC, hot walls (pareti calde), epitassia da fase liquida (LPE), epitassia da fase vapore (MBE, MOCVD). I composti II-VI e IV-VI più interessanti dal punto di vista elettronico sono elencati rispettivamente nelle tabb. VI e VII, assieme ad alcune importanti caratteristiche.
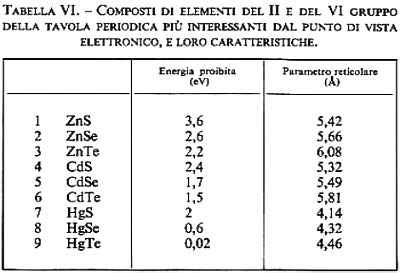
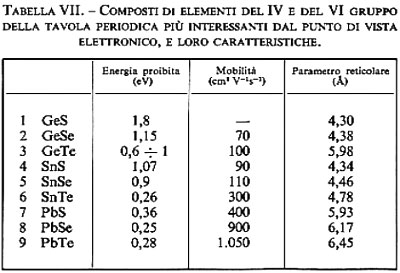
La fig. 12 mostra come esempio la struttura elementare (tipo wurtzite) di un composto come CdS, ZnS, ecc. I materiali 6, 7, 8, 9 della tab, VII, per il loro basso valore di energia proibita, possono essere utilizzati per fabbricare dispositivi per l'infrarosso. Con elementi dei gruppi II-VI e IV-VI possono essere realizzati dei materiali ternari tipo Pb1-xSnxTe o Hg1-xCdxTe, che possono essere ottenuti come leghe dalle seguenti coppie di composti binari: PbTe-SnTe e HgTe-CdTe. Questi materiali sono utilizzati per la fabbricazione di rivelatori per il vicino, medio e lontano infrarosso. Per quanto attiene al PbSnTe, come si evince dalla fig. 13A, la banda di energia proibita è positiva ovunque tranne che per un valore particolare della composizione PbTe-SnTe, in corrispondenza del quale essa vale zero. Ciò consente di fabbricare sensori per l'infrarosso in grado di funzionare quando la lunghezza d'onda della radiazione incidente è maggiore di 4,5 μm. Due caratteristiche fisiche hanno limitato l'uso esteso di questo materiale: la prima porta inevitabilmente ad avere lunghe costanti di tempo (RC) e quindi limitazioni della velocità di funzionamento; la seconda è legata al coefficiente di espansione termica, che è molto maggiore di quello del silicio. Ciò non ha consentito lo sviluppo di strutture ibride PbSnTe-Si, tuttavia questo materiale è molto utilizzato per fabbricare sorgenti laser nell'infrarosso.
Quanto al materiale Hg1-xCdxTe, essendo formato da un semimetallo tipo HgTe, esso può coprire valori di energia proibita da O a circa 1,6 eV (v. fig. 13B). Ciò lo rende di estremo interesse per la fabbricazione di rivelatori per l'infrarosso per lunghezze d'onda maggiori di 0,8 μm. Lo svantaggio che esso presenta è che comincia a perdere mercurio verso, 125 °C, ma malgrado ciò resta il materiale più utilizzato per la larga possibilità di scelta degli intervalli di lunghezze d'onda di lavoro. Fra tutti i materiali per l'infrarosso è quello sul quale ancora oggi sono concentrati molti sforzi per migliorare le tecniche di crescita, in vista della fabbricazione di sistemi lineari e a matrice di rivelatori con caratteristiche ottimizzate soprattutto dal punto di vista dell'uniformità. A proposito di leghe ternarie, va detto che anche il composto InAs1-xSbx dei gruppi III-V può essere utilizzato per fabbricare rivelatori per l'infrarosso, ma solamente nell'intervallo 3-8 μm. La sua energia proibita, funzione della composizione InAs-InSb (v. fig. 13C), non si annulla mai; esso tuttavia ha lo svantaggio di non essere adatto a coprire completamente l'intervallo 8-14 μm, che è quello di maggiore interesse pratico.
n) Nuovi materiali semiconduttori.
Le ricerche teoriche e sperimentali sui semiconduttori sono state fino a oggi imperniate sui materiali ‛tetraedrici' tipo Si, Ge, SiC, composti II-V, composti II-VI, che hanno sostenuto un ruolo dominante in campo scientifico e tecnologico. Esistono tuttavia altre categorie di materiali che presentano proprietà semiconduttrici; per ragioni di completezza è doveroso menzionarli, anche se non hanno raggiunto il successo di quelli già descritti. Essi sono: beegerite (Pb6Bi2S9), berzelianite (Cu2Se), calcopirite (CuFeS2), enargite (Cu3AsSn), galena (PbS), guanaiuatite (Bi2SSe2), guitermanite (Pb3As2S6), pirite (FeS2), tennantite (Cu10Fe2As4S13). Tutti questi minerali sono caratterizzati da intervalli di energia proibita molto piccoli, talvolta inferiori a 1,0 eV. Materiali con Eg superiore e del tipo non tetraedrico stanno suscitando un nuovo interesse per la possibilità di fornire luminescenza nella regione visibile dello spettro. Sono oggetto di studio a questo proposito i materiali SiP oppure SiAs oppure combinazioni tipo SiP-Mg3P2. Infine possono essere segnalati materiali semiconduttori non basati sul ruolo classico di valenza, di difficile preparazione e basati sull'utilizzazione del boro. Essi sono:
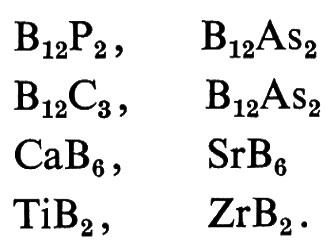
Per esempio, il B12P2 può essere preparato per avere una Eg = 3,3 eV, una mobilità pari a circa 15 cm2 V-1s-1 e una concentrazione di lacune di 1016 cm-3. Questi materiali dovranno essere studiati in modo più approfondito con le moderne tecniche diagnostiche per accertare le loro reali potenzialità nel prossimo futuro.
3. Materiali magnetici.
Le proprietà di maggior rilevanza dei materiali magnetici per l'elettronica sono quelle connesse con la forma, l'estensione e la dipendenza dalla temperatura della loro risposta, sia statica che dinamica, a un segnale elettromagnetico esterno. Naturalmente tutto ciò è correlato strettamente alla struttura cristallina e a eventuali impurezze in essa presenti, le quali possono dar luogo a sensibili variazioni delle proprietà dei materiali. I materiali magnetici per l'elettronica possono essere raggruppati a seconda della composizione o della loro prevalente utilizzazione. Per quanto rignarda l'utilizzazione, una ragionevole elencazione comprende: a) la registrazione magnetica (video e audio); b) le memorie magnetiche; c) i dispositivi magnetoottici; d) i dispositivi a microonde.
Dal punto di vista della composizione occorre invece ricordare: 1) le leghe metalliche (per registrazioni e memorie); 2) gli ossidi (registrazione e dispositivi a microonde); 3) le ferriti (registrazioni, memorie e dispositivi a microonde); 4) i granati (memorie, dispositivi a microonde e magnetoottici). Ci si riferisce qui a materiali e utilizzazioni che sono risultati prevalenti negli ultimi vent'anni, mentre non è possibile, per motivi di brevità, far riferimento ad applicazioni particolari, talune anche di notevole interesse, le quali tuttavia, per una serie di ragioni, non hanno trovato un apprezzabile seguito a livello produttivo.
a) Registrazione magnetica.
Nata alla fine del secolo scorso, la registrazione magnetica, che ora costituisce il principale settore di applicazione dei materiali magnetici (v. Lowman, 1972), conobbe un pieno successo commerciale solo dopo la seconda guerra mondiale. Oggi la registrazione magnetica, analogica e digitale, è la forma più diffusa di registrazione e l'unica ‛semipermanente' (può conservare intatta l'informazione anche in assenza di energia fornita al sistema, ma può anche agevolmente cancellarla per immagazzinare informazioni nuove). Lo schema di un sistema per la registrazione magnetica è illustrato nella fig. 14. L'informazione da registrare, sotto forma di segnale elettrico, viene amplificata e alimenta l'avvolgimento della testa di scrittura, un elettromagnete con traferro molto stretto, generando così un campo magnetico d'ampiezza proporzionale al segnale da registrare. Una piccola parte di questo campo viene dispersa dal traferro, in prossimità del quale scorre il nastro su cui, mediante opportuni collanti, è stato depositato uno strato di particelle magnetiche (v. fig. 15). Il numero di particelle magnetizzate per unità di superficie è strettamente correlato all'intensità del campo disperso, cioè all'ampiezza del segnale da registrare. Se il campo coercitivo del materiale del nastro è sufficientemente elevato, il segnale viene registrato in modo semipermanente. Il processo inverso avviene in fase di lettura. In questo caso il flusso disperso dal nastro penetra nel traferro della testa di lettura: le variazioni di questo flusso generano una tensione nell'avvolgimento concatenato (v. fig. 16), che viene amplificato riproducendo così il segnale di partenza già registrato sul nastro.
Dalla sequenza di operazioni che intervengono nella registrazione magnetica discendono le principali caratteristiche necessarie al materiale magnetico da utilizzare, vale a dire: a) elevata magnetizzazione di saturazione (qualche kgauss) per ottenere un elevato segnale di lettura; b) opportuno valore del campo coercitivo, per consentire a un tempo una buona conservazione della magnetizzazione e un'agevole operazione di riscrittura (qualche centinaio di oersted); c) ciclo d'interessi pressoché rettangolare, per un elevato rapporto fra le magnetizzazioni residua e di saturazione.
Per quanto riguarda la forma di utilizzazione dei materiali, si è ricorso fin dall'inizio ai cosiddetti particulate media, formati da particelle magnetiche aghiformi disperse in collanti organici e orientate parallelamente alla direzione di scorrimento della testina. In prima approssimazione ogni particella costituisce un singolo dominio, il che comporta un'alta anisotropia di forma, un più elevato campo coercitivo e una rapida inversione della magnetizzazione, che in questo caso avviene per simultanea rotazione dei momenti magnetici atomici. I materiali per la registrazione magnetica comprendono prevalentemente ossidi di varia composizione (v. tab. VIII; v. Craik, 1975). I possibili difetti di questi materiali sono il rumore nel segnale di uscita, dovuto principalmente a impurezze; la registrazione passante attraverso strati sottostanti di nastro (print through); la demagnetizzazione spontanea; lo spostamento della posizione dei picchi rispetto alla posizione registrata, a causa della larghezza finita degli impulsi registrati nel passaggio da uno stato di magnetizzazione all'altro (peak shift), come è schematicamente indicato nella fig. 17.
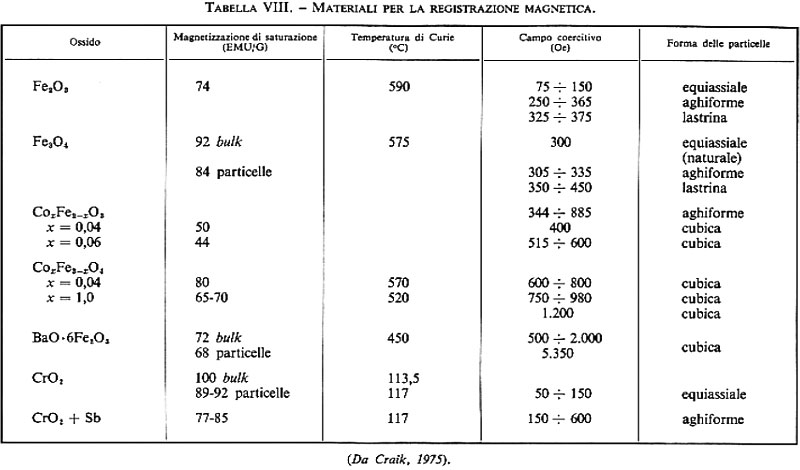
Negli ultimi anni si è cercato di migliorare la qualità della registrazione utilizzando materiali di più elevato campo coercitivo, sotto forma di particelle di minori dimensioni, di forma accuratamente aghiforme e di elevata regolarità. Ciò è stato ottenuto inizialmente modificando la composizione degli ossidi (sostituzione del Fe con il Co) e successivamente realizzando dei nastri con particelle interamente metalliche. I risultati ottenuti indicano un nettissimo miglioramento nei confronti degli ossidi, sia per la registrazione analogica che per la registrazione digitale. Una caratteristica fondamentale della registrazione è fornita dalla direzione della magnetizzazione, che nei sistemi oggi esistenti è diretta parallelamente a quella del moto del nastro e viene appunto chiamata ‛parallela'. Una magnetizzazione giacente nel piano del nastro, ma perpendicolare alla direzione del moto, produrrebbe un minor flusso emergente e sarebbe pertanto di difficile lettura. Con la registrazione parallela sono stati raggiunti elevatissimi valori di densità superficiale d'informazione e ridottissimi tempi d'accesso, a basso costo; al contempo però se ne sono venuti delineando sempre più chiaramente i limiti, prevalentemente di natura magnetostatica, che sono sintetizzati nella fig. 18. Infatti il parallelismo della magnetizzazione al piano del film provoca un accumulo di poli magnetici liberi in corrispondenza della superficie di separazione fra i volumi elementari magnetizzati in verso opposto. Se la lunghezza delle celle viene ridotta, la distribuzione parallela diviene instabile e si avrà una progressiva eliminazione di poli liberi con una forte riduzione dell'ampiezza del segnale di lettura. Per questo motivo si sta lavorando attivamente alla registrazione perpendicolare (v. fig. 19), nella quale non si generano poli magnetici liberi alla superficie di separazione fra un bit e quello successivo e pertanto i corpi demagnetizzanti non raggiungono valori eccessivi. Inoltre si annulla in pratica l'interazione fra bits adiacenti, per cui viene eliminato il fenomeno del peak shift.
