
Molecole
Molecole
Metodi d'indagine strutturale, di Salvatore Califano
Analisi conformazionale delle piccole molecole, di Giancarlo Berti
Analisi conformazionale delle grandi molecole, di John A. Schellman e Charlotte G. Schellman
Metodi d'indagine strutturale
SOMMARIO: 1. Cenni storici. □ 2. Generalità. □ 3. Metodi spettroscopici. □ 4. Metodi diffrattometrici. □ 5. Risultati ottenuti e sviluppi futuri. □ Bibliografia.
1. Cenni storici.
Il livello di conoscenza della struttura molecolare cui è giunta la moderna chimica fisica è direttamente legato, da una parte, alla disponibilità di potenti metodi d'indagine sperimentale e, dall'altra, alla possibilità di sistemare i risultati di queste misure nel quadro di una valida teoria del legame chimico. Allo stato attuale delle conoscenze, il problema della struttura molecolare si presenta con aspetti diversi che, sebbene strettamente connessi, possono essere convenientemente classificati come segue: a) determinazione della disposizione spaziale dei nuclei nella molecola; b) struttura elettronica dei legami chimici; c) informazioni accumulate in una molecola e associate con una data struttura spaziale.
Storicamente questi diversi aspetti si sono sviluppati in tempi diversi, man mano che si scoprivano e si perfezionavano i metodi sperimentali d'indagine e si elaborava la moderna teoria del legame chimico. Se si potesse assegnare una data d'inizio allo studio della struttura molecolare, bisognerebbe probabilmente risalire al 1874, quando J.-A. Le Bel e J. H. van't Hoff formularono, indipendentemente, l'ipotesi che le quattro valenze del carbonio fossero orientate verso i vertici di un tetraedro regolare. La riprova sperimentale della validità dell'ipotesi di Le Bel e van't Hoff venne solo dopo quarant'anni, quando, nel 1914, W. H. e W. L. Bragg, padre e figlio, dimostrarono che nei cristalli di diamante ogni atomo di carbonio è circondato tetraedricamente da altri quattro atomi di carbonio. L'ipotesi di Le Bel e van't Hoff aveva però, nel frattempo, aperto la strada alla stereochimica organica classica e aveva permesso di spiegare in maniera semplice ed elegante l'isomeria ottica, sviluppando in questo modo una teoria modellistica dell'attività ottica.
Per questi quarant'anni il problema della struttura delle molecole si presenta pertanto essenzialmente come problema ‛dell'architettura molecolare', a un livello puramente qualitativo e modellistico. Con la scoperta dei raggi X e con i primi lavori dei Bragg si aprì all'improvviso la possibilità di misurare quantitativamente le distanze interatomiche. Da quel momento la struttura di una molecola diventava ‛misurabile' sperimentalmente, diventava cioè esprimibile in termini di distanze e di angoli di legame. Contemporaneamente allo sviluppo della tecnica dei raggi X si veniva chiarendo, agli inizi del Novecento, il ruolo degli elettroni nella formazione del legame, finché nel 1916 G. N. Lewis pubblicava la sua famosa nota che formerà la base della moderna teoria della valenza.
Con l'avvento della meccanica quantistica e con la formulazione delle teorie del legame di valenza e degli orbitali molecolari la struttura molecolare assume un aspetto nuovo. Si chiarisce che la distribuzione spaziale degli atomi è conseguenza diretta della struttura elettronica e la riflette quindi nei dettagli. In altre parole, la determinazione sperimentale della struttura molecolare e la valutazione quantitativa delle lunghezze e degli angoli di legame diventano fonti importanti di informazioni sulla struttura elettronica. Il periodo dal 1930 alla seconda guerra mondiale è forse, in tutto lo sviluppo della strutturistica chimica, quello più denso di scoperte importanti. La spettroscopia nell'infrarosso, che dal punto di vista tecnico si era sviluppata da lungo tempo, trova nella meccanica quantistica la correlazione con i problemi della struttura molecolare. In meno di dieci anni viene elaborata la teoria degli spettri rotazionali e quella degli spettri vibrazionali. Già nel 1928, sulla guida delle previsioni teoriche di A. Smekal e O. Placzek, Ch. V. Raman aveva scoperto l'effetto che da lui prenderà il nome e che fornisce informazioni complementari a quelle ottenute dagli spettri infrarossi. Nel 1930 viene sviluppata la teoria della diffrazione elettronica da H. Mark e R. F. J. Wierl e già nel 1935 la tecnica relativa e diventata un potente metodo di indagine strutturale per molecole in fase gassosa. Subito dopo la seconda guerra mondiale le microonde fanno il loro ingresso tra i metodi di determinazione della struttura e poco dopo, nel 1946, viene scoperta da due gruppi diversi di ricercatori la risonanza magnetica nucleare.
Dopo la seconda guerra mondiale le tecniche sperimentali di determinazione strutturale escono dall'isolamento dei laboratori specializzati per diventare metodi analitici nella grande industria chimica. Si sviluppa un'industria specializzata, ad altissimo livello tecnologico, per la produzione in serie di apparecchi scientifici completamente automatici capaci di eseguire in pochi minuti e con grande precisione le misure che solo dieci anni prima richiedevano mesi di lavoro di specialisti. Questa rivoluzione tecnologica porta all'accumulo di una massa enorme di dati sperimentali molto accurati e precisi, che sarebbe stato impossibile elaborare nel quadro della teoria, ormai in fase avanzata, se la stessa rivoluzione tecnologica non avesse prodotto lo strumento essenziale dell'elaborazione: il calcolatore elettronico. Lo sviluppo dei calcolatori elettronici produrrà infatti un nuovo salto quantitativo, non tanto a livello della teoria, ormai saldamente organizzata, quanto a livello dell'elaborazione dei dati e delle applicazioni della teoria a sistemi complessi (v. Mizushima, 1954; v. Wheatley, 1958).
Intanto le connessioni della strutturistica molecolare con altre branche della chimica, già ravvisate negli anni precedenti, assumono un significato sempre più preciso e forniscono la chiave per l'interpretazione unificata di una larga parte della fenomenologia chimica. Discipline con una chiara impostazione termodinamica si trasformano con sorprendente rapidità. Lo studio dei meccanismi di reazione in termini di struttura molecolare apre la strada all'interpretazione dei risultati cinetici. Le proprietà meccaniche dei polimeri vengono rapidamente interpretate sulla base delle strutture determinate con i raggi X. Dal meccanismo d'azione dei farmaci a quello dei detersivi, dal colore dei fiori al funzionamento degli enzimi, tutto sembra trovare una spiegazione organica e coerente nella struttura delle molecole. L'influenza sulla biologia delle conoscenze acquisite nel campo della struttura molecolare provoca, in pochi anni, una profonda rivoluzione. Nel 1951 L. Pauling predice, in base a pure considerazioni strutturali, che le proteine potrebbero assumere, sotto certe condizioni, una configurazione stabile a forma di elica, che egli chiama α-elica. La predizione di Pauling viene confermata quasi immediatamente da M. Perutz, che mostra come alcuni polipeptidi sintetici esistano con una struttura ad α-elica. Nel 1953 F. H. C. Crick e J. D. Watson stabiliscono la struttura a doppia elica del DNA. Nasce così la biologia molecolare (v. biologia molecolare), che rapidamente, con la scoperta del codice genetico e dei meccanismi di controllo della catalisi enzimatica, si sviluppa in maniera prodigiosa. Questa nuova frontiera della strutturistica mette ben presto in evidenza un altro importante aspetto della struttura molecolare. Il comportamento chimico-fisico di sistemi così complessi come le macromolecole biologiche, difficilissimo se non impossibile da spiegare entrando nel dettaglio della struttura molecolare, diventa molto più semplice da interpretare associando, a ogni possibile conformazione della molecola o di tratti di essa, una certa ‛quantità di informazione' che ogni molecola porta con sé e che ‛trasmette' nella reazione con altre molecole. La teoria dell'informazione e la strutturistica molecolare si compenetrano così in un processo che, ben lontano per il momento dall'essere sufficientemente chiarito e analizzato, potrebbe avere, negli anni futuri, sviluppi imprevedibili.
2. Generalità.
Per poter ottenere informazioni sulla struttura molecolare, bisogna che le molecole interagiscano con ‛qualcosa' capace di trasportare a un opportuno rivelatore, cioè di tradurre a livello macroscopico, l'informazione ottenuta dalla molecola. Il processo di traduzione è legato al fatto che non si osserva il singolo evento microscopico, ma un numero molto grande di essi, tutti identici e contemporanei. Sia le particelle elementari, soprattutto gli elettroni e i neutroni, sia le onde elettromagnetiche hanno la capacità di interagire con le molecole e possono quindi essere adoperate a questo scopo. Le onde elettromagnetiche sono però di gran lunga la più varia sorgente d'informazione sulla struttura molecolare e sono di fatto alla base della maggior parte dei metodi d'indagine strutturale.
L'interazione di una molecola con un'onda elettromagnetica o con una particella elementare è un fenomeno estremamente complesso, la cui interpretazione dettagliata è al di fuori dei limiti di questa semplice esposizione. Ci accontenteremo pertanto di una descrizione puramente fenomenologica, rimandando il lettore che volesse approfondire l'argomento a testi specifici (v. Herzberg, 1945; v. Heitler, 1954).
Uno dei risultati più importanti della meccanica quantistica è che l'energia di un atomo o di una molecola, a parte l'energia cinetica di traslazione, è quantizzata, non può, cioè, assumere qualsiasi valore, ma solo valori ben determinati. Chiameremo ‛stato stazionario' uno stato della molecola in cui essa assuma uno dei valori quantizzati dell'energia. È conveniente rappresentare schematicamente gli stati stazionari di una molecola sotto forma di livelli di energia in un grafico come quello della fig. 1, relativo a una molecola biatomica. Un altro aspetto importante della meccanica quantistica è che, ai fini della discussione dell'interazione luce-materia, la radiazione elettromagnetica può essere considerata come equivalente a uno sciame di particelle, che prendono il nome di fotoni. Ogni fotone rappresenta un quanto di energia eguale ad hν, dove ν è la frequenza della radiazione e h la costante di Planck (h=6,62517•10-27 erg•s).
Su queste basi possiamo discutere l'interazione di un fotone o di una particella elementare con una molecola, in termini di urti tra particelle. L'urto potrà essere di due tipi, che chiameremo rispettivamente elastico e anelastico, in funzione delle variazioni di energia che subiscono le due particelle. Diremo urto elastico un urto in cui il fotone o la particella elementare incidente sulla molecola non fanno variare l'energia interna di quest'ultima. L'effetto dell'urto è semplicemente una variazione della direzione in cui si propagano il fotone o la particella. Diremo invece anelastico un urto in cui il fotone o la particella incidente cedono o assorbono energia dalla molecola. Ovviamente in un urto anelastico la quantità di energia ceduta o assorbita da una molecola è quantizzata, cioè può essere eguale solo alla differenza tra due stati stazionari.
I metodi d'indagine strutturale in cui si osserva un fenomeno di urto anelastico vanno sotto il nome di metodi spettroscopici, mentre quelli in cui si osservano fenomeni di urto elastico vanno sotto il nome di metodi diffrattometrici o di diffusione, pure se impropriamente viene spesso usato il termine ‛spettroscopia' anche per questi ultimi. I metodi spettroscopici in cui le particelle incidenti sono fotoni si basano per la maggior parte su urti completamente anelastici in cui il fotone incidente viene totalmente assorbito dalla molecola, che passa a un più alto stato stazionario di energia.
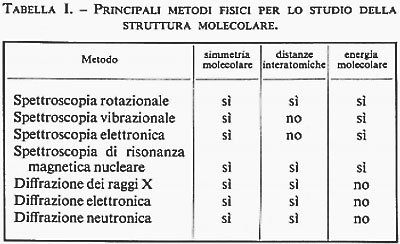
Per la natura stessa del fenomeno su cui sono basati, i metodi spettroscopici danno informazioni primarie sull'energia molecolare, mentre i metodi diffrattometrici, che non coinvolgono variazioni di energia, danno informazioni sulla forma delle molecole. In realtà anche i metodi spettroscopici forniscono informazioni sulla struttura molecolare, attraverso la determinazione dei livelli energetici molecolari. Ciò è dovuto essenzialmente al fatto che l'assorbimento di fotoni da parte di una molecola è completamente regolato dalla simmetria molecolare. Affinché un fotone sia assorbito da una molecola non basta che esso abbia energia hν eguale alla differenza di energia tra due stati stazionari della molecola; infatti, è necessario che la molecola possa trasformare l'energia del fotone in energia propria, il che è possibile solo se la simmetria molecolare lo permette. È chiaro pertanto che i metodi spettroscopici danno informazioni anche sulla simmetria molecolare e quindi sulla disposizione degli atomi nella molecola. Nella tab. I sono elencati i metodi fisici d'indagine usati comunemente nello studio della struttura molecolare e per ognuno di essi sono indicate le informazioni più importanti che si possono ottenere.
3. Metodi spettroscopici.
L'energia di una molecola può essere considerata, con buona approssimazione, come la somma dell'energia Ee degli elettroni all'interno della molecola, dell'energia Ev di vibrazione dei nuclei e dell'energia Er di rotazione della molecola intorno ai suoi assi d'inerzia. Scriveremo quindi
E=Ee+Ev+Er.
Per la quantizzazione dell'energia avremo quindi stati stazionari elettronici, vibrazionali e rotazionali che, in un diagramma come quello della fig. 1, si tradurranno in livelli energetici elettronici, vibrazionali e rotazionali. La separazione tra questi livelli, cioè la differenza in energia tra due di essi, è molto diversa nei tre casi. All'incirca la separazione dei livelli elettronici è 100 volte maggiore della separazione tra livelli vibrazionali, che a sua volta è 100 volte maggiore della separazione tra livelli rotazionali.
Per ragioni di spazio e anche per non complicare inutilmente il diagramma, sono riportati nella fig. 1 due soli livelli elettronici, indicati con E ed E′. Per il livello elettronico E sono poi disegnati i primi tre livelli vibrazionali, numerati da un numero quantico V=0, 1, 2 e per ognuno di essi i primi cinque livelli rotazionali con numero quantico J=0, 1, 2... . Analogamente per il livello E′ avremo livelli vibrazionali V′ e livelli rotazionali J′. Gli spettri molecolari si Originano per transizione tra questi livelli, dovuta ad assorbimento di quanti di radiazione da parte della molecola. A seconda dell'energia hν dei quanti assorbiti, gli spettri cadono in regioni diverse dello spettro elettromagnetico. Nella fig. 2 è riportato lo spettro elettromagnetico con la nomenclatura generalmente usata nelle varie regioni spettrali. Per comodità la scala delle ascisse è logaritmica in lunghezze d'onda. Ricordiamo che la lunghezza d'onda λ di una radiazione è collegata alla frequenza ν dalla relazione λ=c/ν, dove c è la velocità della luce. Per ragioni storiche e di comodità d'uso, nelle varie zone spettrali si adoperano unità di misura diverse. Nella regione delle microonde si usano normalmente le frequenze in kilocicli al secondo o in megacicli al secondo (1 Mc/s=16 c/s) Nella regione dell'infrarosso è più conveniente usare l'inverso della lunghezza d'onda (in cm-1)1/λ, cioè il numero d'onde per centimetro. Nelle regioni del visibile, dell'ultravioletto e dei raggi X si usa invece la lunghezza d'onda in ångström (1 Å=10-10 m) o in nm (1 nm=10-9 m) L'assorbimento della radiazione elettromagnetica da parte delle molecole è regolato da leggi molto precise. In generale si può dire che, affinché un fotone di frequenza ν sia assorbito nella transizione tra due livelli stazionari che differiscano di un'energia pari ad hν, è necessario che nella transizione si produca una variazione in grandezza o direzione del momento di dipolo elettrico o magnetico della molecola. Le leggi che permettono di determinare se in una transizione si può avere assorbimento di radiazione elettromagnetica prendono il nome di ‛regole di selezione' (v. Herzberg, 1945) e sono ricavabili dalla conoscenza della simmetria molecolare nei due stati stazionari interessati dalla transizione.
Transizioni tra livelli rotazionali (v. Weissberger, 1956) che appartengano allo stesso livello vibrazionale e allo stesso livello elettronico originano spettri nella regione delle microonde, detti spettri di rotazione pura. Transizioni tra livelli vibrazionali (ibid.) che appartengano allo stesso livello elettronico originano spettri nell'infrarosso, che prendono il nome di spettri vibrazionali. In realtà ogni transizione vibrazionale è composta dalle possibili transizioni tra i livelli rotazionali dei due stati vibrazionali, così come è indicato nella fig. 1. Le bande vibrazionali di molecole in fase gassosa presentano pertanto una ‛struttura fine' rotazionale e sono quindi bande vibrorotazionali.
Le transizioni tra livelli vibrazionali possono essere studiate, oltre che con la spettroscopia di assorbimento nell'infrarosso, anche con la spettroscopia Raman (v. Gilson e Hendra, 1970), una tecnica completamente diversa, basata anch'essa però sull'urto anelastico di fotoni con molecole. Il meccanismo dell'effetto Raman è mostrato schematicamente nella fig. 1. Se un fotone di energia hv0, diversa dalla differenza tra due stati stazionari, colpisce una molecola, esso non potrà essere assorbito e quindi lo stato non stazionario in cui viene eccitata la molecola avrà un tempo di vita brevissimo. La molecola riemetterà, in una qualsiasi direzione, un fotone della stessa frequenza di quello incidente, ritornando nello stato iniziale. Questo tipo di processo, che è quello più comune, prende il nome di ‛diffusione Rayleigh'. È possibile però che la molecola ritorni in uno stato stazionario di energia più elevata di quello iniziale, emettendo un fotone di energia hν più bassa, di un ammontare eguale alla differenza di energia tra i due stati, come è mostrato nella fig. 1. Se invece la molecola, prima di interagire con il fotone, si trovava già in uno stato eccitato, potrà ritornare in uno stato di energia più basso, emettendo un fotone di energia più elevata di quella del fotone incidente.
Sperimentalmente si illumina un campione con un raggio intenso di luce monocromatica, come quella emessa da un laser, e si analizza la luce diffusa a 90°. Si osserva, oltre a una riga intensa a frequenza ν0, una serie di righe a frequenza più bassa (righe Stokes) e una serie di righe a frequenza più alta (righe anti-Stokes). La differenza in frequenza tra la riga eccitatrice e una riga Raman (Stokes o anti-Stokes) corrisponde a una frequenza vibrazionale delle molecole del campione studiato. Transizioni tra livelli elettronici (v. Weissberger, 1956) danno luogo a bande nella regione dello spettro visibile e soprattutto nell'ultravioletto. A loro volta le transizioni elettroniche sono, in realtà, composte di transizioni tra livelli vibrorotazionali dei due stati elettronici, come è mostrato nella fig. 1.
In presenza di un campo magnetico intenso, molte molecole possono anche assorbire radiazioni elettromagnetiche di grande lunghezza d'onda, come le onde radio. Ciò è dovuto al fatto che alcuni nuclei posseggono un momento magnetico (spin nucleare), cioè si comportano come dei microscopici magneti che, sotto l'azione di un campo magnetico, possono assumere orientazioni diverse, corrispondenti a valori diversi dell'energia. L'effetto del campo magnetico è, quindi, di produrre una separazione dei livelli magnetici nucleari. Transizioni tra questi livelli, che sono molto vicini in energia, originano spettri di assorbimento di onde radio, che prendono il nome di spettri di risonanza magnetica nucleare (NMR) (v. Bovey, 1969; v. Weissberger, 1956).
Anche gli elettroni posseggono un momento magnetico. Normalmente, però, i magnetini elettronici sono accoppiati a due a due, di modo che il momento magnetico risultante in una molecola è nullo. Ma se in qualche modo si riesce a ottenere una molecola con un elettrone spaiato, sia eliminando l'altro partner, sia aggiungendo un elettrone alla molecola, si ottiene quello che si chiama un ‛radicale libero', con un momento magnetico elettronico diverso da zero. In questo caso, in campo magnetico, si verificherà un fenomeno analogo alla risonanza magnetica nucleare e si otterranno quindi transizioni tra livelli magnetici elettronici. Gli spettri corrispondenti prendono il nome di spettri di risonanza elettronica paramagnetica (EPR) (v. Bershon e Baird, 1966). Data la maggiore differenza di energia in campo magnetico dei livelli di spin elettronico, queste transizioni avvengono con assorbimento di microonde. Più recentemente è stata sviluppata una nuova tecnica sperimentale, la spettroscopia ESCA (Electron Spectroscopy for Chemical Analysis) (v. Siegbahn e altri, 1967), basata sull'analisi ad alta risoluzione degli elettroni emessi da un atomo o da una molecola per bombardamento con raggi X. Misurando accuratamente l'energia degli elettroni emessi, si ottengono informazioni preziose sulla loro energia di legame e quindi indirettamente sulla struttura molecolare.
4. Metodi diffrattometrici.
La diffrazione è un fenomeno tipicamente ondulatorio, direttamente connesso all'interferenza delle onde (v. Wheatley, 1958). Se un raggio parallelo di luce monocromatica (cioè di una sola lunghezza d'onda) viene inviato, come nella fig. 3, su un reticolo di punti materiali equidistanti, una parte della radiazione passa indisturbata attraverso il reticolo, mentre un'altra parte viene inviata in altre direzioni (diffratta), qualora la distanza tra i punti sia dello stesso ordine di grandezza della lunghezza d'onda della luce incidente. I raggi diffratti in una certa direzione dai vari punti percorreranno un cammino leggermente diverso e quindi daranno luogo a interferenza, a meno che la differenza di cammino non sia un multiplo intero della lunghezza d'onda. Nella fig. 3 i raggi diffratti BC ed EF si rinforzeranno solo se la loro differenza di cammino ottico EE′=a cos ϕ soddisfa l'equazione
nλ=a cos ϕ,
dove n è un numero intero e λ la lunghezza d'onda.
I raggi X hanno lunghezze d'onda dello stesso ordine di grandezza delle distanze intermolecolari nei cristalli; questi ultimi costituiscono quindi eccellenti reticoli di diffrazione per i raggi X. Dalle figure di diffrazione ottenute si risale alla struttura della cella elementare e si determinano le posizioni degli atomi in essa.
Così come le onde elettromagnetiche, nell'interazione con la materia, si comportano alla stregua di particelle, anche le particelle elementari possono comportarsi come onde elettromagnetiche, nel senso che danno luogo anch'esse a fenomeni di diffrazione. A ogni particella elementare può quindi essere associata una lunghezza d'onda, definita dalla famosa relazione di de Broglie
λ=h/mv,
dove m è la massa della particella, v la sua velocità e h la costante di Planck.
Se un fascio di elettroni è accelerato da un campo elettrico molto intenso, la lunghezza d'onda degli elettroni è dell'ordine di grandezza delle distanze internucleari e di conseguenza il fascio di elettroni sarà diffratto, per esempio, dalle molecole di un gas. Un fascio di neutroni emesso da una pila atomica ha lunghezze d'onda dell'ordine di grandezza delle distanze intermolecolari nei cristalli e quindi i neutroni possono essere usati come i raggi X per lo studio della struttura cristallografica. Quando i raggi X colpiscono le molecole di un cristallo, vengono diffratti essenzialmente dagli elettroni e l'intensità della radiazione diffratta dipende dalla distribuzione degli elettroni nella molecola. In linea di principio, se si conosce la disposizione degli atomi in una molecola e la disposizione delle molecole in un cristallo, si può calcolare la forma e l'intensità del diagramma di diffrazione (v. Guinier, 1956). In realtà ciò che interessa è esattamente il processo inverso, cioè, misurato il diagramma di diffrazione, calcolare la posizione delle molecole nel cristallo e degli atomi nelle molecole. Questo calcolo è possibile grazie a un metodo, ideato dai Bragg, che è alla base di tutta la strutturistica con i raggi X. Il metodo è basato sullo sviluppo in serie di Fourier di grandezze che siano funzioni periodiche delle coordinate. La distribuzione degli atomi in un cristallo è appunto una funzione periodica in tre dimensioni e può essere comodamente espressa con uno sviluppo in serie di Fourier. Con questo metodo è possibile costruire delle mappe di densità elettronica nel cristallo e localizzare gli atomi nelle zone di massima densità elettronica.
Poiché, come abbiamo accennato precedentemente, la diffrazione dei raggi X è dovuta essenzialmente agli elettroni, gli atomi pesanti, che hanno molti elettroni extranucleari, giocano un ruolo predominante nel diagramma di diffrazione. Gli atomi leggeri, soprattutto gli atomi d'idrogeno, che hanno pochi elettroni, contribuiscono invece in misura minima alla diffrazione. La diffrazione dei neutroni, a differenza di quella dei raggi X, non dipende dagli elettroni, ma dai nuclei, tranne che per materiali magnetici. La capacità di diffondere neutroni varia, per i diversi nuclei, al massimo di un fattore 3, per cui tutti i nuclei sono pressoché equivalenti per i neutroni.
La forma dei diagrammi di diffrazione, sia dei raggi X che dei neutroni, è completamente determinata dalla simmetria dei cristalli, allo stesso modo in cui l'assorbimento delle radiazioni elettromagnetiche è regolato dalla simmetria molecolare. La teoria matematica che studia le opera- zioni astratte di simmetria che portano un cristallo infinito a coincidere con se stesso, e cioè la teoria dei gruppi, per- mette una trattazione sistematica e rigorosa delle proprie- tà fisiche dei cristalli e fornisce la chiave per l'interpreta- zione dei diagrammi di diffrazione X o neutronica.
La diffrazione degli elettroni (v. Wheatley, 1958) è in- vece, come metodo di analisi di struttura molecolare, essenzialmente limitata alle molecole in fase gassosa. La diffrazione elettronica dei solidi con elettroni di bassa energia, nota come LEED (Low Energy Electron Diffraction), ha avuto negli ultimi anni un notevole sviluppo, ma non costituisce un metodo d'indagine di struttura molecolare, bensì un potente metodo per lo studio di superfici metalliche e di strati monomolecolari adsorbiti.
I diagrammi di diffrazione elettronica sono complicati dal fatto che i massimi e minimi di diffrazione si sovrappongono a un fondo continuo dovuto alla diffusione elastica e anelastica degli elettroni da parte degli atomi. Questo fondo, che diminuisce molto rapidamente con l'aumentare dell'angolo di diffrazione, deve essere sottratto per ottenere il vero diagramma di diffrazione molecolare.
5. Risultati ottenuti e sviluppi futuri.
I risultati ottenuti finora con l'aiuto delle tecniche sperimentali illustrate precedentemente hanno fornito alla chimica un quadro soddisfacentemente completo della struttura molecolare. Nel valutare il significato e l'importanza di questi risultati conviene distinguere tra quelli relativi alle molecole di piccole e medie dimensioni e quelli relativi alle macromolecole.
Il problema della struttura delle molecole piccole e medie (con pesi molecolari dell'ordine delle decine e anche delle centinaia di unità atomiche) può considerarsi risolto nelle linee generali. Migliaia di strutture molecolari sono state accuratamente determinate con i raggi X. Per molte molecole sono note in dettaglio le frequenze vibrazionali dei nuclei, i livelli elettronici, le interazioni magnetiche all'interno della molecola e spesso sono stati determinati anche effetti del secondo ordine sulle grandezze molecolari. Esistono però in questo campo ancora molti problemi insoluti, soprattutto per quanto riguarda la struttura di molecole instabili e di molecole in stati eccitati. La determinazione della struttura e dei livelli energetici di questi sistemi è il presupposto essenziale per la comprensione profonda dei problemi della catalisi e della cinetica chimica e costituisce una delle direttive di sviluppo più interessanti. La determinazione della struttura di sistemi stabili si pone invece a un livello meno denso di sviluppi per il futuro. I chimici producono continuamente nuove molecole sintetiche, che spesso rivelano strutture complicate e originali. Le ricerche in questa direzione si configurano però essenzialmente come una routine tanto necessaria quanto noiosa, che aggiunge utili ma spesso marginali dettagli a un quadro le cui linee generali sono ben chiarite, confermando continuamente le previsioni teoriche, ritoccando e correggendo i valori numerici delle grandezze molecolari, in un processo di revisione critica e di aggiornamento costante che trova la sua giustificazione solo nella necessità intrinseca della scienza di perfezionare al massimo ogni stadio conoscitivo prima di lanciarsi in una nuova avventura.
Ben diversa è, invece, la situazione per quanto riguarda le macromolecole biologiche, che hanno strutture estremamente complicate, con pesi molecolari dell'ordine di decine e centinaia di migliaia di unità atomiche. In questo caso, sia per le grandi dimensioni delle molecole, sia per la grande variabilità delle strutture in condizioni sperimentali poco diverse, il quadro generale è appena accennato e quindi ben lontano dall'essere sufficientemente definito. I risultati ottenuti in questo campo, soprattutto con la diffrazione dei raggi X, hanno del miracoloso. La struttura spaziale dei polipeptidi e delle proteine (v. proteine), così come quella degli acidi nucleici (v. acidi nucleici), è stata risolta in molti casi con sufficiente accuratezza allo stato solido. Lo studio della conformazione delle proteine in soluzione, anche se non basato su prove sperimentali univoche e decisive, sembra fornire risultati coerenti e inquadrabili, per lo meno a livello qualitativo, in una teoria organica e generale. Poiché il problema della struttura e delle possibili conformazioni delle macromolecole è trattato per esteso in altri articoli (v. polimeri; v. proteine), la nostra discussione sarà limitata soprattutto alle molecole di piccole e medie dimensioni. Analizzeremo, pertanto, i risultati delle singole tecniche sperimentali, illustrandone vantaggi e svantaggi, campi di applicabilità e limitazioni e mettendo in evidenza gli aspetti che sembrano più promettenti per gli sviluppi futuri.
La spettroscopia a microonde è, tra i metodi d'indagine strutturale, quello più preciso e sensibile, ma anche quello con le maggiori limitazioni. Dalla determinazione delle frequenze rotazionali è possibile ottenere i momenti d'inerzia molecolari e da questi le distanze e gli angoli di legame con una precisione di gran lunga superiore a quella delle altre tecniche. Per dare un'idea di questa precisione, riportiamo nella tab. II alcuni dati strutturali ottenuti con le microonde per molecole semplici.

La grave limitazione delle microonde consiste nel fatto che sono studiabili con questa tecnica solo molecole con pochi atomi, dotate di momento dipolare permanente. È possibile inoltre analizzare solo molecole in fase gassosa, il che costituisce un'ulteriore limitazione. L'importanza delle microonde non è, però, legata soltanto alla determinazione accurata di distanze e angoli di legame. Data l'enorme sensibilità e precisione del metodo, è possibile misurare variazioni piccolissime delle distanze di legame, dovute all'effetto di distorsione centrifuga durante la rotazione. Si ottengono così delle ‛costanti di distorsione centrifuga' che rivestono una notevole importanza nel calcolo delle forze che tengono uniti gli atomi delle molecole. Inoltre, con l'aiuto dell'effetto Stark, che utilizza l'azione di un campo elettrico sulle molecole in esame, è possibile calcolare con grande precisione il momento di dipolo elettrico molecolare.
La spettroscopia a microonde non è, in generale, una tecnica molto diffusa. Le difficoltà sperimentali e teoriche ne fanno una branca della strutturistica chimica riservata essenzialmente a specialisti. Recentemente è stato messo in commercio il primo spettrografo a microonde di produzione industriale, che permette di esaminare con maggiore rapidità e con alta precisione gli spettri rotazionali di molecole aventi un più alto numero di atomi rispetto a quelle prima studiabili. La diffusione di apparecchiature completamente automatiche e prodotte in serie aumenterà certamente nel prossimo futuro le ricerche in questo campo. Resta però sempre limitato - sembra a una decina il numero di atomi nelle molecole che si possono oggi studiare con questo metodo. Il futuro dell'impiego delle microonde per questi scopi sembra perciò confinato alle molecole piccole, per le quali si possono avere informazioni molto dettagliate su stati elettronici e vibrazionali eccitati, sulle interazioni vibrorotazionali, su momenti dipolari ecc.
