
Neuropatologia
Neuropatologia
di Fernando De Ritis e Charles E. Lumsden
NEUROPATOLOGIA
Malattie virali del sistema nervoso centrale
di Fernando De Ritis
sommario: 1. Eziologia. 2. Epidemiologia: a) arbovirus: encefalomielite equina occidentale (WEE); b) enterovirus: poliomielite; c) rabdovirus: rabbia; d) paramixo virus: parotite; e) coriomeningite linfocitaria; f) encefalite letargica. 3. Patogenesi generale delle infezioni virali del sistema nervoso centrale (SNC): a) propagazione dei virus nel SNC; b) la barriera ematoencefalica; c) la recettività della cellula nervosa; d) lesioni conseguenti alla penetrazione dei virus nel SNC. 4. Lesioni istologiche: a) quadri istopatologici fondamentali del sistema nervoso: lesioni mesodermiche ed ectodermiche; b) classificazione dei quadri istopatologici delle nevrassiti virali. 5. Lesioni biochimiche. 6. Patologia generale: a) modalità di danneggiamento dei neuroni; b) distribuzione topografica ed esiti delle lesioni. 7. Clinica: a) encefalomieliti equine americane; b) encefalite russa primaverile estiva; c) poliomielite; d) rabbia; e) coriomeningite linfocitaria; f) encefalite letargica; g) meningite da virus coxsackie; h) nevrassite da virus del vaiuolo; i) encefalite morbillosa; l) encefalite postvaccinica; m) nevrassite da virus herpes simplex; n) nevrassite da virus herpes zoster; o) nevrassiti in corso di varicella; p) nevrassiti in corso di polmoniti virali; q) nevrassiti in corso di influenza; r) meningiti da virus ECHO (Enteric Cytopathogenic Human Orpban); s) sintomatologie neurologiche in corso di dengue; t) sintomatologie neurologiche in corso di febbre da flebotomi; u) nevrassite in corso di mononucleosi infettiva; v) nevrassite da virus encefalomiocardite; w) infezioni del SNC da virus lenti: kuru; y) possibile eziologia da virus lenti (non ancora identificati) di alcune malattie neurologiche dell'uomo. 8. Considerazioni conclusive. □ Bibliografia.
1. Eziologia
Secondo l'accezione nosologica più usata si parla oggi non tanto di malattie del sistema nervoso centrale (SNC), quanto di malattie del nevrasse o nevrassiti: ciò perché tutte le strutture istologiche costituenti il nevrasse nel suo insieme (neuroni, glia, meningi, ecc.) e non solo gli elementi neuronali possono essere solidalmente interessate dalle lesioni di vario tipo causate da agenti virali. La dizione e il concetto di virus neurotropi - che lungamente hanno dominato questo capitolo della patologia - si sono venuti nettamente modificando in epoca recente, in conseguenza dell'acquisizione di taluni fatti non compatibili con la concezione che identifica questi agenti in virus capaci di parassitare esclusivamente il tessuto nervoso, e cioè: 1) i cosiddetti virus neurotropi sono in realtà dei virus pantropi, in quanto in determinate circostanze sperimentali (vie di inoculazione ed età adatte dell'ospite) e naturali possono causare lesioni in tessuti extranervosi; 2) molti virus ‛non neurotropi' sono capaci di determinare lesioni del tessuto nervoso (enterovirus, mixovirus, virus parotitico, ecc.); 3) virus considerati neurotropi obbligati sono stati coltivati in vitro su cellule di tessuti extranervosi; 4) virus non neurotropi possono essere sperimentalmente ‛neurotropizzati' (rabbia, febbre gialla, influenza); 5) virus abitualmente agenti di infezioni febbrili indeterminate possono occasionalmente determinare manifestazioni neurologiche (una gran parte degli arbovirus isolati da Artropodi in regioni tropicali).
Attualmente sono denominati neurotropi quei virus capaci di parassitare abitualmente o preferenzialmente il tessuto nervoso, il grado di preferenzialità essendo variabile nei singoli organismi. Oltre a questi, ne esistono altri per i quali la capacità di ledere il tessuto nervoso è un evento possibile ma infrequente o eccezionale. Taluni virus neurotropi hanno perduto la caratteristica di poter migrare dalla periferia al centro attraverso i nervi periferici e in generale, una volta raggiunto il SNC, di propagarvisi raggiungendo le strutture elettivamente recettive, esclusivamente per vie intermediarie, attraverso uno dei meccanismi che vengono di seguito illustrati. Appare dunque solidamente acquisito che manifestazioni morbose neurologiche possono essere provocate da numerosi tipi di virus, molti dei quali solitamente responsabili di altri tipi di malattie non neurologiche; che molti virus, una volta ritenuti strettamente neurotropi, possono invece provocare lesioni extranervose; infine, che nel corso di malattie tipicamente neurologiche sono riscontrabili, anche se clinicamente meno evidenti o inapparenti, lesioni extranervose.
In conclusione, i virus capaci di colonizzare o ledere il sistema nervoso (SN) sono raggruppabili nelle seguenti categorie: 1) virus con alto grado di preferenzialità per il SN, che possono essere a) trasmessi da Artropodi o b) non trasmessi da Artropodi; 2) virus con basso grado di preferenzialità per il SN, i quali pure possono essere a) trasmessi da Artropodi o b) non trasmessi da Artropodi; 3) virus con capacità eccezionale di colonizzare e ledere il SN.
Da un punto di vista epidemiologico essi possono essere classificati meglio così: 1) arbovirus (trasmissione attraverso Artropodi); 2) virus non trasmessi da Artropodi, a meccanismo epidemiologico: a) enterofecale; b) aerogeno; c) sconosciuto.

2. Epidemiologia
Riportiamo le situazioni epidemiologiche più estesamente studiate, relative a cinque gruppi di virus.
a) Arbovirus: encefalomielite equina occidentale (WEE)
Il termine di encefaliti da arbovirus - encefaliti trasmesse da Artropodi (Arthropod born) - fu introdotto da Hammon e Reeves nel 1943 per definire un vasto gruppo di malattie dell'uomo e degli animali, le cui caratteristiche possono essere così schematizzate: malattie umane, a carattere solitamente epidemico, legate a una complessa ecologia che investe i rapporti fra l'uomo e varie specie animali con particolari distribuzioni geografiche e incidenze stagionali, e con la comune caratteristica di essere trasmesse da artropodi ematofagi attraverso un complicato ciclo biologico. Questo comporta una serie di animali ospiti e di animali vettori che assicurano la persistenza del virus nell'ambiente naturale. I criteri che consentono di includere un virus nel gruppo degli arbovirus sono: 1) anzitutto, la dimostrazione della sua trasmissione naturale da Artropodi a Vertebrati e, in mancanza di tale prova, 2) la dimostrazione dell'infettabilità sperimentale degli Artropodi (per alcuni virus nei quali tale ciclo naturale non è noto); 3) l'affinità antigenica con virus per i quali è stata provata una delle precedenti caratteristiche (1, 2); 4) il frequente isolamento da Artropodi, anche in assenza della dimostrazione della trasmissibilità naturale o sperimentale da questi ai Vertebrati.
Questa definizione non è basata su intrinseche caratteristiche dei Virus, ma implica la comunanza di un ciclo biologico che coinvolge, oltre ai Virus, due classi di organismi: Artropodi e Vertebrati. Tale definizione pertanto, anche se tuttora utile e non sostituibile, ha carattere di provvisorietà ed è destinata a mutare quando le conoscenze sui singoli costituenti del gruppo degli arbovirus abbiano raggiunto un tale livello da consentire l'adozione di criteri più validi.
L'infezione dell'uomo è di solito un evento accidentale non necessario e non utile per il perpetuarsi della specie virale.
Gli arbovirus sono trasmessi dai membri di solo quattro famiglie di Artropodi ematofagi (Culicidae, Ceratopogonidae, Psichodidae, Ixodidae). I virioni hanno piccole dimensioni (17-50 mμ in 28 virus, 51-75 mμ in 7 virus, 76-125 mμ in 10 virus, 150 mμ o più in 2 virus), contengono RNA, sono patogeni per il topo per via endocerebrale. Diffusi in tutto il mondo nelle regioni a clima temperato e in quelle a clima tropicale, sono a sviluppo citoplasmatico (nessun arbovirus è stato trovato nel nucleo delle cellule), possiedono probabilmente un involucro lipidico come è suggerito dalla loro sensibilità all'etere e al desossicolato di sodio (120 virus su 125 saggiati), alcuni possiedono una membrana esterna (per es.: EEE, JE, WEE, TBE, Powassan, febbre emorragica di Omsk). Non sufficientemente noto è il tipo di simmetria dei virioni dei singoli virus: cubica per il virus della malattia africana del cavallo e per il virus Kemerovo, elicoidale per il virus della stomatite vescicolare e probabilmente per FEE. Di circa 250 virus noti come arbovirus, 200 sono classificati in 28 gruppi. Di essi almeno 51 sono patogeni per l'uomo. Nella grande maggioranza dei casi le infezioni naturali dell'uomo causate da questo gruppo di virus decorrono in forma inapparente o subclinica. Talvolta hanno il carattere di malattie infettive acute senza manifestazioni neurologiche, che decorrono come febbri indifferenziate benigne, talaltra si presentano con scarse manifestazioni neurologiche non diagnosticabili se non in occasione di episodi epidemici. Solo in una percentuale minima dei casi di infezione si sviluppa la sintomatologia neurologica, con variabile e talora elevata letalità. Ciò è stato fra l'altro dimostrato dalla diffusione di anticorpi sierici, che denuncia un numero di infezioni molto maggiore dei casi clinicamente evidenti.
L'epidemiologia generale delle nevrassiti da arbovirus può essere compendiata nei seguenti termini: nell'uomo le ondate epidemiche di queste malattie possono avere inizio brusco e durare poco tempo, talché il virus può sparire completamente dalle regioni colpite, mentre nei focolai enzootici la malattia può mantenersi nei periodi interepidemici. Secondo la teoria di E. N. Pavlovskij, nello stesso modo in cui un animale tende a vivere in un certo ambiente dove è in associazione con altre specie formando così una biocenosi (insieme di specie viventi fra loro collegate nell'ambito di un ecosistema), una malattia tende ad avere un habitat soprattutto se si tratta di un'affezione la cui trasmissione da un ospite definitivo a un altro non può compiersi se non attraverso l'intervento di un parassita invertebrato. Tali malattie hanno degli habitat naturali in eco- sistemi limitati dove gli ospiti definitivi, i parassiti vettori e gli agenti patogeni stessi fanno parte di una biocenosi nella quale circolano questi ultimi. Pavlovskij parla di una ‛epidemiologia del paesaggio' per sottolineare il fatto che il paesaggio (inteso come struttura geografica di una località) ha un'importanza epidemiologica, perché i suoi caratteri sono quelli di un tipo particolare di ecosistema. L'uomo, quando penetra in un ecosistema dove esiste una zoonosi, può inserirsi nel suo ciclo ed esserne infettato. Molto sovente il suo arrivo modifica l'ambiente in maniera tale da eliminare il focolaio morboso. Al contrario, quando ad esempio stabilisce un sistema di irrigazione, egli rischia di estendere la regione favorevole alla biocenosi nella quale sopravvive la malattia allo stato enzootico.
Particolarmente nota è l'epidemiologia dell'encefalomielite equina occidentale (WEE), che viene riportata come prototipo dell'epidemiologia degli arbovirus encefalitogeni. Le infezioni del cavallo e dell'uomo sono diffuse nelle regioni occidentali degli Stati Uniti con episodi sporadici o epidemici stagionali che ricorrono tra luglio e settembre. Alcuni casi della malattia sono stati segnalati anche in Canada, in Argentina e in vari Stati dell'America Centrale. Il più grave episodio epidemico si è verificato nella regione centrosettentrionale degli Stati Uniti nel 1941: vi furono oltre 3.000 casi diagnosticati con letalità oscillante tra l'8 e il 15%.
La malattia colpisce a preferenza i maschi adulti delle popolazioni rurali, con un piccolo numero di casi tra bambini e infanti. In alcuni episodi epidemici vi è stata però una larga morbosità tra i bambini di età inferiore a un anno; in questi piccoli pazienti la malattia è notevolmente più grave, con una letalità che talvolta supera il 50%.
