
Metabolismo
Metabolismo
Regolazione del metabolismo, di Eric A. Newsholme e Bernard Crabtree
Metabolismo dei carboidrati, di Bernard Axelrod
Metabolismo dei lipidi, di Konrad Bloch
Regolazione del metabolismo
SOMMARIO: 1. Introduzione. □ 2. Aspetti teorici della regolazione metabolica: a) sistemi chiusi e aperti; b) criterio termodinamico e criterio cinetico nello studio del controllo metabolico; c) generazione del flusso in sistemi a stato stazionario; d) regolazione del flusso in sistemi a stato stazionario; e) reazioni reversibili e irreversibili e loro significato nel metabolismo; f) la glicolisi anaerobica nel muscolo. □ 3. Meccanismi fisiologici di regolazione dell'attività enzimatica: a) controllo dell'attività enzimatica mediante grandi variazioni nella concentrazione del regolatore; b) aumento di sensibilità prodotto da più di un regolatore; c) aumento di sensibilità prodotto da cinetiche enzimatiche non iperboliche; d) aumento di sensibilità prodotto da reintroduzione ciclica del substrato; e) aumento di sensibilità prodotto dalla presenza di forme interconvertibili di un enzima. □ Bibliografia.
1. Introduzione.
I progressi fatti durante i secc. XVIII e XIX nella comprensione dei principi delle modificazioni chimiche portarono alcuni scienziati (in particolare A.-L. Lavoisier, L. Pasteur ed E. Buchner) ad applicare questi stessi principi ai sistemi biologici, sottraendo in questo modo tali processi alla sfera del misticismo e iniziando così la scienza della biochimica. Fu, per esempio, assodato che la produzione di etanolo da parte delle cellule di lievito poteva essere descritta dalla seguente equazione:
glucosio→2 etanolo+2CO2.
Analogamente si scoprì che l'ossidazione completa del glucosio negli organismi viventi era descritta dall'equazione
glucosio+6O2→6CO2+6H2O.
Si stabilì inoltre che lo svolgimento di questi processi ‛metabolici' era intimamente correlato con il fenomeno della catalisi. Le reazioni chimiche che costituiscono una via metabolica sono catalizzate ciascuna da catalizzatori biologici specifici noti col nome di enzimi. Ricerche condotte nella prima metà di questo secolo chiarirono sia la natura chimica di queste reazioni sia la sequenza in cui esse sono organizzate in una data via metabolica. I risultati di queste ricerche sono raccolti nei testi di biochimica sotto la denominazione di ‛vie metaboliche'. Queste vie, le rappresentazioni grafiche delle quali danno spesso l'impressione di complicati schemi di impianti elettrici, possono essere considerate l'‛anatomia' del metabolismo, in quanto forniscono un'indicazione delle possibili direzioni che un particolare substrato può seguire. Tuttavia esse non danno alcuna informazione sulla velocità delle singole reazioni e sulla modalità con cui queste velocità sono in relazione fra di loro e vengono regolate. Nondimeno anche i primi ricercatori erano a conoscenza del fatto che la velocità di almeno alcune vie metaboliche era sottoposta a controllo all'interno delle cellule viventi. Per esempio, era stato scoperto che le cellule di lievito producono più etanolo in assenza che in presenza di ossigeno, fenomeno che prende il nome di ‛effetto Pasteur' (v. Pasteur, 1876). Analogamente, fu trovato che il muscolo a riposo produce solo piccole quantità di acido lattico dal glicogeno, ma ne produce grandi quantità durante la contrazione o nel tempo immediatamente successivo (v. Fletcher e Hopkins, 1907). Molti esempi simili potrebbero essere citati; l'assunto di questo articolo è appunto quello di considerare come le velocità dei processi metabolici sono controllate nella cellula vivente. Il chiarimento della natura dei meccanismi di controllo operanti nel metabolismo si è reso possibile solo dopo lo sviluppo di tecniche che hanno permesso il congelamento rapido di tessuti intatti, la successiva estrazione dei metaboliti e il loro dosaggio con metodi enzimatici (v. Wollenberger e altri, 1960; v. Bergmeyer, 1963). Notevoli contributi teorici riguardo al ruolo delle reazioni reversibili e irreversibili nel campo della regolazione metabolica sono stati recati da Krebs (v., 1946 e 1957) e da Bücher e Rüssmann (v., 1964). Un'altra teoria di una certa importanza per la comprensione delle basi molecolari della regolazione metabolica è la teoria ‛allosterica' (v. Monod e altri, 1963 e 1965). Secondo tale teoria (che sarà considerata in seguito) l'attività catalitica di certi enzimi viene modificata da metaboliti che interagiscono con l'enzima in un punto della sua molecola (‛sito allosterico') spazialmente distinto da quello che lega i substrati (‛sito catalitico'). Ciò permette una migliore comprensione, a livello molecolare, della regolazione a retroazione (feedback) delle vie metaboliche.
Gran parte della teoria presentata in questo articolo deriva dai concetti discussi dai suddetti autori, anche se nella presente trattazione viene applicato ai problemi della regolazione metabolica un criterio di studio cinetico piuttosto che termodinamico. Nondimeno i punti di vista cinetico e termodinamico sono connessi tra di loro in maniera diretta, sicché è possibile tradurre gli argomenti derivati da un tipo d'indagine nel linguaggio dell'altro. Gli autori sono convinti che gli argomenti cinetici contribuiscono maggiormente alla comprensione degli aspetti dinamici del metabolismo cellulare. Inoltre essi non presentano le difficoltà concettuali che nascono quando si applica la termodinamica classica al metabolismo (per una stimolante discussione in proposito v. Banks e Vernon, 1970).
2. Aspetti teorici della regolazione metabolica.
a) Sistemi chiusi e aperti.
Tutte le reazioni chimiche (e quindi anche tutte le reazioni enzimatiche) sono fino a un certo grado reversibili (principio della reversibilità microscopica), ma l'esperienza insegna che certe reazioni sono più reversibili di altre. In particolare, nelle vie metaboliche degli organismi viventi la velocità di una reazione in una direzione può essere assai più grande di quella nella direzione opposta, con il risultato che la reazione può essere considerata praticamente irreversibile. Sia data un'ipotetica reazione enzimatica
in cui x e y sono i substrati e v1 e v2 rappresentano le velocità di reazione nei due sensi. Se questa reazione è isolata dall'ambiente circostante, cosicché non c'è produzione o eliminazione di x o di y nel o dal sistema, la concentrazione di queste sostanze si approssimerà a un valore determinato. Questo stato, in cui le velocità di reazione nei due sensi opposti si equivalgono, è denominato ‛equilibrio'. Per un sistema isolato (cioè ‛chiuso') come quello appena descritto, la condizione di equilibrio è l'unica in cui le concentrazioni di x e di y non variano nel tempo. Tuttavia, all'equilibrio il prodotto ‛netto' del processo di interconversione dei reagenti è zero (dato che v1=v2) ed è noto, dai principi della termodinamica classica, che un tale sistema non può compiere un lavoro utile. Perciò un sistema chiuso non fornisce un modello conveniente della situazione metabolica nelle cellule viventi che, come è noto, sono capaci di interconvertire fra di loro le varie sostanze a velocità costante e di effettuare un lavoro nell'ambiente circostante.
I sistemi metabolici sono esempi di sistemi termodinamici ‛aperti', caratterizzati da uno scambio continuo di materia e di energia con l'ambiente circostante. Un sistema aperto può essere esemplificato in questo modo:

Questo sistema racchiude la stessa reazione ipotetica del sistema chiuso sopra descritto, ma stavolta c'è una reazione (A) che rigenera continuamente x dall'ambiente e una reazione (B) che allontana continuamente y dal sistema. Queste due ultime reazioni sono state indicate come irreversibili e la ragione di ciò sarà discussa più avanti. Una proprietà importante di un sistema aperto è che la concentrazione di x e di y può essere indipendente dal tempo anche se la reazione x⇄y è spostata dalla condizione di equilibrio (vale a dire quando v1≠v2). Nell'esempio qui riportato questa situazione si verifica quando la velocità della reazione A è costante e la velocità della reazione B è funzione della concentrazione di y. Questa condizione è denominata ‛stato stazionario' e in essa una sostanza si trasforma in un'altra a velocità costante; la velocità alla quale opera complessivamente l'intero sistema è detta ‛flusso'. Lo stato stazionario fornisce un utile modello per la cinetica enzimatica (v. Dalziel, 1957; v. Cleland, 1967) e servirà nella presente trattazione come modello per descrivere il comportamento delle sequenze metaboliche. Occorre però sottolineare che lo stato stazionario non è l'unica condizione in cui possono esistere i sistemi aperti. Altre condizioni, nelle quali la concentrazione del substrato (o dei prodotti intermedi del metabolismo) è una funzione continua del tempo, sono rappresentate dagli stati transitori (esponenziali), che hanno luogo quando cambia il flusso di un sistema a stato stazionario, e dagli stati oscillatori. Questi ultimi sono stati riscontrati nel sistema glicolitico del lievito (v. Chance e altri, 1964).
Il resto di questo capitolo tratterà l'origine del flusso in sistemi a stato stazionario e quei fattori che influenzano l'entità del flusso stesso (cioè, la sua regolazione). Le conclusioni derivate dall'uso del modello a stato stazionario saranno quindi applicate a un sistema metabolico che è stato oggetto di osservazione sperimentale particolarmente accurata, cioè la glicolisi muscolare. Prima è però necessario esaminare i rapporti tra impostazione termodinamica e impostazione cinetica dello studio della regolazione metabolica.
b) Criterio termodinamico e criterio cinetico nello studio del controllo metabolico.
Molte discussioni sul controllo metabolico si sono finora basate su argomenti termodinamici, mentre la presente trattazione metterà piuttosto in luce il lato cinetico del sistema. Di conseguenza è necessario mostrare le relazioni fra questi due punti di vista, in modo da evitare confusioni in rapporto ad altre trattazioni dello stesso soggetto.
Si consideri la seguente reazione che ha luogo in un ipotetico sistema a stato stazionario:
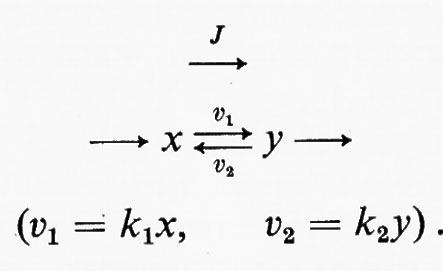
In questo esempio J è il vettore che rappresenta il flusso e k1 e k2 sono le costanti di velocità rispettivamente per le reazioni x→y e y→x. Dalla discussione precedente consegue immediatamente che la reazione x⇄y è spostata dall'equilibrio, in modo tale che (k1x−k2y)=J. L'equazione della termodinamica classica che mette in relazione l'energia libera (ΔG) di una reazione con il suo grado di spostamento dall'equilibrio (equazione dell'energia libera di Gibbs) è:
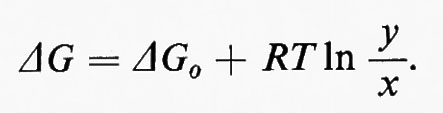
Poiché ΔG0=−RTlnK, dove K è la costante di equilibrio (cioè ye/xe, dove i suffissi indicano le concentrazioni all'equilibrio), questa equazione diventa:
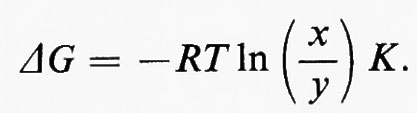
Generalmente il termine (y/x) è chiamato ‛rapporto di azione di massa' e indicato col simbolo Γ. In tal modo l'equazione diventa:
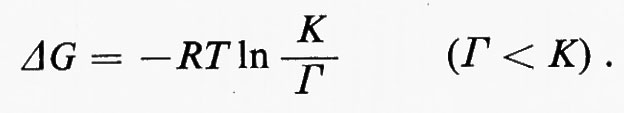
Il rapporto (K/Γ) misura lo spostamento dall'equilibrio: esso è uguale a 1 quando la reazione è all'equilibrio e diventa tanto più grande quanto più la reazione si allontana dall'equilibrio.
In base alla cinetica della reazione le velocità delle reazioni nei due sensi (v1 e v2) possono essere scritte come v1=k1x, v2=k2y. Così

Poiché all'equilibrio k1xe=k2ye (in quanto v1=v2), ne segue che K=ye/xe=k1/k2. Quindi
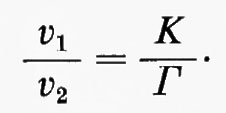
Poiché K/Γ è la misura dello spostamento della reazione dall'equilibrio, anche il rapporto v1/v2 è un'indicazione di questo spostamento. In tal modo viene stabilito un collegamento fra criterio di studio termodinamico e criterio di studio cinetico. È evidente, dalla relazione v1/v2=K/Γ, che una reaziome molto spostata dall'equilibrio (valore elevato di K/Γ) può anche essere descritta come una reazione praticamente irreversibile (valore elevato di v1/v2), mentre una reazione vicina all'equilibrio (K/Γ vicino all'unità) può anche essere descritta come una reazione facilmente reversibile (v1/v2 vicino all'unità). In questo articolo si userà il concetto di ‛reversibilità' di reazione piuttosto che quello di ‛spostamento dall'equilibrio' come base teorica della discussione sulla regolazione metabolica. Nondimeno occorre sottolineare che la maggior parte dei dati sperimentali che hanno permesso la classificazione delle reazioni metaboliche in reversibili e irreversibili è basata sulla relazione fra rapporto di azione di massa e costante di equilibrio (cioè su criteri termodinamici).
Infine, dall'equazione ΔG=−RTln(K/Γ) deriva che la liberazione di energia è tanto più grande quanto più una reazione è spostata dall'equilibrio. Parte di questa energia è liberata come calore, mentre il resto è trasferito ad altre molecole (ATP in particolare) e in questa forma può essere utilizzato per mettere in opera altri sistemi. Purtroppo, l'aspetto quantitativo del discorso sull'energia liberata nelle vie metaboliche rivela l'inadeguatezza della termodinamica classica applicata al metabolismo. Quasi tutte le relazioni termodinamiche che sono state ricavate si applicano soltanto a sistemi che sono nella condizione di equilibrio o molto prossimi a essa. Uno studio termodinamico del metabolismo in termini più realistici dovrebbe nascere dalla considerazione di sistemi aperti, ma una termodinamica di tal genere è molto più complicata di quella classica, così che non è stata ancora applicata con successo al metabolismo intermedio (v. Katchalsky e Curran, 1965).
c) Generazione del flusso in sistemi a stato stazionario.
Le caratteristiche principali di un sistema in condizioni di stato stazionario sono l'indipendenza della concentrazione dei composti intermedi dal tempo e la presenza di un flusso costante. In effetti, la generazione del flusso costante è il fattore principale che stabilisce la presenza di uno stato stazionario. Così un sistema a stato stazionario può essere considerato come composto da due categorie di reazioni: a) quelle che generano il flusso costante; b) quelle che si adattano al flusso, tramite cambi di concentrazione dei loro substrati.
Per esempio, in un sistema a stato stazionario del tipo

un flusso costante è generato a livello della reazione A e la velocità della reazione è data da λ•x. La concentrazione di x allo stato stazionario sarà data dall'equazione
λ•x=flusso
e il flusso è uguale alla velocità della reazione A. Ne consegue che la reazione B si adatta al flusso generato a livello della reazione A. Da queste considerazioni risulta che in ogni sistema a stato stazionario tutte le reazioni, la cui velocità è funzione della concentrazione del substrato, devono conformarsi al flusso. Il flusso è invece generato da una reazione la cui velocità è indipendente dalla concentrazione del suo substrato. Reazioni di questo tipo sono quelle ‛saturate' dal substrato e sono sempre irreversibili. Il seguente esempio ipotetico può essere d'aiuto a chiarire questo punto fondamentale. Sia dato un sistema a stato stazionario simile a quello precedentemente descritto, cioè

nel quale il flusso è generato a livello della reazione A. Se si ammette che la reazione A rappresenta un'unica reazione chimica (o enzimatica), ne consegue che essa non può essere dipendente dal substrato, altrimenti il flusso diminuirebbe continuamente con il diminuire della concentrazione del substrato e uno stato stazionario sarebbe impossibile. Allo stesso modo, se la reazione A consiste in più di una reazione chimica (o enzimatica), nel complesso di reazioni ne deve esistere almeno una che sia indipendente dal substrato.
In conclusione, il flusso attraverso un sistema a stato stazionario è generato a livello di reazioni saturate dal substrato, mentre le reazioni dipendenti dal substrato non fanno altro che adattarsi al flusso. Reazione generatrice di flusso è termine sinonimo di pacemaker e di ‛reazione limitante la velocità' (rate-limiting step). Invece occorre stare attenti all'uso fatto nella discussione precedente del termine ‛substrato'. Il seguente esempio ipotetico
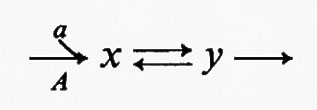
può servire a chiarire tale punto. In questo sistema c'è un cofattore (a) implicato nella reazione generatrice del flusso (A) e questo cofattore è rigenerato a livello di una reazione successiva del sistema. Se il termine ‛substrato di sequenza' è definito come quel substrato che rappresenta il flusso di materia attraverso un sistema aperto, in questo sistema la reazione A deve essere saturata dal substrato di sequenza, ma non necessariamente da a. Quest'ultimo è rigenerato continuamente e perciò si può considerare parte di un sistema ‛conservativo' di metaboliti, in contrapposizione al substrato di sequenza che rappresenta materia in scorrimento attraverso il sistema. Perciò la reazione generatrice del flusso deve essere saturata dal suo substrato di sequenza e ogni reazione che non è saturata dal suo substrato di sequenza deve adattarsi al flusso.
A questo punto occorre chiarire che le reazioni di un sistema metabolico non sono necessariamente confinate in singole cellule o in singoli tessuti, ma il sistema può estendersi a parecchi tessuti collegati fra di loro dal sistema circolatorio. In questi casi la reazione generatrice del flusso può essere presente in un tessuto, mentre molte delle reazioni successive avvengono in altri tessuti. Ecco un esempio ipotetico:
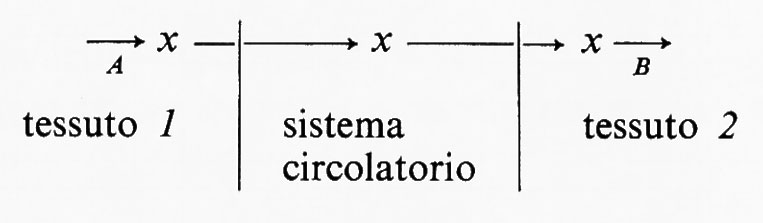
In questo sistema il flusso è generato a livello della reazione A nel tessuto 1 e la velocità di utilizzazione di x da parte del tessuto 2 è funzione della sua concentrazione nel fluido circolante. Un ben noto esempio fisiologico è la produzione di acidi grassi a catena lunga (≡x) da parte del tessuto adiposo (≡tessuto 1) e la loro successiva ossidazione da parte di altri tessuti quali il muscolo (≡tessuto 2).
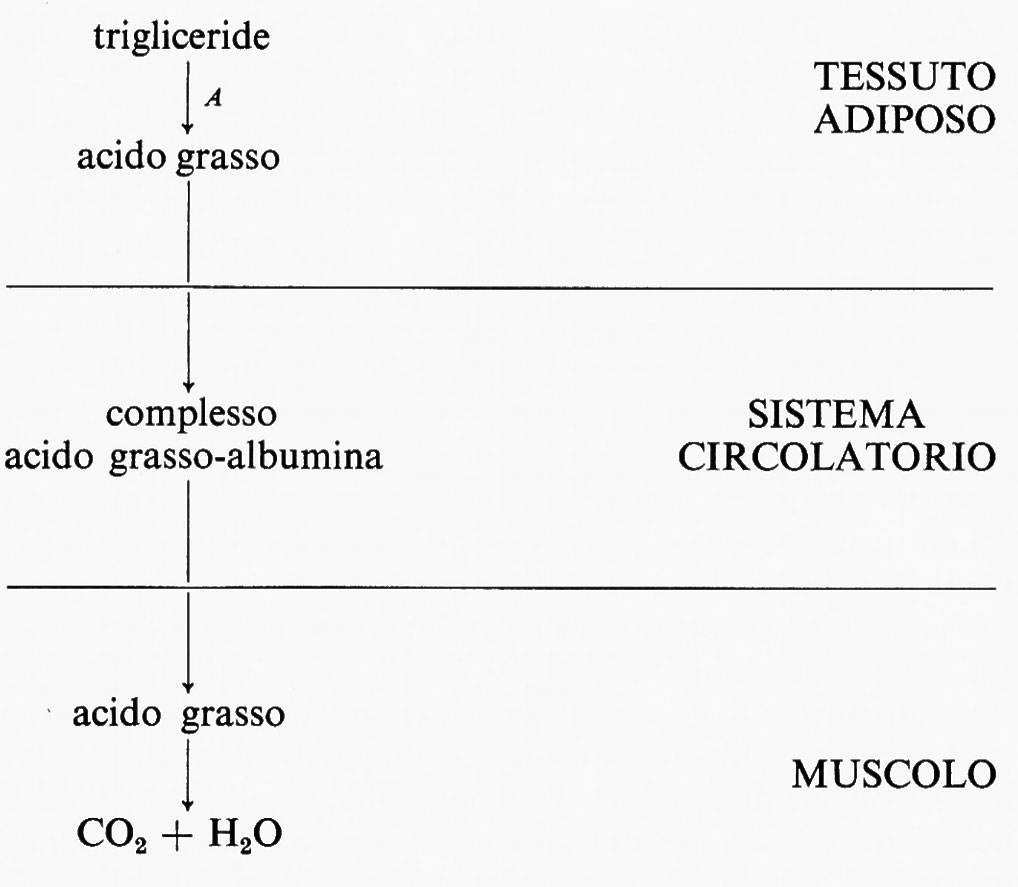
La reazione A è catalizzata dall'enzima trigliceridelipasi, che è sempre saturato con trigliceride, e rappresenta così la reazione generatrice del flusso. L'acido grasso liberato dal tessuto adiposo è trasportato nel sangue sotto forma di un complesso con l'albumina; è noto che la velocità di utilizzazione degli acidi grassi nel muscolo è funzione della concentrazione dell'acido grasso in tale complesso. Questo esempio illustra un altro importante principio che riguarda l'interpretazione di esperimenti su tessuti isolati: il metabolismo di una sostanza in un tessuto rappresenta in genere solo una parte di un sistema metabolico molto più ampio, che può includere molti tessuti.
d) Regolazione del flusso in sistemi a stato stazionario.
A questo punto saranno discussi i fattori che determinano l'entità del flusso attraverso un sistema metabolico, e cioè quei fattori che in definitiva regolano la velocità operativa delle vie metaboliche. Come esempio delle varie possibilità che possono darsi consideriamo dapprima un sistema semplice non ramificato:

L'entità del flusso attraverso questo sistema è determinata dalla reazione A e può essere modificata soltanto da una variazione dell'attività dell'enzima che catalizza la reazione. L'attività di questo enzima, e quindi la velocità della reazione A, può essere modificata sia direttamente (per es., per mezzo dell'inibizione da parte di un metabolita regolatore), sia indirettamente (per es., attraverso modificazioni dell'attività dell'enzima che catalizza la reazione B). Effetti indiretti sulla reazione R possono modificare la reazione generatrice di flusso (A) attraverso variazioni della concentrazione di x. Per esempio, se l'enzima che catalizza la reazione A fosse inibito da x, un aumento della concentrazione di x porterebbe a una riduzione di flusso attraverso il sistema. Così è possibile regolare il flusso attraverso un sistema modificando l'attività di una reazione diversa da quella generatrice del flusso; queste reazioni, insieme a quella generatrice del flusso, sono dette ‛regolatorie'. Si noti che finora niente è stato detto specificamente riguardo alla reversibilità delle reazioni regolatorie, a parte la reazione generatrice del flusso. Si vedrà più avanti che reazioni irreversibili offrono condizioni più favorevoli per la regolazione di reazioni reversibili quando il regolatore è un effettore allosterico. Questa conclusione si fonda sul criterio della sensibilità di una reazione a variazioni della concentrazione di metaboliti potenzialmente regolatori e non esclude possibilità di regolazione per mezzo di effetti sull'attività di reazioni reversibili. Sarebbe comunque errato considerare le reazioni irreversibili come gli unici siti possibili della regolazione.
L'uso del termine ‛regolatorio' può portare confusione se non lo si definisce adeguatamente. Un enzima è definito regolatorio se la sua attività è controllata da fattori diversi dalla concentrazione del substrato (v. Rolleston e Newsholme, 1967): questa è una definizione operativa, e non assoluta, in quanto serve all'identificazione sperimentale di enzimi regolatori. Una generalizzazione acritica di questa definizione potrebbe dare l'impressione che gli enzimi regolatori non sono influenzati dalla concentrazione di substrati e cofattori. Per gli scopi della discussione teorica qui presentata, al termine ‛reazione regolatoria' si darà il significato più ampio, vale a dire che una reazione enzimatica è regolatoria per un dato sistema se: a) una variazione nella sua attività porta in definitiva a una variazione nell'entità di flusso attraverso l'intero sistema o parte di esso; b) tale variazione di attività è un mezzo diretto di interazione con altri sistemi metabolici. Questa definizione include la regolazione per mezzo di substrati, di cofattori o di modificazioni dell'attività enzimatica mediante metaboliti che non partecipano alla catalisi enzimatica (per es. regolatori allosterici). La ragione per includere nella definizione la variazione di flusso attraverso ‛parte di un sistema' può essere chiarita dall'applicazione della definizione stessa a una via metabolica ramificata:
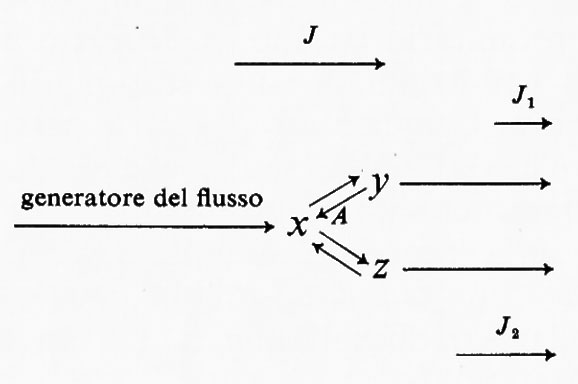
In questo sistema il flusso J si divide al punto di biforcazione in due subflussi J1 e J2 tali che la loro somma è uguale a J. Si ammette che i valori effettivi di J1 e J2 siano determinati dalla cinetica complessiva delle due vie alternative, cioè non c'è altra limitazione imposta dall'una o dall'altra delle vie. In questo sistema è perciò possibile cambiare l'entità del flusso che attraversa ciascuna delle due branche indipendentemente da una variazione del flusso totale J. Questo si può ottenere alterando l'attività di una reazione come A, che produrrà una variazione di J1 più una variazione compensatoria di J2. In questo modo la reazione A può essere considerata regolatoria per questo sistema e mostra perché la definizione del termine regolatorio si applichi a cambiamenti di flusso attraverso tutto un sistema o attraverso parte di esso.
e) Reazioni reversibili e irreversibili e loro significato nel metabolismo.
È noto dai fatti sperimentali che nei sistemi metabolici esistono tre tipi di reazioni, cioè reazioni reversibili, reazioni irreversibili in cui la velocità dipende dalla concentrazione del substrato di sequenza (reazioni irreversibili dipendenti dal substrato) e reazioni irreversibili in cui la velocità non dipende dalla concentrazione del substrato di sequenza (reazioni irreversibili indipendenti dal substrato). I termini ‛reversibile' e ‛irreversibile' si riferiscono alla situazione chimica presente nella cellula vivente come è dedotta dalla comparazione del rapporto di azione di massa con la costante di equilibrio per ogni singola reazione (v. Williamson, 1965; v. Rolleston e Newsholme, 1967). Una reazione in un sistema metabolico è irreversibile in quanto la concentrazione dei prodotti di reazione e l'attività catalitica dell'enzima si mantengono bassi, cosicché la velocità della reazione in senso contrario è evidentemente minore della velocità della reazione ‛in avanti'. Analogamente una reazione è reversibile se la concentrazione dei prodotti e l'attività catalitica dell'enzima sono abbastanza alti da far sì che la velocità della reazione in senso contrario sia molto più grande del flusso nel suo complesso.
Reazioni reversibili. - Dalle discussioni precedenti risulta chiaro che un sistema in stato stazionario ha bisogno di una reazione irreversibile per generare il flusso. D'altra parte un'altra reazione irreversibile è necessaria per eliminare i prodotti terminali, che altrimenti si accumulerebbero con la conseguenza che il processo nel suo insieme tenderebbe a uno stato di equilibrio. In via di principio sarebbe possibile immaginare una via metabolica fatta tutta di reazioni irreversibili, ma è ben noto che le vie metaboliche contengono molte reazioni reversibili. Qual'è la ragione fisiologica di questo fatto? Nel seguente sistema ipotetico contenente una reazione reversibile
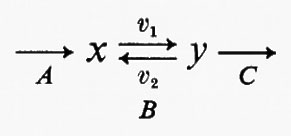
v1 e v2 rappresentano le velocità delle reazioni nei due sensi, che dipendono dalle concentrazioni di x e di y (la reazione è catalizzata dall'enzima B). Poiché, in virtù della sua reversibilità (criterio cinetico), questa reazione è prossima all'equilibrio (criterio termodinamico), il rapporto v1/v2 sarà vicino al valore unitario (v. sopra). Il flusso attraverso la reazione e, in condizioni di stato stazionario, attraverso tutta la sequenza, è uguale a (v1−v2) e perciò, dovendo essere v1/v2 vicino all'unità, sia v1 che v2 devono essere molto più grandi del valore del flusso. Per esempio, se il flusso attraverso la sequenza è 10 unità e v2 è 1 unità, v1 sarebbe uguale a 11 unità e v1/v2 sarebbe uguale a 11. Ma se v2 è 100 unità, v1 sarebbe 110 e v1/v2 1,1. In quest'ultimo caso la reazione è molto più prossima all'equilibrio. Risulta ora chiaro che è la velocità della reazione in senso contrario in rapporto al flusso totale che determina la reversibilità della reazione complessiva e quindi il suo grado di prossimità all'equilibrio. Questo punto verrà ripreso più avanti.
Una conseguenza importante della reversibilità di una reazione in un sistema metabolico è che, essendo v1 e v2 ambedue molto maggiori del flusso (che è uguale a v1− v2), variazioni relativamente piccole di v1 o di v2 producono variazioni di flusso molto grandi. Sia, ad esempio:

Nel primo sistema un aumento di v1 del 10% produce un aumento di flusso appena del 10% circa, mentre nel secondo sistema un alimento di v1 del 10% produce una variazione di flusso totale superiore al 100%. Questi esempi mostrano come reazioni reversibili possano sopportare ampie variazioni di flusso senza subire cambi di concentrazione dei loro reagenti altrettanto ampi. In altre parole, i metaboliti di una sequenza che siano collegati per mezzo di reazioni reversibili sono tamponati contro grosse variazioni della loro concentrazione in presenza di grosse variazioni del flusso. Come sarà discusso in seguito, evitare grossi cambiamenti di concentrazione dei composti intermedi del metabolismo è un vantaggio per la cellula; questa è almeno una ragione per la presenza di reazioni reversibili nei sistemi metabolici.
Tuttavia questa proprietà delle reazioni reversibili riduce la loro utilità come siti potenziali dell'attività regolatoria. Le reazioni reversibili non possono essere saturate da substrati di sequenza e perciò non possono funzionare come reazioni generatrici di flusso. Di conseguenza esse possono regolare il flusso solo per via indiretta, tramite gli effetti che i metaboliti della sequenza hanno sulla reazione generatrice del flusso. In questo caso l'enzima che catalizza una reazione reversibile deve essere regolato da fattori diversi dal suo substrato di sequenza o dal suo prodotto. D'altra parte, per modificare l'attività di una reazione reversibile al fine di produrre una variazione sufficientemente grande della concentrazione del suo substrato, è necessaria una variazione relativa della concentrazione del regolatore ben maggiore che per una reazione irreversibile. A chiarire questo punto può servire l'esempio seguente:
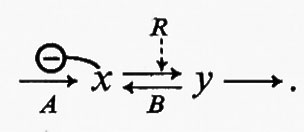
La reazione A genera il flusso ed è inibita da x. La reazione B rappresenta una reazione reversibile la cui velocità è influenzata da un metabolita regolatorio, R. L'attivazione o l'inibizione della reazione B da parte di R equivale a una variazione del flusso attraverso la reazione stessa. È stato però mostrato più sopra che grossi cambiamenti di flusso attraverso reazioni reversibili producono soltanto piccole variazioni della concentrazione dei reagenti (cioè x e y); perciò un grande cambiamento nella concentrazione di R produrrà un grande cambiamento nella velocità della reazione B, ma soltanto una piccola variazione nella concentrazione di x. Ne consegue che, a meno che la reazione A non sia estremamente sensibile a x, la regolazione per mezzo di interazioni a livello di una reazione come B risulterà ben poco efficiente.
Reazioni irreversibili dipendenti dal substrato. - Un sistema metabolico in stato stazionario richiede la presenza di una reazione irreversibile dipendente dal substrato alla fine del sistema o in prossimità di essa. Un esempio è dato dalla glicolisi muscolare con la diffusione del lattato nel sangue. Questo processo è irreversibile in condizioni fisiologiche, poiché la concentrazione di lattato è più alta nel tessuto che nel sangue, a causa dell'utilizzazione di lattato da parte del fegato e di alcuni altri tessuti. Questo fatto impedisce che il lattato si accumuli in grande quantità e mantiene la sequenza in condizioni di stato stazionario.
Un altro ruolo di queste reazioni è quello di mantenere costante la concentrazione dei prodotti delle reazioni reversibili che precedono nel sistema la reazione irreversibile. Nel sistema ipotetico
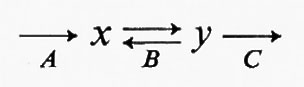
il flusso è generato a livello della reazione A, la reazione B è reversibile e la reazione C è irreversibile dipendente dal substrato. Per ogni dato flusso, la concentrazione di y sarà fissata dall'entità del flusso stesso più i parametri cinetici della reazione C. Poiché la reazione C è molto lontana dall'equilibrio, la concentrazione di y sarà maggiore che se questa reazione fosse vicina all'equilibrio. Quindi la velocità della reazione B in senso contrario, che è funzione della concentrazione di y, sarà anch'essa maggiore e questo contribuirà a mantenere la reazione B prossima all'equilibrio. In altre parole, un ruolo importante delle reazioni irreversibili dipendenti dal substrato è quello di mantenere la concentrazione di alcuni metaboliti intermedi a livelli sufficienti a garantire la reversibilità di altre reazioni. Infine, le reazioni irreversibili dipendenti dal substrato sono siti di regolazione molto migliori delle reazioni reversibili, in quanto per un dato cambiamento di flusso il cambiamento relativo nella concentrazione del substrato è in genere maggiore, come si può vedere dall'esempio seguente:
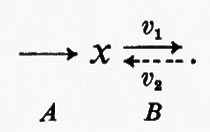
Poiché in una reazione irreversibile la velocità della reazione in senso contrario tende a zero (v2=0), v1 deve essere sempre uguale al valore del flusso; perciò a ogni cambiamento relativo di flusso deve corrispondere un identico cambiamento relativo di v1. Se la cinetica della reazione B non cambia col cambiamento di flusso, allora la concentrazione di x cambierà di tanto quanto è stata la variazione relativa del flusso. Per esempio, nel sistema già descritto

se R interagisce con la reazione C, una variazione relativa di concentrazione di R avrà come conseguenza una variazione relativa di concentrazione di x (e quindi del flusso totale) più grande che se l'interazione avviene con la reazione B. Pertanto le reazioni irreversibili indipendenti dal substrato sono teoricamente dei buoni siti regolatori e, secondo alcune indicazioni sperimentali, spesso servono effettivamente da siti di regolazione nelle cellule viventi.
Reazioni irreversibili indipendenti dal substrato. - Questo tipo di reazioni è quello che genera il flusso nei sistemi a stato stazionario ed è stato discusso precedentemente.
f) La glicolisi anaerobica nel muscolo.
In questo paragrafo i principi teorici discussi più sopra verranno applicati al processo della glicolisi quale si verifica in alcuni muscoli. Nel muscolo, il ruolo della glicolisi è quello di rifosforilare l'ADP formando ATP, che è idrolizzato durante il processo di contrazione. Il controllo del flusso attraverso questa via metabolica è stato studiato in maniera abbastanza particolareggiata da poterne trarre esempi specifici corrispondenti ai molti ipotetici presentati precedentemente.
Le reazioni della glicolisi sono presentate schematicamente nella fig. 1. La tab. I contiene i valori dei rapporti di azione di massa e delle costanti di equilibrio apparenti per alcune reazioni della glicolisi. Paragonando i due valori si può concludere che le reazioni catalizzate dagli enzimi fosfoglucosoisomerasi, aldolasi, triosofosfatoisomerasi, fosfogliceratomutasi ed enolasi sono notevolmente vicine all'equilibrio e quindi sono reversibili. Le reazioni catalizzate dagli enzimi esochinasi, fosfofruttochinasi e piruvatochinasi sono molto spostate dall'equilibrio e perciò sono irreversibili, nella cellula. Il sistema gliceraldeide-3-fosfatodeidrogenasi fosfogliceratochinasi presenta alcune difficoltà riguardo all'interpretazione dei dati. I valori della tab. I indicano che questo sistema è molto spostato dall'equilibrio e quindi irreversibile. Tuttavia, nel fegato, il rapporto di azione di massa per questo sistema è circa 200 M-1 e in questo tessuto il sistema deve essere reversibile dato che partecipa sia alla glicolisi sia alla gluconeogenesi. Ammettendo che la reazione sia reversibile nel fegato, il paragone del rapporto di azione di massa fra fegato e muscolo indica che lo spostamento dall'equilibrio ‛nel muscolo' non è eccessivo (K/Γ è all'incirca 20).
Come per le reazioni riportate nella tab. I, anche per le reazioni catalizzate dalla fosfoglucomutasi e dalla lattato- deidrogenasi si può concludere che esse sono reversibili nella cellula (v. Krebs e Veech, 1969).