La spettroscopia vibrazionale, e specialmente la spettroscopia nell'infrarosso, rappresenta uno dei metodi strutturali più diffusi, sia perché fornisce informazioni molto rapide, sia perché, con apparecchiature relativamente poco costose e senza richiedere un approfondimento della teoria, permette al chimico organico d'individuare con facilità gruppi funzionali caratteristici in molecole complesse. Per avere un'idea della diffusione del metodo, basti pensare che il numero di spettrografi infrarossi installati nei laboratori industriali e di ricerca di tutto il mondo è superiore alle centomila unità.
La spettroscopia vibrazionale fornisce anch'essa informazioni di vario tipo, a differenti livelli di complessità. A differenza della spettroscopia rotazionale, essa non permette di determinare lunghezze e angoli di legame, ma solo la simmetria molecolare. Quest'ultima informazione, però, si ottiene molto rapidamente, specialmente se si dispone contemporaneamente dello spettro infrarosso e di quello Raman. Lo spettro infrarosso e/o Raman di un composto organico permette d'identificare, come abbiamo accennato precedentemente, un gran numero di gruppi chimici, cioè di raggruppamenti atomici con una specifica funzione chimica, come i gruppi CH3, CH2, OH, NH2, CO, fenile ecc., nonché la loro posizione nella molecola e in generale il tipo di composto in esame. Un esempio di spettro infrarosso, con l'indicazione dei più importanti gruppi chimici identificabili dalle bande di assorbimento, è mostrato nella fig. 4. Questo uso, d'impostazione chiaramente empirica, della spettroscopia vibrazionale, anche se largamente diffuso, non sembra destinato a significativi sviluppi, tranne forse una più efficace impostazione metodologica attraverso l'impiego di elaboratori elettronici.
Informazioni molto più importanti per lo studio della dinamica molecolare si possono invece ricavare dall'identificazione dei modi di vibrazione delle molecole e dalla localizzazione nello spettro delle corrispondenti frequenze di vibrazione. Da questi dati si possono calcolare, usando un modello meccanico che approssima i legami chimici a molle elastiche, le forze che tengono uniti gli atomi nelle molecole, gli spostamenti relativi dei nuclei nelle vibrazioni molecolari, il contributo vibrazionale alle funzioni termodinamiche, ecc. Questo indirizzo di ricerca, che ha avuto negli ultimi anni uno sviluppo imprevedibile, grazie all'uso di potenti elaboratori elettronici, ha già dato tutto quello che poteva nell'approssimazione armonica, cioè usando il modello di molle perfettamente elastiche accennato precedentemente. I suoi sviluppi futuri sono perciò essenzialmente legati alla possibilità di calcolare le forze intramolecolari con un modello molto più sofisticato di quello attuale, un modello che tenga conto dell'anarmonicità dei legami chimici e che sia più direttamente collegato alla struttura elettronica delle molecole.
Altrettanto importanti sono le informazioni che si possono avere dalla spettroscopia vibrazionale sulle forze intermolecolari sia in soluzione sia allo stato solido. I risultati ottenuti finora in questo campo sono serviti a mettere in evidenza l'esistenza delle interazioni molecolari piuttosto che a svilupparne una valutazione quantitativa. Recentemente, però, le vibrazioni di cristalli molecolari sono state usate con successo per il calcolo delle interazioni tra molecole in cristalli anche abbastanza complessi.
Queste ricerche, che investono un gran numero di proprietà dei cristalli e parallelamente delle macromolecole, intese come cristalli monodimensionali, sono destinate ad avere un sempre maggiore sviluppo nel prossimo futuro e probabilmente a far affermare la spettroscopia vibrazionale come uno dei metodi chiave per lo studio delle forze intermolecolari nei solidi.
I limiti della spettroscopia nell'infrarosso sono essenzialmente legati alla grande complessità dello spettro, man mano che aumenta il numero di atomi in una molecola. Inoltre quasi tutti i solventi, in particolare l'acqua, sono opachi alle radiazioni in molte zone dello spettro IR. Questo impedisce l'uso di tale tecnica per uno studio dettagliato delle conformazioni in soluzione di macromolecole biologiche. In questa direzione sembra di gran lunga favorita la spettroscopia Raman, specialmente con l'uso di sorgenti laser, poiché nello spettro Raman l'acqua dà relativamente meno fastidio.
La spettroscopia elettronica costituisce il metodo principe per lo studio dell'energia degli elettroni nelle molecole. Anche tale tecnica ha avuto, parallelamente a uno sviluppo teorico e strutturale, uno sviluppo empirico che si è rivelato di grande aiuto in chimica organica e in biologia per l'identificazione di composti, per la loro caratterizzazione anche in tracce minime, per lo studio delle cinetiche in soluzione e in generale per seguire qualsiasi fenomeno riconducibile a una variazione di assorbimento della luce.
La determinazione delle transizioni elettroniche nello spettro visibile e ultravioletto rappresenta il primo stadio per la valutazione della bontà delle autofunzioni molecolari che servono per descrivere il legame chimico nelle molecole. I livelli elettronici di molecole complesse non vengono determinati solamente usando la spettroscopia di assorbimento, ma anche altre tecniche, quali la spettroscopia di emissione, di fluorescenza ecc., che, anche se diverse dal punto di vista sperimentale, danno lo stesso tipo d'informazione. Impiegando poi apparecchiature ad alta risoluzione, capaci cioè di separare frequenze molto vicine, è possibile analizzare la struttura vibrazionale delle bande elettroniche sia allo stato gassoso sia allo stato cristallino e ottenere in questo modo le frequenze di vibrazione, la simmetria e la struttura molecolare in stati elettronici eccitati.
Usando particolari tecniche che consentono la produzione di molecole instabili o di radicali liberi, è possibile con la spettroscopia elettronica studiarne la struttura sia allo stato gassoso, sia addirittura allo stato solido bloccandole in matrici di materiale inerte, alla temperatura dell'elio liquido. Lo sviluppo della spettroscopia elettronica è essenzialmente legato a ricerche ad alta risoluzione e allo studio strutturale di molecole in stati elettronici eccitati, di radicali liberi e di molecole instabili, soprattutto per le implicazioni che tali ricerche possono avere nello studio di meccanismi di reazione e nell'analisi dei processi di trasferimento d'energia.
La risonanza magnetica nucleare (NMR) è, come la spettroscopia vibrazionale, un metodo strutturale notevolmente diffuso, che permette al chimico organico d'individuare con grande rapidità e facilità molti gruppi chimici presenti in molecole complesse. Questa applicazione strutturale dell'NMR è basata sul fatto che un nucleo dotato di momento magnetico assorbe a frequenze diverse o, come si dice tecnicamente, risuona a frequenze diverse, in funzione del tipo di legame che forma con gli altri nuclei e della vicinanza o meno con altri nuclei dotati di momento magnetico. Questo effetto, che prende il nome di shift chimico, è mostrato nella fig. 5, dove è riportato lo spettro di risonanza magnetica nucleare dell'alcool etilico. Dallo spettro si può osservare come i nuclei d'idrogeno risuonino a valori diversi del campo magnetico (per ragioni tecniche, invece di variare la frequenza radio e tenere fisso il campo magnetico, è più conveniente tenere fissa la frequenza radio e variare il campo magnetico) e diano luogo a una struttura fine diversa a seconda del tipo di gruppo funzionale cui appartengono (OH, CH2 e CH3).
La maggior parte delle applicazioni della tecnica NMR allo studio strutturale di molecole complesse è limitata all'analisi delle frequenze di risonanza dell'idrogeno. È però in continuo sviluppo la spettroscopia NMR basata su altri nuclei, come l'azoto, il fluoro, il carbonio, il fosforo ecc., che promette per il futuro sviluppi estremamente interessanti. In questi ultimi anni la risonanza magnetica nucleare è stata usata con qualche successo per lo studio delle conformazioni in soluzione di polipeptidi e proteine. In effetti l'NMR è per il momento la sola tecnica strutturale che permetta d'investigare la struttura di molecole complesse in soluzione e che forse, in un futuro abbastanza prossimo, potrà fornire dati veramente significativi.
La risonanza magnetica nucleare è stata anche usata per la determinazione di distanze di legame in cristalli singoli di molecole contenenti atomi d'idrogeno. In questo caso, dalla misura dell'intensità delle bande d'assorbimento si calcola una grandezza nota come il secondo momento o ampiezza quadratica media, che è collegabile alla distanza tra gli atomi d'idrogeno nella cella elementare. Questa applicazione è abbastanza limitata e certamente non sembra competitiva con la diffrazione neutronica, che dà risultati molto più sicuri.
La risonanza elettronica paramagnetica (EPR) è molto simile, sia teoricamente che sperimentalmente, alla risonanza magnetica nucleare. Nel caso dell'EPR, però, è possibile studiare solo molecole che abbiano elettroni spaiati, cioè radicali liberi o ioni paramagnetici. La spettroscopia EPR si sta diffondendo come un'efficace tecnica per evidenziare la presenza di radicali o di ioni con elettroni spaiati nelle reazioni biologiche e per lo studio della catalisi omogenea ed eterogenea. Parallelamente la spettroscopia EPR si è notevolmente sviluppata dal punto di vista teorico, rivelandosi un metodo molto potente per la valutazione dell'accuratezza delle funzioni d'onda molecolari. Tra i metodi diffrattometrici la diffrazione elettronica è l'unico adatto allo studio di molecole in fase gassosa. Per molecole piccole, con alta simmetria, essa fornisce valori accurati per le distanze di legame, purché la simmetria molecolare sia nota a priori. Man mano che la simmetria diminuisce e aumenta il numero di parametri da determinare, aumenta anche l'errore e i risultati diventano più incerti. In generale è necessario fissare alcune lunghezze di legame e determinare le altre in funzione di queste. Poiché l'errore commesso nello scegliere un parametro può essere compensato dagli errori sui parametri calcolati, il diagramma di diffrazione elettronica di una molecola può corrispondere a tutta una serie di strutture possibili, di poco differenti l'una dall'altra. Per tutte queste ragioni la diffrazione elettronica non è una tecnica molto diffusa e, per lo meno in base alla situazione attuale, difficilmente potrà nel futuro competere con le altre tecniche strutturali.
La diffrazione dei raggi X è invece di gran lunga il più importante e diffuso metodo per la determinazione della struttura molecolare. Più del 90% delle distanze interatomiche finora note sono state ottenute con i raggi X e la percentuale è praticamente del 100% se non si considerano le molecole di piccole dimensioni. In linea di principio la diffrazione dei raggi X può essere usata per studiare molecole sia in fase gassosa, sia liquida e solida. In pratica però, anche se numerose ricerche sono state condotte sui liquidi, essa si è affermata essenzialmente come metodo per lo studio della struttura dei cristalli e come tale ha raggiunto lo sviluppo attuale. Con questa tecnica si possono ottenere due tipi d'informazioni. Dalla forma e dalle proprietà di simmetria del diagramma di diffrazione si possono innanzi tutto conoscere le dimensioni e la simmetria della cella elementare di un cristallo. Successivamente, dalla misura delle intensità delle macchie di diffrazione, si possono localizzare le posizioni degli atomi nella cella elementare e quindi valutare quantitativamente le distanze e gli angoli di legame nelle molecole.
Come accennato precedentemente, nella descrizione generale dei metodi diffrattometrici, i raggi X sono diffratti essenzialmente dagli elettroni. Per ogni atomo di una molecola, l'ampiezza della radiazione diffratta è proporzionale al numero di elettroni presenti, con un coefficiente di proporzionalità che diminuisce al crescere dell'angolo di diffrazione. Pertanto gli atomi dotati di molti elettroni contribuiscono fortemente alle intensità delle macchie di diffrazione, mentre gli atomi con pochi elettroni contribuiscono poco. Poiché, con la sintesi di Fourier, le coordinate atomiche nella cella elementare vengono ottenute dall'intensità del diagramma di diffrazione, è chiaro che le coordinate degli atomi con molti elettroni sono determinabili con maggiore precisione delle coordinate di atomi con pochi elettroni. La difficoltà diventa particolarmente grave nel caso degli atomi d'idrogeno, che hanno la più bassa densità di elettroni, tanto che in molte strutture la posizione degli atomi d'idrogeno è derivata indirettamente dalla configurazione dello scheletro molecolare. Sono state però da tempo messe a punto tecniche particolari di analisi dei dati, come la sintesi differenza di Fourier, che permettono di ridurre notevolmente l'indeterminazione nella posizione degli atomi d'idrogeno e, in casi favorevoli, di localizzarli con sufficiente accuratezza. Ciò nonostante, specialmente se nella molecola vi sono atomi pesanti, la precisione che si raggiunge nella determinazione delle coordinate degli atomi d'idrogeno resta in genere piuttosto bassa. D'altra parte, la presenza di atomi pesanti in una molecola semplifica notevolmente la determinazione della struttura e, soprattutto negli stadi iniziali del processo di raffinamento, può rivelarsi di enorme utilità.
Vi sono poi molti altri fattori che influiscono sulla precisione con cui è possibile determinare le coordinate degli atomi, che vanno dagli errori commessi nella misura delle intensità, alle vibrazioni dei nuclei, a problemi di convergenza nello sviluppo in serie di Fourier, a difficoltà connesse con l'ottenimento di cristalli adatti. Tutti questi fattori possono essere controllati opportunamente e la loro incidenza sulla precisione dei risultati finali può essere ridotta al minimo, aumentando però notevomente il lavoro e il tempo necessario a risolvere una data struttura.
In generale l'analisi con i raggi X non raggiunge mai la precisione dei metodi spettroscopici. Offre però, rispetto a tutte le altre tecniche, l'enorme vantaggio di non avere limitazioni nelle dimensioni delle molecole che possono essere studiate, per cui si presenta oggi come il metodo strutturale con maggiori possibilità di sviluppo nel campo delle grosse molecole. La limitazione più forte resta ancora la quantità di tempo necessaria per ottenere risultati significativi. Per molecole di piccole e medie dimensioni la determinazione della struttura con i raggi X non offre oggi nessuna difficoltà ed è un processo quasi completamente automatico. Per dare un'idea al lettore del tipo di risultato che si ottiene dall'analisi con i raggi X di cristalli molecolari contenenti molecole relativamente semplici, riportiamo nella fig. 6 la struttura della cella elementare di un cristallo di p-clorocinnamammide, vista guardando nella direzione dell'asse cristallografico b nella fig. 6A e nella direzione dell'asse cristallografico a nella fig. 6B. Poiché le molecole non si trovano nello stesso piano, quelle che sono più vicine all'osservatore sono rappresentate con atomi bianchi e quelle più lontane con atomi neri.
La determinazione della struttura di molecole molto complesse, come per esempio una proteina, è invece, a tutt'oggi, un lavoro molto lungo e laborioso. Le poche strutture determinate finora per molecole di grandi dimensioni e bassa simmetria hanno richiesto il continuo e paziente lavoro, spesso per molti anni, di équipes di ricercatori in laboratori specializzati. La diffusione di diffrattometri completamente automatici e di efficienti programmi per elabolatori elettronici ha permesso in questi ultimi anni di ridurre i tempi entro limiti ragionevoli e ha aperto la strada a uno studio su larga scala delle grosse molecole di interesse biologico. La diffrazione dei neutroni è potenzialmente ancora più efficace della diffrazione dei raggi X e la sua relativamente bassa diffusione come metodo d'indagine strutturale è dovuta solo alle limitazioni connesse con la disponibilità di pile atomiche come sorgenti di neutroni.
I principî della tecnica, il trattamento dei dati e le informazioni che si possono ottenere sono del tutto simili, tranne che in ovvi aspetti sperimentali, a quelli della diffrazione dei raggi X. Esistono però alcune differenze tra i due metodi, le quali fanno si che essi non siano l'uno equivalente all'altro, ma che si completino a vicenda. La differenza più importante è collegata al fatto che, mentre i raggi X sono diffratti dagli elettroni, i neutroni sono diffratti dai nuclei. Poiché tutti i nuclei diffrangono i neutroni praticamente allo stesso modo, scompare, nel caso della diffrazione neutronica, la differenza tra atomi pesanti e atomi leggeri che condiziona invece pesantemente la diffrazione dei raggi X. Da questo punto di vista la diffrazione neutronica si presenta come il metodo migliore per la localizzazione degli atomi d'idrogeno nelle molecole e di fatto viene essenzialmente adoperata a questo scopo.
Bibliografia.
Bershon, M., Baird, J. C., An introduction to electron paramagnetic resonance, New York 1966.
Bovey, F. A., Nuclear magnetic resonance spectroscopy, New York 1969.
Gilson, T. R., Hendra, P. J., Laser Raman spectroscopy, New York 1970.
Guinier, A., Théorie et technique de la radiocristallographie, Paris 1956.
Heitler, W., The quantum theory of radiation, Oxford 1954.
Herzberg, G., Molecular spectra and molecular structure, Amsterdam 1945.
Mizushima, S., Structure of molecules and internal rotation, New York 1954.
Siegbahn, K. e altri, ESCA, Uppsala 1967.
Weissberger, A. (a cura di), Technique of organic chemistry, vol. IX, Chemical applications of spectroscopy, New York 1956.
Wheatley, P. J., The determination of molecular structure, Oxford 1958.
Analisi conformazionale delle piccole molecole
SOMMARIO: 1. Introduzione. □ 2. Definizioni. □ 3. Analisi conformazionale di molecole non cicliche. □ 4. Analisi conformazionale di molecole cicliche. □ 5. Metodi per lo studio quantitativo degli equilibri conformazionali. □ 6. Conformazione e reattività. □ 7. Prospettive di sviluppo. □ Bibliografia.
1. Introduzione.
Lo studio della struttura molecolare, che si è andato lentamente sviluppando a partire dagli inizi dell'Ottocento, aveva portato alla fine del secolo all'acquisizione per via puramente empirica di vari concetti fondamentali. La molecola si presentava come un insieme di atomi collegati fra loro con un numero di legami ben definito, caratteristico dell'elemento (valenza), anche se nulla si sapeva sulla natura di tali legami e ben poco sulla disposizione geometrica degli atomi, a parte il fondamentale concetto della distribuzione tetraedrica dei sostituenti intorno al carbonio, intuito da E. Paternò e sviluppato da J.-A. Le Bel e J. H. van't Hoff nel 1874. Solo nel secondo decennio del XX secolo la geniale intuizione di O. N. Lewis sulla natura elettronica del legame chimico e l'introduzione da parte di W. H. e W. L. Bragg della diffrazione dei raggi X per dedurre la geometria molecolare aprirono la via allo studio razionale della struttura e dettero inizio a un intenso periodo di ricerche che nei trent'anni successivi fornirono gli strumenti per una conoscenza molto esatta dei valori delle lunghezze di legame e degli angoli di valenza, cioè dei parametri necessari per definire la geometria molecolare. Era stato però fino ad allora quasi del tutto trascurato, almeno dai chimici organici, un aspetto strutturistico, apparentemente secondario, ma che si è in seguito rivelato molto importante, quello cioè delle differenti forme che una singola molecola pluriatomica può assumere per rotazione interna intorno a uno o più dei suoi legami. Questo disinteresse si giustificava con l'ipotesi che, essendo la rotazione intorno ai legami semplici essenzialmente libera, non avesse interesse pratico il prendere in considerazione singole forme rotazionali. Non mancavano però indicazioni sull'esistenza, in casi particolari, di barriere energetiche anche assai elevate che possono ostacolare tale rotazione, in quanto fin dal 1926 era stato riconosciuto che l'isomeria ottica dei derivati del difenile recanti sostituenti nelle posizioni orto doveva essere attribuita a una dissimmetria legata alla difficoltà per tali molecole di assumere a temperatura ambiente la struttura simmetrica con i due anelli benzenici nello stesso piano. Del resto già nel 1890 H. Sachse, in contrasto con la teoria di A. Baeyer, che assumeva una geometria planare per tutti i cicli, aveva proposto due forme non planari per il cicloesano, in cui gli angoli interni di valenza avevano il normale valore tetraedrico di 109° 28′, anziché quello di 120° prevedibile per un esagono regolare. Tale ipotesi fu a lungo contrastata o ignorata e solo nel 1918 E. Mohr portò nuovi elementi a suo favore.
Furono i chimici fisici, e in particolare K. S. Pitzer e S. Mizushima negli anni trenta, ad affrontare da un punto di vista quantitativo il problema della rotazione interna in molecole semplici (etano, 1,2-dicloroetano) e a stabilire l'esistenza e valutare l'entità di barriere energetiche mediante metodi calorimetrici e spettroscopici. Nel 1943 O. Hassel confermava sperimentalmente con metodi diffrattometrici le ipotesi di Sachse e Mohr sulla forma non planare del cicloesano, ma è stato merito particolare di D. H. R. Barton nel 1950 di indicare la fondamentale importanza della conformazione delle molecole nel determinare le loro proprietà fisiche e chimiche, e in particolare la loro reattività. Lo sviluppo particolarmente fruttuoso degli studi e dei risultati in questo campo è stato uno degli aspetti caratterizzanti della chimica organica del terzo quarto del nostro secolo, un fatto che ha avuto adeguato riconoscimento nel conferimento del premio Nobel per la chimica nel 1969 a Barton e Hassel.
2. Definizioni.
Si definisce ‛conformazione' ognuna delle forme che una singola specie chimica può assumere per rotazione intorno a uno o più dei suoi legami. Scopo dell'‛analisi conformazionale' è studiare gli aspetti strutturali ed energetici delle singole conformazioni di una molecola e della loro interconversione.
Solo molecole estremamente semplici e altamente simmetriche (H2, H2O, H−C≡N) non presentano isomeria conformazionale, in quanto la rotazione interna non porta a forme distinguibili l'una dall'altra.
Di solito vengono considerate ‛isomeri conformazionali' (si usano anche i termini ‛conformeri' e ‛rotameri') solo forme interconvertibili per rotazione intorno a legami covalenti singoli, anche se la differenza fra l'interconversione, ad esempio, di due conformeri dell'1,2-dicloroetano e quella del cis- e trans-1,2-dicloroetilene riguarda essenzialmente l'entità della barriera energetica fra le due forme, molto maggiore nel secondo che nel primo caso. Perciò le forme cis e trans dei derivati etilenici sono normalmente, anche se arbitrariamente, designate come ‛diastereoisomeri' piuttosto che come conformeri. Ancor meno giustificato è l'uso di non considerare come conformeri gli enantiomeri dei derivati difenilici orto-sostituiti, a cui si è accennato nell'introduzione, in quanto essi sono interconvertibili per semplice rotazione intorno a un legame singolo, sia pure a temperature che spesso sono assai più alte di quella ambiente, ciò che permette appunto la separazione di antipodi ottici. Del resto, abbassando sufficientemente la temperatura e quindi l'energia cinetica, qualunque coppia di conformeri diviene in teoria separabile.
Per definire una singola conformazione di una data specie chimica è necessario indicare un numero di angoli pari al numero dei legami la rotazione intorno ai quali produce isomeria conformazionale. Per una struttura del tipo X-A-B-Y viene di solito usato l'angolo diedro τ fra i due piani contenenti l'uno gli atomi X, A e B e l'altro quelli A, B e Y; quest'angolo viene chiamato ‛angolo di torsione'.
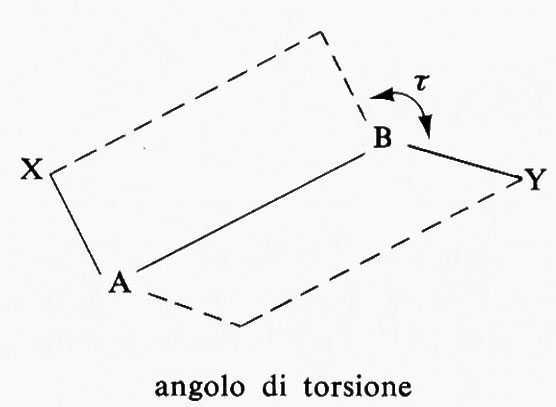
La rappresentazione grafica dei singoli conformeri può essere data mediante formule prospettiche in cui tratti interi indicano legami che si trovano nel piano del foglio, tratti più marcati o cunei legami che proiettano verso l'osservatore, linee tratteggiate legami che stanno al di sotto del piano. Più razionali sono le cosiddette ‛proiezioni di Newman', in cui il legame intorno al quale si svolge la rotazione è immaginato perpendicolare al piano e i tratti che giungono fino al centro del cerchio rappresentano i legami che partono dall'atomo più vicino all'osservatore, quelli che si fermano alla circonferenza i legami che partono dall'atomo più lontano.

Il numero di conformazioni che può assumere una molecola è infinito, in quanto infinitamente piccolo può essere l'angolo di torsione, ma di solito si considerano solo i conformeri che corrispondono a minimi o massimi nelle curve di energia potenziale (v. sotto, cap. 3). Si indicano quindi col termine di ‛sfalsate' (staggered, in inglese) le conformazioni in cui gli angoli torsionali sono di 60°, e con quello di ‛eclissate' quelle in cui essi sono di 0°. Quando i sostituenti sui due atomi interessati dal legame non sono tutti uguali fra loro, sono possibili più conformeri sfalsati; si indica di solito come skew o gauche quello in cui i sostituenti più
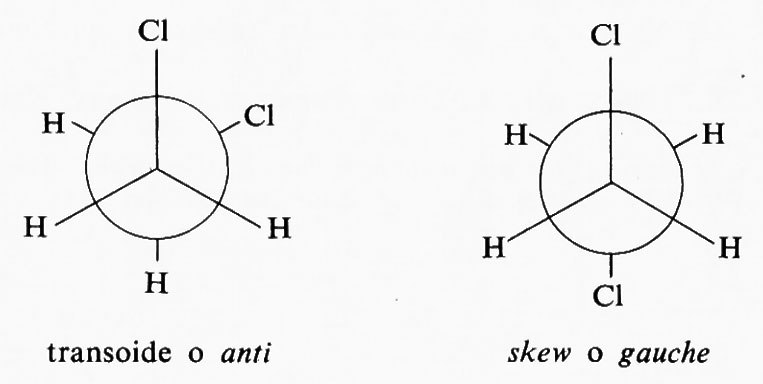
voluminosi sono sfalsati di 60°, come ‛transoide' o anti quello in cui sono sfalsati di 180°.
3. Analisi conformazionale di molecole non cicliche.
Come già accennato nei capitoli precedenti, la rotazione interna intorno a un legame di una molecola non è mai perfettamente libera, cioè è sempre necessario fornire al sistema una certa quantità di energia cinetica perché tale rotazione avvenga. Se consideriamo la molecola dell'etano e, partendo dalla conformazione eclissata (τ=0°) facciamo ruotare uno dei gruppi metilici mantenendo fisso l'altro, notiamo che l'energia potenziale decresce fino a raggiungere un minimo corrispondente a una rotazione di 60°, cioè alla forma sfalsata. Proseguendo nella rotazione, l'energia potenziale cresce fino a ritornare a un valore uguale a quello iniziale dopo 120°. L'andamento dell'energia in funzione dell'angolo di torsione è quindi quello indicato nella fig. 1, ove i minimi corrispondono alle forme sfalsate e i massimi a quelle eclissate. La differenza fra i minimi e i massimi è di circa 3 kcal/mole, ciò che corrisponde appunto alla quantità di energia che dev'essere fornita al sistema perché possa verificarsi la rotazione intorno al legame. In questo caso la barriera energetica è molto al di sotto dell'energia cinetica che l'etano possiede a temperatura ambiente e quindi la rotazione si verifica con alta velocità angolare. Solo al di sotto di 50 °K l'energia cinetica è diminuita sufficientemente da congelare l'etano nella sua conformazione più stabile, quella sfalsata.
Il diagramma dell'energia potenziale si presenta più complesso nel caso di derivati polisostituiti dell'etano. Ad esempio, per il butano esso assume la forma indicata nella fig. 2. Dalla conformazione a più alta energia, corrispondente a quella in cui i sostituenti più ingombranti (i metili) sono eclissati, si scende, dopo una rotazione di 60°, a un primo minimo corrispondente al conformero gauche, per risalire dopo 120° a un secondo massimo, inferiore al primo, per il conformero in cui ognuno dei metili eclissa un idrogeno, e ridiscendere dopo 180° al secondo e più basso minimo che corrisponde alla conformazione transoide, la più stabile di tutte.
I dati sopra esposti possono in una prima analisi essere interpretati secondo il concetto intuitivo che una conformazione eclissata è meno stabile di una sfalsata, a causa della maggior vicinanza fra i sostituenti. Appunto da interpretazioni basate su interazioni repulsive si è sviluppata agli inizi buona parte della teoria dell'analisi conformazionale. Ci si è però ben presto resi conto che tale trattazione, che considera gli atomi come sfere rigide con un diametro fisso, se pure ha dato buoni risultati, almeno dal punto di vista qualitativo, quando venivano considerate interazioni fra gruppi saturi non contenenti atomi elettronegativi, era del tutto inadeguata per ottenere dati quantitativi e poteva portare a previsioni errate nel caso di molecole insature o in presenza di legami molto polarizzati.
Un errore che spesso si fa è quello di considerare tutte le interazioni fra atomi non direttamente legati fra loro in una data molecola come necessariamente repulsive, mentre è ben noto che quando due atomi si avvicinano si ha a una certa distanza un'attrazione, che solo per un ulteriore avvicinamento si trasforma rapidamente in una forte repulsione, come indicato dalla fig. 3, ove sono riportate in ascisse le distanze fra i due atomi e in ordinate le energie potenziali. Che l'interazione sia attrattiva o repulsiva dipenderà quindi, fra l'altro, anche dalla distanza, ma non solo da essa. Sono ormai molti i casi noti in cui l'attrazione supera la repulsione fra gruppi non direttamente collegati; ad esempio quello riportato recentemente da Epiotis (v., 1973) che indica che all'equilibrio l'etere etilpropenilico si trova per l'81% nella forma cis (1), benché una considerazione delle sole dimensioni dei gruppi metilico ed etossilico farebbe prevedere una netta repulsione e quindi una destabilizzazione della forma cis rispetto alla trans (2). Nel caso dei 2-eseni, in cui l'ossigeno dei composti suddetti è sostituito da un CH2 e in cui le dimensioni dei sostituenti sono assai simili, la forma trans (4) è molto più stabile della cis (3). Da
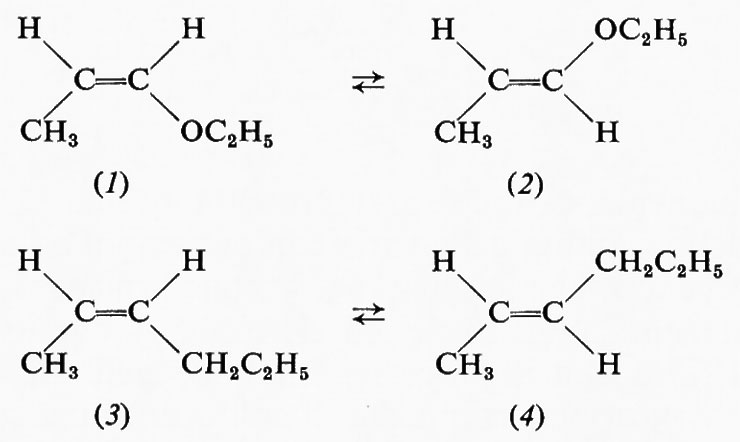
ciò risulta chiaramente che altri fattori, indipendenti dal puro aspetto dimensionale, possono condizionare la stabilità di una forma rispetto all'altra. Occorre sempre tener presente che gli atomi sono costituiti da particelle elementari che interagiscono le une con le altre in maniera assolutamente non razionalizzabile con la meccanica classica e trattabile solo con quella quantistica, ciò che implica calcoli straordinariamente complessi quando si tratti di molecole poliatomiche.