La distribuzione geografica, gli episodi sporadici e gli episodi stagionali della malattia sono legati alla complessa ecologia del virus e dei suoi ospiti. La principale riserva naturale di virus WEE è costituita dagli uccelli selvatici nei quali il virus provoca una malattia inapparente della durata di pochi giorni, che si accompagna a prolungata ed elevata viremia. L'infezione è trasmessa da animale ad animale (ed eventualmente all'uomo) con la puntura di alcuni insetti ematofagi (zanzare dei generi Culex e Anopheles, taluni acari) dai quali è stato frequentemente isolato il virus. Il vettore principale deve essere considerato Culex tarsalis da cui il virus è stato isolato con la massima frequenza in aree epidemiche. La malattia è diffusa principalmente nelle regioni che costituiscono l'habitat di questa specie. Lo studio delle abitudini di Culex tarsalis, condotto con le classiche metodiche dello studio immunologico del sangue raccolto da esemplari catturati nel loro ambiente naturale, ha dimostrato che questa specie di insetto si nutre a preferenza di sangue di uccelli selvatici, ma talora anche di sangue di mammiferi selvatici o domestici o di sangue umano. Una probabile spiegazione della maggior frequenza con la quale Culex tarsalis punge gli uccelli è data dal fatto che questo insetto ha un tropismo positivo verso l'anidride carbonica: l'elevata concentrazione di CO2 tra il fogliame degli alberi, all'imbrunire, quando cessa la fotosintesi, e la contemporanea presenza nello stesso luogo di uccelli in riposo, rende agevole a Culex tarsalis nutrirsi con sangue di uccelli eventualmente infetti. Al pasto infettante segue una diffusione del virus nei tessuti dell'insetto ove si moltiplica fino a raggiungere una concentrazione tale da consentirne, in successive punture, la trasmissione ad animali sani in quantità sufficiente a dare inizio all'infezione (apparente o inapparente).
Solo per evento occasionale (legato di solito alla presenza di un gran numero di esemplari di Culex tarsalis) la malattia viene trasmessa al cavallo e all'uomo. Tanto il cavallo che l'uomo (le uniche specie nelle quali l'infezione con virus WEE trasmessa con la puntura di zanzara infetta provoca talvolta una malattia apparente) debbono essere considerati ospiti terminali: in essi infatti la malattia non si accompagna a una viremia che possa consentire la trasmissione del virus ad altri vettori e quindi ad altri animali. È probabile che gli acari siano responsabili in modo particolare della trasmissione del virus tra uccelli e roditori selvatici, che costituiscono forse un'altra importante riserva del virus. Difatti numerosi acari ematofagi nei mesi invernali si nutrono, allo stato adulto, a spese di roditori selvatici, mentre le loro larve, anch'esse ematofaghe, parassitano in primavera gli uccelli implumi nei nidi: in quest'ultimo caso l'infezione decorre con una viremia più elevata e prolungata che non negli uccelli adulti, così che gli uccelli implumi contribuiscono in maggior misura alla diffusione della malattia, anche perché essendo sprovvisti di piume e dotati di minore mobilità, sono più facilmente punti da Culex tarsalis.
Da quanto è stato esposto, appare evidente che il per- petuarsi del virus in natura è legato a questo complesso ciclo basale di trasmissione (uccelli-zanzare-uccelli-acariroditori-acari-uccelli).
La malattia dell'uomo e del cavallo o l'infezione di altri mammiferi sono eventi accidentali. Perché possano verificarsi episodi epidemici debbono sussistere una serie di concause climatiche e ambientali e in particolare una primavera precoce con un clima notevolmente umido. La primavera precoce provoca un anticipata maturazione delle larve di Culex tarsalis. Le zanzare adulte di questa specie, la sera, attirate tra il fogliame degli alberi dal CO2, trovano nei nidi uccelli ancora implumi, già infettati dalle larve di acari. L'elevata umidità consente alle zanzare una vita più lunga del consueto, con possibilità di portare a compimento più di un ciclo infettante. Se inoltre il numero di Culex tarsalis è notevolmente elevato, esistono maggiori probabilità che esemplari infetti pungano l'uomo o il cavallo provocandone l'infezione o la malattia.
La sopravvivenza del virus nei mesi invernali è probabilmente legata a molteplici fattori. La specie Culex tarsalis si perpetua per sopravvivenza, nei mesi invernali, di alcuni esemplari di femmine adulte. Queste si rifugiano di solito in grotte, cave o miniere buie e riparate dalle intemperie. Se l'insetto subito prima dell'inverno aveva consumato un pasto infetto, può conservare il suo potere infettante per periodi di tempo notevolmente lunghi (oltre 100 giorni). Gli acari infetti possono trasmettere il virus alla progenie per via transovarica. Altre riserve invernali del virus possono essere costituite dai roditori selvatici e dai pipistrelli. Esperienze condotte in laboratorio con altri arbovirus hanno dimostrato che numerose specie di pipistrelli sono suscettibili di infettarsi con la puntura di artropodi infetti. Se subito dopo la puntura infettante il pipistrello è messo in condizioni artificiali di ibernazione, il virus sopravvive allo stato latente per alcuni mesi. La viremia compare quindi al termine dell'ibernazione artificiale, quando la temperatura ambientale diviene elevata. Dev'essere anche prospettata la possibilità che il virus WEE venga importato in primavera da uccelli migratori infetti.
b) Enterovirus: poliomielite
Riportiamo l'epidemiologia della poliomielite come esempio di epidemiologia da enterovirus. Premesso che il virus è eliminato con le feci dai soggetti in fase acuta di infezione e assunto con alimenti comunque contaminati, ne consegue che quella poliomielitica è un'infezione enterica la cui diffusione segue la via oro-fecale.
L'esistenza di tre diversi tipi antigenici consente di considerare diverse almeno sotto certi aspetti (per es. sotto quello immunitario) le malattie determinate da ciascuno di essi, essendo possibile teoricamente che un soggetto guarito da poliomielite si ammali successivamente ancora di poliomielite determinata da un tipo diverso da quello agente della prima infezione. Ciò perché l'immunità che consegue alla malattia è tipo-specifica. Il contagio poliomielitico è diffusissimo, ma nella massima parte dei casi dà infezioni asintomatiche o comunque prive di sintomatologia neurologica.
La malattia poliomielitica si manifesta in un numero minimo di casi. La disseminazione del virus nell'ambiente, con le feci infette, avviene indipendentemente dal fatto che i pazienti presentino localizzazione neurologica: anzi, la difficile riconoscibilità dei soggetti senza malattia neurologica diventa particolarmente pericolosa dal punto di vista epidemiologico, e la loro grandissima prevalenza rispetto ai casi con manifestazioni paralitiche li rende, in pratica, i massimi responsabili della circolazione dei virus poliomielitici nell'ambiente umano. Conseguentemente alla grandissima diffusione delle infezioni da virus poliomielitico e al grandissimo numero di contatti subclinici avvenuti durante l'età infantile, nei paesi a livello igienico-sanitario basso o mediamente elevato, gli individui che hanno superato l'adolescenza sono praticamente immuni, come è dimostrato dal buon livello anticorpale presente nel loro sangue. Nei paesi a livello più elevato, così come nelle classi sociali più elevate di qualsiasi paese, essendo nell'infanzia minori le occasioni di contagio per la minore promiscuità e le più igieniche abitudini di vita, l'età recettiva si sposta oltre l'infanzia fino all'adolescenza e alla giovinezza: è quanto si è verificato in alcuni paesi a elevato tenore sociale ed economico (Stati Uniti, Svezia, ecc.). Per motivi analoghi, mentre nella popolazione rurale, che è più dispersa, l'incidenza della malattia dal secondo anno in poi si mantiene praticamente costante fin verso i 20 anni per poi decrescere gradualmente, nella popolazione urbana, più concentrata, l'acme dell'incidenza si verifica tra il 20 e il 60 anno ed è a partire da tale età che ha inizio il graduale decremento. I soggetti in età fra i e 5 anni costituiscono probabilmente la riserva permanente del virus.
Va tenuto presente che nelle regioni temperate la malattia assume carattere endemico e che l'incidenza della forma paralitica non segue - nonostante la dimostrata eliminazione del virus con le feci da parte dei soggetti infetti - il comportamento stagionale di altre infezioni enteriche come ad esempio la febbre tifoide.
Lo studio dei cosiddetti casi ‛secondari', contagiatisi cioè direttamente dai primi casi epidemici comparsi, per trasmissione diretta del virus, fa rilevare un periodo di incubazione paragonabile a quello durante il quale il virus è presente nella cavità faringea dei casi ‛primari'. Ciò concorda, pertanto, con l'ipotesi che i casi secondari si siano contagiati per trasmissione respiratoria delle secrezioni infette dai casi primari.
c) Rabdovirus: rabbia
La malattia è diffusa in tutto il mondo e colpisce non soltanto il cane ma ogni altro mammifero domestico o selvatico. Al cane spetta però il ruolo di trasmettitore della malattia all'ospite umano, nel quale essa si sviluppa in non più del 10-20% dei casi di morsicature, probabilmente in rapporto alla localizzazione della ferita provocata dal morso. I morsi più pericolosi sono quelli al volto o al collo; meno pericolosi sono quelli agli arti, specie inferiori. Anche l'incidenza della letalità è in rapporto con la sede del morso, perché nei soggetti morsi al collo è 10 volte più frequente che in quelli morsi agli arti superiori e 28 volte più frequente che in quelli morsi agli arti inferiori.
È probabile che esistano portatori di virus clinicamente indenni, come è dimostrato dall'insorgenza della malattia nell'uomo o in animali dopo morsi di pipistrelli e dall'osservazione dei corpi del Negri nel tessuto cerebrale di cani sani morti per incidenti stradali. La mancata insorgenza dell'infezione in uomini morsi da animali sicuramente rabici può essere anche in rapporto alla possibilità che la saliva di questi al momento in cui aggredirono l'uomo non contenesse virus.
La mortalità complessiva per rabbia fra soggetti morsi da animale rabico è del 9% secondo la statistica di Schuder, relativa a circa 15.000 soggetti.
d) Paramixovirus: parotite
La porta d'ingresso del virus è la cavità orale. L'infezione si stabilisce per contatto fra malato e sano mediante l'inalazione di secrezione respiratoria infetta, in sospensione nell'aria degli ambienti abitati da soggetti malati.
La stagione fredda è quella in cui vi sono le maggiori incidenze annuali di morbosità. Si ritiene oggi che la parotite non sia da considerare una malattia localizzata delle ghiandole salivari, ma piuttosto un'infezione generalizzata con stato di viremia e localizzazioni multiple, successive o contemporanee alle ghiandole salivari, come al SNC, al pancreas o al testicolo, anche se talune si svolgono a livello subclinico. È da notare che il SNC può essere colpito isolatamente e indipendentemente da ogni altra localizzazione apparente e che può presentare anche una sintomatologia clinicamente sfumata o inapparente, rivelata solo da note di irritazione meningea o di reazione liquorale. Tali casi costituiscono una parte numericamente importante delle cosiddette meningiti abatteriche o linfocitarie benigne.
e) Coriomeningite linfocitaria
Nell'uomo l'infezione spontanea avviene occasionalmente, per lo più per contatto con materiale murino infetto; infatti il virus, eliminato con le feci e le urine dai topi, può contaminare il cibo e il pulviscolo, cosicché l'uomo può infettarsi per ingestione o per inalazione. Inoltre il virus può penetrare nell'organismo attraverso la cute o la congiuntiva, ad esempio toccando i topi in laboratorio o quelli presi in trappola. Pare che anche il cane sia talvolta responsabile del contagio. Il virus è stato isolato anche da zanzare, pidocchi, zecche e acari; si pensa che questi insetti siano vettori del virus e che lo disseminino nell'ambiente con le feci, ma non lo trasmettano con la puntura. Anche Trichinella spiralis può infettarsi col virus e può infettare l'uomo. Nell'uomo è più frequentemente colpito il sesso maschile nell'età compresa tra i 10 e i 40 anni; non vi sono invece differenze razziali di morbosità. D'estate la frequenza della malattia è inferiore a quella delle altre stagioni.
f) Encefalite letargica
La pandemia dell'encefalite letargica ha avuto inizio da alcuni focolai epidemici in Romania nel 1915, con successive estensioni negli anni 1915-1917 a tutti gli Stati dell'Europa (ove l'acme della morbosità fu raggiunto nel 1919-1921); negli anni 1918-1922 la malattia comparve e si diffuse negli Stati Uniti e nei paesi dell'Estremo Oriente, in modo particolare nel Giappone. La pandemia fu in decremento negli anni 1921-1924, poi vi fu una nuova ondata (particolarmente intensa e prolungata nei paesi nei quali precedentemente l'incidenza della malattia era stata piuttosto bassa), che si andò gradualmente estinguendo fino al 1926. Dal 1926 non si sono più presentati episodi epidemici, e la malattia si è manifestata solo in rari casi sporadici. In Italia la massima morbosità fu raggiunta nel 1920, quindi si è avuto un rapidissimo decremento dal 1921 al 1926; la massima incidenza dei casi si ebbe nei mesi di febbraio-marzo.