La diffusione del lattato dal muscolo è irreversibile in condizioni fisiologiche, poiché la concentrazione di lattato è maggiore nel tessuto che nel sangue. Nella maggior parte dei casi il trasporto del glucosio nel muscolo scheletrico è un processo non-di-equilibrio (cioè il rapporto glucosio extracellulare/glucosio intracellulare è grande) e perciò è irreversibile. Tuttavia il processo può essere reso reversibile se la sua attività è aumentata al di sopra del livello della fosforilazione del glucosio. In questa situazione l'utilizzazione del glucosio è limitata dall'esochinasi (che diviene saturata con glucosio) e si ha accumulo intracellulare di glucosio. Questi fenomeni sono stati descritti nel cuore di ratto isolato e perfuso in presenza di insulina aggiunta per stimolare il trasporto (v. Morgan e altri, 1959).
Reazioni generatrici di flusso nella glicolisi. - Se si ammette che i contenuti tessutali dei composti intermedi della glicolisi determinati sperimentalmente siano distribuiti uniformemente nell'acqua intracellulare, in modo che possano essere tradotti in termini di concentrazione, si può dedurre che le reazioni catalizzate dagli enzimi fosfofruttochinasi, piruvatochinasi ed esochinasi e anche il processo di trasporto del glucosio attraverso la membrana sono, in condizioni fisiologiche, dipendenti dal substrato. Per esempio, la concentrazione stimata di fosfoenolpiruvato nel muscolo è di circa 5•10-5 M, mentre la Km della piruvatochinasi per questo substrato è circa 10-4 M; poiché la concentrazione tessutale non è molto diversa dalla Km, si può concludere che questa reazione nella cellula è dipendente dal substrato (v. fig. 2). Una conclusione analoga si deduce per il sistema di trasporto del glucosio, che ha una Km di circa 9•10-3 M per il glucosio (v. Randle e Morgan, 1962). Poiché la concentrazione ematica del glucosio è circa 5•10-3 M, se ne ricava che questa reazione è dipendente dal substrato nell'organismo animale.
Per quanto sia difficile calcolare con precisione la concentrazione del glicogeno, che è un materiale di riserva a distribuzione diffusa, sembra fuori di dubbio che nel muscolo a riposo la fosforilasi sia saturata con tale sostanza. Per esempio nei muscoli del volo del tafano (Phormia) la concentrazione di glicogeno, a riposo, è circa 5•10-2 M, espressa in equivalenti di glucosio (v. Sacktor e Wormser-Shavit, 1966). La Km per il glicogeno della fosforilasi estratta da questo muscolo è circa 2•10-3 M in condizioni fisiologiche, cioè in presenza di fosfato inorganico 10 mM e di una bassa concentrazione di AMP (v. Childress e Sacktor, 1970). Pertanto in queste condizioni la fosforilasi è saturata con glicogeno e può agire come generatrice di flusso per la glicolisi quando il muscolo usa glicogeno come combustibile per la contrazione. In favore di questo concetto è da tener presente che la fosforilasi è regolata da basse concentrazioni di ioni Ca2+ (circa 10-7 M) che allo stesso tempo stimolano l'ATP-asi miofibrillare dando inizio alla contrazione. È perciò probabile che, quando il combustibile è il glicogeno, il flusso glicolitico sia sincronizzato col ritmo di utilizzazione dell'ATP da parte del processo di contrazione. Tuttavia, il glicogeno può servire da combustibile per l'attività muscolare solo per periodi relativamente brevi, poiché il contenuto in glicogeno del muscolo è relativamente basso rispetto alla velocità della sua utilizzazione (v. Weis-Fogh, 1967; v. Crabtree e Newsholme, 1972). Se la contrazione deve durare per periodi più lunghi di quelli permessi dalla quantità di glicogeno accumulato, il muscolo deve usare un combustibile come il glucosio che è reso continuamente disponibile dal torrente circolatorio. Nel processo di utilizzazione del glucosio extracellulare da parte del muscolo in condizioni normali (glicolisi) non esiste però all'interno del muscolo alcuna reazione saturata con substrato, per cui la reazione generatrice del flusso deve essere presente in qualche altro tessuto. Essa è probabilmente la reazione della fosforilasi nel fegato, che produce glucosio dalle sue relativamente abbondanti scorte di glicogeno.
Controllo del flusso glicolitico a livello della fosfofruttochinasi. - Quando il glucosio è il substrato della glicolisi muscolare, la reazione generatrice del flusso ha sede nel fegato. Nondimeno il muscolo è capace di regolare il flusso glicolitico per adattare la velocità di rifosforilazione dell'ADP ad ATP alla velocità di idrolisi dell'ATP durante l'attività muscolare. Questa regolazione si ottiene tramite l'interazione dei nucleotidi adenilici con l'enzima fosfofruttochinasi, come è mostrato nel diagramma seguente:
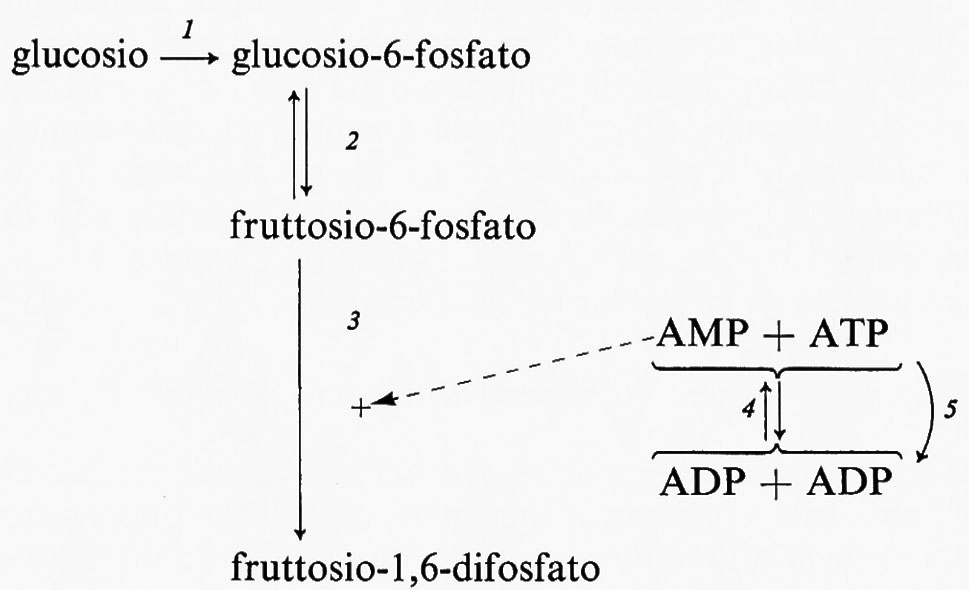
Quando aumenta l'attività dell'ATP-asi miofibrillare (reazione 5), la concentrazione dell'ATP tende a diminuire e quella dell'ADP a salire. D'altra parte la reazione quasi-di-equilibrio catalizzata dall'adenilatochinasi (reazione 4) produce un aumento della concentrazione di AMP molto maggiore rispetto alla diminuzione di quella dell'ATP. L'AMP attiva la fosfofruttochinasi (reazione 3), provocando una diminuzione della concentrazione del fruttosio-6-fosfato e anche del glucosio-6-fosfato a causa della reversibilità della reazione 2, catalizzata dalla fosfoglucosoisomerasi. Poiché il glucosio-6-fosfato inibisce l'esochinasi (reazione 1), una diminuzione della sua concentrazione aumenta l'attività dell'esochinasi. Se il processo di trasporto del glucosio è reversibile, un'accresciuta attività dell'esochinasi porta a una diminuzione della concentrazione del glucosio intracellulare che aumenta la velocità netta di entrata del glucosio nel muscolo. In condizioni in cui il trasporto è irreversibile questo processo è invece stimolato da alcuni regolatori specifici connessi con la concentrazione intracellulare dell'ATP. Purtroppo il processo di trasporto non può essere studiato in vitro, cosicché mancano indicazioni sulla natura del meccanismo di regolazione (v. Randle e Morgan, 1962). La fosfofruttochinasi è dunque un enzima regolatorio per la glicolisi e fornisce il collegamento necessario fra l'utilizzazione dell'ATP durante la contrazione e la sua rifosforilazione da parte del sistema glicolitico. (Per ulteriori notizie su questo sistema si veda la rassegna di Newsholme, 1970).
Ruolo della piruvatochinasi nel muscolo. - Le reazioni glicolitiche comprese tra la fosfofruttochinasi e la piruvatochinasi (v. fig. 1) sono molto probabilmente reversibili e perciò esse rispondono a grandi cambiamenti del flusso glicolitico con minimi cambiamenti nella concentrazione dei loro substrati. La funzione della piruvatochinasi, che catalizza una reazione irreversibile dipendente dal substrato, può essere perciò considerata quella di conservare le concentrazioni dei composti intermedi delle suddette reazioni reversibili, mantenendole quindi prossime all'equilibrio.
3. Meccanismi fisiologici di regolazione dell'attività enzimatica.
Nel cap. 2 si è giunti alla conclusione che le reazioni irreversibili offrono i siti più adatti per la regolazione del flusso attraverso un sistema metabolico. Questa regolazione avviene di norma per mezzo dell'interazione di un metabolita X con l'enzima, secondo il seguente meccanismo:
E+X⇄E*X
dove E è la molecola dell'enzima e X è o il substrato o un regolatore ‛allosterico'. Se X è il substrato, il complesso E*X rappresenta il complesso enzima-substrato che prende parte alla catalisi. In questo caso E può essere considerata la forma inattiva dell'enzima ed E*X la forma cataliticamente attiva. Se X è un regolatore allosterico, E*X rappresenta una forma attiva o inattiva dell'enzima a seconda che X sia un attivatore o un inibitore. In ambedue i casi si ammette che la velocità della reazione enzimatica nel suo complesso sia proporzionale alla concentrazione di E*X. Questo tipo di interazione fra un metabolita (X) e un enzima (E) è denominato ‛legame reversibile' (o ‛di equilibrio') ed è il punto di partenza per considerare la sensibilità della risposta di un enzima a un regolatore. Si ammette anche che la conformazione dell'enzima, quando esso si lega a X, cambi da E a E*. Consideriamo dapprima la situazione più semplice: ogni molecola di E lega soltanto una molecola di X e la concentrazione totale di X è molto più grande di quella di E. In questo caso la frazione di enzima nella forma E*X sarà una funzione iperbolica di X, come mostrato dalla fig. 2. Questo tipo di risposta è la ben nota relazione di Michaelis-Menten dell'enzimologia classica (v. catalisi enzimatica; v. enzimi) ed è analoga all'isoterma di Langmuir della chimica fisica. Le caratteristiche principali di questo tipo di risposta a X sono che essa segue una cinetica di ordine zero (e cioè in condizioni di saturazione) quando la concentrazione di X diventa molto grande, mentre la cinetica è di primo ordine a concentrazioni molto basse di X. La risposta a X nel suo complesso può essere descritta da due parametri (v. fig. 2), la velocità massima (V) e la costante di Michaelis (Km). La velocità della reazione (che è proporzionale a E*X) è maggiormente sensibile a cambiamenti di concentrazione di X quando la concentrazione iniziale di X è bassa relativamente a Km, cioè in condizioni di cinetica di primo ordine. La sensibilità della risposta a X cala continuamente fino ad annullarsi, a mano a mano che aumenta la concentrazione di X, come può dedursi intuitivamente osservando (v. fig. 2) l'appiattimento dell'iperbole al crescere della concentrazione di X. Si può calcolare che un cambiamento di velocità della reazione dall'1 al 95% del valore massimo richiederebbe un cambiamento di 1881 volte nella concentrazione di X. Questo enorme cambiamento nella concentrazione del regolatore potrebbe essere contenuto in limiti più ridotti se la massima attività catalitica dell'enzima (E) fosse molto maggiore del flusso massimo attraverso la sequenza a cui l'enzima appartiene. In tal caso la risposta dell'attività enzimatica a X potrebbe aver luogo interamente nella parte iniziale (quasi lineare) dell'iperbole, dove la sensibilità della risposta è massima. Tuttavia, anche nel caso di cinetica del primo ordine, il cambiamento relativo nella concentrazione di X non potrebbe mai essere inferiore al cambiamento relativo nel flusso. Per esempio, se il flusso aumentasse di 100 volte, il cambiamento minimo nella concentrazione di X sarebbe anch'esso di 100 volte. Per la maggior parte dei composti intermedi del metabolismo, anche un cambiamento di 100 volte è assai problematico. Per esempio, alte concentrazioni di metaboliti possono dare origine a reazioni collaterali non desiderabili (come decarbossilazioni spontanee, formazione di complessi con ioni metallici, formazione di complessi con enzimi a effetto inibitore). Vi possono anche essere effetti indiretti su altre vie metaboliche alle quali sono interessati metaboliti collegati a uno che subisca un grande cambiamento di concentrazione. In effetti i risultati sperimentali indicano che la concentrazione della maggior parte dei composti intermedi del metabolismo subisce soltanto minime variazioni, anche in presenza di grandi variazioni di flusso attraverso la sequenza. Per esempio, nel tafano (Phormia regina), il flusso glicolitico nei muscoli del volo aumenta di circa 100 volte quando l'insetto vola, ma la concentrazione di tutti i composti della glicolisi in questo tessuto rimane relativamente costante (v. Sacktor e Wormser-Shavit, 1966). È particolarmente interessante la costanza della concentrazione del fosfoenolpiruvato, poiché esso è il substrato di una reazione irreversibile dipendente dal substrato (piruvatochinasi): questo fa pensare che la sensibilità della piruvatochinasi alla concentrazione di fosfoenolpiruvato sia accresciuta al di là del limite prevedibile in base a una risposta iperbolica. Conclusioni analoghe valgono anche per il fruttosio-6-fosfato e la fosfofruttochinasi.
Ricapitolando, considerazioni teoriche e risultati sperimentali portano a concludere che la sensibilità di reazioni irreversibili a cambiamenti di concentrazione di metaboliti regolatori (substrati inclusi) è spesso maggiore di quanto prevedibile in base a una semplice risposta iperbolica. I meccanismi con cui si ottiene questo risultato formano l'oggetto dei paragrafi successivi.
a) Controllo dell'attività enzimatica mediante grandi variazioni nella concentrazione del regolatore.
Benché sia stato sottolineato che variazioni molto grandi nella concentrazione di un regolatore metabolico sono poco probabili nella cellula, esiste un buon esempio di questo tipo di regolazione. Esso sarà discusso abbastanza estesamente, perché chiarisce parecchi punti importanti che riguardano la regolazione metabolica.
Nel citoplasma di un muscolo a riposo la concentrazione dello ione Ca2+ libero è circa 10-8 M. Una concentrazione così bassa è il risultato del fatto che la maggior parte del Ca2+ cellulare è confinato in un sistema di membrane noto come reticolo sarcoplasmatico e così non è disponibile per il citoplasma. Per iniziare la contrazione, Ca2+ è liberato dal reticolo sarcoplasmatico nel citoplasma e la concentrazione dello ione Ca2+ libero nel citoplasma aumenta rapidamente fino a circa 10-6 M, attivando in tal modo il meccanismo della contrazione (per maggiori notizie v. tessuto muscolare e le rassegne di Pringle, 1967, e di Ebashi ed Endo, 1968). Questo stesso intervallo di concentrazione del Ca2+ serve anche ad attivare gli enzimi chinasi della fosforilasi b e glicerolo-3-fosfatodeidrogenasi mitocondriale (v. Hansford e Chappell, 1967; v. Hansford e Sacktor, 1970); questi due enzimi sono implicati nell'utilizzazione dei carboidrati al fine di rigenerare l'ATP idrolizzato dal meccanismo della contrazione.
Perciò nel muscolo c'è un cambiamento di circa 100 volte nella concentrazione del Ca2+ citoplasmatico nel passaggio dallo stato di riposo a quello di attività. Questa grande variazione relativa di concentrazione è possibile perché il Ca2+ non ha altre funzioni metaboliche all'interno della cellula oltre a quella regolatoria e la sua concentrazione può cambiare indipendentemente dalla concentrazione di altri metaboliti. Una concentrazione di base intorno a 10-8 M non potrebbe realizzarsi per un composto intermedio di una via metabolica come la glicolisi, non soltanto perché sarebbe inferiore a quella di molti enzimi, ma anche perché richiederebbe la presenza di enzimi con attività catalitica estremamente alta e con alte affinità per il substrato per poter garantire un flusso sufficiente attraverso la sequenza metabolica. Essa inoltre implicherebbe un intervallo di concentrazioni a livelli altrettanto bassi per i prodotti della reazione (e perciò delle altre reazioni della sequenza), data l'esigenza dell'irreversibilità della via metabolica nel suo complesso. Il vantaggio di una concentrazione molto bassa di Ca2+ nel muscolo a riposo sta nel fatto che è possibile aumentarla di 100 volte e che malgrado ciò la concentrazione finale è soltanto 10-6 M, un valore che non provoca interferenze indirette con il metabolismo. Le concentrazioni nella cellula di molti composti intermedi del metabolismo sono dell'ordine di 10-4 M, e se aumentassero di 100 volte diventerebbero 10-2 M. A questa concentrazione il Ca2+, per esempio, reagirebbe con il fosfato inorganico all'interno della cellula e precipiterebbe come fosfato di calcio; per questo il meccanismo di controllo della concentrazione del Ca2+ citoplasmatico è stato specificamente realizzato in modo da operare a basse concentrazioni, ma a questo fine è necessaria la presenza del reticolo sarcoplasmatico con la sua considerevole complessità strutturale, capace di un meccanismo di controllo tanto specifico.
b) Aumento di sensibilità prodotto da più di un regolatore.
La sensibilità della risposta iperbolica di un enzima a variazioni della concentrazione di un regolatore può essere migliorata se alla regolazione partecipa più di un metabolita regolatorio. Si consideri, per esempio, un enzima con due substrati che catalizzi una reazione irreversibile nella cellula. Se il legame di un substrato all'enzima non ha effetto sul legame dell'altro, la velocità della reazione, espressa come frazione della velocità massima, sarà data dall'equazione:
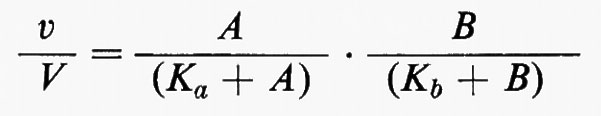
dove A e B sono le concentrazioni dei due substrati, Ka e Kb rappresentano le costanti di Michaelis rispettivamente per A e per B, v rappresenta la velocità effettiva e V la velocità massima della reazione (v. Dalziel, 1957). Questa relazione è il prodotto di due funzioni iperboliche e, perciò, per un dato cambiamento relativo di v, ammettendo che le concentrazioni di A e di B cambino ambedue nella stessa direzione, le variazioni relative delle concentrazioni di A e di B sono minori che se fossero responsabili della variazione di flusso ciascuna da sola. Questo si può valutare meglio se si semplifica l'equazione considerando Ka>A e Kb>B, così che essa può essere espressa nella forma approssimativa

Una variazione del 20% in v/V richiederebbe quindi una variazione del 20% nella concentrazione di A o di B se si modificasse la concentrazione di un substrato soltanto. Ma se si modifica la concentrazione di ambedue i substrati nella stessa direzione, allora basta una variazione del 10% nella concentrazione e di A e di B per produrre la variazione richiesta del 20% nel valore di v (una variazione relativa di v è la somma delle variazioni relative delle concentrazioni di A e di B, solo se le variazioni relative sono piccole e Ka>A, Kb>B). Analogamente al caso precedentemente discusso di reazione con un substrato, si può concludere che le condizioni specificate dall'equazione in forma semplificata rappresentano la massima sensibilità possibile per questo tipo di interazione e che, a mano a mano che le concentrazioni dei due substrati approssimano e poi superano i valori delle rispettive costanti di Michaelis, la sensibilità della risposta enzimatica diminuisce continuamente fino ad annullarsi.
Nella discussione precedente si è assunto che Ka e Kb non siano funzioni della concentrazione rispettivamente di A e di B. Tuttavia può accadere che Ka e Kb siano modificate da variazioni nella concentrazione del substrato (v. Dalziel, 1957). Per esempio, un aumento della concentrazione di A può abbassare la Km per l'altro substrato, B, e questo fa aumentare la risposta dell'enzima a cambiamenti della concentrazione di A in maggior misura di quanto descritto nell'esempio riportato più sopra.
L'aspetto più sfavorevole di questo tipo di interazione in rapporto alla regolazione dell'attività di un enzima è che esso richiede coordinazione fra almeno due regolatori (A e B). Se nell'esempio di prima la concentrazione di A e di B fosse cambiata in opposte direzioni, la sensibilità della risposta a ciascun substrato sarebbe stata inferiore a quella della risposta iperbolica. Quindi il prezzo da pagare per questo aumento di sensibilità di un enzima attraverso tale meccanismo è il grado di organizzazione necessario per coordinare i cambiamenti dei metaboliti regolatori. L'esperienza generale indica che questo tipo di interazione è importante per ridurre al minimo le variazioni di concentrazione dei substrati di sequenza in certe reazioni irreversibili dipendenti dal substrato, cioè quando la concentrazione di uno dei substrati (A o B) è determinata dal flusso e dalla risposta cinetica dell'enzima di cui esso è il substrato. Poiché la risposta cinetica può dipendere dalla concentrazione del substrato non di sequenza (per es. un cofattore), una variazione della sua concentrazione può diminuire la Km per il substrato di sequenza e perciò il flusso può aumentare senza che la concentrazione di stato stazionario del substrato di sequenza cambi notevolmente. Nel seguente sistema ipotetico
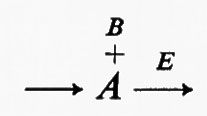
l'enzima E rappresenta una reazione irreversibile dipendente dalla concentrazione del substrato di sequenza A, e B rappresenta un cofattore. La concentrazione di A allo stato stazionario è funzione del flusso e della risposta cinetica di E. Se la concentrazione di B non cambia, ed E non è soggetto ad alcuna regolazione specifica, il flusso attraverso la reazione può aumentare solo se aumenta la concentrazione di A. Se invece l'aumento di concentrazione di B portasse a una diminuzione della Km dell'enzima per A, il flusso potrebbe aumentare senza cambiamenti notevoli nella concentrazione di A. Un esempio possibile per un sistema del genere è dato, nella glicolisi, dall'enzima piruvatochinasi, che catalizza la seguente reazione irreversibile:
fosfoenolpiruvato+ADP→piruvato+ATP.
Nel muscolo la risposta cinetica di quest'enzima è iperbolica sia verso il fosfoenolpiruvato (PEP) sia verso l'ADP e quindi un aumento del flusso attraverso la glicolisi di 100 volte potrebbe elevare la concentrazione del PFP di almeno 100 volte. Tuttavia i risultati sperimentali indicano che, per variazioni del flusso glicolitico fra 20 e 100 volte, la concentrazione del PFP non varia più di 4 volte (v. Williamson, 1965; v. Sacktor e Wormser-Shavit, 1966; v. Wilson e altri, 1967). Questa limitazione dell'aumento di concentrazione del PEP può essere dovuta al fatto che, quando aumenta il flusso attraverso la glicolisi, aumenta la concentrazione di ADP nel compartimento citoplasmatico della cellula.
c) Aumento di sensibilità prodotto da cinetiche enzimatiche non iperboliche.
In una reazione irreversibile con due substrati, il cambiamento relativo (o percentuale) di concentrazione di ciascun substrato può essere inferiore al cambiamento relativo del flusso che attraversa la reazione, purché la concentrazione di ambedue i substrati cambi nella stessa direzione. Questo tipo di sistema sarà ora esteso al caso in cui i due substrati siano rappresentati dallo stesso composto (S). L'equazione data precedentemente diventa:

dove Ka=Kb=K. La funzione espressa da questa equazione è continua per tutti i valori positivi di S e non possiede punti di flesso in questa regione. v/V si avvicina a 1 quando S diventa molto grande. Quando invece S piccolo rispetto a K, l'equazione diviene approssimativamente:
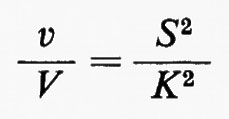
che esprime una relazione cinetica di secondo ordine rispetto a S. La risposta a S è mostrata nella fig. 3: il grafico di v in funzione di S è sigmoide (è ovviamente iperbolico quando una sola molecola di S si lega all'enzima). La deviazione dalla cinetica iperbolica è tanto più grande quanto maggiore è il valore di K, specialmente a basse concentrazioni di substrato.
I risultati di questo tipo di analisi possono essere estesi a reazioni in cui tre o più molecole di S si legano all'enzima. Se, per esempio, le molecole che si legano all'enzima sono tre, la parte iniziale della curva sigmoide sarà cubica in accordo con l'equazione:
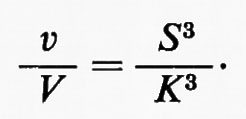
Per n molecole leganti di S la parte di destra dell'equazione diviene Sn/Kn. La ‛ripidità' della curva che segue all'‛appiattimento' iniziale dell'iperbole (analogo al periodo di ritardo - lag - di alcune reazioni) è pertanto funzione del numero di molecole che si legano a una molecola di enzima. Anche la sensibilità della risposta a S può essere accresciuta in questo modo, purché le costanti di affinità (Ks) per le differenti molecole di substrato non siano identiche, ma siano funzioni della concentrazione di S (cioè il legame di una molecola di substrato accresca l'affinità per la molecola successiva). In questo caso l'equazione che mette in relazione v/V e S è più complessa, ma dello stesso tipo dell'equazione presentata all'inizio di questo paragrafo. La sensibilità della risposta può essere così molto più grande di quella di una risposta iperbolica o lineare (cioè del primo ordine), come è illustato nella fig. 4 dove si può vedere che, se occorre modificare la velocità della reazione totale da v1 a v2, con una variazione relativa uguale a (v2−v1)/v1, le concentrazioni finali di S (c e d rispettivamente) sono simili nella risposta iperbolica e nella risposta sigmoide, ma nella risposta sigmoide la concentrazione iniziale di S (b) è maggiore che nella risposta iperbolica (a) e perciò la variazione relativa della concentrazione di S è minore. Dalla fig. 4 si può calcolare che, per una data variazione relativa della velocità di reazione (v2−v1)/v1, la variazione relativa di S è 44 e 3,7 per le risposte iperbolica e sigmoide rispettivamente. In altre parole, la sensibilità della risposta dell'enzima a S si è accresciuta rispetto alla risposta iperbolica. Qualitativamente, la transizione dalla risposta iperbolica a quella sigmoide può essere vista come una riduzione dell'attività enzimatica a basse concentrazioni di substrato, mentre essa non è modificata a concentrazioni alte. In questo modo la concentrazione del substrato non deve ridursi a livelli eccessivamente bassi quando è richiesta una bassa attività enzimatica. La variazione relativa di concentrazione del substrato necessaria per produrre una data variazione del flusso è così più limitata e la sensibilità della risposta al substrato (o al regolatore) nella regione di basse concentrazioni dello stesso è aumentata oltre quella della risposta iperbolica.
Ci sono molti esempi di enzimi che rispondono a un regolatore in modo sigmoide invece che iperbolico, per es. la fosfofruttochinasi (v. Monod e altri, 1965; v. Whitehead, 1970). Esistono anche molti modelli matematici per spiegare il comportamento sigmoide (v. Whitehead, 1970). La maggior parte di questi modelli sono basati su principi analoghi a quelli esposti all'inizio di questo paragrafo, cioè il legame di più di una molecola di regolatore per molecola di enzima, e sulla presenza di una qualche interazione fra i siti di legame. Uno di questi modelli è quello di Monod e altri (v., 1965), che nella sua forma più semplice dà origine all'equazione:

dove n rappresenta il numero di molecole di regolatore che si legano all'enzima, L (la costante allosterica) è in relazione all'interazione fra i siti di legame e α è un parametro direttamente proporzionale alla concentrazione del substrato o del regolatore. La forma di questa equazione è simile a quella data al principio del paragrafo (con n=2): la principale differenza consiste nella presenza di un termine aggiuntivo al numeratore, che però non modifica le caratteristiche qualitative della risposta.
d) Aumento di sensibilità prodotto da reintroduzione ciclica del substrato.
La natura sigmoide della risposta di una reazione enzimatica alla concentrazione di un regolatore aumenta la sensibilità della risposta stessa. Questa risposta si può considerare come una ‛deformazione' della risposta iperbolica, in cui l'attività enzimatica a basse concentrazioni di regolatore è ridotta. Tuttavia è probabile che esista un limite all'entità di questa deformazione e quindi alla sensibilità al regolatore di questo sistema. In teoria sarebbe possibile produrre una risposta sigmoide come quella illustrata nella fig. 5, e quindi una reazione ‛a scatto' nei riguardi della concentrazione del substrato o del regolatore, ma questo richiederebbe valori molto alti di n e di L. Secondo il modello semplificato di Monod e altri (v. sopra, cap. 3, È c), n dovrebbe essere all'incirca 60 (il che significa 60 moli di regolatore per mole di enzima) e L circa 1050, vale a dire valori impossibili per un semplice enzima globulare. Ci sono limiti superiori posti al valore di n da fattori come l'impedimento sterico e le grandi dimensioni e la complessità della molecola proteica; in pratica n ha un valore fra 2 e 6 moli per mole di enzima in un gran numero di enzimi. Limiti superiori esistono probabilmente anche per il grado d'interazione fra i siti di legame, che dipende dalla chimica fisica dei cambiamenti conformazionali della molecola proteica.
C'è però un altro mezzo per accrescere la sensibilità della risposta di un enzima alla concentrazione di un regolatore, senza che la cinetica dell'enzima venga alterata. Supponiamo che nella seguente reazione ipotetica catalizzata da un enzima E
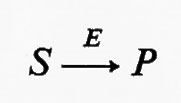
l'attività dell'enzima risponda a S in maniera sigmoide (v. fig. 6, curva 1). Se esiste una reazione irreversibile completamente differente, catalizzata dall'enzima F, che trasforma P in S, e se ambedue gli enzimi operano simultaneamente, si stabilisce fra S e P un ‛ciclo del substrato', cioè
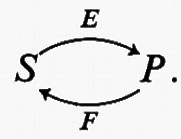
Se supponiamo inoltre che l'attività dell'enzima F sia costante e sia circa il 10% dell'attività massima di E (v. fig. 6, linea 3), la presenza di F cambierà la risposta della reazione netta nella direzione S→P rispetto a S (paragonare le curve 1 e 2 della fig. 6). L'effetto di F è di spostare in basso la curva di risposta, cosicché la velocità della reazione netta S→P è zero a concentrazioni finite di S. Qualitativamente è un effetto analogo alla ‛deformazione' dell'iperbole prodotta dalla risposta sigmoide.
Tuttavia questo mezzo di esaltazione della risposta al regolatore ha due svantaggi: il primo è che l'operazione del ciclo porta a liberazione continua di energia, essendo ambedue le reazioni irreversibili, e questa energia nella cellula vivente è fornita dal metabolismo; il secondo è che, nel sistema descritto, la velocità massima della reazione globale nella direzione S→P sarà più bassa del 10% rispetto all'attività massima dell'enzima E (v. fig. 6). Il primo di questi svantaggi non è molto grave se la velocità di operazione del ciclo è relativamente bassa, così che il ritmo di utilizzazione dell'energia non sia eccessivo e costituisca un prezzo ragionevole per l'accresciuta sensibilità di risposta. Il secondo svantaggio può essere completamente eliminato nel caso che l'attività dell'enzima F sia inibita dall'aumento di concentrazione di S (v. fig. 7). In questa situazione si ottiene ancora un'accresciuta sensibilità rispetto alla risposta di E da solo, ma il ciclo opera soltanto a basse concentrazioni di S, mentre ad alte concentrazioni di S non c'è riduzione del flusso totale a causa della presenza del ciclo. La somiglianza con la ‛deformazione' discussa in precedenza è qui anche più stretta: la velocità di reazione è ridotta a basse concentrazioni di S, ma è inalterata ad alte concentrazioni.
Un esempio fisiologico di ‛ciclo del substrato' è il sistema fosfofruttochinasi(PFK)-fruttosiodifosfatasi(FDP-asi), che in parte è responsabile della regolazione della glicolisi muscolare. Il sistema è il seguente:
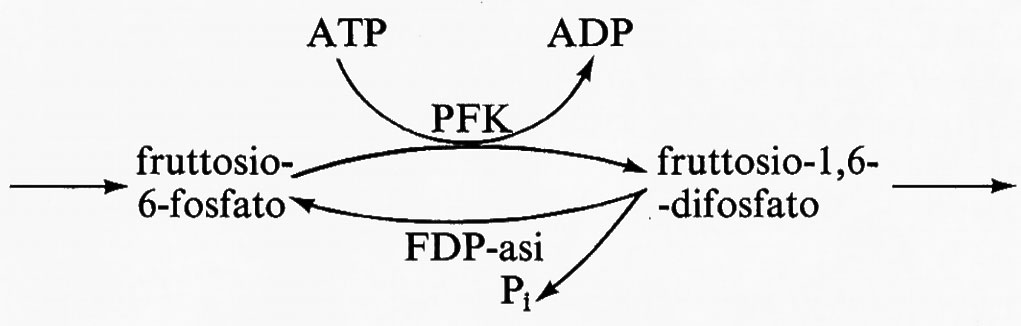
La massima attività catalitica dell'FDP-asi (equivalente all'enzima F) è in molti muscoli fra il 2 e il 10% di quella della PFK (equivalente all'enzima E). La PFK è attivata dal regolatore AMP, mentre la FDP-asi è inibita dall'AMP. Perciò la risposta all'AMP del flusso totale che attraversa questo sistema sarà simile a quella data nella fig. 7 piuttosto che a quella della fig. 6. Il costo energetico di questo ciclo sarà considerato nel paragrafo seguente.
L'importanza nella glicolisi di questo ‛ciclo del substrato' a livello della reazione della PFK è che il flusso netto da fruttosio-6-fosfato a fruttosio-difosfato diventa regolatorio per tutta la glicolisi e si adatta, mediante le variazioni di concentrazione dell'AMP, alla velocità di utilizzazione di energia (ATP) da parte della cellula (v. sopra, cap. 2). Grandi variazioni relative della concentrazione di AMP sono però ritenute impossibili per il loro effetto negativo sul rapporto (ATP)/(ADP) del citoplasma (v. Newsholme, 1970). I dati sperimentali indicano d'altra parte che, quando il flusso che attraversa la glicolisi cambia da 5 a 100 volte, la massima variazione nella concentrazione di AMP è soltanto di circa 4 volte (v. Opie e altri, 1971; v. Sacktor e Hurlbut, 1966). Perciò la risposta del flusso glicolitico a variazioni di concentrazione dell'AMP deve essere molto sensibile e si pensa che questa alta sensibilità all'AMP sia dovuta alla presenza di un ‛ciclo del substrato' a livello della fosforilazione del fruttosio-6-fosfato. Nella tab. II è presentata un'analisi teorica di questo sistema secondo il modello semplificato di Monod e altri. Da questa analisi si può vedere che un aumento di circa 5 volte della concentrazione di AMP (da 2,5 a 12 unità) aumenta il flusso di circa 435 volte (da 0,2 a 87,9 unità), rispetto all'aumento di circa 10 volte (da 9,3 a 89 unità) che si ha con la sola PFK in assenza di ciclo. Notizie più estese sul ruolo dei ‛cicli del substrato' nel metabolismo possono trovarsi nelle rassegne di Newsholme e Oevers (v., 1967) e di Newsholme e Start (v., 1972).