Del resto anche per una molecola molto semplice, come quella dell'etano, una valutazione delle repulsioni di van der Waals fra gli atomi di idrogeno vicinali nella conformazione eclissata è del tutto inadeguata a spiegare la differenza di energia di circa 3 kcal/mole che essa presenta rispetto alla forma sfalsata. Difatti, essendo gli idrogeni eclissati distanziati di 2,3 Å, pari a circa il doppio del raggio atomico, la repulsione è assai debole, cioè circa 0,12 kcal/mole per ogni coppia. In tutto, quindi, le tre interazioni giustificherebbero solo 0,36 kcal/mole di destabilizzazione, cioè poco più di un decimo del valore sperimentalmente osservato. Questa difficoltà interpretativa ha stimolato negli ultimi dieci anni i chimici teorici a studiare a fondo il problema, affrontandolo con vari metodi di calcolo più o meno raffinati. Sono state date molte interpretazioni, alcune in buon accordo con il dato sperimentale, ma tutte inficiate dalla necessità di introdurre semplificazioni o premesse non rigorosamente dimostrate, così che purtroppo anche per il semplice caso della barriera rotazionale nell'etano manca ancora oggi una spiegazione veramente completa e ineccepibile.
Occorre tener presente che l'energia totale di una molecola è scomponibile in vari termini: accanto alla repulsione reciproca fra nuclei e a quella fra elettroni di atomi differenti, c'è l'attrazione reciproca fra nuclei ed elettroni. La differenza di stabilità fra due conformazioni della stessa molecola è quindi determinata dal differente peso che in essa hanno i termini repulsivi e attrattivi.
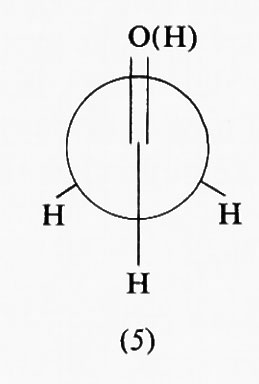
Una barriera alla rotazione non è quindi necessariamente legata a una repulsione fra atomi o orbitali e perciò a una destabilizzazione di una conformazione rispetto all'altra, ma può essere dovuta a un'interazione attrattiva che ne rende una più stabile dell'altra. Fra le varie teorie avanzate per spiegare la barriera alla rotazione nell'etano ve n'è appunto una che la interpreta in base a una stabilizzazione della forma sfalsata rispetto alla eclissata, piuttosto che alla destabilizzazione della seconda rispetto alla prima. Analoga spiegazione viene data del fatto che nell'acetaldeide la conformazione più stabile è quella in cui uno degli idrogeni del metile è eclissato con il doppio legame carbonilico (5), conformazione che in base all'approccio intuitivo basato sulle dimensioni atomiche si potrebbe ritenere come la meno stabile.
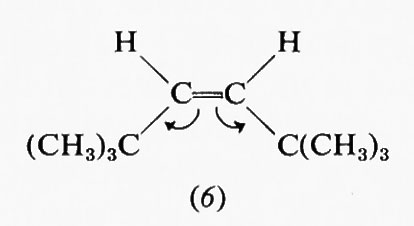
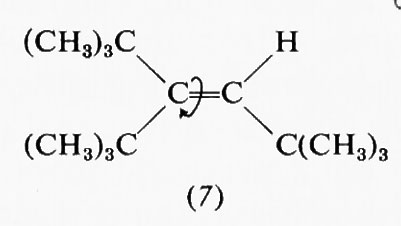
Estremamente complesso si presenta quindi il calcolo teorico della stabilità delle varie conformazioni e delle barriere energetiche che le separano, particolarmente in molecole contenenti molti nuclei ed elettroni. A questo va aggiunto il fatto che il processo dinamico dell'interconversione dei conformeri comporta di solito non una semplice rotazione intorno a uno o più legami, ma tutta una serie di altre modificazioni nella geometria molecolare, come allungamento di legami e allargamento o restringimento di angoli di valenza. Una forte interazione repulsiva in una forma gauche o eclissata potrà essere in parte compensata da deformazioni di questo tipo, di cui un'analisi conformazionale quantitativa dovrebbe tener conto, ma che rendono il calcolo ancora più complesso. Deviazioni dalla normale geometria molecolare sono facilmente evidenziabili in derivati etilenici con due sostituenti ingombranti in cis. Per esempio, secondo recenti dati di Ermer e Lifson (v., 1974), nel cis-diterz-butiletilene (6) gli angoli fra i gruppi sostituenti e il doppio legame assumono il valore di 135°, anziché quello normale di 120°, a causa della reciproca repulsione fra tali gruppi, mentre per il triterz-butiletilene (7) questo allargamento è ostacolato dal fatto che avvicinerebbe i due sostituenti legati allo stesso atomo di carbonio etilenico. In quest'ultima molecola l'interazione repulsiva viene quindi diminuita attraverso una torsione di 16° intorno al doppio legame, in contrasto con la completa coplanarità normalmente osservata nei sistemi olefinici.
4. Analisi conformazionale di molecole cicliche.
Di particolare interesse è lo studio delle conformazioni dei composti aliciclici. In essi l'equilibrio conformazionale è determinato essenzialmente da due esigenze, spesso in competizione fra loro: da un lato quella di minimizzare la tensione angolare, cioè di avere gli angoli interni al ciclo più vicini possibile al valore normale di 109,5°, dall'altro quella di rendere minime le interazioni torsionali repulsive fra atomi non direttamente legati fra loro, cioè di avvicinarsi il più possibile a conformazioni sfalsate.
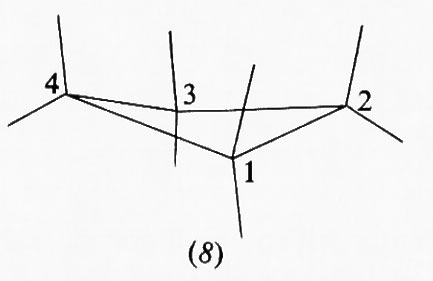
Se consideriamo il ciclobutano, una forma in cui i quattro atomi di carbonio si trovano tutti nello stesso piano sarebbe la più favorita dal punto di vista della tensione angolare, in quanto in essa gli angoli interni avrebbero il valore massimo possibile (90°) e quindi quello che più si avvicina al valore tetraedrico, ma la meno favorita dal punto di vista dell'energia torsionale, in quanto gli atomi di idrogeno sarebbero a quattro a quattro eclissati fra loro. In effetti, tecniche diffrattometriche e spettroscopiche mostrano che l'anello del ciclobutano non è piano, ma preferisce una conformazione (8) in cui il piano contenente i carboni 1, 2 e 3 e quello contenente i carbonî 1, 4 e 3 formano fra loro un angolo diedro di circa 160° che, seppure comporta angoli interni inferiori a 90°, non ha legami C−H eclissati. Con questa deformazione dell'anello la molecola guadagna circa 2 kcal/mole di stabilizzazione.
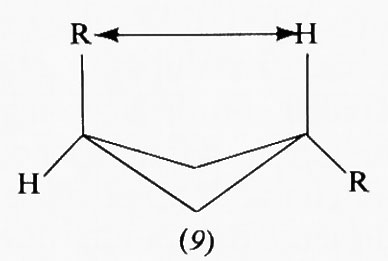
Una conseguenza di questa situazione è che nei ciclobutani 1,3-disostituiti è di solito più stabile il diastereoisomero cis di quello trans. Ciò non sarebbe spiegabile con una conformazione planare dell'anello, ma lo è con quella non planare, in quanto, mentre nel composto trans (9) uno dei sostituenti viene necessariamente a trovarsi assai vicino all'idrogeno in posizione 3 rispetto a esso, quello cis (10) può assumere una conformazione in cui entrambi i sostituenti sono relativamente poco impediti.

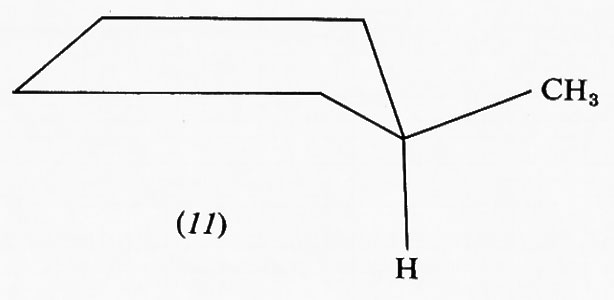
Nel ciclopentano la forma planare in cui gli atomi di carbonio sono situati ai vertici di un pentagono regolare sarebbe praticamente priva di tensione angolare avendo angoli interni di 108°, ma l'eclissamento reciproco porterebbe a una tensione torsionale di almeno 14 kcal/mole. Un'opportuna deviazione dalla planarità dell'anello, che aumenta la tensione angolare di circa 4 kcal/mole, può diminuire quella torsionale fino a 8 kcal/mole, di modo che la tensione complessiva viene ridotta a circa 10 kcal/mole nella conformazione più stabile. In effetti, nel ciclopentano non sostituito non si hanno dei minimi pronunciati nella curva dell'equilibrio conformazionale, ma uno spostamento continuo della deformazione intorno all'anello, senza che si passi mai attraverso la forma planare. Questo processo, in cui l'energia interna della molecola varia solo pochissimo (meno di 600 kcal/mole fra il massimo e il minimo) viene chiamato ‛pseudorotazione'. La presenza di sostituenti sull'anello ciclopentanico può però ostacolare parzialmente o totalmente la pseudorotazione e una conformazione può divenire nettamente favorita; è questo il caso del metilciclopentano, che assume di preferenza la conformazione cosiddetta ‛a busta' (11), in cui il metile è legato nella posizione meno impedita.
La situazione conformazionale nel cicloesano e nei suoi derivati è di gran lunga quella più studiata e meglio nota, sia perché questo sistema si trova in numerosi e importanti composti diffusi in natura, sia perché esso presenta aspetti
strutturali particolarmente interessanti dal punto di vista interpretativo. Com'è accennato nell'introduzione, proprio sul cicloesano Sachse fece nel 1890 le prime proposte di carattere conformazionale, e le osservazioni di Barton sui derivati cicloesilici aprirono nel 1950 il capitolo della moderna analisi conformazionale.
Varie sono le conformazioni non planari che un anello a sei termini può assumere conservando angoli interni di 109,5°. Di queste, quella che viene designata come forma ‛a sedia' (12) è la più stabile, in quanto tutti gli idrogeni vicinali sono sfalsati l'uno rispetto agli altri, ciò che corrisponde a un minimo nell'energia torsionale. Esiste poi un gruppo costituito da un numero infinito di conformazioni, tutte prive di tensione angolare, in quanto aventi angoli interni tutti tetraedrici, e interconvertibili in maniera continua attraverso un processo di pseudorotazione simile a quello sopra discusso per il ciclopentano.

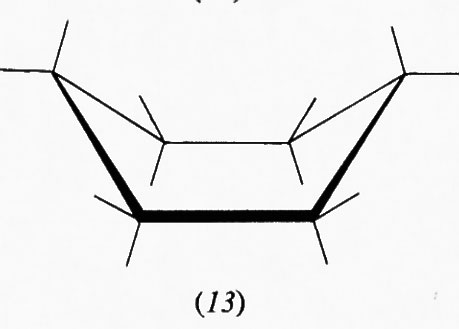
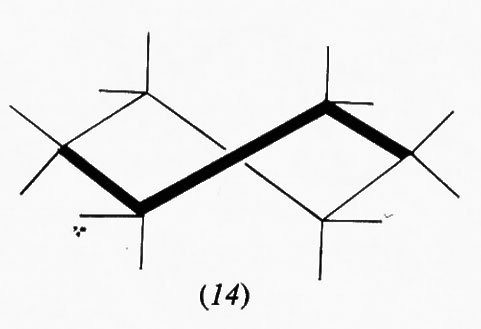
Questo gruppo di conformazioni viene spesso designato impropriamente come forme ‛a barca', ma tale termine andrebbe riservato solo a una conformazione (13) che corrisponde al punto di massima energia potenziale e quindi di minima stabilità nel processo di pseudorotazione; essa è di almeno 7 kcal/mole meno stabile della conformazione a sedia, in quanto comporta quattro coppie di legami C−H eclissati e due atomi di idrogeno in posizione 1,4 particolarmente vicini (1,8 Å). Attraverso la pseudorotazione queste interazioni repulsive diminuiscono fino a raggiungere un minimo nella forma cosiddetta twist (intrecciata) (14), che è comunque sempre meno stabile di circa 5 kcal/mole rispetto alla forma a sedia. Perciò, salvo casi particolari, si può considerare che i derivati del cicloesano si trovino in pratica esclusivamente nella conformazione a sedia, in quanto una differenza di energia di 5 kcal/mole fra due specie comporta all'equilibrio un rapporto fra di esse superiore a 99,90:0,10.
Nella forma a sedia del cicloesano i legami C−H possono essere suddivisi in due gruppi: sei disposti parallelamente all'asse del ciclo e sei disposti radialmente, detti rispettivamente ‛assiali' (a) ed ‛equatoriali' (e). Pur essendo tutti i legami sfalsati gli uni rispetto agli altri, il sistema non è però del tutto privo di tensione interna, come dimostrato, ad esempio, da precise determinazioni della geometria molecolare con metodi diffrattometrici, che indicano un leggero appiattimento dell'anello e allargamento degli angoli interni, rispetto alla conformazione teorica con angoli esattamente tetraedrici. I legami assiali non sono quindi esattamente paralleli fra loro, ma inclinati di circa 7° verso l'esterno. L'origine di questa tensione interna è stata attribuita al fatto che i legami assiali, essendo a tre a tre paralleli fra loro, tengono gli atomi di idrogeno a essi legati a una distanza di 2,5 Å, appena superiore alla somma dei raggi atomici, il che causa un sia pur piccolo effetto repulsivo, che può aumentare notevolmente quando uno degli idrogeni viene sostituito da un atomo o gruppo di atomi più ingombrante. Da ciò deriva una regola abbastanza generale per cui un sostituente sull'anello cicloesanico occupa di preferenza una delle posizioni equatoriali. Un alchilcicloesano, per esempio, esiste come una miscela in equilibrio delle due conformazioni a sedia (15a) e (15b), la cui composizione è determinata dalle dimensioni del gruppo R; nel derivato metilico la forma equatoriale è presente per circa il 90%, in quello terz-butilico per il 99,99%, cioè in pratica
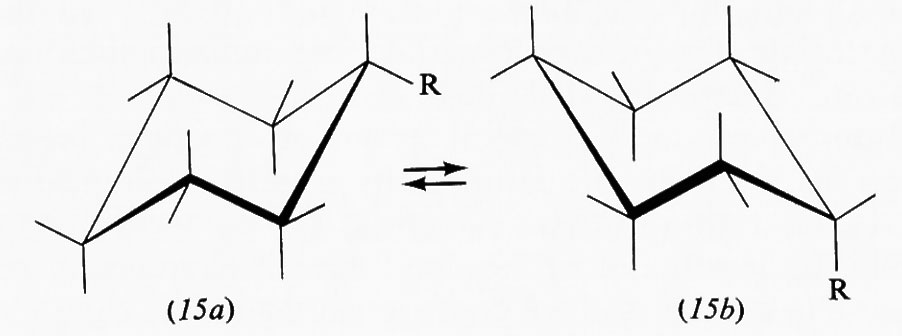
esiste all'equilibrio un solo conformero, e si può quindi considerare la molecola come conformazionalmente rigida.
Nel caso di derivati cicloesilici conformazionalmente non rigidi i due conformeri a sedia sono facilmente rivelabili con metodi fisici, ma non sono separabili, almeno a temperatura ambiente, in quanto la barriera all'interconversione è dell'ordine di una diecina di kcal/mole, insufficiente per impedire una rapida equilibrazione, ma assai più elevata di quella che esiste normalmente in molecole non cicliche. Difatti il passaggio da una conformazione a sedia all'altra comporta quali stadi intermedi forme, come quella completamente planare o più probabilmente altre in cui quattro o cinque dei carboni del ciclo sono coplanan, con energia potenziale assai superiore a quella delle conformazioni a sedia.
È da tener presente che anche per i composti aliciclici, come per quelli aciclici, l'interpretazione degli equilibri conformazionali sulla sola base di effetti sterici repulsivi può condurre a previsioni del tutto errate. Così, ad esempio, nei monoalogenocicloesani la preferenza per la conformazione con il sostituente equatoriale è assai più piccola che nel caso del metilcicloesano e circa uguale (intorno al 65%) per il fluoro-, il cloro-, il bromo- e lo iododerivato, benchè il metile abbia dimensioni vicine a quelle del bromo e assai minori di quelle dello iodio. Inoltre il trans-1, 2-dibromoci-cloesano preferisce la conformazione diassiale (16) e dati recenti di F.A.L. Anet mostrano che per l'acetato di cicloesilmercurio (17), in cui le dimensioni del sostituente sono particolarmente rilevanti, all'equilibrio il rapporto fra conformeri assiale ed equatoriale è di 60 : 40. Questi dati, incompatibili con un'interpretazione che tenga conto solo delle dimensioni del sostituente, possono essere spiegati in
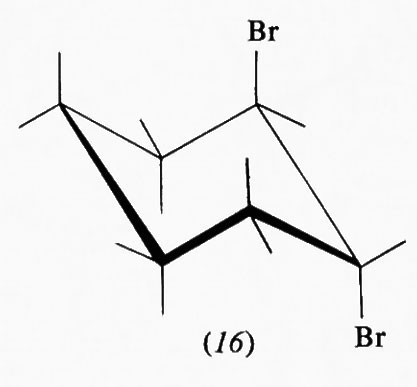

parte col fatto che con l'aumento del raggio atomico dell'elemento aumenta anche la lunghezza del legame e quindi la distanza fra il sostituente e i due atomi di idrogeno assiali a esso paralleli (sin-assiali), ciò che può portare a una diminuzione delle interazioni repulsive. Inoltre, nel caso di sostituenti elettronegativi, come gli alogeni, la polarizzazione del legame C−X provoca una maggiore densità elettronica sull'atomo X, che può perciò esercitare una certa attrazione sul debole dipolo C−H, di cui l'idrogeno è l'estremità positiva. Infine, nel caso del dibromocicloesano, una repulsione fra i dipoli C−Br può favorire la conformazione diassiale in cui gli atomi di alogeno sono più lontani. Peraltro anche queste considerazioni piuttosto empiriche non sono chiaramente sufficienti a dare un'interpretazione quantitativa della situazione, ed è in atto una revisione di molti dei concetti tradizionali su base più strettamente teorica, che sta mostrando quanto siano inadeguati i modelli finora adottati. È stato addirittura proposto che la preferenza di sostituenti alchilici per la disposizione equatoriale non sia dovuta solo a effetti sterici repulsivi nel conformero assiale ma, almeno in parte, anche a interazioni attrattive in quello equatoriale, fra l'idrogeno assiale, geminale al sostituente, e atomi di carbonio dell'anello.
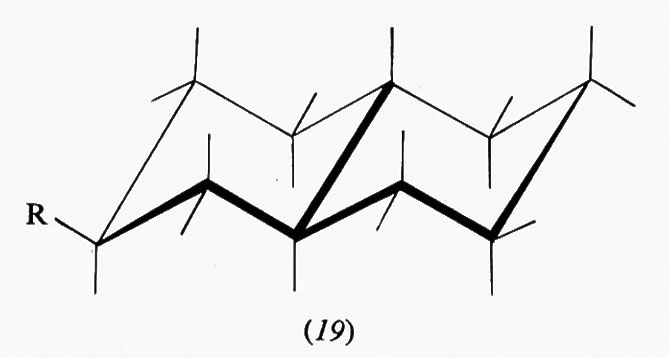
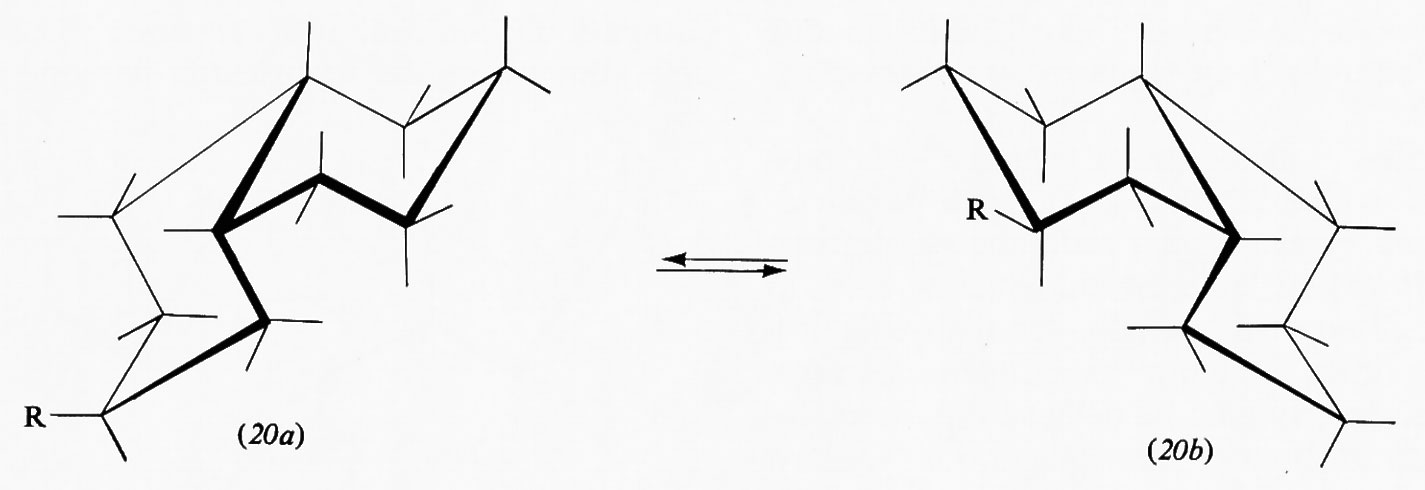
Interessanti sono i sistemi in cui più anelli cicloesanici sono fusi fra loro, in quanto molti prodotti biologicamente attivi rientrano in questa categoria. Del composto con due anelli, la decalina, esistono due forme diastereoisomere, la trans e la cis. La prima è conformazionalmente rigida, cioè può esistere in una sola conformazione, in quanto il secondo anello può legarsi al primo solo utilizzando due legami equatoriali di questo; l'inversione conformazionale non può avvenire poiché richiederebbe il passaggio a una forma in cui il secondo anello sarebbe fuso al primo attraverso due dei suoi legami assiali vicinali diretti in senso opposto l'uno rispetto all'altro, ciò che comporterebbe un'enorme tensione interna. Ne consegue che su uno qualunque degli atomi di carbonio della trans-decalina un sostituente R può trovarsi in posizione assiale (18) o equatoriale (19). Le due forme sono diastereoisomere e non interconvertibili attraverso processi conformazionali. Diverso è il caso della cis-decalina, in cui la fusione dei due anelli cicloesanici interessa un legame assiale e uno equatoriale e l'inversione conformazionale contemporanea dei due anelli è possibile, dato che anche nella forma invertita sono interessati alla fusione anulare due legami dello stesso tipo. I derivati della cis-decalina possono quindi presentare un equilibrio conformazionale in cui un sostituente R può passare dalla posizione assiale (20a) a quella equatoriale (20b) e viceversa.

Tipici della classe dei composti policiclici sono gli steroidi, di cui alcuni, quali ormoni, provitamine, tensioattivi, ecc., sono di grande importanza biologica. Una struttura fondamentale di questa serie è quella del colestano, in cui sono presenti tre anelli cicloesanici nella forma a sedia e uno ciclopentanico nella forma a busta. Di esso esistono in natura derivati di due forme diastereoisomere, differenziate dal fatto di avere i primi due anelli fusi in trans, 5α-colestano (21), o in cis, 5β-colestano o coprostano (22).
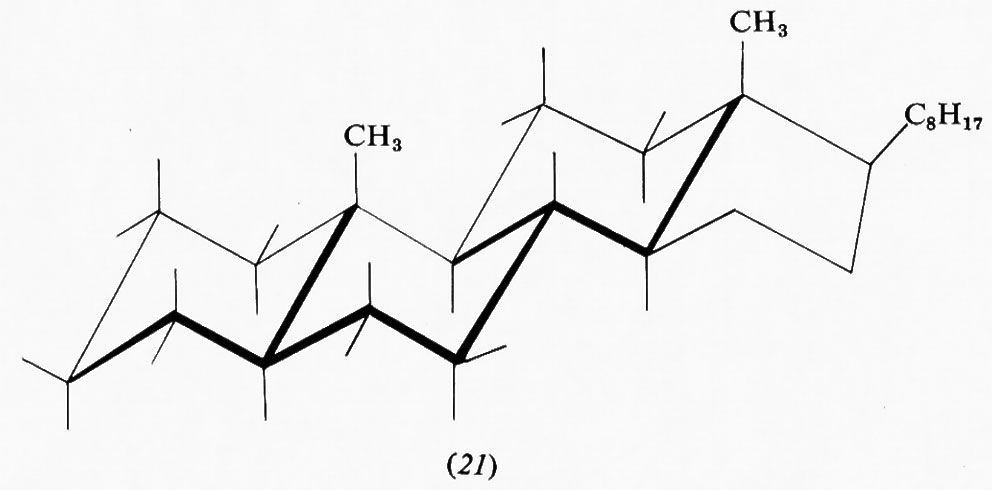

La regola che derivati del cicloesano mono- o policiclici si trovino esclusivamente nelle conformazioni a sedia presenta qualche eccezione per casi in cui interazioni repulsive particolarmente rilevanti fra sostituenti destabilizzino tale conformazione sufficientemente da rendere competitiva quella flessibile. Ad esempio, nella forma a sedia del trans- 1,3-diterz-butilcicloesano (23a) uno dei voluminosi sostituenti dovrebbe stare necessariamente in posizione assiale, ciò che comporterebbe una destabilizzazione di almeno 6 kcal/mole. La molecola assume quindi una conformazione twist (23b) in cui le interazioni repulsive fra gruppi terz-butilici e idrogeni sono minori.
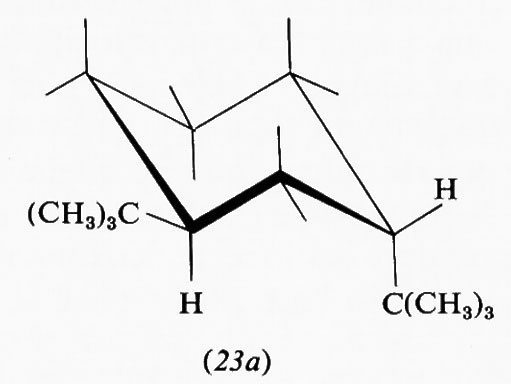

Un altro caso in cui l'anello cicloesanico si trova in una conformazione non a sedia è quello del biciclo[2,2,1]eptano (24), in cui la presenza di un gruppo metilenico che fa da ponte fra le posizioni 1 e 4 dell'anello a sei termini costringe questo ad assumere una conformazione a barca. Tale sistema biciclico si trova in vari composti naturali, ad esempio nella canfora.
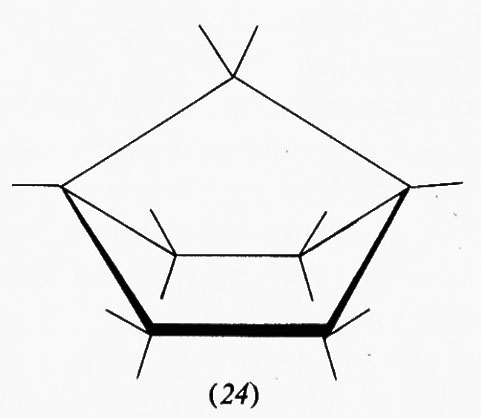
Anche per anelli a sette o più termini esistono sempre più forme non planari prive di tensione angolare, che però per quelli a 7-11 termini presentano una sensibile tensione interna dovuta soprattutto a effetti torsionali e di repulsione fra atomi di idrogeno che vengono a trovarsi assai vicini nello spazio. È quindi particolarmente difficile ottenere anelli di questo tipo per ciclizzazione di precursori non ciclici. Solo per anelli più grandi (12 o più termini) questi effetti repulsivi si attenuano fino a scomparire del tutto. Riportiamo come esempio la conformazione più stabile del ciclodecano (25).
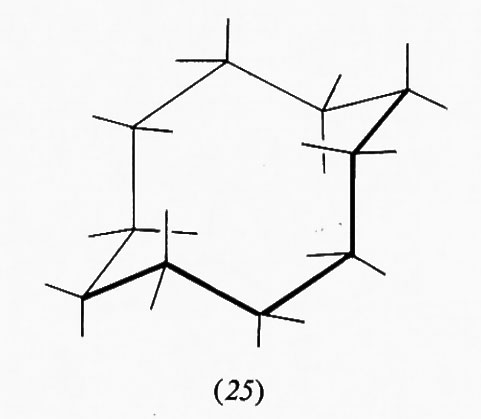
Le considerazioni sopra fatte sulle conformazioni dei composti aliciclici valgono in buona parte anche per gli analoghi composti eterociclici, poiché la sostituzione di uno o più atomi di ossigeno, zolfo, azoto, ecc. a gruppi metilenici modifica solo di poco la geometria dell'anello. Di particolare interesse biologico è la struttura degli zuccheri, che si trovano in soluzione come miscele in equilibrio di più forme emiacetaliche cicliche con anelli a cinque (furanosi) o sei termini (piranosi). La composizione di tali miscele dipende sia dalla configurazione relativa dei vari carboni chirali, sia dalla conformazione più stabile dei vari composti in equilibrio. Cosi il glucosio esiste per più del 99% quale equilibrio delle due forme piranosiche α (26) e β (27) in rapporto di circa 40 : 60. Quest'alta preferenza per le forme con anello a sei termini è determinata dal fatto che esse possono assumere conformazioni in cui 5 (forma β) o 4 sostituenti (forma α) si trovano in posizione equatoriale; la forma β è più stabile della α a causa del maggior numero di sostituenti equatoriali. Il maggiore eclissamento fra sostituenti presente nelle forme furanosiche del glucosio le rende non competitive rispetto a quelle piranosiche. Diversa è la situazione per altri aldoesosi. Per esempio, nell'idosio è presente all'equilibrio un 25% di forme furanosiche (28c) poiché in quelle piranosiche almeno tre degli idrossili (28a) o il più ingombrante gruppo CH2OH (28b) devono essere in posizione assiale.