L'encefalite letargica compare, anche durante gli episodi epidemici, con casi apparentemente isolati e non collegati da un rilevabile nesso epidemiologico. Rarissime sono le segnalazioni di più casi in una stessa famiglia, rari i casi di dimostrato contagio diretto interumano. Si ritiene che la durata media del periodo di incubazione sia di una o due settimane; in qualche caso è possibile che il periodo di incubazione sia stato più lungo, di uno o due mesi.
La malattia colpisce prevalentemente individui giovani senza preferenza di sesso, di razza, di professione, di costituzione. Non sono note le modalità di trasmissione del presunto agente infettante che, ritenuto un virus, non è stato peraltro mai isolato, né coltivato, né identificato.
3. Patogenesi generale delle infezioni virali del sistema nervoso centrale (SNC)
a) Propagazione dei virus nel SNC
Come già si è detto, la vecchia concezione di virus capaci di proliferare e di ledere elettivamente il SNC risparmiando tessuti extranervosi è stata eliminata dall'accertata possibilità di lesioni nervose (meningiti ed encefaliti) causate da virus solitamente non neurotropi (ECHO, coxsackie, herpes simplex, arbo, ecc.) e dall'opposta osservazione di lesioni extranervose in corso di infezioni da virus una volta ritenuti tipicamente neurotropi (polio, ecc.), così come dalla loro crescita in colture di tessuti non nervosi.
Il concetto di neurotropismo obbligato è pertanto da tempo divenuto insostenibile e attualmente il problema del neurotropismo riguarda piuttosto la ricerca dei meccanismi e delle vie attraverso i quali i virus possono raggiungere il SNC e quindi propagarvisi. Tale propagazione neurotropa, anche se in base a un complesso di fatti sperimentali e clinici non si può respingere, risulta tuttavia incomprensibile quando si consideri la struttura fisica dei nervi che non sembra consentire alcuna possibilità di movimento ai virioni.
Due opposte interpretazioni del fenomeno dettero E. W. Goodpasture (propagazione del virus erpetico per moltiplicazione lungo gli assoni) e A. B. Sabin (propagazione del virus B della scimmia e pseudorabbia lungo i cilindrassi, fino al nucleo, prima che si stabilisca la replica dei virioni).
La possibilità che i virus procedano lungo i linfatici sarebbe apparentemente esclusa dal fatto che questi sono ubicati nel perineurio e non consentono comunicazione con gli spazi sottoaracnoidei se non in condizioni antifisiologiche.
Secondo Wright, essendo precluse sia la via degli assoni, a causa dell'elevata viscosità dell'assoplasma, sia quella dei linfatici, a causa della direzione centrifuga della linfa, tranne che in condizioni altamente antifisiologiche, non resta da prendere in considerazione che la possibilità della propagazione lungo gli spazi interneuronali. Contrasta con questa ipotesi, ma solo in apparenza, l'osservazione che l'obliterazione di tali spazi provocata da un agente sderosante conduce a un'accelerazione dell'insorgenza della mielite dopo inoculazione periferica del virus: infatti è possibile che la propagazione del virus avvenga non attraverso un ‛condotto' dello spazio interneurale, ma per moltiplicazione lungo le cellule endoneurali (meccanismo compatibile con l'osservazione di inclusi di virus della pseudorabbia nelle cellule di Schwann dei nervi periferici, oppure nel connettivo proliferato in seguito all'azione sderosante). L'introduzione delle metodiche di immunofluorescenza ha confermato questa possibilità, in assenza di virus nel sangue e nei visceri. Questa via non può essere tuttavia considerata unica; è anzi ammissibile che i virus si propaghino attraverso i nervi, in quanto è stato dimostrato che il taglio di nervi infettati distalmente previene l'infezione mielitica, mentre a nervo intatto i gangli delle radici dorsali sono i primi a essere colpiti, pur in assenza di infezione endoneurale ascendente.
Una via alternativa a quella della propagazione per infezione delle cellule neuronali deve perciò implicare la propagazione lungo un qualche spazio ascendente. E tuttavia improbabile che la via nervosa, la cui importanza è stata affermata sulla base di ricerche sperimentali compiute con virus altamente neurotropizzati, sia quella abitualmente seguita in natura, a eccezione delle due infezioni trasmesse da morsi (rabbia e virus B) nelle quali la compromissione mielitica iniziale si verifica al livello midollare corrispondente ai nervi spinali innervanti il territorio sede della lesione infettante.
Anche nell'infezione da virus varicella-zoster la distribuzione dermatomerica delle lesioni dell'herpes zoster de- pone per una probabile diffusione del virus lungo i tronchi nervosi (verosimilmente dai gangli delle radici dorsali raggiunti per via ematogena, dove si presume sia rimasto latente a seguito di un'infezione varicellosa infantile) in senso centrifugo verso la cute.
Una seconda via di diffusione del virus al SNC è la via olfattoria, bene stabilita per i virus polio, herpes simplex, arbo. In queste infezioni è possibile isolare il virus dai bulbi olfattori e dai lobi piriformi, prima che da qualsiasi altra zona cerebrale ; per converso, l'asportazione dei bulbi olfattori e il loro danneggiamento, insieme a quello della mucosa olfattoria, con agenti chimici, prevengono la diffusione del virus.
La stessa impervietà della barriera ematoencefalica ai virus ha indotto a considerare la viremia di certe encefalomieliti da arbovirus come la manifestazione del trasporto del virus dai siti di moltiplicazione periferica alla mucosa nasale, dalla quale è possibile l'invasione successiva del SNC.
Fondamentalmente il meccanismo di diffusione del virus dalla mucosa nasale al SNC può essere considerato identico a quello già descritto per la via nervosa, ma con alcune differenze in rapporto alla particolare anatomia della regione. È dimostrata, ad esempio, una diffusione di particelle di carbone o di proteine marcate presenti nel liquido cefalorachidiano dai manicotti di aracnoide che si estendono a formare dei culs-de-sac attraverso la piastra cribiforme e la zona intorno ai nervi.
È d'altra parte noto che la comunicazione indiretta della sottomucosa con i linfatici consente una diffusione del liquido cerebrospinale, attraverso l'area olfattoria, circa 10 volte maggiore di quella possibile attraverso le radici dei nervi spinali nei linfatici paravertebrali. Ma, oltre a tale via essenzialmente linfatica, è da tener presente che i recettori olfattori, almeno in certe specie animali, si estendono fino all'epitelio olfattorio e oltre, tanto che particelle di colorante ivi depositate possono raggiungere i bulbi olfattori in un'ora, presumibilmente lungo gli spazi interstiziali delle guaine perineurali dei nervi olfattori. In tal caso la penetrazione si può compiere senza la preventiva infezione della mucosa, ma per diretto contatto fra virus e nervi olfattori.
Questi meccanismi sono stati confermati in ricerche sperimentali con il virus ectromelico, con il virus erpetico e con il virus West Nile.
Quale il significato della via olfattoria per le infezioni virali umane? Mentre è certo che nelle infezioni da arbovirus la via naturale di diffusione è quella ematogena, è ben noto che in laboratorio alcune infezioni si sono propagate per inalazione di aereosol infetti; d'altra parte, e con riserva della diversa suscettibilità delle cellule costituenti una popolazione di neuroni, nell'encefalite da herpes simplex dell'adulto le prime zone colpite sono state quelle orbitofrontale e del lobo temporale, le zone cioè più facilmente aggredibili attraverso la mucosa olfattoria.
La via ematogena è certamente operante per alcune infezioni sperimentali (per l'encefalomielite equina orientale, lesioni cerebrali diffuse nella cavia inoculata per via extraneurale; per la febbre gialla, lesioni simultanee nelle principali zone cerebrali dopo inoculazione extraneurale). Per il virus polio le opinioni contrastanti, ma non inconciliabili nè reciprocamente incompatibili, di Bodian e Sabin affermano che dai siti intestinali di moltiplicazione il virus raggiungerebbe il SNC, rispettivamente per via ematica e per via nervosa. Per via ematica si diffondono i virus coxsackie, ECHO, della meningite coriolinfocitaria, della parotite, herpes simplex, della rosolia e citomegalico (nei feti) e probabilmente gli arbovirus.
È certo che la barriera ematoencefalica rappresenta un consistente ostacolo alla penetrazione dei virus nel SNC per via ematica: il periodo di incubazione di virus encefalitogeni inoculati per via endovenosa si avvicina a quello rilevato nell'infezione per via periferica extraneurale o olfattoria; ma ciò potrebbe anche essere interpretato come la necessità che successivamente a un'attiva moltiplicazione periferica extraneurale - la quale naturalmente richiede un certo tempo - siano i virioni così replicati a raggiungere i neuroni, superando la barriera ematoencefalica attraverso un persistente mantenimento della viremia. A tale riguardo dev'essere tenuto presente che i virus introdotti per via endovenosa sono allontanati dal torrente circolatorio dalle cellule del sistema reticoloendoteliale (SRE), ma che esiste un equilibrio fra potere rimuovente del SRE e capacità moltiplicativa dei virus; infatti, a misura che questi sono rimossi dal plasma sanguigno, vi viene introdotto altro virus proveniente dalla moltiplicazione negli endoteli vasali e nei linfatici, col risultato di una viremia persistente. Ciò si verifica soprattutto per i virus più piccoli (foresta di Semliki e polio nelle infezioni sperimentali).
Altre volte i virus sono adsorbiti alla membrana delle emazie e si moltiplicano nell'interno dei leucociti, e la viremia è conseguentemente mantenuta dalla circolazione degli elementi corpuscolati del sangue. Il virus erpetico e quello morbilloso sono stati isolati da leucociti ed è possibile che la loro presenza nell'interno di tali elementi li protegga dall'azione fagocitaria delle cellule reticoloendoteliali e dagli anticorpi circolanti. Pertanto, le dimensioni dei virioni, la loro sede intraleucocitaria, l'età dell'ospite, le infezioni intercorrenti, lo stato immunitario e l'integrità del SRE dell'ospite sono fattori che, determinando la durata della viremia, condizionano l'accesso dei virus al SNC.
Associata alla viremia, e come elemento capace di condizionarne l'intensità e la durata, è la capacità di replicazione dei virus in tessuti extranervosi (epitelio respiratorio in topi infettati con aereosol di virus West Nile; epitelio intestinale, tonsille o placche di Peyer, in uomini e in scimmie infettati con virus poliomielitico; molti tessuti extraneurali, ma soprattutto epiteli vascolari per i virus Sindbis e West Nile).
Fra i siti di moltiplicazione secondaria capaci di mantenere elevate viremie è compreso il grasso bruno delle ghiandole di ibernazione, che consente la moltiplicazione di molti virus neurotropi coxsackie, vaiuolo, polio, reovirus, rabbia). È stato anche affermato che la preliminare moltiplicazione del virus polio nel grasso bruno ne aumenta il potere patogeno per il tessuto nervoso.
b) La barriera ematoencefalica
L'accessibilità del SNC a sostanze o patogeni corpuscolati provenienti dal sangue è dipendente dalla possibilità che questi fattori possano attraversare la cosiddetta ‛barriera ematoencefalica' (BEE), termine con il quale si esprime non già una determinata formazione anatomica, bensì una particolare situazione fisiologica. Non è noto tuttavia a quale livello essa precisamente operi né se consista in un unico meccanismo o struttura o in un insieme di meccanismi e strutture anatomiche da individuare fra le seguenti: a) parete capillare (cellule endoteliali, sostanza interstiziale cementante); b) membrana piogliale (invaginazione della pia che penetra nel SNC al seguito dei vasi sanguigni che vi si approfondano, con la cui parete esterna forma gli spazi di Virchow-Robin); c) processi basali delle cellule astrogliali.
Per raggiungere il SNC i virus devono attraversare dunque questo spazio o struttura che, in qualsiasi senso sia intesa, protegge a livello dei capillari sanguigni il tessuto nervoso dall'invasione di elementi estranei. I virus possono poi penetrare nel SNC sia attraversandolo, sia moltiplicandosi nelle cellule del plesso coroideo, che per essere dotato di un epitelio poroso circondante uno stroma di connettivo lasso ha una struttura anatomica diversa da quella dei capillari cerebrali.
Studi condotti a tale riguardo con un virus (batteriofago) incapace di essere replicato nel cane hanno dimostrato che quando a questo animale sia stata iniettata una grossa dose di virus per via endovenosa, piccole quantità se ne ritrovano nel liquor cerebrospinale (LCS). Sembra dunque che la BEE sia operante nello schermare il SNC dall'ingresso di questo virus, ma non completamente. Dopo la penetrazione nel liquor, lo spostamento dei virioni attraverso le meningi e le cellule ependimali può avvenire per diffusione attraverso gli elementi cellulari costitutivi di queste strutture o per infezione e moltiplicazione in esse.