Naturalmente pochi materiali consentono di ottenere strati sottili con la magnetizzazione normale al piano dello strato. Attualmente si usano leghe metalliche (per esempio Co-Cr) depositate mediante sputtering, che danno luogo a una struttura microscopica assimilabile a particelle aghiformi ortogonali allo strato (v. fig. 20). La registrazione e la lettura vengono effettuate mediante testine che forniscono e rilevano campi perpendicolari particolarmente concentrati.
b) Memorie magnetiche.
La forma e le dimensioni del ciclo d'isteresi accoppiate a un'elevata resistività sono state per lunghi anni alla base dell'utilizzazione dei materiali magnetici nelle memorie per calcolatori. Una classe di composti che presentano queste caratteristiche in grado soddisfacente sono le ferriti cubiche di formula MFe2O4, dove M è di solito un metallo bivalente, e di struttura del tipo degli spinelli. Tipici materiali utilizzati per le memorie sono le ferriti di manganese e di magnesio con varie aggiunte di altri ioni. La tecnologia e le prestazioni di tali materiali non hanno subito sostanziali cambiamenti dalla fine degli anni sessanta, mentre gli anni settanta sono stati caratterizzati dal tentativo di utilizzare in maniera selettiva altri parametri magnetici, vale a dire l'anisotropia e la dinamica dei domini. Si è così assistito a un notevole impegno scientifico e tecnico nei confronti delle memorie a bolle magnetiche (v. Bobeck e Della Torre, 1975), per le quali sono state utilizzate prima le ortoferriti e quindi i granati. Il principio su cui si basa il loro funzionamento è conseguente alla possibilità di formare, magnetizzando piastrine sottili o film di materiale magnetico con anisotropia uniassiale, diretta perpendicolarmente al piano della lastrina, dominî cilindrici che risultino stabili in un intervallo abbastanza ampio di campo applicato (v. fig. 21; v. Paoletti, 1978). È possibile far muovere questi domini mediante un opportuno gradiente di campo (v. fig. 22) ed è possibile visualizzarli mediante l'effetto Faraday, se il materiale è trasparente. Infine il loro passaggio attraverso una certa posizione può essere rilevato mediante misure di magnetoresistenza. Ogni bolla corrisponde a un bit e la sua presenza o assenza in un dato punto corrisponde alle due cifre del codice binario. Particolari circuiti consentono di creare o annichilare le bolle (v. Watson, 1980). In questo modo si possono realizzare registri a scorrimento, i quali a loro volta possono essere organizzati mediante varie architetture a costituire la memoria a bolle. Nonostante alcune allettanti caratteristiche (come la non volatilità) e l'impegno veramente notevole delle industrie elettroniche, le memorie a bolle non hanno trovato sul mercato uno spazio apprezzabile, anche se il loro sviluppo prosegue, sia pure in misura ridotta (v. Eschenfelder, 1980).
c) Dispositivi magnetoottici.
I dispositivi magnetoottici sono basati principalmente su due effetti magnetoottici: la birifrangenza magnetica lineare (effetto Cotton-Monton: v. fig. 23 A) e la birifrangenza magnetica circolare (effetto Faraday: v. fig. 23 B). L'effetto Cotton-Mouton si verifica quando un fascio di luce polarizzata linearmente attraversa un mezzo in cui il vettore magnetizzazione M sia perpendicolare alla direzione di propagazione della luce. Il fascio può essere considerato decomposto in due fasci linearmente polarizzati parallelamente e perpendicolarmente a M, i quali si propagano nel mezzo con velocità diversa, dando cosi luogo, in uscita, a una differenza di fase fra i due fasci e quindi a polarizzazione ellittica. Nell'effetto Faraday la magnetizzazione M è invece parallela alla direzione di propagazione di un fascio polarizzato linearmente e ne risulta una rotazione sul piano di polarizzazione.
I granati sono gli unici materiali noti che danno luogo a effetti magnetoottici in una regione dello spettro ove l'assorbimento è nullo. Inoltre la presenza di tre sottoreticoli (v. fig. 24) comporta che la dipendenza dell'effetto Faraday dalla temperatura risulti da una combinazione lineare della dipendenza da T delle rispettive magnetizzazioni (v. fig. 25). Un notevole ampliamento delle prospettive di utilizzazione dei dispositivi magnetoottici è stato determinato dalla rilevazione che l'inserimento di ioni Bi3+ nei siti dodecaedrici porta a un incremento di dieci volte della figura di merito nel visibile, definita come rapporto fra il potere specifico di rotazione F e il coefficiente di assorbimento. Occorre a questo punto ricordare che i film di granati magnetici, cresciuti epitassialmente, soddisfano pienamente i requisiti dei materiali per ottica integrata, in quanto possiedono un indice di rifrazione n = 2 che risulta maggiore di quello dei rispettivi substrati, oltre a una cifra di merito che risulta assai elevata alla lunghezza d'onda di minimo assorbimento da parte delle fibre ottiche utilizzate per trasmettere segnali ottici a lunghe distanze. Queste proprietà del granato di Bi consentono la realizzazione di particolari dispositivi, quali modulatori di polarizzazione, isolatori, memorie magnetoottiche.
Per quanto riguarda i visualizzatori si fa ancora uso dell'effetto Faraday. Si riesce a ottenere, con granati contenenti Bi e luce bianca, un contrasto medio nella regione del visibile di 20 : 1. Mediante opportune combinazioni di campi magnetici in visualizzatori a film multipli è possibile inoltre ottenere colori diversi, dal blu scuro al rosso. Questo tipo di visualizzatore è basato sulla possibilità di dare a un dominio magnetico o a una serie di dominî di una certa polarità, contenuti in un film magnetico trasparente, la forma di un simbolo grafico, mentre il resto del film resta polarizzato in verso opposto. La luce polarizzata linearmente che attraversa il simbolo sarà allora ruotata in verso opposto rispetto a quella che attraversa la parte del film non scritta. Mediante l'analizzatore viene bloccata la luce in uno dei due strati di polarizzazione, in modo da far apparire sullo schermo la forma del simbolo desiderato. Per ottenere dominî della forma desiderata si possono utilizzare gruppi ordinati di bolle magnetiche. Per esempio, utilizzando un granato di composizione BiTm2Fe4GaO12, in cui possono generarsi bolle di 7 μm di diametro che possono muoversi con il consueto meccanismo del campo ruotante su circuiti T and bar, sono stati scritti caratteri alfanumerici in un tempo di circa 3 ms. Altri metodi di scrittura utilizzano le proprietà di film di granato (GdBi)3 (FeGa)5O12 aventi un elevatissimo coefficiente di Faraday e una Tcomp molto vicina a una Tamb. Si realizzano così valvole ottiche' comandate dalle variazioni di temperatura, le quali vengono accoppiate con la piastra fotosensibile di una normale copiatrice. La stessa famiglia di granati è stata impiegata per ottenere deflettori di luce bidimensionale.
d) Dispositivi a microonde.
Tra le più interessanti proprietà dei granati occorre ricordare quelle che si manifestano alla frequenza delle microonde e che sono normalmente studiate mediante esperienze di risonanza ferromagnetica. L'YIG (yttrium iron garnet, granato di ittrio e ferro) cubico, magneticamente ordinato e ottimo isolante, è il sistema determinante per la comprensione della risonanza ferromagnetica che si verifica appunto alla frequenza delle microonde. In particolare si è dovuto mettere in evidenza il ruolo delle onde di spin e delle impurezze nella comprensione dei fenomeni di rilassamento. Per quanto riguarda i modi che possono essere eccitati in un materiale magnetico, essi possono essere classificati a seconda dell'approssimazione usata nella descrizione delle interazioni di dipolo e di scambio. Nella maggior parte delle situazioni di interesse applicativo i campi di dipolo vengono trattati in approssimazione quasi statica, secondo la quale i campi indotti dalla magnetizzazione si propagano istantaneamente a tutte le parti del campione. Un'altra approssimazione trascura i campi di dipoli generati dalla discontinuità della magnetizzazione alla superficie del campione. I modi con i quali si ha a che fare in un materiale magnetico sono soprattutto le onde di spin. La possibilità di crescere film epitassiali di granati magnetici, di notevole regolarità e grande spessore (fino a 100 μm), ha avuto come naturale conseguenza l'inizio di un'attività nel settore dei dispositivi a microonde. Infatti, quando la lunghezza d'onda delle onde di spin è confrontabile con le dimensioni lineari del campione, occorre tener conto che al crescere dello spessore gli effetti di demagnetizzazione di origine dipolare diventano prevalenti rispetto agli effetti di scambio. In questa situazione si manifestano i cosiddetti moti magnetostatici (MSW), i quali possono essere calcolati tenendo presenti le condizioni di ancoraggio degli spins agli estremi della superficie del film e assumono particolare rilevanza per lunghezze d'onda millimetriche.
I relativi dispositivi (v. fig. 26) possono essere basati sul comportamento non simmetrico delle ferriti rispetto al verso di propagazione delle onde (isolatori e circolatori) o all'azione che sulla propagazione delle microonde può esercitare un campo magnetico variabile (commutatori, sfasatori, filtri accordabili). Occorre disporre di materiali a basse perdite dielettriche e magnetiche con stretti picchi di assorbimento di risonanza alla frequenza delle microonde e grande stabilità termica per applicazioni a potenze elevate. I granati magnetici costituiscono una classe di materiali che presenta queste caratteristiche. Inoltre è possibile ottenere risonanza ferromagnetica alla frequenza delle microonde con l'applicazione di campi magnetici relativamente bassi e pertanto ottenibili facilmente con magneti permanenti, mentre la struttura con vari tipi di siti a simmetria tetraedrica, ottaedrica e dodecaedrica, che possono ospitare una gran varietà di ioni, consente di realizzare una vera e propria ingegneria magnetica per ottenere caratteristiche di magnetizzazione, anisotropia e rilassamento, ottimali per il dispositivo in progetto (v. ‟Thin solid films", 1985).
Bibliografia.
Abricosov, N. Kh., Bankina, V. F., Poertskaya, L.V., Schelinova, L.E., Skudnova, E. V., Semiconducting II-VI, IV-VI and V-VI components, vol. III, New York 1969.
Bobeck, A. H., Della Torre, E., Magnetic bubbles, Amsterdam 1975.
Craik, D. J. (a cura di), Magnetic oxides, New York 1975.
Esaki, L., Large scale integrated circuits technology: state of the art and prospect, NATO advanced study institutes, series E, n. 55, 1958.
Eschenfelder, A. H., Magnetic bubbles technology, Berlin-New York 1980.
Gooch, C. H., Gallium arsenide laser, New York 1969.
Goodman, C. H. L., New semiconductors, vol. XX, New York 1985.
Grasserbauer, M. (a cura di), Progress in material analysis, voll. I - II, Berlin-New York 1985.
Hummel, R.E., Electronic properties of materials. An introduction for engineers, Berlin-New York 1985.
?????? ‟IEEE transactions. Magnetics", 1984, XLII, p. 286; 1985, XLVI, p. 274, XLIX, pp. 93, 195.
Kaldis, E., Crystal growth of electronic materials, Amsterdam 1985.
Kasprzak, L.A., Gaind, A. K., Near-ideal Si-SiO2 interface, in ‟IBM journal of research and development", 1980, XXIV, 3.
Kittel, C., Introduction to solid state physics, New York 19765.
Lark-Horowitz, K., The new electronics. Present state of physics, Washington 1954.
Lowman, C.E., Magnetic recording, New York 1972.
Murarka, S. P., Silicides for VLSI applications, New York 1983.
Nagels, P., Electronic transport in Amorphous semiconductors, (a cura di M. H. Brodsky), Berlin-New York 1979.
Oppolzer, H., Neppl, F., Hieber, K., Huber, V., Influence of slight deviations from TaSi2 stoichiometry on the high-temperature stability of tantalum silicide/silicon contact, in ‟Journal of vacuum science and technology, B", 1984, II, 4.
Ottaviani, G., Review of binary alloy formation by thin film interactions, in ‟Journal of vacuum science and technology", 1979, XVI, 5, pp. 1112-1119.
Ottaviani, G., Mayer, J. W., Mechanisms and interfacial layers in silicide formation, in Reliability and degradation: semiconductor devices and circuits (a cura di M. J. Howes e D. V. Morgan), New York 1981, cap. 2.
Pantelides, S. T., The physics of SiO2 and its interface, in Proceedings of International topical conference Yorktown Heights, N. Y. - March 22-24, 1978. New York 1978.
Paoletti, A. (a cura di), Physics of magnetic garnets, Amsterdam 1978.
Sze, S. M., Physics of semiconductor devices, New York 1976.
‟Thin solid films", n. speciale Magnetic films, 1985.
Watson, J. K., Applications of magnetism, New York 1980.
Willardson, R. K., Semiconductors and semimetals, 2 voll., New York 1966-1968.