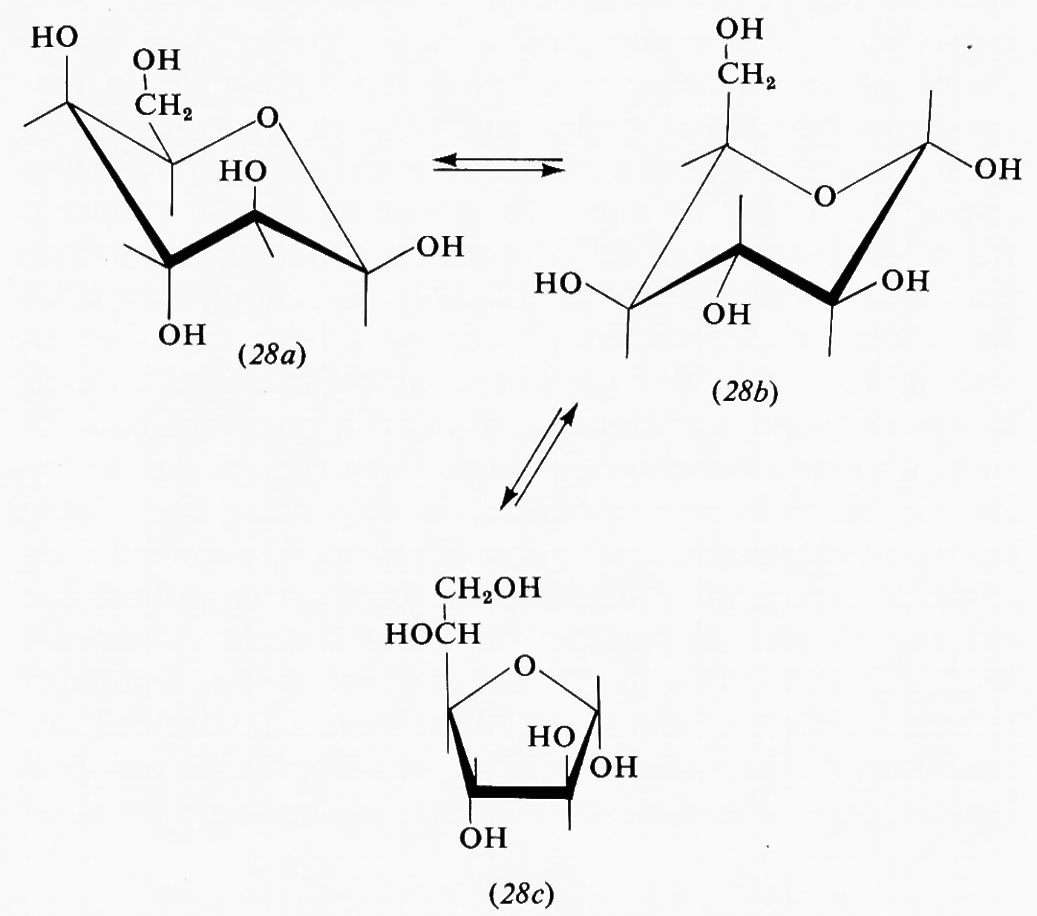
5. Metodi per lo studio quantitativo degli equilibri conformazionali.
L'analisi conformazionale quantitativa si propone da un lato di accertare l'energia e quindi la stabilità relativa delle varie conformazioni di una molecola, dall'altro di valutare le barriere all'interconversione di tali conformazioni. L'approccio ideale sarebbe quello del calcolo teorico di questi dati energetici, ma purtroppo lo stato attuale della chimica teorica, nonostante gli enormi progressi compiuti nell'ultimo ventennio e la disponibilità di grandi elaboratori elettronici, è ancora ben lontano dalla risoluzione completa del problema per molecole che non siano delle più semplici. Un calcolo ab initio, anche per una molecola molto semplice come quella dell'etano, comporta un'analisi di tutte le possibili interazioni fra 8 nuclei e 18 elettroni, considerando non solo i livelli occupati dagli elettroni nello stato fondamentale, ma anche quelli assai più elevati, almeno fino all'11s e al 7p per il carbonio e al 6s per l'idrogeno, il che rende il calcolo praticamente proibitivo. Si usano perciò di solito metodi di calcolo semiempirici (CNDO, INDO, MINDO, ecc.) che trascurano gli elettroni dei gusci interni e introducono varie altre semplificazioni. Divengono quindi accessibili anche molecole assai complesse, ina i risultati che si ottengono vanno presi con molta cautela, in quanto anche una buona corrispondenza col dato sperimentale potrebbe essere puramente casuale. Per queste ragioni buona parte dei dati disponibili sugli equilibri conformazionali sono basati su metodi sperimentali.
Metodi calorimetrici possono fornire dati piuttosto accurati sulle barriere rotazionali in molecole semplici, in base a misure di entalpia e di entropia. Se il valore dell'entropia così ottenuto coincide con quello dedotto con tecniche spettroscopiche o da calcoli di meccanica statistica, si può concludere che la molecola esiste essenzialmente in una sola conformazione; se invece l'entropia calorimetrica è inferiore, cio e indizio dell'esistenza di più conformeri separati da una barriera di energia, di cui è possibile calcolare l'entità appunto in base a questa differenza. Basandosi su tale metodo già nel 1935 E. Teller e B. Topley proposero un valore di circa 3 kcal/mole per la barriera dell'etano, valore rimarchevolmente accurato anche alla luce dei dati più recenti.

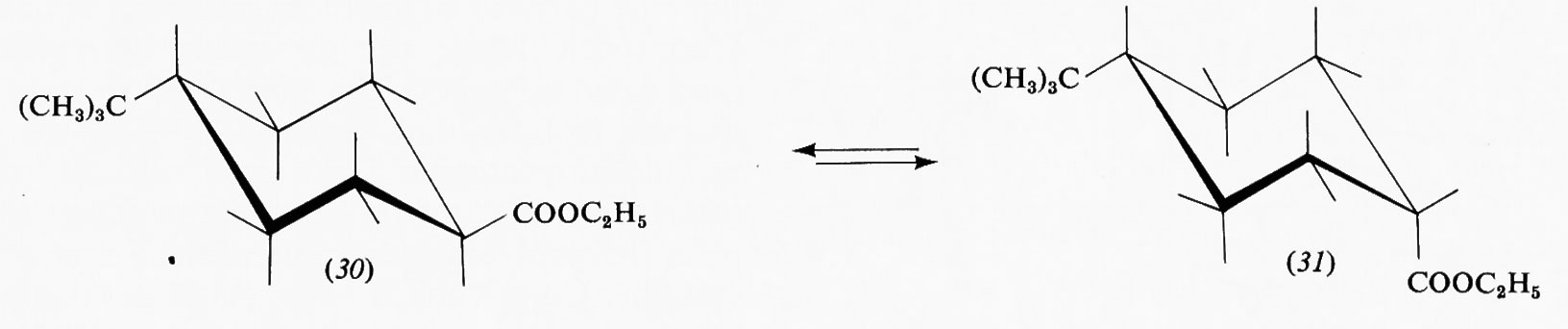
Per molecole complesse è più facile ricorrere a metodi di analisi più diretti, che diano la composizione della miscela di conformeri all'equilibrio, per esempio con le tecniche spettroscopiche più sotto discusse. Dato il valore di solito basso delle barriere energetiche fra conformeri, che hanno perciò vita media breve, non ne è di solito possibile un'analisi chimica o cromatografica diretta. Il problema può però spesso essere risolto ricorrendo a modelli conformazionalmente rigidi. Per esempio, volendo conoscere il rapporto fra le concentrazioni dei conformeri equatoriale e assiale del cicloesancarbossilato di etile (29), si prendono come loro modelli rispettivamente i corrispondenti trans- e cis-terz-butilderivati (30) o (31), in cui la presenza del voluminoso sostituente terz-butilico, fisso nella posizione equatoriale, impone all'anello cicloesanico una rigidità conformazionale praticamente completa. È da notare che (30) e (31) sono diastereoisomeri, non conformeri, e come tali facilmente separabili e analizzabili. La loro equilibrazione avviene a opera di basi per dare una miscela contenente l85% di (30) e il 15% di (31), ciò che corrisponde a una differenza di energia libera di 1,2 kcal/mole a favore dell'isomero con il sostituente COOC2H5 equatoriale. Si può pertanto ritenere che la stessa differenza esista anche fra i due conformeri del cicloesancarbossilato di etile e che quindi anch'essi siano presenti all'equilibrio in un rapporto di circa 85 : 15; ciò è stato confermato mediante analisi con la risonanza magnetica nucleare. Una variante del suddetto metodo termodinamico è utile quando sia difficile equilibrare i composti rigidi presi come modello; ci si può basare cioè su misure cinetiche, purché si trovi una reazione in cui il gruppo funzionale reagisca con velocità differente a seconda che esso sia assiale o equatoriale. Una volta determinate le costanti di velocità (ke e ka) per i due isomeri rigidi con sostituente equatoriale e assiale e quella (k) del composto non rigido di cui si vuole conoscere l'equilibrio conformazionale, si possono dedurre le frazioni molari dei due conformeri (Ne e Na) in base al semplice sistema di equazioni:
k=Neke+Naka
Ne+Na=1.
Naturalmente, per la validità di questi due metodi occorre ammettere che all'equilibrio siano presenti solo due conformeri e inoltre che il gruppo terz-butilico non abbia altro effetto che quello di tenere la molecola in una conformazione rigida. In effetti, studi più recenti hanno indicato che la seconda ipotesi non è rigorosamente esatta, in quanto il sostituente terz-butilico provoca una certa deformazione nella geometria dell'anello cicloesanico per effetti sterici e torsionali. Ciò può modificare apprezzabilmente sia la posizione dell'equilibrio, sia l'energia di attivazione e quindi la velocità di reazione nei confronti di quella relativa al composto di cui si vuole determinare l'equilibrio conformazionale. Perciò i dati ottenuti con questi due metodi, pur utili da un punto di vista qualitativo, non possono essere considerati rigorosamente quantitativi.
Di particolare importanza per lo studio degli equilibri conformazionali sono le tecniche spettroscopiche. Qualunque tipo di spettrometria di assorbimento potrebbe dare informazioni sulla struttura e quindi anche sulle conformazioni di una molecola, purché la zona spettrale esaminata sia sufficientemente ampia da fornire un quadro completo delle interazioni fra un dato tipo di radiazione elettromagnetica e i nuclei o gli elettroni della molecola e purché la teoria sia sufficientemente raffinata da permettere di tradurre i dati spettrali in dati strutturali. Non sempre queste condizioni si verificano.
I metodi più diretti per ottenere dati strutturali sono quelli diffrattometrici, che la disponibilità di diffrattometri automatici e di calcolatori rapidi ha reso facilmente accessibili, almeno per quanto riguarda la diffrazione dei raggi X, la quale dà un'immagine molto precisa della posizione dei nuclei e quindi della geometria molecolare. Una limitazione di questa tecnica, come di quella più recente della diffrazione neutronica, è che esse sono applicabili solo ai solidi cristallini e possono quindi indicare soltanto un'unica conformazione di un dato composto, quella cioè che esso assume nel reticolo del cristallo, ma non la situazione conformazionale allo stato liquido, gassoso o in soluzione. Invece la diffrazione elettronica è applicabile a molecole allo stato di gas, cioè nelle condizioni ideali per un equilibrio conformazionale, ma le informazioni da essa fornite sono assai più difficili da interpretare, almeno per molecole complesse. Questa tecnica soffre quindi per ora di notevoli limitazioni, anche se ha fornito utili e precise indicazioni in casi particolari, come, per esempio, quelle sull'esatta geometria del cicloesano.
Gli spettri elettronici di assorbimento (U. V. -visibile) non permettono di solito un'analisi strutturale completa, soprattutto per la grande difficoltà tecnica di seguirli fino al lontano ultravioletto (al di sotto di 150 nm), dove assorbono i legami singoli; viene quindi normalmente presa in esame solo la zona al di sopra di 180 nm, che fornisce informazioni su sistemi insaturi, di limitata utilità per l'analisi conformazionale. In casi particolari, specialmente quelli riguardanti sistemi coniugati, in cui si alternano legami singoli a legami multipli, gli spettri elettronici possono però dare utili indicazioni sulla conformazione, poiché la coniugazione comporta spostamento dei massimi di assorbimento verso lunghezze d'onda maggiori (effetto batocromico) e aumenta di solito anche l'intensità dell'assorbimento (effetto ipercromico). Questi effetti richiedono che il sistema coniugato sia coplanare e sono tanto minori quanto più esso devia dalla coplanarità. Per esempio, nei due stereoisomeri dello stilbene si può constatare che il trans (32) è completamente planare, mentre nel cis (33) la presenza di un massimo di assorbimento a minore lunghezza d'onda e di minore intensità mostra chiaramente che i due anelli benzenici si trovano su piani differenti.
Più facilmente accessibile dal punto di vista tecnico è invece l'intera gamma degli spettri infrarossi e Raman, che forniscono gran copia di informazioni strutturali su ogni molecola. Ma è proprio l'eccessivo numero d'informazioni che costituisce la limitazione maggiore di queste tecniche, rendendo oltremodo difficile per molecole complesse la traduzione di dati spettroscopici in dati strutturali. Per composti semplici, come l'1,2-dicloroetano, è stato comunque possibile fare un'analisi completa degli spettri, mettendo in evidenza e attribuendo tutte le bande corrispondenti sia alla forma gauche che a quella anti, ambedue presenti nel composto allo stato liquido o di soluzione. Per molecole più complesse è comunque spesso possibile dedurre informazioni almeno parziali sulla conformazione dagli spettri infrarossi e Raman. Per esempio è regola abbastanza generale che la frequenza della banda di stretching del legame C−X di un cicloesano sostituito sia maggiore quando X è equatoriale che quando esso è assiale. Inoltre è facile dedurre dall'assorbimento infrarosso se un idrossile forma un legame a idrogeno con un altro gruppo funzionale della stessa molecola, attraverso la posizione e la forma della banda di stretching O−H; si può così studiare l'equilibrio conformazionale in composti come il trans-cicloesan-1,2-diolo, basandosi sul fatto che solo nella forma diequatoriale (34a), e non in quella diassiale (34b), i due idrossili sono sufficientemente vicini da poter formare il legame a idrogeno intramolecolare.
Anche gli spettri di microonde possono in teoria fornire dati utili sugli equilibri conformazionali, ma la loro importanza è limitata a molecole semplici e facilmente trasformabili in gas.
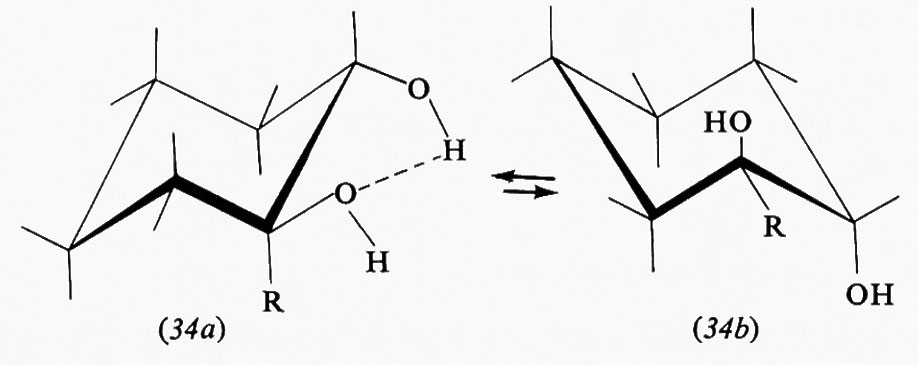
La tecnica spettroscopica di gran lunga più usata per lo studio conformazionale è senz'altro quella della risonanza magnetica nucleare (RMN), che fornisce informazioni di solito facilmente interpretabili su tutti i nuclei con spin differente da zero. Nella gran maggioranza dei casi viene limitata all'analisi dei nuclei d'idrogeno, in quanto essa è tecnicamente più semplice e la definizione della posizione reciproca degli atomi d'idrogeno in una molecola organica è di solito sufficiente per determinarne la conformazione.
Alcune informazioni possono essere dedotte dal valore dello spostamento chimico (chemical shift) dei singoli atomi d'idrogeno, cioè dalla differenza tra la frequenza di risonanza di questi atomi e quella degli atomi d'idrogeno di una sostanza di riferimento, che di solito è il tetrametilsilano. Per esempio, nel caso dei derivati del cicloesano questa differenza è maggiore per un atomo d'idrogeno nella posizione equatoriale che per uno in quella assiale, ciò che permette di distinguere i due conformeri di un cicloesano monosostituito in base al fatto che quello con X equatoriale (35a) ha l'idrogeno geminale a X assiale e quindi con spostastamento chimico minore rispetto all'analogo idrogeno della forma con X assiale (35b).
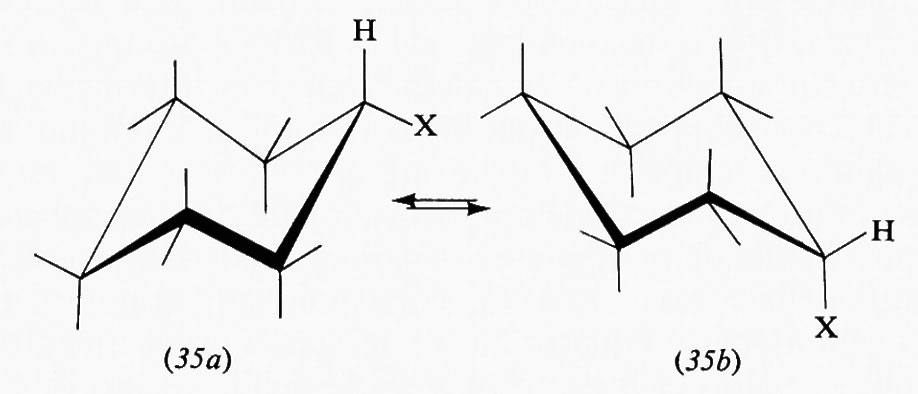
Assai più utili ai fini dell'assegnazione delle conformazioni sono però le costanti di accoppiamento (J) fra i vari atomi d'idrogeno della molecola, cioè le interazioni fra gli spins dei singoli nuclei, che producono una caratteristica molteplicità nei corrispondenti segnali di risonanza, strettamente dipendente, fra l'altro, dalla distanza fra i nuclei accoppiati e dall'orientamento dei legami. Nel caso di atomi d'idrogeno legati a carbonî vicinali c'è una relazione, dovuta a Karplus, fra la J (espressa come differenza di frequenza in Hz fra i componenti del multipletto) e l'angolo torsionale (τ), che nella forma più semplice è: J=a cos2 τ, ove a è una costante caratteristica del sistema in esame. Per esempio, in un cicloesano 1 ,2-disostituito nella forma a sedia è facile distinguere la disposizione diassiale (36) da quella diequatoriale (37), in quanto nella prima i due idrogeni geminali sono in posizione gauche (τ=60°), il che corrisponde a una J di 2-3 Hz, nella seconda in quella anti (τ=180°), che comporta una J di 10-12 Hz.
Mentre la tecnica suddetta si presta molto bene allo studio di composti conformazionalmente rigidi o comunque equilibrabili solo lentamente, che danno gruppi di segnali separati e riconoscibili per i differenti conformeri o diastereoisomeri, essa presenta alcune limitazioni per equilibri fra conformeri rapidamente interconvertibili, per cui si vedono solo segnali il cui spostamento chimico corrisponde alla media ponderale di quelli relativi ai due conformeri.
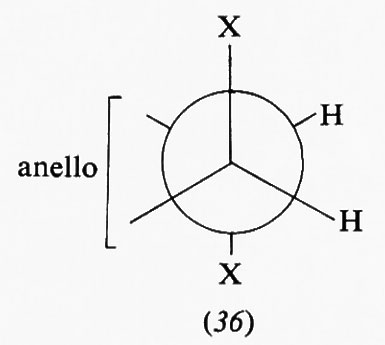
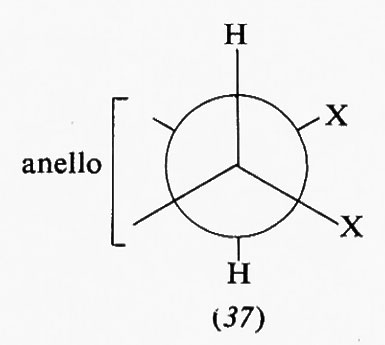
Ciò è dovuto al fatto che con una data tecnica spettroscopica varie specie in equilibrio possono essere osservate come entità separate solo quando il tempo di vita media di ogni specie sia sufficientemente superiore all'inverso della frequenza del tipo di radiazione usata per la misura. Ciò si verifica nel caso della spettroscopia elettronica e infrarossa, ove le frequenze sono molto elevate, ma di solito non per quella di RMN, dove esse sono assai minori. Quest'ultima è quindi una tecnica troppo lenta per fissare come immagini separate due forme che si trasformano rapidamente l'una nell'altra; per usare un efficace paragone proposto da J. D. Roberts, è come se volessimo fotografare una ruota che gira rapidamente usando un tempo di esposizione troppo lungo: anziché vedere nella fotografia i singoli raggi della ruota si osserverà un cerchio continuo che rappresenta la posizione media dei raggi stessi. Essi diverranno invece visibili singolarmente solo diminuendo la velocità di rotazione o il tempo di esposizione.
Per esempio, il cicloesano dà a temperatura ambiente uno spettro di RMN costituito da una singola linea, nonostante che nella conformazione a sedia esistano due tipi di atomi d'idrogeno, quelli assiali e quelli equatoriali, che risuonano a frequenze differenti. Il fatto è che, a causa della rapida inversione anulare, un idrogeno che a un dato istante è in posizione assiale subito dopo passerà a quella equatoriale e viceversa. Con la tecnica lenta della RMN potremo quindi vedere un solo tipo di segnale a frequenza intermedia fra quelle corrispondenti ai due tipi di atomi. Comunque, abbassando la temperatura del campione si arriva a un punto in cui l'interconversione conformazionale diviene sufficientemente lenta da permettere l'osservazione di due segnali separati della stessa intensità, corrispondenti appunto agli idrogeni assiali o equatoriali. Se lo stesso procedimento si applica al clorocicloesano, il segnale dell'idrogeno in α al cloro, unico a temperatura ambiente, si sdoppia a bassa temperatura, dandone due, le cui intensità, differenti, sono proporzionali alle concentrazioni dei conformeri assiale ed equatoriale. È questo uno dei metodi più precisi per la determinazione dell'equilibrio conformazionale, che ha il notevole vantaggio di fornire anche dati sull'energia di attivazione, cioè sull'entità della barriera all'interconversione, in base alla temperatura a cui si verifica lo sdoppiamento dei segnali.
Un esempio dell'applicazione di questi concetti a sistemi non ciclici si ha nello studio della rotazione intorno al legame C−N delle ammidi, particolarmente importante per la struttura delle proteine. In una semplice ammide, come la dimetilformammide, i due metili danno, anche a temperatura ambiente, due segnali distinti nello spettro di RMN, ciò che indica che la rotazione intorno al legame C−N è sufficientemente lenta da non permettere ai due metili, magneticamente non equivalenti in quanto situati in posizione differente rispetto al gruppo C=O, di apparire equivalenti. L'equivalenza per mediazione dei segnali si verifica infatti solo al di sopra dei 100 °C, ciò che comporta una barriera rotazionale di più di 10 kcal/mole. Questa resistenza alla rotazione, del tutto insolita per legami singoli, è dovuta a un parziale carattere di doppio legame per il C−N ammidico, dovuto al contributo della forma di risonanza (38b).
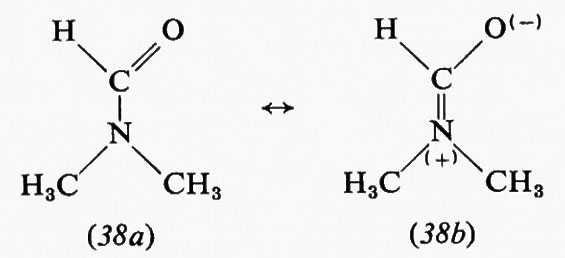
Informazioni utili sugli equilibri conformazionali di molecole contenenti legami polarizzati si possono dedurre anche dai momenti dipolari. Per esempio, nell'1,2-dicloroetano i due dipoli uguali C−Cl si annullano nella conformazione anti, in quanto diretti in senso opposto, e quindi il momento complessivo dev'essere zero. Nella conformazione gauche si può invece calcolare un momento di 3,2 debye. Il valore sperimentale varia a seconda del solvente e ha valori intermedi fra O e 3,2 debye; da esso si può facilmente calcolare il rapporto fra le concentrazioni dei due conformeri, che sono presenti in quantità circa equivalenti nel liquido puro, mentre in soluzione e allo stato gassoso predomina la conformazione anti.
Numerose altre tecniche chimico-fisiche, in pratica tutte quelle in cui si misura una quantità strettamente dipendente dalla struttura molecolare, possono fornire informazioni sugli equilibri conformazionali. Fra queste, la dispersione rotatoria (variazione del potere rotatorio con la lunghezza d'onda della luce), l'effetto Kerr (doppia rifrazione causata da una differenza di potenziale), il comportamento alla riduzione polarografica, l'assorbimento del suono, la basicità e l'acidità, ecc.
6. Conformazione e reattività.
Una delle ragioni per cui è necessaria un'approfondita conoscenza degli aspetti conformazionali della struttura molecolare è la stretta connessione fra conformazione e reattività. Di solito il raggiungimento dello stato di transizione, con il conseguente compimento della reazione chimica, richiede una ben definita disposizione geometrica degli atomi e gruppi che partecipano alla reazione stessa; la reattività sarà quindi fortemente condizionata dalla facilità con cui le molecole dei reagenti possono disporsi nella conformazione più favorevole per realizzare la geometria migliore dello stato di transizione.
Per studiare effetti di questo tipo si prestano particolarmente composti conformazionalmente rigidi, come i derivati del terz-butilcicloesano, della trans-decalina e degli steroidi, in cui, come abbiamo accennato, ogni sostituente si trova fissato in posizione assiale o equatoriale, senza che possa verificarsi l'inversione conformazionale. Per esempio, la reazione di esterificazione di un alcool o quella di idrolisi di un suo estere sono di norma più veloci quando il sostituente si trova in posizione equatoriale, in quanto l'avvicinamento del reagente è meno impedito dall'ingombro causato dagli atomi vicini: per il 4-terz-butilcicloesanolo la forma trans (OH equatoriale) si esterifica con anidride acetica quattro volte più velocemente che la forma cis (OH assiale).
In altri tipi di reazioni la velocità è maggiore quando il sostituente è assiale che quando esso è equatoriale: per esempio nell'ossidazione di cicloesanoli a cicloesanoni; così l'ossidazione cromica del cis-4-terz-butilcicloesanolo è 3,2 volte più veloce di quella dell'isomero trans, quella del cis-2-terz-butilcicloesanolo è addirittura 60 volte più veloce; la velocità è cioè tanto maggiore quanto più grande è l'ingombro sterico. Ciò può spiegarsi col fatto che il passaggio dall'alcool al chetone, comportando la trasformazione di un carbonio tetraedrico in uno trigonale, diminuisce la tensione sterica nel composto e quindi il guadagno energetico sarà tanto maggiore, quanto più l'idrossile è impedito.
Le reazioni di eliminazione richiedono di solito che la molecola si disponga nella conformazione (39) in cui i gruppi da eliminare siano in anti; tutti gli effetti che ostacolano il raggiungimento di tale conformazione riducono la velocità di queste reazioni o possono impedirle del tutto.
Così la velocità di eliminazione di HBr dal 4-terz-butil-l-bromoci- cloesano è 65 volte maggiore per l'isomero cis (bromo assiale) che per quello trans (bromo equatoriale), in quanto solo quando l'alogeno è assiale un atomo d'idrogeno può trovarsi in anti a esso. Analogamente la formazione dell'epossido (41) a opera di basi è molto più veloce con il composto (40) che con quello (42), mentre l'isomero cis (43) non fornisce affatto l'epossido, ma solo il chetone (44). Mentre in (40) il bromo e l'idrossile sono già disposti in maniera ideale per l'eliminazione di HBr, in (42) la disposizione anti può realizzarsi solo con un inversione conformazionale, estremamente sfavorevole poiché porterebbe i gruppi terz-butile e OH nelle posizioni assiali, o meglio con il passaggio a una forma twist (42a) anch'essa energeticamente sfavorevole. Nel caso del composto cis (43) l'anello a sei termini impedisce del tutto il passaggio a una conformazione in cui OH e Br siano anti e la reazione deve seguire necessariamente un decorso differente, cioè un'eliminazione di HBr in cui l'idrogeno viene fornito dal C−H, favorevolmente situato, anziché dall'O−H.
7. Prospettive di sviluppo.
Lo studio dei vari aspetti conformazionali della struttura molecolare ha avuto nell'ultimo ventennio uno sviluppo eccezionalmente vigoroso. Migliaia di pubblicazioni hanno approfondito l'argomento sia dal punto di vista teorico, sia da quello sperimentale, e fornito un quadro piuttosto completo di molti dei fattori che condizionano gli equilibri conformazionali. Ci possiamo quindi chiedere se questo sia un campo di ricerca ormai sufficientemente chiarito, e se sia ancora giustificato continuare a dedicare a esso una così rilevante quantità di sforzi intellettuali e di risorse finanziarie. E questo un problema che i ricercatori dovrebbero porsi per ogni nuovo campo di studio, quando, superato il periodo più proficuo e più originale della scoperta, si passa a quello del consolidamento e approfondimento delle nuove acquisizioni, periodo altrettanto importante, ma che rischia poi di trasformarsi in una routine non più produttiva, per la pur comprensibile riluttanza del ricercatore ad abbandonare un campo che gli ha dato tante soddisfazioni.
L'opinione di chi scrive è che per l'analisi conformazionale questo secondo periodo non sia ancora giunto, anche se è indubbiamente finito quello delle scoperte più importanti. Occorre perciò fare delle scelte, concentrando gli sforzi su determinate aree ancora poco chiare dal punto di vista teorico, o suscettibili di particolari sviluppi di carattere applicativo. La disponibilità, da un lato, di apparecchiature e di elaboratori elettronici sempre più perfezionati, che permettono con un minimo sforzo di ottenere una messe copiosa di dati, e quella, dall'altro, di un numero praticamente illimitato di molecole organiche su cui eseguire misure e calcoli possono favorire un tipo di ricerca basata sull'accumulo di sempre più numerosi dati strutturali, molti dei quali ben scarso contributo possono apportare al vero progresso scientifico. Non è quindi in questa direzione che dovrebbero essere incoraggiate ricerche di analisi conformazionale.
Vale invece certamente ancora la pena di approfondire e migliorare i metodi di calcolo, in modo da renderli capaci di prevedere in maniera accurata gli equilibri conformazionali e le barriere alla rotazione. La progressiva presa di coscienza delle gravi limitazioni dell'approccio empirico ha reso evidente il ruolo indispensabile del chimico teorico nello studio e nell'interpretazione dei fenomeni conformazionali. Nonostante le grandi difficoltà, cui si è sopra accennato, a trattare molecole complesse con metodi di calcolo ab initio, è quindi indubbio che una collaborazione fra le profonde conoscenze matematiche dei chimici teorici e chimici fisici e la maggiore sensibilità per il dato sperimentale dei chimici organici è altamente desiderabile e può essere veramente produttiva. Pur essendo ancora lontano il giorno in cui sarà possibile fare a meno dello sperimentatore e prevedere tutte le proprietà di una molecola a tavolino, col solo ausilio del terminale di un calcolatore, è innegabile che notevoli progressi in questo senso sono già stati fatti ed è facilmente prevedibile che di molto maggiori ne potranno seguire in un futuro non lontano.
Dal punto di vista sperimentale un settore ancora relativamente poco studiato è quello degli equilibri conformazionali in molecole non cicliche. Una parte nettamente preponderante dei dati di cui disponiamo si riferisce a un numero assai limitato di tipi di composti, e fra questi dominano di gran lunga i sistemi aliciclici ed eterociclici con anelli a sei termini, che hanno goduto di una particolare attenzione da parte dei ricercatori, in quanto particolarmente facili da studiare dal punto di vista conformazionale. Lo studio degli equilibri rotamerici in molecole non cicliche, oltre a fornire stimolanti difficoltà sperimentali, comunque in buona parte superabili con le tecniche attualmente disponibili, può portare a risultati di notevole utilità e interesse.