e) Aumento di sensibilità prodotto dalla presenza di forme interconvertibili di un enzima.
Un ‛ciclo del substrato' è capace di accrescere la sensibilità di un enzima al cambiamento di concentrazione di un regolatore. Per quanto teoricamente la sensibilità potrebbe essere accresciuta indefinitamente aumentando il ritmo del processo ciclico (v. Newsholme e Start, 1972), in pratica esistono dei limiti a questo meccanismo. La limitazione principale consiste nel consumo di energia durante l'operazione del ciclo. Nell'esempio illustrato dalla fig. 8, con una velocità di ciclo pari al 10% dell'attività massima dell'enzima, un flusso netto uguale a zero è prodotto con (S)=a; la variazione relativa di (S) necessaria per cambiare il flusso da zero a v2 (che è vicino al valore di V) sarà (c−a)/a. Se ci fosse bisogno di accrescere la sensibilità al di sopra di questo intervallo di flusso, occorrerebbe aumentare la velocità di operazione del ciclo. Se, per esempio, ci fosse bisogno di una variazione relativa di (S) pari a (c−b)/b (v. fig. 8), occorrerebbe produrre un flusso zero per (S)=b. In questo caso la velocità di ciclo sarebbe circa il 50% dell'attività massima dell'enzima e il consumo di energia potrebbe essere eccessivo. Nel muscolo pettorale di fagiano l'attività massima della fosfofruttochinasi a 35 °C è circa 280 μmoli/min/g di muscolo, cosicché a una velocità di ciclo pari al 50% di questa attività, la velocità di idrolisi dell'ATP, solamente ai fini del ‛ciclo del substrato', è 140 μmoli/min/g. Per fornire al ciclo energia sufficiente sarebbe necessario ossidare glucosio alla velocità di circa 4 μmoli/min/g. Poiché il peso totale del muscolo pettorale di fagiano è circa 200 g, l'uccello dovrebbe utilizzare 800 ‛moli di glucosio per minuto, a riposo, per mantenere il ciclo del substrato, mentre la quantità totale di glucosio nel sangue del fagiano è meno di 800 μmoli. L'attività effettiva dell'FDP-asi (e quindi la massima velocità del ‛ciclo del substrato') in questo muscolo è solo 9 μmoli/min/g (a 35°), il che significa una velocità di consumo di glucosio nell'intero animale di circa 40 μmoli/min/g al fine di mantenere il ciclo: questo valore incide in misura notevolmente inferiore sulle riserve di carboidrati. Il ‛ciclo del substrato' è perciò inadeguato se è necessaria una sensibilità molto alta e in particolare se l'attività enzimatica deve passare da livelli molto bassi a livelli molto alti. Un altro sistema per accrescere la sensibilità della risposta di un enzima a un regolatore sfrutta l'esistenza di forme interconvertibili di enzima. È noto che alcuni enzimi esistono in due forme, attiva e inattiva, che possono trasformarsi l'una nell'altra per azione catalitica di enzimi specifici, come illustrato nello schema seguente:

In questo sistema l'enzima E esiste in due forme interconvertibili, una enzimaticamente attiva e l'altra inattiva o molto meno attiva. La conversione di una forma nell'altra è catalizzata da due distinte reazioni enzimatiche (enzimi F e G). Ognuna delle due reazioni è irreversibile e catalizza una conversione quasi totale di una forma di E nell'altra. L'operazione simultanea delle reazioni catalizzate da F e da G è perciò un processo che consuma energia, analogo a un ciclo del substrato. Ma la differenza importante rispetto al ciclo del substrato sta nel fatto che le attività massime di F e di G non dipendono dal flusso che attraversa la via metabolica regolata dall'enzima E. Perciò le attività di F e G possono essere sufficienti a produrre una rapida conversione di una forma di E nell'altra (e così attivare o inattivare l'enzima) e allo stesso tempo abbastanza basse da esigere un rifornimento continuo, ma non eccessivo, di energia; un ciclo di questo tipo potrebbe operare con le attività degli enzimi F e G vicine o superiori al 50% dell'attività massima.
In questo tipo di sistema di controllo, appena l'attività dell'enzima F sopravanza quella dell'enzima G, E viene trasformato nella forma inattiva e viceversa. Come nel caso di ciclo del substrato, se l'enzima F fosse inibito e l'enzima G fosse attivato da un regolatore R (v. lo schema precedente), una variazione molto piccola di concentrazione del regolatore (anche solo del 10%) nella regione in cui le attività di F e di G sono simili potrebbe produrre un'attivazione o un'inibizione quasi totali dell'enzima E. In questo modo il sistema offre un mezzo assai sensibile di regolazione dell'attività enzimatica. Il limite superiore alla sensibilità del sistema è fissato dalla ‛velocità netta' di interconversione delle due forme. Nell'esempio precedente questa velocità è data dalla differenza fra le attività di F e di G; essa (e quindi anche la variazione di R necessaria a produrla) deve essere sufficiente a trasformare fra di loro le due forme in un tempo ragionevolmente breve. Un esempio di questo sistema è la glutamminasintetasi di Escherichia coli (v. Holzer, 1969). Questo enzima esiste in una forma attiva e in una molto meno attiva: la forma inattiva è prodotta dall'adenilazione (addizione di AMP con formazione di legame ‛covalente') della forma attiva, mentre la sua attivazione richiede la deadenilazione. Le attività degli enzimi dell'interconversione (equivalenti agli enzimi F e G dell'esempio ipotetico) sono influenzate dalla glutammina in maniera analoga all'ipotetico effetto di R su F e G. Così un aumento di concentrazione della glutammina relativamente piccolo può produrre un'attivazione pressoché totale della glutamminasintetasi, portando all'arresto della sintesi di questo metabolita. Questo esempio sottolinea anche una caratteristica importante dei sistemi con forme interconvertibili di enzima: l'interconversione implica la formazione di legami ‛covalenti' nella molecola proteica, e questo processo è reversibile solo per mezzo di una diversa reazione chimica. Questa situazione contrasta con i sistemi a legame reversibile che sono stati considerati finora. Altri enzimi che esistono in due forme interconvertibili sono la glicogenosintetasi, la lipasi e la piruvatodeidrogenasi (v. Villar-Palasi e Larner, 1968; v. Reed, 1969; v. Corbin e altri, 1970).
C'è un'altra proprietà estremamente importante dei sistemi regolatori a forme interconvertibili di enzima, che interviene specialmente quando la concentrazione del regolatore è paragonabile o è molto minore rispetto a quella dell'enzima regolatorio. Con un meccanismo di legame reversibile del tipo
E +X⇄EX
la concentrazione totale di X deve essere più grande o almeno eguale rispetto a quella di E perché si abbia un'attivazione o un'inibizione massima di E. Questa condizione è stata presupposta durante tutte le discussioni precedenti. Va da sé che in un sistema a legame reversibile un effetto massimo di X sull'attività di E sarebbe impossibile qualora la concentrazione di X fosse inferiore a quella di E. Nei sistemi di controllo a forme enzimatiche interconvertibili la concentrazione del regolatore può essere invece molto più bassa di quella dell'enzima regolatorio perché modifica l'attività degli enzimi d'interconversione.
Se la concentrazione totale di X fosse uguale o leggermente superiore a quella di E, in modo da rendere teoricamente possibile una risposta massima a X, resterebbe ancora un problema collegato alla regolazione dell'attività di E da parte di X, in conseguenza del fatto che la costante di affinità di E per X dovrebbe essere in questo caso molto grande. Newsholme e Start (v., 1972) hanno discusso l'influenza della costante di affinità di un sistema a legame reversibile, cioè il rapporto

sul tempo necessario a trasformare EX in E, in conseguenza di un abbassamento di concentrazione di X, in condizioni in cui le concentrazioni di X ed E siano simili. Questi autori hanno mostrato che il tempo necessario alla trasformazione di EX in E aumenta notevolmente all'aumentare del valore della costante di affinità. Un sistema simile sarebbe perciò inadeguato in un sito di regolazione del flusso, poiché l'inerzia di un sistema metabolico così regolato farebbe durare la sua operazione oltre il cambiamento dello stimolo regolatore, rendendo pertanto instabile l'intero sistema. Perciò esiste un limite superiore alla costante di affinità in un sistema regolatorio a legame reversibile e quindi un limite inferiore alla concentrazione del regolatore rispetto a quella dell'enzima. In una cellula la concentrazione di un enzima è determinata dal flusso massimo che attraversa la via metabolica di cui esso è parte e dal meccanismo chimico del processo catalitico; la combinazione di questi due parametri si riflette nell'attività specifica dell'enzima. In effetti, si conoscono molti esempi in cui la concentrazione dell'enzima è maggiore di quella del regolatore e quindi non è possibile un semplice sistema di controllo a legame reversibile, ma è necessario un sistema a forme enzimatiche interconvertibili.
Se in un sistema di controllo dipendente da enzimi interconvertibili la concentrazione del regolatore è superiore - o simile - a quella dell'enzima di interconversione, il sistema di controllo può essere esteso a includere forme interconvertibili degli enzimi F e G, come mostrato nello schema seguente:
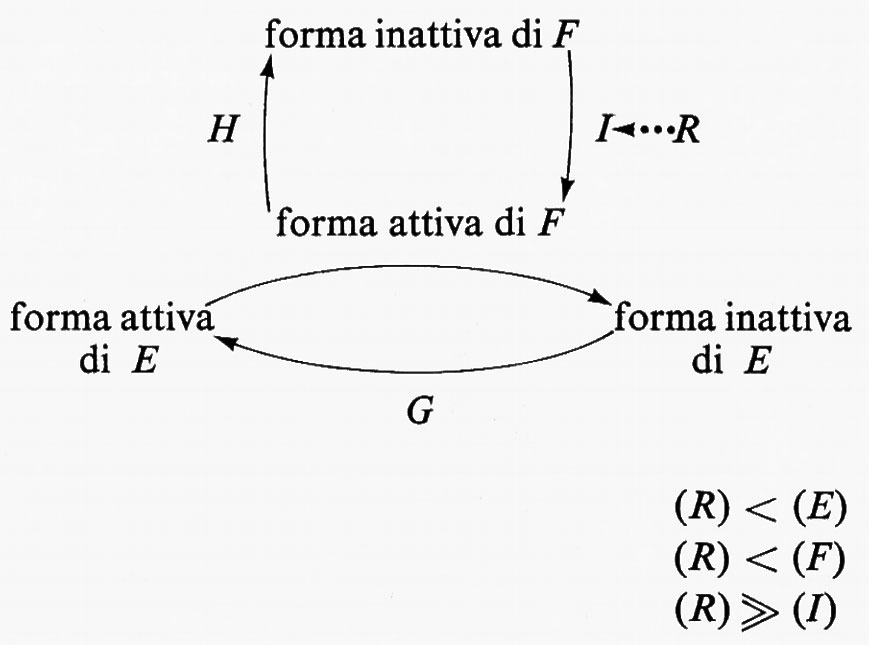
In questo sistema la concentrazione del regolatore R è minore di quella dell'enzima E o di quella del suo enzima d'interconversione F, ma è molto più grande di quella dell'enzima I. R interagisce con I mediante legame reversibile per produrre effetto massimo sull'attività di I; questo a sua volta produce effetto massimo nell'attivazione di F che provoca inattivazione massima di E. Questo sistema è del tipo ‛a cascata' poiché in esso una bassa concentrazione di regolatore può attivare o inattivare una concentrazione di enzima molto più alta. Il primo esempio che sia stato scoperto di un effetto ‛a cascata' è il sistema della coagulazione del sangue, in cui una quantità molto piccola del fattore coagulante iniziale porta alla conversione di quantità molto più grandi di fibrinogeno in fibrina (v. MacFarlane, 1964). C'è soltanto un altro esempio specifico di regolazione enzimatica per mezzo di un sistema di forme interconvertibili disposte ‛a cascata'. Si tratta della glicogenofosforilasi (v. Fischer e altri, 1970). Questo sistema ‛a cascata' è regolato da piccolissime variazioni di concentrazione assoluta di AMP ciclico. Per garantire un meccanismo rapido e reversibile di regolazione da parte dell'AMP ciclico, un sistema ‛a cascata' è essenziale (v. Newsholme e Start, 1972).
Bibliografia.
Banks, B. E. C., Vernon, C. A., Ressessment of the role of ATP in vivo, in ‟Journal of theoretical biology", 1970, XXIX, pp. 301-326.
Bergmeyer, H. U. (a cura di), Methods of enzymatic analysis, New York-London 1963.
Bücher, Th., Rüssmann, W., Equilibrium and nonequilibrium in the glycolysis system, in ‟Angewandte Chemie. International edition", 1964, III, pp. 426-439.
Chance, B., Eastbrook, R. W., Ghosh, A., Damped sinusoidal oscillations of cytoplasmic reduced pyridine nucleotide in yeast cells, in ‟Proceedings of the National Academy of Sciences", 1964, LI, pp. 1244-1251.
Childress, C. C., Sacktor, B., Regulation of glycogen metabolism in Insect flight muscle. Purification and properties of phosphorylases in vitro and in vivo, in ‟Journal of biological chemistry", 1970, CCXLV, pp. 2927-2936.
Cleland, W. W., Enzyme kinetics, in ‟Annual review of biochemistry", 1967, XXXVI, pp. 77-112.
Corbin, J. D., Reinman, E. M., Walsh, P. A., Krebs, E. G., Activation of adipose tissue lipase by skeletal muscle cyclic adenosine 3′, 5′-monophosphate-stimulated protein kinase, in ‟Journal of biological chemistry", 1970, CCXLV, pp. 4849-4851.
Crabtree, B., Newshome, E. A., The activities of hexokinase, phosphorylase, phosphofructokinase, lactate dehydrogenase and the glycerol 3-phosphate dehydrogenases in muscles from Vertebrates and Invertebrates, in ‟Biochemical journal", 1972, CXXVI, pp. 49-58.
Dalziel, K., Initial steady-state velocities in the evaluation of enzyme-coenzyme-substrate reaction mechanisms, in ‟Acta chemica scandinavica", 1957, XI, pp. 1706-1723.
Ebashi, S., Endo, M., Calcium ion and muscle contraction, in ‟Progress in biophysic and molecular biology", 1968, XVIII, pp. 123-183.
Fischer, E. H., Pocker, A., Saari, J. C., The structure, function and control of glycogen phosphorylase, in Essays in biochemistry (a cura di P. N. Campbell e F. Dickens), vol. VI, London-New York 1970, pp. 23-68.
Fletcher, W. M., Hopkins, F. G., Lactic acid in Amphibian muscle, in ‟Journal of physiology", 1907, XXXV, pp. 247-309.
Hansford, R. G., Chappell, J. B., The effects of Ca2+ on the oxidation of glycerol phosphate by blowfly flight muscle mitochondria, in ‟Biochemical and biophysical research communications", 1967, XXVII, pp. 686-692.
Hansford, R. G., Sacktor, B., Regulation of glycogen metabolism in Insect flight muscle. Activation of phosphorylase ‛b' kinase by calcium and inorganic phosphate, in ‟FEBS letters", 1970, VII, pp. 183-187.
Holzer, H., Regulation of enzymes by enzyme-catalyzed chemical modification, in Advances in enzymology (a cura di F. F. Nord), vol. XXXII, New York 1969, pp. 297-326.
Katchalsky, A., Curran, P. F., Nonequilibrium thermodynamics in biophysics, Cambridge, Mass., 1965.
Krebs, H. A., Cyclic processes in living matter, in ‟Enzymologia", 1946, XII, pp. 88-100.
Krebs, H. A., Control of metabolic processes, in ‟Endeavour", 1957, XVI, pp. 125-132.
Krebs, H. A., Veech, R. L., Pyridine-nucleotide interrelations, in The energy level and metabolic control in mitochondria (a cura di S. Papa, J. M. Tager, E. Quagliariello ed E. C. Slater), Bari 1969, pp. 329-382.
MacFarlane, R. G., An enzyme cascade in the blood clotting mechanism and its function as a biochemical amplifier, in ‟Nature", 1964, CCII, pp. 498-499.
Monod, J., Changeux, J.-P., Jacob, Fr., Allosteric proteins and cellular control systems, in ‟Journal of molecular biology", 1963, VI, pp. 306-329.
Monod, J., Wyman, J., Changeux, J.-P., On the nature of allosteric transitions. A plausible model, in ‟Journal of molecular biology", 1965, XII, pp. 88-118.
Morgan, H. E., Randle, P. J., Regen, D. M., Regulation of glucose uptake by muscle. The effects of insulin, anoxia, salicylate and 2:4-dinotrephenol on membrane transport and intracellular phosphorylation of glucose in the isolated rat heart, in ‟Biochemical journal", 1959, LXXIII, pp. 573-579.
Newsholme, E. A., Theoretical and experimental considerations on the control of glycolysis in muscle, in Essays in cell metabolism (a cura di W. Bartley, H. L. Kornberg e J. R. Quayle), New York 1970, pp. 189-223.
Newsholme, E. A., Gevers, W., Control of glycosis and gluconeogenesis in liver and kidney cortex, in Vitamins and hormones, vol. XXV (a cura di R. S. Harris e I. G. Wool), New York 1967, pp. 1-87.
Newshome, E. A., Start, C., Insulin and the regulation of enzyme activity, in Handbook of physiology: the endocrine pancreas (a cura di D. F. Steiner, N. Frainkel), Washington 1972.
Opie, L. H., Mansford, K. R. L., Owen, P., Effects of increased heart work on glycolysis and adenine nucleotides in the perfused heart of normal and diabetic rats, in ‟Biochemical journal", 1971, CXXIV, pp. 457-490.
Pasteur, L., Études sur la bière, Paris 1876.
Pringle, J. W. S., The contractile mechanism of Insect fibrillar flight muscle, in ‟Progress in biophysics and molecular biology", 1967, XVII, pp. 1-60.
Randle, P. J., Morgan, H. E., Regulation of glucose uptake by muscle, in Vitamins and hormones, vol. XX (a cura di R. S. Harris, D. J. Ingle e J. H. Loraine), New York 1962, pp. 199-249.
Reed, L. J., Pyruvate dehydrogenase complex, in Current topics in cellular regulation (a cura di B. L. Horecker ed E. R. Stadtman), vol. I, New York-London 1969, pp. 233-296.
Rolleston, F. S., Newsholme, E. A., Control of glycolysis in cerebral cortex slices, in ‟Biochemical journal", 1967, CIV, pp. 524-533.
Sacktor, B., Hurlbut, E. C., Regulation of metabolism in working muscle in vivo: concentration of adenine nucleotides, arginine phosphate and inorganic phosphate in Insect flight muscle during flight, in ‟Journal of biological chemistry", 1966, CCXLI, pp. 632-634.
Sacktor, B., Wormser-Shavit, E., Regulation of metabolism in working muscle in vivo: concentration of some glycolytic, tricarboxylic acid cycle and amino acid intermediates in Insect flight muscle during flight, in ‟Journal of biological chemistry", 1966, CCXLI, pp. 624-631.
Villar-Palasi, C., Larner, J., The hormonal regulation of glycogen metabolism in muscle, in Vitamins and hormones, vol. XXVI (a cura di R. S. Harris, I. G. Wool e J. H. Loraine), New York 1968, pp. 65-118.
Weis-Fogh, T., Metabolism and weight economy in migrating animals, particularly Birds and Insects, in Insects and physiology (a cura di J. W. Beament e J. H. Trehearne), Edinburgh 1967, pp. 143-159.
Whitehead, E. P., The regulation of enzyme activity and allosteric transitions, in ‟Progress in biophysics and molecular biology", 1970, XXI, pp. 321-397.
Williamson, J. R., Glycolytic control mechanism. I. Inhibition of glycolysis by acetate and pyruvate in the perfused rat heart, in ‟Journal of biological chemistry", 1965, CCXL, pp. 2308-2321.
Wilson, J. E., Sacktor, B., Tiekert, C. G., In situ regulation of glycolysis in tetanized cat skeletal muscle, in ‟Archives of biochemistry and biophysics", 1967, CXX, pp. 542-546.
Wollenberger, A., Ristau, O., Schoffa, G., Eine einfache Technik der extrem schnellen Abkühlung grösserer Gewebestücke, in ‟Pflügers Archiv", 1960, CCLXX, pp. 399-412.
Metabolismo dei carboidrati
SOMMARIO: 1. Introduzione. □ 2. Glicolisi. □ 3. La via dei pentosofosfati. □ 4. Gluconeogenesi. □ 5. Monosaccaridi. □ 6. La via dei glucuronato. □ 7. Oligo- e polisaccaridi. □ 8. Polisaccaridi complessi. □ 9. Aspetti immunologici dei carboidrati. □ 10. Diabete mellito. □ 11. Digestione. □ Bibliografia.
1. Introduzione.
La dimestichezza dell'uomo con il metabolismo dei carboidrati deve risalire a tempi molto antecedenti alla storia scritta, in quanto il lievito, e quindi la fermentazione, è stato sempre usato. Sappiamo poi che i medici dell'antica Grecia erano in grado di riconoscere un alterato metabolismo dei carboidrati in base alla circostanza che le urine dei diabetici sono dolci.
Solo raramente è possibile indicare nella storia della scienza un singolo evento che abbia determinato un progresso sensazionale in un'importante branca del sapere; tale è stato l'esperimento fatto da F. Buchner nel 1897. In quell'anno egli triturò con sabbia cellule di lievito e ne ottenne un materiale solubile, privo di cellule, capace di fermentare il glucosio con produzione di CO2 e di etanolo. Noi sappiamo, oggi, che la cosiddetta ‛zimasi', l'enzima della fermentazione, è formata in realtà da parecchi enzimi e cofattori, tutti solubili e capaci di operare anche a prescindere dalla integrità dell'organizzazione cellulare.
Disponendo di questo sistema sperimentale ideale, A. Harden e W. J. Young scoprirono che nel corso della reazione scompariva del fosfato inorganico, il che indicava la formazione di esteri organici dell'acido fosforico.
Fu una felice coincidenza il fatto che fosse disponibile un metodo eccellente, benché, misurato col metro di oggi, complicato, per la determinazione dei fosfati. Così cominciò l'isolamento di molti composti intermedi lungo il percorso metabolico del glucosio fino ai suoi prodotti di fermentazione.
Per menzionare solo alcuni dei pionieri che eseguirono queste ricerche ci limiteremo a ricordare R. Robinson, O. Meyerhof, C. Neuberg, G. Embden, J. K. Parnas, O. Warburg e i coniugi C. F. e G. T. Cori.
Man mano che i prodotti intermedi venivano scoperti e messi in ordine di successione, la ‛zimasi' si andò frazionando nei suoi enzimi costitutivi, mentre venivano identificati e collocati al loro posto i vari cofattori. Nel 1940 il mosaico era terminato, con l'eccezione di dettagli riguardanti la cinetica, la termodinamica e la regolazione, lasciati alle indagini dei ricercatori successivi.
Questa impresa è stata veramente notevole, perché compiuta prima dell'avvento delle tecniche radioisotopiche e cromatografiche, e delle sofisticate attrezzature attuali. In onore dei principali scopritori di questa via metabolica - Embden, Meyerhof e Parnas - tale sequenza di reazioni divenne nota come via di EMP.
Benché la fermentazione, o glicolisi anaerobica, sia stata studiata prevalentemente nel lievito, essa è presente, con piccole modificazioni, in ogni essere vivente: ciò non deve sorprendere, se si considera l'universalità del glucosio come alimento. È vero tuttavia che l'etanolo può essere sostituito da altri composti ridotti, come l'acido lattico nell'uomo e nella maggior parte degli animali: si vedrà in seguito che gli esseri aerobi formano di solito come composto finale della glicolisi l'acido piruvico, che viene metabolizzato attraverso il ciclo degli acidi tricarbossilici.
Il metabolismo glucidico comprende naturalmente, oltre al metabolismo del glucosio e dei suoi derivati, anche le reazioni anaboliche e cataboliche degli altri carboidrati. Esso riguarda i monosaccaridi, compresa una serie di isomeri degli esosi, e meno frequentemente i pentosi e gli eptosi. Si conoscono, come si vedrà, numerosi zuccheri sostituiti, coniugati con fenoli, fiavonoidi, amminoderivati, fosfato, solfato, basi degli acidi nucleici, lipidi, proteine, inositolo, steroidi e molti altri agliconi. Gli zuccheri possono trovarsi come disaccaridi, costituiti da due unità eguali o diverse, trisaccaridi e così via, fino ai polisaccaridi a elevato peso molecolare, come il glicogeno, l'amido, la chitina e la cellulosa. I carboidrati possono anche essere legati o mescolati con proteine e lipidi in importanti costituenti metabolici e come componenti delle complesse pareti cellulari e delle membrane degli organuli subcellulari.
Di solito si verifica che, se un organismo è in grado di sintetizzare uno zucchero o un suo derivato, esso stesso o un altro organismo sono capaci di degradarlo.
Lo studio del metabolismo dei carboidrati comprende la loro formazione, l'utilizzazione, la regolazione e l'integrazione nei fabbisogni dell'organismo. La regolazione nell'uomo è importantissima per la medicina ed è oggetto di studio sempre più attento.
Intimamente connesso col problema del metabolismo è il campo, in rapida espansione, del trasporto di membrana.
2. Glicolisi.
Lo schema della glicolisi secondo Embden-Meyerhof-Parnas viene riportato nella pagina seguente (v. schema 1). Si noti che il piruvato può trasformarsi in etanolo, via acetaldeide, o alternativamente in acido lattico, in entrambi i casi con ossidazione del NADH (nicotinammide-adenindinucleotide ridotto) a NAD+: la prima reazione avviene nel lievito, la seconda nei muscoli. Il modo in cui sono riossidati i coenzimi piridinici varia a seconda delle specie, come varia la specificità per i coenzimi. Inoltre vari zuccheri, oligo- e polisaccaridi, possono rifornire di monosaccaridi il sistema. I passaggi principali sono stati studiati nel lievito, nei Mammiferi, nelle piante superiori e in molti microrganismi.
Esochinasi e fruttochinasi. - Benché, come da tutti accettato, le esochinasi siano enzimi che catalizzano la fosforilazione del glucosio tramite l'ATP (adenosintrifosfato), alcune di esse sono poco specifiche: infatti l'esochinasi del lievito può fosforilare, oltre al glucosio, la fruttosammina, il fruttosio e il mannosio. Tutte le esochinasi richiedono Mg2+; per mezzo di substrati marcati con 18O si è potuto stabilire che esse agiscono sul legame P−O piuttosto che su quello C−O.
Nel fegato dei Mammiferi vi sono due tipi diversi di enzimi fosforilanti il glucosio: oltre alla classica esochinasi, che svolge il ruolo indicato nello schema precedente, vi è un secondo enzima, detto glucochinasi. Una delle funzioni del fegato è l'accumulo del glucosio in eccesso come glicogeno; si suppone che la glucochinasi abbia la funzione di bloccare il glucosio libero sotto forma di glucosio-6-fosfato, prima tappa della sua conversione a glicogeno attraverso il glucosio-1-fosfato e l'uridindifosfoglucosio (v. sotto).
Questa ipotesi del ruolo della glucochinasi nella glicogenesi è basata sui punti seguenti: a) a differenza dell'esochinasi, non è inibita dal glucosio-6-fosfato; b) ha una costante di Michaelis (Km) per il glucosio molto più alta di quella dell'esochinasi e quindi diventa più attiva quando il livello intracellulare di glucosio aumenta. Per contro, l'esochinasi, avendo una bassa Km, può competere bene per il glucosio quando questo è in basse concentrazioni per soddisfare il normale fabbisogno glicolitico della cellula: questo fabbisogno deve ovviamente avere la precedenza rispetto all'accumulo quando il livello di glucosio è basso; c) il contenuto di glucochinasi è basso nel fegato del feto, in cui non si ha eccesso di glucosio; d) trattamenti ormonali che causano ipoglicemia producono una diminuzione della glucochinasi, ma non dell'esochinasi; e) la glucochinasi si trova in quei tessuti che formano glicogeno, ma è assente in quelli in cui si ha solo la glicolisi.
Vi sono malattie genetiche dell'uomo legate a una deficienza di esochinasi negli eritrociti: ciò comporta una precoce distruzione di tali cellule e provoca anemie emolitiche croniche.
Benché gli enzimi della glicolisi siano generalmente di- sciolti nel citoplasma, in alcuni casi le esochinasi sembrano legate a organuli, e precisamente ai mitocondri.
Le esochinasi dei Mammiferi si trovano in tipi multipli isolabili mediante elettroforesi e sono presenti in tutti i tessuti esaminati. Benché la specificità delle esochinasi sia abbastanza ampia da riguardare anche il fruttosio, i tessuti dei Mammiferi contengono una fruttochinasi specifica che catalizza la formazione di fruttosio-1-fosfato, a differenza dell'esochinasi che forma fruttosio-6-fosfato. Un difetto genetico della fruttochinasi può talvolta provocare, in caso di alimentazione con fruttosio, la fruttosuria. È quindi probabile che in vivo non sia l'esochinasi ad attaccare il fruttosio. La deficienza di fruttochinasi non causa comunque disturbi apprezzabili.
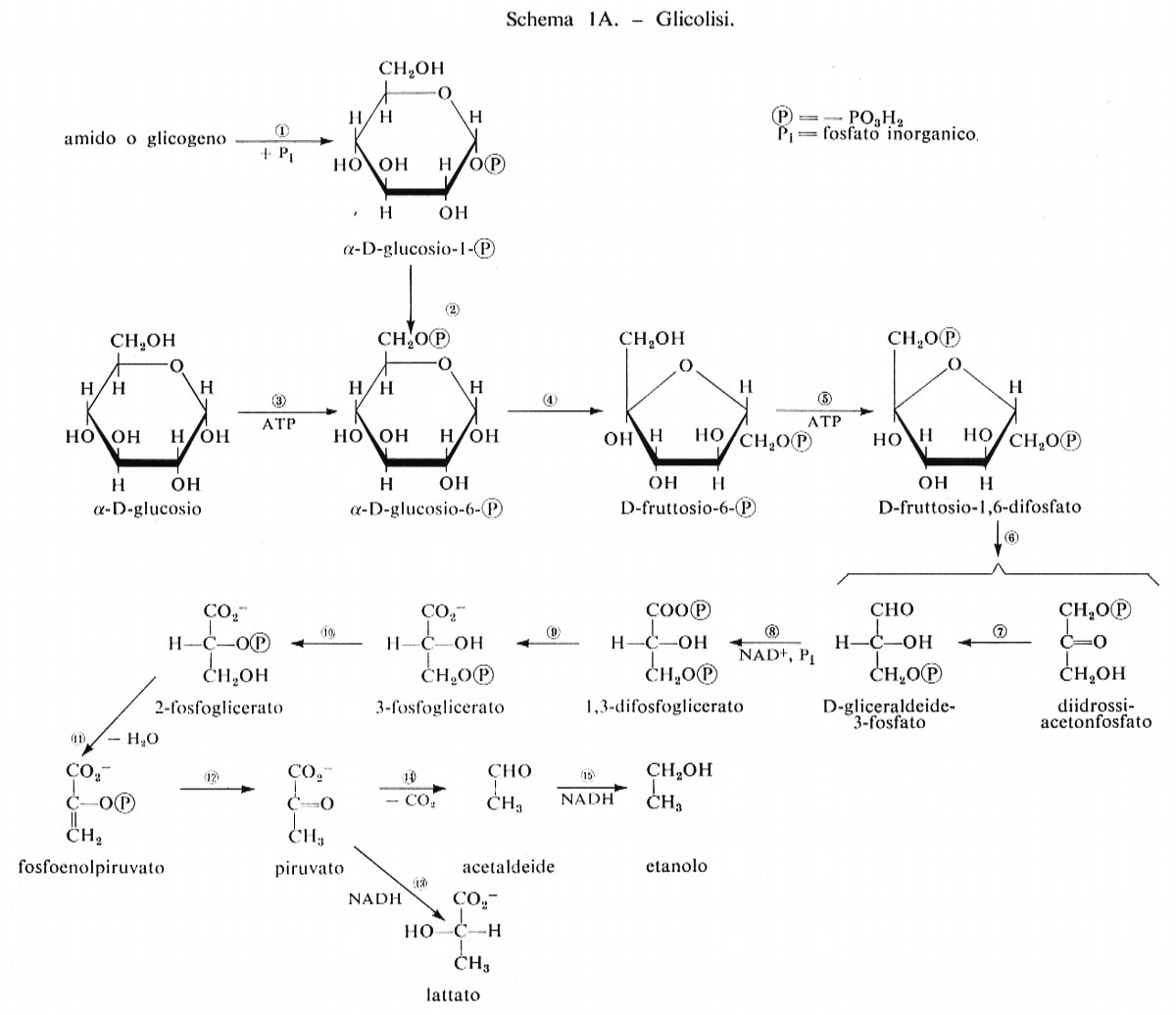

Fosfoglucomutasi. - La fosfoglucomutasi ha un ruolo fondamentale nell'introdurre nella via metabolica di EMP il glucosio-1-fosfato, che deriva dalla fosforolisi del glicogeno negli animali o dell'amido nelle piante; l'enzima catalizza anche la reazione inversa, formando glucosio-1-fosfato dal glucosio-6-fosfato. Come si vedrà in seguito, il glucosio-1-fosfato è il precursore dei derivati nucleotidici del glucosio per la formazione del glicogeno, dell'amido, e di molti altri glucosidi. L'enzima, che richiede Mg2+, è stato purificato e ampiamente studiato: il meccanismo della reazione comporta trasferimento di fosforo tra il substrato e l'enzima nel modo qui illustrato:

in cui E−P indica l'enzima fosforilato. Quindi si formano, come prodotti collaterali, piccole quantità di glucosio-1,6-difosfato e di enzima defosforilato.
Il glucosio-1,6-difosfato stimola la reazione, se vi è enzima defosforilato, causando la sua rifosforilazione, e quindi si comporta da cofattore. È largamente provato, per fosfoglucomutasi di diversa provenienza, che nell'enzima fosforilato l'acido fosforico è esterificato con un residuo di senna.
Tre genotipi comuni e indipendenti determinano le forme di fosfoglucomutasi presenti nell'uomo: l'elettroforesi rivela forme molecolari multiple dell'enzima, con fenotipi mostranti da 5 a 7 isoenzimi, tutti con proprietà enzimatiche molto simili. Ulteriori varianti derivano dalle diverse possibilità alleliche per ciascuno di questi geni. Non vi sono condizioni patologiche associate con varianti genetiche, probabilmente a causa della grande molteplicità di forme enzimatiche riscontrabili in ciascun individuo.
Fosfoesosoisomerasi. - La formazione reversibile di fruttosio-6-fosfato a partire dal glucosio-6-fosfato è catalizzata dalla glucosio-6-fosfatoisomerasi; tale reazione è comune alla glicolisi e alla glucogenesi. Analoga è la mannosio-6-fosfatoisomerasi, che utilizza il mannosio-6-fosfato; benché questi enzimi agiscano su di un substrato comune, il fruttosio-6-fosfato, mostrano di distinguere l'uno dall'altro, a seconda della configurazione spaziale, i due idrogeni legati al carbonio 1 (v. schema 2).
In esperimenti a breve durata si è visto che l'atomo di idrogeno rimosso dal carbonio i viene trasferito al carbonio 2. Nell'uomo si trovano varianti della glucosio-6-fosfato-isomerasi: è noto che una variante difettiva, controllata geneticamente, causa anemia emolitica interferendo con la glicolisi negli eritrociti.
Fosfofruttochinasi. - Questo enzima, responsabile della trasformazione del fruttosio-6-fosfato in fruttosio-1,6-difosfato, occupa una posizione chiave nella glicolisi, in quanto catalizza una reazione irreversibile ed è quindi un enzima esclusivamente glicolitico. I coniugi Cori, trent'anni or sono, studiando il tessuto muscolare, indicarono la fosfofruttochinasi come enzima vitale di controllo, avendo osservato che grandi variazioni nella concentrazione di esosomonofosfato non erano accompagnate da equivalenti variazioni del prodotto della glicolisi, l'acido lattico. Qualche tempo dopo H.A. Lardy scoprì che un eccesso di uno dei substrati, l'adenosintrifosfato, inibiva l'enzima. Oggi si sa che l'enzima ha un ruolo regolatore e che è controllato allostericamente (v. enzimi). Le fosfofruttochinasi di diversa origine esistono in una forma attiva, associata, e una inattiva, dissociata: mettendo in grafico l'attività enzimatica in funzione della concentrazione di fruttosio-6-fosfato si ottiene una curva sigmoide.
Il fosfato inorganico e l'acido 5′-adenilico, le cui concentrazioni intracellulari sono alte quando l'adenosintrifosfato è diminuito, stimolano l'enzima. Viceversa alti livelli di adenosintrifosfato, indicanti notevoli riserve di energia, fanno diminuire ulteriormente la glicolisi. Anche il citrato inibisce questa chinasi, collegando così la regolazione della glicolisi al ciclo degli acidi tricarbossilici di Krebs.
L'effetto stimolatore della serotonina sulla glicolisi in omogenati del parassita epatico Fasciola hepatica può essere attribuito anch'esso all'attivazione della fosfofruttochinasi: l'effetto è mediato dalla produzione indotta di acido adenilico 3′-5′-ciclico. Questo composto è stato scoperto da E. W. Sutherland come intermediario dell'attivazione, da parte dell'adrenalina, della fosforilasi epatica e in seguito è stato indicato come mediatore tra diversi ormoni ed enzimi di cui è necessaria la regolazione.
Aldolasi. - La fruttosiodifosfatoaldolasi ha un duplice ruolo, partecipando sia alla glicolisi sia alla glucogenesi; essa catalizza la scissione reversibile del fruttosio-1,6-difo- sfato in D-gliceraldeide-3-fosfato e diidrossiacetonfosfato. L'enzima è largamente diffuso; solo alcuni batteri eterofermentativi, non possedendolo, debbono portare gli esosi a un grado maggiore di ossidazione prima di scinderli.
In natura sono presenti due tipi di aldolasi: quelle di tipo I si trovano negli animali, nelle piante superiori e nelle alghe verdi, mentre le aldolasi di tipo II si trovano nel lievito e nei Procarioti, Batteri e alghe azzurre. I protozoi fotosintetici le contengono entrambe, il che fa supporre che i loro cloroplasti siano vestigia di alghe azzurre.
Gli enzimi di tipo I, a differenza di quelli di tipo II, non richiedono Zn2+ e sono tipicamente inattivati per riduzione con boroidruro di sodio in presenza di fruttosiodifosfato o diidrossiacetonfosfato. Esaminando il meccanismo della reazione, si comprende questo comportamento: il diidrossiacetonfosfato forma un complesso reversibile con l'enzima sotto forma di base di Schiff (I); dopo riduzione si forma un'ammina sostituita (II) che non si può dissociare:

Si è stabilito che alla reazione partecipa il gruppo amminico ε di un residuo di lisina.
Nella normale reazione enzimatica la molecola di diidrossiacetonfosfato può semplicemente staccarsi dall'enzima o può andare incontro alla condensazione aldolica con una aldeide: mentre vi è una certa possibilità di usare aldeidi diverse (benché la D-gliceraldeide-3-fosfato sia di gran lunga preferita dall'enzima del muscolo), vi è una specificità obbligatoria per il diidrossiacetonfosfato.
Il meccanismo delle aldolasi di tipo II non comporta una base di Schiff: il metallo, lo Zn2+, che può essere però sostituito da altri metalli di transizione, partecipa probabilmente a formare un chelato con il substrato. È da segnalare il fatto che la natura abbia sviluppato due meccanismi così diversi per svolgere la medesima funzione.
Nell'uomo si trovano tre aldolasi strutturalmente e immunologicamente diverse; l'aldolasi A è l'unica significativamente presente nel muscolo; nel fegato si trovano la A e la B, con predominanza della seconda; l'aldolasi C si trova nel cervello. L'aldolasi A è la classica fruttosio-1,6-difosfatoaldolasi, che però è capace di agire con minore efficienza anche sul fruttosio-1-fosfato catalizzando la seguente reazione:
fruttosio-1-fosfato???01???diidrossiacetonfosfato+D-gliceraldeide.
Così, gli estratti di muscolo mostrano un rapporto tra l'attività sul fruttosio-1,6-difosfato e quella sul fruttosio- 1-fosfato di 50 a 1; negli estratti di fegato il rapporto è di 1 a 1, in quanto l'aldolasi B è relativamente attiva sul monofosfato. Nell'intolleranza ereditaria al fruttosio si trova grande riduzione dell'aldolasi B, mentre l'aldolasi A non è alterata; campioni bioptici di fegato di pazienti con questo difetto genetico mostrano un rapporto maggiore di 6 a causa della scarsezza in aldolasi B. Le conseguenze di questa alterazione diventano gravi quando si ingerisce fruttosio, a causa dell'accumulo di fruttosio-1-fosfato. I soggetti affetti da questa malattia possono evitare situazioni patologiche con la semplice eliminazione del fruttosio dalla dieta.
Triosofosfatoisomerasi. - Il passaggio successivo nello schema EMP riguarda la D-gliceraldeide-3-fosfato. I tessuti che glicolizzano sono di solito ben forniti dell'enzima che catalizza la rapida interconversione dei triosofosfati. Quando bisogna invertire il ciclo si deve produrre diidrossiacetonfosfato dalla D-gliceraldeide-3-fosfato. Si noti che il diidrossiacetonfosfato è anche il precursore dell'α-L-glicerofosfato tramite l'azione dell'α-glicerofosfatodeidrogenasi.
Studi effettuati con substrati marcati con 18O hanno dimostrato che la triosofosfatoisomerasi opera con lo stesso meccanismo impiegato dalla glucosio-6-fosfatoisomerasi.
Un errore congenito del metabolismo, ereditato come carattere recessivo autosomico, è responsabile della deficienza di questo enzima, che è presente a livelli intermedi in condizioni eterozigotiche; gli individui omozigoti presentano un'anemia emolitica e, in alcuni casi, disturbi neurologici.
D-gliceraldeide-3-fosfatodeidrogenasi. - Questo è un interessante enzima bifunzionale che catalizza un'ossidoriduzione e una fosforilazione.
La reazione è reversibile entro ampi limiti, in quanto deve permettere, in direzione inversa, la glucogenesi. Particolarmente interessante è il fatto che è necessaria la fosforilazione perché la reazione proceda. L'arseniato può sostituire il fosfato con formazione di acido 1-arseno-3-fosfoglicerico, che si idrolizza istantaneamente rendendo la reazione essenzialmente irreversibile.
Si è potuto stabilire che il sito catalitico dell'enzima contiene cisteina; il potenziale ossidante del NAD+ è mediato da questa cisteina, che reagisce con l'aldeide formando un derivato tioacilico legato; il gruppo acilico viene quindi rimosso dall'enzima come acilfosfato, per fosforolisi.
L'enzima isolato dai più svariati organismi ha sempre uguali dimensioni e consiste di quattro subunità; il frammento peptidico che contiene la cisteina essenziale è simile in molti di questi enzimi.
Nelle piante superiori vi è una specifica triosofosfatodeidrogenasi, associata con l'apparato fotosintetico, presumibilmente per permettere la formazione di esosi a partire dai triosi generati dalla fissazione del CO2. A differenza degli enzimi del lievito e dei Mammiferi, l'enzima della fotosintesi richiede NADP+.
Fosfogliceratochinasi. - La reazione catalizzata dalla fosfogliceratochinasi è il primo passaggio nella glicolisi in cui si forma l'ATP, la valuta liberamente negoziabile dell'energia cellulare. Per la sua attività è richiesto Mg2+, che può però essere sostituito da Mn2+. Come si verifica abitualmente per le vere chinasi, l'enzima, nel trasferire il gruppo fosforico, attacca il legame tra P e O.
Fosfogliceratomutasi. - L'isomerizzazione del 3-fosfoglicerato a 2-fosfoglicerato è formalmente e forse meccanicisticamente analoga alla reazione della fosfoglucomutasi. Tuttavia è ancora da provare l'esistenza della forma fosforilata dell'enzima, forse perché l'equilibrio favorisce il composto intermedio 2,3-difosfoglicerato. L'enzima isolato dal muscolo o dal lievito richiede il 2,3-difosfoglicerato come cofattore; esso mostra anche una certa attività fosfatasica verso il difosfato.
Enolasi. - Formalmente questo enzima catalizza una semplice disidratazione, però con l'importante conseguenza di trasformare un estere fosforico a bassa energia di idrolisi (ΔG0′=−3,1 kcal) in uno ad alta energia (ΔG0′=−14,8 kcal). Questo enzima richiede Mg2+ o altri metalli divalenti. La vecchia osservazione che la fermentazione è inibita dal fluoruro è probabilmente correlata alla sensibilità dell'enolasi verso il fluorofosfato di magnesio.
Piruvatochinasi. - La reazione catalizzata da questo enzima è il secondo punto nel corso della glicolisi in cui si ottiene ATP. Poiché il ΔG0′ di idrolisi dell'ATP, pur essendo alto (−7,2 kcal), è considerevolmente più basso del valore per il fosfoenolpiruvato (−14,8 kcal), la reazione è praticamente irreversibile. Questo fatto richiede la presenza nell'organismo di un diverso meccanismo per la sintesi del glucosio dal piruvato. La piruvatochinasi richiede non solo Mg2+, ma anche uno ione monovalente, come K+.
Nell'uomo sono normalmente presenti due distinte forme di piruvatochinasi, una delle quali si trova solamente negli eritrociti e nel fegato, l'altra nel fegato e in tutti gli altri tessuti. Si conoscono difetti, trasmessi geneticamente, dell'enzima eritrocitario ed è possibile riscontrare casi nei quali l'enzima eritrocitario è difettoso nel fegato, mentre il tipo generale è normale. Negli eterozigoti si trovano livelli intermedi di tale enzima eritrocitario; negli omozigoti la deficienza enzimatica causa un alterato metabolismo del globulo rosso, con conseguente anemia emolitica.
Anche nel ratto il fegato contiene due forme genetica- mente distinte, la M, che è del resto ubiquitaria, e la L, che si trova nel fegato e negli eritrociti; la concentrazione della seconda nel fegato è influenzata dalla dieta e da ormoni. Infatti l'attività piruvatochinasica del fegato di ratto diminuisce notevolmente in animali a dieta povera di carboidrati e aumenta molto al di sopra dei livelli normali a dieta ricca di carboidrati. Ratti diabetici hanno livelli di enzima bassi, che possono essere elevati iniettando insulina.
La tendenza della piruvatochinasi a trasformare il fosfoenolpiruvato in piruvato può essere autocontrollata quando è necessaria la glucogenesi a partire dal piruvato, quindi è importante notare che la piruvatochinasi, proteina tetramerica, è un enzima regolatore; l'enzima generalmente è inibito dall'ATP e la forma L è attivata dal fruttosio-6-fosfato.
Destino del piruvato. - In condizioni anaerobiche è necessario rigenerare il NAD+ dal NADH formato nella deidrogenazione della D-gliceraldeide-3-fosfato; negli animali superiori ciò viene effettuato tramite la lattatodeidrogenasi:
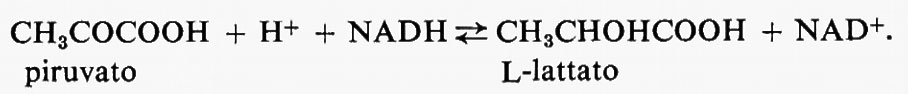
La fermentazione di una mole di glucosio può pertanto essere schematizzata nel modo seguente:
C6H12O6+2Pi+2ADP→2CH3CHOHCOOH
+2ATP+2H2O.
Si noti la formazione di due moli di ATP, composto ‛ricco di energia'. Se il piruvato fosse stato ossidato totalmente dal sistema mitocondriale a CO2 e H2O, si sarebbero formate altre 28 moli di ATP:
C6H12O6+30Pi+30ADP→6CO2+6H2O+30ATP.
L'estrazione di energia dal glucosio in assenza d'ossigeno è quindi un processo poco redditizio; però nella logistica del rifornimento di energia, la glicolisi anaerobica può essere utile: la capacità dei tessuti di ricavare energia quando l'O2 è limitante, è particolarmente importante quando il muscolo opera in condizioni difficili. Né del resto il metabolismo anaerobio è uno spreco irrimediabile, in quanto con limitato consumo energetico il lattato può essere riconvertito a piruvato; questo può essere quindi consumato, quando il fabbisogno di O2 non è più critico, per totale ossidazione nei mitocondri. Nei Mammiferi, molto dell'acido lattico formato in anaerobiosi è liberato nel sistema circolatorio e raggiunge il fegato, dove viene ritrasformato in glucosio e glicogeno.
In alcuni organismi, tra cui il lievito è l'esempio storicamente importante, la via anaerobia termina con la formazione di etanolo con il seguente meccanismo:
CH3COCOOH→CH3CHO+CO2
CH3CHO+NADH+H+⇄CH3CH2OH+NAD+.
In molti organismi è la 3-fosfogliceratodeidrogenasi a rigenerare il NADH in questo modo:
2-O3POCH2CHOHCOOH+NAD+⇆
⇄2-O3POCH2COCOOH+NADH+H+.
La decarbossilazione richiede come cofattore l'estere difosforico della tiamina (vitamina B1). Questa scoperta storica, effettuata da K. Lohmann e Ph. Schuster nel 1937, fu la prima dimostrazione del fatto che una vitamina potesse agire da gruppo prostetico di un enzima.
3. La via dei pentosofosfati.
La via dei pentosofosfati è una via metabolica alternativa del glucosio; essa normalmente non sostituisce del tutto la glicolisi e il ciclo degli acidi tricarbossilici nell'ossidazione completa del glucosio: quantità variabili di questo sono indirizzate nella via dei pentosi a seconda delle necessità e del tipo di tessuto o di organismo. La via, chiamata anche del fosfogluconato, è un'importante fonte di NADPH e di pentosi; il primo è di vitale importanza per la sintesi di molti composti, tra cui lipidi e steroidi; interviene inoltre in molte reazioni delle monoossigenasi; negli eritrociti è necessario perché vi sia un ambiente riducente per proteggere l'integrità strutturale della cellula e probabilmente per mantenere il ferro dell'emoglobina allo stato ferroso. I pentosi sono necessari per la sintesi di nucleotidi e di acidi nucleici.
Nonostante che questa via metabolica (v. schema 3, A e B).
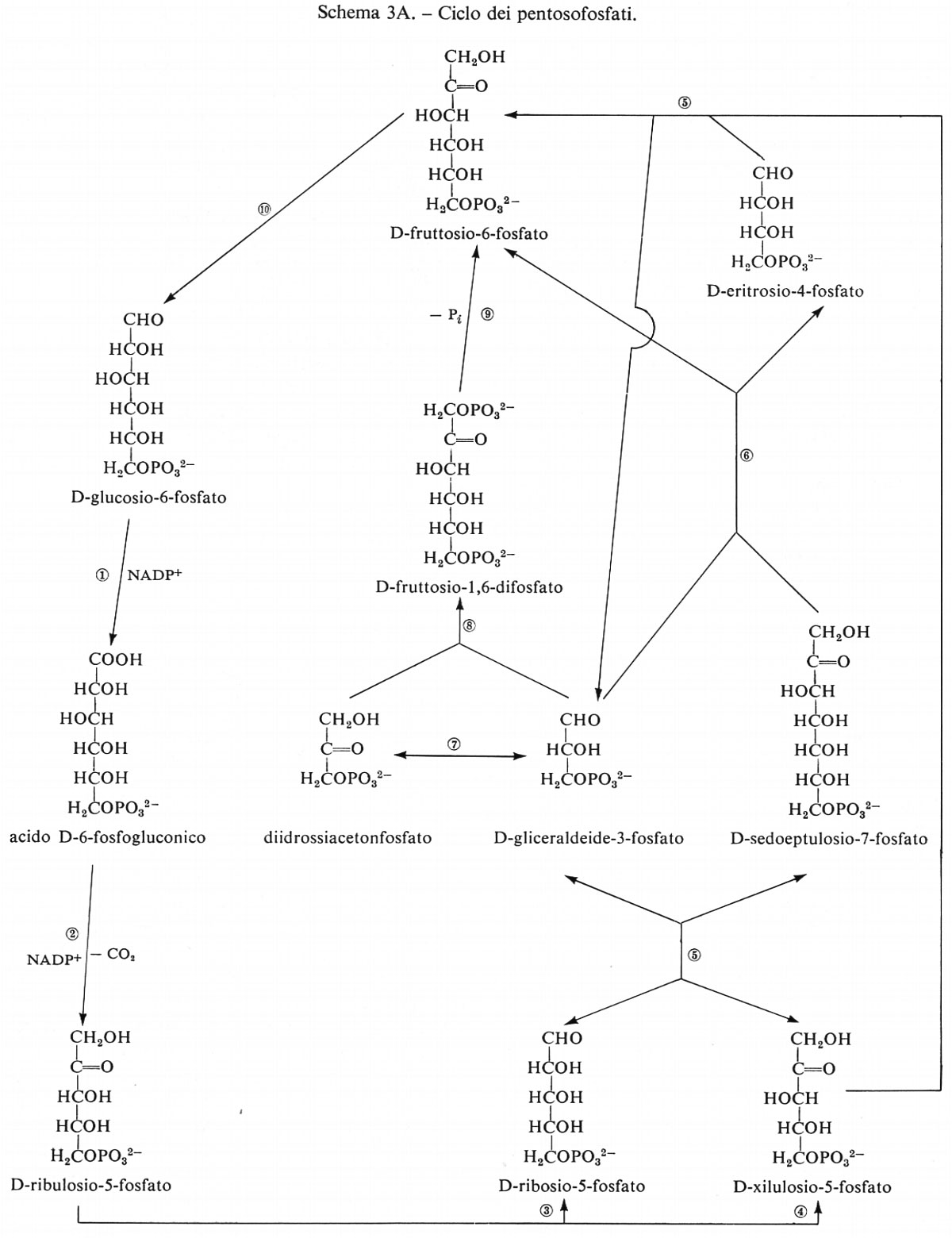
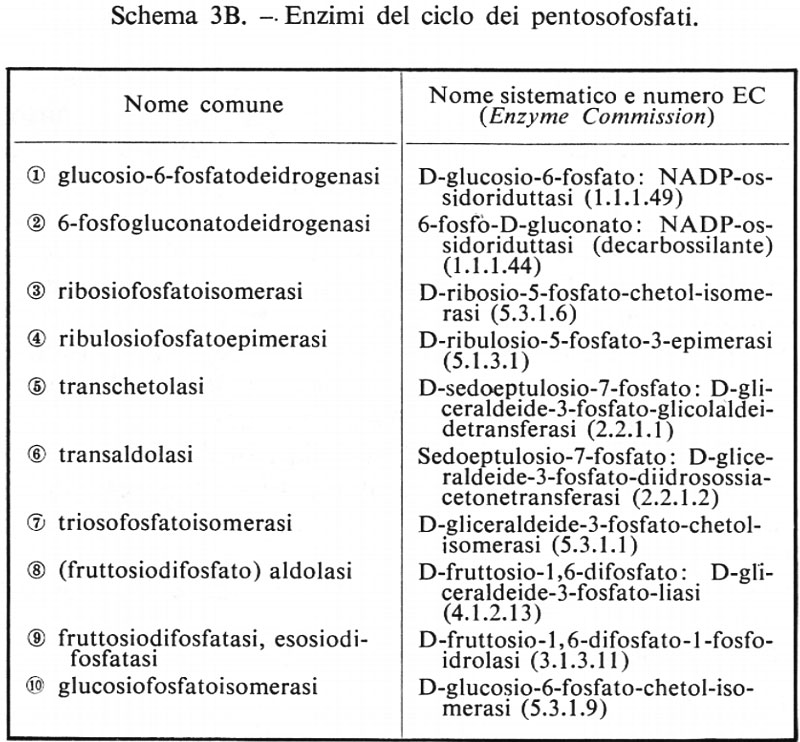
cominci con l'ossidazione del glucosio-6-fosfato, seguita dalla decarbossilazione ossidativa del 6-fosfogluconato, si deve notare la possibilità di effettuare interconversioni, senza ossidazione, tra esteri fosforici di zuccheri a 3, 4, 5, 6 e 7 atomi di carbonio.
Glucosio-6-fosfatodeidrogenasi. - Questo enzima fu scoperto da O. Warburg nel 1931, mentre era alla ricerca di una connessione tra la deidrogenazione dei carboidrati e il consumo di O2; nel corso di questi studi egli scoprì un enzima che chiamò Zwischenferment a causa del suo ruolo intermediario. Notò che per la sua attività era necessario un cofattore contenente l'anello della piridina; chiarì la struttura di questo e lo chiamò ‟coenzima II"; ora esso è conosciuto come NADP (nicotinammide-adenin-dinucleotide-fosfato).
Mentre in alcuni batteri il NAD può sostituire il NADP, gli enzimi dei Mammiferi sono specifici per il secondo; in qualche caso essi possono utilizzare il NAD quando questo è a concentrazioni elevate: è stata avanzata l'ipotesi, a tale proposito, che questo coenzima abbia un ruolo regolatore.
Gli enzimi di questo tipo fin qui purificati sono enzimi regolatori, capaci di associarsi e dissociarsi reversibilmente; in qualche caso la loro attività è funzione del rapporto NADP+/NADPH e della presenza di AMP e ATP, di ormoni steroidi e di acil-CoA a lunga catena.
Questi enzimi sono stati molto studiati nell'eritrocita umano perché correlati a uno stato patologico; oggi se ne conoscono molte varianti genetiche: alcune di queste non hanno rilievo clinico; molte, invece, sono causa di anemie emolitiche. Quando il danno genetico produce un enzima poco efficiente o instabile, le manifestazioni cliniche possono presentarsi solo se gli eritrociti sono attaccati da fattori tossici. La somministrazione in massa di farmaci antimalarici, come la primachina, a soldati americani inviati durante la seconda guerra mondiale nel Pacifico sudoccidentale, provocò la scoperta, particolarmente tra i soldati negri, di numerose varianti responsabili di anemie. Una malattia simile, ma non identica, il favismo, è conosciuta sin dal tempo dei Romani; essa incide particolarmente sulle popolazioni del Mediterraneo centro-settentrionale.
Il favismo deve il suo nome al fatto che la malattia è scatenata dall'ingestione di fave (Vicia faba) da parte di individui che non hanno l'enzima normale. Da sempre nelle regioni interessate dal favismo sono esistiti tabù contro le fave, benché probabilmente solo una piccola percentuale della popolazione fosse sensibile. In ogni caso le fave sono sempre state coltivate in queste regioni; si dice che Archimede andò incontro alla morte, a Siracusa, facendosi catturare dai soldati invasori piuttosto che tentare di fuggire attraverso un campo di fave.
Le varianti presenti nei Negri e nei Caucasici sono legate al sesso, e associate al cromosoma X; i portatori maschi mostrano quindi una grave deficienza d'enzima, le femmine eterozigoti ne hanno livelli intermedi in quanto posseggono due cromosomi X: poiché uno dei due cromosomi X nelle cellule somatiche è soppresso in modo casuale, ne risulta un effetto misto, in quanto metà delle cellule sono normali e metà patologiche.
Nella malattia che colpisce i Negri la deficienza riguarda solo gli eritrociti; nelle altre, la deficienza si estende anche ad altre cellule.
L'anemia che ne risulta può essere attribuita alla diminuita sopravvivenza degli eritrociti; in condizioni normali essi vivono 100-120 giorni e sono sostituiti dopo la usi. Durante il tempo di vita sono esposti a condizioni ossidanti che possono generare radicali perossido e H2O2, i quali contribuiscono a danneggiare la membrana e a formare ferriemoglobina, che non ha funzione fisiologica. Le cellule sono fornite di catalasi e perossidodismutasi per proteggersi dai pericoli dell'ossidazione, però può avvenire la perossidazione; si pensa che l'alta concentrazione di glutatione ridotto presente nelle cellule possa subire un processo durante il quale il glutatione ridotto è ossidato a disolfuro, che può essere ripristinato nella forma sulfidrilica per azione della glutationereduttasi richiedente NADPH:
GSSG+NADPH+H+→2GSH+NADP+.
La rigenerazione del NADPH è effettuata dalla glucosio-6-fosfatodeidrogenasi. La capacità del sistema eritropoietico di formare eritrociti è di solito sufficiente a prevenire deficienze nel trasporto di ossigeno anche se gli eritrociti hanno vita più breve, salvo che intervengano fattori esterni come farmaci, tossine o infezioni (v. sangue: Anemie emolitiche).
6-fosfogluconatodeidrogenasi. - Il prodotto immediato della deidrogenasi precedente è, come si vede dalla reazione seguente, un lattone:
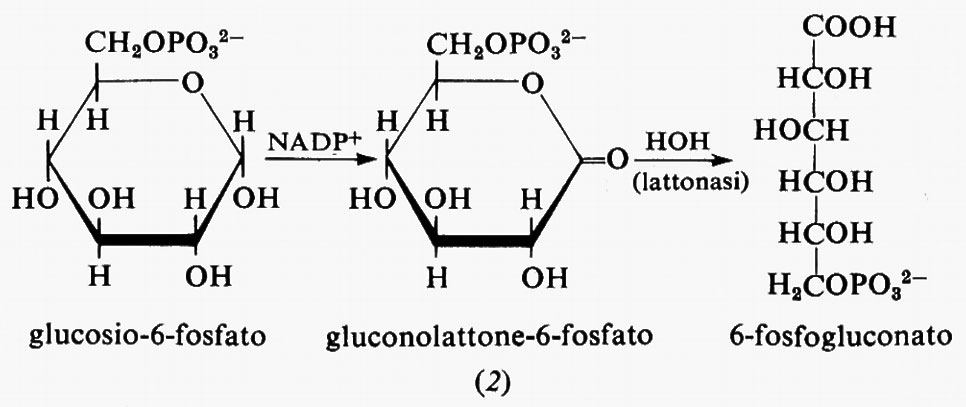
La 6-fosfogluconatodeidrogenasi catalizza la decarbossilazione ossidativa solo della forma aperta e per questo tra le due deidrogenasi interviene una lattonasi. Anche la seconda deidrogenasi è specifica per il NADP+. Si conoscono nell'uomo forme alleliche dell'enzima: poiché esso è un dimero, negli individui eterozigoti mostra un polimorfismo, con tre bande distinguibili elettroforeticamente.
Ribosiofosfatoisomerasi e D-ribulosio-5-fosfatoepimerasi. - Il prodotto della reazione precedente, il ribulosio-5-fosfato, è in rapido equilibrio, per azione dell'isomerasi, con il ribosio-5-fosfato; questo può essere usato per la sintesi dei nucleotidi. D'altro canto, il ribulosio-5-fosfato è in equilibrio, tramite l'epimerasi, con lo xilulosio-5-fosfato.
Transchetolasi. - La transchetolasi trasferisce un gruppo chetolico da un donatore specifico a un accettore specifico; le esigenze strutturali sono illustrate qui di seguito:

Questo enzima contiene tiamindifosfato, cui si lega il radicale chetolico, e richiede la presenza di Mg2+. L'addizione del gruppo chetolico avviene sempre in configurazione trans:
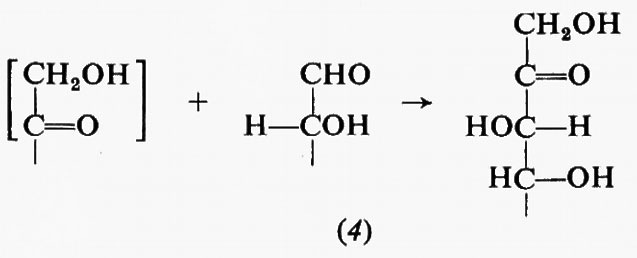
quindi diversi carboidrati possono fungere da accettori, benché i derivati fosforilati, D-ribosio-5-fosfato, D-gliceraldeide-3-fosfato e D-eritrosio-4-fosfato, siano di gran lunga preferiti ai composti non fosforilati.
La deficienza alimentare di tiamina si riflette in una diminuita attività transchetolasica degli eritrociti: l'aumento di attività che risulta dall'aggiunta di tiamindifosfato in vitro può essere usato per determinare il grado di deficienza di questa vitamina.
Transaldolasi. - La transaldolasi, che si trova sempre unita alla transchetolasi, trasferisce un residuo tricarbonioso da chetosi con una configurazione degli atomi di carbonio 1-3 uguale a quella del sedoeptulosio-7-fosfato o del fruttosio-6-fosfato; questo enzima rende possibile la formazione degli esosi nella via dei pentosofosfati.
Quando non si ha storno di carboidrati verso altre reazioni, il bilancio del ciclo può essere indicato nel modo seguente, ammettendo che siano presenti anche l'aldolasi, la fruttosio-1,6-difosfatasi e la esosofosfatasi:
6C6H12O6+12NADP++6H2O→
→5C6H12O6+12NADPH+6CO2+12H+.
Via del carbonio nella fotosintesi. - La via del carbonio nella fotosintesi, detta anche via di Calvin-Benson, è ora del tutto chiarita, benché si sia prospettata la possibilità che vi possano essere, in alcune piante, altre vie per la fissazione del CO2. La via, che coincide essenzialmente con la parte non ossidativa della via dei pentosofosfati, è rappresentata nello schema 4.
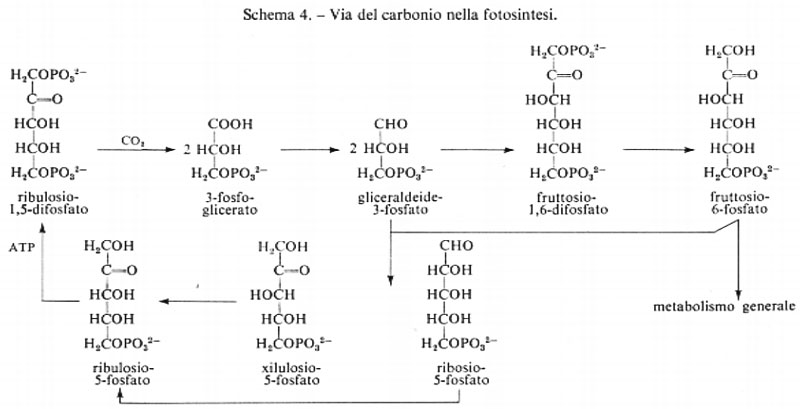
Nei tessuti fotosintetici ci sono tuttavia due enzimi particolari che interessano i fosfopentosi: il primo è la D-ribulosio-1,5-difosfatocarbossilasi, che catalizza la seguente reazione:
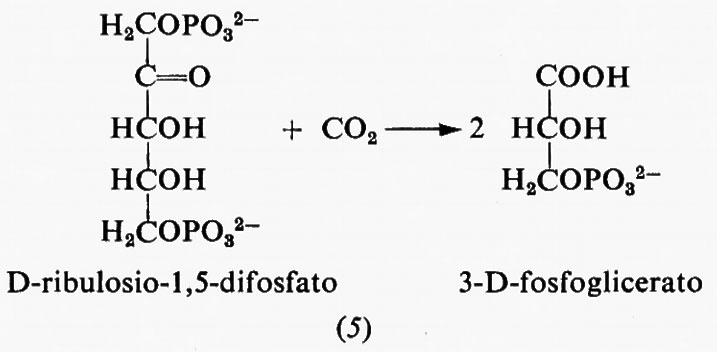
Questo enzima, almeno nelle piante superiori, catalizza anche una reazione ossigenasica:
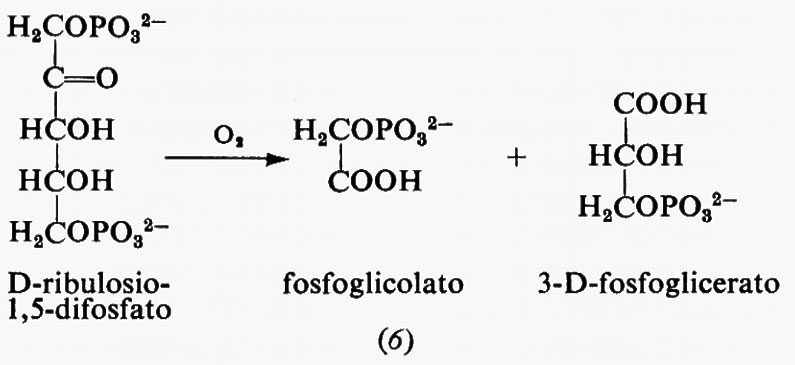
Quindi Q2 e CO2 sono substrati in competizione; questa proprietà può spiegare il ben noto effetto inibitore dell'O2 sulla fotosintesi in molte piante superiori.
Il secondo enzima, necessario per la sintesi del D-ribulosio-1,5-difosfato, è una chinasi.
4. Gluconeogenesi.
La capacità di sintetizzare glucosio a partire da piccole molecole originatesi nella glicolisi e da altri metaboliti, come amminoacidi e acidi carbossilici, è indispensabile per l'economia di moltissimi organismi; alcune piante e alcuni microrganismi possono sintetizzare carboidrati anche dagli acidi grassi, ma nè l'uomo nè gli altri animali sono in grado di farlo.
Una visione superficiale dello schema della glicolisi potrebbe condurre all'erronea conclusione che una semplice inversione della glicolisi possa formare glucosio. Ciò non è possibile e per motivi termodinamici e per motivi cinetici. Non ci si può attendere una significativa inversione di un metabolismo energetico se non fornendo energia chimica. Si è già detto che la reazione piruvatochinasica è essenzialmente irreversibile: non solo l'enzima non è responsabile della reazione inversa, ma potrebbe addirittura eliminare tutto il fosfoenolpiruvato eventualmente formatosi dal piruvato o dai suoi precursori. Molto opportunamente la piruvatochinasi è inibita da alte concentrazioni di ATP, condizione che si verifica proprio quando si rende necessario ricostituire o conservare le riserve di carboidrati. Come detto in precedenza, il fegato, che è il principale deposito di carboidrati, perde molta della sua piruvatochinasi quando in esso diminuiscono i carboidrati; d'altra parte l'accumulo di fruttosio-1,6-difosfato tende ad attivare l'enzima.
Poiché anche l'attività fosfofruttochinasica è praticamente irreversibile, deve esistere una via alternativa per trasformare il fruttosio-1,6-difosfato in fruttosio-6-fosfato: questa è costituita dalla fruttosiodifosfatasi, che è attivata dall'ATP e inibita dall'AMP.
La fosfofruttochinasi occupa una posizione chiave nella glicolisi; è un enzima regolatore e, come notato sopra, viene attivata quando il fabbisogno energetico aumenta, il che viene segnalato dall'aumentata concentrazione degli attivatori 3′-5′-AMP ciclico, 5′-AMP e Pi, mentre viene inattivata da concentrazioni elevate di ATP e citrato.
Prima di partecipare alla gluconeogenesi il piruvato deve essere trasformato in fosfoenolpiruvato; nei tessuti animali la conversione avviene secondo lo schema 5.