Materiali polimerici di Paolo Corradini e Luigi Nicolais
SOMMARIO: 1. Considerazioni introduttive. □ 2. Aspetti della chimica e struttura chimica dei materiali polimerici: a) introduzione; b) esempi di polimerizzazione a catena; c) esempi di polimerizzazione a stadi; d) ulteriori aspetti della chimica e struttura chimica dei polimeri. □ 3. Aspetti della struttura fisica e proprietà dei materiali polimerici: a) introduzione; b) lo stato amorfo (viscoso e vetroso); c) lo stato semicristallino; d) lo stato mesomorfo; e) alcuni polimeri con proprietà fisiche speciali; f) le leghe e i sistemi eterofasici. □ 4. Classi di polimeri in relazione alla lavorazione e all'uso: a) termoplastici; b) termoindurenti; c) elastomeri e gomme; d) fibre; e) vernici, rivestimenti e adesivi; f) film e membrane; g) compositi. □ 5. Tecnologie di lavorazione dei materiali polimerici: a) introduzione; b) estrusione; c) stampaggio; d) filatura di fibre; e) soffiaggio di film; f) altre tecnologie. □ 6. Applicazioni: a) generazione, distribuzione e conservazione dell'energia; b) i materiali polimerici nell'industria dei trasporti; c) i materiali polimerici nell'industria edile; d) i materiali polimerici nell'agricoltura e nell'industria alimentare; e) i materiali polimerici in medicina; f) considerazioni conclusive. □ Bibliografia.
1. Considerazioni introduttive.
Se dovessimo definire la nostra epoca in base ai materiali che la caratterizzano, dovremmo chiamarla ‛civiltà dei materiali polimerici', o della plastica. Infatti, anche se la cosiddetta ‛plastica' ha assunto talvolta, ma a torto, un significato deteriore, intesa come sostituto più economico di materiali tradizionali in applicazioni a basso valore aggiunto, emerge sempre più il suo impiego in aree tecnologiche molto avanzate. Ciò è possibile grazie alla gamma praticamente illimitata di proprietà che si possono ottenere da questi materiali, sia da soli che mescolati tra di loro o con altri materiali organici o inorganici.
Per la loro ottimizzazione i materiali polimerici richiedono però la convergenza di competenze disparate, che vanno dalla chimica alla fisica, dall'ingegneria alla matematica. Infatti la scelta di un materiale per una specifica applicazione richiede l'analisi di diverse caratteristiche, quali le sue proprietà fisiche (meccaniche, di trasporto di materia e di energia), la stabilità nel tempo e nell'ambiente in cui verrà utilizzato, la tecnologia di fabbricazione e l'economicità. Nel caso dei polimeri particolare importanza assume il processo con cui viene ottenuto il manufatto, perché spesso, utilizzando opportune tecnologie e condizioni di lavorazione, è possibile indurre nel materiale proprietà particolari. Purtroppo, però, nel passato spesso ci si è soffermati solo su aspetti formulativi, sottovalutando le variabili di processo o il design, con il risultato che i materiali plastici non sono stati usati sempre al meglio.
Ad esempio, polimeri altamente orientati posseggono valori di resistenza meccanica e di rigidezza molto elevati e vicini a quelli dei materiali metallici utilizzati come materiali da costruzione. Analogamente i compositi costituiti da resine polimeriche rinforzate con fibre presentano una così ampia gamma di proprietà da permettere di soddisfare le più esigenti richieste dell'utenza. Nella tabella sono riportati i valori tipici delle proprietà meccaniche a trazione di alcuni dei più importanti materiali per applicazioni strutturali sia metallici che a base polimerica. Dati i bassi valori della densità dei polimeri e dei compositi a matrice polimerica, le proprietà meccaniche specifiche di questi materiali (valori della proprietà divisi per la densità) sono in genere molto elevate e in alcuni casi superiori a quelle dei metalli. Bisogna tuttavia osservare che le informazioni riportate nella tabella sarebbero incomplete se dovessero essere utilizzate per una progettazione strutturale, dal momento che le proprietà dei materiali polimerici sono spesso dipendenti, oltre che dalla temperatura, anche dall'ambiente in cui si trovano e dal tempo.
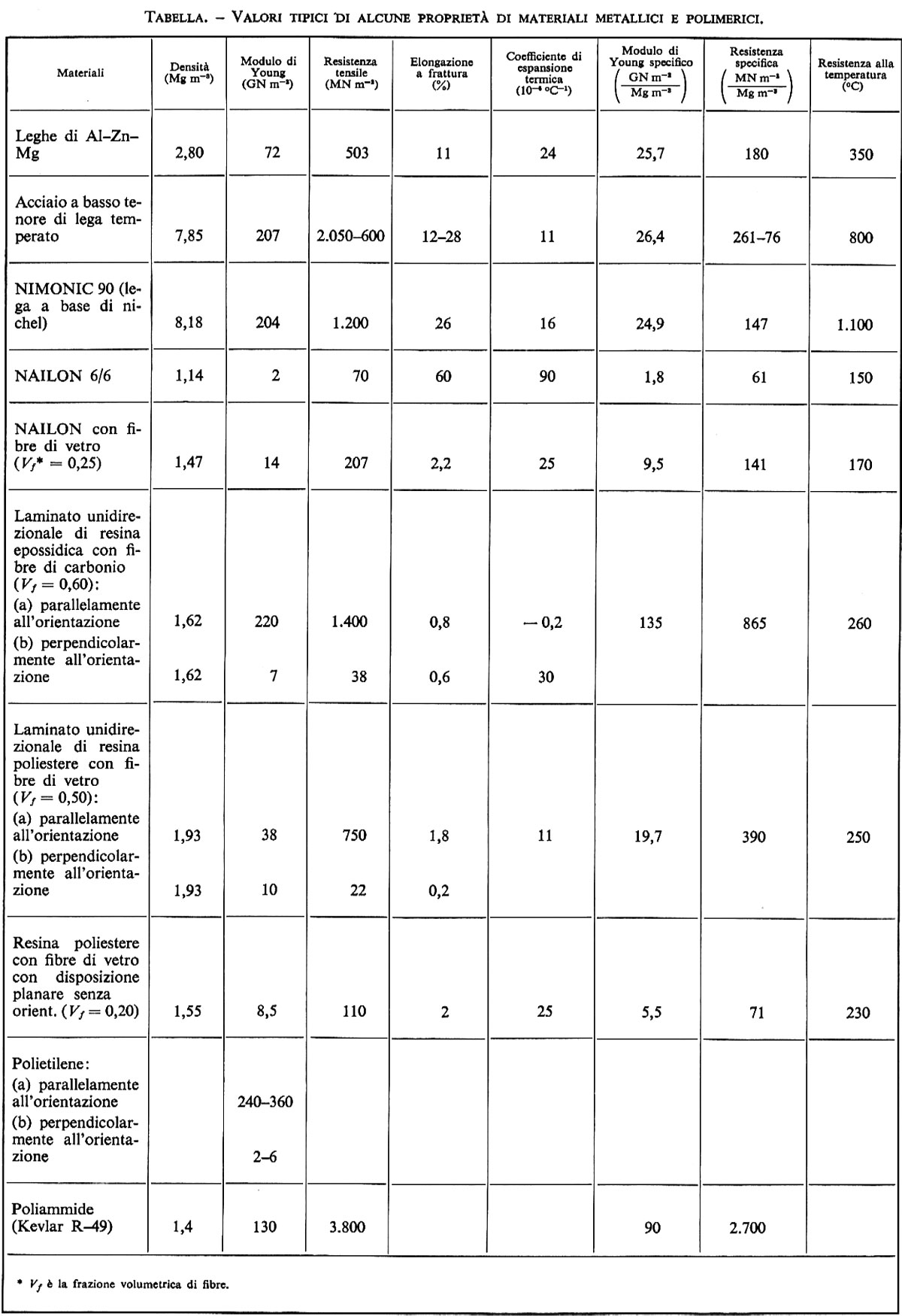
Sempre più frequentemente materiali leggeri e resistenti, sia meccanicamente che agli agenti aggressivi, sono richiesti in sostituzione dei metalli per molte applicazioni strutturali. L'uso di materiali a base polimerica spesso determina un risparmio energetico indipendentemente dal fatto che i polimeri stessi derivino dal petrolio. Questo risparmio può essere dovuto non solo a metodi efficienti di polimerizzazione e trasformazione, ma anche alla durabilità del prodotto e alla sua più efficiente utilizzazione.
Compositi a matrice polimerica contenenti grafite, vetro o fibre polimeriche ad alta resistenza e ad alto modulo offrono degli evidenti vantaggi grazie al loro alto rapporto resistenza/densità (v. tabella). Per questo motivo, poiché, ad esempio, il peso di un aeroplano è critico sia per la sua capacità di trasporto che per le altre prestazioni richieste, un grosso sforzo di ricerca e di sviluppo è stato effettuato negli anni ottanta per sostituire l'alluminio, il titanio e altri materiali metallici con materiali plastici rinforzati con fibre, come i compositi di matrice epossidica con fibre di carbonio.
Sugli attuali aeroplani Boeing 767, ad esempio, la percentuale di materiali compositi è pari al 3% della massa totale; predominano ancora i metalli, con in testa l'alluminio (81%); per la prossima generazione di aeroplani è però previsto un uso molto più massiccio di materiali compositi, fino a coprire - secondo alcune previsioni - oltre la metà della massa totale dell'aereo (v. fig. 1).
La ragione di questa forte tendenza all'uso di materiali plastici rinforzati va ricercata nell'alto costo del carburante, che richiede strutture sempre più leggere, ma anche progettazioni sempre più sofisticate.
Anche nell'industria automobilistica è prevista una sempre più ampia utilizzazione di polimeri e compositi. La tendenza alla sostituzione dei materiali metallici con materiali plastici e compositi è anche in questo caso legata alla riduzione dei costi e del peso dell'autovettura, con un conseguente risparmio energetico durante la sua utilizzazione.
La sostituzione di materiali tradizionali con materiali innovativi è, però, spesso ritardata da considerazioni economiche e tecnologiche: per esempio, la sostituzione dell'acciaio e dell'alluminio con compositi avanzati in componenti strutturali di aeromobili o di autoveicoli richiede lo sviluppo di nuove tecniche di progettazione che si adattino alle particolari proprietà dei compositi o dei polimeri altamente orientati.
Inoltre è in genere necessario sviluppare nuove tecnologie di lavorazione, che possono richiedere uno sforzo economico ingente. Già la semplice sostituzione di un polimero con un altro in un processo tradizionale di formatura a iniezione richiede la messa a punto di nuovi stampi che tengano conto dei diversi coefficienti di espansione termica del nuovo materiale.
Nonostante queste limitazioni, l'uso dei polimeri in sostituzione dei materiali tradizionali è sempre più esteso.
2. Aspetti della chimica e struttura chimica dei materiali polimerici.
a) Introduzione.
I materiali polimerici sono costituiti esclusivamente o in prevalenza da polimeri. I polimeri sono sostanze costituite da molecole molto grandi (‛macromolecole') risultanti dal concatenamento di numerose unità chimiche (‛unità costituzionali', o ‛unità costituzionali ripetitive', se il polimero, detto allora ‛regolare', può considerarsi formato da unità costituzionali tutte di uno stesso tipo).
Le reazioni chimiche di ottenimento di un polimero a partire da una o più sostanze costituite da molecole piccole (‛monomeri') sono dette ‛reazioni di polimerizzazione'; le unità costituzionali di un polimero, cioè le molecole di monomero, sono dette anche ‛unità monomeriche'.
Le unità costituzionali di un polimero si collegano in catene che possono essere lineari o ramificate, ed essere eventualmente legate ‛a ponte' ad altre catene. In quest'ultimo caso le catene danno luogo a un materiale polimerico reticolato.
Alcuni materiali polimerici, utilizzati nel passato e in alcuni casi tuttora in uso, derivano dalla modificazione chimica di polimeri esistenti in natura (per es. il cuoio conciato, la gomma vulcanizzata, l'acetato di cellulosa).
Il maggior interesse industriale è rivolto tuttavia a quei materiali polimerici che vengono ottenuti per sintesi chimica a partire da monomeri.
Per essere incorporate nelle catene macromolecolari le molecole del monomero devono essere capaci di reagire tra loro legandosi a più (di norma due) molecole. Ciò può avvenire in tre modi: 1) per apertura di un legame multiplo (generalmente doppio, come, ad es., nei monomeri vinilici o dienici); 2) per apertura di un ciclo (come, ad es., nei monomeri epossidici); 3) per collegamento attraverso due o più gruppi funzionali reattivi (come, ad es., nel caso che i monomeri siano una diammina e un acido bicarbossilico).
Un esempio di monomero vinilico è il cloruro di vinile (CH2=CHCl); l'unità costitutiva ripetitiva del polimero (polivinildoruro) è −CH2−CHCl−.
Un esempio di monomero dienico è il butadiene (CH2=CH−CH=CH2); il polimero (polibutadiene) ha unità costitutive del tipo −CH2−CH=CH−CH2− e/o
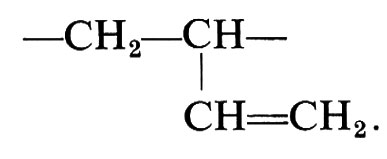
Un esempio di monomero epossidico è l'etilenossido
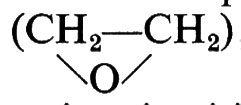
il polimero (polietilenossido) ha unità costitutive ripetitive del tipo −CH2−CH2−O−.
L'esametilendiammina (H2N−(CH2)6−NH2) e l'acido adipico (HOOC−(CH2)4−COOH) esemplificano il caso in cui i due monomeri sono una diammina e un acido bicarbossilico; il polimero è una poliammide (nailon) con unità costitutive ripetitive del tipo
−HN−(CH2)6−NH−CO−(CH2)4−CO−.