A parte gli aspetti strutturali delle singole conformazioni e delle loro interconversioni, è auspicabile un approfondimento degli studi sulle relazioni fra conformazione e reattività, in quanto proprio in questo campo possono prevedersi i risultati più rilevanti dal punto di vista applicativo. Per una completa interpretazione dei meccanismi di reazione e per la messa a punto di metodi di sintesi altamente specifici, è essenziale una perfetta conoscenza degli aspetti conformazionali dei reagenti, estesa anche alle conformazioni che essi assumono nello stato di transizione della reazione. Uno dei problemi più affascinanti, attuali e impegnativi che si propongono al chimico è quello di conoscere come si svolgono le sintesi e le trasformazioni metaboliche all'interno della cellula, di spiegare l'enorme efficienza della catalisi enzimatica, d'interpretare il meccanismo d'azione di vitamine, ormoni, farmaci, ecc. a livello dei rispettivi recettori biologici. Nonostante gli straordinari progressi compiuti dalla biologia molecolare, ben poco sappiamo ancora su questi argomenti, ma è comunque chiaro che la conformazione molecolare giuoca un ruolo fondamentale nei processi biologici: da un lato la struttura estremamente complessa delle macromolecole dell'enzima e del recettore deve assumere una sola, ben definita conformazione fra le innumerevoli possibili, dall'altro la piccola molecola del substrato può essere accolta dal sito attivo solo se risponde a rigide esigenze strutturali e conformazionali. Il verificarsi di ambedue queste condizioni è premessa essenziale perché la reazione biologica possa svolgersi con la selettività praticamente totale, che è essenziale per la conservazione e la riproduzione della specie. Fondamentali sono quindi un impegno profondo e una stretta collaborazione del chimico con il biologo per gettare luce sui processi fisiologici e per combattere quelli patologici mediante la messa a punto di farmaci altamente selettivi. In questo quadro, essenziale può essere il contributo del chimico che si dedica allo studio dei fattori conformazionali necessari per il favorevole svolgimento delle singole reazioni.
Una più profonda conoscenza dei fattori che condizionano l'alta specificità della catalisi enzimatica può d'altra parte essere di grande utilità per perfezionare la teoria e la pratica della sintesi di laboratorio e industriale, che, di fronte alla biosintesi, è di solito estremamente meno efficiente e specifica, in quanto la resa e la selettività di una reazione organica sono spesso considerate come molto soddisfacenti se si aggirano sull'80-90%. Questo è dovuto a un approccio ancora essenzialmente empirico alla catalisi e a una scarsa conoscenza dei relativi meccanismi. Solo un'adeguata valutazione e comprensione dei fattori che condizionano l'interazione fra reagenti e catalizzatore può permettere la previsione di condizioni di reazione tali da favorire in maniera netta il raggiungimento del particolare stato di transizione che dia il prodotto desiderato rispetto a quello di altri stati di transizione che porterebbero a prodotti differenti. E anche in questo campo è essenziale un'esatta conoscenza degli aspetti conformazionali.
Si può quindi concludere che lo studio conformazionale delle molecole è ancora ben lungi dall'aver esaurito i suoi problemi e dall'essere entrato nella fase di routine. I campi da esplorare e le mete da raggiungere sono ancora molteplici e vari, ed è quindi da ritenere che la ricerca in questo settore continuerà vigorosa e proficua e terrà impegnati molti studiosi almeno per la parte restante di questo secolo.
Bibliografia.
Barton, D. H. R., Cookson, R. C., The principles of conformational analysis, in ‟Quarterly reviews", 1956, X, pp. 44-82.
Binsch, G., The study of intramolecular rate processes by dynamic nuclear magnetic resonance, in Topics in stereochemistry (a cura di E. L. Eliel e N. L. Allinger), vol. III, New York 1968, pp. 98-192.
Chiordoglu, G., Conformational analysis, New York 1971.
Eliel, E. L., Allinger, N. L., Angyal, S. J., Morrison, G. A., Conformational analysis, New York 1965.
Epiotis, N. D., Attractive nonbonded interactions in organic molecules, in ‟Journal of the American Chemical Society", 1973, XCV, pp. 3087-3096.
Ermer, O., Lifson, S., On the release of strain in overcrowded substituted ethylenes, in ‟Tetrahedron", 1974, XXX, pp. 2425-2429.
Kellie, G. M., Riddel, F. G., Non-chair conformations of six-membered rings, in Topics in stereochemistry (a cura di E. L. Eliel e N. L. Allinger), vol. VIII, New York 1974, pp. 225-269.
Liberles, A., Greenberg, A., Eilers, J. E., Attractive steric effects, in ‟Journal of chemical education", 1973, L, pp. 676-678.
Lowe, J. P., The barrier to internal rotation in ethane, in ‟Science", 1973, CLXXIX, pp. 527-532.
Sandstrom, J., Dynamic nuclear magnetic resonance spectroscopy, in ‟Endeavour", 1954, XXXIII, pp. 111-118.
Thomas, W. A., Nuclear magnetic resonance spectroscopy in conformational analysis, in ‟Annual review of NMR spectroscopy", 1968, I, pp. 43-89; 1970, III, pp. 92-147.
Analisi conformazionale delle grandi molecole
SOMMARIO: 1. Introduzione. □ 2. Teoria della conformazione. □ 3. Blocchi costitutivi delle macromolecole: a) polipeptidi e proteine; b) blocchi costitutivi dei polisaccaridi; c) blocchi costitutivi degli acidi nucleici. □ 4. Macromolecole informazionali. □ 5. Molecole elicoidali: a) polipeptidi in conformazione elicoidale stabile; b) eliche multiple; c) polisaccaridi. □ 6. Molecole stocastiche. □ 7. Strutture tridimensionali: a) proteine; b) transfer RNA; c) strutture di ordine superiore. □ 8. Orientamenti attuali. □ Bibliografia.
1. Introduzione.
Il XX secolo è stato affascinante come altre epoche della storia della scienza. I concetti fondamentali delle scienze fisiche sono stati rielaborati e si e così giunti a un gran numero di nuove idee e di scoperte. Come è normale durante periodi di notevoli innovazioni, l'interesse è andato frequentemente al di là dei confini della scienza, sollecitando l'immaginazione popolare. La relatività, le sorprendenti predizioni della meccanica quantistica, la scissione degli atomi, il potenziale dell'energia nucleare hanno avuto tutti il loro spazio sulla scena contemporanea. Non ultimo fra tali argomenti di largo interesse è il campo della struttura delle molecole di grandissime dimensioni o macromolecole. I relativi principi strutturali da una parte trovano applicazione nell'industria delle materie plastiche e delle fibre, e dall'altra servono a spiegare il meccanismo dei processi vitali: non c'è dubbio, quindi, che essi abbiano una grandissima importanza per l'umanità, sia agli effetti pratici che scientifici. In conseguenza della notevole mole di lavoro sia sperimentale sia teorico, di grande eleganza e profondità, la concezione di tali molecole ha subito una completa rivoluzione.
Gli interrogativi fondamentali sul meccanismo dei processi vitali, ad es. l'azione degli enzimi e la genetica, sono racchiusi nelle proprietà strutturali delle macromolecole. Inoltre gran parte dei più interessanti lavori recenti nei campi della meccanica quantistica, della meccanica statistica e della spettroscopia sono stati ispirati dai particolari problemi delle grandi molecole che, per la loro difficoltà, hanno costretto i ricercatori a superare frontiere che erano state considerate soddisfacenti nel caso delle piccole molecole. La presente trattazione è concentrata su un aspetto ben definito di questo argomento, chiamato analisi conformazionale, che si occupa del modo in cui i fattori energetici e geometrici determinano le conformazioni stabili delle macromolecole.
Per lungo tempo si è ritenuto che le grandi molecole fossero costruite su principi diversi da quelli delle piccole molecole. L'idea generale era che le molecole fossero fondamentalmente piccoli aggregati di atomi. La scoperta di particelle di grandi dimensioni nelle proteine, nei polisaccaridi, nella gomma naturale e nei polimeri sintetici, ebbe scarsa influenza su questo punto di vista. Questi aggregati furono considerati come insiemi di piccole molecole tenute unite da modeste forze fisiche. Anche E. Fischer, che aveva sintetizzato diversi polipeptidi di considerevole lunghezza, suppose che trenta amminoacidi costituissero il limite massimo di lunghezza per una catena polipeptidica, immaginando evidentemente che il problema della sintesi fosse in natura altrettanto complicato che nel laboratorio del chimico organico. In conseguenza di ciò molti sistemi macromolecolari furono posti nella categoria dei colloidi. La chimica dei colloidi è un campo in cui i sistemi vengono trattati in base a grossolane proprietà fenomenologiche, cosicché il problema della struttura dettagliata fu perso di vista. Vi furono in realtà alcune determinazioni strutturali e furono avanzate delle proposte, ma queste si riferivano esclusivamente alle mitiche piccole molecole nello stato aggregato colloidale.
La cellulosa offre un esempio delle difficoltà che furono incontrate. L'esame di fibre di cellulosa mediante i raggi X rivelò l'esistenza di regioni cristalline e di regioni amorfe. Le regioni cristalline hanno dimensioni dell'ordine di centinaia di ångström; le corrispondenti celle elementari sono lunghe 10,3 Å. Fu molto difficile per i primi ricercatori comprendere che nessuna di queste dimensioni aveva relazione alcuna con la grandezza delle molecole di cellulosa, che sono lunghe parecchie migliaia di ångström. Questa proprietà di essere cristallini in alcune regioni, amorfi in altre, è ora nota come una proprietà comune degli alti polimeri allo stato solido.
Un'altra caratteristica delle macromolecole biologiche, che fu completamente oscurata dal concetto di colloide, è l'unicità delle strutture per quanto riguarda sia la grandezza sia la conformazione. Benché alcuni sperimentatori interpretassero la formazione di cristalli come dimostrazione di un'unica struttura, in generale tale opinione non fu in alcun modo accettata. Notevole luce venne fatta in questo campo dopo la prima guerra mondiale, con il lavoro di S. P. L. Sørensen e Th. Svedberg sulle proteine e quello di H. Staudinger sui polimeri sintetici. Svedberg riuscì a dimostrare che le proteine si muovevano nell'ultracentrifuga dando luogo a segnali netti, indicando con ciò un unico peso molecolare o almeno un intervallo molto ristretto. I polimeri sintetici, d'altra parte, davano luogo a curve di sedimentazione molto diffuse, indicando un ampio intervallo di pesi molecolari, in completo accordo con le moderne teorie sul meccanismo statistico della loro formazione. Parallelamente al lavoro di Svedberg sulle proteine, Staudinger dimostrò che i polimeri sintetici e naturali come la gomma erano vere e proprie grandi molecole tenute insieme da una rete continua di legami chimici covalenti. Non appena l'idea di molecole molto grandi ebbe superato i pregiudizi del tempo, il concetto di colloide scomparve e da allora non vi sono stati più seri dubbi sulla natura macromolecolare dei biopolimeri, benché il tema dei colbidi sia riapparso sporadicamente agli inizi degli anni trenta.
L'esistenza di polimeri ordinati in cui gli atomi hanno un definito ordine ripetitivo nello spazio fu scoperta per la prima volta mediante studi sulla diffrazione dei raggi X su fibre, particolarmente fibre proteiche. Queste poterono essere osservate direttamente nella loro forma naturale, in penne, capelli, tendini, ecc. Le indagini di W. Th. Astbury alla fine degli anni venti e all'inizio dei trenta mostrarono che vi erano piccole distanze ripetitive in tali fibre, indicanti una continua replicazione, lungo l'asse della fibra, di situazioni conformazionali uguali. Due di tali modi di ripetizione con distanze ripetitive di 5,1 e 3,4 Å furono chiamati rispettivamente la forma α e la forma β della catena polipeptidica, in conformità con l'uso comune di usare l'alfabeto greco per etichettare situazioni sperimentali individuate, ma non comprese. La ricerca della vera natura delle strutture che mostrano il diagramma delle fibre α e β richiese vent'anni (nel 1954 L. Pauling vinse il premio Nobel per la chimica per la sua scoperta dell'α-elica). Tutto il successivo lavoro sulle strutture dei biopolimeri è stato basato sui principi stabiliti da Pauling e da R. B. Corey per le strutture α e β. Questi principi, che saranno discussi nel cap. 2, hanno dovuto essere perfezionati e migliorati per affrontare i più difficili problemi e discutere le più sottili questioni della moderna analisi conformazionale, ma costituiscono tuttora la spina dorsale di tutto il lavoro in questo campo.
2. Teoria della conformazione.
La conformazione di una molecola è una specificazione delle posizioni relative di tutti i suoi atomi. Le rotazioni o le traslazioni di una molecola come un tutto non cambiano le posizioni relative dei suoi atomi e perciò non cambiano la conformazione. Una ‛conformazione di equilibrio' è una conformazione in cui tutti gli atomi sono nella posizione di minima energia (energia libera) rispetto al movimento di un dato atomo. Il minimo non è necessariamente, anzi spesso non lo è, il minimo assoluto di energia per la molecola. Le vibrazioni, sempre presenti, delle molecole intorno alle loro conformazioni d'equilibrio non sono considerate come cambiamenti conformazionali. Una ‛conformazione dinamica', d'altra parte, è una conformazione nella quale tutte le posizioni relative degli atomi sono fissate, ma l'energia non è al minimo. Le forze non sono equilibrate nelle conformazioni dinamiche, cosicché esse normalmente corrispondono a istantanee di molecole in movimento.
Un modo per specificare una conformazione è dare le coordinate di ogni atomo nella molecola rispetto a un sistema di assi esterni. Questa è la specificazione più assoluta della conformazione ed è il mezzo con il quale sono riportate le strutture dai cristallografi che operano con i raggi X. Il metodo ha due svantaggi. In primo luogo, tutte le coordinate sono cambiate dalle rotazioni e traslazioni della molecola; in secondo luogo, i chimici amano pensare in termini di legami chimici, mentre l'immagine dei legami è oscurata in un sistema di coordinate esterne. Coordinate basate sulla lunghezza e sulla giustapposizione dei legami sono chiamate ‛coordinate interne' e saranno discusse brevemente. Queste coordinate sono pienamente adeguate per una comprensione intuitiva della conformazione di piccole molecole, ma una tabella di coordinate interne non dà un quadro adeguato degli effetti a lunga distanza nelle macromolecole. Trattandosi di macromolecole, è molto più vantaggioso usare le coordinate interne per discutere la conformazione locale, le coordinate esterne per una descrizione quantitativa dell'intera struttura, e modelli tridimensionali che forniscono una buona rappresentazione delle proprietà a lunga distanza delle molecole e dei legami locali.
È possibile basare la teoria della conformazione proprio su tre variabili: lunghezza del legame, angolo di legame e angoli di torsione o diedri. La lunghezza del legame è la distanza tra due atomi uniti da un legame chimico. L'angolo di legame richiede la specificazione della posizione relativa di tre atomi ed è l'angolo sotteso da due atomi ciascuno dei quali connesso a un terzo atomo da un legame chimico. L'angolo di torsione richiede la specificazione e le posizioni relative di quattro atomi. A scopo illustrativo usiamo la molecola dell'1,2-difluoroetano (v. fig. 1). La conformazione di questa molecola può essere specificata dalla sequenza dei quattro atomi F−C−C−F. Nella fig. 1A è rappresentata la configurazione eclissata, cioè la conformazione dove gli atomi sostituenti sono l'uno sovrapposto all'altro guardando lungo il legame C−C. I quattro atomi sono in un piano e l'angolo di torsione ϑ è per definizione uguale a zero. Un modo più quantitativo di rappresentare gli angoli di torsione è il diagramma di Neumann, mostrato nella fig. 1B. La conformazione è ora quella in cui i gruppi vicini sono stati ruotati di 60° rispetto alla conformazione eclissata. Conformazioni in cui i sostituenti sono disposti come nella fig. 1B sono chiamate conformazioni sfalsate. Di tali conformazioni ne esistono tre,ϑ=60°, 180° e 300°. È da notare che ci sono nove differenti raggruppamenti di quattro atomi che possono essere scelti per definire ϑ e che questi potrebbero essere equivalenti solo se ciascuna metà della molecola avesse simmetria trigonale, cosa che non si verifica. Tale ambiguità deve essere eliminata da convenzioni, che sono state messe a punto per ognuno dei molti campi dell'analisi conformazionale. Per esempio, nel difluoroetano si usano i due atomi di fluoro per determinare l'angolo di torsione.
L'insieme delle lunghezze di legame, degli angoli di legame e degli angoli diedri è sufficiente per definire l'intera conformazione di una molecola. Per esempio, in F−C−C−F le dodici coordinate richieste per specificare le posizioni (x, y, z) dei quattro atomi sono sostituite da sei coordinate interne (tre lunghezze di legame, due angoli di legame, un angolo di torsione), più le sei coordinate esterne (tre traslazioni e tre rotazioni dell'intera molecola), che non influenzano la conformazione. Una completa specificazione delle coordinate interne è perciò una completa specificazione delle posizioni relative di tutti gli atomi e una rappresentazione totale della conformazione. L'analisi conformazionale raramente va così lontano con molecole relativamente grandi e mai con macromolecole. Fortunatamente le lunghezze di legame variano ben poco e gli angoli di legame solo di pochi gradi. Il più comune tipo di analisi conformazionale trae vantaggio da queste proprietà assumendo valori standard di lunghezze di legame e angoli di legame e considerando l'intero problema conformazionale solamente in termini di angoli di torsione. Questo approccio, seguito nella maggior parte della letteratura, sarà adottato anche in questo articolo (v. tuttavia il paragrafo conclusivo di questo capitolo).
I polipeptidi costituiscono un ottimo esempio. La struttura di base di un polipeptide dipende in primo luogo dalla conformazione della catena peptidica e, in misura minore (ma essenziale), dalla conformazione delle catene laterali. Moltissima attenzione è stata rivolta alle conformazioni favorite delle catene polipeptidiche a causa della loro relazione con la struttura delle proteine. Un segmento di una catena polipeptidica è illustrato nella fig. 2, dove si può vedere che ci sono tre angoli di torsione per unità peptidica. Pauling fu il primo a capire che il problema della conformazione di un peptide è considerevolmente più semplice di quanto appaia. Egli osservò che il legame centrale C−N del gruppo peptidico è un parziale doppio legame. Questo automaticamente rende planare l'intero gruppo, cosicché le posizioni di tutti e quattro gli atomi periferici sono fissate. Ci sono due possibili disposizioni cis e trans, che sono interconvertibili mediante rotazione di 180°. Nella conformazione cis, gli atomi di carbonio in α sono entrambi dalla stessa parte rispetto al legame C−N e ω=0°. Nella forma trans gli atomi di carbonio sono da parti opposte e ω=180°. Praticamente tutti i residui peptidici sono trans nelle molecole naturali, eccetto che per l'amminoacido prolina, che può essere cis o trans. Ne segue che, con la sola eccezione della prolina, solo due angoli conformazionali per unità peptidica devono essere specificati. Questo conduce alla semplice rappresentazione bidimensionale della conformazione peptidica mostrata nella fig. 3. Ogni punto del piano ϕ, ψ rappresenta una particolare conformazione. I biopolimeri si possono trovare in tre tipi di conformazioni: conformazioni regolari, in cui tutte le unità si ripetono secondo un semplice schema; schemi irregolari, dove la catena principale è fissata, ma con un ordine soltanto parziale, come nelle proteine globulari e negli acidi ribonucleici transfer (tRNA); molecole stocastiche (v. sotto, cap. 6), che cambiano continuamente struttura cosicché la conformazione è descrivibile soltanto in termini di funzioni statistiche di distribuzione. Alcune strutture regolari di polipeptidi sono riportate nella fig. 3.
Sfortunatamente, non sempre è possibile specificare la conformazione di un'unità costitutiva di un biopolimero soltanto con due angoli, e il metodo grafico è insufficiente. Con tre angoli è possibile fissarne uno come parametro e usare un diagramma bidimensionale per gli altri due. Tuttavia negli acidi nucleici vi sono, in un nucleotide, cinque angoli di torsione della catena principale e un angolo di torsione della catena laterale, che sono di fondamentale importanza per specificare la struttura (v. fig. 9). Una rappresentazione grafica delle strutture diviene impossibile in queste circostanze ed è necessario ricorrere a numerose elaborazioni mediante un calcolatore elettronico per una dettagliata e completa analisi conformazionale. È possibile fissare quattro degli angoli e rappresentare la variazione degli altri due su un grafico; questo sta diventando una pratica comune. Nei paragrafi seguenti saranno principalmente usati come esempi i polipeptidi poiché essi esemplificano tutti i principi dell'analisi conformazionale e ne permettono una rappresentazione semplice e diretta.
Energia conformazionale. - Se una conformazione è possibile o no dipende dalla sua energia ε attraverso il fattore di probabilità di Boltzmann exp[−ε/kT]. Il calcolo di ε per grandi molecole che possono assumere molte conformazioni è un lavoro al di là delle possibilità umane e i progressi secondo queste linee sono dovuti per la maggior parte all'avvento dei calcolatori numerici. Nei più semplici casi di calcolo sono considerati solo due tipi di energia: barriere intrinseche alla rotazione e interazioni tra gruppi non legati, cioè
ε=ε (di torsione) +ε(di non legame). (1)
Barriere intrinseche alla rotazione furono scoperte per la prima volta nell'etano, dove si trovò che la configurazione eclissata (v. fig. 1A) aveva un'energia di circa 3 kcal/mole maggiore di quella della conformazione sfalsata (v. fig. 1B). Questa barriera ha un'origine quantomeccanica e la comprensione del suo meccanismo è stata raggiunta proprio in questi ultimi anni. L'energia associata all'angolo di torsione ϑ, nel caso dell'etano, è data con sufficiente accuratezza dalla formula
ε(ϑ)=1,5 kcal (1+cos 3ϑ).
Questo è valido per jl legame trigonale C−C. Funzioni empiriche sono disponibili per molti altri tipi di legami, sicché è possibile calcolare l'energia di torsione per la maggior parte dei casi che interessano.
Le interazioni tra gruppi non legati sono quelle tra atomi che non sono connessi da un legame chimico, cioè interazioni a una certa distanza all'interno di una molecola. Tre tipi di interazioni di questo genere sono prevalenti, secondo calcoli recenti: l'interazione di cariche, l'attrazione di van der Waals e la repulsione. Per una coppia di atomi, i e j, l'energia può essere rappresentata dall'espressione
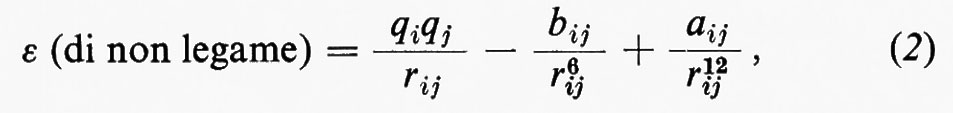
dove qi e qj sono le cariche sugli atomi polari, aij e bij sono costanti empiriche che sono state stabilite per i tipi di atomi i e j, rij è la distanza tra gli atomi i e j. Il primo termine rappresenta l'attrazione delle cariche elettriche (o la repulsione, secondo il segno delle cariche), il secondo l'attrazione di van der Waals e il terzo la repulsione che due atorni esercitano l'uno sull'altro quando si vengono a trovare troppo vicini fra di loro. Il primo e il secondo termine hanno una base teorica; il terzo è puramente empirico.
In un tipico calcolo di energia conformazionale le lunghezze del legame e gli angoli fissati sono posti inizialmente con tutti gli angoli di torsione a valori opportuni. Il calcolatore poi ruota gli angoli a valori specificati, un legame per volta, finché si è ottenuta la conformazione desiderata. Le coordinate sono poi trasferite di nuovo nella forma cartesiana per facilitare il calcolo di rij per tutte le coppie di atomi. Si calcola infine l'energia usando l'equazione (1) e introducendo i parametri appropriati per ogni coppia di atomi i e j e ciascun angolo di torsione ϑ. Se si fa questo per molte centinaia di punti nello spazio delle coordinate interne, si possono disegnare le curve di energia costante. Per esempio, la fig. 4A mostra tali curve per una coppia di gruppi peptidici che fiancheggiano un residuo di alanina. La molecola si troverà per la maggior parte del tempo in conformazioni che differiscono di poche centinaia di calorie dal valore più basso dell'energia e per pochissimo tempo in conformazioni con energie più grandi di 2RT (≈1 kcal). L'interpretazione di mappe come quelle delle figg. 4A e 4B è uno dei principali problemi di questo campo.
Le mappe per gli altri amminoacidi sono qualitativamente simili alle mappe per l'alanina, eccetto che per la prolina, per l'idrossiprolina e per la glicina. L'anello nei derivati della prolina costringe l'angolo ϕ ad avvicinarsi a −60°. Ne risulta che sono possibili solamente conformazioni vicine ad αD e C di fig. 3. Inoltre il passaggio da trans a cis scambia atomi di carbonio alifatici simili (v. fig. 5). La differenza di energia è poca e i residui di cis-prolina esistono sia nelle proteine, sia nei polimeri sintetici. L'assenza di una catena laterale nella glicina elimina molte delle restrizioni conformazionali che sono presenti nell'alanina. L'intero centro della mappa ϕ, ψ risulta permesso. Ne segue che la glicina ha un ruolo strutturale speciale in molti sistemi proteici. Benché questo sia il modo in cui viene eseguita una gran parte dei calcoli dell'energia conformazionale, vi sono tre perfezionamenti che vengono comunemente praticati.
1. Sono stati sviluppati procedimenti di minimizzazione in cui il calcolatore esplora lo spazio delle coordinate e localizza i minimi di energia conformazionale. Questo va bene per piccole molecole, ma comporta dei problemi nel caso di grandi molecole a causa delle difficoltà che si incontrano quando si ha a che fare con uno spazio a molte dimensioni.
2. Vengono fatti calcoli in cui angoli e lunghezze di legame sono variabili, cosi come gli angoli di torsione. Non c'è dubbio che questo fornisca una rappresentazione migliore delle proprietà conformazionali di una molecola, ma i calcoli sono complessi e costosi anche per piccole molecole.
3. In un solvente è l'energia libera piuttosto che l'energia che deve essere messa nel fattore di Boltzmann. Benché la valutazione dell'energia libera non sia molto accurata, alcuni ricercatori hanno tentato di aggiungere un'energia libera di solvatazione nei calcoli di energia. Questo è un punto importante poiché accade che la solvatazione sia uno dei fattori determinanti per la conformazione. I più trovano buone conformazioni ‛nel vuoto' usando il procedimento sopra detto e poi tentano di correggere le loro previsioni in base a considerazioni qualitative circa l'effetto della solvatazione.
3. Blocchi costitutivi delle macromolecole.
Quando diventano disponibili campioni purificati di varie macromolecole naturali, si rende possibile determinare come sono costituite. In generale il primo passo in questa direzione richiede la degradazione dei polimeri in unità più piccole, possibilmente nei blocchi costitutivi da cui essi sono sintetizzati in natura. Molti di tali polimeri (specialmente biopolimeri) sono risultati sensibili agli agenti idrolitici, cioè a quelli che scindono i legami aggiungendo una molecola di acqua a ogni subunità liberata. Viceversa tali lunghe catene possono essere costruite eliminando acqua o qualche altra piccola molecola dai loro blocchi costitutivi bifunzionali, come mostra la fig. 6. Questo tipo di polimerizzazione è chiamato ‛polimerizzazione per condensazione'. Un altro tipo di polimerizzazione, chiamato ‛per addizione', ha luogo quando i monomeri si collegano insieme senza dover espellere alcuno dei loro atomi.
In generale le catene principali (o ‛scheletri') delle macromolecole sono formate dal ripetersi di unità identiche, che possono essere rappresentate dai rettangoli della fig. 6; la diversità viene determinata dalla variazione dei sostituenti nelle ‛catene laterali', rappresentate dai circoli. Il numero di specie differenti dei possibili gruppi R varia da un tipo di polimero all'altro. Per esempio, gli acidi nucleici ne contengono normalmente cinque specie, mentre le proteine ne contengono una ventina.
La conformazione tridimensionale complessiva delle macromolecole è determinata dalle conformazioni locali dei componenti sia della catena principale sia di quelle laterali. Come si vedrà, la maggior parte dei blocchi costitutivi della catena principale possiede un numero relativamente piccolo di possibili conformazioni stabili o preferite, con solo pochi legami singoli intorno ai quali è possibile una rotazione relativamente libera. In una molecola complessa la più accurata informazione concernente la posizione dei singoli atomi è ottenuta con la tecnica della cristallografia mediante raggi X. Questa tecnica richiede che ogni molecola abbia un gran numero di blocchi costitutivi disposti nella stessa conformazione che si ripete o che un gran numero di molecole identiche (ognuna con la stessa conformazione interna) siano disposte in modo ordinato in un cristallo.
Pauling e Corey proposero di ricavare informazioni dettagliate sulla struttura di unità costitutive relativamente piccole o di composti modello, ottenibili in forma cristallina, e di utilizzare poi tali dati per prevedere come le suddette unità debbano legarsi per dare lo spettro di diffrazione ottenuto dai polimeri. A partire da questo loro lavoro pionieristico sulle strutture amminoacidiche e sulla loro applicazione alla struttura delle proteine, sono stati effettuati numerosi studi, basati sullo stesso principio, per altre sostanze polimeriche. Le strutture cristalline possono fornire valori precisi per le distanze interatomiche e gli angoli di legame e spesso mostrano la conformazione più favorita intorno a tali legami.
a) Polipeptidi e proteine.
I blocchi costitutivi elementari da cui sono sintetizzati proteine e polipeptidi (v. biologia; v. proteine) e a cui essi possono essere ridotti per idrolisi sono gli amminoacidi che, come dice il loro nome, hanno un gruppo amminico e un gruppo acido (carbossile) legato a un atomo di carbonio centrale indicato con Cα.
Le macromolecole proteiche si formano per il distacco di un −OH del gruppo carbossilico di un amminoacido e di un −H del gruppo amminico del successivo amminoacido con formazione di un legame ammidico ovvero peptidico. Lo scheletro di questi polimeri è costituito da una catena di atomi di carbonio α legati da gruppi ammidici planari (v. fig. 2). La diversità di struttura dipende dalle varie catene laterali degli amminoacidi (gruppi R della fig. 2) unite agli atomi Cα. Poiché ci sono venti amminoacidi comunemente ricorrenti (più pochi altri in particolari proteine), il numero di sequenze specifiche possibili è praticamente infinito. Le abbreviazioni a tre e a una lettera per i blocchi costitutivi delle comuni proteine e le formule delle relative catene laterali sono riportate nella tab. I. La parte di ogni amminoacido che rimane dopo aver tolto l'acqua per formare il legame ammidico è normalmente chiamata residuo di amminoacido. Per certi scopi è più conveniente considerare un gruppo −CαH(R)− e il gruppo peptidico dalla parte del suo carbossile come un'unità, che viene indicata come ‛unità peptidica' (v. fig. 2A). Eccetto che per la glicina, che ha due atomi d'idrogeno legati al Cα, tutti i residui di amminoacidi posseggono un atomo Cα asimmetrico, poiché tutti e quattro i suoi sostituenti, disposti secondo i vertici di un tetraedro, sono differenti. Questo vuol dire che ci sono due possibili configurazioni dei quattro sostituenti, l'una immagine speculare dell'altra. Questi isomeri ottici sono chiamati forma L (Levo) e forma D (Destro). Gli L-amminoacidi sono i soli che si trovino di solito nelle proteine naturali ; la configurazione L è quella mostrata nella fig. 2.