In altri organismi vi sono piccole variazioni o addirittura vie alternative; così in alcuni batteri e in alcune erbe si trova un interessante enzima, la piruvatofosfodichinasi, che provoca la fosforilazione diretta del piruvato:
piruvato+ATP+Pi⇄fosfoenolpiruvato+AMP+PPi.
La reazione è accoppiata a una pirofosfatasi che la rende unidirezionale idrolizzando irreversibilmente il pirofosfato (difosfato) inorganico:
PPi→2Pi.
Tutti i metaboliti che possono dar luogo al piruvato o agli acidi a quattro atomi di carbonio biologicamente importanti, cioè malico, succinico, fumarico e ossalacetico, sono in potenza glucogenici; lo sono largamente la maggior parte degli amminoacidi; lo è la parte alcolica dei trigliceridi, cioè il glicerolo, mentre gli acidi grassi che producono acetato sono chetogenici, ma non glucogenici, eccetto in quegli organismi che possiedono gli enzimi del ciclo del gliossilato. In tale ciclo (v. schema 6) due molecole di acetil-CoA possono in definitiva dar luogo ad acido succinico.
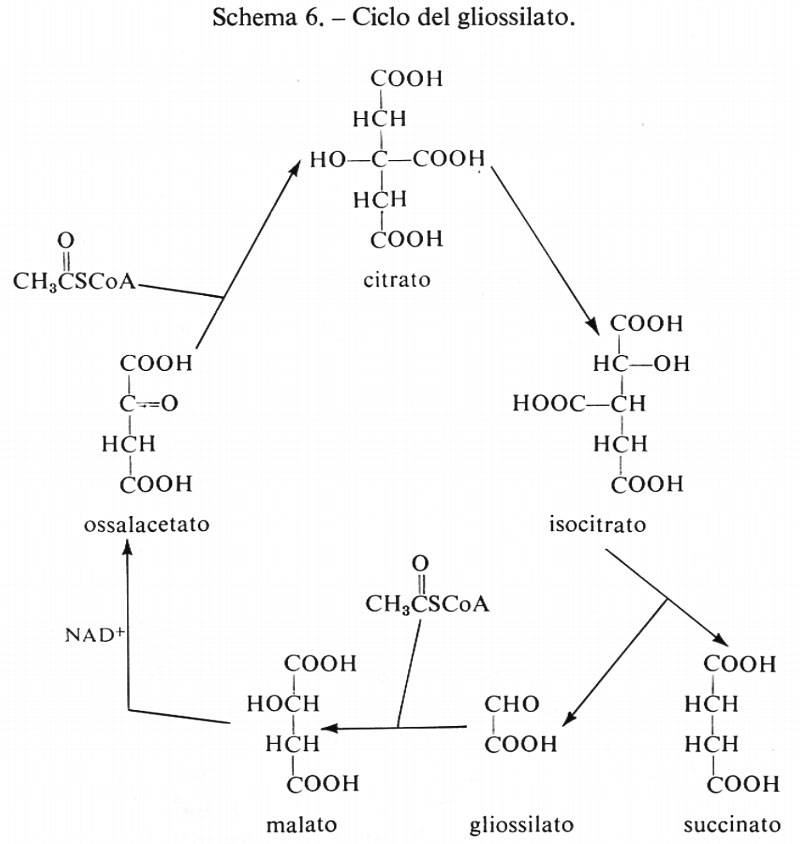
Il bilancio netto del ciclo è il seguente:
2 acetil-CoA+NADH+H++2H2O→succinato
+2CoA+NAD+.
L'acido succinico passa ad acido malico nel seguente modo:
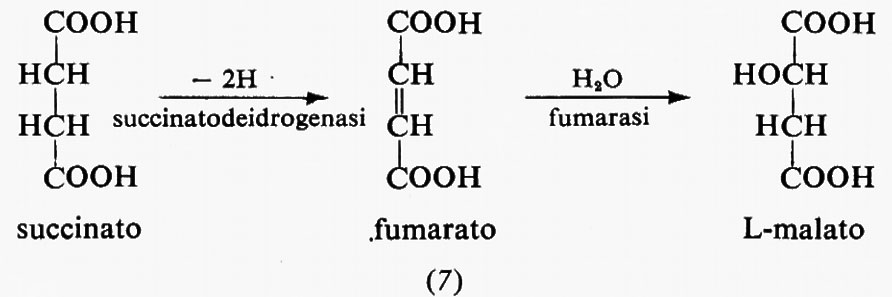
Quest'ultimo a sua volta diviene fosfoenolpiruvico, come già visto nello schema 5.
5. Monosaccaridi.
Benché in natura s'incontrino liberi in discreta quantità solo due monosaccaridi, nell'ambito della chimica se ne conosce un notevole numero.
Il glucosio è il più importante zucchero libero in natura; con poche eccezioni esso rappresenta la fonte di carbonio e di energia ricercata da ogni cellula di ogni organismo. Il perché di questo onore non è chiaro: si può solo considerare che esso nella sua forma termodinamicamente più stabile (8) presenta tutti gli idrossili in posizione equatoriale e quindi è più disponibile per ogni reazione chimica. L'H e l'OH del carbonio glucosidico, segnato con un asterisco, sono ovviamente interscambiabili:
Il secondo posto tra i monosaccaridi più comuni e quasi universalmente adoperati è tenuto dal D-fruttosio (9):

Generalmente gli zuccheri liberi devono essere fosforilati prima di essere utilizzati, tuttavia questa non è una necessità assoluta: alcuni monosaccaridi possono andare incontro a trasformazioni enzimatiche anche in forma non fosforilata. Ad esempio, molti funghi producono un enzima che catalizza la seguente ossidazione:

La conversione del gluconolattone in acido gluconico è operata da una lattonasi. Un enzima analogo si trova in alcuni funghi: esso ossida specificamente il D-galattosio con formazione della corrispondente 1,6-dialdeide. Mentre il glucosio si trova libero in tutti gli organismi viventi, il fruttosio si trova prevalentemente nelle piante.
Particolarmente interessante è il comportamento metabolico del fruttosio nel liquido seminale umano, in quanto rappresenta un esempio, in tessuti animali, di trasformazione diretta di un esosio in un altro senza intermediari fosforilati; la reazione è la seguente:
Si presume che il fruttosio, che rappresenta la sorgente di energia degli spermatozoi, sia assorbito meno facilmente del glucosio, dai tessuti circostanti, in competizione con gli spermatozoi; questi ultimi contengono molta esochinasi per fosforilare il fruttosio.
Interconversione tra galattosio e glucosio. - Il galattosio, che forma la metà della quota glucidica nella dieta dei neonati, viene convertito a glucosio per mezzo delle seguenti reazioni enzimatiche:
1) galattosio+ATP→galattosio-1-fosfato+ADP;
2) galattosio-1-fosfato+uridindifosfoglucosio
⇄uridindifosfogalattosio+glucosio-1-fosfato;
3) uridindifosfogalattosio⇄uridindifosfoglucosio;
4) uridintrifosfato+glucosio-1-fosfato
⇄uridindifosfoglucosio+PPi;
5) uridintrifosfato+galattosio-1-fosfato
→uridindifosfogalattosio+PPi.
Si noti che la somma delle reazioni 2) e 3) produce l'interconversione del glucosio-1-fosfato e del galattosio-1-fosfato, mentre l'uridindifosfoglucosio, formato nella reazione 4), ha un ruolo catalitico.
Galattosemia. - Come conseguenza della mancanza di galattosio-1-fosfatouridiltransferasi, l'enzima che catalizza la reazione 2), compare nei neonati una grave malattia, la galattosemia congenita: il difetto si trasmette come un carattere autosomico recessivo, onde la malattia si manifesta solo negli omozigoti. Gli eterozigoti posseggono diminuite quantità di enzima, ma non hanno problemi di tolleranza del galattosio. La deficienza enzimatica produce aumenti del galattosio e del galattosio-1-fosfato; si pensa che quest'ultimo possa interferire con reazioni della glicolisi che interessano composti fosforilati. Alti tassi di galattosio producono eccessivi livelli del suo prodotto di riduzione, il galattinolo (12), che, non essendo facilmente eliminabile, causa seri inconvenienti osmotici.
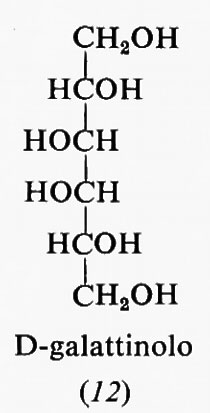
I bambini malati crescono poco, presentano cataratta, fegato cirrotico e ritardo mentale. L'eliminazione dalla dieta del latte e di altre fonti di galattosio, se effettuata precocemente, permette un normale sviluppo. Perfino animali da esperimento normali mostrano risposte patologiche alla somministrazione sperimentale di grandi quantità di galattosio.
La determinazione del livello di galattosio-1-fosfatouridiltransferasi rende possibile scoprire, in futuri genitori, lo stato eterozigote. Una malattia genetica alquanto meno comune è legata alla deficienza di galattochinasi, l'enzima che catalizza la reazione 1); in questa condizione si ha ancora galattosemia e cataratta; mancano invece gli altri segni patologici, come il ritardo mentale, probabilmente perché, in assenza di galattochinasi, non si formano grandi quantità di galattosio-1-fosfato.
Altri monosaccaridi. - Nella pagina seguente sono riportati altri monosaccaridi (13) che non si trovano comunemente liberi in natura, se non del tutto transitoriamente, e che svolgono ruoli importanti in associazione ad altre molecole. Ciascuno di questi zuccheri ha, negli organismi che li producono, un proprio anabolismo e un proprio catabolismo; solo catabolismo negli organismi che li utilizzano. Questi monosaccaridi, e alcuni importanti loro derivati, sono qui rappresentati con le formule a catena aperta.
6. La via del glucuronato.
Il glucuronato partecipa al metabolismo animale dei carboidrati come intermediario tra il glucosio e l'acido ascorbico, a eccezione, ovviamente, di quegli animali che, come l'uomo, non sono in grado di sintetizzare questa vitamina. Il glucuronato è anche un intermedio in una via alternativa tra glucosio e pentosi e nel catabolismo dell'inositolo. Nello schema 7 è rappresentata la via del glucuronato a partire dal glucosio e dall'inositolo; si noti che la deidrogenazione con sottrazione di 4 elettroni del glucosio avviene mentre esso è legato all'uridindifosfato.
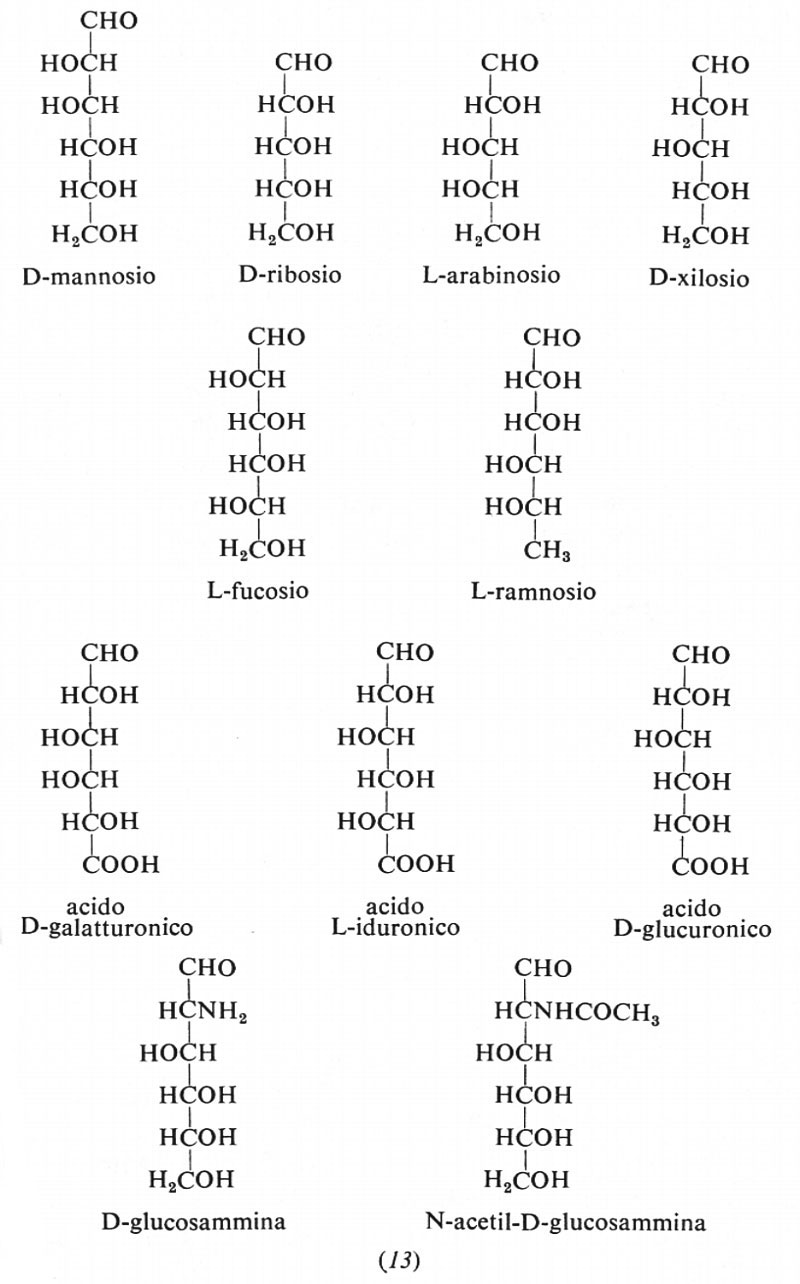
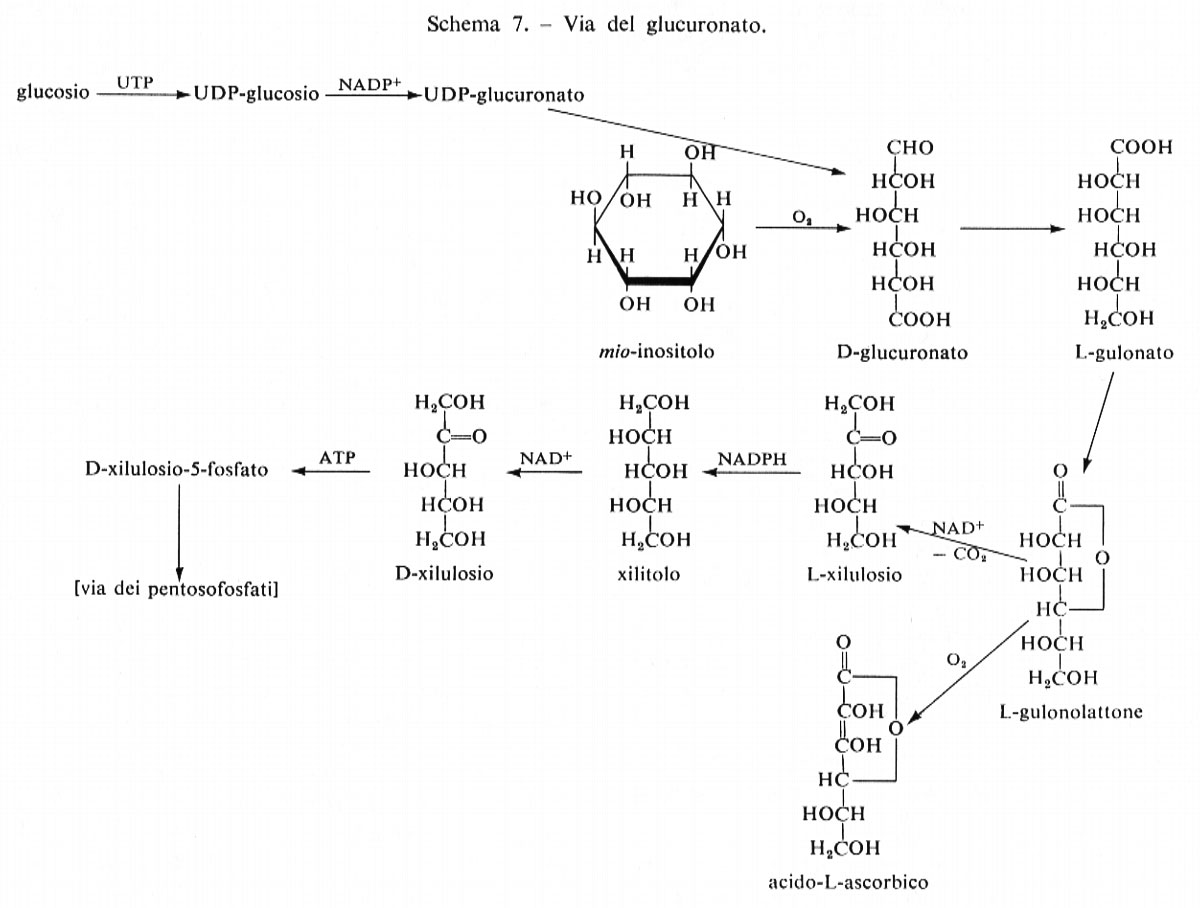
Il risultato finale di questa via metabolica è la produzione di una molecola di D-xilulosio e di CO2 per molecola di glucosio. La fosforilazione del D-xilulosio per mezzo della xilulochinasi produce D-xilulosio-5-fosfato, che è rapidamente metabolizzato nella via dei pentosofosfati.
Nello stesso schema si vede la biosintesi dell'acido Lascorbico, che costituisce una diramazione della via del glucuronato a livello dell'L-gulonolattone. I Primati, ivi compreso l'uomo, e la cavia devono assumere acido ascorbico (v. vitamine) con la dieta perché mancano dell'enzima L-gulonolattoneossidasi.
La pentosuria congenita è una condizione benigna in cui manca la L-xilulosioriduttasi; ne risulta una notevole escrezione (2-4 g al di) di L-xilulosio nelle urine. Questo fatto, unitamente alla conoscenza che l'uomo può eliminare più di 1 grammo al giorno di glucuronato come glucuronide, rende evidente l'importanza della via metabolica in oggetto.
Il glucuronato forma coniugati idrosolubili con materiali estranei e con metaboliti naturali da eliminare. Uronidi si formano con fenoli e acidi carbossilici: per esempio fenilglucuronidi e benzoilgiucuronidi; se ne formano anche con composti amminici. Parte degli ormoni steroidi endogeni sono escreti nelle urine come glucuronidi; anche la bilirubina, prodotto catabolico insolubile dell'eme, è resa solubile nel fegato per coniugazione con il glucuronato ed escreta nel sistema biliare. La coniugazione enzimatica richiede che il glucuronato sia sotto forma di uridindifosfoglucuronato.
7. Oligo- e polisaccaridi.
Nel 1951 considerevoli progressi erano già stati fatti nel descrivere la struttura di molti saccaridi, dai di- e oligosaccaridi fino agli alti polimeri come l'amido, il glicogeno, la cellulosa, le gomme, le emicellulose e altri. Il sistema enzimatico con cui i gruppi glucosidici vengono legati ad altri carboidrati oppure a molecole non glucidiche era per lo più sconosciuto per la maggior parte dei composti di interesse biologico. La scoperta che la reazione catalizzata dalla glucanofosforilasi

era reversibile fornì una traccia sbagliata, in quanto presto fu chiaro che la reazione inversa non ha significato fisiologico.
Nel 1951 L. F. Leloir scoprì che per la sintesi del trealosio, un disaccaride del glucosio, era necessaria la presenza di uno zucchero legato a un nucleotide, aprendo così nuovi orizzonti in questo campo. Benché la formazione di disaccaridi tramite composti gluconucleotidici sia la modalità più comune, in natura esistono altri metodi di glucosilazione, tra cui: 1) trasferimento da glucosidi preesistenti, catalizzato da glucosidasi, come si vedrà in seguito nella sintesi di glicogeno e amido; 2) trasferimento da glucosilfosfati, come avviene nella reazione inversa della glucanofosforilasi di cui si è detto.
Un gluconucleotide è formato da un purina- (o pirimidina-) ribosilfosfato, cioè un nucleotide, legato con legame difosforico al radicale fosforico di un glucosilfosfato. Esempio tipico è quello dell'uridindifosfoglucosio (UDPG), di cui si riporta qui la formula:
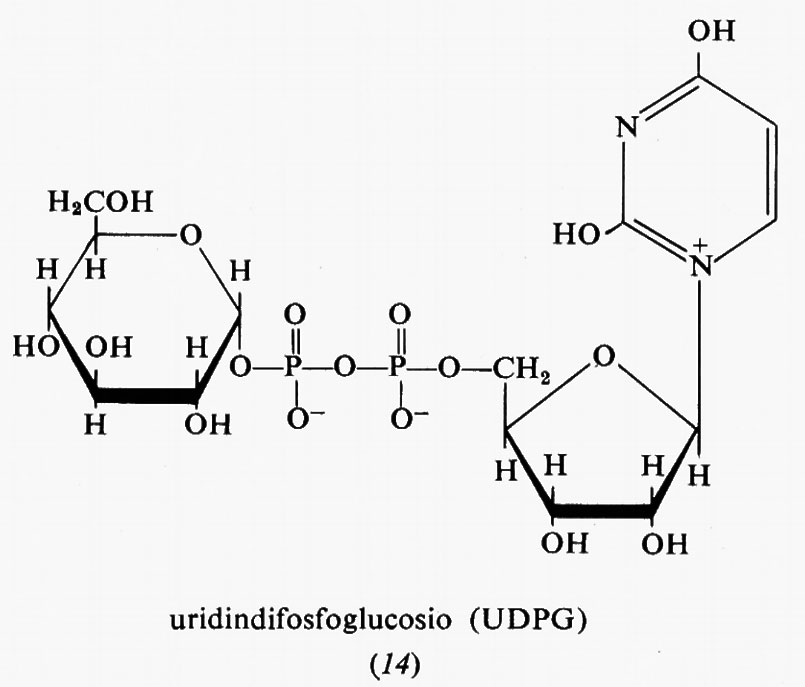
Nei gluconucleotidi che si trovano in natura sono presenti svariati nucleosidi: tra essi l'uridina, la citidina, la guanidina, la timidina, la desossiuridina. La parte glucosidica può essere formata in pratica da uno qualunque dei monosaccaridi conosciuti, e anche da loro derivati, quali desossiammine, derivati acetammidici, acidi uronici, solfati e, sorprendentemente, anche polialcoli. I gluconucleotidi sono necessari non solo per la sintesi di glucosidi: spesso alterazioni chimiche della parte glucidica avvengono solo se essa è legata al nucleosidedifosfato.
Disaccaridi. - È possibile trovare in natura una grande varietà di polisaccaridi, compresi i disaccaridi, risultanti dall'azione di glucosidasi su alti polimeri del glucosio. I disaccaridi possono formarsi anche per trasferimento di un monosaccaride su di un altro per azione di glucosidasi operanti su polisaccaridi o altri disaccaridi. Il significato fisiologico di queste tracce transitorie di disaccaridi non è chiaro. Di converso lattosio, saccarosio e trealosio sono esempi di disaccaridi formati specificamente e in grandi quantità per assolvere precise funzioni fisiologiche negli organismi che li elaborano.
Saccarosio. - Il saccarosio è di gran lunga il più comune dei disaccaridi presenti in natura, essendo sintetizzato, in pratica, da tutte le piante fotosintetiche. Le principali fonti commerciali, la canna e la barbabietola da zucchero, ne producono circa 85 milioni di tonnellate per anno. Nella maggior parte delle piante superiori il saccarosio rappresenta il tipo principale di carboidrato trasportato dai tessuti fotosintetici alle altre parti della pianta. Nei semi di Ricinus communis il grasso, che è la riserva predominante di carbonio e di energia, viene improvvisamente trasformato in saccarosio quando comincia la germinazione.
Il saccarosio, disaccaride non riducente in cui i gruppi riducenti del glucosio e del fruttosio sono legati fra loro, presenta la seguente struttura:

Esso viene sintetizzato attraverso le seguenti reazioni:
1) uridindifosfoglucosio+fruttosio-6-fosfato
⇄uridindifosfato+saccarosio-6-fosfato;
2) saccarosio-6-fosfato→saccarosio+Pi.
Un'altra via mediante la quale si può formare saccarosio è la seguente:
3) uridindifosfoglucosio+fruttosio⇄uridindifosfato
+saccarosio.
Si pensa che la prima via (reazioni 1 e 2) sia importante fisiologicamente per la sintesi del saccarosio, mentre la seconda (reazione 3) sia importante per la sua demolizione. Quando si somministra CO2 radioattivo a una pianta che fotosintetizza, il primo zucchero libero, cioè non fosforilato, che si forma è il saccarosio. Storicamente la prima sintesi enzimatica del saccarosio osservata fu quella operata da un enzima batterico che catalizzava la sua produzione da glucosio-1-fosfato e fruttosio; è evidente tuttavia che questa reazione è metabolicamente significativa solo nella direzione opposta e rappresenta il primo passo del catabolismo del saccarosio in quel batterio.
Lattosio. - Con l'eccezione della sapotiglia (Achros sapota), la produzione di lattosio sembra prerogativa esclusiva delle femmine adulte dei Mammiferi. Il lattosio, disaccaride riducente,
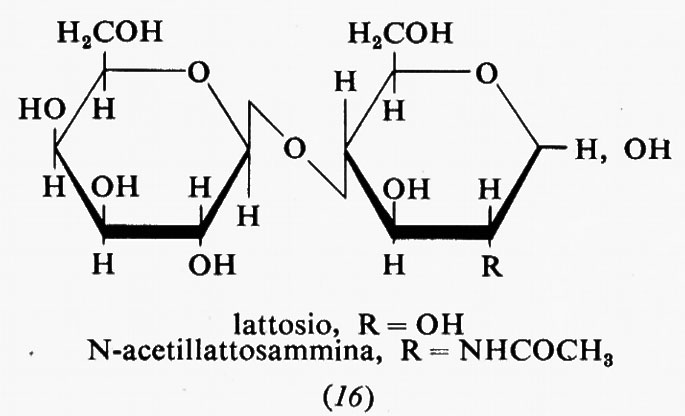
è il principale alimento glucidico per i piccoli di quasi tutti i Mammiferi, dal formichiere alla balena. È interessante notare che esso fu scoperto fin dal 1615 da Fabrizio Bartoletti.
La lattosiosintetasi è un enzima complesso formato da due unità dissociabili, le cosiddette proteina A e proteina B. Quest'ultima, nota per lungo tempo come a-lattalbumina, uno dei normali costituenti del latte, non ha attività enzimatica conosciuta. La proteina A, in assenza della B, si comporta come una N-acetillattosamminasintetasi e catalizza la seguente reazione:
uridindifosfogalattosio+N-acetil-D-glucosammina⇄
⇄uridindifosfato+N-acetillattosammina.
In presenza, tuttavia, della proteina B, il comportamento della proteina A è modificato in modo da non usare più l'acetilglucosammina, ma piuttosto il glucosio, con conseguente sintesi del lattosio. La proteina A è essenziale sia per organismi lattanti che per non lattanti, poiché la N-acetillattosammina e i suoi derivati sono indispensabili per molte attività; la α-lattalbumina, invece, è peculiare della femmina che allatta, e la sua sintesi è regolata dall'ormone lattogenico, la prolattina. La α-lattalbumina, che è sintetizzata in grande quantità e secreta nel latte, costituisce una parte del nutrimento proteico del poppante.
Benché il lattosio, nel bambino normale, sia efficientemente metabolizzato, vi sono casi di intolleranza congenita a questo zucchero. È anche possibile che una madre che allatta possa presentare transitoriamente alti livelli di lattosio nel sangue, con conseguente comparsa di questo zucchero nelle urine, ma questa condizione non ha serie conseguenze.
Trealosio. - Questo disaccaride non riducente
viene formato dalla trealosiosintetasi attraverso le seguenti reazioni:
1) uridindifosfoglucosio+glucosio-6-fosfato
⇄uridindifosfato+trealosio-6-fosfato
2) trealosio-6-fosfato+H2O→trealosio+Pi.
Il trealosio ha un ruolo importante nel mantenere un livello costante di glucosio nell'emolinfa degli Insetti; quando la concentrazione di glucosio-6-fosfato aumenta, si forma trealosio come materiale primario di accumulo. Anche il glicogeno si forma a partire dal glucosio-6-fosfato, specialmente quando la concentrazione del fosfato aumenta. Un'elevata concentrazione di trealosio inibisce la trealosio-6-fosfatosintetasi, dirigendo l'uridindifosfoglucosio verso la sintesi del glicogeno. Il trealosio è idrolizzato dalla trealasi quando vi è bisogno fisiologico di glucosio.
Polisaccaridi. - La distinzione tra oligosaccaridi e polisaccaridi è necessariamente arbitraria e talvolta entrambi i termini sono usati indifferentemente per molecole formate da 10-15 residui di monosaccaridi. Descriveremo più in dettaglio i più comuni polisaccaridi, che sono polimeri del glucosio, cioè l'amido, il glicogeno e la cellulosa.
Vi sono altri polisaccaridi, noti come ‛omoglicani', che sono composti da un solo tipo di zucchero. Per esempio l'inulina, un polisaccaride di riserva del tubero della dalia o del carciofo di Gerusalemme, è composta di fruttosio. Polisaccaridi formati da diversi tipi di monosaccaridi sono indicati col nome di ‛eteroglicani': tra di essi vi sono le gomme, le pectine, le emicellulose, le gomme dei lieviti e dei Batteri, che contengono i comuni monosaccaridi così come loro derivati carbossilici quali gli acidi glucuronico e galatturonico, o sostituiti con gruppi metilici, solforici, amminici, ecc. Gli zuccheri che si trovano di solito nei polisaccaridi, oltre a glucosio e fruttosio, possono essere galattosio, mannosio, L-arabinosio e D-xilosio.
Amido. - L'amido si trova come polisaccaride di riserva nelle piante sotto forma di amilosio e di amilopectina. L'amilosio è un polimero lineare del glucosio, senza ramificazioni, costituito da catene formate da circa 200-1.000 unità legate fra di loro con legami (α-1→4)-glucosidici.
L'amilopectina è formata anch'essa da catene di glucosio con legami (α-1→4), ma contiene molte ramificazioni, che avvengono con legame (α-1→6). L'amilosio si colora in blu con lo iodio, mentre l'amilopectina si cobra in rosso. L'amido si trova isolato in granuli che generalmente contengono una miscela di poliglucani lineari e ramificati; alcuni cereali presentano tuttavia tipi genetici che producono prevalentemente una delle due forme.
Le piante che sintetizzano l'amido formano anche enzimi che lo degradano. Si conoscono due amilasi: la α e la β; ambedue sono specifiche per i legami (α-1→4), ma l'α-amilasi è un endoenzima che attacca questi legami indiscriminatamente all'interno della molecola, mentre la β-amilasi è un esoenzima che attacca l'amido a partire dall'estremità non riducente idrolizzando il penultimo legame e liberando il disaccaride maltosio. Poiché questo secondo enzima non può agire oltre i punti di diramazione, l'amilopectina viene da esso idrolizzata fino a una destrina limite, che può essere ulteriormente idrolizzata per mezzo del- l'α-amilasi.
I semi che accumulano amido, quando sono messi a germinare, sintetizzano grandi quantità di α-amilasi, che diffonde nell'endosperma per digerire il nutrimento accumulato necessario per l'embrione in sviluppo: la sintesi è scatenata dall'ormone gibberellina. Contemporaneamente si ha anche un aumento di β-amilasi.
L'amilofosforilasi, anch'essa presente nelle piante, attacca l'amido nel modo seguente:
Questo enzima catalizza esattamente la stessa reazione della glicogenofosforilasi dei Mammiferi ma, a differenza di quest'ultima, non presenta interconversione tra forma attiva e inattiva.
Amilosintetasi. - La sintesi dell'amido avviene secondo la reazione
ADPG+GGGn→ADP+GGGn+1
in cui ADPG è l'adenosindifosfoglucosio e GGGn rappresenta un (α-1→4)-glucano a catena lineare o ramificata. Il gruppo glucosidico si lega all'estremità non riducente; è necessario un iniziatore, benché anche piccoli oligosaccaridi possano servire allo scopo; l'UDPG può rimpiazzare l'ADPG.
Per determinare una ramificazione è necessario un secondo enzima, il cosiddetto enzima ramificante o Q, che catalizza uno spostamento intramolecolare di una parte del polisaccaride con idrolisi di un legame (α-1→4) e formazione di un legame (α-1→6).
Cellulosa. - Tutte le piante superiori contengono cellulosa, benché esse non siano le uniche depositarie di questa sostanza, il più abbondante dei materiali biologici, che si trova anche nelle piccole piante inferiori, in alcuni batteri e perfino in alcuni invertebrati marini, come i Tunicati. La cellulosa si trova nelle pareti cellulari di tutte le piante superiori e contribuisce alla rigidità così caratteristica delle cellule vegetali.
La cellulosa, come l'amilosio, è un polimero lineare di unità di glucosio unite con legame (1→4); a differenza dell'amilosio, questo legame ha configurazione β invece che α. La configurazione β produce catene perfettamente orientate che si dispongono a formare microfibrille cristalline. Una singola catena è formata da circa 4.000-5.000 residui. L'effetto cooperativo di molti legami a idrogeno dà luogo alla salda unione delle catene; a loro volta le microfibrille si organizzano in fibre, che possono contenere anche altre sostanze.
La cellulosa si forma da un gluconucleotide, il guanosindifosfoglucosio (ODPG), per mezzo di una specifica glucosiltransferasi:
GDPG+(glucosio-β-1→4)n→
→guanosindifosfato+(glucosio-β-1→4)n+1.
Il modo in cui i gluconucleotidi a basso peso molecolare o i loro precursori sono trasportati dall'interno della cellula alle catene di cellulosa in accrescimento, disposte al di là della membrana, rimane un problema irrisolto.
Glicogeno. - Il glicogeno, il carboidrato di riserva degli animali, viene sintetizzato tramite la glicogenosintetasi in modo molto simile a quello dell'amido (v. sopra). Il gluconucleotide specifico per la glicogenosintetasi, l'UDPG, si forma per azione dell'uridindifosfoglucosiopirofosforilasi:
UTP+glucosio-1-fosfato→UDPG+PPi.
Vi è anche un enzima ramificante, la (1→4)-α-D-glucano: (1→6)-α-D-6-glucosiltransferasi, che stacca frammenti lineari formati da circa sette unità glucosidiche della catena lineare e li riattacca, come ramificazioni della catena principale, con legami (1→6).
Il glicogeno è soggetto a costante parziale demolizione e risintesi ed esiste come materiale polidisperso dal peso molecolare tra 100.000 e parecchi milioni. Il poliglucano è molto ramificato, con lunghezza media delle catene inferiore a 20 unità glicosidiche.
Benché la sintesi del glicogeno avvenga in piccole quantità in molti tessuti animali, la maggior parte di esso si trova nei muscoli e nel fegato. Il glicogeno muscolare funge da riserva di glucosio per casi di assoluta emergenza. Anche il fegato necessita di una scorta di glucosio prontamente disponibile perché esso è chiamato a mantenere adeguati livelli di glucosio ematico. D'altra parte il fegato deve anche impedire l'accumulo di glucosio nel sangue sintetizzando glicogeno; questo organo è anche il sito più importante della gluconeogenesi, con utilizzazione di materiali metabolici e alimentari non glucidici prelevati dal circolo sanguigno. Non sorprende quindi il fatto che la sintesi e la degradazione del glicogeno debbano essere sottoposte a un controllo metabolico e ormonale finemente regolato.
La glicogenosintetasi si trova in due forme, una attiva e l'altra inattiva; il meccanismo di regolazione consiste nell'interconversione di queste due forme. La forma inattiva è l'enzima fosforilato; la rimozione del fosfato per mezzo di una fosfatasi specifica lo rende attivo; la rifosforilazione, operata da una chinasi che richiede ATP, riporta l'enzima alla sua forma inattiva. La chinasi stessa è a sua volta attivata dal 3′-5′-AMP ciclico formato dall'adenilatociclasi secondo la reazione:
ATP→AMP ciclico+PPi.
L'attività adenilatociclasica è stimolata dalla presenza dell'ormone adrenalina, liberato dalla midollare dei surreni per stimolo nervoso causato da richiesta energetica. Così il sistema forma un sensibile circuito a cascata che permette di amplificare gli effetti di piccole quantità di ormone. L'AMP ciclico ha una breve esistenza, in quanto viene idrolizzato da una diesterasi specifica; deve quindi essere costantemente rigenerato sotto stimolo ormonale continuo per prolungarne l'effetto. Il glucagone agisce come l'adrenalina nel fegato, ma non ha azione nel muscolo.
Glicogenolisi. - La glicogenofosforilasi, il primo enzima della mobilizzazione del glucosio dal glicogeno, agisce in modo simile alla fosforilasi delle piante, già descritta. L'enzima delle piante non è però regolatore.
Come la sintetasi, anche la glicogenofosforilasi può presentare una forma attiva e una inattiva; a differenza però della sintetasi, l'enzima fosforilato è attivo mentre quello defosforilato è relativamente inattivo. L'enzima del muscolo, che è un tetramero, quando è defosforilato tende a dissociarsi in dimeri inattivi; anche l'enzima del fegato viene inattivato dalla defosforilazione, ma non si dissocia. Analogamente allo schema regolatorio della sintetasi, anche la fosforilasi può essere attiva o inattiva; l'interconversione delle due forme è operata da una chinasi e da una fosfatasi specifiche; anche questa chinasi è attivata dall'AMP ciclico, il cui livello è controllato come descritto in precedenza. Quindi nello stesso tessuto un singolo ormone ha effetti opposti su due differenti processi, stimolando la scissione del glicogeno e inibendone la sintesi.
Bisogna ricordare che le fosforilasi e le sintetasi dei diversi tessuti possono variare nella struttura e nel modo di regolazione; bisogna anche notare che l'azione di questi enzimi può essere influenzata dalla concentrazione di glucosio, dai glucocorticoidi e dall'insulina, e in alcuni tessuti dagli ormoni ipofisari.
L'esistenza di ramificazioni con legame (α-1→6) evita l'idrolisi completa del glicogeno da parte della sola fosforilasi: l'azione di questa si ferma a circa 4 unità glucosidiche di distanza da un punto di ramificazione. Un enzima deramificante stacca un trisaccaride da questo troncone e allunga una vicina catena lineare, lasciando un solo gruppo glucosidico al punto di ramificazione; esiste una specifica (α-1→6)-glucosidasi che stacca questo residuo come glucosio libero.
8. Polisaccaridi complessi.
Col termine ‛polisaccaridi complessi' ci riferiamo a macromolecole contenenti carboidrati uniti ad altro materiale, come lipidi o proteine. Poco interessanti in genere sono le miriadi di polisaccaridi vegetali - la cui struttura è stata chiarita nei primi due terzi del XX secolo - perché la maggior parte di essi non ha grande importanza fisiologica. Al contrario, i polisaccaridi complessi di origine animale sono di grande interesse e valore in quanto ciascuno di essi ha molto probabilmente un'attività fisiologica essenziale: o nella struttura delle membrane o in quanto implicato in certi meccanismi quali il trasporto, i fenomeni immunologici e le malattie connesse, la fecondazione, la divisione cellulare, la formazione del tessuto connettivo, la coagulazione del sangue, ecc. I polisaccaridi complessi sono importanti anche come costituenti delle pareti e delle membrane cellulari dei Batteri. Solo di recente sono stati applicati a questo difficile campo tecniche, ingegnosità e impegno in misura adeguata, e già se ne vedono i frutti. Si può dire veramente che la scienza oggi si prepara a entrare nell'età dell'oro della struttura e del metabolismo dei polisaccaridi complessi.
Glicoproteine. - Nella terminologia usata da alcuni, l'espressione ‛glicoproteina' indica qualsiasi molecola contenente proteine e carboidrati uniti covalentemente; tuttavia vi è una certa tendenza, ancora però non generalizzata, a restringere il termine alle proteine contenenti piccole quantità di carboidrati. Esempi di glicoproteine in senso stretto sono i seguenti: sieroalbumina, globuline, immunoglobuline, fibrinogeno, transferrina, tireoglobulina, collageno, ovalbumina, invertasi del lievito, perossidasi vegetali, ribonucleasi pancreatica di maiale. Nel caso di alcuni enzimi, esempio tipico le perossidasi vegetali, la proteina è legata con quantità variabili di carboidrati oppure è possibile staccare enzimaticamente parte di essi senza alterare l'attività enzimatica.
I carboidrati possono essere legati agli amminoacidi delle glicoproteine in due modi: con legame O-glucosidico o con legame N-glucosidico. Il legame con l'idrossilisina è del tipo O-glucosidico e ricorre solo nel collageno; questo legame è molto importante negli animali superiori, nei quali il collageno, che fa parte del tessuto connettivo, costituisce più di un quarto di tutte le proteine del corpo; in esso vi sono unità D-galattosidiche legate in configurazione β con i residui di 5-idrossi-L-lisina che si trovano nella catena polipeptidica; si può trovare anche glucosio unito al galattosio con legame (β-1→2):