Il ritrovamento nel SNC di virioni di varianti neurotrope di virus influenzale, e non del ceppo non neurotropizzato, tende a far ammettere che la penetrazione del virus avvenga non attraverso un processo di diffusione meccanica, ma attraverso un'attiva replica.
Nell'uomo la replica dei virus nel plesso coroideo e la loro diffusione nel liquor con iniziale moltiplicazione nelle cellule delle meningi può spiegare la facilità con la quale alcuni patogeni (ECHO e coxsackie) possono essere isolati dal liquor in corso di localizzazioni nel SNC, in contrasto con la rarità di isolamento di altri virus (herpes simplex e polio) che probabilmente raggiungono il SNC attraverso altre vie.
In conclusione, a livello dei capillari sanguigni la BEE sembra consistere nell'associazione di una membrana basale dello spessore di circa 450 Å e delle cellule (astrociti) strettamente giustapposte.
Meccanismi alternativi a quello del superamento della barriera sono la penetrazione mediante trasporto in cellule leucocitarie fagocitanti, la replica dei virioni nelle cellule endoteliali e nelle strutture della barriera (possibilità di dimostrare alcuni virus - herpes simplex, encefalite delle volpi, influenza, West Nile, Sindbis - nelle cellule endoteliali prima che nel tessuto nervoso), la diffusione passiva dei virioni (virus poliomielitico) attraverso le cosiddette zone di permeabilità (in contrasto con l'indimostrabilità di altri virus, come nel caso dell'encefalite da zecche) nelle cellule endoteliali dei capillari e la loro presenza nelle cellule nervose.
Altri modelli sperimentali (inoculazione intracerebrale di virus) hanno portato alla conclusione che la diffusione di questi patogeni nel tessuto nervoso, una volta che vi siano pervenuti, può avvenire attraverso vari meccanismi: per contiguità interneuronica reale o inapparente, o per diffusione lungo gli spazi intercellulari esistenti fra i plasmalemmi delle cellule. Tutti questi modelli sperimentali, quali che siano la via seguita e il meccanismo in causa, dimostrano pertanto che i virus possono effettivamente passare dal sangue periferico al SNC.
c) La recettività della cellula nervosa
Quale che sia la via seguita dai virus per raggiungere le cellule, l'infezione del SNC è resa possibile dalla loro recettività, così come la malattia è dipendente dalla lesione funzionale che la replica virale induce nella cellula.
La tab. II riporta la suscettibilità delle diverse componenti cellulari del SNC a vari virus capaci di aggredirlo.
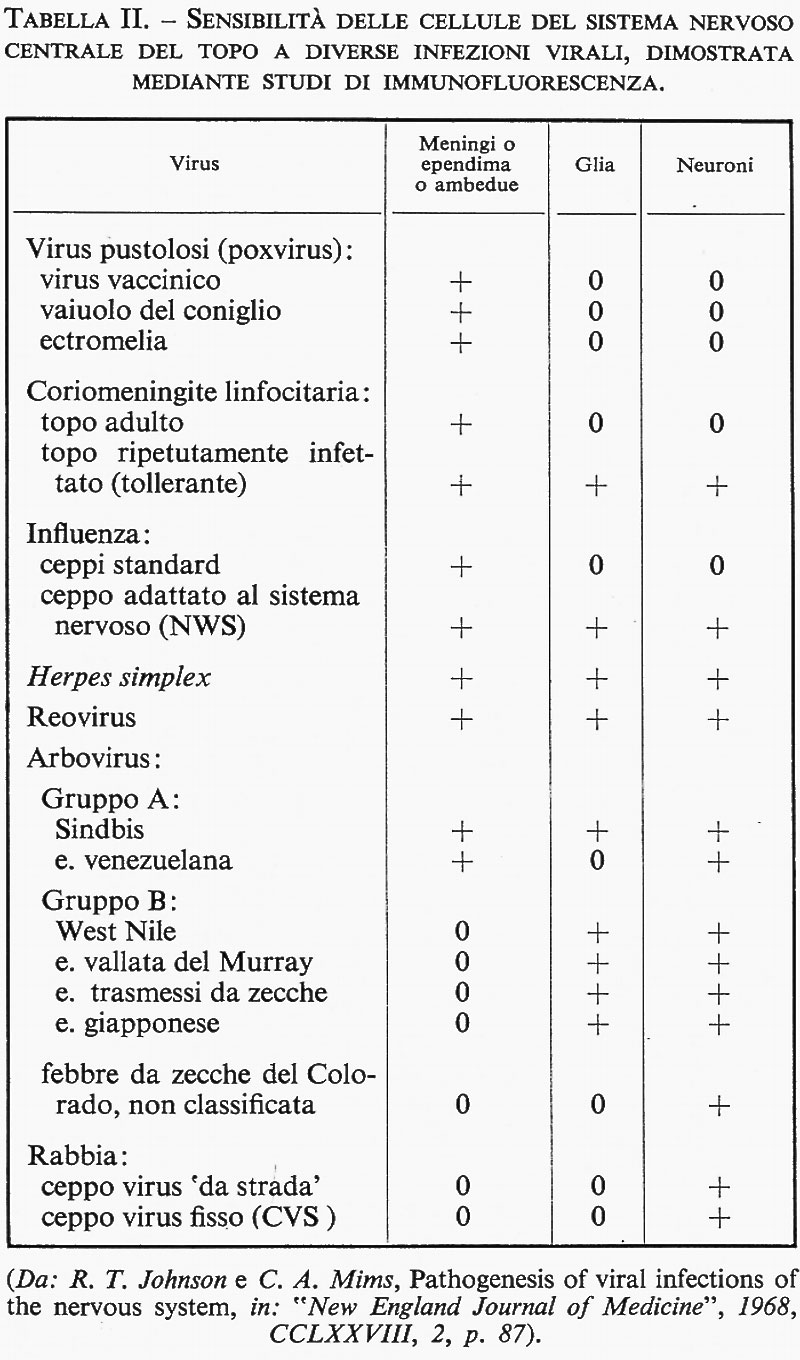
Pur nell'ambito del tessuto nervoso diversa è la recettività ai singoli virus delle varie zone del tessuto stesso (presenza di virus della febbre gialla negli astrociti, di reovirus nei neuroni, di virus fisso della rabbia nelle cellule di Purkinjie con rispetto degli altri elementi cellulari del cervelletto del topo, di virus polio nei motoneuroni umani, di virus delle encefalomieliti umane nella materia grigia e di virus herpes simplex per tutti indistintamente i costituenti del SNC). Ed è più probabile che ciò si verifichi non solamente per la facilità dei singoli virus di raggiungere determinate strutture nervose, in rapporto alle diverse vie e meccanismi di accesso, ma anche per una specifica e selettiva affinità di certi virus per determinate cellule e strutture cellulari.
La recettività selettiva di certi tipi cellulari e delle cellule di certe specie animali a determinati virus è in funzione di vari meccanismi ed è identificabile a vari livelli cellulari. Un locus importantissimo è rappresentato dalla presenza di recettori specifici sulla membrana esterna della cellula che vi consenta l'iniziale attacco del virus, la cui penetrazione risulterebbe altrimenti impossibile. Al riguardo è di particolare interesse l'osservazione sperimentale che l'inoculazione di acido nucleico infettante di virus polio ricoperto da un mantello di proteine del virus coxsackie in tessuto di topo normalmente recettivo al virus C ma resistente al virus polio, è seguita dalla replica del virus polio: la penetrazione del genoma infettante di quest'ultimo è resa possibile dalla capacità delle proteine del virus coxsackie di fissarsi ai recettori cellulari, tappa preliminare obbligata per la colonizzazione virale della cellula.
Variazioni nella presenza di recettori specifici per un determinato virus possono verificarsi in rapporto a una specificità di specie, all'età (come nel caso dei virus coxsackie e Sindbis) e alla eterogeneità della popolazione neuronica. L'ulteriore penetrazione dei virioni avverrebbe mediante il processo di viropessi, non specifico ma comune anche ad altre particelle corpuscolate ; infine la capacità della cellula di liberare il virione dal suo guscio proteico o lipoproteico consentendo al genoma virale, costituito da acido nucleico (DNA o RNA), di inserirsi, dopo il periodo di eclisse, nei meccanismi biochimici cellulari e di assumere il controllo delle sintesi, sia proteica sia nucleoproteica, così da orientarle verso la replica virale, costituisce l'evento essenziale di tale processo patologico.
Tale capacità può esistere solo in determinate parti della struttura cellulare, come è dimostrato dalla presenza di virioni herpes simplex nel pericario ma non negli assoni, e ciò in rapporto a una presumibile specializzazione degli organelli endocellulari.
d) Lesioni conseguenti alla penetrazione dei virus nel SNC
La replica virale nelle cellule del SNC non comporta necessariamente uno stato di malattia, potendo essere compatibile con l'integrità anatomica e funzionale dei neuroni: è noto, ad esempio, che il virus rabico viene replicato in colture di tessuti senza produrre lesioni cellulari.
Le lesioni - anatomiche e funzionali - si verificano a seguito sia dell'azione citolesiva diretta del virus, sia dell'infiammazione secondaria, sia infine dello stato di autoimmunità scatenato da un virus che potrebbe, senza l'intervento di questo meccanismo, essere non lesivo (v. immunologia e immunopatologia: Malattie autoimmuni).
Le lesioni cellulari sono primitivamente degenerative, come poté dimostrare Bodian mettendo in evidenza nei motoneuroni infettati con il virus polio fenomeni di cromatolisi e di neuronofagia, con possibilità, quando il danno neuronale sia contenuto entro certi limiti, di recupero anatomico delle strutture e della restitutio ad integrum del neurone.
Sotto questo punto di vista è certo un errore confondere l'azione citolesiva primaria dei virus, che è tipicamente induttrice di fenomeni di degenerazione-necrosi, con l'infiammazione polimorfonucleare che è un fenomeno consequenziale, satellite e patogeneticamente meno rilevante, anche se clinicamente importante (v. cellula: Patologia della cellula; v. infiammazione).
Ciò è stato anche recentemente confermato dalle conoscenze acquisite nella patologia da virus lenti (scrapie e visna) capaci di determinare fenomeni di degenerazione neuronale e demielinizzazione senza fenomeni infiammatori.
Infezioni persistenti senza lesioni delle strutture cellulari possono portare a stati morbosi anche mortali attraverso profonde lesioni funzionali dei neuroni; in alcuni casi, tuttavia, possono essere completamente privi di effetti patogeni, come nell'infezione congenita del topo da conomeningite linfocitaria, o a proposito della probabile persistenza del virus varicella-zoster a livello dei gangli delle radici dorsali e di quella del virus roseolico nei neonati durante i primi tre mesi di vita dopo l'infezione contratta in sede intrauterina.
Per converso, la reazione mononucleare-linfocitaria a sede perivascolare può intervenire in assenza di lesioni delle cellule parenchimali, come nel caso dell'infezione da virus parotitico: questo tipo di infiammazione naturalmente deve essere valutato diversamente da quella polimorfonucleare para- o postnecrotica, e il suo significato è tuttora controverso.
Infine, controversa è l'importanza dei fattori immunitari, in quanto si è dimostrato che la loro inibizione, a opera dell'irradiazione e della timectomia neonatale o del trattamento immunosoppressivo, impedisce la comparsa di determinate manifestazioni morbose virali di comune osservazione nei soggetti integri, pur persistendo la replica virale: è quanto si verifica nelle infezioni croniche inapparenti da virus della coriomeningite linfocitaria del topo.
Presenza o assenza di recettori cellulari, velocità e tipo di risposta immune e di sintesi di interferon - sostanza che senza neutralizzare i virus interferisce con la loro replica - idoneità dei neuroni ai complessi meccanismi biochimici condizionanti la replica virale, e probabilmente l'attività di allontanamento delle particelle virali svolta dai macrofagi, costituiscono un complesso di fattori i quali possono difendere il SNC dalle infezioni virali o per converso, quando non siano operanti, consentirle.
4. Lesioni istologiche
Solo eccezionalmente è possibile una diagnosi istologica delle singole nevrassiti virali che consenta di precisarne anche l'eziologia, perché il tessuto nervoso nei confronti dei vari agenti virali presenta solo un numero limitato di tipi di reazione istologicamente documentabili; la prevalenza di un tipo piuttosto che di un altro dipende più da particolari caratteristiche del tessuto ospite, e in particolare delle sue diverse zone, che da quelle di patogenicità del virus.