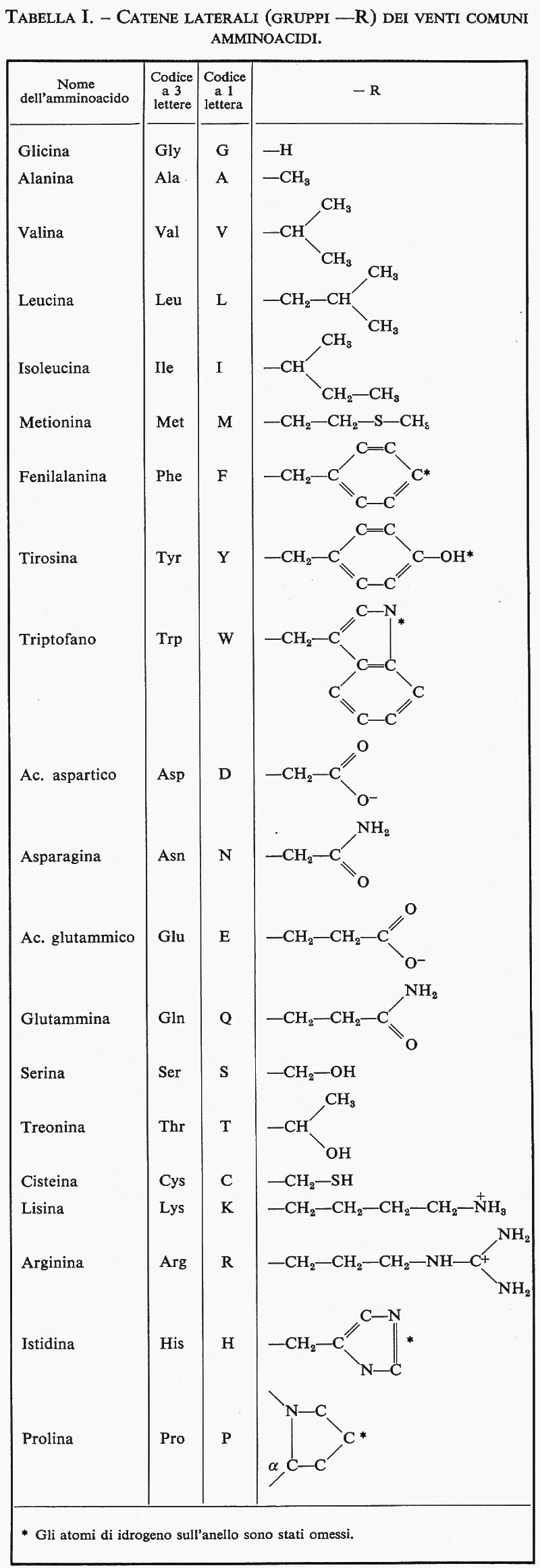
Come abbiamo detto, Pauling e Corey determinarono le strutture cristalline di numerosi amminoacidi e composti strettamente affini per ottenere il miglior valore delle distanze interatomiche e degli angoli di legame nelle proteine. I loro risultati, in parte modificati da più recenti studi, sono mostrati nella fig. 2 per un segmento poli- peptidico generico. Tali studi non indicano le conformazioni intorno ai legami singoli che uniscono due gruppi ammidici e una catena laterale a ogni atomo Cα, poiché c'è una considerevole libertà di rotazione intorno a questi legami a causa di fattori che saranno discussi più avanti.
b) Blocchi costitutivi dei polisaccaridi.
I blocchi costitutivi per i polisaccaridi sono zuccheri semplici (monosaccaridi). Questi ultimi sono poludrossialdeidi (aldosi) o -chetoni (chetosi) con la formula generale Cn(H2O)m. La maggior parte degli atomi di carbonio hanno legato un gruppo −OH. I polimeri si formano per eliminazione di una molecola d'acqua da una coppia di gruppi −OH, ciascuno di due diverse unità monomeriche, per formare ponti eterei (glicosidici) tra essi (v. fig. 6). Poiché di solito ci sono più di due gruppi −OH in ogni monomero, sono possibili anche catene ramificate. La grande varietà dei polisaccaridi è dovuta, più che alla diversità degli zuccheri semplici che li costituiscono, alle numerose forme isomere per ciascuno di questi.
La maggior parte dei polisaccaridi dei quali è nota la conformazione sono derivati da zuccheri aldeidici contenenti cinque atomi di carbonio (aldopentosi) o sei atomi di carbonio (aldoesosi). I principali pentosi sono i ribosi, che sono importanti costituenti dei polinucleotidi e saranno discussi nel È c. La discussione qui sarà concentrata sull'aldoesosio ‛glucosio', poiché sia esso sia i suoi derivati sono i più comuni costituenti dei polisaccaridi naturali. La fig. 7 mostra sia i diversi modi di disegnare la struttura del glucosio, sia i vari isomeri strutturali.
A ogni formula di zucchero con un dato numero di atomi di carbonio corrisponde in realtà un gruppo di zuccheri costituito da più isomeri strutturali, poiché la molecola contiene vari atomi di carbonio asimmetrici. Ognuno di tali gruppi comprende coppie di zuccheri, isomeri ottici fra loro, le cui formule di struttura sono immagini speculari l'una dell'altra, come gli isomeri del glucosio D e L mostrati nella fig. 7. La maggior parte degli zuccheri trovati in natura hanno configurazione D.
Benché gli zuccheri possano esistere in soluzione transitoriamente sotto forma di strutture a catena lineare aperta, come la formula di Fischer per il glucosio mostrata nella fig. 7, i pentosi e gli esosi esistono normalmente sotto forma di struttura ad anello di 5 (furanosi) o 6 (piranosi) termini, come mostrato nella stessa figura. I furanosi hanno nell'anello quattro atomi di carbonio e uno di ossigeno, mentre i piranosi hanno cinque atomi di carbonio e uno di ossigeno. La chiusura dell'anello fa scomparire il gruppo riducente =CO, creando un altro gruppo −OH, che può assumere due diverse posizioni rispetto al piano dell'anello. Queste altre due forme isomeriche, chiamate anomeri α e β, differiscono per la configurazione all'atomo di carbonio i (v. fig. 7). In soluzione sono presenti ambedue queste forme, che si trovano in equilibrio tra loro. Tuttavia in natura la polimerizzazione è catalizzata da enzimi che preferenzialmente scelgono l'una o l'altra forma. La principale modalità di variazione in diversi polisaccaridi derivati dal glucosio è basata su queste due configurazioni.
Gli anelli dello zucchero sono relativamente rigidi ma non planari, cosicché ne sono possibili parecchie conformazioni interconvertibili. La costruzione di modelli molecolari e studi conformazionali indicano che i piranosi possono esistere in due forme ‛a sedia' (v. fig. 7D), così come nelle forme meno stabili ‛a barca' e ‛distorte' (skew). Le due forme a sedia Cl e lC (C da chair=sedia) sono più stabili perché i sostituenti sugli atomi dell'anello possono assumere posizioni più sfalsate. Un sostituente che abbia il legame che lo collega all'anello pressoché parallelo all'asse dell'anello stesso, così da essere rivolto verso l'alto o verso il basso rispetto a questo, viene indicato come ‛assiale' (a).
Un sostituente che abbia questo legame approssimativamente perpendicolare all'asse, in modo da essere rivolto fuori dal piano dell'anello, viene chiamato ‛equatoriale' (e). Questi due tipi sono mostrati nella fig. 7, nelle rappresentazioni delle due possibili forme a sedia.
Grossi sostituenti in posizioni assiali hanno più probabilità di trovarsi vicini altri gruppi in analoga situazione. Perciò, in generale, una conformazione che ha il massimo numero di gruppi ingombranti in posizioni equatoriali sarà la più stabile. Per il β-D-glucosio la conformazione Cl ha tutti i gruppi −OH e il gruppo −CH2OH sull'atomo di carbonio 5 in posizioni equatoriali e dovrebbe perciò essere favorita rispetto alla conformazione lC in cui questi stessi gruppi sono assiali. Questo è confermato dai calcoli conformazionali e dagli studi cristallografici con i raggi X. Le poche conformazioni stabili possibili per gli anelli conferiscono una considerevole rigidità allo scheletro delle macromolecole dei polisaccaridi in un modo alquanto simile ai gruppi planari peptidici nei polipeptidi. Le sole rotazioni relativamente libere lungo le catene sono attorno ai due legami che congiungono gli anelli essenzialmente rigidi all'atomo di ossigeno glicosidico che li collega.
A causa del numero di gruppi −OH disponibili per formare connessioni fra unità di zuccheri e a causa della esistenza delle due forme monomeriche α e β all'atomo di carbonio 1, anche un singolo zucchero come il glucosio può dare origine a numerose conformazioni polimeriche con proprietà del tutto differenti, come sarà mostrato nella discussione sui polisaccaridi.
Dal punto di vista chimico e cristallografico molti polimeri sono catene formate da unità dimere. Mediante adatta digestione dei polisaccaridi si possono ottenere spesso frammenti disaccaridici che danno considerevoli informazioni sulla struttura dei polimeri da cui si sono formati. Alcuni esempi sono dati nella fig. 8. Il maltosio (dall'amido) è costituito da due unità di glucosio con un legame α(1,4), mentre il cellobiosio (dalla cellulosa) ha un legame β(1,4). Queste differenze conducono a profonde diversità nei polimeri da essi formati. La chitina, un importante costituente del materiale scheletrico di alcuni invertebrati, produce chitobiosio, che è un derivato del cellobiosio con un −OH su ogni atomo di carbonio 2 rimpiazzato da un gruppo N-acetilamminico. Il cell wall (parete cellulare) di molti batteri contiene dimeri simili al chitobiosio, con una catena laterale di acido lattico sull'atomo di carbonio 3 di una delle unità di N-acetilglucosammina (NAG); questo componente del dimero è chiamato acido N-acetilmurammico (NAM).
c) Blocchi costitutivi degli acidi nucleici.
I polinucleotidi (acidi nucleici), così importanti per i processi vitali, risultano da unità nucleotidiche che si susseguono, costituite da tre diverse specie chimiche (v. fig. 9). I gruppi che costituiscono lo scheletro o catena principale delle macromolecole sono un pentosio e un gruppo fosfato, mentre le catene laterali sono formate da quattro basi organiche, appartenenti due alla famiglia della purina e due alla famiglia della pirimidina.
Nell'acido desossiribonucleico (DNA) l'unità che costituisce lo scheletro è il fosfato del 2-desossiribosio, mentre nell'acido ribonucleico (RNA) lo zucchero è il ribosio. I gruppi fosfati sono legati agli anelli dello zucchero nelle posizioni C(3′) e C(5′), mentre le basi sono legate al C(1′) nella configurazione β. In entrambi i tipi di acidi nucleici la specificità è ottenuta variando la sequenza delle basi delle catene laterali. Il DNA contiene le purine adenina (A) e guanina (O), mentre le pirimidine sono la timina (T) e la citosina (C). Nell'RNA si trova l'uracile (U) al posto della timina. Le strutture di queste basi sono schematizzate nella fig. 9.
Le più dettagliate informazioni concernenti le strutture del DNA e dell'RNA sono state ottenute dagli studi conformazionali su singoli nucleotidi e su dinucleotidi, che sono stati utilizzati come modelli per gli studi sui polimeri. Negli acidi nucleici gli anelli dello zucchero hanno struttura furanosica, cioè hanno un anello a cinque membri. Inizialmente si era supposto che gli anelli dello zucchero fossero essenzialmente planari, ma gli studi ai raggi X e i calcoli conformazionali indicarono che essi sono piegati. Ci sono due conformazioni estreme possibili per l'anello furanosico, chiamate ‛a busta' e ‛a mezza sedia' (o twist), ma sono state trovate anche molte altre forme intermedie. I vari sostituenti sugli anelli stabilizzano le forme a mezza sedia, in cui il C(2′) e il C(3′) sono fuori dal piano definito dai tre atomi dell'anello. Sono possibili quattro principali tipi di tali conformazioni a seconda di quale dei due atomi, C(2′) o C(3′), sia spostato più lontano dal piano e a seconda che tale atomo di carbonio e il C(5′) siano sullo stesso lato del piano (endo) o su quello opposto (eso), come è mostrato nella fig. 10. La forma C(2′)-eso non è stata fin qui osservata negli studi sui cristalli. A causa del gran numero di legami singoli nello scheletro della catena, è possibile una libertà di rotazione considerevolmente maggiore lungo la catena stessa rispetto a quanto accade nelle proteine e nei polisaccaridi. Inoltre ci sono legami singoli che uniscono le basi della catena laterale agli anelli dello zucchero attraverso gli atomi di ossigeno sul C(1′). I vari angoli di rotazione intorno a questi legami sono mostrati nella fig. 9. I sistemi ad anello delle basi sono tutti planari a causa degli estesi sistemi di doppi legami in ognuno di essi (v. acidi nucleici; v. biologia).
4. Macromolecole informazionali.
Il più semplice tipo di polimero consiste nella uniforme ripetizione di un singolo blocco costitutivo. La maggior parte dei polimeri sintetici e le materie plastiche sono di questo tipo, cosi come lo sono le gomme naturali, la cellulosa, il glicogeno, l'amido, ecc. Qualche volta l'unità ripetuta contiene due blocchi costitutivi, come la coppia acido adipico-esametilendiammina del nailon 6-6 o l'N-acetilglucosammina-acido N-acetil-murammico del cell wall batterico (v. fig. 8). Tali polimeri sono chiamati omopolimeri se c e solo un tipo di blocco costitutivo, e polimeri alternati se c'è una semplice ripetizione di sequenza di due o più blocchi costitutivi. Nei sistemi biologici tali molecole sono importanti poiché costituiscono materiale di riserva (glicogeno, amido, caseina) o materiale di sostegno (cellulosa, collageno, chitina, ecc.) in quanto dotate di proprietà meccaniche adatte. Anche le industrie delle fibre sintetiche e delle materie plastiche fanno assegnamento sulle utili proprietà di semplici macromolecole.
Una delle caratteristiche peculiari delle proteine e degli acidi nucleici, che conferisce loro importanti ruoli funzionali in biologia, è che essi sono copolimeri di un insieme di blocchi costitutivi disposti con una precisa sequenza. I blocchi costitutivi per le proteine sono i venti L-amminoacidi riportati nella tab. I; per l'RNA i ribosonucleotidi r-A, r-U, r-G e r-C; per il DNA i quattro desossiribosonucleotidi d-A, d-T, d-G e d-C. Le molecole con sequenze specifiche non periodiche sono chiamate ‛molecole informazionali'. Sono informazionali nel senso della teoria dell'informazione, cioè consistono in una riconoscibile sequenza di simboli non casuali (gli stessi gruppi chimici o le lettere usate per designarli), che perciò costituiscono un messaggio. Sono informazionali in senso fisico perché conducono alla replica di strutture fisiche o chimiche o sono capaci di riconoscere altre strutture mediante interazione fisica o chimica.
L'importanza di questo aspetto delle macromolecole biologiche non sarà mai troppo sottolineata. Essa è meglio illustrata da pochi esempi. Il DNA a doppia elica si auto- riconosce, cioè la doppia elica intatta non può formarsi senza che ogni adenina o guanina di una catena venga a trovarsi strutturalmente appaiata con una timina o citosina dell'altra catena (v. fig. 16). Il risultato è che in un sistema vivente capace di sintetizzare il DNA, una catena ‛genitrice' detta la sequenza dell'altra catena ‛figlia'. Questa sequenza è l'eredità genetica dell'organismo in oggetto e si ritiene che tutte le informazioni (istruzioni biochimiche) necessarie per lo sviluppo del singolo individuo fin dagli inizi (uovo fertilizzato, spora, ecc.) siano contenute nella sequenza del suo DNA. Qualche volta sistemi specializzati come i Virus utilizzano per questo scopo l'RNA.
Come ulteriore esempio si può ricordare che la sequenza specifica di una proteina è originata, com'è noto, da una sequenza corrispondente di triplette nucleotidiche dell'RNA messaggero, la quale è a sua volta correlata con una sequenza di un segmento di DNA. Così l'informazione viene passata attraverso gli stadi DNA→RNA→polipeptide sequenziale. La storia tuttavia non finisce a questo punto, dal momento che il polipeptide risultante ha la proprietà di autoriconoscimento strutturale. Questo deriva dal fatto che tutte le proteine richiedono in pratica, per essere funzionali, non solo una specifica sequenza, ma anche una struttura tridimensionale altamente elaborata (v. fig. 20), in cui la maggior parte degli atomi occupano posizioni specifiche. È ora noto che molte proteine formano la loro struttura tridimensionale spontaneamente e si suppone che questo valga per tutte le proteine. Si pensa che l'energia libera della proteina sia più bassa quando essa è ripiegata nella sua struttura specifica che quando si trova in quella specie di stato disordinato che normalmente ci si attenderebbe per macromolecole a catena lunga. Questa bassa energia libera è la conseguenza della geometria altamente specifica delle interazioni nella struttura avvolta e questa a sua volta dipende dalla sequenza. Così l'informazione per l'avvolgimento in una forma vitale e attiva è contenuta nella sequenza.
Tutti gli enzimi e la maggior parte delle proteine possiedono questa proprietà di una struttura tridimensionale altamente specializzata ottenuta in seguito a interazioni che si autoriconoscono. Tali interazioni avvengono internamente alla struttura della proteina. Per poter essere funzionali molte proteine devono inoltre possedere punti di riconoscimento esterni grazie a interazioni favorevoli delle loro parti esterne con altre molecole. Un enzima deve interagire con il suo substrato o col coenzima in maniera specifica e caratteristica, altrimenti non può catalizzare reazioni chimiche. Gli enzimi sono caratterizzati da un alto grado di specificità nel corso della loro azione catalitica. Una molecola di anticorpo deve riconoscere il suo antigene specifico e interagire con esso, altrimenti non può avvenire l'immunizzazione. Le molecole del muscolo (miosina, actina e altre) devono avere siti esterni di riconoscimento, in modo che le singole unità proteiche vadano a costituire spontaneamente la complessa architettura della fibra muscolare, ecc.
La biologia molecolare ha conseguito brillanti successi nel correlare eventi biologici e biochimici con il contenuto informazionale di macromolecole. Uno degli argomenti di maggior interesse è oggi il tentativo di chiarire il meccanismo chimico-fisico che è alla base di questo codice biologico. La connessione con le proprietà conformazionali delle molecole è chiara e costituisce uno dei maggiori incentivi per lo sviluppo delle ricerche in questo campo.
5. Molecole elicoidali.
Una delle più affascinanti proprietà dei biopolimeri è la loro tendenza a formare strutture ordinate, cioè strutture rispondenti a semplici regole che determinano la giustapposizione fra gli atomi di un'unità monomerica e quelli delle unità vicinali. Queste regole sono manifestazioni di simmetria. Le unità monomeriche stesse possono avere simmetria, chiamata simmetria di punto, che è descrivibile in termini di piani di riflessione e assi di rotazione (per es. H2O con due piani di simmetria e un asse binario, benzene con un asse esagonale e molti piani di simmetria). Le molecole di un polimero possono avere un ulteriore tipo di simmetria, chiamata simmetria traslazionale, determinata dal fatto che una traslazione, caratteristica della molecola (spesso unita a una rotazione o a una riflessione), conduce a un identico intorno atomico nella successiva unità monomerica.
Uno dei più semplici e importanti tipi di simmetria traslazionale si ha quando tutte le unità monomeriche sono equivalenti le une alle altre. È facile vedere come ciò possa accadere. Consideriamo due identiche unità monomeriche A e B. Fra tutti i modi in cui esse possono esercitare un'azione reciproca, c'è normalmente una conformazione relativa che è la più favorevole, cioè quella di più bassa energia. Se l'azione reciproca fra A e B non interferisce con un'identica azione reciproca fra B e una terza unità monomerica C, si può verificare di nuovo la conformazione più favorevole e per induzione può essere propagata lungo l'intera catena. Questo è il tipo di simmetria e di stabilità che si trova nei muri di mattoni, nei pavimenti di piastrelle e nei cristalli. Un mattone è un oggetto altamente simmetrico e anche le strutture costruite con mattoni hanno un'elevata simmetria. Consideriamo il più comune tipo di muro di mattoni. Non c'è nessuna intrinseca differenza tra un lato e l'altro o, ignorando la gravità, tra la cima e il fondo. Come si è detto nel capitolo 3, riguardante i blocchi costitutivi, la maggior parte delle unità strutturali dei biopolimeri sono intrinsecamente asimmetriche e perciò tendono a formare strutture polimeriche asimmetriche. Il principio degli intorni equivalenti è stato analizzato nel caso di molecole asimmetriche da F. H. C. Crick e J. D. Watson, e più tardi da A. Klug e altri. Essi hanno mostrato che un modo particolarmente efficace per soddisfare questo principio è la formazione di una struttura a elica. Un'elica è una spirale uniforme intorno a un asse ed è intrinsecamente asimmetrica: sinistrorsa o destrorsa.
Dal punto di vista matematico un'elica può essere generata combinando una rotazione di un angolo ϑ intorno all'asse con una traslazione lungo lo stesso asse di una distanza d. L'operazione combinata, rotazione più traslazione, è detta operazione di avvitamento e l'asse di rotazione è detto asse di avvitamento dell'elica. In una struttura a elica, ciascun atomo equivalente forma un'elica classica e uniforme con una distanza fissa dall'asse e lo stesso spostamento angolare ϑ e assiale d fra unità successive. L'intero insieme di atomi connessi da legami covalenti è indicato poco esattamente come elica. In pratica si è soliti specificare le eliche mediante lo spostamento assiale d e il numero n di residui per spirale dell'elica, dove n=360/ϑ.
Lo studio dei cristalli ha permesso di ottenere soddisfacenti conoscenze di base per comprendere la diffusione dei principi di simmetria in natura. Sfortunatamente esso ha portato anche a conclusioni alquanto inesatte. I cristalli molecolari sono insiemi di unità legate solo da forze attrattive fisiche. Poichè le molecole sono separate e indipendenti, esse non sono costrette nei tipi di orientazioni che possono assumere le une rispetto alle altre, anche se ovviamente alcune orientazioni risultano energeticamente favorite. Inoltre esse si devono assestare in una struttura tridimensionale, cioè avere tre operazioni di simmetria traslazionale indipendenti. Si può provare che in cristalli tridimensionali l'elica può avere soltanto assi binari, ternari, quaternari e senari, cioè n=2, 3, 4 e 6. I primi studi sui biopolimeri si servirono di questo principio non appropriato per eliche unidimensionali e portarono a strutture che attualmente si sa essere poco stabili. Furono Pauling e Corey che per primi delinearono il modo più convincente di derivare le strutture teoriche per le macromolecole. Essi attribuirono grande importanza a due postulati fondamentali: le singole unità devono assumere nel poli mero conformazioni stabili e si deve formare il massimo numero di legami a idrogeno. Come è già stato detto, il primo principio fu suggerito dagli studi sulla diffrazione dei raggi X in piccoli peptidi, che fornirono informazioni sulle lunghezze dei legami stabili e sugli angoli delle unità peptidiche. Questi valori (v. fig. 2, A e B) furono poi impiegati nella costruzione di modelli. Inoltre, i suddetti autori osservarono che la risonanza costringe il gruppo peptidico ad assumere una struttura planare.
Sulla base di questi cinque principi (migliore conoscenza delle lunghezze di legame, migliore conoscenza degli angoli di legame, planarità delle unità peptidiche, formazione del numero massimo di legami a idrogeno ed eliminazione di contatti atomici sfavorevoli), il problema di predire la conformazione di un peptide diviene abbastanza semplice. Considerando solamente peptidi trans, i gradi di libertà sono forniti dagli angoli ϕ e ψ della fig. 2. Con tutte le restrizioni imposte, ci sono soltanto poche conformazioni stabili che possono formare legami a idrogeno lungo la catena; queste conformazioni furono studiate sistematicamente da Pauling e Corey già agli inizi degli anni cinquanta. La più stabile conformazione di catena singola con legami a idrogeno è l'α-elica (v. fig. 11), che fu intuita da Pauling arrotolando una striscia di carta per passare il tempo in una camera d'albergo. Affrontando il problema da questo punto di vista, la nozione di numero intero di residui per spira è superflua, e infatti la maggior parte di eliche a catena singola non ne contengono un numero intero (v. tab. 11). D'altra parte sono state scoperte eliche a più catene con asse binario, ternario e quaternario.
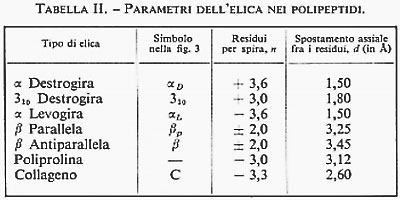
La mappa ϕ, ψ offre un metodo molto efficace per esplorare le strutture a elica, come fu messo in rilievo da J. Ramachandran e dai suoi collaboratori. Nel caso di un polipeptide a elica con un solo residuo per unità costitutiva, ogni residuo ha gli stessi angoli ϕ e ψ. Viceversa ogni coppia di valori di ϕ e ψ, cioè ogni punto del piano ϕ, ψ, rappresenta un'elica potenziale. Noi possiamo pensare a un'elica definita dagli angoli ϕ e ψ dei residui, oppure dai parametri dell'elica d ed n già discussi. Si può andare anche oltre disegnando, sul diagramma ϕ, ψ, dei punti caratteristici (loci) di tutte le eliche binarie, ternarie, quaternarie, ecc. e di tutte le distanze traslazionali d. Questo diagramma è mostrato nella fig. 12.1 contorni per eliche non contenenti un numero intero di unità per spira possono essere stimati mediante interpolazione visuale. I valori negativi di n rappresentano le eliche sinistrorse. Notiamo che α-eliche sinistrorse e destrorse si trovano alle intersezioni della curva per d=1,5 Å con quelle per n=±3,6. Notiamo anche che i punti singolari di tutte le eliche binarie sono molto vicini a una diagonale che attraversa la mappa.
Abbiamo già veduto come la mappa ϕ, ψ possa essere usata per rappresentare l'energia di una coppia di unità peptidiche adiacente a un atomo di carbonio α (v. fig. 4A). Essa può anche essere usata per rappresentare l'energia (per residuo) di eliche in funzione di ϕ e ψ. Si assuma che il polipeptide contenga N atomi di carbonio α ed N+1 unità di peptide. Ci sono pertanto N(ϕ, ψ) coppie che possono variare. I valori di ϕ e di ψ di tutti gli N residui sono fissati a un dato valore e l'energia è calcolata in conseguenza. Risultati tipici sono mostrati nella fig. 4B. I minimi sono molto più bassi perché interazioni diverse da quelle con le unità più vicine contribuiscono alla stabilizzazione. In particolare, i legami a idrogeno nella conformazione ad a-elica portano a un considerevole aumento della stabilità.
a) Polipeptidi in conformazione elicoidale stabile.
L'α-elica destrorsa. - La struttura con ϕ=−57,4° e ψ=−47,5° è la più stabile catena singola a conformazione elicoidale per la maggior parte dei polipeptidi. Tale conformazione è risultata essere un'importante unità strutturale negli enzimi e in altre proteine globulari. Essa è contenuta in molte proteine fibrose, come la miosina, e costituisce la base dell'unità strutturale delle fibre α di Astbury. I polipeptidi sintetici formano spesso spontaneamente α-eliche in appropriati solventi. A causa della facilità di preparazione e del loro interesse intrinseco, i sistemi ad α-elica sono stati oggetto di migliaia di pubblicazioni. Come risultato di tali ricerche, le proprietà dell'α-elica (conformazionali, termodinamiche, spettroscopiche, ecc.) sono servite da modello per gli studi di tutti gli altri sistemi macromolecolari. L'α-elica destrorsa comparirà di nuovo nella trattazione degli enzimi, delle proteine globulari e di quelle fibrose (strutturali).
α-Eliche sinistrorse. - Analisi sperimentali e teoriche hanno mostrato che l'α-elica destrorsa è la forma più stabile per gli L-amminoacidi. Al contrario i D-amminoacidi formano α-eliche sinistrorse, dal momento che la riflessione in immagine speculare non varia l'energia di una conformazione. Tuttavia alcuni L-amminoacidi, a causa delle caratteristiche peculiari della catena laterale, hanno minimi di energia inferiori per l'elica destrorsa che per quella sinistrorsa. Un esempio molto studiato è quello del polibenzilaspartato. α-Eliche sinistrorse non sono state trovate nelle molecole biologiche e quando esistono come polimeri sintetici sono meno stabili delle forme destrorse.
Conformazione β. - Un'altra regione del piano ϕ, ψ che si trova a energia relativamente bassa è quella nel quadrante in alto a sinistra lungo la linea dell'asse binario. Poiché un asse binario implica che gruppi >C=o e >N−H alternati vengano proiettati dalla catena ad angoli di 1800 l'uno rispetto all'altro, questa conformazione è la più logica per strutture che si estendono su di un piano. Tale conformazione è adottata dalla natura per le proteine fibrose e globulari e sarà discussa con il nome di conformazione β (a foglio pieghettato) parlando di strutture bidimensionali e tridimensionali.
Conformazione del collageno. - La regione vicina a ϕ=−60° e ψ=150° è una regione di bassa energia che è molto utilizzata nelle proteine e nei polipeptidi sintetici e naturali. La sua importanza come conformazione locale nelle proteine naturali e nei polipeptidi random sarà discussa in seguito; per esempio essa è la conformazione naturale della poliprolina. I polipeptidi della prolina non possono formare α-eliche, dal momento che l'atomo di idrogeno del gruppo >N−H è sostituito dal più ingombrante δ-C dell'anello a 5 membri. Studi sulla diffrazione dei raggi X e l'analisi conformazionale mostrano che la regione (−60°, 150°) è eccezionalmente stabile per questo polipeptide, cosicché si forma un'elica (© nella fig. 3) anche in assenza delle interazioni stabilizzanti dei legami a idrogeno. La proteina strutturale collageno ha un alto contenuto di prolina e di idrossiprolina e utilizza questa regione di spazio conformazionale per formare lunghe solide molecole che sono l'ideale per il loro ruolo strutturale negli organismi animali. Per questa ragione tale conformazione è chiamata ‛spirale del tipo collageno'. Il collageno ha la struttura di un'elica multipla e sarà discusso nel prossimo paragrafo. Anche la poliglicina forma eliche con questa conformazione.
Come è stato detto prima, anche la prolina e l'idrossiprolina si comportano diversamente dagli altri amminoacidi, in quanto nel passaggio dalle strutture trans alle cis avviene l'interscambio tra due atomi di carbonio quasi equivalenti. Ne segue che non c'è nessuna marcata preferenza per la forma trans, benché nei solventi polari questa sia la forma più stabile. E nota una struttura di poliprolina in cui tutti i residui hanno una conformazione cis; essa forma un'elica quasi rigida anche a (−60°, 150°). Nelle proteine globulari sono state trovate unità di prolina sia cis sia trans.
b) Eliche multiple.
Benché le conformazioni a spirale α, β e del tipo collageno siano i modelli basilari secondo cui si formano le più importanti eliche di polipeptidi, le molecole naturali e biologiche non esistono quasi mai come semplici eliche. Una simile situazione si verifica invece nella struttura degli acidi nucleici. Per poter fare una discussione più generale, è utile considerare a questo punto il concetto di eliche multiple, cioè l'avvolgimento di un'elica intorno a un'altra nello stesso modo in cui fasci di fibre sono intrecciati a formare una fune. Questo espediente è così frequentemente utilizzato in natura che si deve concludere che molecole costituite in tal modo sono più forti, più stabili e più funzionali di quelle formate da eliche singole.
Polipeptidi a eliche multiple. - La struttura della miosina, una delle principali componenti del muscolo, dà una dimostrazione del modo in cui α-eliche possono avvolgersi le une alle altre per offrire una struttura più forte in una situazione biologica dove sia richiesta una buona resistenza meccanica. Come si può vedere nella fig. 13, la molecola è come una lunga bacchetta con una doppia testa alla fine. È stato mostrato che l'intera molecola contiene due sub-unità, ognuna con peso molecolare uguale alla metà di quello complessivo.
In base alle conoscenze attuali, si ritiene che la parte lunga della molecola consista in due α-eliche attorcigliate una intorno all'altra (ogni elica derivata dalla metà delle molecole componenti) e terminanti in due teste che sono più o meno separate. La funzione enzimatica della miosina risiede nelle due teste, sulla cui struttura attualmente si possono fare soltanto congetture.