In genere il glucosio ricorre raramente nelle glicoproteine. La maggior parte delle glicoproteine sopra elencate sono esempi del legame tipo asparagina che s'istituisce tra la glucosammina e il gruppo ammidico dell'asparagina, con configurazione β come qui mostrato:
L'amminoacido terminale con il gruppo carbossilico libero è la serina; talvolta si può trovare la treonina.
Le mucine formano soluzioni molto viscose contenenti quantità relativamente elevate di carboidrati e di conseguenza minore quantità di proteine; loro caratteristica è inoltre quella di contenere elevate quantità di acido sialico. Esempio tipico di esse è la glicoproteina della ghiandola submascellare di bovino, che contiene il 42% di carboidrati, di cui circa un terzo è rappresentato dall'acido O, N-diacetilneuramminico:
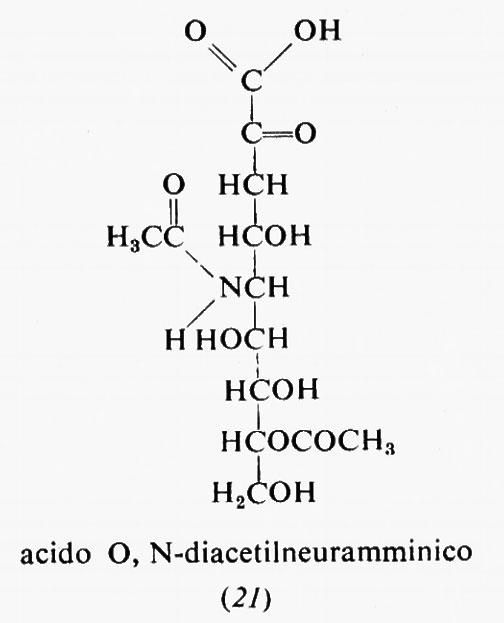
L'estremità riducente dell'oligosaccaride è di solito la D-galattosammina unita con legame glucosidico al gruppo −OH della serina o della treonina.
Mucopolisaccaridi. - I mucopolisaccaridi, che comprendono, tra l'altro, il dermatano, l'acido ialuronico, l'eparina e l'acido controitinsolforico, si trovano in vari tessuti strutturali, nell'umor vitreo e nel liquido sinoviale. Sono costituiti da lunghe catene polisaccaridiche formate da unità di un acido uronico alternantesi con unità di una esosammina, alle quali sono legate generalmente con legami (β-1→3). Per esempio, il dermatansolfato è composto prevalentemente di acido L-iduronico e di N-acetilgalattosammina-4-solfato. L'eparina contiene acido D-glucuronico e glucosammina-O, N-solfato. Queste descrizioni sono semplificate, in quanto si trovano anche altri componenti: i mucopolisaccaridi hanno di solito un componente proteico. Si conosce un gruppo di malattie ereditarie, indicate come mucopolisaccaridosi, che comportano una vasta gamma di disturbi a carico dello scheletro, del sistema nervoso e di numerosi organi, le quali sono state attribuite a un alterato metabolismo dei mucopolisaccaridi: in una di queste, la sindrome di Hurler, il difetto è stato collegato all'assenza di una glicosidasi specifica.
Pareti delle cellule batteriche. - Le pareti delle cellule batteriche contengono, oltre ai carboidrati, quantità variabili di lipidi e protidi; la complessità della loro struttura e della loro composizione è affascinante e presenta miriadi di varietà. È importante citare alcuni aspetti della loro struttura per avere un'idea della grande varietà di differenti molecole e combinazioni che si intrecciano nelle coperture esterne dei Batteri. Bisogna peraltro distinguere tra parete cellulare (cell wall), che conferisce rigidità alle cellule stesse, e membrana, che è parte integrante della cellula metabolizzante: infatti è possibile allontanare le pareti cellulari di alcuni batteri senza alterare i loro processi vitali, mentre, naturalmente, le cellule animali non hanno affatto pareti.
Le pareti dei batteri gramnegativi (particolarmente studiate sono state quelle di Salmonella ed E. coli) sono ricche di lipopolisaccaridi. Il polimero è formato da una serie di acidi grassi, tra cui predomina l'acido 3-idrossitetradecanoico, disposti in modo particolare, e da una grande varietà di zuccheri, alcuni poco comuni, largamente sostituiti, che presentano in alcuni punti legami incrociati formati da esteri fosforici. Le regioni periferiche contengono polimeri di tetra- o pentasaccaridi ramificati responsabili del peculiare potere antigene di alcuni tipi batterici, come l'antigene O di Salmonella. L'analisi della parte interna dei lipopolisaccaridi ha evidenziato la presenza di D-glucosio, D-galattosio, D-glucosammina, acido 3-desossi-D-mannoottulosonico e L-glicero-D-mannoeptosio; inoltre, nel sito antigenico di un certo tipo si trovano D-mannosio, abequosio, L-ramnosio e D-galattosio, in un altro D-ribosio e D-galattosio.
Riportiamo le formule di alcuni di tali zuccheri poco comuni:
È stata dimostrata la partecipazione di un alcool isoprenoide a 55 atomi di carbonio nella sintesi del lipopolisaccaride. L'estere fosforico di questo alcool, l'undecaprenilfosfato, funge da accettore dei glucosidi che costituiscono la caratteristica unità periodica della catena periferica; in seguito la catena preformata viene trasferita all'esterno. La partecipazione di alcoli polusoprenoidi alla sintesi dei polisaccaridi è stata descritta in altri casi nei microbi; ne è stata prospettata la partecipazione anche alla sintesi della cellulosa nelle piante.
Un materiale strutturale comune a tutte le pareti delle cellule batteriche è il peptidoglicano, polisaccaride lineare costituito di una lunga catena delle seguenti unità disaccaridiche formate dall'alternarsi di acido N-acetilmurammico e di N-acetil-D-glucosammina:
Diramazioni polipeptidiche formano legami crociati tra le varie catene polisaccaridiche per unione del gruppo carbossilico dell'acido N-acetilmurammico con il gruppo amminico terminale di una diramazione polipeptidica.
L'effetto è la produzione di una gigantesca molecola che assomiglia a una rete all'interno di un sacco completamente chiuso.
La struttura polisaccaridica del peptidoglicano è suscettibile all'azione del lisozima, che idrolizza i legami β dell'acido N-acetilmurammico; il lisozima si trova nelle lacrime, nel muco nasale, nel latte, in vari tessuti e nel bianco d'uovo a protezione contro i Batteri. I grampositivi sono particolarmente sensibili all'enzima e vanno incontro a lisi quando perdono la rigidità della loro parete cellulare. Benché anche il peptidoglicano dei batteri gramnegativi sia attaccato, questi resistono alla lisi, probabilmente a causa della presenza del lipopolisaccaride e forse di un ulteriore polimero strutturale.
Glicosfingolipidi. - Si conosce una serie di glicolipidi che sono glicosidi di cerammidi; queste ultime sono acilsfingosine in cui il gruppo acilico può variare, ma spesso è costituito dall'acido nervonico C23H45COOH. Qui si riporta un esempio di glicosfingolipide: il cerebroside nervone, N-derivato della psicosina con l'acido nervonico.
In questo gruppo sono compresi: i cerebrosidi, che hanno un gruppo glucosidico; i cerebrosolfatidi, che presentano il galattosiosolfato; i gangliosidi, in cui è presente l'acido neuramminico, come in questo esempio:
galattosio-(β-1→3)-N-acetil-D-galattosammina-(β-1→4)-[acido N-acetilneuramminico (α-2→3)]-D-galattosio-(β-1→4)-α-D-glucosio.
Nel corso normale del metabolismo un ganglioside che porta un tale gruppo polisaccaridico va incontro gradualmente alla glicolisi. Sono conosciuti diversi difetti genetici collegati alla deficienza di qualcuno degli enzimi implicati in tali processi idrolitici: ne risulta una serie di malattie caratterizzate dall'accumulo di qualcuno dei prodotti di idrolisi parziale.
Malgrado il loro nome e la loro ben nota prevalenza nel tessuto nervoso, i glicolipidi sono ubiquitari nelle membrane cellulari e i disturbi del loro metabolismo causano seri danni a molti tessuti dell'organismo, compreso, ben s'intende, il tessuto nervoso.
9. Aspetti immunologici dei carboidrati.
I carboidrati possono fungere da antigeni, benché molecole relativamente piccole di essi siano apteni deboli.
Due classi di sostanze, con specificità isoantigenica corrispondente ai gruppi sanguigni A e B, sono presenti nei globuli rossi: una classe è alcool-solubile, viene facilmente allontanata per estrazione ed è una macromolecola glicosfingolipidica; una seconda diversa sostanza con la stessa specificità antigenica è legata tenacemente alla cellula, per cui è molto difficile estrarla e può essere studiata solo indirettamente; fortunatamente si può isolare dalle secrezioni (saliva, muco gastrointestinale, latte) degli stessi individui una sostanza, di natura glicoproteica, con la stessa specificità antigenica. Le specificità di entrambe le sostanze antigeniche del sangue, alcool- e idrosolubile, è determinata da una specifica disposizione strutturale di un piccolo gruppo di molecole glucidiche; l'idrolisi di queste macromolecole glicoproteiche produce: L-fucosio, D-galattosio, N-acetil-D-glucosammina ed N-acetil-D-galattosammina.
Il tipo antigenico è stato correlato a specifiche sequenze di 4 o 5 di questi zuccheri.
La specificità H è prevalente in individui con sangue tipo O, cioè mancanti di entrambe le specificità A e B; essa si trova anche, in minor misura, in individui con specificità A, B e AB. Sono noti altri tipi di specificità. I tipi di glucidi terminali in questi gruppi sono illustrati negli schemi 8, 9 e 10.
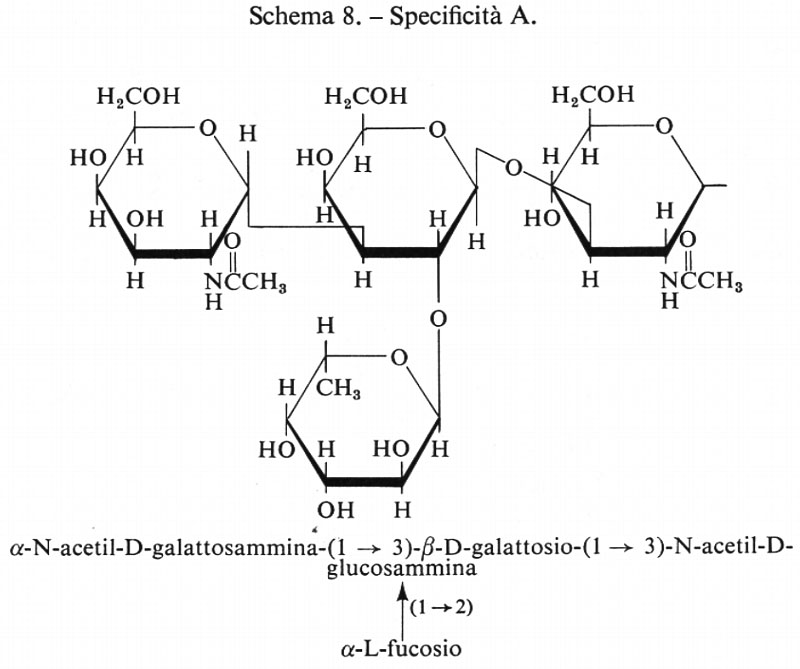
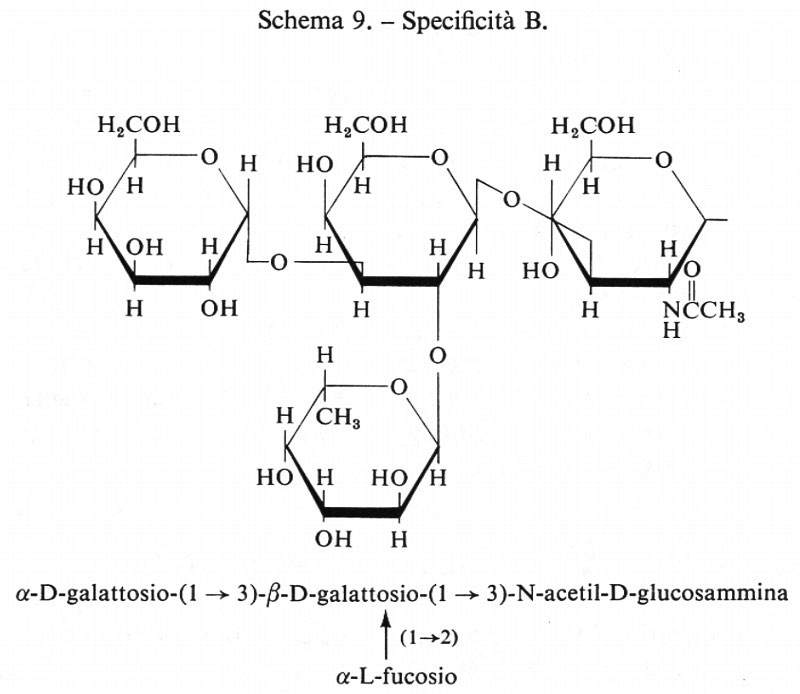
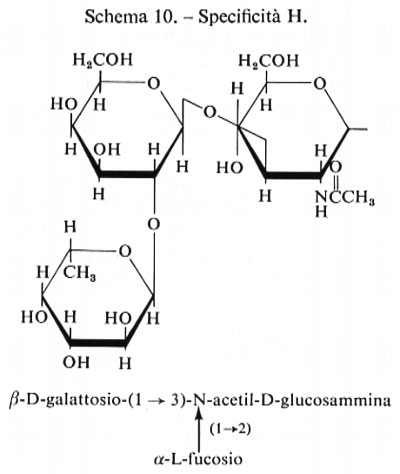
Sono stati identificati enzimi che partecipano all'addizione graduale di specifici gruppi glucosidici. Di converso, è possibile rimuovere specificamente residui di carboidrati usando appropriate glucosidasi e trasformare così un gruppo in un altro: sostanza A→sostanza H+N-acetilgalattosammina; sostanza B→sostanza H+galattosio. Si noti come l'aggiunta di un solo gruppo glucosidico sia sufficiente a cambiare la specificità.
Le sostanze glicolipidiche o glicoproteiche contengono molte copie della struttura determinante, ed essendo così plurivalenti, reagiscono energicamente con l'anticorpo corrispondente.
10. Diabete mellito.
L'uomo, come del resto la maggior parte degli animali, è particolarmente predisposto a mantenere nel torrente sanguigno un livello di glucosio pressoché costante. Grazie a un'intricata interazione di ormoni, di controlli nervosi, di enzimi regolatori e di membrane selettive, le variazioni in contenuto di glucosio nel siero, benché transitoriamente risentano del ritmo di ingestione e di utilizzazione, sono mantenute entro limiti ben definiti. Nell'uomo il contenuto ematico del glucosio varia tra 60 e 90 mg per 100 ml, benché sia transitoriamente più alto dopo i pasti. Quando il livello si eleva al di sopra di 170 mg per 100 ml, viene superata la capacità ritentiva del rene per il glucosio, che comincia a questo punto a comparire nelle urine. Come prevedibile, il contenuto in glucosio del sangue arterioso è un po' maggiore di quello del sangue venoso.
Il diabete mellito, la più importante malattia metabolica che colpisca l'uomo, è collegato a un disordine del metabolismo glucidico e può avere un effetto primario praticamente su tutte le cellule. Le vie metaboliche dei lipidi, delle proteine e dei carboidrati sono così strettamente connesse che, come effetto secondario, si possono verificare seri inconvenienti in ogni fase del metabolismo. Nel 1889 J. von Mering e O. Minkowski scoprirono che nel cane la rimozione del pancreas provocava gravi alterazioni diabeto-simili del metabolismo glucidico; l'ipotesi secondo cui si era in tal modo rimossa una ghiandola producente ormoni fu clamorosamente confermata nel 1921 da J. J. R. Mac Leod, F. G. Banting e C. H. Best: la causa primaria della malattia e un insufficiente produzione di insulina da parte delle isole di Langerhans del pancreas.
Malgrado la straordinaria complessità del problema dell'azione dell'insulina, e malgrado il sovrapporsi di effetti secondari, vi sono chiare dimostrazioni che l'insulina è necessaria per permettere l'assorbimento del glucosio da parte delle cellule. Ciò non significa che, indipendentemente, non sia alterato anche l'assorbimento, per esempio, degli amminoacidi: ma problema principale deve essere considerata l'interazione con il trasporto del glucosio. Non tutti i tessuti necessitano dell'insulina, per es. quello cerebrale, quello renale e la mucosa intestinale.
È interessante il fatto che il pancreas secerna sia l'insulina sia il glucagone: la prima provoca flusso di glucosio dal sangue alle cellule, il secondo, invece, stimola il fegato a liberare glucosio nel circolo (v. ormoni nei vertebrati).
Il glucosio raggiunge nel sangue dei diabetici concentrazioni elevate in quanto è impedito il suo passaggio nei muscoli e in altri tessuti. Il sangue circolante è filtrato nei reni, attraverso i glomeruli, e normalmente il glucosio è completamente riassorbito dai tubuli; nel diabetico, invece, la sua concentrazione è così alta da superare la capacità di riassorbimento, e pertanto il glucosio compare nelle urine.
11. Digestione.
La digestione dell'amido e del glicogeno comincia appena essi si mescolano nella bocca con la secrezione salivare; l'enzima che la causa è un'α-amilasi che attacca indiscriminatamente, all'interno del polisaccaride, legami (α-1→4)-glucosidici; questo enzima si trova anche, oltre che negli animali, nelle piante, nei Batteri e nei Funghi, mentre la β-amilasi non si trova mai negli animali.
L'azione dell'amilasi salivare cessa quando il cibo ingerito viene fortemente acidificato nello stomaco, benché ivi possa aver luogo un'idrolisi acida, specialmente di fruttosidi. L'attività amilasica riprende vigorosamente nell'intestino grazie alla copiosa secrezione di α-amilasi pancreatica. La degradazione del polisaccaride e degli oligosaccaridi formatisi per azione dell'amilasi continua per azione di (α-1→4)-glucosidasi che idrolizzano i residui glucosidici terminali, mentre la amilo-(1→6)-glucosidasi, che si trova nella mucosa intestinale, attacca i legami (1→6) dei punti di ramificazione del glicogeno e dell'amilopectina.
Il lattosio e il saccarosio non sono attaccati da enzimi secreti nel lume intestinale, ma da idrolasi presenti nella membrana delle cellule epiteliali a colonna dell'intestino tenue. I vari monosaccaridi di importanza nutritiva sono infine assorbiti con specifici meccanismi comportanti il trasporto attivo.
Benché il tratto intestinale dell'uomo non sia privo di microrganismi, la digestione dei carboidrati ‛facilmente digeribili' è solo scarsamente influenzata dai Batteri. Tuttavia i pentosani e alcuni altri polisaccaridi, per i quali non vengono prodotti, nell'uomo, appropriati enzimi digestivi, possono arrivare nei tratti bassi dell'intestino, dove l'anaerobiosi e la presenza di grandi concentrazioni batteriche ne facilitano la degradazione fermentativa: ciò spiega la ben nota flatulenza che si accompagna all'ingestione di fagioli.
Gli animali generalmente non possono digerire la cellulosa direttamente, in quanto non producono cellulasi: tutti i Ruminanti, e in qualche caso anche i non Ruminanti come il cavallo, e i parassiti del legno come i tarli e le termiti, dipendono dalla simbiosi con i loro microrganismi intestinali.
Lattosio e saccarosio devono essere idrolizzati prima della loro utilizzazione da parte dell'uomo: se assorbiti o iniettati direttamente nel torrente circolatorio, vengono escreti. Nel sangue c'è una piccola quantità di α-glucosidasi e quindi il maltosio può essere parzialmente idrolizzato e utilizzato. I pentosi, se iniettati, vengono scarsamente utilizzati e sono escreti attraverso i reni.
Bibliografia.
Axelrod, B., Glycolysis, in Metabolic pathways (a cura di D. M. Greenberg), vol. I, New York 19673, pp. 112-145.
Axelrod, B., Other pathways of carbohydrate metabolism, in Metabolic pathways (a cura di D. M. Greenberg), vol. I, New York 19673, pp. 271-306.
Bondy, P. K., Disorders of carbohydrate metabolism, in Duncan's diseases of metabolism, vol. I, Philadelphia 19696, pp. 199-294.
Hassid, W. Z., Biosynthesis of complex saccharides, in Metabolic pathways (a cura di D. M. Greenberg), vol. I, New York 19673, pp. 307-393.
Heath, E. C., Complex polysaccharides, in ‟Annual review of biochemistry", 1971, XL, pp. 29-56.
Nikaido, H., Hassid, W. Z., Biosynthesis of saccharides from glycopyranosyl esters of nucleoside pyrophosphates (sugar nucleotides'), in ‟Advances in carbohydrate chemistry and biochemistry", 1971, XXVI, pp. 351-483.
Pontremoli, S., Grazi, E., Hexose-monophosphate oxidation, in Comprehensive biochemistry (a cura di M. Florkin e E. H. Stotz), vol. XVII, Carbohydrate metabolism, Amsterdam 1969, pp. 163-189.
Spiro, R. G., Glycoproteins, in ‟Annual review of biochemistry", 1970, XXXIX, pp. 599-638.
Touster, O., Aldonic and uronic acids, in Comprehensive biochemistry (a cura di M. Florkin e E. H. Stotz), vol. XVII, Carbohydrate metabolism, Amsterdam 1969, pp. 219-240.
Villar-Palasi, C., Larner, J., Glycogen metabolism and glycolytic enzymes, in ‟Annual review of biochemistry", 1970, XXXIX, pp. 639-672.
Metabolismo dei lipidi
SOMMARIO: 1. Classificazione. □ 2. Acidi grassi: a) struttura chimica; b) catabolismo degli acidi grassi, β-ossidazione; c) altri meccanismi di ossidazione degli acidi grassi; d) biosintesi degli acidi grassi; e) controllo della sintesi degli acidi grassi; f) biosintesi degli acidi grassi monoinsaturi; g) biosintesi degli acidi grassi polinsaturi. □ 3. Biosintesi dei gliceridi: a) trigliceridi; b) fosfolipidi; c) glicolipidi. □ 4. Steroli e terpeni: a) struttura; b) biosintesi del colesterolo; c) funzione del colesterolo; d) composti poliisoprenoidi; e) regolazione del metabolismo del colesterolo. □ Bibliografia.
1. Classificazione.
I lipidi, come le proteine, gli acidi nucleici e i carboidrati, sono componenti fondamentali della cellula che si trovano in tutte le forme di vita. Come implica lo stesso nome (dal greco λίπος, grasso), i lipidi hanno consistenza grassa, in genere basso punto di fusione e non sono miscibili con l'acqua (per es. il burro, l'olio d'oliva, il sego bovino). Essi possono essere separati dai tessuti mediante estrazione con vari solventi organici, fra i quali l'etere e miscele di metanolo e cloroformio sono i più efficaci. Ma al di là di questa classificazione operativa fondata su alcune proprietà fisiche, i lipidi sono più difficili da definire, sia dal punto di vista chimico sia da quello funzionale. Affermare che le funzioni cellulari dipendono dall'esistenza di queste molecole insolubili in acqua definisce il problema, ma non lo risolve. Inoltre, appare sempre più evidente che i lipidi hanno differenti funzioni, alcune generali e altre più specializzate.
La presenza nei lipidi di lunghe catene idrocarburiche immiscibili con l'acqua (idrofobiche) non solo spiega le loro caratteristiche di solubilità, ma è anche la più tipica proprietà chimica che tutti i lipidi hanno in comune. Ma nemmeno questo attributo strutturale può essere definito con precisione. Le catene polimetileniche dei lipidi possono essere brevi o lunghe, lineari o ramificate, sature o insature, cicliche o acicliche. Inoltre un lipide può essere costituito solo di una porzione idrocarburica (idrocarburi lineari, colesterolo, battoprenolo, carotenoidi) o può contenere uno o più acidi grassi legati a molecole alcoliche per mezzo di legami estere (gliceridi, fosfogliceridi, esteri sterolici, cere). Alcuni lipidi meno comuni contengono acidi grassi uniti con legame ammidico (sfingolipidi). Gli esteri degli acidi grassi sono i lipidi più comuni e fra essi i fosfogliceridi (fosfolipidi) meritano un'attenzione speciale. Essi sono localizzati per lo più nelle membrane cellulari e sono, insieme alle proteine, i principali elementi strutturali degli involucri che racchiudono le cellule e i compartimenti subcellulari.
La diversità strutturale e la varia nonché variabile composizione in lipidi di cellule e organismi diversi sono le proprietà più caratteristiche e interessanti dei lipidi cellulari. Finora lo sforzo più rilevante nel campo dei lipidi, in gran parte coronato da successo, è stato rivolto alla caratterizzazione chimica e alla chiarificazione delle sequenze biosintetiche e cataboliche. La correlazione fra struttura delle diverse molecole lipidiche e funzione biologica sarà l'impegnativo traguardo della ricerca futura.
2. Acidi grassi.
a) Struttura chimica.
Acidi grassi di lunghezza compresa fra 4 e 26 atomi di carbonio sono stati isolati come costituenti dei lipidi naturali. Fra gli acidi saturi, i più comuni sono formati da molecole a numero pari di atomi di carbonio, con catena lineare; gli acidi C16 e C18 sono i più diffusi di questa classe (v. tab. I). Il punto di fusione di un acido grasso cresce con il numero di atomi di carbonio della catena, e nello stesso modo si comporta il punto di fusione del lipide di cui l'acido grasso in questione è un costituente. Il burro deve la sua consistenza molle, almeno in parte, al suo contenuto relativamente elevato di acidi grassi con meno di 16 atomi di carbonio, mentre il sego bovino, che è un grasso molto più duro, ha un alto contenuto di acido stearico (C18).
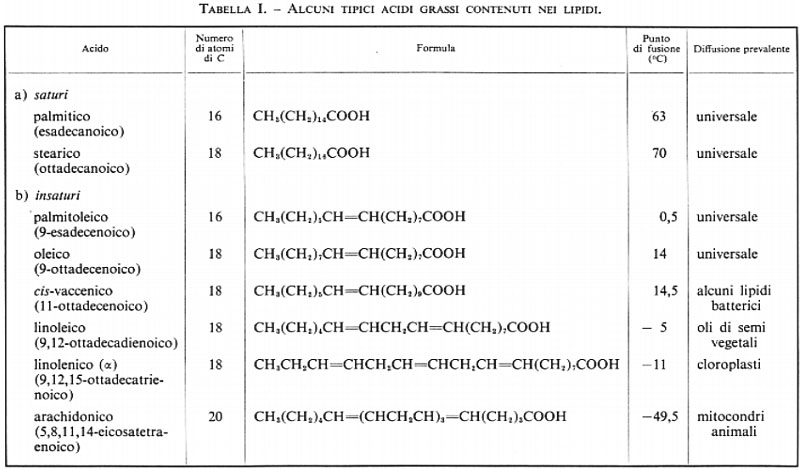
Anche gli acidi olefinici, contenenti un doppio legame, come l'acido palmitoleico (cis-9-esadecenoico) e l'acido oleico (cis-9-ottadecenoico), sono ampiamente diffusi. Il doppio legame è situato tipicamente in posizione 9-10 e ha configurazione cis. Questo carattere strutturale deriva dalla specificità dell'enzima (desaturasi) che trasforma il palmitato in palmitoleato e lo stearato in oleato, che sono rispettivamente precursori e prodotti con lo stesso numero di atomi di carbonio. Al contrario dei tessuti animali, delle piante e della maggior parte dei microbi, i Batteri non contengono acido oleico, ma il suo isomero, acido cis-vaccenico (acido cis-11-ottadecenoico), prodotto da essi con uno speciale meccanismo biosintetico.
L'insaturazione abbassa notevolmente il punto di fusione di un acido grasso, per es., per un acido a 18 atomi di carbonio, da 70 a 14 °C. Perciò l'insaturazione influenza la consistenza di un grasso non meno del numero di atomi di carbonio. L'olio di oliva deve il suo basso punto di fusione a un alto contenuto di acido oleico e l'olio di semi di lino alla preponderanza in esso degli acidi linoleico e linolenico, rispettivamente con 2 e 3 doppi legami. L'insaturazione influenza non solo il punto di fusione di un acido grasso, ma anche la sua configurazione geometrica, due fenomeni strettamente connessi fra di loro. Un acido grasso saturo è essenzialmente una molecola lineare fatta a zig-zag. Un doppio legame cis deforma nettamente questa linearità della catena idrocarburica sia introducendo una protuberanza sia piegando la catena stessa. Più doppi legami ci sono nella molecola, più essa si allontana da una forma lineare (v. formule 1, 2 e 3).
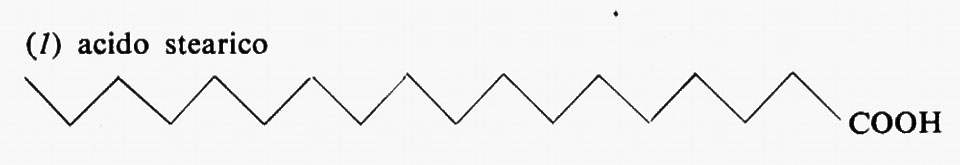
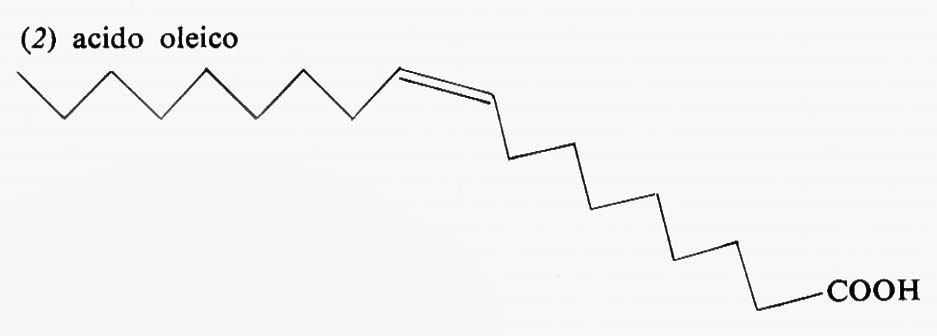
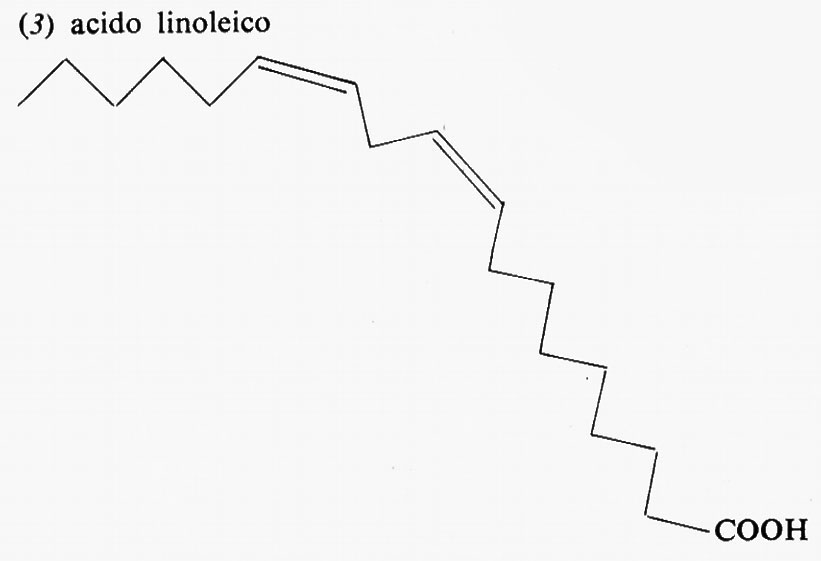
Queste molecole insature si avvicinano fra di loro molto meno intimamente delle molecole sature (per l'indebolimento delle interazioni idrofobiche che esistono fra due catene parallele) e tale fatto accresce la loro mobilità. Questo è un fattore importante per la stabilità e per diverse altre proprietà della membrana cellulare che dipendono in maniera critica dal grado di insaturazione degli acidi grassi contenuti nei fosfolipidi della membrana stessa.
Un ulteriore mezzo usato in natura per liquefare una catena idrocarburica, oltre alla lunghezza e al grado di insaturazione, è la ramificazione. Per esempio, l'acido ante-iso-14-metilesadecanoico (v. tab. II) fonde a una temperatura di 25 °C più bassa di quella del corrispondente acido C17 a catena lineare. Vari batteri grampositivi producono acidi grassi con catene ramificate al posto di catene olefiniche. La ramificazione vicino al centro di un acido grasso, tanto nella forma di un anello ciclopropanico (acido lattobacillico) che nella forma di un gruppo metilico (acido tubercolostearico), ha effetti fisici similari. La tab. II mostra questi acidi a catena ramificata e annche alcuni acidi grassi contenenti ossigeno che hanno una distribuzione speciale e un'attività fisiologica sconosciuta.
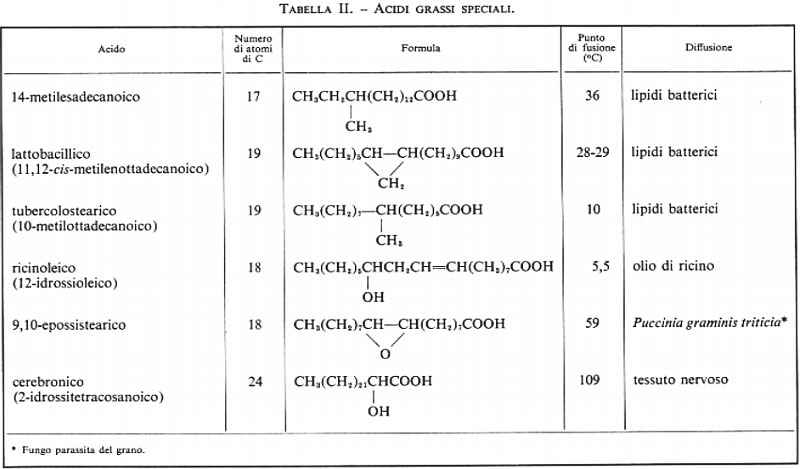
b) Catabolismo degli acidi grassi, β-ossidazione.
Nei tessuti animali, nelle piante e, più raramente, nei microrganismi, gli acidi grassi servono da principale riserva di combustibile, sotto forma di trigliceridi. Essi sono immagazzinati in cellule grasse specializzate (il tessuto adiposo) del corpo animale e nei semi delle piante. Sotto lo stimolo di segnali ormonali (adrenalina dalla midollare surrenale) avviene la liberazione dell'AMP ciclico (3′-5′-AMP), messaggero secondario che a sua volta attiva la scissione idrolitica dei trigliceridi del tessuto adiposo a glicerolo e ad acidi grassi liberi. Questi ultimi sono catturati dall'albumina del siero e sono trasportati in questa forma dalla corrente sanguigna verso il fegato e il muscolo per esservi demoliti.
Prima della demolizione ossidativa, gli acidi grassi liberi (o non esterificati) subiscono un complesso processo di attivazione che ha due finalità: a) trasformare la funzione carbossilica nel più attivo derivato tioestere del coenzima A (CoA); b) facilitare l'entrata dell'acido grasso nei mitocondri del fegato e del muscolo, dove sono localizzati gli enzimi ossidanti. Questo processo preparatorio consta di tre stadi:
1. acido grasso+CoA+ATP→acil-CoA
2. acil-CoA+carnitina→acilcarnitina
3. acilcarnitina+CoA→acil-CoA.
La reazione 1 avviene al di fuori dei mitocondri ed è catalizzata dall'enzima tiochinasi dell'acido grasso. La reazione 2 trasferisce il residuo di acido grasso al gruppo idrossilico della molecola trasportatrice carnitina (acido β-idrossi-γ-trimetilamminobutirrico). L'estere dell'acido grasso con la carnitina è la forma di trasporto dell'acido grasso attraverso la membrana mitocondriale. Nella reazione 3, che avviene all'interno del mitocondrio, il gruppo dell'acido grasso ritorna dalla carnitina al CoA. La catena dell'acido grasso è ora attivata (dal legame tioestere ‛ricco di energia') e collocata a stretto contatto degli enzimi ossidanti mitocondriali (v. bioenergetica). Si rigenera la carnitina, che può così rientrare nel ciclo del trasporto. Il processo della β-ossidazione trasforma ossidativamente un acido grasso contenente n atomi di carbonio in n/2 frammenti a due atomi di carbonio (acetil-CoA), come appare nello schema 1.
La sequenza delle 4 reazioni che sono rappresentate nello schema 1 accorcia la catena dell'acido grasso di due atomi di carbonio per volta, producendo CH3(CH2)n-2 COSCoA, acetil-CoA e 1 mole di ciascuno dei trasportatori di elettroni ridotti FADH2 e NADH secondo l'equazione complessiva:
CH3(CH2)nCOSCoA+CoASH+FAD+NAD++H2O
→CH3(CH2)n-2COSCoA+acetil-CoA+FADH2
+NADH+H+.