Le modalità di risposta della cellula nervosa nel cui interno abbia luogo la moltiplicazione del virus sono essenzialmente la degenerazione e la lisi, fenomeni per i quali la denominazione di reazione è per lo meno impropria.
Fra i numerosi tipi di encefaliti virali, soltanto in pochi casi sono note lesioni istologiche patognomoniche, la cui dimostrazione consente pertanto di risalire all'eziologia: la presenza di corpi inclusi intracitoplasmatici (corpi di Negri nella rabbia) o intranucleari (corpi di Lipschütz nell'encefalite erpetica) o intranucleari e intracitoplasmatici contemporaneamente (encefalite a inclusioni).
Una distinzione orientativa può essere piuttosto fatta, entro certi limiti, sulla base della distribuzione topografica delle lesioni nelle varie zone del sistema nervoso centrale.
a) Quadri istopatologici fondamentali del sistema nervoso: lesioni mesodermiche ed ectodermiche
I quadri istopatologici fondamentali del sistema nervoso rientrano nei gruppi distinti delle lesioni degenerative e di quelle infiammatorie. La coesistenza dei due tipi di lesioni pone la questione della loro interdipendenza causale, nel senso della possibilità che le prime producano nei tessuti circostanti una reazione infiammatoria o che questa si manifesti indipendentemente da quelle, come è da taluni sostenuto (Scheinker).
Le lesioni mesodermiche sono espressione di un pro- cesso infiammatorio senza alcuna nota di specificità e consistono in manicotti di infiltrazione, attorno ai piccoli vasi del cervello e delle meningi, prevalentemente linfocitari, meno frequentemente polinucleari o plasmacellulari, oppure ancor più raramente di elementi della microglia o di fibroblasti, sovente scarsissimi, raccolti negli spazi perivascolari di Virchow-Robin.
Gli elementi cellulari di infiltrazione sono osservabili non solo intorno ai vasi, ma anche nello spessore della parete vasale stessa e nel tessuto nervoso adiacente, che suole presentare anche una diffusa reazione della microglia.
Le lesioni ectodermiche sono strettamente degenerative e consistono in: a) rigonfiamento e sporgenza di determinati settori del contorno del corpo cellulare; b) maggiore tingibilità dei nuclei con minore nettezza del contorno nucleare; c) frammentazione della sostanza di Nissì e sua dislocazione a ridosso della membrana citoplasmatica, interpretabile come l'espressione iniziale di un danno neuronale che può sfociare in quadri più conclamati, nella completa distruzione della cellula nervosa, con successiva invasione di cellule gliali; in altri casi si nota un altro tipo di lesione locale, circoscritta, consistente nella trasformazione spugnosa e nella completa distruzione del tessuto nervoso (cellule, fibre nervose e guaine mieliniche) con reazione gliale relativamente modesta, senza rapporti topografici con i vasi sanguigni, simile alle lesioni descritte da Zimmerman come ‛placche acellulari'; d) alterazione degenerativa delle guaine mieliniche fino alla loro frammentazione e scomparsa, con eventuale integrità delle fibre nervose, indipendentemente dall'entità del danno neuronale.
Accanto alle lesioni degenerative ectodermiche, e cioè concomitantemente con la distruzione dei neuroni, si nota una marcata proliferazione di elementi della microglia, che può indurre, se circoscritta a foci isolati, la formazione di noduli o granulomi cellulari composti da un gran numero di cellule microgliali proliferate, con notevole polimorfismo nucleare e irregolarità morfologiche del contorno, talora somiglianti ai linfociti, e da un piccolo numero di grandi cellule mononucleate con nucleo rotondo o ovale e abbondante citoplasma.
b) Classificazione dei quadri istopatologici delle nevrassiti virali
La variabile commistione dei diversi tipi di lesioni istologiche, la varia intensità con cui ciascuna di esse evolve, non solo nella stessa forma morbosa ma nei singoli individui che ne sono affetti, e la differente prevalenza dell'una o dell'altra in rapporto ai vari stadi del processo spiegano sufficientemente il polimorfismo dei quadri istopatologici delle nevrassiti da virus e i numerosi tentativi di classificarli.
Secondo la classificazione di Woll, i principali quadri istopatologici - quelli meglio conosciuti anche dal punto di vista eziologico - sono suddivisi in tre gruppi fondamentali a seconda della localizzazione delle lesioni. Prototipi del I gruppo sono l'encefalite rabica e l'encefalite letargica, rispettivamente encefalomielite acuta ed encefalite subacuta o cronica, localizzate la prima prevalentemente nell'estremità occipitale, la seconda nell'estremità frontale dell'encefalo. Prototipi del II gruppo sono le cosiddette encefaliti da Artropodi (St. Louis, encefalomieliti equine orientale e occidentale, encefalite giapponese tipo B, encefalite venezuelana) in cui le lesioni sono preminenti nella corteccia cerebrale e nelle strutture della base del cranio. Prototipi del III gruppo sono le encefaliti cosiddette ‛a inclusioni' (erpetica e a inclusioni) in cui sono interessati preminentemente la corteccia cerebrale, la materia bianca e i gangli della base. Nei primi due gruppi di nevrassiti esiste un cointeressamento di grado variabile del midollo spinale, mentre nel III gruppo tale interessamento è praticamente assente.
Di questa classificazione delle lesioni nevrassitiche conviene accettare transitoriamente la schematizzazione e la praticità, con la consapevolezza della sua insufficienza di fronte a quadri istologici, per la loro specifica natura, diffusi e non localizzati. D'altra parte la limitazione concettuale che è implicita può essere in parte tollerata in considerazione del vantaggio pratico che tale suddivisione offre; essa infatti favorisce l'orientamento dello studioso nel complesso dei quadri istopatologici in cui i tipi di lesioni sono fondamentalmente sempre gli stessi, seppure commisti in vario grado fra di loro, e in cui conseguentemente viene a mancare la possibilità di ogni ulteriore differenziazione istologica.
Risultano, in conclusione, i seguenti fatti: 1) il danno cellulare può raggiungere in singoli elementi diversi stadi, dei quali alcuni coincidono con la distruzione definitiva del neurone e altri, essendo regredibili, possono essere seguiti dalla riparazione anatomica delle cellule, cui consegue talvolta, con un certo ritardo, il ripristino delle normali funzioni biologiche; 2) alla distruzione cellulare, e solo a questa, seguono i fenomeni di neuronofagia e di infiltrazione cellulare avventiziale e gliale; 3) altre volte il danno cellulare, oltre a non essere incompatibile con la guarigione anatomo-funzionale successiva, non è neppure incompatibile con una certa conservazione della funzione anche durante la fase più acuta del processo, come è presumibile che avvenga nel caso dei neuroni corrispondenti ai muscoli non paralizzati ma più o meno indeboliti nella forza contrattile.
5. Lesioni biochimiche
L'esistenza di disordini muscolari in distretti funzionalmente dipendenti da motoneuroni risultati non irreversibilmente e non apparentemente lesi da un punto di vista microscopico ha favorito il concetto che lesioni reversibili e/o inapparenti possano alterare la funzione ma non - almeno in modo evidente - la struttura dei motoneuroni; si determinerebbe tuttavia un deficit funzionale tale da produrre le anomalie meccaniche ed elettriche della contrazione dei muscoli dipendenti.
I tentativi di identificare tali lesioni hanno dato finora risultati scarsamente attendibili, in considerazione dell'inevitabile commistione delle anomalie biochimiche direttamente indotte dalla replica virale con quelle derivanti dalla necrosi in quanto tale, e cioè come evento aspecifico, producibile da parte di qualsiasi noxa patogena non necessariamente virale.
6. Patologia generale
a) Modalità di danneggiamento dei neuroni
Le sintomatologie cliniche che nel corso delle nevrassiti compaiono, evolvono, regrediscono, si associano, si separano, sfociano in esiti permanenti o nella guarigione completa, sono la conseguenza di lesioni che l'infezione virale produce direttamente o indirettamente nel SNC, a carico dei neuroni e della glia.
La replica virale può provocare, anche direttamente, danni di vario grado ai neuroni. Talora essa si stabilisce parzialmente e reversibilmente senza che la lesione biochimica conseguente induca la necrosi cellulare. Altre volte la replica virale intracellulare non è compatibile con la sopravvivenza del neurone, ma ne determina la degenerazione e la necrosi: ciò avviene perché l'acido nucleico del genoma virale viene accettato dalla cellula e il suo codice genetico si sostituisce a quello cellulare; il controllo delle sintesi proteiche del neurone viene così deviato dai suoi obiettivi fisiologici - sintesi delle proteine cellulari - e orientato verso l'obiettivo patologico della sintesi delle nucleoproteine virali, cioè della replica virale.
In alcuni casi la replica virale è contenuta entro grandezze compatibili con la sopravvivenza nei neuroni, ma non compatibili con la loro integrità funzionale; altre volte la replica avviene con velocità tale da non consentire la sopravvivenza cellulare, onde consegue la necrosi dei neuroni. Il tessuto gliale e i vasi sanguigni presentano vistose manifestazioni infiammatorie consistenti in edema, congestione vasale, essudazione, infiltrazioni cellulari, che danneggiano indirettamente i neuroni, aggravandone il grado di sofferenza, mediante fenomeni meccanici di compressione o l'azione chimica citolesiva di sostanze delle quali è provocata la liberazione (v. infiammazione).
Un terzo meccanismo di danno è rappresentato dalla possibilità che si produca uno stato autoimmunitario, in quanto antigeni virali o provenienti dalla cellula parassitata sono in grado di indurre la produzione di anticorpi diretti non solo verso il virus, ma anche verso la stessa cellula nervosa, determinandosi conseguentemente manifestazioni di danno neuronale e di demielinizzazione (v. immunologia e immunopatologia: Immunologia generale e Malattie autoimmuni; v. neuropatologia: Ricerche immunologiche).
Tale condizione autoimmunitaria può stabilirsi nei seguenti modi fondamentali: 1) il virus ha antigeni comuni con il citoplasma dei neuroni, vi è cioè una situazione di reattività crociata; 2) i protidi del neurone denaturati dalla replica virale diventano essi stessi antigeni, in quanto risultano così profondamente alterati da non essere più riconosciuti dal sistema immunocompetente; 3) l'infezione virale può esercitare uno stimolo capace di indurre alcune cellule immunocompetenti a formare anticorpi attivi verso i motoneuroni occupati dai virus.
È possibile che un virus sia attivamente lesivo del SNC, agendo attraverso diversi e simultanei meccanismi e attaccando i neuroni sia direttamente sia indirettamente attraverso meccanismi autoimmunitari o infiammatori. Quale che sia il meccanismo direttamente o indirettamente responsabile del danno neuronale, è a questo ed esclusivamente a questo che è imputabile la sintomatologia clinica corrispondente. A tale danno il tessuto connettivo e la rete vasale partecipano come coproduttori ma non sono direttamente responsabili della sintomatologia.
In base a quanto si è detto, la denominazione di encefalite, che sottintende il carattere infiammatorio del processo, deve essere considerata impropria in certi casi, in altri incompleta, in altri ancora, infine, errata, in rapporto alla varia entità della componente flogistica.
b) Distribuzione topografica ed esiti delle lesioni
Circa il danno neuronale direttamente indotto dalla replica virale, occorre considerare che anche nell'ambito di una ristretta area di motoneuroni non tutte le unità cellulari si comportano nella stessa maniera di fronte all'offesa virale: situazioni biochimiche cellulari individuali, un metabolismo locale proprio di singole aree cerebrali corrispondono a strutture morfologiche individuali e a particolari organizzazioni sinaptiche dei singoli nuclei grigi. Ciò spiega come il tessuto nervoso non sia uniformemente colpito dall'offesa virale, ma risponda all'aggressione in maniera notevolmente variabile in intensità e distribuzione topografica configurando il quadro istologico di una poliencefalite disseminata con distribuzione delle lesioni a focolai o lungo sistemi funzionali. La distribuzione topografica può essere caratteristica di ciascun virus e la sindrome cliniconeurologica che ne risulta è conseguentemente determinata dal particolare tipo di virus, dall'intensità delle lesioni, dal tipo prevalente di queste, dal numero e dal tipo dei neuroni che sono stati o distrutti, o danneggiati ma sopravviventi, o non danneggiati affatto. L'evoluzione nel tempo delle varie componenti lesive condiziona l'evoluzione e la trasformazione del quadro clinico.