Dal punto di vista del meccanismo chimico, le reazioni di sintesi dei materiali polimerici (‛polimerizzazioni') possono essere classificate in reazioni di polimerizzazione a catena e in reazioni di polimerizzazione a stadi.
In una polimerizzazione a catena le molecole del monomero si attaccano in successione al terminale di una catena polimerica crescente - terminale che può essere un radicale o uno ione o può essere legato chimicamente (per es. coordinato a un metallo) in modo tale che ne sia accentuata la reattività - finché non interviene una reazione di terminazione della crescita. In una polimerizzazione a stadi le molecole di monomero possiedono almeno due gruppi funzionali reattivi e, a differenza del caso della polimerizzazione a catena, le molecole polimeriche crescono, oltre che per reazione con le molecole di monomero, anche per reazione tra di loro.
La sintesi in quantità sempre maggiori di materiali polimerici ha stimolato, dagli anni cinquanta in poi, lo sviluppo di una grande industria petrolchimica per la produzione dei monomeri necessari.
Monomeri prodotti in grande quantità e a basso prezzo per dar luogo a polimeri di larghissimo uso come materie plastiche (commodity polymers) sono l'etilene, il propilene, il cloruro di vinile e lo stirene. Altri monomeri vengono prodotti in quantità ragguardevoli per la sintesi di materiali polimerici costituenti di fibre e gomme.
Numerosissimi altri monomeri vengono prodotti in quantità più ridotte e a costi talvolta notevolmente più elevati, per dar luogo a polimeri di qualità, con una costituzione chimica progettata in modo da soddisfare le più specifiche e diversificate esigenze applicative (engineering e specialty polymers); la produzione di questi polimeri ha presentemente un elevato tasso di crescita.
b) Esempi di polimerizzazione a catena.
Le polimerizzazioni a catena possono essere iniziate da radicali o da ioni, o avvenire su catalizzatori di coordinazione.
La polimerizzazione radicalica è di particolare importanza. Un tipo di polimero dell'etilene (il cosiddetto polietilene a bassa densità, Low Density Polyethylene, sigla LDPF), i polimeri dello stirene (polistirene, sigla PS) e i polimeri del cloruro di vinile (polivinilcloruro, sigla PVC) sono prodotti su larga scala attraverso una polimerizzazione a catena iniziata da radicali liberi. Altre resine termoplastiche ottenute con processi radicalici sono il polivinilacetato, il polimetilmetacrilato, il copolimero acrilonitrile/butadiene/stirene (sigla ABS) e il politetrafluoroetilene. La polimerizzazione radicalica è usata anche per la sintesi di gomme (copolimeri stirene-butadiene; policloroprene).
La generazione di radicali (schematizzati con X•) avviene, ad esempio, per decomposizione delle molecole di una sostanza opportuna, denominata ‛iniziatore', aggiunta al monomero in piccola concentrazione. Un radicale reagisce con una molecola di monomero (tipicamente una olefina CH2=CHR o un diene) per dar luogo a una specie radicalica attivata, alla quale si vengono addizionando successivamente altre unità monomeriche.
Nel caso di un monomero vinilico si ha:
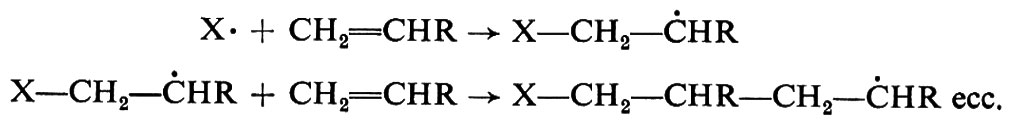
La terminazione di catena può avvenire, ad esempio, per reazione tra due radicali polimerici, dando luogo a somma (1) o disproporzione (trasferimento di idrogeno) (2):
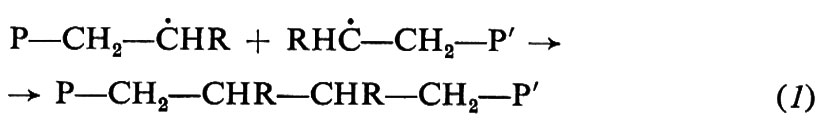
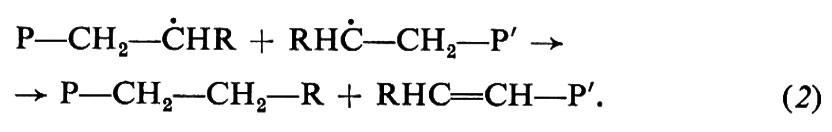
I polimeri dei monomeri vinilici, ottenuti con questi processi, hanno una costituzione ‛regolare', nel senso che le macromolecole sono prevalentemente costituite da unità −CH2−CHR− in successione ‛testa-coda':
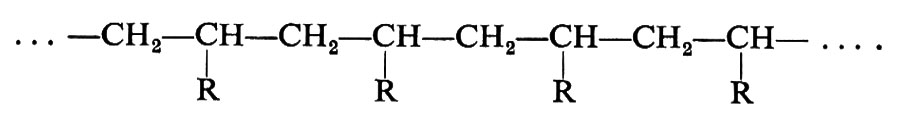
La configurazione spaziale relativa dei gruppi R risulta invece più o meno casuale nei tipi che sono stati definiti come meso (m)

e racemico (r)
per cui i polimeri hanno una configurazione spaziale sostanzialmente irregolare (non stereoregolare). Questa è una caratteristica pressoché generale dei polimeri ottenuti per via radicalica.
Oltre che da radicali liberi, la polimerizzazione a catena può essere promossa da cationi, da anioni o da catalizzatori di coordinazione. In particolare questi ultimi, sviluppati negli ultimi trent'anni, consentono una regolazione assai migliore sia della costituzione che della configurazione spaziale delle catene risultanti.
I catalizzatori di coordinazione più importanti furono scoperti da K. Ziegler e collaboratori, alla fine del 1953, per la sintesi del polietilene lineare, e da G. Natta e collaboratori, nel 1954 e anni successivi, per la sintesi di tutta una serie di polimeri di olefine e diolefine a concatenamento stereoregolare. Per questa scoperta Natta e Ziegler ricevettero il premio Nobel nel 1963.
Alla fine del 1953 era già noto il metodo per ottenere polimeri dall'etilene con processi radicalici: tali processi sono tuttora in uso e danno luogo a prodotti con una struttura chimica della catena contenente ramificazioni lunghe e corte (le ramificazioni corte sono prevalentemente costituite da gruppi n-butilici). Tali ramificazioni costituiscono delle irregolarità; conseguentemente i polimeri dell'etilene che le contengono hanno cristallinità non elevata, punto di fusione e densità bassi (donde la denominazione low density polyethylene). La realizzazione di un metodo di pratica applicabilità per la sintesi di polimeri dell'etilene completamente lineari si deve a Ziegler. Questi identificò un catalizzatore risultante dalla combinazione di un composto di un metallo di transizione (tipicamente tetracloruro di titanio) con un metalbalchile (per es. alluminiotrietile). Il polietilene che si ottiene ha elevata cristallinità e densità e punto di fusione sensibilmente più alti del corrispondente polimero radicalico, avente catena ramificata (donde la denominazione High Density Polyethylene, sigla HDPE).
Il meccanismo di polimerizzazione comporta con tutta probabilità la crescita della catena polimerica −CH2−CH2−Pn per coordinazione dell'olefina al metallo di transizione (Me) e successivo suo inserimento su un legame metallo-carbonio:

Natta e collaboratori, agli inizi del 1954, trovarono che i catalizzatori di ‛coordinazione' di Ziegler potevano dare polimeri cristallini anche di 1-alcheni, e in particolare del propilene; i catalizzatori che all'epoca furono trovati più adatti a tale scopo sono eterogenei (per es. titanio tricloruro solido e un alluminiotrialchile, di cui il primo insolubile nel mezzo di reazione). Le catene di polipropilene ottenute in presenza di questi catalizzatori hanno una costituzione regolare, risultante dal concatenamento testa-coda di unità monomeriche
Ma la caratteristica strutturale essenziale, che conferisce al polimero le sue eccellenti proprietà d'uso, è, oltre a una costituzione regolare, una configurazione spaziale altamente regolare delle catene di polipropilene (queste sono, cioè, stereoregolari). È stato dimostrato che l'elevata cristallinità del polimero si deve associare al fatto che le configurazioni spaziali relative dei gruppi
sono in larghissima prevalenza del tipo meso:

Un modello della catena risultante è schematizzato nella fig. 2a. Una catena di tale tipo è stata denominata ‛isotattica'.
La polimerizzazione del propilene a polimero isotattico costituì la prima importante polimerizzazione stereospecifica, in cui cioè le unità monomeriche risultano avere una specifica, ordinata configurazione sterica, attuata per sintesi. È da notare che reazioni stereospecifiche avvengono in natura nella sintesi delle proteine e di altri polimeri di origine biologica, come la gomma naturale e la guttaperca (con strutture dei doppi legami, rispettivamente, cis e trans reciprocamente stereoisomeriche).
Opportuni catalizzatori Ziegler-Natta hanno consentito l'ottenimento di polimeri stereoregolari da monomeri vinilici (per esempio 1-alcheni), da monomeri dienici (come il butadiene e l'isoprene) e da altri monomeri.
Nel caso degli 1-alcheni, come il propilene, che verrà trattato con maggior dettaglio, il meccanismo che consente di orientare le molecole di monomero in modo che il concatenamento delle unità monomeriche sia stereoregolare (isotattico nel caso discusso) comporta il coordinamento delle molecole di monomero su un sito del catalizzatore, prima che esse si inseriscano nella catena crescente. Si pensa che il sito catalitico abbia una forma spaziale tale che l'orientamento delle molecole di monomero risulti vincolato a essere sempre lo stesso. L'attacco susseguente della molecola di monomero alla catena crescente avviene poi per apertura cis del doppio legame, come è già stato schematizzato per il caso della polimerizzazione dell'etilene. Il soddisfacimento delle condizioni suesposte è sufficiente a dar luogo all'elevata stereoregolarità del polimero.
La produzione di polipropilene (sigla PP) isotattico con catalizzatori del tipo Ziegler-Natta è andata continuamente aumentando nel tempo, per le eccellenti proprietà del polimero, in relazione al non elevato costo del monomero e del processo di polimerizzazione, e per la possibilità di effettuare opportune modifiche del prodotto (ad es. per copolimerizzazione) onde migliorarne le prestazioni per usi specifici. Un recentissimo abbattimento dei costi di produzione ha fatto seguito alla scoperta di nuove classi di catalizzatori eterogenei a base di magnesio e titanio, detti ‛ad alta resa', con produttività di oltre una tonnellata di polimero per grammo di titanio.
c) Esempi di polimerizzazione a stadi.
Attraverso procedimenti di polimerizzazione a stadi vengono sintetizzati le poliammidi o nailon (caratterizzate dal legame ammidico −CO−NH− nella catena), i poliesteri (caratterizzati dal legame estereo −CO−O− nella catena) e molti dei polimeri con struttura chimica progettata per impieghi speciali, come l'uso continuato a temperature molto elevate.
A titolo di esempio, viene descritta la sintesi del poliestere per antonomasia, il polietilentereftalato (sigla PET), di larghissimo uso, dopo trasformazione in fibra tessile, per policondensazione da glicole etilenico e dimetiltereftalato. La polimerizzazione avviene, tipicamente, in presenza di catalizzatori debolmente basici, come gli alcossidi di titanio, e comporta dapprima la reazione del dimetiltereftalato con un eccesso di glicole etilenico, con eliminazione di metanolo (CH3OH). Si ottiene una miscela di esteri del glicole monomerici (n = 1 nello schema sottostante) e oligomerici (n = 2, 3, 4, e comunque piccolo, nello schema sottostante):

Alla temperatura di reazione tutto il metanolo formatosi viene allontanato; la temperatura è poi gradatamente innalzata in modo da portarla al di sopra della temperatura di ebollizione del glicole etilenico e infine di quella di fusione del polimero (Tf = 260 °C). In questo modo viene allontanato il glicole etilenico e il valore medio di n aumenta finché la massa media delle molecole di polimero raggiunge il valore ottimale per l'applicazione desiderata (prevalentemente produzione di fibre e film).
La distribuzione delle masse molecolari risultanti nel caso di una reazione di ‛policondensazione' del tipo descritto, a differenti gradi di conversione, è mostrata nella fig. 3; tipi di distribuzione diversi risultano specificamente da altri processi di polimerizzazione.
d) Ulteriori aspetti della chimica e struttura chimica dei polimeri.
Le proprietà di lavorazione e d'uso dei materiali polimerici vengono in larga misura determinate dal procedimento di preparazione adottato. In particolare dipendono dal metodo di preparazione: la composizione chimica (il materiale polimerico può essere costituito da un omopolimero, da un copolimero, da una miscela di polimeri); la costituzione (le unità che si ripetono lungo la catena polimerica possono avere tutte la stessa costituzione e lo stesso modo di concatenamento oppure, secondo certe proporzioni, costituzioni e modi di concatenamento differenti); la distribuzione delle masse molecolari (a differenza di quanto accade nel caso di composti chimici costituiti da molecole piccole, in un materiale polimerico non sono definibili e misurabili altro che valori medi delle masse delle singole macromolecole); la stereochimica (i doppi legami in catena possono essere cis o trans, il posizionamento relativo di due unità monomeriche olefiniche in una catena può essere di tipo meso o racemico); la topologia delle molecole (le catene possono essere lineari, ramificate o reticolate).