Il processo si ripete finché l'intero acido grasso è scisso in frammenti C2. In questo modo 1 mole di acido palmitico dà 8 moli di acetil-CoA e 7 moli sia di FADH2, sia di NADH. L'acetil-CoA entra nel ciclo degli acidi tricarbossilici per essere bruciato completamente a CO2 e H2O. Questo processo genera 12 moli di ATP per ogni unità C2 o 96 moli per ogni mole di palmitato. A queste si devono aggiungere 35 moli di ATP generate dalla fosforilazione ossidativa durante il trasporto di elettroni dalle 7 moli di FADH2 e di NADH all'ossigeno. Dal totale di 96+35=131 moli di ATP per mole di acido palmitico ossidato si deve sottrarre 1 mole di ATP, dato che la reazione iniziale di attivazione del palmitato a palmitil-CoA richiede il consumo di 1 ATP. Così l'ossidazione totale di 1 mole di acido palmitico a CO2 e H2O mette a disposizione un prodotto netto di 130 moli di ATP, equivalenti a circa 1.040 kcal.
c) Altri meccanismi di ossidazione degli acidi grassi.
Alcune cellule hanno altri meccanismi per ossidare gli acidi grassi, ma essi servono a scopi speciali e non sono necessariamente accoppiati alla produzione di ATP. Per esempio il processo dell'ω-ossidazione, presentato più sotto, introduce ossigeno molecolare nella posizione terminale di una catena idrocarburica per mezzo di una delle cosiddette reazioni ossigenasiche:
CH3(CH2)nCOOH+O2+NADPH+H+→
CH2OH(CH2)nCOOH+NADP++H2O.
Nell'ω-ossidazione i substrati sono gli acidi grassi liberi, non i loro tioesteri con il CoA. L'ω-idrossiacido può essere ossidato e demolito ulteriormente in rapporto alla lunghezza della catena carboniosa. Un meccanismo di questo tipo si ritrova anche nell'ossidazione di certi idrocarburi (esano, ottano) che possono servire da fonte di carbonio per alcune specie di Pseudomonas e di lieviti aerobi.
Un altro meccanismo non molto diffuso è l'ossidazione di acidi grassi a catena lunga nella posizione α, con trasformazione in α-idrossiacidi. Non si conosce il significato fisiologico dell'α-ossidazione.
d) Biosintesi degli acidi grassi.
L'acetil-CoA è non solo il prodotto di scissione nell'ossidazione degli acidi grassi, ma serve anche da pietra costitutiva per l'edificio della biosintesi degli acidi grassi. Si potrebbe perciò supporre che gli acidi grassi possano essere biosintetizzati percorrendo in senso opposto la via della β-ossidazione. I primi tre passaggi della β-ossidazione (v. schema 2) possono in realtà procedere in ambedue i sensi, ma la reazione catalizzata dalla β-chetotiolasi (reazione 4) è essenzialmente irreversibile. Perciò un'inversione totale della via catabolica è sfavorevole dal punto di vista energetico. Per superare questa barriera energetica e facilitare la formazione del legame carbonio-carbonio, una molecola di acetil-CoA viene ‛iperattivata' nella reazione seguente, catalizzata dalla acetil-CoA-carbossilasi:


In questo modo un gruppo metilico relativamente inerte è trasformato nel più reattivo gruppo metilenico del malonil-CoA.
Prima che avvenga la condensazione fra le unità acetilica e malonilica, i loro CoA-derivati devono essere trasferiti su una molecola chiamata ACP (Acyl Carrier Protein, proteina trasportatrice di acile). L'ACP è una proteina a basso peso molecolare (circa 8.000) che contiene la 4-fosfopanteteina del coenzima A e, al pari di questo coenzima, possiede un gruppo solfidrilico a cui può attaccarsi un gruppo acilico.
All'inizio, quindi, acetil-CoA e malonil-CoA donano i loro gruppi acilici all'ACP per formare i derivati acil-tioesteri della proteina trasportatrice (v. schema 2, reazioni 1 e 2).
Nel passaggio successivo di allungamento della catena (reazione 3), la condensazione fra le unità acetilica e malonilica legate alla proteina è accoppiata con la rimozione dall'unità malonilica del gruppo carbossilico libero, che ha anche la funzione di metterla in moto. È questo il passaggio che distingue chimicamente la sintesi dall'ossidazione dell'acido grasso.
Il composto intermedio così prodotto, cioè il derivato acetoacetilico o β-chetoacilico, è ridotto nelle reazioni 4, 5, 6 in modo da formare una catena satura. In questo modo le reazioni 1-6 producono l'allungamento della catena carboniosa da acetato a butirrato e nel contempo trasformano molecole più ossidate in molecole più ridotte. Come molti altri processi biosintetici, anche la formazione di acidi grassi richiede NADPH come fonte di potere riducente.
La butirril-ACP prodotta dalla reazione 6 rientra nel ciclo al punto 1 e si condensa con un'altra molecola di malonil-ACP per formare una catena a 6 atomi di carbonio, che sarà poi modificata come sopra attraverso le reazioni 4 e 6 per formare esanoil-ACP. Il medesimo processo ciclico viene ripetuto finché la catena dell'acido grasso è lunga 16 o 18 atomi di carbonio. Normalmente il ciclo di allungamento è concluso dal trasferimento della catena completa o al CoA (reazione 7a) o all'acqua (7b). Quando palmitato e stearato escono dal ciclo dell'acido grasso sotto forma di CoA-derivati, possono essere utilizzati direttamente in reazioni di esterificazione per formare lipidi più complessi (v. sotto). Queste reazioni avvengono nel lievito, nelle piante e nei Batteri. Nei tessuti animali il sistema della sintetasi dell'acido grasso scarica acidi grassi liberi nella corrente sanguigna perché siano trasportati dal luogo di sintesi ad altri organi o tessuti. Nello stesso tempo, si libera CoA, che diventa disponibile per altri processi metabolici.
A un esame superficiale, la β-ossidazione e la biosintesi dell'acido grasso appaiono come processi molto simili. Invece sono due sequenze completamente indipendenti, come mostrato dalla tab. III.
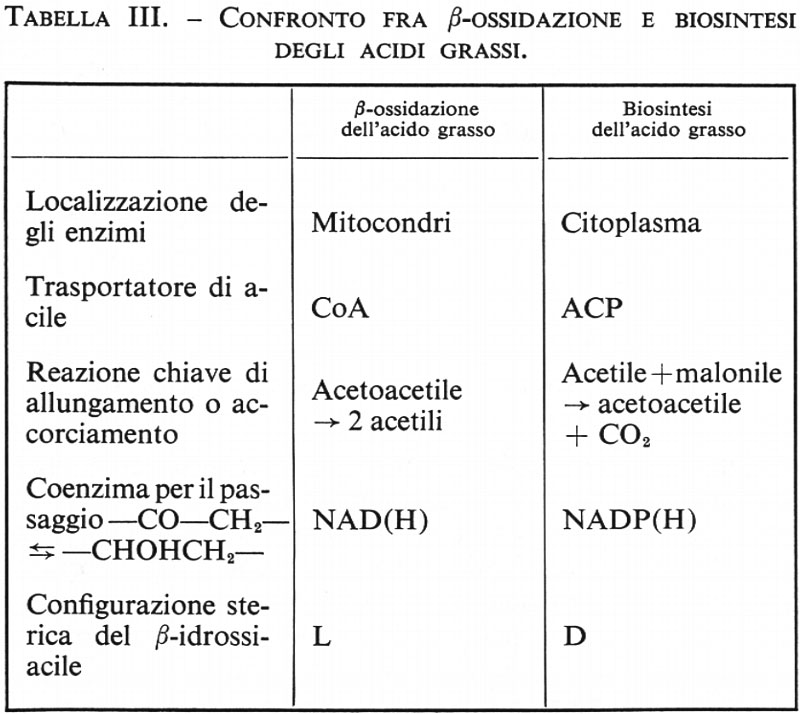
Benché le sintetasi degli acidi grassi catalizzino in tutte le cellule e in tutti gli organismi la medesima serie di reazioni - formazione di acidi grassi a catena lunga da acetil-CoA e malonil-CoA - esse sono notevolmente differenti fra di loro per proprietà molecolari e idrodinamiche. Questi sistemi enzimatici appartengono a due tipi principali. Nel lievito e nei tessuti animali le reazioni 1-7 dello schema 2 sono catalizzate da enzimi associati fra di loro in un complesso a elevato peso molecolare e si comportano come una singola entità fisica. Il peso molecolare è 2×106 per il sistema del lievito e 0,5×106 per quello del fegato. Probabilmente questi sistemi multienzimatici contengono al loro interno qualcosa di più di una semplice copia dei sette enzimi interessati e dell'ACP. Gli enzimi che catalizzano i singoli passaggi sono tenuti insieme così strettamente da legami idrofobici ed elettrostatici che non possono essere separati l'uno dall'altro e non sono capaci di attività individuale. Questi complessi multienzimatici si formano probabilmente per un processo di autoaggregazione dei singoli componenti catalitici, ACP inclusa, in un sistema cooperativo capace di funzionare efficacemente. Questa coordinazione fisica di più attività metaboliche destinate a operare consecutivamente permette di ottenere un alto grado di efficienza e risulta pertanto di reale beneficio per la cellula.
In molti batteri e piante il sistema denominato sintetasi dell'acido grasso è di un altro tipo. Gli enzimi che catalizzano i passaggi 1-7 e anche l'ACP sono stati isolati da queste fonti come unità discrete, ognuna delle quali catalizza uno solo dei sette passaggi suddetti. Il peso molecolare di questi enzimi è inferiore a 100.000. In effetti quello che già si conosce sui particolari del meccanismo chimico della sintesi degli acidi grassi è stato appreso principalmente dallo studio dei sette enzimi della sintetasi dell'acido grasso purificati individualmente dal batterio Escherichia coli.
Il fatto che per la sintetasi dell'acido grasso esistano nei diversi organismi sistemi non organizzati e sistemi organizzati (più efficienti?) e gli schemi secondo i quali essi sono distribuiti sono fenomeni curiosi che hanno forse un significato filogenetico.
Un altro aspetto poco chiaro della biosintesi degli acidi grassi è la terminazione della catena a una certa lunghezza. Perché in certe cellule il processo ripetitivo si arresta dopo 8 cicli (C16), in altre dopo 9 (C18) e in altri casi isolati dopo 13 (C26)? Non si comprendono al momento attuale né il meccanismo che controlla la terminazione della catena, nè i motivi per cui le cellule hanno bisogno di acidi grassi di lunghezza particolare.
e) Controllo della sintesi degli acidi grassi.
La velocità delle varie sequenze metaboliche può essere controllata, in senso negativo o positivo, modulando la velocità di un enzima chiave nel processo complessivo. Questo avviene in seguito a segnali nervosi, ormonali o chimici. Questi enzimi regolatori sono in genere responsabili di reazioni irreversibili e limitanti la velocità complessiva. Nella sintesi di un acido grasso il controllo fisiologico della velocità non è esercitato su nessuno dei sette enzimi del ciclo di allungamento. L'enzima regolatore è invece, in questo processo, l'acetil-CoA-carbossilasi, che produce malonil-CoA, il substrato essenziale per la sintesi degli acidi grassi. Come molti altri enzimi regolatori, l'acetil-CoA-carbossilasi esiste in due forme 6 dimensioni molecolari, una inerte dal punto di vista catalitico e un'altra cataliticamente attiva.
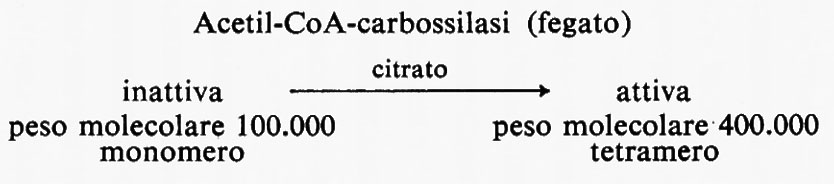
Il citrato è una delle piccole molecole che provocano la transizione monomero-tetramero dell'acetil-CoA-carbossilasi del fegato, ‛innescando' in tal modo il potere catalitico. Non è noto perché l'enzima aggregato o polimerico è cataliticamente attivo mentre quello monomerico non lo è. Tuttavia si pensa che la transizione dalla forma inattiva monomerica a quella attiva polimerica sia associata con cambiamenti nella struttura tridimensionale (conformazione). Questi cambiamenti possono esporre o scoprire le regioni dell'enzima che sono cataliticamente attive e renderle così più accessibili al substrato.
La scelta particolare dell'acido citrico come attivatore dell'acetil-CoA-carbossilasi può essere razionalizzata nel modo seguente. Il citrato è il primo substrato del ciclo degli acidi tricarbossilici, processo con il quale i metaboliti sono trasformati ossidativamente in CO2 e H2O, con produzione concomitante di ATP. Quando le richieste cellulari di energia sotto forma di ATP sono soddisfatte, il ciclo degli acidi tricarbossilici rallenta e il citrato si accumula. In questo caso la cellula tenderà a non sciupare il materiale combustibile presente in eccesso rispetto ai bisogni energetici e a immagazzinarlo per una futura utilizzazione. La sintesi di grassi è il sistema più efficace per questo scopo e perciò elevati livelli di citrato costituiscono un segnale idoneo a mettere in moto il processo di immagazzinamento di energia sotto forma di grasso. Il processo di controllo che regola la sintesi di grasso tramite l'acetil-CoA-carbossilasi è stato finora dimostrato solo per la sintesi degli acidi grassi nei tessuti animali. Non si sa come questo problema sia risolto dalle piante e dai microrganismi.
f) Biosintesi degli acidi grassi monoinsaturi.
Sembra che per la funzione cellulare sia necessario un appropriato rapporto fra acidi grassi saturi e insaturi (olefinici) contenuti nei trigliceridi e nei fosfolipidi. Perciò esistono dei meccanismi anche per la sintesi biologica degli acidi grassi insaturi. Vi sono due modi per introdurre un doppio legame olefinico negli acidi grassi saturi. Secondo un meccanismo, la catena dell'acido grasso saturo è desaturata per mezzo della rimozione ossidativa di due atomi di idrogeno da due atomi di carbonio adiacenti; secondo l'altro, un legame olefinico si stabilisce durante la crescita della catena idrocarburica, cioè mentre essa è ancora in fase di allungamento. Il primo meccanismo è il più comune. Il legame olefinico prodotto secondo l'uno o l'altro dei due meccanismi ha sempre configurazione cis.
Desaturazione ossidativa. - La formazione di olefine tramite desaturazione è descritta dalla seguente equazione generale:
CH3(CH2)nCH2CH2(CH2)mCOR+O2+NADPH+H+
→CH3(CH2)nCH=CH(CH2)mCOR+2H2O
+NADP+.
Nella maggior parte dei casi, il substrato per l'enzima desaturante è l'acido palmitico, o l'acido stearico, nella forma di tioestere del coenzima A (R=coenzima A). Il doppio legame viene generalmente introdotto fra il nono e il decimo atomo di carbonio e assume sempre la configurazione cis. Gli atomi di idrogeno vengono staccati in maniera stereospecifica dal C9 e dal C10 della catena carboniosa e vengono trasferiti alla forma ossidata di un accettore. Infine gli elettroni passano all'ossigeno formando acqua. Nel caso della desaturasi dell'alga Euglena gracilis la catena di trasporto degli elettroni per la desaturazione ossidativa degli acidi grassi è composta di tre enzimi separati, NADPH-ossidasi, una proteina a ferro non eminico (ferredossina) e la vera e propria desaturasi:
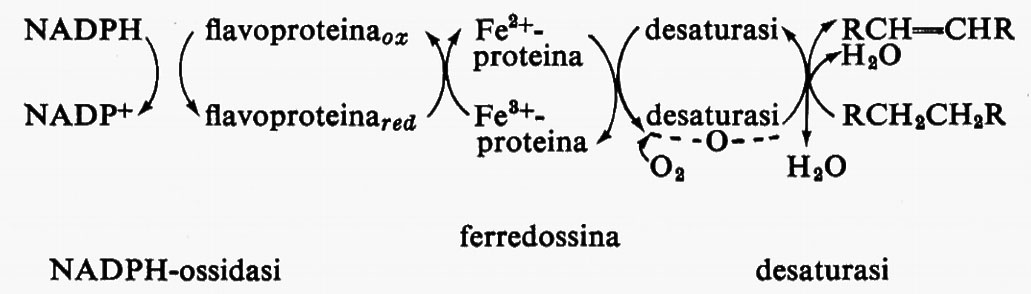
L'aspetto caratteristico della desaturazione ossidativa è l'esigenza di ossigeno molecolare come accettore di elettroni. Questo processo è biosintetico e deve essere distinto dalla reazione di rimozione di idrogeno che avviene quando i CoA-derivati degli acidi grassi saturi sono deidrogenati ad α, β-trans-olefine durante la β-ossidazione.
A parte il sistema enzimatico di sintesi delle olefine nell'Euglena gracilis, le acil-CoA-desaturasi sono poco caratterizzate perché esse non possono essere dissociate dal reticolo endoplasmatico e da altre frazioni cellulari particolate. Una possibile spiegazione del fatto per cui il substrato della reazione desaturante è l'acil-CoA piuttosto che l'acido grasso libero è la seguente. In alcune sintetasi dell'acido grasso gli acidi grassi a catena lunga vengono prodotti dal processo di allungamento sotto forma di tioesteri del CoA. Una fase ulteriore della biosintesi dei lipidi, l'esterificazione con il glicerolo, richiede anch'essa acil-CoA invece che acido grasso libero. Mantenendo il legame tioestere dell'acido grasso durante la desaturazione, la cellula risparmia l'energia (ATP) che sarebbe altrimenti necessaria per trasformare l'acido grasso libero in derivato del CoA. Così, l'uso di derivati del CoA nella desaturazione degli acidi grassi può essere spiegato in base al principio di conservazione dell'energia piuttosto che con necessità meccanicistiche.
Disidratazione durante la sequenza di allungamento. - Lo schema 3 mostra come si formano gli acidi palmitoleico e vaccenico in Escherichia coli e in alcune altre specie batteriche.

Questa via metabolica è ramificata, e fornisce acidi grassi sia saturi sia insaturi. La catena carboniosa viene prima costruita fino allo stadio di un β-idrossiacido a C10, per mezzo delle reazioni già discusse (v. schema 2). A questo punto la via si divide e produce acidi saturi a lunga catena nella maniera solita (diramazione a) o alternativamente imbocca la strada degli acidi olefinici (diramazione b). Quest'ultima è iniziata da un enzima specifico ed esclusivamente batterico che elimina acqua dal β-idrossiacido a C10 formando un composto acilico cis-β-γ (o 3-4) olefinico. Il doppio legame presente in questo composto non può essere ridotto e si conserva perciò in tutte le successive reazioni di allungamento. Il risultato di questa via è la produzione degli acidi cis-olefinici palmitoleico e vaccenico. In questa sequenza esclusivamente batterica di produzione degli acidi grassi insaturi, i legami olefinici sono generati da una reazione di disidratazione invece che dalla deidrogenazione ossidativa discussa in precedenza. Perciò viene denominata via anaerobica per gli acidi grassi insaturi. Dato che si pensa che le primissime forme di vita si siano sviluppate in un'atmosfera completamente priva di ossigeno, la sequenza anaerobica batterica potrebbe rappresentare un meccanismo biosintetico più primitivo.
La metilazione del doppio legame olefinico degli acidi palmitoleico e vaccenico porta alla formazione dei derivati ciclopropanici, cioè, rispettivamente, l'acido 9,10-metilene-sadecanoico e l'acido 11,12-metilenottadecanoico (acido lattobacillico, v. tab. II), due composti abbastanza comuni. Queste reazioni di ramificazione avvengono soltanto nei Batteri.
g) Biosintesi degli acidi grassi polinsaturi.
L'inserimento di ulteriori doppi legami nella catena di un acido grasso avviene di solito a partire dall'acido oleico mediante desaturazione ossidativa, secondo lo schema 4.

L'inserimento del secondo doppio legame, che ha anch'esso configurazione cis, avviene fra gli atomi di carbonio terzo e quarto dal doppio legame 9-10, o in direzione del gruppo carbossilico o verso il gruppo metilico terminale della catena. La struttura dei due acidi isomeri, 9,12-ottadecadienoico (linoleico) e 6,9-ottadecadienoico, è detta struttura divinilmetanica. I numerosi acidi poliinsaturi con 3, 4, 5, o 6 doppi legami sono tutti costruiti in base allo stesso principio. Il sostituente R del gruppo carbossilico può essere il coenzima A o un'altra molecola capace di combinarsi mediante un legame estere. È stata avanzata l'ipotesi che il substrato per le successive desaturazioni sia il fosfolipide oleil-fosfatidilcolina.
Il corredo naturale di acidi grassi poliinsaturi varia notevolmente da organismo a organismo. Nei Batteri gli acidi poliinsaturi sono eccezionalmente rari e non sembrano essenziali per la crescita o il metabolismo. Questa non essenzialità è chiaramente dimostrata in certi mutanti di Escherichia coli, che sono incapaci di sintetizzare qualsiasi acido grasso olefinico. Per crescere, tali mutanti devono essere coltivati in terreni addizionati di acidi grassi poliinsaturi. Sono sufficienti a questo scopo gli acidi oleico e vaccenico; dato che essi non sono desaturati ulteriormente, ne consegue che gli acidi grassi a più alto grado di insaturazione non sono essenziali per la cellula batterica. La stessa indipendenza nutrizionale è mostrata da alcuni lieviti che derivano la propria energia da processi fermentativi. Per esempio, si può far crescere il Saccharomyces cerevisiae in ambiente completamente anaerobico, purché il mezzo di crescita contenga acido oleico. Questo lievito, come Escherichia coli, non possiede gli enzimi per l'ulteriore desaturazione dell'acido oleico. Un acido grasso provvisto di un solo doppio legame è perciò sufficiente per la sua crescita e il suo mantenimento.
Al contrario, lieviti obbligatoriamente aerobi, come Torulopsis utilis, producono grandi quantità di acidi linoleico e γ-linolenico (6,9, 12-ottadecatrienoico). Ciò indica che questi composti hanno una funzione necessaria nel metabolismo energetico ossidativo. In effetti i mitocondri, la sede dei meccanismi ossidativi, sono particolarmente ricchi in acidi grassi poliinsaturi.

La tab. IV descrive alcune delle sequenze conosciute per la formazione degli acidi grassi poliinsaturi nei tessuti animali e nelle piante. È interessante notare l'incapacità di tutte le specie animali studiate a trasformare l'oleato in linoleato. L'enzima desaturante specifico per l'introduzione del secondo doppio legame deve essere stato perduto precocemente per mutazione nell'evoluzione delle specie animali. In seguito alla carenza di questo enzima, gli animali hanno bisogno di introdurre con l'alimentazione acidi grassi poliinsaturi, che sono perciò chiamati anche acidi grassi essenziali. Questa esigenza dietetica è soddisfatta dall'acido linoleico e dagli acidi C18 e C20 a più alto grado di insaturazione.
Gli acidi poliinsaturi sono essenziali per i Vertebrati. Non risulta che l'acido linoleico o acidi a più alto grado di insaturazione siano sintetizzati o necessari in Insetti o altri Invertebrati.
L'acido linoleico alimentare è trasformato dagli animali in acido 6,9,12-ottadecatrienoico, noto anche come acido γ-linolenico, mediante inserimento del terzo doppio legame dalla parte del carbossile (v. tab. IV). A questo punto la catena carboniosa si allunga per dare l'acido trienoico a C20, che per ulteriore desaturazione dà l'acido tetraenoico (arachidonico), uno dei principali acidi grassi dei mitocondri epatici e surrenali. Nella maggior parte delle piante la sequenza di desaturazione si svolge in maniera differente. Anzitutto, le piante contengono l'enzima desaturante acido oleico-acido linoleico, che manca negli animali. In secondo luogo, l'ulteriore desaturazione dell'acido linoleico nelle piante produce l'acido 9,12,15-ottadecatrie- noico (α-linolenico), isomero dell'acido 6,9, 12-trienoico prodotto dai tessuti animali. L'acido α-linolenico è particolarmente abbondante nei tessuti verdi delle piante, ma non ha efficacia alimentare per gli animali come fonte di ‛acidi grassi essenziali'. Benché l'acido α-linolenico sia metabolizzato ulteriormente nell'animale ad acidi più lunghi e più insaturi (tetra-, penta- ed esaenoico), queste strutture si sono rivelate inadatte come sostituti dell'acido arachidonico. Una delle possibili ragioni di questo fatto è stata messa in evidenza di recente. L'acido arachidonico è il precursore di una classe di sostanze ormonali altamente attive, note come prostaglandine.
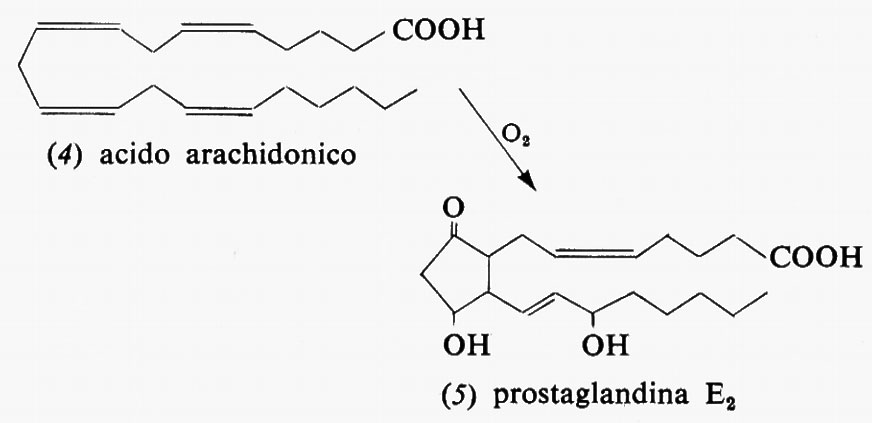
Esse sono formate e si ritrovano nella prostata e in minor grado in molti altri tessuti animali. Fra i numerosi effetti esercitati dalle prostaglandine sono la stimolazione della contrazione della muscolatura liscia, l'abbassamento della pressione sanguigna e l'antagonismo verso alcuni ormoni (vasopressina, adrenalina). Queste sostanze sono attive a concentrazioni minime. Oltre alla formazione delle prostaglandine, che è una funzione accertata dell'acido arachidonico, è probabile che esistano altri ruoli per questo tipo di molecole. Si è accennato all'alta concentrazione di acidi grassi poliinsaturi presente nei fosfolipidi mitocondriali. Nelle piante, invece, ricche di acido a-linolenico, questo tipo di acido polunsaturo si accumula prevalentemente nei lipidi dei cloroplasti.
3. Biosintesi dei gliceridi.
Gli acidi grassi sono presenti nei tessuti e nei liquidi dell'organismo prevalentemente sotto forma di esteri. Al pH fisiologico, gli acidi grassi esistono per lo più come anioni carbossilato, che hanno azioni inibitrici o denaturanti su molti enzimi e membrane. Anche quando gli acidi grassi non sono esterificati, come avviene nel torrente circolatorio, essi sono fortemente legati a una proteina serica (albumina). In questa forma complessata gli acidi grassi sono relativamente non tossici. Per lo più gli acidi grassi si ritrovano in natura come esteri del glicerolo, del glicerofosfato e di suoi derivati, di zuccheri, steroli e alcoli alifatici a lunga catena. Più recentemente sono stati descritti derivati di acidi grassi con il glicole etilenico o glicerofosfonati.
La natura endoergonica della formazione di esteri da acidi e alcoli richiede che in qualche stadio del processo venga fornita energia. Questa richiesta è soddisfatta dall'uso di derivati tioesteri degli acidi grassi come reagenti. In genere essi sono derivati tioesteri del coenzima A o, in qualche caso, della proteina trasportatrice di acile:
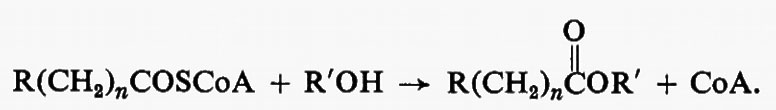
a) Trigliceridi.
I trigliceridi, che servono come principali lipidi di deposito nelle piante e negli animali, sono sintetizzati essenzialmente con la sequenza indicata nello schema 5.
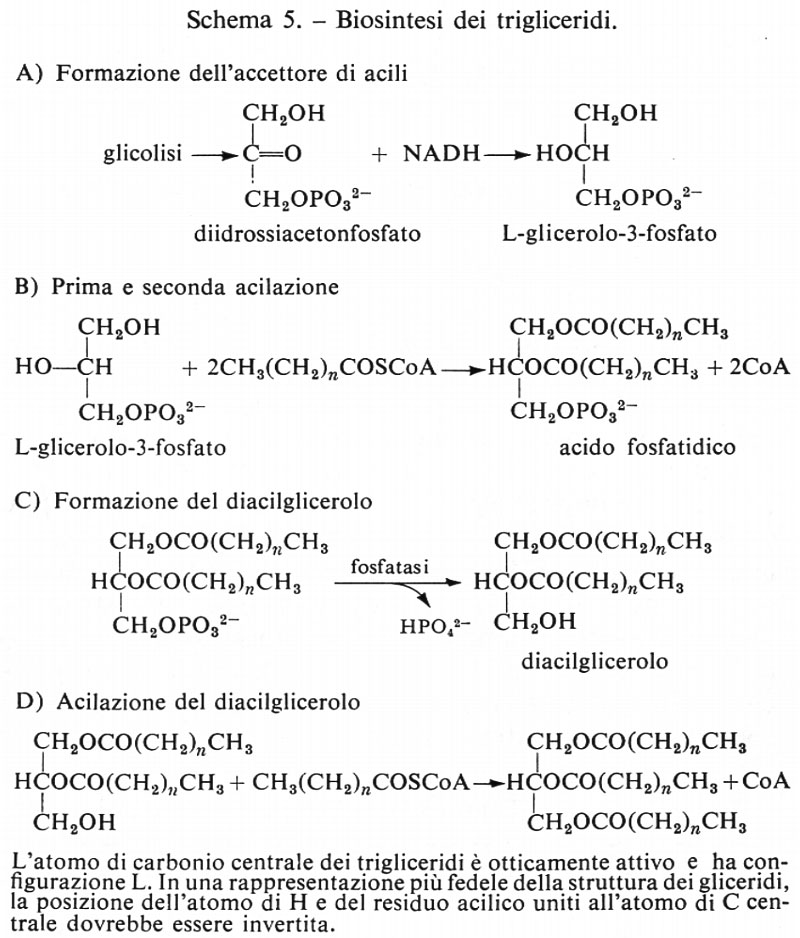
La loro formazione è intimamente legata al metabolismo dei carboidrati, dato che l'L-glicerolo-3-fosfato, il primo accettore di radicali acilici, si forma per riduzione del diidrossiacetonfosfato, composto intermedio della glicolisi (v. metabolismo dei carboidrati). Nello schema 5 l'esterificazione dell'L-glicerolo-3-fosfato a derivato diacilico è rappresentata come un passaggio singolo, ma è probabile che il processo avvenga per stadi, con l'introduzione di un residuo acilico per volta. Il primo prodotto di transacilazione sarebbe perciò un monoacilglicerofosfato o acido lisofosfatidico. Poiché gli enzimi esterificanti sono legati alle membrane, è difficile dimostrare l'esistenza di due enzimi separati. Prima della terza reazione di acilazione il gruppo fosfato in posizione 3 è staccato da una specifica fosfatasi dell'acido fosfatidico.
Come s'è già detto, i trigliceridi isolati dalle diverse fonti naturali variano ampiamente nella loro composizione in acidi grassi, principalmente per quanto riguarda la lunghezza della catena e il grado di insaturazione dei residui di acido grasso. Spesso, ma non sempre, il residuo in posizione 2 del glicerolo è più insaturo di quelli in posizione 1 e 3.
I Batteri costituiscono un'eccezione, in quanto sintetizzano o accumulano pochissimo grasso sotto forma di trigliceridi.
b) Fosf0lipidi.
I fosfolipidi sono derivati di acidi fosfatidici in cui il gruppo fosfato è esterificato con uno dei seguenti composti alcolici: le basi azotate etanolammina e colina, l'amminoacido serina, lo zucchero ciclico inositolo, il glicerolo; oppure è legato a una seconda molecola di acido fosfatidico. Poiché varia anche la natura dei residui di acido grasso nella parte fosfatidica, si può immaginare un numero pressoché infinito di strutture fosfolipidiche Si riportano qui le formule di alcuni gruppi di fosfolipidi (v. formule 6).
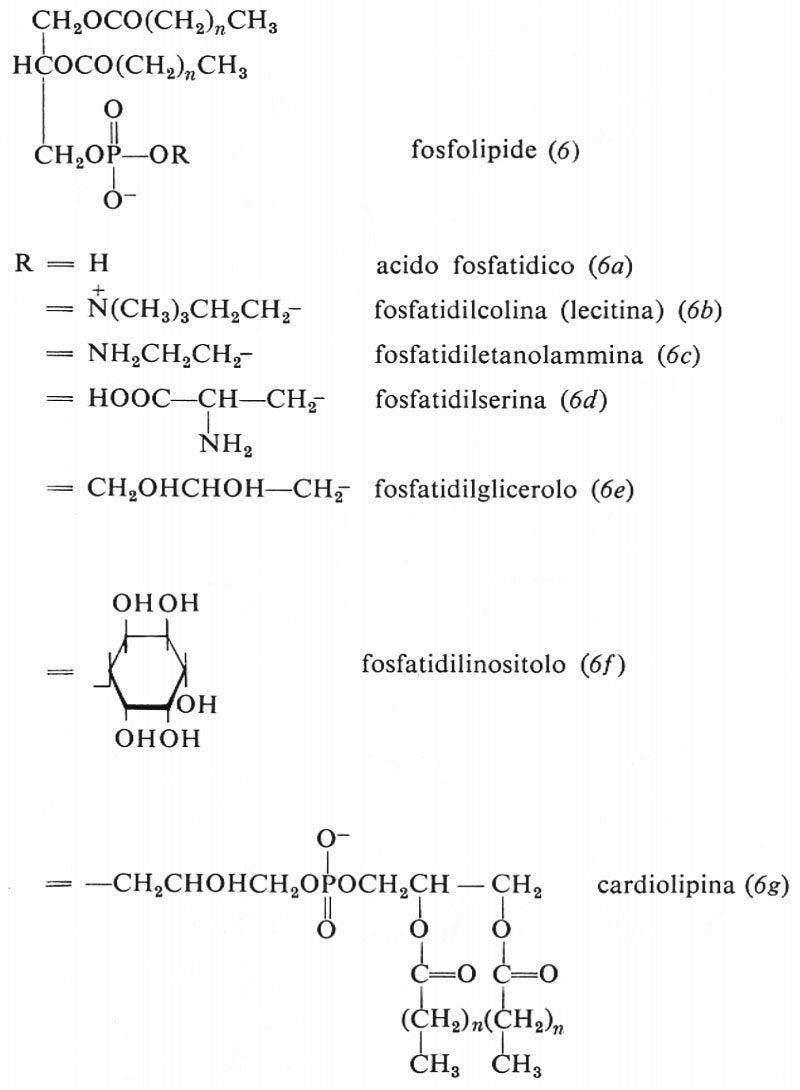
I plasmalogeni rappresentano un'altra variante abbastanza comune della struttura fondamentale dei fosfolipidi. In queste molecole uno dei legami esteri fra il glicerolo e l'acido grasso è sostituito da un legame etere vinilico:
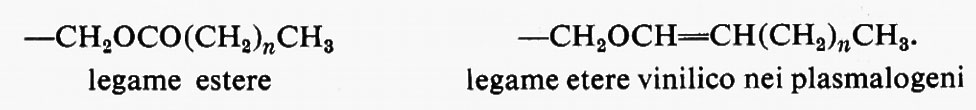
La catena idrocarburica attaccata al glicerolo per mezzo del legame etere ha la stessa lunghezza (16 e 18 atomi di carbonio) della catena dei fosfolipidi a struttura estere.
Le sfingomieline (v. formula 7) sono fosfolipidi con uno schema strutturale differente dai derivati dell'acido fosfatidico discussi finora. Comunque, le sfingomieline hanno in comune con gli altri fosfolipidi due lunghe appendici idrocarburiche, una sotto forma di ammide di un acido grasso, l'altra rappresentata dalla sfingosina, una base a catena lunga. Un gruppo idrossilico della sfingosina è legato alla fosforilcolina e il gruppo amminico della stessa
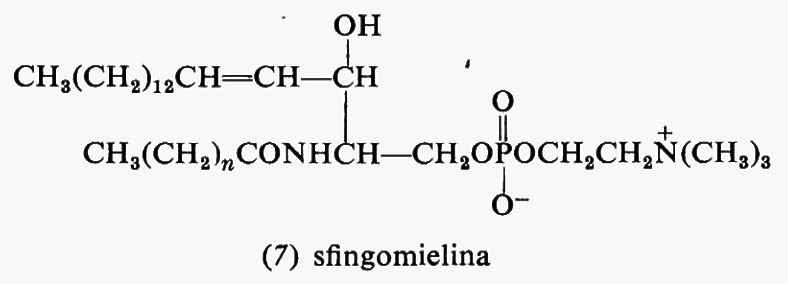
sostanza costituisce il punto di attacco per un residuo di acido grasso.
Ci sono alcuni elementi strutturali comuni che sembrano essere importanti per la funzione dei fosfolipidi. Essi sono i cosiddetti gruppi a testa polare (che comprendono il gruppo fosfato e il componente alcolico R a esso attaccato) e le due catene idrocarburiche non polari. A seconda della natura del gruppo R, la carica netta del fosfolipide può essere negativa (fosfatidiletanolammina, fosfatidilgucerolo, fosfatidilinositolo), negativa bivalente (cardiolipina) o anfionica (sfingomielina). Un'altra caratteristica importante della struttura fosfolipidica appare all'ispezione dei modelli a spazi pieni. Essi mostrano che in tutti i derivati dell'acido fosfatidico e nella sfingomielina le due catene idrocarburiche sono parallele fra di loro e strettamente accostate. Questo accostamento è dovuto alle forze di van der Waals. Più lunghe e più sature sono le catene idrocarburiche, più forti sono le interazioni. Poiché contengono sia un gruppo a testa polare che catene idrocarburiche non polari, le molecole di fosfolipidi sono denominate ‛anfipatiche'. Il ruolo speciale esercitato dai fosfolipidi come costituenti delle membrane è probabilmente dovuto a questo carattere anfipatico.
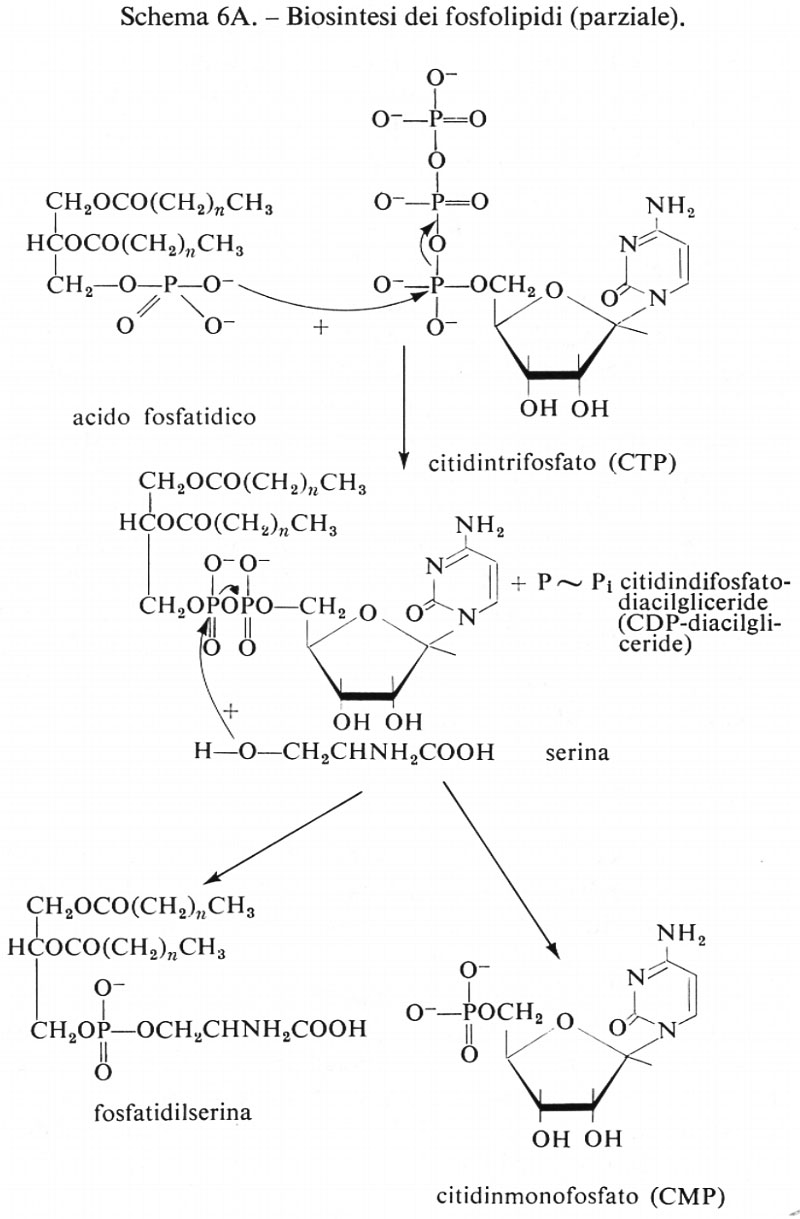
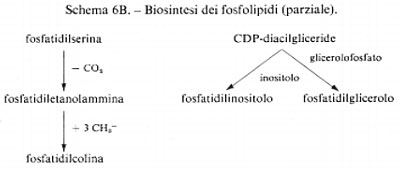
L'acido fosfatidico è il precursore comune dei trigliceridi e anche di tutti i fosfolipidi, eccetto le sfingomieline. L'acido fosfatidico è situato perciò al punto di biforcazione delle due più importanti vie biosintetiche dei lipidi. Come illustrato nello schema 6A, l'attacco dei vari residui R al fosfato dell'acido fosfatidico è realizzato con l'intervento del citidintrifosfato per mezzo dell'intermediario chiave citidindifosfato-diacilgliceride. In questo passaggio viene applicato un principio comune dei processi biosintetici. Un legame pirofosforico è scisso e questa scissione è accoppiata alla sintesi di un nuovo legame pirofosforico. Nel composto intermedio CDP-diacilgliceride, un fosfato del ponte pirofosforico (difosforico) di nuova formazione deriva dal CTP e l'altro dall'acido fosfatidico. Nel passaggio successivo, l'ossigeno idrossilico dell'amminoacido serina si inserisce nel ponte pirofosforico del CDP-diacilgliceride, portando alla formazione di fosfatidilserina con eliminazione di CMP. Questo primo prodotto fosfolipidico è poi modificato per decarbossilazione della serina a fosfatidiletanolammina (v. schema 6B). Per metilazione di quest'ultima, si forma fosfatidilcolina. Come mostrato nello schema 6B, il fosfatidilinositolo e il fosfatidilglicerolo si formano in maniera analoga, dal CDP-diacilgliceride, per reazione con l'inositolo e il glicerolofosfato rispettivamente. È chiaro il ruolo fondamentale del citidintrifosfato, uno dei precursori degli acidi nucleici, nella biosintesi dei fosfolipidi. Il CTP è altamente specifico e non può essere sostituito da altri nucleosidi-trifosfati.
Funzione dei fosfolipidi. - Tutte le membrane cellulari contengono fosfolipidi. Quando questi fosfolipidi vengono rimossi per idrolisi enzimatica (fosfolipasi) o per estrazione con solventi organici, si perde l'organizzazione strutturale delle membrane. Perciò i fosfolipidi sono importanti per l'integrità strutturale della membrana cellulare. Inoltre i fosfolipidi si rivelano essenziali per l'attività catalitica di parecchi enzimi che sono essi stessi parte della struttura della membrana. Per mezzo di tecniche speciali, soprattutto l'uso di agenti tensioattivi (detergenti), spesso si riesce a ottenere questi enzimi in forma solubile. In molti casi questi enzimi solubilizzati derivanti da membrane sono notevolmente attivati dall'aggiunta di un fosfolipide. Alcuni esempi di attivazione da fosfolipidi sono presentati nella tab. V. In genere un dato enzima mostra preferenza, ma non specificità assoluta, per un particolare fosfolipide.