Uno degli esiti dell'infezione acuta può essere l'atrofia del parenchima, nel caso di neuroni che abbiano subito un danno non così grave da determinarne la necrosi e che siano quindi sopravvissuti all'infezione: probabilmente con tale meccanismo si manifestano la progressiva atrofia della substantia nigra che dopo un'encefalite di von Economo conduce al parkinsonismo postencefalitico; la progressiva atrofia dei muscoli spinali dopo una poliomielite; la sindrome di sclerosi laterale amiotrofica che può conseguire in esito a un'encefalite da arbovirus.
Difficile è l'interpretazione di questi fatti: un'ipotesi che li interpreti come conseguenza dell'alterato metabolismo degli acidi nucleici, che nel normale è geneticamente determinato, dovrebbe implicitamente ammettere la possibilità di una mutazione biologica, finora non dimostrata, indotta dal virus a livello di tale metabolismo. Accettando tale interpretazione, comunque, un simile esito dovrebbe essere considerato come un evento ben diverso dalle usuali sequele di processi patologici, che non sono progressive ma semplicemente statiche.
Nelle polioencefaliti il danno neuronale è esclusivamente o preminentemente provocato dall'offesa direttamente recata ai neuroni dal virus, con nessuna o scarsa o minima compartecipazione dei fenomeni autoimmunitari. L'inserzione di quest'ultimo meccanismo potrebbe anche dipendere dalla quantità del virus presente, ovvero dalla velocità della sua replica. Infatti anche se è comunemente ammesso che un solo virione, che penetri in una cellula recettiva, è in grado di scatenare una malattia virale, bisogna tuttavia tener presente che solo un virus di scarsa virulenza può raggiungere nelle cellule ospiti una concentrazione tale da determinare un'abnorme risposta immunitaria.
Ben diverso dal quadro, precedentemente descritto, delle polioencefaliti disseminate a focolai è quello di un secondo tipo di encefalite, la perivenosa focale: questa si identifica con talune encefaliti para- o postinfettive, postvaccinali o insorgenti in corso di reazioni da sensibilizzazione. Si tratta di processi di leucoencefalite localizzati intorno alle venule della sostanza bianca, con rottura della barriera ematica, parziale ma continua necrosi mielinica e intensa reazione istiocitica microgliale.
Diversamente dalle polioencefaliti, infatti, nelle encefaliti focali perivenose diffuse non può essere identificata alcuna distribuzione topografica caratteristica di singoli virus o malattie. Può invece probabilmente esser preso in considerazione il fattore cronologico: sembra infatti che siano colpiti solo individui di una determinata età, al disotto della quale l'immaturità della risposta immunitaria impedisce lo stabilirsi delle lesioni da sensibilizzazione.
Esistono infine quadri nei quali sono variabilmente commiste in senso topografico e quantitativo le lesioni derivanti dalla diretta offesa virale a livello dei neuroni (tipiche delle polioencefaliti acute disseminate della sostanza grigia) e le lesioni derivanti da meccanismi autoimmunitari o allergici scatenati dal virus, quali si riscontrano nelle encefaliti morbillose, nelle encefaliti rubeoliche, in quelle da herpes simplex: infiltrati cellulari, reazione gliale, neuronofagia, inclusioni intranucleari. L'equilibrio fra i due tipi di lesioni e quindi di meccanismi patogeni può per uno stesso virus variare nelle diverse epidemie e, naturalmente, durante la stessa epidemia nei diversi soggetti.
L'entità e il tempo della possibile inserzione di uno stato autoimmunitario nei meccanismi patogenetici di una nevrassite virale varia da individuo a individuo: in alcuni può completamente mancare, in altri essere presente a basso livello, in altri ancora essere preminente, almeno in certe fasi della malattia.
7. Clinica
La concezione secondo la quale il nevrasse costituisce un'unità anatomo-funzionale è in accordo con l'acquisizione che i vari tessuti che lo compongono possono essere isolatamente e globalmente, in varia associazione fra di loro, sede di colonizzazione di virus neurotropi e conseguentemente di lesioni e sintomatologie cliniche dipendenti. Ciò spiega come i virus neurotropi possano di volta in volta essere agenti di encefaliti, mieliti, encefalomieliti, meningiti, meningoencefaliti, neuriti radicolari, ecc. Nei singoli casi, inoltre, le sedi di colonizzazione virale e di localizzazione delle lesioni possono variare nel corso della malattia. Le sintomatologie cliniche meglio studiate, o perché più frequenti - per certi virus - o perché più gravi e come tali più frequentemente causa di ricovero in ospedale, sono quelle delle encefalomieliti o meningoencefalomieliti.
a) Encefalomieliti equine americane
Le sintomatologie cliniche riscontrabili nei vari tipi di malattie neurologiche da arbovirus non presentano caratteristiche peculiari in rapporto alle singole eziologie. Questa sostanziale uniformità clinica ne consiglia una trattazione clinica unica, mentre si rimanda ai trattati di neurologia per le localizzazioni minori o più distrettuali, quali meningiti, radicoliti, neuriti, ecc.
La malattia può insorgere acutamente o essere preceduta da un periodo prodromico della durata di 2-4 giorni caratterizzato da febbre, astenia, cefalea, fotofobia, dolori alla regione nucale, rachialgia, nausea, vomito, dolori addominali, irregolarità dell'alvo e talvolta capogiri o vertigini; frequentemente vi sono anche dolori oculari e segni di flogosi delle prime vie respiratorie. In questo stadio l'esame obiettivo non mette in evidenza segni di compromissione neurologica, non si rilevano altri segni meningei e il quadro clinico non è differenziabile da quello di una malattia infettiva acuta similinfluenzale. Dopo questa prima fase vi può essere un periodo transitorio di remissione, cui seguono una brusca ripresa della febbre e della sintomatologia generale e la comparsa di una complessa sintomatologia neurologica. La febbre, accompagnata da sensazione di freddo e brividi, raggiunge rapidamente il suo nuovo acme (39-41 °C), e nella maggior parte dei casi cade dopo alcuni giorni per rapida lisi; l'apiressia viene di solito raggiunta entro 7-10 giorni, ma in alcuni casi, sia pure in assenza di complicazioni, anche dopo un tempo molto più lungo, fino a 4-6 settimane. La frequenza del polso è di solito proporzionale alla temperatura; talvolta vi è marcata bradicardia, raramente tachicardia. La pressione arteriosa è normale.
La sintomatologia neurologica è caratterizzata da cefalea insopportabile, dolori nucali, rachialgia, fotofobia. Molto frequenti sono i tremori alla lingua e alle estremità, capogiro e vertigine. In un gran numero di pazienti vi è difficoltà di parola, che nei casi più gravi può giungere fino all'afasia. Caratteristiche sono le alterazioni del sensorio: sono evidenti quasi sempre disorientamento nel tempo e nello spazio, frequentemente stupore o sonnolenza che nei casi gravi giunge fino al coma; di solito, se stimolato, il paziente risponde alle domande con difficoltà, a monosillabi o con poche parole stentate e mal pronunciate, per ricadere subito dopo nel suo stato stuporoso. Solo in una minoranza di casi è possibile rilevare agitazione di grado variabile, insonnia, delirio con verbigerazione e frequenti movimenti incoordinati. In una limitata percentuale di pazienti adulti si manifestano convulsioni epilettiformi, presenti invece nella quasi totalità dei bambini nei quali rappresentano anzi il sintomo dominante. La rigidità nucale, pressoché costante, si accompagna ai segni di Kernig e Brudzinsky. I riflessi muscolari sono irregolarmente e incostantemente modificati, potendo presentarsi accentuati o indeboliti; anche nelle fasi precoci di malattia i riflessi addominali sono frequentemente assenti. Se pure non costantemente, possono essere messi in evidenza i segni di compromissione piramidale di Oppenheim, di Gordon, di Chaddock, di Babinski, il cui comportamento può comunque variare nello stesso paziente nelle varie fasi della malattia. Solo in una minoranza di casi, di regola durante la fase acuta della malattia e solitamente prima della apiressia, si osserva la comparsa di paralisi: queste interessano quasi sempre le estremità, hanno varia estensione e possono quindi avere carattere di monoplegie, emiplegie, tetraplegie, e sono nella maggioranza dei casi di tipo spastico, eccezionalmente di tipo flaccido. Le pupille appaiono isoconche, miotiche, normoreagenti all'accomodazione, spesso iporeagenti alla stimolazione luminosa; le oftalmoplegie sono rare, il nistagmo - comunque di breve durata - è infrequente; l'esame oftalmoscopico non mette in evidenza alterazioni del fondo oculare. Frequentissimi la nausea e il vomito, che in alcuni casi, tormentoso e incoercibile, può provocare notevole disidratazione con disturbi dell'equilibrio idro-salino. L'alvo è spesso irregolare, di solito stitico, talvolta diarroico con emissione di feci liquide che possono eccezionalmente essere commiste a muco e a sangue. In alcuni casi vi è incontinenza delle feci; frequente è la ritenzione urinaria con iscuria paradossa da sovradistensione vescicale, mentre più raramente si osserva vera incontinenza delle urine spesso associata all'incontinenza delle feci. In un discreto numero di casi vi sono disturbi della salivazione e della sudorazione.
La rachicentesi dà luogo a un liquor limpido, lievemente iperteso; non è dimostrabile ipoglicorrachia, la iperproteinorrachia è modesta; vi è quasi sempre pleiocitosi di lieve o media entità, da 20 a 200 cellule per mm3 rappresentate prevalentemente da linfociti e mononucleati, raramente da polimorfonucleati.
Talvolta è possibile rilevare una modesta epatosplenomegalia. La velocità di sedimentazione è frequentemente elevata, mentre le modificazioni della crasi ematica sono incostanti: il reperto più frequente è quello di una leucocitosi con neutrofilia, raramente si osserva leucopenia con linfocitosi o linfomonocitosi. Da questo quadro clinico, in linea di massima comune a tutte le encefaliti sia pure con ampia variabilità, per presenza e intensità, delle singole sintomatologie, si distacca per particolari caratteristiche quello di determinate forme.
b) Encefalite russa primaverile estiva
Questa forma si può presentare clinicamente come una polioencefalomielite paralitica o come una meningoencefalite bifasica. La polioencefalomielite paralitica mostra un quadro clinico che ricorda sostanzialmente quello delle encefaliti conclamate precedentemente descritto, ma è caratterizzato dall'elevata e precoce incidenza di paralisi flaccide localizzate quasi sempre ai muscoli della nuca, del cingolo scapolare, delle braccia.
La meningoencefalite bifasica, diffusa soprattutto nelle regioni occidentali della Russia e nei paesi dell'Europa centrale, è caratterizzata da un tipico decorso febbrile bifasico. La malattia inizia con cefalea, vertigini, nausea, vomito, febbre moderata, iperemia congiuntivale, dolori muscolari, stipsi, lievissimi segni di compromissione meningea: questa prima fase, della durata di 4-7 giorni, è seguita da un periodo di 5-8 giorni di remissione quasi completa della sintomatologia. Compare quindi febbre alta, fino a 39-40 °C, che raggiunge l'acme dopo circa due giorni e cede quindi per lisi; vi si accompagnano fotofobia, iperestesia e cefalea tormentosa, che si esacerba con il movimento del capo. Vi è una caratteristica congestione del volto e delle sclere, gli occhi sono fissi, i movimenti oculari dolorosi. Sono presenti i segni di Kernig e di Brudzinsky, e i sintomi di una lieve compromissione encefalica (diminuzione dell'attenzione e della memoria, raramente eccitazione psicomotoria, frequenti disturbi del sonno). In alcuni casi sono rilevabili anche modesti segni piramidali, extrapiramidali o cerebellari. Frequentemente si osservano anisocoria, miosi, difficoltà alla convergenza. Talvolta vi è scialorrea. Il liquor è iperteso, vi è pleiocitosi e iperproteinorrachia. La meningoencefalite bifasica ha decorso benigno e volge di regola a guarigione senza reliquati. La letalità è irrilevante.
Varianti cliniche del quadro morboso fondamentale delle encefaliti sono: 1) i casi di infezione clinicamente inapparenti, dimostrabili solo con dati sierologici, la cui enorme diffusione è documentata dall'elevatissima incidenza, fino al 90%, di individui adulti viventi in aree endemiche che pur con anamnesi neurologica negativa, presentano anticorpi sierici N o FC per i virus encefalitogeni diffusi nella regione; 2) i casi di malattia infettiva acuta similinfluenzale con assoluta mancanza di localizzazioni neurologiche, che presentano un quadro clinico caratterizzato da febbre talora associata a flogosi delle prime vie respiratorie, cefalea, dolori oculari, fotofobia, nausea, vomito, talvolta lieve rigidità nucale; 3) i casi decorrenti come meningiti asettiche, nei quali si osservano rigidità nucale, i segni di Kernig e di Brudzinsky, ipertensione liquorale con pleiocitosi e iperproteinorrachia, senza alcuna manifestazione di encefalomielite; 4) i casi caratterizzati da particolare gravità del decorso per ipertermia marcata, convulsioni generalizzate o corna profondo, talvolta evolventi verso l'exitus molto precocemente, già al 1°-2° giorno di malattia, specialmente nei bambini nei quali la sintomatologia convulsiva è dominante.
c) Poliomielite
La malattia nella maggior parte dei casi decorre in modo clinicamente inapparente (infezione e malattia inapparente); altre volte il suo decorso è così poco tipico (fase sistemica) che solo il criterio epidemiologico può far sospettare un'infezione in atto.