L'insieme delle caratteristiche chimiche appena citate influenza in modo significativo lo stato fisico e le proprietà d'uso del materiale polimerico risultante. Il procedimento sintetico può essere perciò indirizzato in modo da ottenere le proprietà maggiormente desiderabili per ogni specifica applicazione.
La chimica dei materiali polimerici, d'altro canto, non riguarda solo gli aspetti sintetici, ma anche le modificazioni chimiche che intervengono successivamente, talvolta indesiderate, talaltra fatte avvenire di proposito. Esse comprendono la degradazione, la stabilizzazione, l'additivazione, la modificazione strutturale dei materiali polimerici.
In particolare tutti i polimeri, esposti alle condizioni ambientali e non adeguatamente protetti, subiscono, in varia misura, cambiamenti fisici e chimici graduali (aging), che possono peggiorarne le caratteristiche fisiche fino ad alterarli in modo irreparabile.
Alcuni di questi effetti sono dovuti all'ossidazione termica o alla fotoossidazione. L'ossidazione termica di un materiale plastico può avere inizio nel fuso durante la lavorazione ad alta temperatura e a livelli di ossigeno molto bassi e continuare lentamente all'aria anche nell'intorno della temperatura ambiente. Nella preparazione di manufatti di lunga durabilità, a quasi tutti i polimeri è necessario aggiungere antiossidanti ed eventualmente stabilizzatori verso la luce ultravioletta.
In altri casi, specie nelle fibre, può essere utile addizionare prodotti chimici che ne riducano l'infiammabilità.
La degradazione dei polimeri può tuttavia essere utilizzata con vantaggio in molte aree problematiche. Essa può essere usata, ad esempio, per evitare che i polimeri diano luogo ad accumuli indesiderati di rifiuti; per facilitare il riciclo di materie prime; per la messa a punto di sintesi a erogazione controllata di prodotti chimici o farmaceutici; per la fabbricazione di circuiti nell'industria dei semiconduttori.
A conclusione di questo capitolo, in cui sono stati descritti alcuni aspetti della chimica dei materiali polimerici, è opportuno sottolineare l'importanza della loro caratterizzazione (per alcuni aspetti della quale v. polimeri).
Tra i più importanti metodi fisici di indagine strutturale e chimico-fisica vanno citati la risonanza magnetica nucleare (RMN), la spettroscopia infrarossa (IR) e Raman, l'analisi con i raggi X e la cromatografia di permeazione su gel (GPC) (v. chimica: Analisi chimica strumentale, suppl.).
La conoscenza delle strutture dettagliate di omopolimeri e copolimeri è necessaria per avanzare nella comprensione delle relazioni proprietà-struttura, per progettare la sintesi di polimeri con proprietà particolari e per migliorare la vita utile dei materiali.
Tecniche strumentali potenti e versatili come quelle sopra riportate permettono di affrontare con successo molti problemi di caratterizzazione chimico-fisica e strutturale dei polimeri; le informazioni dettagliate così ottenute portano a loro volta ad approfondire le ricerche sia sulla sintesi che sulle proprietà dei materiali polimerici.
3. Aspetti della struttura fisica e proprietà dei materiali polimerici.
a) Introduzione.
Le complessità inerenti alla lunghezza delle catene macromolecolari si riflettono nelle peculiari caratteristiche fisiche dei materiali polimerici, che sono per molti aspetti differenti da quelle che si riscontrano in materiali costituiti da molecole piccole.
Già a livello della caratterizzazione della struttura chimica si è vista la notevole diversità che intercorre tra molecole piccole e macromolecole; in particolare, a differenza da quanto accade nel caso di sostanze costituite da molecole piccole, tutte uguali, nel caso dei polimeri, che sono costituiti da un insieme di molecole grandi, o macromolecole, aventi di norma diversa grandezza, le caratteristiche chimiche sono definibili e misurabili solo come valori medi.
Lo stato cristallino dei polimeri mostra localmente lo stesso modo regolare di impacchettamento che caratterizza le sostanze con massa molecolare piccola, ma almeno in una direzione, quella dell'asse delle catene, si è trovato che raramente i cristalli si estendono al di là di alcune decine di nanometri. Nei materiali polimerici i cristalliti si formano di solito a temperature notevolmente inferiori a quella di fusione, in modo veloce anziché lento, e quindi in condizioni di notevole lontananza dall'equilibrio.
Una singola catena macromolecolare può far parte di più cristalli. Inoltre tutti i materiali polimerici cristallini contengono una significativa frazione (spesso dell'ordine del 5o% o più) di fase amorfa, e andrebbero più appropriatamente designati come ‛semicristallini'.
Differenze significative rispetto ai sistemi di molecole piccole si osservano anche nel caso di materiali polimerici completamente nello stato amorfo. Tali possono essere i polimeri incapaci di cristallizzare (come quasi tutti i polimeri vinilici non stereoregolari), oppure i polimeri capaci di cristallizzare, ma al di sopra della temperatura di fusione.
Per le molecole piccole, lo stato amorfo è normalmente uno stato fluido o ‛liquido' caratterizzato da scorrimento viscoso. I materiali polimerici amorfi possono essere fluidi, ma, a causa delle grandi dimensioni molecolari e dei conseguenti aggrovigliamenti tra molecole, tali fluidi hanno un'alta viscosità e un comportamento significativamente viscoelastico, con un ‛modulo elastico' anche a basse frequenze. Inoltre, piccoli cambiamenti nella struttura locale (tipicamente il cambiamento topologico associato alla formazione di legami a ponte) possono dar luogo a solidi, con il comportamento caratteristico delle gomme.
Al di sotto di una determinata temperatura (denominata ‛temperatura di transizione vetrosa', Tg) un materiale polimerico amorfo può subire una transizione a uno stato cosiddetto ‛vetroso': si tratta di una condizione di non equilibrio - che nel caso di sostanze non polimeriche è assai meno comune - in cui il materiale è caratterizzato da una rigidità analoga a quella del vetro.
Molti materiali polimerici amorfi vengono usati a temperatura ambiente nel loro stato vetroso (per es. il polistirene, il polimetilmetacrilato e numerosi polimeri reticolati).
Alcuni dei più interessanti materiali polimerici sono le leghe e i compositi.
Le leghe tra due polimeri possono essere miscele, nel senso termodinamico di soluzioni o fini dispersioni; tali miscele, monofasiche o eterofasiche che siano, possono mostrare proprietà migliori, per un determinato uso, rispetto a quelle dei componenti. Ad esempio, un materiale vetroso, e perciò fragile, può essere reso resistente all'urto attraverso l'inclusione di materiale gommoso, purché disperso sotto forma di particelle molto piccole.
Il termine ‛materiale composito' si applica a sistemi eterofasici, comprendenti due o più materiali. Anche in questo caso l'unione di due materiali (per es. polipropilene e fibre di vetro) è intesa a migliorare talune caratteristiche d'uso del polimero.
Nel seguito di questo capitolo verranno descritte, con un certo dettaglio, la morfologia e le proprietà fisiche che caratterizzano nel caso specifico dei materiali polimerici: a) lo stato amorfo (viscoso o vetroso); b) lo stato semicristallino e mesomorfo; c) le leghe e i sistemi eterofasici.
b) Lo stato amorfo (viscoso e vetroso).
Lo stato amorfo dei polimeri è caratterizzato dall'assenza di ordine tridimensionale a lunga distanza. Come detto, esso può riguardare l'intera massa di un materiale polimerico o solo una parte; quest'ultimo è il caso dei polimeri semicristallini, che presentano regioni di ordine cristallino, separate da dominî amorfi.
Ad alte temperature, quando le molecole o i segmenti molecolari mantengono una sufficiente mobilità relativa, un materiale polimerico amorfo può comportarsi come un fluido viscoso (o, meglio, viscoelastico), mentre al di sotto di una determinata temperatura (temperatura di transizione vetrosa, Tg) esso può assumere le caratteristiche di un vetro.
I polimeri amorfi trovano usi assai importanti e diffusi sia come elastomeri sia come materie plastiche; inoltre i polimeri cristallizzabili e usati in manufatti nello stato semicristallino vengono quasi sempre lavorati nello stato amorfo, al di sopra della temperatura di fusione.
Il modo di variare della viscosità con la temperatura, nel limite della velocità di scorrimento tendente a zero (il che garantisce, per semplificare l'illustrazione del fenomeno, un comportamento newtoniano del fluido), è mostrato nella fig. 4 per vari polimeri. La pendenza delle rette che esprimono il logaritmo della viscosità in funzione dell'inverso della temperatura varia in funzione della struttura chimica e della conseguente flessibilità della catena.
Nella fig. 5 è riportato l'andamento del logaritmo della viscosità (sempre per velocità di scorrimento tendente a zero) in funzione del logaritmo della massa molecolare media per polimeri con catena flessibile; il cambiamento di pendenza della curva si deve collegare con l'instaurarsi di aggrovigliamenti di catene, quando la lunghezza delle stesse supera un certo valore critico (M-W > M-WC). L'inibizione al movimento delle catene che ne consegue è locale, nel senso che i movimenti laterali rispetto al contorno della catena sono molto più fortemente inibiti di quanto non lo siano i movimenti lungo il contorno. Da questi ultimi movimenti consegue una residua capacità di movimento delle macromolecole, movimento denominato ‛reptazione', perché in esso è stata vista un'analogia con il modo di muoversi dei rettili.
Nelle applicazioni bisogna bilanciare il decadimento delle caratteristiche di lavorabilità che si ha nel caso di alte viscosità, cioè di alte resistenze al fluire del polimero a caldo (decadimento che peraltro si verifica sempre in regime di comportamento non newtoniano) con le proprietà meccaniche dello stato solido: la lavorabilità migliora infatti al diminuire della massa molecolare media delle macromolecole, mentre decadono le proprietà meccaniche.
Le proprietà caratteristiche che le sostanze macromolecolari possiedono allo stato amorfo possono essere spiegate con la conformazione raggomitolata delle catene, estremamente lunghe in rapporto alle dimensioni trasversali. Recenti studi hanno mostrato la sostanziale analogia tra la conformazione e l'ingombro complessivo delle macromolecole nello stato amorfo e in soluzione diluita (alla cosiddetta temperatura Θ), confermando l'assenza di ogni ordine a lunga distanza. La distanza media tra i terminali delle catene per una data massa molecolare della catena è un indice del suo grado di raggomitolamento: sembra che tale distanza si mantenga invariata anche nel passaggio di un materiale polimerico dallo stato amorfo (fuso) allo stato semicristallino.
La maggior parte dei polimeri privi di costituzione e/o di configurazione regolari risultano incapaci di cristallizzare e, al diminuire della temperatura, possono passare allo stato amorfo vetroso. La transizione allo stato vetroso è evidenziata dal cambiamento della pendenza delle curve secondo cui variano, in funzione della temperatura, certe grandezze termodinamiche (volume, energia o entropia).
A livello molecolare il passaggio dallo stato amorfo viscoso allo stato amorfo vetroso corrisponde al ‛congelamento' delle macromolecole, cioè a una brusca riduzione della loro mobilità.
Molti polimeri amorfi, sia lineari (ad es. polistirene, polimetilmetacrilato) sia reticolati, vengono usati a temperature al di sotto di Tg (che può essere anche sensibilmente più alta della temperatura ambiente) per la notevole rigidità dei manufatti risultanti. Si noti peraltro che un polimero amorfo opportunamente reticolato, se usato al di sopra di Tg, potrebbe avere invece il comportamento di una gomma.
c) Lo stato semicristallino.
Lo stato semicristallino implica un ordine tridimensionale a lunga distanza; tuttavia, nel caso dei polimeri, esso può estendersi al massimo fino ad alcune decine di nanometri lungo l'asse delle catene. Anche nelle direzioni trasversali l'ordine di grandezza delle dimensioni delle zone d'ordine cristallino è analogo, tranne che per i cristalli singoli ottenuti da soluzioni.
La caratteristica principale che regola la capacità di cristallizzare di un polimero è la microstruttura delle catene macromolecolari; per poter cristallizzare le macromolecole devono avere sia una costituzione sia una configurazione prevalentemente regolari. Un polipropilene a concatenamento regolare, per esempio, non è capace di cristallizzare, a meno che non sia isotattico o sindiotattico (v. fig. 2). Solo in rari casi eventuali imperfezioni strutturali non impediscono la cristallizzazione.
I materiali polimerici presentano morfologie diversificate a seconda delle condizioni di cristallizzazione.
Per cristallizzazione da soluzioni diluite si formano cristalli lamellari, con lati dell'ordine di alcuni micrometri e con spessori dell'ordine di 5-20 nm, tanto più grandi quanto più alta è la temperatura di cristallizzazione. Le molecole (molto più lunghe dello spessore) attraversano la lamella, si ripiegano più o meno irregolarmente, riattraversano la lamella da una faccia all'altra, si ripiegano ancora e così via per molte volte. Nelle ripiegature sono coinvolti molti più atomi concatenati di quelli strettamente necessari, talché sulle due facce delle lamelle si forma un cospicuo strato di materiale polimerico nello stato amorfo.
Nel raffreddamento dallo stato fuso la cristallizzazione avviene in un intervallo di temperatura, di solito parecchi gradi al di sotto della temperatura di fusione. Alla fine della cristallizzazione il materiale polimerico appare, di norma, al microscopio come un insieme composto di ‛sferuliti' di diametro generalmente compreso fra 1 e 100 μm. Si tratta di aggregati di fibrille lunghe e sottili che si irradiano da un centro evolvendo da precursori lamellari. Le fibrille sono a loro volta costituite da lamelle cristalline accartocciate, nelle quali le macromolecole sono orientate come nei cristalli cresciuti da soluzione. Lamelle vicine sono collegate da macromolecole che passano da una lamella all'altra ‛cementando' i cristalliti.