Questo tipo di struttura è chiamato ‛super-elica'. Un modo di raffigurarsi questa struttura è quello d'immaginare che gli assi delle due α-eliche siano attorcigliati in lente spirali intorno a un asse comune. I residui di ognuna delle α-eliche sono attorcigliati intorno a queste spirati in una versione distorta dell'elica usuale, leggermente compressa all'interno, leggermente stirata al di fuori. L'intera struttura è tenuta insieme da interazioni di contatto fra le due eliche.
Qualcosa di simile a questa superstruttura si trova nel- l'α-cheratina dei capelli, della lana e di altre fibre naturali. Secondo le attuali vedute, le singole eliche dell'α-cheratina sono attorcigliate in una superelica tripla di 20 Å di diametro. Le triple eliche sono poi raggruppate in una microfibrilla derivante da undici eliche. Un gran numero di microfibrille sono raggruppate insieme per dare le fibre caratteristiche di questi materiali.
Il collageno, una delle più importanti proteine strutturali dei Vertebrati, è formato da eliche triple di molecole disposte secondo le spirali tipiche del collageno. Le singole eliche di questa struttura sono sinistrorse, con 3,3 unità peptidiche per spira e uno spostamento assiale (passo) di 8,58 Å per giro. Le supereliche sono larghe spirali destrogire con un passo di 86 Å (v. fig. 14). Proprietà speciali sono richieste da un polipeptide se è usato per formare una spirale di collageno. Ogni terzo residuo della catena dev'essere un residuo di glicina poiché ogni terza posizione della struttura è costretta in uno spazio talmente limitato nell'interno della superelica che non ci sarebbe posto per una catena laterale. Inoltre, i residui di prolina e di idrossiprolina non solo si adattano bene nella struttura, ma la stabilizzano. Gli organismi traggono vantaggio da questa proprietà, in quanto il contenuto di prolina e di idrossiprolina del collageno aumenta con la temperatura ambiente. Così, a causa di un più basso contenuto di prolina, il collageno degli animali che vivono in acqua fredda è instabile alla temperatura dei Mammiferi, animali a sangue caldo, ecc. Chiaramente, la particolare spirale del collageno impone sequenze ordinate particolari per i polipeptidi costituenti. Molecole simili al collageno sono state preparate sinteticamente da polimeri alternanti del tipo (-Gly-Pro-Pro-)n o (-Gly-Pro-X-)n, dove X è un amminoacido arbitrario.
Un'altra struttura di grande importanza nasce dal pieghettamento di tipo β. Come è già stato detto prima, in questa struttura le catene hanno assi binari, cosicché i gruppi >N−H e >C=O, che formano legami a idrogeno, puntano fuori dall'elica, alternativamente, a 180° l'uno dall'altro. Così, se la catena C è legata alla catena B allo stesso modo in cui la catena B è legata alla catena A, come richiesto dal principio di equivalenza, il risultato è un foglio bidimensionale. Tali fogli possono essere costruiti in due diversi modi, uno con tutte le catene parallele, cioè che procedono tutte nello stesso modo cominciando dall'estremità col gruppo amminico libero, e l'altro con catene alternate antiparallele le une alle altre. Queste sono chiamate rispettivamente strutture β parallele e antiparaltele e sono raffigurate nella fig. 15. Esse sono anche chiamate strutture a fogli pieghettati a causa della caratteristica corrugazione causata dall'inclinazione del gruppo peptidico planare rispetto al piano del foglio. La struttura anti- parallela del foglio pieghettato è la base della struttura β trovata nella seta e in altre fibre naturali. Sezioni distorte di strutture β sono state trovate in molte proteine globulari e in enzimi. Ciò sarà discusso in seguito.
Acidi nucleici. - A prima vista il problema dell'analisi conformazionale degli acidi nucleici appare molto più complesso di quello delle proteine e dei polipeptidi. Come si può vedere nella fig. 9, lungo la catena ci sono cinque angoli per nucleotide che possono variare per rotazione e a questi si deve aggiungere l'angolo di torsione associato con il collegamento della base al ribosio e la possibilità di piegamento dell'anello del ribosio stesso. Ma, come vedremo brevemente, ci sono caratteristiche limitative anche per il problema dell'acido nucleico che permettono predizioni conformazionali di grande importanza e interesse. Non è possibile entrare negli aspetti quantitativi di questo problema, come abbiamo fatto con i polipeptidi, senza una lunga e complicata discussione tecnica, ma i fattori qualitativi che determinano la struttura degli acidi nucleici possono essere esaminati piuttosto facilmente.
In primo luogo, tutti i legami lungo la catena principale hanno triple barriere di rotazione (v. la trattazione del- l'etano nel cap. 2). Si sa bene che le tre conformazioni sfalsate, che sono a 120° l'una rispetto all'altra, sono fortemente favorite in una situazione di questo tipo. Restringendo le nostre considerazioni alle conformazioni sfalsate, ci sarebbero così 35=243 conformazioni per unità, contando la stretta regione angolare intorno a ogni minimo d'energia come una conformazione singola. Il lavoro con modelli e i calcoli di energia conformazionale mostrano che la maggior parte di queste possibilità non sono permesse poiché sono bloccate da interferenze tra vari gruppi di notevoli dimensioni (ribosio, fosfato, basi, ecc.). I ricercatori in questo campo non sono interamente d'accordo su tale punto, ma ammettono tutti che sono permesse non più di dieci conformazioni della catena principale per unità di nucleotide. L'angolo di torsione che unisce la base all'anello del ribosio è anch'esso molto limitato (specialmente per le basi pirimidiniche), come è stato mostrato studiando la diffrazione dei raggi X per i nucleosidi e l'analisi conformazionale. Il sorprendente risultato è che anche una catena di acido nucleico ‛senza struttura' è fortemente tensionata.
Un altro fattore semplificante nelle analisi conforma- zionali degli acidi nucleici viene dal principio di complementarità formulato da Watson e Crick per il DNA e ora generalizzato per un gran numero di altre applicazioni strutturali e meccaniche. Per comprendere questo principio è istruttivo paragonare gli acidi nucleici con le proteine e i polipeptidi. Le strutture che sono state sopra discusse per i polipeptidi (con eccezioni minori) sono state tutte basate sul principio del riconoscimento dello scheletro essenziale (catena principale). Questo significa che le forze fisiche che sono responsabili del modello molecolare base (α, β, ecc.) tendono a collegare lo scheletro di un poli- peptide con se stesso (α-elica) o con altri scheletri poli- peptidici (struttura β, collageno). Il punto è che, a parte la prolina che non può formare legami a idrogeno e la glicina che riveste un ruolo speciale nel collageno, la natura della catena laterale dell'amminoacido ha solo un ruolo secondario. Le catene laterali degli amminoacidi certamente influenzano la stabilità delle α-eliche e delle strutture β, ma si tratta di perturbazioni che non alterano la struttura di base, determinata dalle proprietà dello scheletro. L'aspetto più importante degli acidi nucleici è che sono costituiti sul principio del riconoscimento ‛delle catene laterali'; le loro eliche sono distanziate all'interno rispetto a quelle dei polipeptidi.
Coppie complementari planari legate per mezzo di legami a idrogeno possono essere formate tra adenina e timina (o uridina) e tra guanina e citosina (v. fig. 16) e possono così collegare due eliche di DNA. Purché le catene vadano in direzioni opposte, una delle conformazioni che lo scheletro della catena assume facilmente è quella in cui ogni base su una catena può collegarsi a una base complementare sull'altra catena se, e soltanto se, la complementarità è soddisfatta. La geometria della base accoppiante è tale che la sostituzione di una coppia GC con AT può essere effettuata senza distorcere la struttura geometrica fondamentale della catena. Il risultato è la struttura di Watson e Crick (v. fig. 17), in cui le basi di spirali complementari di DNA sono ordinatamente connesse in una spirale che contiene dieci coppie di base per spira. Attualmente l'energia ditale connessione, più che i legami a idrogeno, è considerata la causa principale di stabilità della struttura.
Non è questa la sede per entrare nell'argomento della complementarità e delle numerose implicazioni relative, importanti per i processi vitali (v. acidi nucleici). Limiteremo la discussione agli aspetti strutturali del problema.
Le forme a elica degli acidi nucleici sono più flessibili delle eliche polipeptidiche. Questo provoca una variazione della struttura con il cambiare dell'intorno dovuto al solvente. In una soluzione salina diluita il DNA assume la struttura di Watson-Crick-Wilkins, in cui i piani delle basi sono quasi perpendicolari all'asse dell'elica. Ci sono dieci coppie di basi per spira e uno spostamento assiale di 3,37 Å fra una coppia di basi e l'altra. Questa forma è chiamata forma B (v. fig. 17B). A bassissima umidità si produce la forma A, che ha 20° d'inclinazione delle basi e undici coppie di basi per spira (v. fig. 17A). Sotto forma di sale di litio e in determinate condizioni saline il DNA è convertito in una forma C, in cui ci sono 9 e ⅓ residui per spira e un'inclinazione delle basi di −6° (un angolo negativo vuol dire un'inclinazione che simula la superficie di contatto di una vite sinistrorsa). La forma C è correlata piuttosto da vicino alla forma B. Le tre forme sono facilmente distinguibili in soluzione in virtù del loro spettro di dicroismo circolare a circa 260 nm. La tab. III elenca i parametri dell'elica delle forme A, B e C.
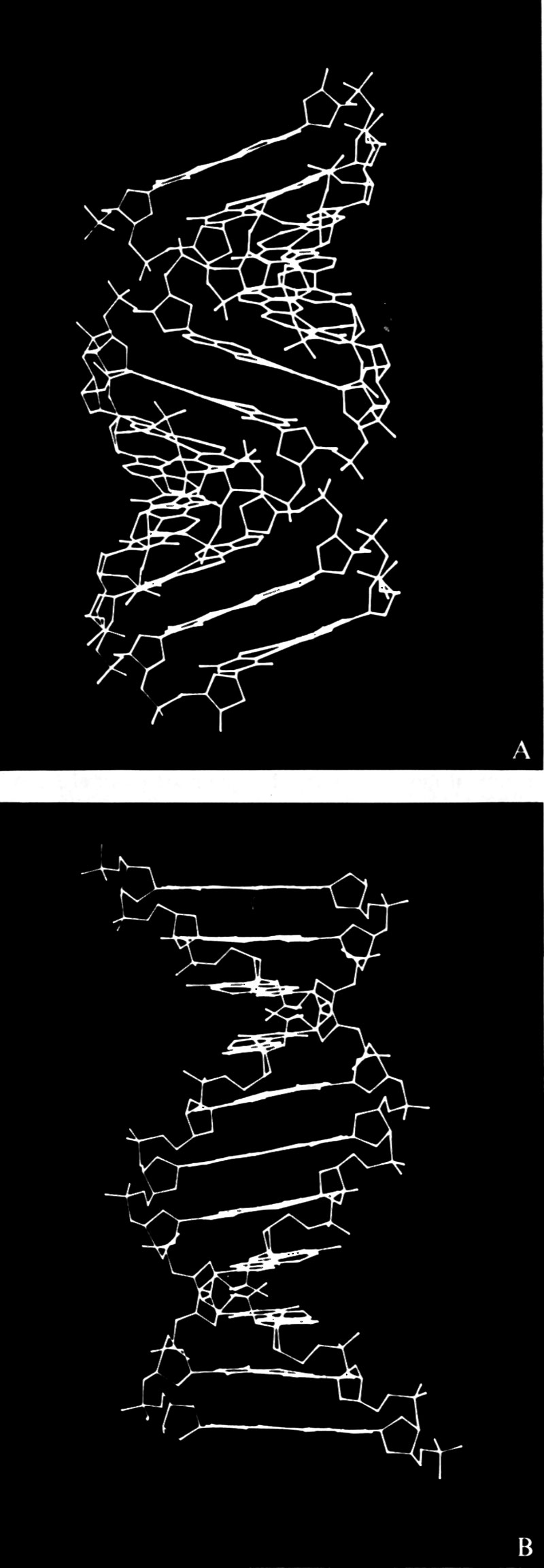
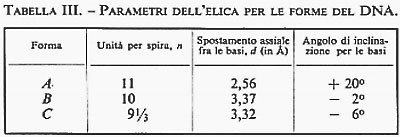
Gli acidi ribonucleici (RNA) si trovano sotto forma di spirali singole (RNA messaggero), di doppie spirali e di complesse strutture tridimensionali (RNA di trasferimento o transfer). Queste ultime saranno discusse nel cap. 6. Oltre a queste, sono state preparate un gran numero di strutture di RNA a spirale singola, doppia e tripla, alcune con sequenze ingegnosamente pianificate. La forma naturale delle spirali doppie di RNA è molto simile alla forma A del DNA (v. fig. 17A). Dal punto di vista chimico gli RNA sono distinti dai DNA per la presenza di un gruppo idrossilico sull'atomo di carbonio 2 dell'anello dello zucchero e per l'assenza del gruppo metilico che distingue la timina dall'uracile. A causa dei ruoli biologici fondamentalmente differenti svolti dall'RNA e dal DNA, la razionalizzazione delle differenze strutturali elaborate da queste due alterazioni di modesta entità costituisce uno dei maggiori problemi insoluti dell'analisi conformazionale degli acidi nucleici.
Entro i limiti del metodo dell'esame di fibre ai raggi X, le strutture delle forme A, B e C degli acidi nucleici a doppia spirale si possono ritenere conosciute. Fino al periodo in cui è stato scritto questo articolo, la maggior parte delle indagini sull'analisi conformazionale di strutture sconosciute sono state effettuate in modo qualitativo mediante un uso ‛impavido' di modelli molecolari. Il procedimento è all'incirca il seguente. Si formano, ove possibile, doppie spirali dall'accoppiamento di basi complementari usando la geometria standard. Le basi sono accostate il più possibile. L'uso di modelli spaziali compatti elimina automaticamente i cattivi contatti atomici. Poste queste tre condizioni, non rimane molta flessibilità nel modello costitutivo. Questo tipo di procedimento viene ora usato nei progetti per predire la geometria dei sistemi ripetitivi, il modo di legarsi di leganti complessi al DNA e all'RNA, il meccanismo dei ribosomi e le strutture del transfer RNA. Un recentissimo argomento è ‛la formazione a quadrifoglio' nel DNA. Se una sequenza di DNA è seguita dalla sua sequenza complementare in ordine inverso, la struttura a doppia elica può essere rappresentata, molto schematicamente, come segue,
con parziale appaiamento fra le due catene. In tale rappresentazione A e B sono sequenze date, A′ e B′ le loro complementari. Con una piccolissima perdita di struttura a elica, un tal tratto di DNA può trasformarsi nella struttura
con parziale appaiamento all'interno di ognuna delle due catene. Normalmente si pensa che strutture come questa possano servire da indicatori per l'interazione del DNA con proteine che sono coinvolte in processi di replicazione.
c) Polisaccaridi.
Nei sistemi biologici i polisaccaridi sono usati come unità strutturali meccaniche o per immagazzinamento d'energia. Codificazioni sequenziali elaborate come quelle esistenti nei polipeptidi e negli acidi nucleici non avrebbero nessuno scopo in tali funzioni e non si trovano in natura. Al contrario, le proteine strutturali sono codificate, ma spesso con semplici strutture sequenziali. Tuttavia i polisaccaridi raggiungono una grande varietà di espressioni strutturali in virtù di: 1) la disponibilità di numerosi pentosi ed esosi come blocchi costitutivi; 2) la varietà di possibili posizioni a ponte di etere che possono essere usate per prolungare la catena; 3) la possibilità di ramificarsi formando tre ponti di etere da un'unità di zucchero; 4) l'alternativa di collegamenti glicosidici α o β; 5) la presenza di sostituenti sugli anelli dei pentosi o degli esosi.
I dati strutturali sono stati meno facili da ottenere per i polisaccaridi che per le proteine e gli acidi nucleici e vi è stata una notevole dipendenza dalle analisi conformazionali. Molte delle più importanti molecole tra i polisaccaridi sono polimeri del glucosio o di derivati del glucosio. Le proprietà conformazionali delle catene di unità di glucosio sembrano oggi comprese abbastanza bene e la trattazione che segue sarà limitata a quella classe di polimeri che comprende cellulosa, glicogeni, amidi, chitina e polisaccaridi delle pareti cellulari dei Batteri. Benché le proprietà conformazionali di queste catene siano conosciute abbastanza a fondo, rimangono ancora molti problemi insoluti concernenti la loro struttura nello stato biologico. Ciò è dovuto al fatto che le molecole solubili sono generalmente molto ramificate, cosicché i problemi principali hanno a che fare con la struttura covalente piuttosto che con la conformazione, mentre d'altro canto le molecole allo stato solido come la cellulosa, la chitina e i polisaccaridi delle pareti cellulari dei Batteri pongono molti problemi circa il modo in cui le catene si combinano per formare un'unità strutturale. I metodi di diffrazione dei raggi X non sono stati così chiari nell'interpretazione come lo sono stati invece per le proteine e gli acidi nucleici.
Cellulosa. - La cellulosa consiste di catene non ramificate di D-glucosio con legami β-glucosidici colleganti le posizioni 1 e 4 delle unità di glucosio adiacenti, cioè β (1, 4). La molecola può essere considerata un polimero del dimero cellobiosio, come è indicato nella fig. 8B. Numerosi calcoli conformazionali e determinazioni della struttura hanno indicato che la più stabile forma dell'anello è la struttura Cl indicata nella flg. 7D. Ignorando le possibili fluttuazioni da questa conformazione dell'anello, ci sono solo due gradi conformazionali di libertà per residuo nella catena. Questi sono costituiti dagli angoli di torsione ϕ e ψ associati con i due legami del ponte etereo (v. fig. 8B). La fig. 18 rappresenta una mappa di esclusione conformazionale per questo sistema. Tutte le conformazioni fuori del piccolo spazio centrale danno cattivi contatti di van der Waals e possono essere escluse. La struttura con ϕ=−30°, ψ=150° è chiamata struttura di Herman. Questa conformazione genera un'elica binaria, è libera da forti tensioni e può formare un legame a idrogeno tra l'idrossile sull'atomo di carbonio 3 di un anello e l'ossigeno 5 dell'anello adiacente (v. fig. 8B, dove l'−OH dell'atomo di carbonio 3 è rappresentato da R2).
I parametri dell'elica per la cellulosa sono dati nella tab. IV. Le molecole della cellulosa hanno evidentemente solo un piccolo grado di libertà nelle regioni amorfe delle fibre. La struttura della cellulosa nelle regioni cristalline non è interamente determinata. La struttura oggi più accettata è quella in cui le singole catene sono nella struttura di Herman; le catene non sono parallele le une alle altre nella cella elementare e sono presenti legami a idrogeno intermolecolari tra i gruppi −OH degli atomi di carbonio 6 su una catena e gli atomi di ossigeno dei ponti eterei su un'altra catena.

Amilosio. - L'amilosio, che costituisce una frazione dell'amido, è un polimero del glucosio con legami glucosidici α (1, 4). Esso è stato oggetto di molti studi poiché è formato da catene singole anziché da catene ramificate, come quelle dell'amilopectina e del glicogeno. A differenza delle catene con legami β (1, 4), c'è una considerevole libertà nella mappa ϕ, ψ. La catena può formare una gran varietà di eliche destrorse e sinistrorse con n≥5. Le conformazioni con n=2 sono rigorosamente escluse. Ne risulta che l'amilosio è, in soluzione, una molecola abbastanza flessibile, i cui contorni tendono a rassomigliare ad ampie spirali, a differenza delle molecole di cellulosa, che sono più simili a nastri in tensione attorcigliati. L'amilosio forma fibre di diversa struttura, in dipendenza dalle condizioni di solvatazione. Sono state proposte eliche con n da 4 a 8. Una delle forme più cristalline è quella chiamata forma V, che è stata trovata in presenza di agenti complessanti, come lo iodio, ed è un'elica con 6 unità per spira sinistrorsa. I parametri dell'elica per la forma V sono dati nella tab. IV. La variabilità delle forme cristalline è d'accordo con la già detta libertà conformazionale di queste molecole. Sono disponibili in letteratura mappe dell'energia potenziale in funzione di ϕ e ψ.
Amido e glicogeno. - Gli amidi sono polisaccaridi costituenti una riserva alimentare del regno vegetale; generalmente sono miscele di amilosio e amilopectina. Questa ultima è costituita prevalentemente, come l'amilosio, da unità di glucosio legate con legami α (1, 4), ma le catene sono altamente ramificate, e utilizzano, nei punti di ramificazione, legami α (1, 6). L'equivalente polisaccaride che costituisce una riserva alimentare del regno animale è il glicogeno, che è notevolmente simile all'amilopectina nel suo disegno strutturale. Le principali differenze consistono nella presenza di alcuni legami α (1, 3) e nella maggiore ramificazione delle catene. Per esempio, il complesso intensamente colorato dell'amido con lo iodio non si forma con glicogeno a causa della brevità delle catene altamente ramificate. Le molecole come l'amido e il glicogeno sono probabilmente stocastiche sia nella loro struttura covalente (lunghezza delle catene e punti di ramificazione) sia nella loro conformazione. Come tali, esse hanno una forte rassomiglianza con i polimeri sintetici ramificati e sono soggette allo stesso tipo di analisi statistica che ha avuto un notevole successo nel caso di queste ultime molecole.
Chitina e polisaccaridi delle pareti cellulari. - La chitina è un polisaccaride che ha una stretta correlazione con la cellulosa. La sua molecola è una catena lineare di unità di glucosio con legami glucosidici β (1, 4). A prescindere dalle possibili differenze nella distribuzione dei pesi molecolari, la sola differenza consiste nella sostituzione dell'−OH dell'atomo di carbonio 2 con il gruppo acetammidico −NHCOCH3. Questa sostanza ha, nel mondo degli Invertebrati, lo stesso ruolo della cellulosa nel mondo dei vegetali. Essa è un importante componente strutturale dell'esoscheletro dei Crostacei, della cuticola degli Insetti, nonché delle pareti cellulari di alcuni Funghi e Batteri. Il gruppo acetammidico causa differenze considerevoli nel- l'impacchettamento delle catene nelle regioni cristalline. Una forma cristallina, l'α-chitina, è stata particolarmente ben caratterizzata. Essa contiene catene non parallele e ha due assi binari a vite perpendicolari all'asse binario della catena principale. I parametri dell'elica sono dati nella tab. IV. La chitina ha l'interessante proprietà di formare miscele strutturali con le proteine. È proprio in questa forma che si trova frequentemente negli Insetti e nei Crostacei. Questi complessi chitino-proteici danno spettri di diffrazione dei raggi X poco risolti; solo in questi ultimi anni sono stati compiuti progressi nel chiarimento della loro struttura.
I polisaccaridi che costituiscono il cell wall batterico consistono in una matrice covalente di catene polisaccaridiche e polipeptidiche. I legami glucosidici sono β (1, 4) come nella cellulosa e nella chitina. Come indicato nella fig. 8, la struttura covalente è simile a quella della chitina, salvo che un residuo di acido lattico ha sostituito l'−OH dell'atomo di carbonio 3 su una ogni due unità di glucosio. L'unità che si ripete è generalmente simboleggiata con NAG-NAM invece dell'ingombrante nome N-acetilglucosammina-acido-N-acetilmurammico. I polipeptidi (che sono miscele di D- e L-amminoacidi) si estendono come brevi catene che legano fra loro le catene laterali di acido lattico. La conformazione di questa struttura reticolata è sconosciuta, come lo sono certe configurazioni della struttura covalente. Quando quest'ultimo problema sarà risolto, tale sistema costituirà un importante campo per l'applicazione dell'analisi conformazionale di sistemi complessi. Non sappiamo se si potranno ottenere chiare informazioni strutturali da tecniche come la diffrazione dei raggi X. In compenso, le analisi conformazionali del componente polipeptidico e dei componenti polisaccaridici sono state sviluppate a uno stadio abbastanza avanzato. Appare certo che la catena di polisaccaridi è distorta rispetto alla struttura di Herman in corrispondenza di unità alternate di glucosio perché la presenza di acido lattico elimina la possibilità di legami a idrogeno nell'interno della catena.
6. Molecole stocastiche.
Le molecole senza conformazione fissa, cioè le molecole la cui conformazione è determinata solo come una distribuzione probabilistica, saranno denominate, per lo scopo di questa trattazione, molecole ‛stocastiche'. Questa non è la comune terminologia ed è impiegata solo per evitare la parola random (a caso), che è usata in modi diversi da vari ricercatori. Una molecola conformazionalmente stocastica in soluzione libera cambia continuamente la sua conformazione locale, la sua forma globale e le sue dimensioni. Essa è meglio descritta dalle sue proprietà medie e queste possono essere pensate come determinate osservando la condotta di una singola molecola per un lungo periodo di tempo, ovvero scattando un'istantanea di un grande numero di molecole e facendo la media della distribuzione. Per il teorema ergodico questi procedimenti per mediare sono equivalenti. Meccanismi statistici convenzionali fanno uso di questo teorema per fornire descrizioni accurate di proprietà molecolari usando medie che non dipendono dal tempo.
Prendiamo come esempio un polipeptide. Supponiamo dapprima che la conformazione descritta dagli angoli ϕ, ψ di ogni residuo sia indipendente dalla conformazione di tutti gli altri residui. Dunque c'è la probabilità che la molecola abbia una conformazione globale semplicemente uguale al prodotto delle probabilità locali per ogni coppia ϕ, ψ. Fin dalla fine degli anni quaranta sono disponibili metodi per compiere medie statistiche nel caso di questo tipo di polimeri. Per i polipeptidi l'indipendenza locale delle conformazioni non costituisce una cattiva approssimazione e sono stati fatti considerevoli progressi, particolarmente dal gruppo di P. J. Flory, nella comprensione delle proprietà fisiche di polipeptidi random. Per determinare la probabilità locale di determinate funzioni si usano i contorni dell'energia potenziale su una mappa ϕ, ψ e si calcola poi la media sull'intera catena usando metodi sofisticati ma ben collaudati.
L'ipotesi di conformazioni locali indipendenti è lontana dall'essere generalmente vera. Essa non è valida per catene alifatiche o polinucleotidiche (a meno che tutti e cinque gli angoli siano usati per specificare una conformazione locale). I residui di prolina nella poliprolina sono fortemente influenzati dagli altri vicini. Se un residuo di prolina è nella conformazione cis, i suoi vicini tendono a essere nella conformazione cis. Anche le conformazioni trans sono fra loro correlate in maniera piuttosto forte. Se un polipeptide si trova in condizioni in cui assume in parte una conformazione a elica, l'ipotesi di conformazioni indipendenti non può essere fatta. Ciò perché la formazione di eliche è cooperativa. Un residuo ha una tendenza molto più forte ad assumere una conformazione ad α-elica se può aggiungersi sopra un'elica esistente piuttosto che se deve agire isolato. Questi casi, benché meno random del caso delle unità indipendenti, possono anche essere compresi in base a teorie esistenti.
Un'altra situazione stocastica si verifica se immaginiamo macromolecole che tornano a orientarsi continuamente in una soluzione e sono poi congelate di colpo nelle loro conformazioni istantanee. Di conseguenza abbiamo un sistema che è distribuito statisticamente, ma è costante nel tempo. Ogni molecola ha una conformazione fissa e le proprietà stocastiche sono solo apparenti se si prende in considerazione tutto l'insieme di molecole. Infatti non occorre che il processo sia veloce. Il solo requisito è che le proprietà di distribuzione dell'insieme ‛fissato' siano le stesse di quelle del sistema di molecole che si riorientano liberamente. Condizioni come questa sono state incontrate negli studi sui gel, sulle pellicole, sui sistemi polimerici amorfi, come in ogni fase solida non cristallina di polimeri.
Questo ci conduce a un concetto finale, le cosiddette regioni random di molecole a struttura complessa, quali le proteine globulari e i tRNA. L'analisi strutturale di una tipica proteina globulare mostra che questa può essere divisa in regioni alle quali è assegnabile una struttura definita. Ci sono regioni ad α-elica e strutture a foglietto con legami a idrogeno che possono essere riconosciute come forme β distorte. Oltre a queste ci sono regioni che non posseggono caratteristiche percettibili di simmetria. Tali regioni sono state chiamate random, amorfe, non ordinate, disordinate, ecc. Non esiste una terminologia accettata universalmente.
Si deve riconoscere che non c'è niente di casuale nell'organizzazione di queste regioni della molecola. Eccetto che per piccole regioni terminali di catena che pendono dalla zona reticolata principale, gli atomi delle regioni random sono tanto ben definiti strutturalmente quanto ogni altra parte della molecola. É stato detto che i numeri interi richiesti per specificare il numero irrazionale π sembrano trovarsi in una sequenza random, mentre nello stesso tempo la sequenza, ovviamente, è unica. L'analogia con i campioni strutturali è molto grande. Le regioni random delle proteine globulari non sono stocastiche nel tempo né sono statisticamente distribuite su differenti molecole di proteine. Esse sono stocastiche solo in quanto mostrano un'immagine non ovvia per la mente umana.
Come è stato detto sopra, le conformazioni locali di un polipeptide hanno la stessa forma di distribuzione probabilistica dei contorni dell'energia in una mappa ϕ, ψ. È comprensibile che l'evoluzione delle molecole di proteine globulari abbia proceduto lungo linee che mantengono l'energia conformazionale di regioni casuali entro fluttuazioni moderate al di sopra di valori minimi. Se così è, la frequenza di popolazione di conformazioni locali deve cadere in una regione formata come i contorni di energia più bassa della mappa ϕ, ψ. Ciò risulta vero, come mostra la fig. 19, dove l'incidenza di conformazioni locali in regioni non ordinate è sovrapposta a una mappa dei contorni di energia standard. La correlazione è inequivocabile. La presenza di regioni di molecole proteiche aventi le distribuzioni conformazionali rassomiglianti a catene stocastiche è stata stabilita da misure di rotazione ottica tra il 1950 e il 1955.
7. Strutture tridimensionali.
La struttura delle macromolecole può essere molto complessa. La fig. 20, per esempio, mostra la struttura di una proteina relativamente piccola e semplice, la ribonucleasi. All'inizio degli anni cinquanta K. Linderstrøm-Lang introdusse una divisione euristica della struttura proteica in aspetti primari, secondari e terziari. Questa terminologia è stata estesa ad aspetti di ordine superiore della struttura molecolare ed è ancora usata estensivamente come aiuto concettuale.
La struttura primaria di un polimero specifica la natura chimica dei blocchi costitutivi e la loro sequenza nella catena o nelle catene covalenti. La struttura secondaria specifica il tipo di periodicità che è presente nella struttura. Per esempio, specificazioni comuni di strutture secondarie nelle proteine sono l'α-elica, la struttura β a foglio pieghettato e quella non ordinata o random. Questo linguaggio ci permette di applicare il concetto importante di ordine elicoidale a strutture più complesse. La struttura terziaria riguarda la giustapposizione di unità secondarie nello spazio. Queste definizioni sono esemplificate nella discussione sulle proteine e sul tRNA riportata sotto. Esse si riferiscono a molecole singole che possono contenere una o più catene. La struttura quaternaria va un gradino al di là e tratta la relazione geometrica tra una macromolecola e l'altra. Esempi di struttura quaternaria si trovano nei Virus, nelle subunità di enzimi (importanti nel controllo biologico), nei ribosomi, ecc. L'argomento di un più alto ordine di struttura sarà trattato solo superficialmente nel paragrafo che segue.
Nella discussione sulla struttura terziaria, per la prima volta veniamo alle prese con il problema di strutture che devono presentare una disposizione soddisfacente nelle tre dimensioni. Le eliche multiple e le strutture β discusse sopra imposero semplicemente delle restrizioni sul tipo di passo elicoidale permesso. La formazione di un'elica è essenzialmente un processo lineare. Il salto a considerazioni tridimensionali aggiunge complessità al problema.