Solo i casi paralitici, la cui incidenza è di 1/1000-1/3000 di tutti i casi di infezione, presentano un quadro clinico riconoscibile e caratteristico. Il periodo di incubazione è asintomatico, della durata di circa 3-15 giorni e di 6-36 giorni relativamente alla comparsa delle manifestazioni paralitiche: in media 17 giorni. Nello stadio iniziale prodromico nel 25% dei casi può essere presente un transitorio rialzo termico; questo è talora seguito, dopo un intervallo di apiressia, da un secondo rialzo termico, conseguente a una fase viremica e associato alla localizzazione del virus nel SNC. In questa fase il liquido cefalorachidiano è normale e nessun altro sintomo è presente: conseguentemente la diagnosi clinica è impossibile e la malattia, come già si è detto, può solo essere sospettata esclusivamente in periodo di epidemia. Il processo morboso può anche esaurirsi in tale fase. A questa può invece far seguito, non obbligatoriamente, lo stadio sistemico (preparalitico), caratterizzato da sintomi non neurologici e non patognomonici: febbre generalmente moderata e raramente al disopra dei 38,5 °C, più elevata e persistente nelle forme bulbari; cefalea, di solito non intensa; vomito, che si accompagna a sensazione di nausea e a malessere indefinito. A questi si associa un variabile corteo di altri sintomi meno costanti: lombalgia, angina rossa, disturbi intestinali (stipsi, diarrea), lieve rigidità nucale, dei quali solo l'ultimo può richiamare l'attenzione del medico su un possibile interessamento del SNC. Il sintomo, tuttavia, è di comune rilievo in numerose infezioni acute, specie nei bambini, onde il sospetto di poliomielite può essere confermato soltanto dalla dimostrazione di reali modificazioni patologiche del liquido cefalorachidiano. D'altra parte, il rilievo di alterazioni liquorali non consente la discriminazione diagnostica con altre meningiti virali: anche in questi casi sono infatti presenti nel liquor modesti segni infiammatori, consistenti in aumento delle proteine, particolarmente delle globuline, e in pleiocitosi linfocitaria; l'unico elemento differenziale è costituito dal fatto che nella poliomielite prevalgono inizialmente i neutrofili, sostituiti nelle fasi ulteriori dai linfociti. La rigidità nucale tuttavia è in genere meno pronunciata che nelle altre meningiti virali: si può mettere in evidenza invitando i pazienti a sedere sul letto, manovra nella quale essi procedono con cautela poggiando il palmo delle mani a piatto sul letto, dietro la schiena, in maniera da non essere costretti a flettere in avanti la colonna vertebrale per conservare in tale posizione l'equilibrio, altrimenti instabile. Invitato a toccare con il mento la regione sternale, il paziente, per evitare di flettere il collo in avanti, tenta di eseguire la manovra aprendo la bocca e abbassando così la mandibola.
Inoltre, sostenuto per le spalle egli abbandona la testa indietro per ottenere il rilassamento dei muscoli della nuca, la cui contrazione spastica gli riesce dolorosa, senza peraltro che i movimenti di lateralità del collo ne siano impediti, come avverrebbe in caso di paralisi dei muscoli di tale segmento. In generale, trattandosi di piccoli pazienti, i segni meningei più facilmente rilevabili sono quelli che richiedono minore cooperazione dei soggetti e che si rivelano in manovre passive, quali ad esempio la flessione anteriore del collo, ecc.
Se la sintomatologia non si risolve, la malattia può progredire verso lo stadio paralitico. È presente un vivace dolore alle masse muscolari, prevalentemente degli arti inferiori o superiori o del tronco, che viene denunciato dal paziente o rilevato obiettivamente nei tentativi di flessione passiva compiuti dall'osservatore; i piccoli pazienti, per motivi antalgici, si astengono da movimenti attivi.
Ma l'interessamento del SNC è dimostrato dalla repentina comparsa di paralisi flaccida, mentre, a prescindere dal dolore muscolare in rapporto allo stato di shock spinale e forse anche al cointeressamento dei cordoni posteriori, le vie sensitive sono abitualmente indenni. Le paralisi intervengono fra il 2° e il 4° giorno dopo la comparsa dei segni meningei, con estremi da poche ore a 5-6 giorni; esse nel periodo acuto devono essere accuratamente ricercate, specie nei piccoli pazienti nei quali la sintomatologia dolorosa aveva potuto nasconderne la presenza per la conseguente immobilizzazione antalgica del tronco e degli arti. D'altra parte, occorre tener presente che la dolorabilità di un gruppo muscolare non implica la sua paralisi in atto o imminente. Le paralisi poliomielitiche colpiscono singoli gruppi muscolari degli arti piuttosto che gli arti in toto: ciò dipende dal fatto che i grandi tronchi nervosi diretti agli arti sono costituiti da fibre provenienti da vari livelli delle corna grigie anteriori, solo alcuni dei quali possono essere stati colpiti dalla localizzazione virale. Tale caratteristica anatomo-patologica spiega anche come le paralisi possano colpire gruppi muscolari diversi nell'ambito di uno stesso arto, prevalentemente e con maggiore precocità i grandi muscoli degli arti piuttosto che i piccoli muscoli delle mani e dei piedi. Le paralisi poliomielitiche si stabiliscono nel giro di 12-24 ore e sono di tipo periferico, contrassegnate quindi da flaccidità e abolizione dei riflessi osteoperiostei, tendinei e cutanei. Più frequenti sono le paralisi degli arti inferiori (mono- e paraplegie), meno quelle dei superiori o quelle associate degli arti superiori e inferiori (tetraplegie) o degli arti e del tronco. Prevalentemente colpiti negli arti inferiori sono i glutei, gli adduttori, il quadricipite crurale, il gastrocnemio, il tibiale anteriore, il peroniero; negli arti superiori il deltoide e il tricipite omerale, mentre relativamente risparmiati sono i bicipiti e i flessori dell'avambraccio.
La lesione disseminata dei motoneuroni, alcuni dei quali, nell'ambito di un segmento midollare, possono essere totalmente distrutti e altri rispettati o danneggiati solo lievemente, tanto che non ne consegue cessazione della funzione, si traduce in una distribuzione apparentemente illogica e capricciosa delle paralisi che possono colpire i singoli muscoli o, nel contesto di questi, gruppi isolati di fibre muscolari; talché, specie in quest'ultimo caso, solo uno studio semeiologico particolarmente accurato e completo, comprendente anche l'esame elettromiografico, può rilevare deficit isolati e selettivi che non compromettono apparentemente la funzione dell'arto e che sfuggirebbero a un'osservazione più superficiale. In pratica, devono essere esplorati sistematicamente oltre un centinaio di diversi e separati fasci muscolari.
Precocemente subentrano i fenomeni degenerativi, che sono rivelati dall'ipotonia, dall'atonia, dal raffreddamento e dalla progrediente atrofia, nonché dalla reazione elettrica di tipo degenerativo.
Il liquor non mostra in questa fase caratteri sostanzialmente diversi rispetto alla fase precedente, mentre è solo in quella successiva, dell'atrofia - quando cioè la malattia come infezione si è conclusa e solo ne persistono i danni indotti nel sistema neuro-muscolare - che può presentare il quadro di una dissociazione albumino-citologica.
L'estensione topografica delle paralisi tende successivamente a ridursi rispetto a quella iniziale in parte per il riassorbimento degli essudati flogistici, in parte per la rigenerazione dei neuroni non irreversibilmente lesi, e infine anche per l'intervento di fenomeni di ipertrofia compensatoria di fibre muscolari indenni.
I quadri bulbari, la cui incidenza è dell'1-2% rispetto a tutti i casi paralitici, sono caratterizzati da deficit dei muscoli del palato molle, della faringe e della laringe con conseguenti disordini della deglutizione e della fonazione: rigurgito nasale di cibi liquidi, loro aspirazione nelle vie aeree, voce stridula o bitonale, afonia. Di particolare importanza clinica sono i disturbi della deglutizione in quanto per l'aspirazione dei liquidi ingenti nelle vie aeree si possono verificare episodi acuti di asfissia, aggravati da spasmi riflessi dei muscoli indenni, e infezioni polmonari da aspirazione. La temperatura febbrile è più alta e persistente che nelle forme non bulbari, e la frequenza cardiaca assai elevata negli stadi preterminali; sono anche descritti disordini pressori di difficile interpretazione nel senso sia di iper- sia di ipotensione, nonché disordini del ritmo cardiaco e di quello respiratorio fino alle loro più gravi espressioni.
Abitualmente il sensorio non appare compromesso, i malati sono vigili, coscienti e ben orientati; meno frequentemente si osserva un certo grado di obnubilamento. Sintomatologie con marcati difetti intellettivi e con segni di lesione dei moteneuroni superiori (paralisi spastiche), così come quadri a tipo di progressivo cointeressamento nevrassiale ascendente o anche discendente, una volta attribuiti al virus poliomielitico, sono attualmente valutati con prudente riserva in relazione al possibile ruolo eziologico di virus nevrassitici non poliomielitici.
L'esito letale che si verifica in un certo numero di casi è in rapporto all'insufficienza respiratoria, che si può stabilire per meccanismi patogenetici diversi: 1) centrale, da lesioni dei nuclei del IX, X, XI, XII paio dei nervi cranici (paralisi linguale e spostamento posteriore della lingua, paralisi faringea con accumulo di secreto mucoso nella faringe, aspirazione di contenuto gastrico nella trachea, accumulo di secrezione bronchiale per perdita della capacità di tossire, spasmo riflesso dei muscoli laringei o paralisi dei muscoli adduttori della laringe); 2) bulbare, da lesione dei nuclei del respiro, con totale perdita della capacità di controllo del ritmo del respiro (irregolare, superficiale, aritmico) cui frequentemente si accompagnano frequenti turbe circolatorie (tachicardia, aritmia, ipertensione) da lesione del vicino centro circolatorio bulbare e/o da iperpnea per iperventilazione; 3) spinale, per paralisi dei muscoli respiratori (diaframma, intercostali, addominali) mono- o bilaterali; la sintomatologia che ne consegue, talora all'inizio non facilmente riconoscibile, è contrassegnata da ansietà, alitazione del naso, cianosi, e segni premonitori ne sono la riluttanza alla conversazione e il discorso monosillabico riflettenti l'autoprotezione inconsciamente esercitata dai malati nei riguardi di episodi acuti di insufficienza respiratoria che anche la sola fonazione potrebbe scatenare. L'esame del paziente per la ricerca di paralisi respiratorie si esegue esercitando una manovra di compressione e immobilizzazione dell'addome e del torace, alternativamente, e osservando attentamente la muscolatura della parte non compressa e la funzione respiratoria durante ciascuna delle due alterne fasi. Espressione di paralisi bilaterale diaframmatica è il segno di Bériel-Durand, consistente nella comparsa di un movimento alternato a basculla toraco-addominale; naturalmente l'esame radioscopico chiarisce lo stato funzionale della muscolatura diaframmatica. Espressivi della paralisi dei muscoli intercostali sono l'immobilità del torace con depressione degli spazi intercostali e la convessità globosa inspiratoria dell'addome, con respiro costale superiore per intervento degli sternocleidomastoidei e scaleni a testa iperestesa; 4) broncopneumonico, per ostruzione bronchiale con conseguente atelettasia da broncopolmoniti batteriche; frequente è l'associazione di varie componenti dell'insufficienza respiratoria, che si rivela quando vi è diminuzione del 35% della capacità respiratoria normale.