Nei cristalliti che si formano dal fuso il numero dei ripiegamenti delle catene è dunque inferiore che nei cristalliti che si formano da soluzioni diluite, ma in entrambi i casi le superfici estese delle lamelle risultano coperte di materiale amorfo. La frazione di materiale amorfo presente in un polimero semicristallino (pur con alte caratteristiche di regolarità costituzionale e configurazionale) si attesta normalmente intorno al 50%, anche se in taluni casi, dipendentemente dalla natura del polimero e dalla storia della cristallizzazione, può ridursi fino al 10% (ad es. nei cristalli singoli di polietilene, cresciuti da soluzioni).
I polimeri semicristallini possono essere lavorati in modo da ottenerne fibre o film orientati. Nelle regioni cristalline delle fibre le molecole a catena sono orientate preferenzialmente nella direzione dell'asse della fibra. La morfologia risultante è costituita da un insieme di cristalliti orientati, separati da regioni amorfe che possono presentare un notevole grado di orientamento molecolare, soprattutto per quanto riguarda le numerose catene di connessione tra i cristalliti. La periodicità con cui si alternano regioni cristalline e regioni amorfe nelle fibre è dell'ordine dei 10 nm. Morfologie analoghe possono riscontrasi nei film ‛stirati' in una sola direzione, mentre sono più complesse nei film prodotti con stiro biassiale.
L'alternarsi di strati amorfi e cristallini dà luogo a materiali con moduli elastici non grandi, dell'ordine di 10 GPa. Se però le fibre o i film vengono stirati in modo che i cristalliti risultino collegati da numerose catene (nelle zone amorfe) quasi completamente estese, si può arrivare a materiali fibrosi con moduli elastici assiali di oltre 100 GPa per il polietilene e di oltre 40 GPa per il polipropilene (per questi due polimeri i valori limite teorici calcolabili dalla struttura cristallina sono rispettivamente ~320 GPa e ~90 GPa).
Fibre ad altissimo modulo, di polietilene ma soprattutto di altri polimeri (come il Kevlar), sono prodotte industrialmente per sofisticate applicazioni.
Lo studio sperimentale della struttura cristallina dei polimeri, della loro morfologia e delle relazioni intercorrenti tra proprietà e struttura ha avuto un enorme sviluppo negli ultimi tre decenni.
Per quanto riguarda lo stato cristallino dei polimeri, numerosi studi hanno consentito di determinarne le conformazioni macromolecolari e di giustificarle sulla base di principi di strutturistica molecolare.
Per un polimero di data struttura chimica è possibile prevedere la struttura della catena allo stato cristallino attraverso una minimizzazione dell'energia interna, sotto il vincolo della ripetizione lungo un asse. Togliendo questo vincolo, è possibile altresì prevedere quale possa essere la distribuzione delle conformazioni sia in soluzione che allo stato fuso. Così, allo stato cristallino, la catena del polipropilene isotattico ha la conformazione di un'elica ternaria (v. fig. 6), mentre allo stato fuso la conformazione corrisponderebbe a una successione di piccoli tratti spiralizzati in senso alternatamente opposto.
Anche se non è possibile in questa sede entrare in maggiori dettagli sulla conformazione spaziale delle catene polimeriche, il cui studio ha ricevuto un impulso notevole dalla scoperta dei polimeri stereoregolari, e sulle implicazioni che l'organizzazione molecolare ha sulle proprietà fisiche, è interessante citare al riguardo due frasi significative della conferenza per il premio Nobel tenuta da P. J. Flory: ‟La comprensione delle relazioni spaziali tra gli atomi di una molecola è un prerequisito universale per connettere la formula grafica con le proprietà della sostanza così costituita. Se le marcate differenze fra le proprietà che distinguono la grande varietà di sostanze polimeriche, sia naturali sia sintetiche, devono essere razionalmente capite in termini fondamentali molecolari, questo deve essere il fulcro della futura ricerca".
d) Lo stato mesomorfo.
Alcune sostanze consistenti di molecole piccole possono dar luogo a uno stato liquido mesomorfo (‛cristalli liquidi'), in cui manca sia il disordine completo di un fluido normale sia l'ordine tridimensionale di un solido cristallino. Le molecole, dette ‛mesogeniche', hanno di solito un asse molecolare più lungo degli altri due e hanno la forma di una bacchetta; lo stato liquido mesomorfo è caratterizzato da una tendenza alla parallelizzazione tra loro degli assi molecolari più lunghi, per domini molto più grandi delle dimensioni molecolari.
Anche alcuni polimeri, le cui macromolecole hanno una catena rigida, come il Kevlar,
formano soluzioni liquido-cristalline; nel liquido vi sono domini ordinati degli assi molecolari.
Le soluzioni liquido-cristalline possono essere usate per la fabbricazione di fibre orientate con ottime proprietà meccaniche, quali un'elevata resistenza a rottura e un elevato modulo elastico, pur avendo una densità molto più bassa di quella dei materiali metallici.
Polimeri mesogenici, che danno fasi liquido-cristalline anche in assenza di solvente, si formano altresì da macromolecole con catena principale flessibile e con gruppi laterali lunghi e rigidi, oppure da macromolecole la cui catena principale è costituita da un alternarsi di gruppi rigidi (a bacchetta) e di gruppi o snodi più flessibili.
Applicazioni dello stato mesomorfo dei materiali polimerici nella tecnologia sono in corso di sviluppo.
e) Alcuni polimeri con proprietà fisiche speciali.
Può essere interessante passare in rassegna alcuni esempi di polimeri, in cui peculiari proprietà fisiche risultano connesse a una particolare struttura chimica.
Il politetrafluoroetilene (−CF2−CF2−)n è un polimero di altissima inerzia chimica, alto punto di fusione e coefficiente di frizione eccezionalmente basso.
Il polivinilidenfluoruro (−CH2−CF2−)n è un polimero che presenta un forte effetto piezoelettrico.
Il polidimetilsilossano (−O−Si(CH3)2−)n dà luogo a oli molto fluidi quando la massa molecolare è piccola; a massa molecolare alta dà luogo a materiali gommosi resilienti con la più bassa Tg (− 120 °C) nota.
Quasi tutti i polimeri - e tipicamente il polietilene - costituiscono dei buoni e talora eccellenti isolanti elettrici. È perciò interessante notare che in tempi recenti si è riusciti invece a ottenere conduttività dell'ordine di grandezza di quelle dei metalli con polimeri come il poliacetilene (−CH=CH−)n, attraverso l'incorporazione di ‛droganti' come I2 e SbF5.
Sono stati infine sviluppati numerosi polimeri con eccellenti proprietà ingegneristiche in estesi campi di temperatura. Uno di questi è il poli(etere-etere-chetone) (sigla PEEK), che ha la struttura:
Esso presenta una Tg = 143 °C e una temperatura di fusione Tf = 334 °C. Le sue proprietà meccaniche di resistenza, rigidità e tenacità risultano eccellenti anche a temperature elevate e si mantengono tali per tempi molto lunghi.
f) Le leghe e i sistemi eterofasici.
Di notevole interesse applicativo si sono rivelati sistemi polimerici microeterogenei, che consistono di due o più fasi con un grado di dispersione maggiore di quello molecolare (1 nm), ma minore di quello macroscopico (0,1 mm). La loro morfologia è determinata largamente da fattori cinetici. In questo paragrafo descriveremo in primo luogo alcuni casi particolarmente significativi: quello dei ‛copolimeri a blocchi' o ‛aggraffati' e quello degli ‛ionomeri'; completeremo poi il discorso con una breve discussione sulle leghe polimeriche.
Nei copolimeri a blocchi o aggraffati le macromolecole consistono di due o più segmenti costituiti da sequenze piuttosto lunghe di unità costituzionali ripetitive differenti. Se il copolimero è a blocchi, le diverse sequenze si succedono nella catena principale; nei copolimeri aggraffati, alla catena principale di un dato tipo sono ‛appese' una o più catene laterali con sequenze di un tipo differente. Se le sequenze sono lunghe e sufficientemente diverse dal punto di vista chimico, nel materiale polimerico si può avere separazione in microfasi, le cui dimensioni sono peraltro limitate - com'è intuibile - dalle dimensioni medie delle sequenze. Le microfasi danno luogo a svariate morfologie, dipendenti in parte dalle condizioni di ottenimento. In qualche caso si hanno separazioni tanto regolari da dar luogo a un macroreticolo evidenziabile, oltreché con la microscopia elettronica, anche con i caratteristici fenomeni di diffrazione dei raggi X o della luce.
Sistemi microeterogenei costituiti da copolimeri a blocchi amorfi hanno spesso le proprietà delle gomme termoplastiche. Ciò avviene nel campo di temperature al di sopra di Tg per i blocchi di un tipo (blocchi ‛molli') e al di sotto di Tg per i blocchi dell'altro (blocchi ‛duri'). Si consideri, ad esempio, un polimero a tre blocchi D-M-D, dove M rappresenta un blocco molle e D un blocco duro. A temperature superiori alla Tg dei blocchi D, l'intero materiale è allo stato amorfo viscoso e può dunque fluire ed essere lavorato; a temperature inferiori alla Tg dei blocchi D e superiori alla Tg dei blocchi M, solo questi ultimi restano allo stato amorfo viscoso e - se ne costituiscono la componente maggioritaria - il materiale si comporta come una gomma reticolata. Se invece la componente gommosa del copolimero è minoritaria, questo avrà le proprietà di un materiale polimerico vetroso, dotato però di elevata resistenza all'urto: le microfasi gommose disperse nel materiale hanno infatti una notevole capacità di fermare la propagazione di ‛cricche' (incrinature), che potrebbero portare a una rottura del manufatto.
Gli ionomeri sono tipi speciali di copolimeri, a blocchi o aggraffati, nei quali un componente ha gruppi ionizzabili (ad es. −COOH o −SO3H). I corrispondenti sali hanno una più elevata tenacità e bassa distorsione al calore. Ciò è associato - dal punto di vista strutturale - alla separazione di microfasi a carattere ionico. Può essere interessante notare come, nel caso di ionomeri che si possono considerare derivati da un polimero cristallino come il polietilene (per esempio il polimero che si ottiene copolimerizzando l'etilene con acido metacrilico), la ionizzazione dia luogo a una sostanziale diminuzione di cristallinità e di velocità di cristallizzazione; la formazione di sferuliti è soppressa e questo porta tra l'altro a una migliore trasparenza dei prodotti.
Le leghe sono miscele intime di due o più polimeri differenti. Esse si dicono compatibili se danno luogo a sistemi monofasici in cui i componenti risultano dispersi a livello molecolare. Tuttavia, a causa della bassa entropia di mescolamento tra macromolecole, le leghe compatibili rappresentano piuttosto un'eccezione che la regola; per quanto si cerchi di mescolare il più intimamente possibile due materiali polimerici diversi, il risultato sarà in generale una miscela microeterogenea.
Per le leghe polimeriche le relazioni tra composizione e proprietà dipendono molto dalla morfologia. La rottura dei manufatti è spesso localizzata sulle superfici di separa zione tra le fasi; le caratteristiche meccaniche sono perciò fortemente dipendenti dal grado di adesione reciproca tra queste. L'adesione può essere incrementata attraverso l'aggiunta di opportuni copolimeri a blocchi, in cui i due diversi tipi di segmenti siano compatibili ciascuno con un diverso componente della lega. Un polimero rigido, quindi con alto modulo o alta Tg, disperso in una matrice polimerica più molle, aumenta la rigidità complessiva della lega polimerica risultante.
Dispersioni di polimeri con bassa Tg (a comportamento gommoso) in matrici con alta Tg (a comportamento vetroso) danno luogo a un miglioramento della resistenza all'urto.
Anche in questo caso la coerenza tra le fasi può essere promossa, come accennato in precedenza, attraverso l'aggraffaggio o l'aggiunta di opportune quantità di copolimeri a blocchi.
È opportuno infine notare che in talune applicazioni vengono usate miscele di polimeri gommosi con polimeri cristallini (per es. copolimeri etilene-propilene con polipropilene isotattico) per migliorarne la resistenza all urto. In questo caso la coerenza tra le diverse microfasi può essere aumentata dalla cristallizzazione di segmenti del copolimero con la matrice.
4. Classi di polimeri in relazione alla lavorazio ne e all'uso.
a) Termoplastici.
I termoplastici sono polimeri la cui forma può essere cambiata ripetutamente per deformazione meccanica dopo riscaldamento. Essi sono costituiti da catene lineari e/o ramificate, non reticolate.
Le deformazioni macroscopiche provocano un riarrangiamento molecolare senza apprezzabili alterazioni dei legami chimici. Al di sopra del punto di fusione cristallino e della temperatura di transizione vetrosa i termoplastici so no fusi viscosi aventi comportamento viscoelastico a causa dei lunghi tempi di rilassamento associati alla riorganizzazione molecolare. Per la maggior parte dei polimeri semicristallini esiste un intervallo di temperatura in cui le regioni amorfe sono sopra la loro temperatura di transizione vetrosa, mentre le regioni cristalline sono sotto il loro punto di fusione. Tale intervallo di temperatura può essere ampio e comprendere anche la temperatura di uso di molti materiali.