c) Glicolipidi.
I glicolipidi costituiscono una classe di diacilgliceridi non contenenti fosforo in cui un idrossile del glicerolo è legato con legame glucosidico a uno o più residui glucidici, in genere esosi (v. formule 8-11).

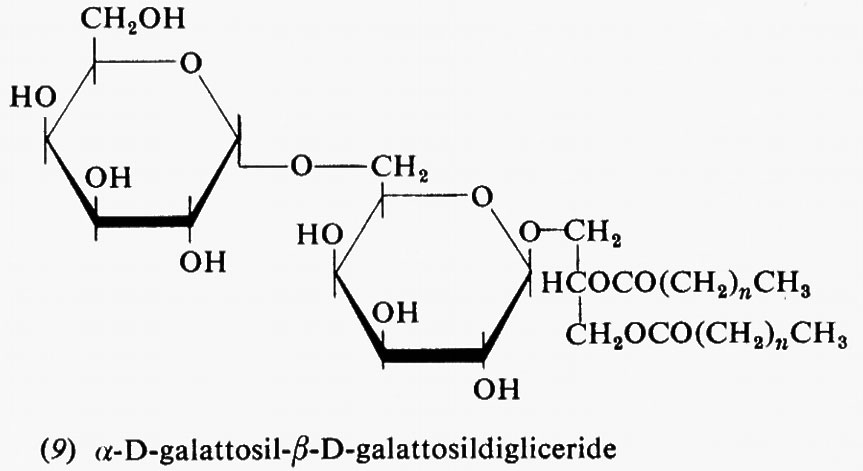

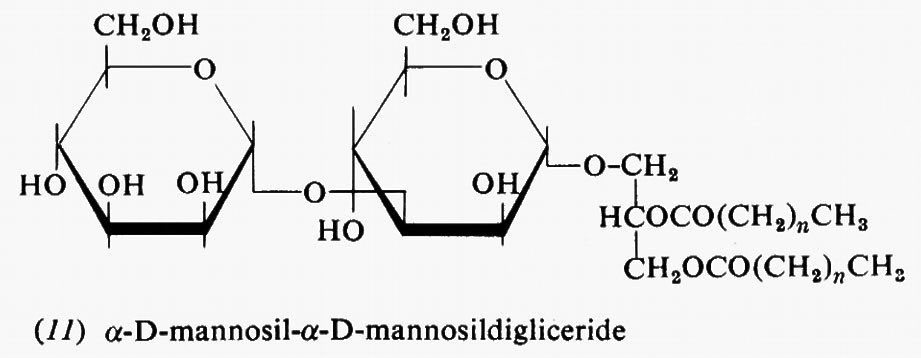
Queste molecole sono molto polari, data l'abbondanza di gruppi idrossilici liberi nella porzione glucidica. Esse non portano carica netta, se si eccettua il solfochinovosildigliceride. La distribuzione naturale dei glicolipidi è un po' più specifica di quella dei fosfolipidi. I due galattosilgliceridi e il solfochinovosildigliceride sono i più importanti lipidi delle lamelle dei cloroplasti tanto nelle Alghe che nelle piante superiori. Caratteristica di questi glicolipidi vegetali è l'elevata concentrazione dell'acido poliinsaturo α-linolenico. Un glicolipide batterico comune è l'α-D-mannosil-α-D-mannosildigliceride.
I gangliosidi sono i glicolipidi più complessi finora conosciuti (v. formula 12). Queste molecole si trovano in maniera specifica sulla superficie delle membrane delle cellule nervose.
Un carattere generale della biosintesi dei glicolipidi è l'utilizzazione di un nucleotide di un esoso, e precisamente un uridindifosfoesoso, come donatore della molecola di esoso per la glicosilazione dell'α, β-diacilglicerolo. Per esempio,
UDP-galattosio+α, β-diacilglicerolo
→galattosildiacilgliceride+UDP.
4. Steroli e terpeni.
a) Struttura.
Tutti gli animali e le piante e molti microrganismi contengono steroidi. Questa classe speciale di lipidi non ha alcuna relazione strutturale con i gliceridi o i fosfolipidi. Nondimeno gli steroidi possono essere classificati fra i lipidi poiché sono solubili in solventi orgarnci e insolubili in acqua, e per lo più sono untuosi al tatto.
Gli steroidi contengono molti gruppi metilenici disposti in una struttura tetraciclica nota come scheletro del ciclopentanoperidrofenantrene. Nel colesterolo (v. formula 13), che è il rappresentante più comune di questa classe di composti, una catena laterale alifatica a 8 atomi di carbonio (il gruppo isoottilico) è attaccata all'anello pentagonale dello scheletro steroideo. L'ergosterolo (v. formula 14), lo sterolo principale dei lieviti, contiene una catena laterale ramificata a 9 atomi di carbonio, mentre nel β-sitosterolo (v. formula 15), sterolo comune nelle piante, questa porzione è composta da 10 atomi di carbonio.

b) Biosintesi del colesterolo.
La sequenza di biosintesi del colesterolo (v. schema 7) si può per convenienza dividere in quattro stadi: 1) la trasformazione dell'acido acetico nell'unità isoprenoide a 5 atomi di carbonio isopentenilpirofosfato (IPP): 2) la trasformazione dell'IPP nell'idrocarburo squalene; 3) la ciclizzazione dello squalene a lanosterolo; 4) la trasformazione del lanosterolo in colesterolo.
Stadio 1. - Nel primo passaggio specifico della biosintesi del colesterolo (v. schema 7A), i due comuni metaboliti acetil-CoA e acetoacetil-CoA (provenienti dall'ossidazione di grassi e carboidrati) si condensano per formare il composto a catena ramificata a 6 atomi di carbonio β-idrossi-β-metilglutaril-CoA. La riduzione del gruppo carbossilico tioesterificato di questa molecola da parte di 2 equivalenti di NADPH produce acido mevalonico (denominazione corrente dell'acido 3-metil-3,5-diidrossivalerianico). Questo importante composto intermedio della biosintesi dei terpeni e degli steroli fu scoperto come fattore ‛sostitutivo dell'acetato' per certi ceppi di Lactobacillus acidophilus.
La trasformazione dell'acido mevalonico in isopentenilpirofosfato (IPP) avviene attraverso il mevalonato-5-fosfato e il mevalonato-5-pirofosfato (v. schema 7B). Ciò si ottiene per mezzo di tre tappe di fosforilazione, la terza delle quali è accoppiata all'eliminazione di acqua e diossido di carbonio dalla struttura a 6 atomi di carbonio. L'immissione di tre molecole di ATP genera un'unità isoprenoide ‛attivata', che è capace di formare legami carbonio-carbonio producendo catene polusoprenoidi di composizione generale (C5H5)n.
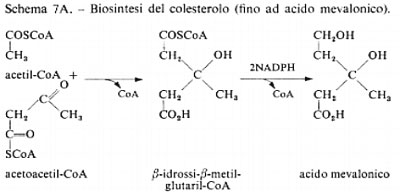

Stadio 2. - Prima della condensazione delle unità a C5, una molecola di IPP si isomerizza a dimetilallilpirofosfato, per spostamento in posizione allilica del doppio legame posto esternamente (v. schema 7C). Quindi si ha l'interazione di una molecola di IPP e di una di dimetilallilpirofosfato per mezzo del legame del carbonio C-1 di una delle due unità isopenteniliche con il carbonio C-5 dell'altra (condensazione testa-coda). L'energia per la formazione del legame carbonio-carbonio è fornita dall'eliminazione del pirofosfato. Il prodotto di condensazione a 10 atomi di carbonio, il geranilpirofosfato, addiziona ora un'altra unità a C5 in una condensazione testa-coda analoga alla prima unione C5+C5. Ne risulta il composto a C15 farnesilpirofosfato.
Il precursore aciclico della struttura steroidea è l'idrocarburo squalene (C30). Questa molecola simmetrica si origina per congiunzione coda-coda di due molecole di farnesilpirofosfato in presenza di NADPH. In questo processo si eliminano due molecole di pirofosfato e ancora una volta questa eliminazione fornisce l'energia necessaria per la formazione del legame carbonio-carbonio.
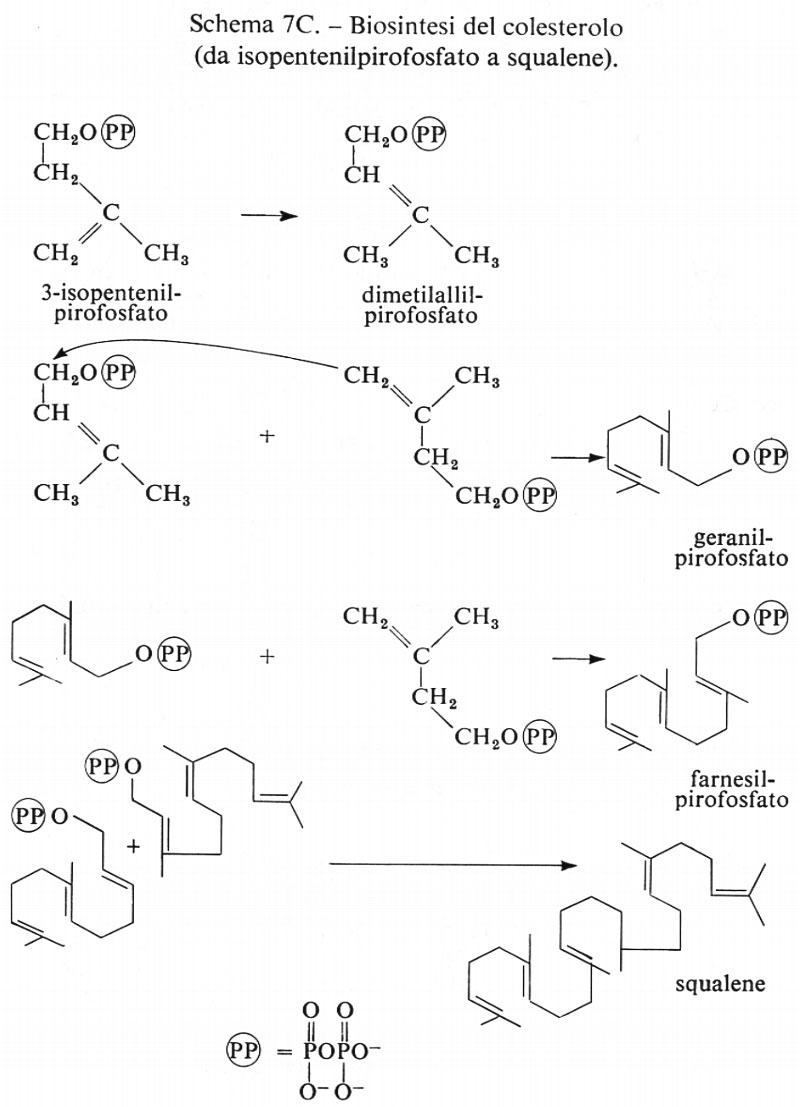
In genere lo squalene non si accumula nei tessuti animali. Fa eccezione il fegato di pescecane, che ne può contenere fino al 30% del suo peso.
Stadio 3. - Il lanosterolo (4,4, 14-trimetil-8,24-colesta- diene-3-olo), uno sterolo assai simile allo squalene per composizione elementare, è biosintetizzato in base all'equazione seguente:
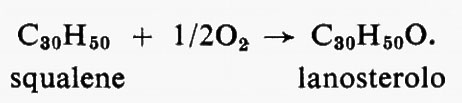
La reazione nel suo complesso consiste: a) nell'introduzione di 1 atomo di ossigeno nell'idrocarburo; b) nella trasformazione di una molecola aciclica in una molecola tetracidica (v. schema 7D).
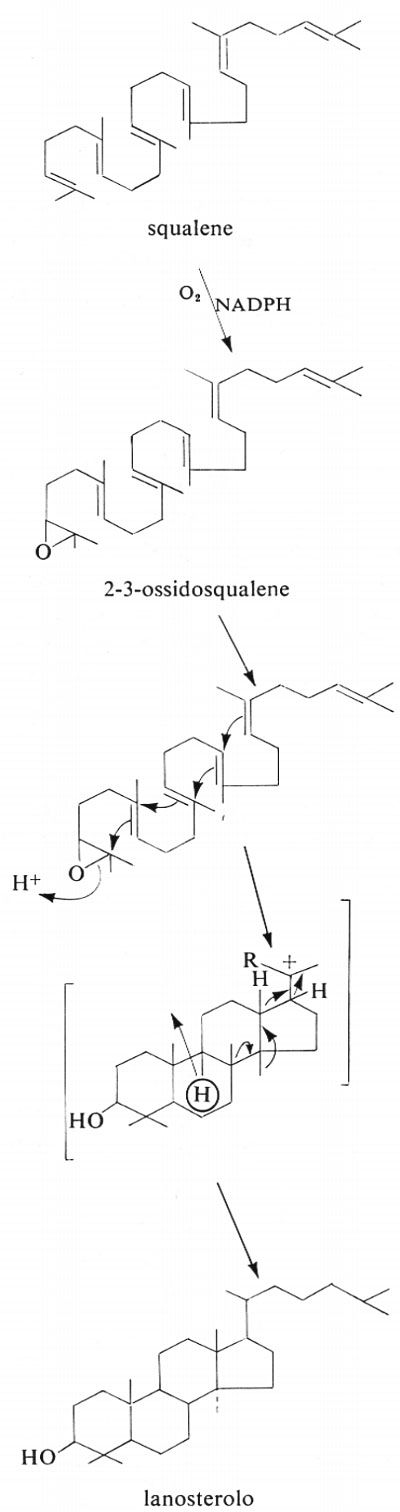
Queste trasformazioni sono catalizzate da due diversi enzimi:

La squalenepossidasi è una cosiddetta ossigenasi o ossidasi a funzione mista. Essa appartiene a una vasta categoria di enzimi che catalizzano l'inserimento di ossigeno molecolare in composti carboniosi in presenza di un agente riducente (NADPH).
La 2-3-ossidosqualene-lanosterolo-ciclasi trasforma la molecola aciclica dello squalene instaurando la struttura tetraciclica degli steroli attraverso una serie di spostamenti elettronici sincronizzati. Nel contempo molti atomi di H e gruppi metilici si spostano in posizione adiacente. Non si conoscono altri enzimi capaci di provocare cambiamenti così numerosi e complessi.
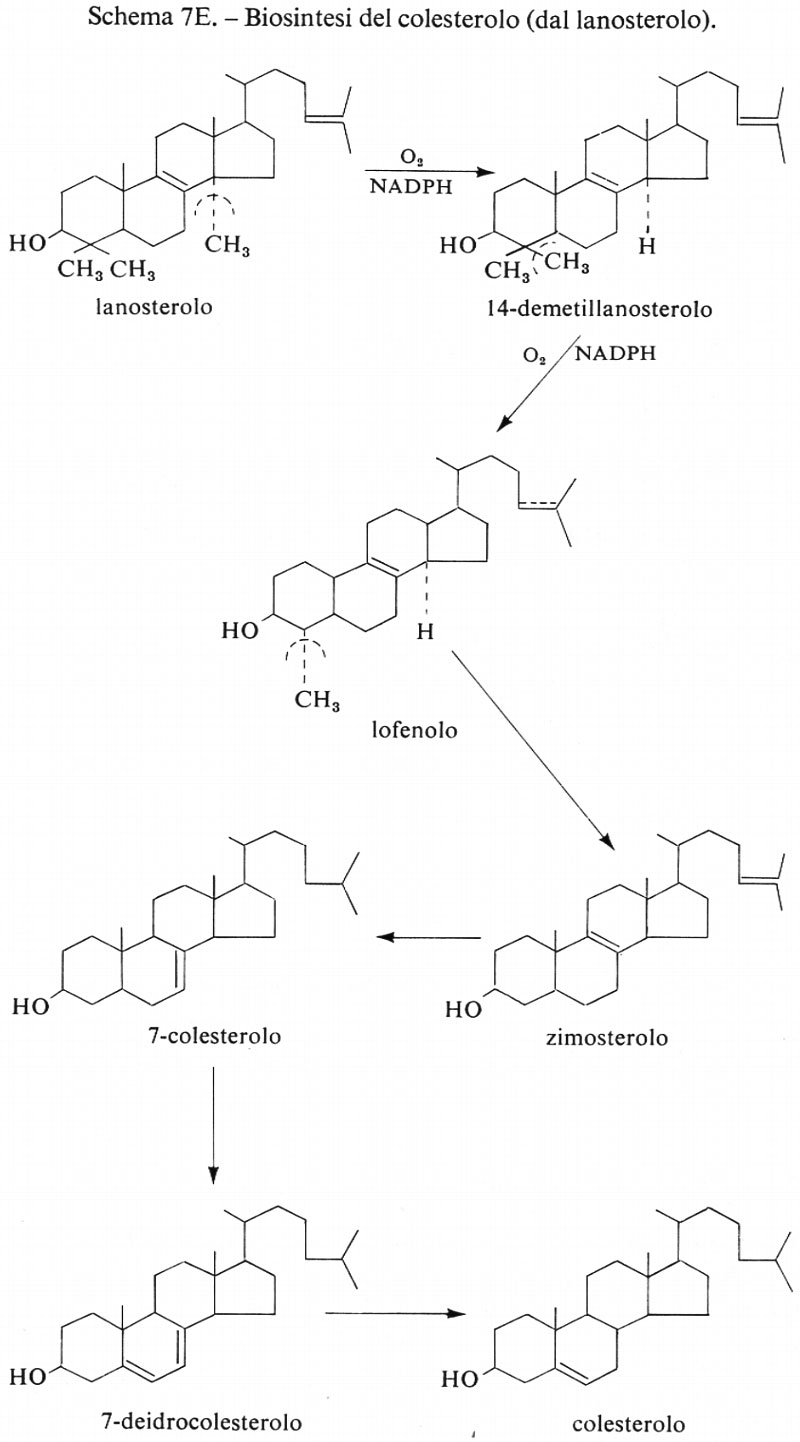
Stadio 4. - Nella maggior parte dei tessuti, specialmente il fegato, il lanosterolo ha vita breve ed è rapidamente trasformato in colesterolo (v. schema 7E), che è la forma metabolicamente più stabile. In queste fasi terminali della biosintesi degli steroli, tre gruppi metilici sono staccati dal sistema ciclico per ossidazione, il doppio legame della catena laterale è ridotto e il doppio legame nel nucleo cambia posizione. Non si conosce il meccanismo particolareggiato di queste reazioni, né sono stati isolati gli enzimi che le catalizzano. Il distacco di ciascuno dei tre gruppi metilici del nucleo ha bisogno di ossigeno molecolare e NADPH e avviene probabilmente in questi quattro passaggi distinti:
1) RCH3+O2+NADPH→RCH2OH;
2) RCH2OH+NAD→RCHO;
3) RCHO+NAD→RCOOH;
4) RCOOH→RH+CO2.
È stato calcolato che per tutta la sintesi del colesterolo servono circa 30 enzimi. Circa metà di essi sono implicati nella trasformazione di 18 molecole di acetil-CoA in lanosterolo, e il resto nella trasformazione del lanosterolo in colesterolo. Gli enzimi e i substrati di tutti i passaggi da acetil-CoA a farnesilpirofosfato sono solubili in acqua, mentre lo squalene e tutti i composti fra lo squalene e il colesterolo non lo sono (molecole idrofobiche) e gli enzimi che agiscono su di essi sono strettamente legati alle membrane di organelli intracellulari (reticolo endoplasmatico). L'ambiente naturale in cui agiscono questi enzimi sembra essere una specie di interfase lipido-proteica. In effetti è stato dimostrato che il fosfolipide fosfatidilserina è necessario per l'attività della squalenepossidasi, uno di questi enzimi.
c) Funzione del colesterolo.
L'attività fisiologica del colesterolo nel corpo animale è duplice, metabolica e strutturale. Il ruolo metabolico del colesterolo consiste nell'essere il precursore universale degli acidi biliari, dei vari ormoni steroidi (cortisolo, aldosterone, ecc.) della corteccia surrenale, dell'estradiolo follicolare, del progesterone del corpo luteo e dell'ormone testicolare testosterone (v. formule 16-21). Tutte queste trasformazioni, che avvengono nel fegato o nelle ghiandole endocrine, implicano l'accorciamento o l'eliminazione completa della catena laterale isoottilica, mentre il sistema tetraciclico rimane intatto. Sembra che non esistano nei tessuti animali enzimi per la demolizione completa del nucleo steroideo. La maggior parte del colesterolo tessutale è escreto nell'intestino crasso ed eliminato o come tale o dopo riduzione al derivato dudrogenato coprosterolo.
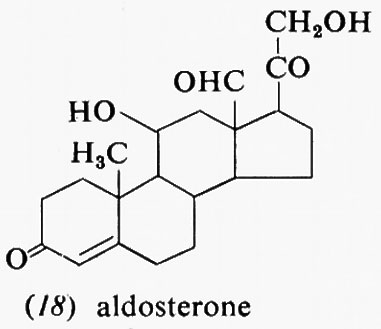
Gli Invertebrati non hanno nessuno degli enzimi della sintesi del colesterolo a partire da molecole piccole. Ciò nondimeno gli steroli sono essenziali al funzionamento del loro organismo. Gli Insetti, per esempio, e probabilmente altri Invertebrati, si procurano queste molecole da adatte sorgenti vegetali o animali. L'ecdisone (v. formula 22), un ormone dello sviluppo che controlla i processi di metamorfosi negli Insetti, deriva dal colesterolo (v. ormoni negli invertebrati). È interessante notare che l'ecdisone ha lo stesso numero di atomi di carbonio del colesterolo, ma non ha alcuna parentela chimica con gli ormoni steroidi dei Vertebrati. L'organismo degli Invertebrati non compie le reazioni di degradazione della catena laterale del tipo di quelle che avvengono nei Vertebrati e che portano alla formazione degli acidi biliari, del cortisone o degli ormoni sessuali.

Gli steroli sono ubiquitari fra le specie viventi, ma sono scarsamente presenti nei Batteri. La cellula batterica (procariotica) differisce dalle cellule degli organismi superiori (eucariotici) in quanto possiede un'organizzazione morfologica più rudimentale. Nei Batteri non esistono mitocondri, reticolo endoplasmatico e un nucleo definito con una sua membrana. Invece gli steroli degli Organismi superiori sono associati quasi esclusivamente con gli elementi delle membrane degli organelli eucariotici, insieme a proteinè e fosfolipidi. L'inserimento di steroli in una membrana lipoproteica sembra conferire robustezza meccanica e una forma definita alla parete degli organelli. Questa proprietà strutturale è dovuta probabilmente alla rigidità e alla planarità del nucleo sterolico, che è tenuto insieme dalle forze di connessione interna del sistema tetraciclico. Questo ruolo di rinforzo esercitato dagli steroli nelle membrane è illustrato dal modo d'azione dei cosiddetti antibiotici polienici (nistatina, acido stipitatico), che sono emolitici e inibiscono la crescita dei Funghi ma non dei Batteri. Queste sostanze, in altre parole, agiscono solo sugli organismi contenenti steroli. Esse esercitano la loro azione combinandosi specificamente con la componente sterolica della membrana eucariotica, sovvertendo in tal modo il delicato equilibrio delle interazioni lipoproteiche necessario per la stabilità della membrana.
La quantità eccezionale di colesterolo presente nel cervello e nei nervi serve probabilmente non solo come elemento strutturale della guaina mielinica, ma anche come isolante elettrico (v. neurone e impulso nervoso).
d) Composti poliisoprenoidi.
La struttura di molti prodotti naturali, lineari e ciclici, obbedisce alla ‛regola dell'isoprene' (v. biosintesi); vale a dire essa può essere fatta derivare dalla giustapposizione di elementi costitutivi isopentenilici, assumendo che si formi per condensazioni testa-coda o coda-testa o entrambe. La regola dell'isoprene è suffragata dall'esistenza di sistemi enzimatici capaci di trasformare l'acido mevalonico e l'isopentenilpirofosfato in gomma, carotenoidi, retinale, vitamina A, fitolo, catene laterali polusoprenoidi dell'ubichinone e del battoprenolo e molti altri composti. Probabilmente il meccanismo di condensazione per la sintesi di questi derivati terpenici è analogo alla formazione del geranilpirofosfato da isopentenilpirofosfato e dimetilallilpirofosfato o alla condensazione coda-coda di 2 moli di farnesilpirofosfato per formare squalene. Riportiamo la struttura chimica e la funzione di alcune di queste molecole poliisoprenoidi e di loro derivati (formule 23-28).
e) Regolazione del metabolismo del colesterolo.
In alcune specie animali la velocità di biosintesi del colesterolo è regolata, almeno in parte, con un meccanismo di controllo a retroazione (feedback). L'essenza di questo meccanismo di controllo consiste nell'effetto inibitore esercitato dall'ultimo prodotto di una via biosintetica sulla sintesi o l'attività di uno degli enzimi iniziali nella sequenza che porta al prodotto terminale. Un prodotto terminale che si accumula in eccesso rispetto alla richiesta arresterà o rallenterà in questo modo la sua stessa sintesi finché il suo livello non sarà ritornato normale. In genere è il primo passaggio specifico di una data via biosintetica e non altri a rispondere al controllo a retroazione. Perciò, l'arresto dell'enzima controllato produce un controllo metabolico altamente selettivo.
L'alimentazione di animali con colesterolo inibisce l'incorporazione di acido acetico marcato nella sintesi del colesterolo epatico, ma non influenza la trasformazione dell'acido mevalonico nel prodotto terminale. Perciò, il punto sotto controllo a retroazione deve essere situato nel passaggio dall'acetato all'acido mevalonico e non in uno dei passaggi dell'ulteriore trasformazione dell'acido mevalonico. L'enzima sensibile al colesterolo sembra difatti essere la β-idrossi-β-metilglutaril-CoA-riduttasi, che catalizza la prima reazione specifica della biosintesi del colesterolo; in conformità al principio del controllo a retroazione la sua attività è ridotta dal colesterolo alimentare.
I fattori che controllano la velocità di biosintesi del colesterolo nell'organismo animale hanno ricevuto attenzione particolare perché l'aterosclerosi è attribuita, almeno in parte, a un eccesso d'introduzione e di produzione endogena di colesterolo e di altri lipidi. È assodato che questa malattia vascolare - che consiste essenzialmente nell'indurimento delle arterie - è strettamente correlata con la deposizione di lipidi sulla parete dei grossi vasi sanguigni. Tuttavia, il contributo relativo dei lipidi alimentari ed endogeni all'aterosclerosi è assai poco chiaro. Studi statistici hanno mostrato che popolazioni con alto livello di vita consumano quantità relativamente alte di grasso animale, con un elevato contenuto in colesterolo. Anche sperimentalmente, la concentrazione lipidica del sangue può essere aumentata da una forte introduzione di grasso e, a loro volta, questi alti tassi lipidemici possono essere correlati fino a un certo punto con l'incidenza dell'aterosclerosi. Per quanto non esistano finora prove convincenti che i lipidi siano il principale fattore eziopatogenetico dell'aterosclerosi nell'uomo, la possibilità di controllare con successo la biosintesi lipidica appare potenzialmente di grande valore chemioterapico.
Il fenomeno del controllo a retroazione della biosintesi del colesterolo fornisce una guida assai promettente alla realizzazione di preparati terapeutici. Una sostanza chimica che interferisce con la sintesi dell'acido mevalonico competendo con il normale substrato dell'enzima dovrebbe essere capace di ridurre la produzione di colesterolo e di conseguenza il tasso ematico di esso. Con questo intento sono stati sintetizzati composti a struttura rassomigliante a quella dell'acido β-idrossi-β-metilglutarico, che, introdotti nella pratica clinica, si sono rivelati abbastanza efficaci come agenti capaci di abbassare il colesterolo del sangue. Uno di questi composti è mostrato qui sotto, accanto all'acido β-idrossi-β-metilglutarico:

Tuttavia non è sicuro che questo composto inibisca effettivamente la biosintesi del colesterolo con il meccanismo previsto, cioè inibendo l'enzima che riduce il β-idrossi-β-metilglutaril-CoA ad acido mevalonico.
Bibliografia.
Ansell, G. B., Hawthorne, J. N., Phospholipids chemistry, metabolism and function, New York 1964.
Bressler, R., Physiological-chemical aspects of fatty acid oxidation, in Lipid metabolism (a cura di S. J. Wakil), New York 1970, pp. 44-73.
Clayton, R. B., Biosynthesis of sterols, steroids and terpenoids. Part I. Biogenesis of cholesterol and the fundamental steps in terpenoid biosynthesis, in ‟Quarterly review", 1965, XIX, pp. 168-200.
Deenen, L. L. M. van, Phospholipids and biomembranes, in ‟Progress in the chemistry of fats and other lipids", 1966, VIII, pp. 1-127.
Fieser, L. F., Fieser, M., Steroids, New York 1959.
Gunstone, F. D., An introduction to the chemistry and biochemistry of fatty acids, London 1967.
Paoletti, R., Kritchevsky, D. (a cura di), Advances in lipid research, voll. I ss., New York 1963 ss.
Wakil, S. J., Fatty acid metabolism, in Lipid metabolism (a cura di S. J. Wakil), New York 1970, pp. 1-44.
Wakil, S. J. (a cura di), Lipid metabolism, New York 1970.