Un'idea della complessità della situazione si può avere come segue. Supponiamo una catena polipeptidica di lunghezza standard, circa 150 unità, e supponiamo inoltre che ogni unità possa avere un ordine di quattro conformazioni per unità, corrispondenti alle quattro regioni maggiormente permesse nella mappa ϕ, ψ. Ignorando le conformazioni escluse dove le unità si sovrappongono, si ha un totale di 4150≈2•1090 conformazioni. Se questa catena polipeptidica ha la sequenza adatta per formare una proteina globulare, la sua struttura naturale sarà una sola (o al massimo poche) di queste strutture potenziali. Se la molecola cambia conformazione alla velocità di una vibrazione molecolare, cioè ~1013 volte per secondo, e le conformazioni sono scelte a caso, un semplice calcolo mostra che essa impiegherà un tempo dell'ordine di 1069 anni per raggiungere la corretta struttura ‛nativa'. Questo sorpassa l'età stimata dell'universo di parecchie dozzine di ordini di grandezza. Chiaramente le cose non vanno in questo senso, dato che proteine di questa misura hanno mostrato di trovare la loro conformazione nativa in tempi dell'ordine di millisecondi. Un chimico teorico che voglia prevedere la struttura di una macromolecola tridimensionale e che non abbia idee chiare su come selezionare le strutture che stanno per raggiungere la struttura finale, deve affrontare questo tipo di situazione. L'opinione corrente su questo problema è che il ripiegamento della catena segua un percorso cinetico rapido determinato dalla struttura primaria.
Le proteine globulari e i tRNA costituiscono due esempi in cui la struttura tridimensionale è di importanza dominante. Il primo campo ha raggiunto un grado di sviluppo notevole, mentre il secondo è ancora agli inizi.
a) Proteine.
Le proteine globulari e gli enzimi costituiscono una delle più importanti classi di macromolecole biologiche. I fattori che determinano la struttura tridimensionale di queste molecole sono forse i più complessi di tutta la chimica strutturale. La situazione potrebbe essere senza speranza se non ci fosse il fatto davvero rimarchevole che molte di queste molecole formano cristalli che sono ordinati fino al livello delle dimensioni atomiche. Questo fu rilevato per la prima volta nel 1934 da J. D. Bernal e D. Crowfoot, che immediatamente ne trassero due deduzioni che avrebbero portato lontano: 1) le proteine globulari hanno gli atomi disposti in un unico disegno tridimensionale; 2) questa struttura tridimensionale può in linea di principio essere rilevata mediante la tecnica basata sulla diffrazione dei raggi X. Questa meta fu raggiunta circa venticinque anni più tardi da M. Perutz, J. Kendrew e i loro collaboratori con le molecole, simili fra loro, dell'emoglobina e della mioglobina.
Da un punto di vista superficiale lo studio della conformazione delle proteine può essere diviso in tre gruppi: studi di diffrazione dei raggi X su cristalli di proteine; applicazione dei principi dell'analisi conformazionale a peptidi e proteine; elaborazione di tecniche sperimentali (dicroismo circolare, risonanza magnetica nucleare, fluorescenza, spettrofotometria nell'infrarosso, ecc.) per determinare le proprietà conformazionali dei sistemi in soluzione. Quest'ultimo argomento è troppo ampio per essere discusso qui, ma una delle sue principali motivazioni è il problema dei cambiamenti conformazionali che accompagnano i fenomeni biologici, mentre le tecniche di diffrazione dei raggi X si applicano soltanto a sistemi statici.
Dopo l'originale determinazione della struttura della mioglobina effettuata da Kendrew e collaboratori, è stata determinata la struttura di circa 35 altre proteine con il metodo della diffrazione dei raggi X. La struttura di una di queste (la ribonucleasi) è riportata nelle figg. 20A e 20B. Benchè molte caratteristiche di questa struttura siano correlate in maniera specifica con l'attività biologica della molecola, vi è un certo numero di aspetti conformazionali generali che sembrano essere comuni a molte proteine. Alcuni di questi possono essere così elencati.
1. Nella struttura sono presenti α-eliche che tendono a essere brevi e distorte nelle regioni terminali. Le eliche sembrano terminare per rilassamento della simmetria rigorosamente elicoidale. La frazione di residui che è avvolta in α-eliche varia da quasi zero a circa il 75%.
2. Strutture a fogli bidimensionali (strutture β) esistono nelle proteine, ma sono generalmente molto piccole e irregolari. Inoltre, invece di essere planari come nelle fibre β, di solito si presentano come fogli distorti.
3. Essenzialmente tutte le conformazioni locali cadono entro le regioni permesse del diagramma ϕ, ψ.
4. Cambiamenti nella direzione della catena sono compiuti, di norma, solamente da poche conformazioni locali della catena stessa. Una completa inversione di direzione nella catena può avvenire quando sono fissati ϕ e ψ propri di due residui consecutivi. Se sono correttamente collocati, questi angoli orientano tre gruppi peptidici in modo che risulta possibile un legame a idrogeno tra il >C=O del primo e l'>N−H del terzo. Tali spire sono talvolta indicate come spire β perché si manifestano frequentemente in lamelle β antiparallele.
5. Le interazioni di catene laterali l'una con l'altra e con lo scheletro peptidico sono molto complicate. Le cariche sono essenzialmente all'esterno della molecola, con i gruppi polari che tendono a essere esterni e i gruppi idrofobici, invece, interni. I donatori per il legame a idrogeno nell'interno della molecola sono generalmente legati tra loro mediante legame a idrogeno. I residui aromatici tendono a essere vicini gli uni agli altri. Praticamente tutto lo spazio nell'ambito molecolare è riempito di componenti proteici, cioè c'è poco spazio per il solvente o altre occlusioni.
È consigliabile mettere in correlazione la sequenza di amminoacidi di una proteina con le strutture secondaria e terziaria della molecola. Questo può essere effettuato con modelli, se sono disponibili, oppure parzialmente con foto o con disegni di modelli, o con una mappa di struttura del tipo mostrato nella fig. 21A per la ribonucleasi. Queste ‛mappe a ‛triscia' (strip maps) danno una pronta rappresentazione del ruolo che ogni amminoacido della sequenza riveste nella struttura globale, benché vadano perduti gli aspetti spaziali, ricavabili dalla mappa ϕ, ψ della fig. 21B.
La massa di informazioni complesse contenute nella struttura dettagliata delle numerose proteine che sono state risolte è veramente imponente e passerà molto tempo prima che ne vengano ricavate tutte le possibili importanti conseguenze. Parecchi ricercatori, che inizialmente erano preoccupati per la determinazione della struttura, fine a se stessa, ora si stanno ponendo importanti domande circa la funzione delle proteine esaminate, studiando cristalli che non solo contengono molecole di proteine, ma substrati, inibitori, coenzimi, attivatori e simili.
Nel frattempo, l'analisi conformazionale teorica ha fatto grandi passi. Iniziato con i calcoli sui dipeptidi e sulle eliche visti sopra, il lavoro si è esteso a strutture meno regolari. Ci sono numerosi peptidi ciclici naturali relativa- mente piccoli che rivestono svariati ruoli biologici. Essi presentano un problema di ordine intermedio tra quelli di un dipeptide e di una proteina e non solo sono interessanti per se stessi, ma costituiscono anche un terreno di prova per metodi che è sperabile possano poi applicarsi alle proteine. I risultati per tali molecole sono stati finora deludenti, eccetto quando le strutture erano già state parzialmente determinate per via sperimentale. C'è, infatti, un gran numero di minimi di energia per una molecola con una decina di coppie di angoli ϕ, ψ e i programmi per calcolatori redatti finora possono trovare i minimi, ma non hanno la capacità di trovarli tutti nè di paragonarli fra loro.
Gli studi sulla diffrazione dei raggi X con cristalli di una proteina e le analisi conformazionali sono stati più strettamente collegati di quanto non sia stato indicato fin qui. I risultati ottenuti dai raggi X sono stati la fonte tradizionale dei dati numerici di partenza per i calcoli conformazionali. Inoltre, un esteso confronto di predizioni conformazionali con accurate informazioni cristallografiche ha condotto a un continuo miglioramento per quanto riguarda l'accuratezza dei campi di forza intramolecolari e intermolecolari. Infatti uno degli argomenti più studiati è ora l'analisi conformazionale dei cristalli. La struttura dei cristalli è generalmente nota in tutti i suoi particolari; ora è oggetto di studio lo sviluppo di funzioni potenziali molto accurate che possano render conto dell'intera struttura geometrica della cella elementare. Queste funzioni possono essere successivamente applicate a sistemi incogniti. Nello stesso tempo le determinazioni con i raggi X sono molto avvantaggiate dai risultati dell'analisi conformazionale. Ciò è dovuto al fatto che i risultati degli studi della diffrazione sulle proteine globulari sono meno precisi di almeno un ordine di grandezza di quelli sulle piccole molecole. Ne segue che di solito ci sono regioni nella struttura di una proteina, particolarmente nelle regioni meno ordinate, dove le principali caratteristiche della catena polipeptidica sono discernibili, ma non lo è la conformazione locale. In questo caso la struttura è spesso stabilita entro limiti ragionevoli costruendo modelli, dove si collocano quanti più residui è possibile in regioni di bassa energia del piano ϕ, ψ. Questo ottimizza la selezione degli angoli di torsione. Recentemente al problema dei cristalli di proteine globulari sono state applicate tutte le risorse dell'analisi conformazionale. Si determina inizialmente la struttura nel modo consueto, costruendo un modello. Le strutture così determinate contengono numerosi cattivi contatti a causa dell'uso di modelli reticolari. Per la rappresentazione dei raggi di van der Waals sono richiesti modelli tridimensionali, ma sarebbe impossibile lavorare con questi nella costruzione del modello iniziale. Inoltre sono state imposte numerose restrizioni artificiali: l'assoluta planarità del legame peptidico, tutti gli angoli e la lunghezza del legame al loro valore standard o vicino a esso, ecc. L'energia dell'intera struttura è formulata usando le equazioni (1) e (2) e la struttura può rilassarsi (nel calcolatore) fino a un minimo di energia. Questo fornisce una struttura rifinita che mantiene le caratteristiche del modello originario, ma con le numerose incongruenze interne eliminate o almeno diminuite. Il problema dei minimi multipli di energia non è così difficile come nel caso generale, poiché una buona struttura iniziale è già presumibilmente nella zona di bassa energia limitata inferiormente dal minimo di energia che si ha nel cristallo. Questo sistema altamente sofisticato può condurre non solo al perfezionamento della struttura determinata ai raggi X, ma anche a una migliore comprensione dell'energetica delle proteine. Qualche volta, però, la struttura calcolata non corrisponde perfettamente alla mappa delle densità elettroniche mentre si abbassa l'energia. Ciò accade perché le funzioni potenziali sono solo approssimate. L'ultimo approccio consiste in un procedimento di minimizzazione dell'energia che usa la mappa delle densità elettroniche per imporre delle limitazioni, ma questo costituisce un enorme problema di calcolo.
Gli sviluppi nella comprensione della conformazione delle proteine hanno preso di recente una direzione insospettata. Alcune notizie storiche possono essere di aiuto per valutare la nuova situazione. Nella concezione originale di Pauling, le strutture secondarie tipo α-elica o quelle β planari avevano una grande stabilità grazie all'esteso numero di legami a idrogeno all'interno delle molecole. È noto tuttavia che strutture con legami a idrogeno sono solo limitatamente stabili o addirittura instabili in acqua. La ragione è che i legami a idrogeno che si spezzano nella rottura di una struttura secondaria vengono sostituiti da legami a idrogeno con le molecole di acqua fortemente polari, e questi nuovi legami sono stabili all'incirca quanto quelli originali. A causa della bassa stabilità termodinamica del legame a idrogeno in acqua, al loro posto si considerano oggi come stabilizzanti delle proteine certi legami idrofobici. Un legame idrofobico è costituito dall'interazione di due gruppi non polari in soluzione acquosa. La forza che ne determina la formazione è data dal vantaggioso abbassamento del contatto tra acqua e gruppi non polari. Dal momento che tutti i gruppi idrofobici nelle proteine si trovano nelle catene laterali, ne deriva che la stabilità strutturale delle proteine globulari è connessa principalmente con interazioni terziarie. Se ne concluse, in un primo tempo, che l'analisi conformazionale di una proteina globulare dovesse necessariamente comprendere l'intera molecola proteica per poter rendere conto della rete tridimensionale di interazioni all'interno delle unità strutturali secondarie e fra di esse.
Recenti lavori hanno mostrato che possono essere fatti progressi considerevoli in questo problema ignorando le interazioni terziarie e concentrandosi sulle sequenze locali nella catena polipeptidica. Ci sono molti modi in cui questo può essere fatto, ma il seguente procedimento è di provata efficacia. Conoscendosi le strutture di circa 35 proteine globulari, è possibile fare un'analisi statistica della presenza dei vari amminoacidi nelle regioni elicoidali, β, random, ecc. Sulla base di queste analisi per ogni amminoacido, possono essere assegnati ‛pesi' statistici con riferimento alla tendenza a dare strutture elicoidali, planari, ecc. Chiaramente questi pesi non hanno valore deduttivo se gli amminoacidi in una sequenza sono osservati uno alla volta, poiché la formazione di un'unità della struttura secondaria richiede la partecipazione di un certo numero di residui di amminoacidi. Per esempio, un residuo con forte tendenza alla formazione di una struttura elicoidale sarà completamente annullato se è circondato da residui che sono riluttanti a formare eliche. Di conseguenza devono essere fissate le regole per includere il peso statistico dei residui vicini. Un mezzo per fare questo è di aggiungere i pesi statistici dei residui vicini da entrambi i lati, così da permettere a essi di rinforzare o annullare la tendenza strutturale del residuo mediano. Le regole per trattare il problema dei residui vicini e i pesi statistici stessi vengono variate per dare una corrispondenza ottimale con una serie di proteine; queste regole e questi parametri sono poi applicati per la predizione di strutture secondarie di un'altra serie di proteine conosciute. La corrispondenza tra i risultati così previsti e quelli sperimentali è straordinariamente buona. Le unità strutturali che sono riferibili a questo tipo di analisi sono l'α-elica, le strutture β planari e β piegate. Le regioni che non rientrano in una di queste categorie sono classificate come non ordinate o random. Il metodo è sufficientemente semplice da permettere a un ricercatore di prevedere, da una sequenza primaria, le unità secondarie in un'ora di lavoro. Il procedimento può, naturalmente, essere applicato a proteine sconosciute. Per esempio, il metodo fu applicato al lisozima del fago T4 prima che ne fosse determinata la struttura ai raggi X. La corrispondenza fra i residui previsti e quelli osservati nello stato elicoidale fu di circa il 70%. L'accordo è di solito migliore di questo per le eliche, ma non per le strutture β planari. Questo tipo di previsione della struttura riguarda soltanto gli aspetti più grossolani della struttura proteica. Poiché la struttura terziaria non è inclusa e le deduzioni circa la struttura secondaria sono statistiche, le previsioni sono di poco aiuto, per esempio, a un enzimologo che stia cercando di stabilire un meccanismo sulla base della struttura di un centro attivo. D'altro canto, i risultati sono di grande importanza per comprendere le forze che mantengono le strutture delle proteine globulari e forse il modo in cui queste strutture sono formate. In primo luogo, questo tipo di analisi non potrebbe funzionare se le conformazioni non fossero determinate sia localmente sia globalmente. Sembra che i requisiti per la più favorevole struttura secondaria e terziaria si rafforzino l'uno con l'altro. Questa situazione può essere chiarita con un esempio. Supponiamo che in un'elica di un enzima sia richiesta una determinata sequenza di amminoacidi. Il requisito locale per questa struttura sarebbe che il tratto di catena fosse costituito prevalentemente da amminoacidi che tendono verso l'elica. Poiché ci sono circa dieci ditali amminoacidi, c'è un gran numero di sequenze che soddisfarebbero questa richiesta. I requisiti per la struttura terziaria sono associati con le interazioni delle catene laterali: legami idrofobici, legami a idrogeno delle catene laterali, tutti gli ioni all'esterno piuttosto che all'interno della proteina dove provocherebbero instabilità, ecc. Il successo ditali schemi di previsione indica che ambedue le specie di restrizioni sono soddisfatte allo stesso tempo. Anche dopo aver soddisfatto questi due gruppi di restrizioni, rimane ampio spazio per variazioni, come è messo in evidenza dal gran numero di specie e di varianti mutanti della struttura primaria che producono la stessa struttura dello scheletro principale della molecola.
Questo risultato potrebbe significare che le proteine si sono evolute in modo tale da dare un'ottimizzazione sia della stabilizzazione locale sia di quella globale. Alcuni ricercatori pensano che ciò sia una manifestazione del meccanismo di ripiegamento delle proteine, che è generalmente rapido. Un interessante meccanismo proposto per un rapido ripiegamento è costituito dalla nucleazione da parte di unità di struttura secondaria formatesi transitoria- mente. In questo modo e richiesta una stabilità locale per la formazione dei nuclei e le strutture secondarie risultanti vengono incorporate nella struttura finale perché esse sono un gradino del più efficiente meccanismo di ripiegamento. Se quest'ultima congettura è vera, potrebbero esserci altre strutture di proteine più stabili delle molecole attive, ma che sono irrilevanti perché realizzate attraverso percorsi cinetici troppo lenti per essere produttivi.
Questi problemi sono oggi ancora lontani dall'essere risolti. In ogni caso la dipendenza della struttura secondaria dalla struttura primaria è stata un dono inaspettato che semplifica considerevolmente quello che potrebbe essere il problema conformazionale più complicato nei sistemi biologici.
b) Transfer RNA.
Le strutture degli acidi nucleici discusse precedentemente sono tutte connesse con molecole che formano strutture fibrose, come il DNA, e queste risultano essere eliche complementari doppie o triple. Ci sono altre forme di acidi nucleici per cui strutture elicoidali semplici o doppie non sembrano appropriate. Queste sono l'RNA messaggero, l'RNA ribosomiale e quello transfer (o tRNA). L'RNA messaggero è evidentemente una molecola stocastica a elica singola, senza un frequente appaiamento delle basi. La struttura dell'RNA ribosomiale non è conosciuta, ma esiste come parte di un elaboratissimo complesso nucleoproteico. D'altra parte il tRNA ha una struttura terziaria e in qualche modo assomiglia a una proteina globulare nelle sue caratteristiche strutturali e nel sistema di supporto. Fin dall'inizio si sospettò che il tRNA avesse una struttura tridimensionale a causa delle sue proprietà fisiche (che indicavano una molecola compatta con un frequente appaiamento delle basi) e del suo ruolo biologico consistente nel presentare una tipica tripletta (‛anticodone') complementare a un'altra presente nell'RNA messaggero sul ribosoma. I processi simultanei di trasferimento e di riconoscimento da esso esplicati richiedono un ordine strutturale. In particolare, una semplice struttura a doppia elica nasconderebbe l'anticodone, mentre una struttura a elica singola presenterebbe molte triplette che sarebbero viste dal ribosoma come anticodone.
Come è stato detto prima, le regole per le previsioni conformazionali degli acidi nucleici a un livello sommario sono piuttosto semplici. Si considera la struttura primaria e si avvolge la catena per formare il maggior numero possibile di regioni complementari a doppia elica. Questo è un tentativo come la previsione conformazionale per le proteine, con la differenza che la base è considerevolmente più sicura. In primo luogo la formazione della doppia elica da parte degli acidi nucleici è una forza termodinamica molto forte in acqua (una catena di 6 unità formerà una doppia elica se può trovare la sua complementare) e, inoltre, la formazione dell'elica implica requisiti specifici piuttosto che tendenze statistiche. Nella loro pubblicazione originale sulla prima struttura primaria per un tRNA, R. W. Holley e altri (1965) delinearono tre possibili topologie, una delle quali (v. fig. 22), si è dimostrata essenzialmente corretta. Notiamo che questa struttura indica doppie eliche, sacche, codoni, siti per l'attacco di amminoacidi, ecc., ma non dà una chiara idea della forma complessiva della molecola, nè alcun dettaglio tridimensionale, eccetto quelli imposti dall'accoppiamento delle basi. Nonostante ciò le possibilità di previsione sono notevoli e hanno guidato una gran parte delle successive ricerche chimiche in questo campo.
Dopo molti tentativi è stata trovata la possibilità di cristallizzare alcuni tRNA. Da allora la previsione della struttura tridimensionale di un tRNA è stata soltanto questione di tempo, benché il problema tecnicamente fosse di un'estrema difficoltà. Strutture tridimensionali quasi identiche del tRNA della fenilalanina sono state riportate da gruppi di ricercatori (S. H. Kim e altri, J. A. Robertus e altri) quasi simultaneamente (estate 1974) e sono mostrate nella fig. 23. La struttura è essenzialmente quella a quadrifoglio della fig. 22, sebbene ciò non si veda facilmente paragonando le due figure. Questa nuova informazione aprirà senza dubbio una nuova era nell'analisi conformazionale degli acidi nucleici.
c) Strutture di ordine superiore.
La gerarchia delle caratteristiche strutturali va ancora oltre per le molecole biologiche. Ci sono molti esempi in cui una molecola con le sue proprie strutture primaria, secondaria e terziaria è semplicemente un'unità di una struttura più altamente ordinata. Le molecole di collageno e di miosina, discusse nel paragrafo sulle eliche multiple, sono combinate per formare le fibrille del tessuto strutturale e gli spessi filamenti delle cellule del muscolo. I virus come il virus del mosaico del tabacco e il virus fd (un batteriofago) consistono di gruppi di singole unità contenenti proteine globulari e RNA o DNA, avvolte in spirali. I fagi sono strutture molto intricate costituite da molte proteine diverse, ognuna delle quali ha uno specifico ruolo strutturale. Gli enzimi, specialmente quelli implicati in una serie di reazioni con un meccanismo di controllo, spesso non funzionano come singole molecole, ma come aggregati, strutturati con precisione, di 2, 3, 4, 6, ecc. molecole di enzima singolo, che possono essere identiche o non esserlo nella struttura primaria o secondaria. Il ribosoma, che sintetizza le proteine utilizzando un RNA messaggero per il codice della sequenza e gli amminoacidi legati ognuno al proprio tRNA, contiene almeno 50 diverse proteine.
Questi elementi strutturali che descnvono le posizioni relative delle già complesse macromolecole sono chiamati struttura quaternaria. Attualmente c'è un evidente tenta- tivo di unire le serie ascendenti del chimico atomo, blocco costitutivo, struttura primaria, struttura secondaria, strutture terziaria e quaternaria con le serie discendenti del biologo: organismo, organo, cellula, organello. Per la maggior parte dei sistemi la struttura quaternaria può essere discussa soltanto dal punto di vista concettuale, a causa della mancanza di dettagliate informazioni strutturali. Un'eccezione è l'emoglobina, la cui molecola fu una delle prime per cui si ottenne la struttura tridimensionale. L'emoglobina è un tetramero e il meccanismo per un'efficiente assunzione e cessione dell'ossigeno richiede la azione cooperativa di tutt'e quattro le subunità. I particolari del meccanismo molecolare di questo processo operativo sono attualmente in corso di elaborazione. Un altro caso è costituito dagli enzimi ‛deidrogenasi', che cristallizzano come interi aggregati piuttosto che come singole molecole. Gli studi sulla diffrazione dei raggi X stanno iniziando a rivelare le relazioni conformazionali delle molecole di monomero di enzima tra di loro e il modo con cui esse interagiscono con substrato, coenzimi, inibitori, ecc. Il ruolo della struttura quaternaria nei meccanismi di feedback e di controllo delle reazioni enzimatiche è attualmente uno degli argomenti più interessanti dell'enzimologia. Benché durante gli studi della struttura quaternaria siano stati chiariti aspetti molto interessanti della conformazione delle macromolecole, il soggetto è troppo ampio per essere trattato a fondo nel presente contesto.
8. Orientamenti attuali.
Questo articolo sarà concluso con un'esposizione dello stato attuale dell'analisi conformazionale dei biopolimeri e degli orientamenti della ricerca futura. A causa della sua relativa semplicità concettuale e della grande quantità di dati sperimentali, il campo dell'analisi conformazionale dei peptidi viene fatto a mano a mano progredire fino ai livelli più sofisticati. Il lavoro negli anni sessanta fu in gran parte limitato allo studio di modelli in cui la lunghezza dei legami e gli angoli erano fissati nel calcolo e soltanto gli angoli di torsione potevano variare. Inoltre l'angolo di torsione del legame peptidico era fissato in posizione cis o trans. Recentemente ci sono stati tentativi per sviluppare un campo di forza generalizzato (‛superficie di Born-Oppenheimer') per il movimento dei nuclei di piccole molecole senza nessuna restrizione. I campi dell'analisi conformazionale e dell'analisi vibrazionale sono in questo modo combinati. Questo, sebbene aggiunga complessità ai calcoli, apporta anche una notevole potenzialità poiché si possono usare come dati sperimentali non solo quantità strutturali (lunghezze di legame, angoli, ecc.), ma anche interi spettri vibrazionali, come quelli determinati dalla spettroscopia Raman e infrarossa. Questo convincente avvicinamento ha avuto un marcato successo con le piccole molecole. I problemi inerenti alla sua applicazione alle grandi molecole sono costituiti dal tempo eccessivo richiesto dai calcolatori e dalla necessità di costituire gradualmente uno schedario di parametri per il calcolo basati su dati sperimentali, sia conformazionali che vibrazionali.
Come si è detto prima, l'analisi conformazionale è stata usata anche per costruire i modelli finali di strutture di proteine globulari determinate mediante diffrazione dei raggi X. Questo è un mezzo sistematico per ottenere dettagli strutturali al di là della risoluzione data dagli spettri di diffrazione. Sono stati fatti progressi lungo queste linee, ma il procedimento è oggi ancora lontano dalla routine.
I calcoli discussi in questo articolo sono essenzialmente classici, nel senso che per calcolare la forma della superficie dell'energia potenziale vengono usate funzioni empiriche. Allo stesso tempo si tenta attivamente di determinare la superficie dell'energia mediante la soluzione approssimata dell'equazione di Schrödinger come una funzione della conformazione nucleare. I ricercatori in questo campo sono orientati o verso il metodo quantistico o verso il metodo fenomenologico, e ci sono stati pochi tentativi di comparare realmente i due metodi per una ben determinata situazione sperimentale. Per la nostra trattazione abbiamo scelto il metodo fenomenologico, perché è oggi più familiare e più semplice.
Gli acidi nucleici stanno attraversando ora il loro secondo stadio di sviluppo. Appaiono regolarmente nella letteratura contorni di energia determinati accuratamente come funzione di una coppia di angoli diedri. La soluzione di questo problema è stata un po' ritardata dal gran numero di angoli diedri nella catena principale e dal problema della flessibilità dell'anello del ribosio. E stato calcolato che lo spazio conformazionale disponibile per una catena di poliribosofosfato è molto minore di quanto ci si aspetterebbe intuitivamente, in conformità con i risultati sperimentali sulla rigidità della catena. Recentemente sono state sviluppate buone teorie sulla flessibilità dell'RNA e del DNA e sono state scoperte numerose interessanti restrizioni strutturali sugli angoli di torsione. Questo argomento è troppo lungo perché possa essere trattato qui.
Come è stato detto prima, è stata compiuta recentemente la determinazione della struttura cristallina del tRNA della fenilalanina. L'analisi dettagliata di questa struttura darà alle analisi conformazionali degli acidi nucleici lo stesso incentivo dato allo studio dei polipeptidi dalla struttura delle proteine globulari.
S. Arnott e i suoi collaboratori sono passati ad analizzare la struttura di tutti gli importanti sistemi di fibre (α-eliche, strutture β, A-DNA, B-DNA, RNA, ecc.) usando tecniche di ottimizzazione simultanea per l'accordo con lo spettro di diffrazione della fibra e con le costrizioni molecolari stabilite. Le strutture così determinate differiscono nei dettagli da quelle delle prime ricerche e sono oggi considerate le più attendibili.
C'è un grandissimo interesse nell'applicazione di analisi conformazionali ai sistemi di proteine e di enzimi aventi struttura quaternaria. I più grandi progressi sono stati fatti con l'emoglobina. Il meccanismo dell'anemia a cellule falciformi e di malattie analoghe (v. sangue: Anemie emolitiche ed Emoglobina) è stato ora messo in relazione con le anomalie conformazionali nell'emoglobina causate da cambiamenti, indotti da mutazioni, nella struttura primaria. Questo tipo di lavoro può procedere soltanto con buoni dati diffrattometrici sulle proteine. Tali dati stanno ora divenendo disponibili per gli enzimi allosterici, cioè quelli che mostrano controllo, feedback e cooperatività. Questi processi vitali sono manifestazioni di cambiamenti nella struttura terziaria e quaternaria innescati da molecole chiave (substrati, coenzimi, inibitori, induttori). Oggi è quasi impossibile leggere una pubblicazione sul meccanismo d'azione degli enzimi senza incontrare il concetto di variazione conformazionale.
Infine si sta facendo una gran quantità di lavoro per stabilire le relazioni tra misure spettrali e conformazione, e ciò porta a una combinazione di teorie spettrali e conformazionali che va al di là dei limiti di questa rassegna. Citiamo un esempio da un recente lavoro per dimostrare il tipo di diagrammi di correlazione che sono ora prodotti e usati. Un diagramma del genere è mostrato nella fig. 24, che riproduce il grafico dell'entità del dicroismo circolare della banda d'assorbimento tipica dei peptidi, alle lunghezze d'onda più elevate, in funzione degli angoli conformazionali, ϕ e ψ.
Non c'è nessuna tecnica fisica per la determinazione della struttura che non abbia aumentato la propria potenza e la propria raffinatezza in seguito ai progressi nelle analisi conformazionali delineati sopra.
Bibliografia.
Arnott, S., Hukins, D. W. L., Optimised parameters for A-DNA and B-DNA, in ‟Biochemical and biophysical research communications", 1972, XLVII, pp. 1504-1509.
Dickerson, R. E., Geis, I., The structure and action of proteins, New York 1969 (tr. it.: Struttura e funzione delle proteine, Bologna 1973).
Holley, R. W., Apgar, J., Everett, G. A., Madison, J. T., Marquisee, M., Merrill, S. H., Penswick, J. R., Zamir, A., Structure of a ribonucleic acid, in ‟Science", 1965, CXLVII, pp. 1462-1465.
Kim, S. H., Suddath, F. L., Quigley, G. J., McPherson, A., Sussman, J. L., Wang, A. H. J., Seeman, N. C., Rich, A., Three-dimensional tertiary structure of yeast phenylalanine transfer RNA, in ‟Science", 1974, CLXXXV, pp. 435-440.
Marchessault, R. H., Sarko, A., X-ray structure of polysaccharides, in Advances in carbohydrate chemistry (a cura di W. W. Pigman e M. L. Wolfrom), vol. XXII, New York 1967, pp. 421-482.
Marvin, D. A., Spencer, M., Wilkins, M. H. F., Hamilton, L. D., The molecular configuration of deoxyribonucleic acid. III. X-ray diffraction study of the C form of the lithium salt, in ‟Journal of molecular biology", 1961, III, pp. 547-565.
Ramachandran, G. N., Sasisekharan, V., Conformation of polypeptides and proteins, in Advances in protein chemistry (a cura di M. L. Anson e J. T. Edsall), vol. XXIII, New York 1968, pp. 283-437.
Robertus, J. A., Ladner, J. E., Finch, J. T., Rhodes, D., Brown, R. S., Clark, B. F. C., Klug, A., Structure of yeast phenylalanine t. RNA at 3 Å resolution, in ‟Nature", 1974, CCL, pp. 546-551.
Yathindra, N., Sundaralingam, M., Conformational studies on pyrimidine 5′-monophosphates and 3′,5′-diphosphates. Effect of the phosphate groups on the backbone conformation of polynucleotides, in ‟Biopolymers", 1973, XII, pp. 2261-2277.