Alla classe dei termoplastici appartengono i polimeri di maggiore rilevanza commerciale, come il polietilene, il polipropilene, il cloruro di polivinile e il polistirene, che coprono una vastissima gamma di applicazioni; i tecnopolimeri, quali le poliammidi, i policarbonati, i poliacetali, i poliacrilati, caratterizzati da proprietà meccaniche molto buone e adatti quindi a impieghi ingegneristici in alternativa a materiali tradizionali; i polimeri speciali, quali i fluorurati, adatti per impieghi particolari.
b) Termoindurenti.
I termoindurenti rappresentano un'ampia classe di materiali polimerici costituiti essenzialmente da reticoli molecolari tridimensionali derivanti da reazioni di polimerizzazione fra opportune specie reattive di massa molecolare bassa. Essi sono pertanto insolubili e infusibili.
I termoindurenti trovano applicazioni molto diversificate, sia in sostituzione di termoplastici che come adesivi, vernici e matrici per materiali compositi. Le resine sviluppate in questi ultimi anni presentano delle velocità di reticolazione molto elevate accoppiate a migliori caratteristiche alle alte temperature e sotto sforzo. Ciò ha permesso un rapido sviluppo di questi materiali anche nell'industria automobilistica.
Nella classe dei termoindurenti sono compresi i poliuretani e il policaprolattame, trasformati con la tecnologia di iniezione con reazione (Reaction Jnjection Molding, RIM), e la grande famiglia delle resine epossidiche e poliestere. Tra i polimeri reticolati stanno inoltre acquistando interesse i cosiddetti ‛sistemi reticolati interpenetrati' (reticoli di una composizione, dispersi nel reticolo di una matrice polimerica di composizione diversa).
c) Elastomeri e gomme.
La reversibilità di deformazioni anche cospicue è la proprietà che caratterizza gomme ed elastomeri; essa è ottenuta tramite reticolazione fisica o chimica di macromolecole lineari.
In questa classe di materiali ricadono tutti i termoindurenti la cui temperatura di esercizio è al di sopra della loro temperatura di transizione vetrosa.
Partendo dalla gomma naturale e da quella stirenebutadiene, la resistenza ai solventi e alla temperatura è stata fortemente aumentata introducendo sostituenti inorganici, come ad esempio il cloro (gomme neopreniche) o l'azoto (copolimeri butadiene-acrilonitrile) o il fluoro (gomme al fluoruro di vinilidene), in polimeri con catene principali di atomi di carbonio, oppure il silicio in polimeri con catena principale costituita da silicio e ossigeno (polisilossani).
d) Fibre.
Potenzialmente tutte le macromolecole sintetiche possono essere orientate in uno stato fibrillare, che è fortemente anisotropo. In pratica però solo alcune di esse riescono ad avere delle proprietà tali da renderle utilizzabili industrialmente. Basti pensare al poliestere o al nailon, utilizzati in sostituzione delle fibre naturali, o alle fibre arammidiche, utilizzate come rinforzo sia di gomme, nei pneumatici, che di resine epossidiche, nei compositi avanzati. Le tecnologie di produzione di queste fibre sintetiche sono ormai così sofisticate da permettere la produzione di filamenti con rigidità e resistenza vicine ai limiti teorici del legame covalente. Inoltre, partendo da soluzioni polimeriche con comportamento liquido-cristallino è possibile ottenere fibre con altissima stabilità termica e ottime proprietà meccaniche.
e) Vernici, rivestimenti e adesivi.
Quasi tutti i materiali sono soggetti a degradazione o ad attacco chimico, a causa di fattori ambientali quali l'acqua, il sale, i gas industriali, la luce ultravioletta, l'ossigeno, l'ozono, ecc., e pertanto è spesso indispensabile proteggerli per aumentarne la durata in servizio. I rivestimenti polimerici svolgono un ruolo importantissimo al riguardo e riescono a raggiungere lo scopo in maniera eccellente nei settori più svariati, che vanno dall'elettronica alle costruzioni in ferro. In questi ultimi anni si sono poi sviluppati nuovi tipi di rivestimenti e vernici: alcuni hanno un più alto contenuto di solidi, altri utilizzano l'acqua come fluido sospendente, altri ancora si presentano in forma di polveri senza solvente, che vengono depositate elettrostaticamente sulla superficie da proteggere.
Anche nel caso degli adesivi i materiali e la tecnologia esistenti al giorno d'oggi ne permettono l'utilizzazione nei campi più sofisticati della tecnica. Basti pensare che molte strutture aeronautiche sono incollate con sistemi adesivi sviluppati ad hoc.
f) Film e membrane.
Molti film polimerici sono utilizzati come contenitori nell'industria alimentare e in altre applicazioni; generalmente hanno spessori molto bassi (anche minori di 10 μm) e sono orientati mono- o biassialmente, per aumentare la tenacità. Poiché devono possedere numerose proprietà, che vanno dall'inerzia chimica all'inchiostrabilità, dall'impermeabilità a certi gas e permeabilità ad altri a buone caratteristiche meccaniche, che difficilmente un solo materiale riesce ad avere, spesso vengono utilizzati film multistrato, ottenuti con tecnologie molto avanzate che ne permettono anche la produzione a costi contenuti. Inoltre membrane polimeriche con proprietà di permeabilità selettiva sia per liquidi che per gas sono prodotte in quantità sempre crescente in conseguenza del loro utilizzo in processi chimici, biochimici e per il trattamento delle acque e dei gas di scarico.
g) Compositi.
Quando due o più materiali sono mescolati assieme, il materiale ‛composito' risultante ha molto spesso proprietà fisiche che sono considerevolmente diverse dalle proprietà dei singoli costituenti.
In questi ultimi anni si è avuta una forte richiesta di materiali strutturali capaci di accoppiare ad alti valori della resistenza a rottura e della rigidità per unità di peso un'elevata resistenza a fatica. Come riportato precedentemente nella tabella, questa richiesta è ampiamente soddisfatta dai materiali compositi fibrosi. Infatti le fibre, poiché sono molto più resistenti degli ordinari materiali polimerici, normalmente aumentano la resistenza di qualsiasi matrice in cui sono incluse. Quando le fibre sono tutte allineate in una direzione, si ottiene il massimo valore della resistenza nella direzione delle fibre. Sistemandole opportunamente, è possibile aumentare la resistenza del composito in direzioni particolari e quindi soddisfare i requisiti di progetto di una struttura soggetta a sforzi multiassiali. È precisamente questa possibilità di ‛progettare' un materiale capace di soddisfare complessi requisiti, accoppiata all'aumento delle proprietà specifiche, che rende i materiali compositi così interessanti.
A parte i compositi di materiali polimerici con fibre continue, generalmente indicati come ‛compositi ad alta prestazione', un grosso sviluppo hanno avuto anche i compositi di materiali polimerici con fibre corte o con particelle. Infatti questi ultimi, anche se non possono competere con i precedenti per le prestazioni meccaniche, sono senz'altro molto meno costosi e presentano proprietà meccaniche decisamente superiori a quelle dei polimeri non rinforzati.
La differenza fra i costi di queste due classi di materiali è principalmente dovuta alle differenti tecnologie utilizzate per la loro produzione. Infatti nel primo caso i prodotti finiti vengono ottenuti per laminazione di strati orientati, e ciò comporta una forte incidenza della manodopera e una bassa produttività, mentre i secondi sono generalmente trasformati con le stesse tecnologie sviluppate per i polimeri non rinforzati, tecnologie altamente automatizzate e che consentono alte velocità di produzione.
5. Tecnologie di lavorazione dei materiali polimerici.
a) Introduzione.
Raramente un polimero è impiegato tal quale come esce dall'impianto di sintesi. Generalmente la trasformazione definitiva a manufatto avviene attraverso una sequenza più o meno complessa di operazioni di formulazione, additivazione, miscelazione e finalmente stampaggio, durante le quali il polimero è in genere allo stato fuso. È pertanto evidente che le proprietà finali di un manufatto di materiale plastico non solo dipendono dalla sua struttura chimica, ma sono anche fortemente condizionate dal processo di lavorazione, e ciò soprattutto per i sistemi polimerici multicomponenti.
In questi ultimi anni un crescente interesse è stato rivolto alla possibilità di ottimizzare le proprietà di determinati manufatti tramite orientazioni indotte dalla specifica tecnologia di lavorazione utilizzata (orientazioni molecolari e morfologia dei cristalli).
Le tecnologie principalmente usate per lavorare i materiali polimerici sono: l'estrusione, lo stampaggio, la filatura di fibre, il soffiaggio di film.
b) Estrusione.
L'estrusione è senz'altro la più importante fase di lavorazione dei termoplastici. Essa consiste nel fondere e spingere il materiale attraverso un ugello di forma opportuna, dove il materiale viene formato. Contemporaneamente vengono effettuate anche altre operazioni, come la miscelazione, l'omogeneizzazione, la devolatilizzazione ed eventualmente il mescolamento con cariche inerti o rinforzi fibrosi.
Nel processo di estrusione con viti il polimero in granuli viene immesso in una tramoggia, da cui cade su una vite che, ruotando, lo trasporta attraverso un tubo riscaldato (v. fig. 7). Il materiale riscaldato fonde e, in conseguenza della speciale forma della vite (v. fig. 8), sul polimero fuso viene esercitata un'elevata pressione che spinge il materiale attraverso un ugello avente la forma desiderata.
Molto lavoro è stato fatto per ottimizzare la forma della vite, che varia da polimero a polimero, ma ancora oggi non esistono validi criteri di progettazione del profilo della vite, specialmente nella zona di carico, dove il materiale è ancora freddo.
Oltre il 90% dei processi di estrusione vengono effettuati con estrusori a vite; il rimanente 10% è effettuato con estrusori a pistoni. Questo processo consiste nel riscaldare il polimero in un contenitore chiuso e quindi spingerlo attraverso un ugello con un pistone. Questo tipo di estrusore è sempre più utilizzato per lavorare compositi rinforzati con fibre corte.
c) Stampaggio.
La trasformazione del granulo di resina base in oggetto finito avviene con vari sistemi, a seconda del materiale e della forma dell'oggetto. Lo stampaggio a compressione (v. fig. 9) consiste nel sistemare il materiale, a volte preformato, in uno stampo cavo riscaldato, chiudere lo stampo e spingere il polimero a fluire e ad assumere la forma voluta, stringendo le due parti dello stampo con una pressa.
Lo stampaggio a compressione richiede cicli produttivi lenti, economicamente vantaggiosi solo nel caso dei termoindurenti, che non richiedono il raffreddamento dello stampo fra un ciclo e l'altro. Lo stampaggio a iniezione, invece, realizza cicli di stampaggio più rapidi, ma utilizza anche macchinari più sofisticati di quelli impiegati nelle tecnologie a compressione. Infatti in questo caso il materiale è riscaldato in un cilindro, iniettato in uno stampo chiuso raffreddato, in cui assume la forma finale, e quindi espulso dallo stampo, che a questo punto si apre (v. fig. 10). Questa tecnologia è usata principalmente per termoplastici. Lo stampaggio a iniezione con reazione (RIM) implica la rapida miscelazione del prepolimero liquido con l'agente reticolante e quindi l'iniezione della miscela nello stampo dove si completa la reazione di reticolazione. I vantaggi di questo processo sono: l'eliminazione del riscaldamento e della fusione prima della fase di iniezione e, data la bassa viscosità del prepolimero, la possibilità di operare con basse pressioni e quindi di risparmiare energia. In una versione modificata di questa tecnologia (Reinforced Reaction Injection Molding, RRIM), lo stampo è preventivamente riempito di fibre di vetro lunghe e successivamente riempito con resine reattive. In questo modo è possibile ottenere oggetti in composito a costi economicamente vantaggiosi.
d) Filatura di fibre.
Le fibre sintetiche sono prodotte con tre diversi procedimenti di filatura: a umido, a secco, allo stato fuso. Nel primo, una soluzione contenente il polimero è forzata sotto pressione attraverso una filiera in un liquido non solvente che fa precipitare la fibra. Nel secondo, la soluzione è forzata attraverso la filiera in una camera riscaldata, dove il solvente è allontanato per evaporazione. Nel terzo, infine, la fibra è ottenuta per estrusione del fuso attraverso una filiera a luci molto sottili e quindi raffreddata ad aria.
e) Soffiaggio di film.
La maggior parte dei film plastici è prodotta per soffiaggio interno di tubi estrusi a pareti sottili. Il polimero fuso è spinto con un estrusore attraverso un ugello a sezione anulare in modo da formare un tubo a pareti sottili. Questo tubo è tirato longitudinalmente e contemporaneamente gonfiato con aria pressurizzata (v. fig. 11). L'aumento dell'area superficiale e il raffreddamento derivante dall'aria soffiata determinano la solidificazione del film, che viene pertanto raccolto e tagliato nelle dimensioni desiderate.
Variando opportunamente le condizioni di soffiaggio è possibile produrre film biorientati o con proprietà speciali; utilizzando più estrusori che alimentino diversi polimeri alla testa di soffiaggio è possibile ottenere film multistrato coestrusi.
f) Altre tecnologie.
Oltre alle tecnologie precedentemente decritte, ne esistono molte altre che sono state sviluppate o specificamente studiate per un manufatto o per un materiale. Tra di esse ricordiamo, per i compositi, la laminazione, la poltrusione, l'avvolgimento filamentare, la centrifugazione; per i polimeri la formatura rotazionale, il soffiaggio, la calandratura, la formatura sotto vuoto, ecc.
In particolare, la laminazione consiste nel predisporre lamine di fibre preimpregnate con resine termoindurenti non ancora reticolate, secondo le direzioni previste dal progettista su di una forma. Le lamine vengono coperte da un sacco flessibile in cui viene fatto il vuoto per eliminare l'aria inglobata. Il tutto viene quindi introdotto in un'autoclave dove la pressione e la temperatura sono programmate in modo da ottenere la completa reticolazione delle resine e l'eliminazione della resina in eccesso.
La poltrusione è un metodo di produzione in continuo simile all'estrusione. In particolare, le fibre vengono tirate attraverso un bagno di resina e quindi attraverso una forma riscaldata. Il profilato così ottenuto, dopo essere passato eventualmente attraverso un forno per completare la reticolazione, viene tagliato alla lunghezza desiderata (v. fig. 12).
Nella tecnologia dell'avvolgimento filamentare (v. fig. 13) le fibre, dopo essere state bagnate nella resina, sono avvolte su un mandrino rotante a velocità prefissata. I due diversi moti, di rotazione del mandrino e di traslazione del distributore di fibre, permettono di disporre le fibre in maniera programmata per soddisfare le più varie esigenze di progettazione.
La centrifugazione è una tecnologia usata per ottenere tubi in plastica rinforzata con fibre di vetro di grossi spessori. In questo caso la resina contenente fibre corte e sabbia viene deposta all'interno di una forma cilindrica rotante ad alta velocità e riscaldata alla temperatura di reazione.
La formatura rotazionale (v. fig. 14) e il soffiaggio sono due diverse tecnologie usate per produrre oggetti di plastica cavi. Nella prima la polvere di polimero è sistemata in una metà della forma metallica. Quindi le due metà sono fissate insieme e riscaldate in un forno. Durante questo stadio di riscaldamento la forma ruota attorno a due assi ortogonali tra di loro. Ciò determina la distribuzione uniforme della polvere polimerica sulle pareti della forma, dove fonde. L'oggetto formato viene successivamente raffreddato ed estratto dalla forma. Questa tecnologia, anche se un po' lenta, offre la possibilità di ottenere oggetti cavi di grosse dimensioni e con spessori elevati e molto uniformi, a costi relativamente bassi. Il soffiaggio, invece, consiste in due stadi. Nel primo un tubo di plastica chiamato parison viene estruso per mezzo di un estrusore. Quindi una forma blocca il parison ancora caldo e aria in pressione viene inviata all'interno del tubo, che si deforma assumendo la forma voluta (v. fig. 15).
La calandratura è un metodo per produrre film e lastre di plastica e consiste nel ridurre lo spessore di lastre preformate costringendole a passare attraverso un'intercapedine ricavata tra due rulli riscaldati ruotanti in senso inverso; è un procedimento utilizzato specialmente per polimeri poco resistenti alle alte temperature (v. fig. 16).
Infine, nel processo di formatura sotto vuoto, una lastra di materiale termoplastico viene riscaldata mediante lampade a raggi infrarossi e termoformata mediante riduzione della pressione dell'aria interposta fra essa e la forma (v. fig. 17).
6. Applicazioni.
a) Generazione, distribuzione e conservazione dell'energia.
Gli attuali impianti generatori di energia elettrica già utilizzano in grandi quantità i materiali polimerici. Infatti le eccellenti proprietà elettriche e meccaniche e la facilità di fabbricazione di questi materiali assicurano loro un ruolo centrale nella costruzione di apparecchiature elettriche. In particolare, i generatori tradizionali richiedono isolanti con elevate proprietà dielettriche, che solo alcune resine epossidiche modificate posseggono.
I cavi per la distribuzione di energia sono isolati con polimeri di alta qualità (polietileni reticolati), che risultano molto affidabili nel tempo sia dal punto di vista meccanico sia da quello elettrico. La costruzione di trasformatori è un'altra area in cui la versatilità della tecnologia di lavorazione dei polimeri è estremamente utile. In questo caso la reticolazione di speciali formulazioni di resine epossidiche deve essere ottenuta con un basso ritiro ed evitando la formazione di bolle, che potrebbero dar luogo a fenomeni di arborescenza, scariche parziali e fratture. Le celle elettrochimiche (batterie) utilizzano piatti separatori e contenitori costituiti da polimeri; le celle a combustibile sono in genere contenute in recipienti polimerici e fanno uso di membrane polimeriche.
I sistemi di sfruttamento dell'energia solare utilizzano in larga misura componenti polimerici sia per i vantaggi di fabbricazione che per la loro stabilità nel tempo e la loro leggerezza. Infine stanno emergendo nuovi polimeri, che presentano alta conduttività elettrica e che, anche se è prematuro prevederne applicazioni specifiche, sembra possano trovare degli spazi interessanti nelle tecnologie avanzate.
Anche i polimeri piezoelettrici e piroelettrici, attualmente utilizzati come trasduttori e sensori termici, suscitano un crescente interesse in vista di applicazioni legate alla trasformazione dell'energia.
b) I materiali polimerici nell'industria dei trasporti.
Il continuo aumento del costo del carburante ha profondamente inciso sulla fabbricazione dei mezzi di trasporto, specialmente per quel che riguarda i materiali utilizzati. Infatti, la riduzione del peso delle autovetture mediante la sostituzione di materiali metallici con materiali più leggeri, come le plastiche e i compositi a matrice polimerica, è diventata uno degli obiettivi principali delle strategie di innovazione attuate dai costruttori d'auto. Basti ricordare che la General Motors ha recentemente sostituito balestre metalliche del peso di circa 20 kg l'una con balestre in composito pesanti circa 3,5 kg, con conseguente risparmio di peso netto di 16,5 kg a balestra.
Nell'industria aeronautica la riduzione del peso delle strutture risulta ancor più vantaggiosa. Si può calcolare che in questo caso, per ogni chilogrammo di peso risparmiato, si ha un calo netto dei costi d'esercizio dell'aeromobile di circa 200 dollari per kg e per anno, e pertanto questa industria può permettersi di utilizzare in modo massiccio compositi avanzati, anche se i loro costi sono ancora molto sostenuti (v. fig. 1). Ad esempio, l'intera struttura del Lear Fan Jet è stata costruita in composito, limitando così il peso totale dell'aeromobile (motori esclusi) a circa 600 kg.
Come già accennato precedentemente, molte società costruttrici di aeroplani prevedono un uso esteso di compositi con fibre di grafite e poliarammidiche nella prossima generazione di aeromobili commerciali.
c) I materiali polimerici nell'industria edile.
I materiali polimerici sono usati nel settore delle costruzioni civili per fabbricare finestre, porte, tubazioni, isolanti termici ed elettrici, coperture, componenti dei servizi igienici, rivestimenti, pavimenti, pareti di separazione, ecc. È stato notato che, se l'industria delle costruzioni continuasse a usare i materiali polimerici allo stesso tasso di incremento che ha caratterizzato il mercato in questi anni, essi diventerebbero i materiali da costruzione più usati nel XXI secolo. I tipi di polimeri utilizzati in questo settore sono i più vari e vanno dalle resine fenoliche, ureiche e melamminiche, per i laminati plastici, alle resine acriliche e ai policarbonati, per pannelli trasparenti in sostituzione del vetro; dalle schiume poliuretaniche e polistireniche, per l'isolamento, ai copolimeri acrilonitrile-butadiene-stirene (ABS), al cloruro di polivinile (PVC), al polietilene e ai poliesteri per le tubazioni e così via.
Per quel che riguarda questi materiali, molti dei problemi che in passato ne avevano limitato l'utilizzazione come materiali da costruzione sono stati superati o fortemente ridotti. In particolare, la resistenza agli agenti esterni di alcuni di essi può oggi essere considerata eccellente e la loro resistenza alla fiamma è stata notevolmente aumentata grazie ad additivi e rivestimenti protettivi.
d) I materiali polimerici nell'agricoltura e nell'industria alimentare.
Una delle più interessanti applicazioni dei polimeri nell'agricoltura riguarda la tecnologia del rilascio controllato di pesticidi e agenti stimolatori della crescita. In questo caso i polimeri fissano l'agente attivo e lo rilasciano in maniera continua, a velocità e concentrazione prefissate, per il tempo desiderato. Ciò è possibile sia mediante la degradazione del polimero, sia mediante il rilascio per diffusione della specie attiva attraverso il film polimerico che la contiene.
Inoltre, va sempre più sviluppandosi, specialmente nelle zone molto fredde, l'uso di rivestimenti polimerici per semi. Questa tecnica permette una migliore stabilità e germinazione dei semi, la possibilità di incorporare, durante l'incapsulamento, sostanze nutrienti e agenti protettivi per il seme e una maggiore facilità di semina.
Inoltre, film e tubazioni in plastica sono ampiamente utilizzati in agricoltura già da molti anni. Recentemente film plastici con proprietà ottiche particolari sono stati utilizzati in serricultura per l'accrescimento selettivo di specie vegetali.
Anche nell'industria alimentare l'utilizzazione dei materiali plastici è molto sviluppata: basta pensare all'impacchettamento di cibi sia solidi sia liquidi.
e) I materiali polimerici in medicina.
In medicina l'uso dei materiali polimerici ha avuto, negli ultimi anni, uno sviluppo eccezionale: il medico e il chirurgo non possono più fare a meno di oggetti di plastica come i tubi endotracheali per l'anestesia, i contenitori di sangue, i tubi per le trasfusioni, le siringhe per le iniezioni, i guanti chirurgici, i bendaggi, i cateteri, ecc.
I requisiti richiesti a un materiale per la sua utilizzazione in medicina sono molteplici e vanno dalle adeguate proprietà meccaniche, chimiche e fisiche alla resistenza all'ambiente in cui sono inseriti. I materiali più utilizzati sono i siliconi, gli acrilati, gli uretani, i polimeri fluorurati, il polietilene, il polipropilene e molti altri. Nel campo delle protesi e degli organi artificiali i materiali polimerici vengono impiegati per fabbricare lenti a contatto, morbide o rigide, ossigenatori di sangue, macchine per la dialisi, protesi ortopediche e odontointriche e cuori artificiali. In tali applicazioni è fondamentale il requisito della biocompatibilità.
f) Considerazioni conclusive.
I materiali polimerici sintetici e i compositi nella cui formulazione essi intervengono - vengono usati in quantità sempre maggiori al posto di prodotti naturali o di materiali metallici e ceramici, ottenendo spesso migliori prestazioni.
Attualmente, per la maggiore consapevolezza dei limiti delle risorse e nell'intento di conferire un maggior valore aggiunto alla produzione, l'attenzione dei produttori di materiali polimerici si sta spostando dall'obiettivo della quantità a quello della qualità e dell'idoneità del prodotto a usi specifici.
Lo studio delle interrelazioni tra proprietà, struttura e tecnologie di lavorazione - che si può considerare il fulcro della scienza dei materiali polimerici - non può non acquisire un ruolo centrale nella realizzazione di nuovi materiali e nel processo di miglioramento della qualità e dell'idoneità di prodotti tradizionali, destinati a un impiego sempre più sofisticato e diffuso in settori di importanza cruciale, come l'industria dei mezzi di trasporto, l'edilizia, l'agricoltura, la medicina.
Bibliografia.
Bever, M. B. (a cura di), Encyclopedia of materials science and engineering, Oxford 1985.
Billmeyer, F. W., Text-book of polymer science, New York 1984.
Birley, A. W., Scott, M. J. (a cura di), Plastic materials: properties and applications, New York 1982.
Blumstein, A. (a cura di), Liquid crystalline order in polymers, New York 1978.
Boor, J. Jr., Ziegler-Natta catalysts and polymerizations, New York 1979.
Bovey, F. A., Winslow, F. H., Macromolecules, New York 1982.
Brandrup, J., Immergut, E. H. (a cura di), Polymer handbook, New York 19752.
Carver, C. D. (a cura di), Polymer characterization: spectroscopic, chromatrographic and physical instrumental methods, in Advances in chemistry series, vol. CCIII, Washington 1983.
Ciardelli, F. (a cura di), Macromolecole, scienza e tecnologia, 2 voll., Pisa 1983.
Crawford, R. J., Plastics engineering, in Progress in polymer science, vol. VII, Oxford 1981.
Elias, H. G., Macromolecules, 2 voll., New York 1984.
Encyclopedia of polymer science and engineering, 19 voll., New York 19852.
Flory, P. J., Principles of polymer chemistry, Ithaca, N. Y., 1953.
Gjachter, R., Müller, H., Platics adhesives handbook, München 1984.
Harper, C. A. (a cura di), Handbook of plastics and elastomers, New York 1975.
Hiemenz, C., Polymer chemistry: the basic concepts, New York 1984.
Kaufman, H. S., Falcetta, J. J., Introduction to polymer science and technology, New York 1977.
Kelly, A., Rabotnov, Y. N., Handbook of composites, Amsterdam 1985.
Kronenthal, R. L., Oser, Z., Martin, E., Polymers in medicine and surgery, New York 1975.
Mark, J. E., Eisenberg, A., Graessley, W. W., Mandelkern, L., Koenig, J. L., Physical properties of polymers, Washington1984.
Mascia, L., Thermoplastics: materials engineering, Essex 1982.
Middleman, S., Fundamentals of polymer processing, New York 1977.
Scheldom, R. P., Composite polymeric materials, Amsterdam 1982.
Schildnecht, C. E., Skeist, I., Polymerization processes, New York 1977.
Sechting, A., International plastics handbook, München 1983.
Szycher, M., Robinson, W J., Synthetic biomedical polymers: concepts and applications, Westport, Conn., 1980.
Tadmor, Z., Gogos, C. G., Principles of polymer processing, New York-Toronto 1979.