d) Rabbia
L'incubazione ha una durata variabilissima, da 10 giorni a due anni dopo il morso infettante: secondo Webster, nell'uomo l'insorgenza della malattia si verifica nel 20% dei casi entro 30 giorni, nell'80% entro 60 giorni, mentre è eccezionale dopo il quarto mese. Durante il periodo di incubazione si possono avere disturbi non caratteristici della cenestesi: irrequietudine, tristezza, depressione, malinconia, allucinazioni olfattive, visive, uditive, spasmi, eccitazione generale. Precocemente, nell'80% dei casi, sono percepite sensazioni anormali intorno alla zona della ferita conseguenti alla stimolazione di vari gruppi di neuroni sensitivi, quindi compaiono manifestazioni di aumentata attività motoria quali tic, iperriflessia, ipertonia. Il polso è frequente, vi possono essere midriasi, lacrimazione, sudorazione, salivazione; compare febbre, che rapidamente raggiunge alti livelli (39-40 °C), e si manifestano cefalea e frequentemente vomito. Ben presto si rendono evidenti i due sintomi patognomonici: l'aerofobia, la cui comparsa è spesso precedente, consistente nel fatto che una corrente o un soffio d'aria diretti sul volto del malato provocano un'inspirazione laboriosa e penosa e senso di asfissia; e l'idrofobia, consistente nell'insorgenza di penosi spasmi laringo-faringei conseguenti al contatto delle labbra con l'acqua o, nei casi più gravi e avanzati, alla semplice vista di un liquido o di un recipiente destinato a contenerlo, o al rumore stesso dell'acqua erogata da un rubinetto, cioè a ogni percezione in grado di evocare il ricordo della deglutizione dei liquidi. L'idrofobia è forse il sintomo più penoso, e l'insorgenza dei suoi accessi provoca gravi sintomatologie cardiache e respiratorie riflesse. L'aerofobia e l'idrofobia segnano nel corso della malattia una tappa irreversibile e ne preannunciano l'esito costantemente infausto in quanto segnalano l'invasione del bulbo da parte del virus. Nella maggior parte dei casi sono presenti agitazione motoria ed eccitazione psichica, iperestesia, spasmi muscolari; l'eccitazione si trasforma in crisi di furore, e il paziente può divenire aggressivo e tentare di percuotere o di mordere le persone che gli sono vicine. Frequentemente, inoltre, si manifestano ipereccitazione sessuale, satiriasi, eiaculazioni spontanee. È presente scialorrea caratterizzata da emissione frequentissima di saliva, prima fluida e schiumosa e poi mucosa, che viene sparsa all'intorno. Dispnea e ansietà aumentano la sensazione di soffocamento dei malati; tremori, brividi, voce rauca e convulsa, sibilante attraverso la glottide contratta, completano il tragico quadro.
Mentre all'inizio della malattia è caratteristico l'alternarsi di fasi eccitative e di fasi depressive, in prosieguo compaiono manifestazioni paralitiche all'inizio generalmente agli arti inferiori, premonitrici dell'exitus che si verifica in uno stato progressivo di apatia, stupore e infine coma.
In altri malati le manifestazioni eccitative e perfino l'aerofobia e l'idrofobia mancano, e la malattia ha un decorso tranquillo per la prevalenza delle manifestazioni paralitiche a carattere generalmente ascendente (sindrome di Landry).
I quadri di rabbia eccitativa, dalla comparsa dei sintomi bulbari alla morte, hanno un decorso di 4-5 giorni, mentre quelli di rabbia paralitica durano più a lungo, fino a 10-12 giorni.
Vi può essere leucocitosi con valori di 15-30.000 elementi per mm3 costituiti prevalentemente da granulociti e da mononucleati. Il liquor è generalmente normale.
e) Coriomeningite linfocitaria
La malattia può assumere tre diverse forme cliniche: sistemica, meningitica ed encefalomielitica (Top, 1955).
La forma sistemica, preceduta da un periodo di incubazione di durata ignota, ha un esordio brusco, con febbre tra i 37,5-39,5 °C, cefalea, malessere, diffusi dolori muscolari, rinofaringite, tosse, astenia. Non si evidenziano localizzazioni a organi o apparati. Questo quadro similinfluenzale può subire diverse evoluzioni: risoluzione e guarigione entro pochi giorni; evoluzione lenta verso la guarigione con periodi di remissione e ricadute; evoluzione verso la forma meningitica o verso quella encefalomielitica; evoluzione verso un grave quadro di polmonite interstiziale.
La forma meningitica raramente si presenta come manifestazione primitiva della malattia; generalmente insorge dopo una forma sistemica non guarita, in diretta continuazione della sintomatologia, o qualche settimana dopo l'apparente guarigione di tale forma. L'esordio è simile a quello degli altri tipi di meningite, ma meno acuto di quello delle meningiti purulente, contrassegnato da intensa cefalea, dolore nucale, modica febbre, malessere, nausea e vomito; sono presenti i segni di Kernig e di Brudzinsky, mentre il segno di Babinski è raro. I pazienti non presentano turbe psichiche, tranne una certa sonnolenza. Nel liquor si rilevano pressione normale o poco aumentata, pleiocitosi con forte prevalenza dei linfociti (80-95%), aumentata concentrazione proteica ma clorurorrachia e glicorrachia normali o inferiori alla norma. Il virus è presente nel liquor nei primi 7-10 giorni di malattia. Entro una-due settimane il quadro clinico evolve verso la guarigione, con una convalescenza che spesso dura parecchie settimane.
Anche la forma encefalomielitica, più rara della precedente, può comparire come manifestazione primitiva o seguire alla fase sistemica. È caratterizzata da febbre che può raggiungere i 40,5 °C, cefalea intensa e persistente, difficoltà di parola che nei casi più gravi può giungere fino all'afasia; sono presenti frequentemente diplopia e paralisi dei muscoli oculari e del facciale, talvolta emiplegie. Si osservano inoltre disturbi della sensibilità, talvolta anche anestesia alle estremità che, per la contemporanea presenza delle paralisi, possono simulare un quadro di poli omielite: il criterio discriminativo è rappresentato però dall'assenza di atrofia muscolare. Il paziente può essere agitato o sonnolento; talora avverte dolore e rigidità al collo e alla schiena. In alcuni casi si osserva iperreflessività tendinea con ipertono muscolare, in altri invece i riflessi tendinei scompaiono conseguentemente alla predominanza del quadro mielitico. I reperti liquorali sono simili a quelli della forma meningitica.
Bisogna infine considerare la forma inapparente della malattia, che decorre senza alcuna sintomatologia clinica ed è svelata solo dalla presenza nel sangue di anticorpi neutralizzanti quale segno di pregressa infezione.
Le complicanze sono rare: è stato segnalato un caso di aracnoidite cronica e uno di orchite e parotite. Da notare a tale proposito che alcuni autori considerano le forme meningitica ed encefalomielitica come complicanze della forma sistemica.
f) Encefalite letargica
La forma più comune e più caratteristica della malattia, la letargico-oftalmoplegica, esordisce con i segni di un processo infettivo acuto: febbre sui 38-40 °C continua o continuo-remittente, frequentemente angina, dolori oculari, talvolta cefalea e rachialgia. Dopo alcuni giorni, mentre la febbre può anche scomparire per lisi, si manifesta e progressivamente si estende la sintomatologia neurologica. Inizialmente si osservano di solito turbe del sonno di vario tipo, caratterizzate in alcuni casi da episodi frequenti e ricorrenti di sonno profondo della durata di 2 o 3 ore interrotti da brevi periodi di risveglio spontaneo, in altri da ipersonnia ininterrotta che può avere la durata di alcuni giorni o anche di una o più settimane; in altri casi, infine, il sonno appare superficiale e ininterrotto, associato a stato confusionale e talvolta a delirio onirico. Trattasi sempre di sonno nel quale le funzioni vegetative (respirazione, deglutizione, ecc.) si svolgono di solito senza turbe notevoli; inoltre, stimolazioni fisiche o sonore di entità variabile possono talvolta provocare un risveglio completo, altre volte un risveglio parziale con persistenza di uno stato crepuscolare della conoscenza e limitato al periodo in cui agisce la stimolazione, cessata la quale il malato ricade in breve tempo nello stato di sopore in cui si trovava. Alle turbe del sonno già descritte si associano, nella maggioranza dei casi in un secondo tempo, solo raramente fin dall'inizio, caratteristiche alterazioni della motilità oculare sostenute da paralisi del III, del IV e del VI paio di nervi cranici: nelle forme più lievi queste possono essere denunciate da diplopia senza evidente strabismo; nelle forme conclamate, nelle quali è più frequente la compromissione del VI paio, si possono invece osservare le più diverse forme di strabismo, monoculare o bioculare, che variamente associate costituiscono uno dei segni caratteristici della malattia. In alcuni casi si osserva midriasi paralitica reazionale; il riflesso fotomotore è spesso indebolito e si svolge incompletamente e tardivamente anche dopo intensa stimolazione luminosa, e talvolta è del tutto abolito. Assai vario appare anche il comportamento del riflesso all'accomodazione: può essere conservato pur essendo abolito il riflesso fotomotore, può essere abolito insieme al riflesso fotomotore, può infine essere abolito pur essendo conservato il riflesso fotomotore. Spesso vi è ptosi di una o entrambe le palpebre superiori che solo incompletamente, e con estrema difficoltà e lentezza, possono essere sollevate dal paziente, il cui aspetto sonnolento risulta pertanto accentuato. Caratteristica peculiare di queste alterazioni della motilità oculare intrinseca ed estrinseca è la loro variabilità nel tempo, legata al carattere parcellare e transitorio delle lesioni con rapide puntate di estensione e regressione. Ai descritti deficit della motilità oculare da lesioni nucleari si possono associare disturbi da lesioni delle vie sopranucleari e internucleari: nistagmo, perdita dei movimenti combinati, ecc.
Oltre alle turbe oculari in alcuni casi si osservano deficit periferico del facciale da lesione nucleare e disturbi della fonazione e della deglutizione da lesioni dei nuclei del IX, XI e XII paio di nervi cranici. Raramente si rilevano segni di compromissione piramidale o cerebellare. Sono invece frequenti alcuni disturbi vegetativi: iperidro si, scialorrea, ipersecrezione delle ghiandole sebacee del volto, probabilmente legati a compromissione diencefalica. Sono talvolta evidenti anche lievi mioclonie. Il comportamento dei riflessi muscolari è variabile; la loro totale scomparsa è comunque segno di notevole gravità della malattia. Incostanti e di lieve entità sono i segni di compromissione meningea.
La forma amiostatico-ipertonica è di solito caratterizzata da un inizio brusco della sintomatologia, con febbre alta (40-41 °C) che si accompagna a profonda compromissione dello stato generale, cefalea, rachialgia, vomito, precoce evidenza dei segni di compromissione meningea. Nel giro di alcuni giorni si assiste a una progressiva attenuazione della febbre e all'accentuarsi della compromissione neurologica, che è caratterizzata da ipertonia muscolare diffusa con amiostasi, profonda amimia e gravi alterazioni del sonno consistenti talvolta in profonda sonnolenza o vero sonno, talvolta in invincibile agripnia. In un'elevata percentuale di casi sono anche presenti disturbi vegetativi: scialorrea, iperidrosi, seborrea. Anche la forma ipercineticomioclonica è caratterizzata da brusca insorgenza con febbre molto alta (40-41 °C), cefalea, rachialgia, vomito, segni precoci ed evidenti di compromissione meningea; in rari casi possono manifestarsi anche convulsioni epilettiformi.
Alla diminuzione e alla scomparsa di questa sintomatologia infettiva si accompagna l'insorgenza di movimenti involontari, accessionali, del più vario tipo: sono di solito prevalenti le mioclonie, ma in alcuni casi possono manifestarsi corea, spasmi muscolari, singhiozzo, che si accompagnano frequentemente a episodi paretici di breve durata a carico dei muscoli maggiormente colpiti. Tali disturbi motori sono di solito associati ad agripnia invincibile e a un notevole stato di ipereccitazione psichica che, nei casi più gravi, può giungere a uno stato maniacale. Solo in una minoranza di casi i disturbi motori sono associati a sonnolenza o a inversione del ciclo nictemerale e a uno stato di depressione psichica. Frequenti i disturbi vegetativi: iperidrosi, scialorrea, seborrea.
In diversi focolai epidemici si è osservata tra i colpiti l'evidentissima predominanza numerica di una delle forme cliniche.
L'encefalite letargica, come del resto le nevrassiti a eziologia virale, è una malattia proteiforme, e quindi accanto alle forme tipiche già descritte esiste un'infinita varietà di singoli casi nei quali ai differenti sintomi neurologici già descritti, che si intrecciano e prevalgono nei più vari modi, si associano numerosi altri segni di compromissione neurologica di varia natura ed entità, che testimoniano il carattere di nevrassite a localizzazioni multiple. Prevalgono generalmente le sintomatologie dolorose dipendenti da nevriti, radicoliti e sindrome talamica, contrassegnate da atroci dolori resistenti a ogni tipo di terapia e in alcuni casi accompagnati da emiparesi.
In altre forme dell'encefalite letargica è prevalente la compromissione del midollo: predominano allora i segni di una mielite, radicolite o radicoloneurite. Forme atipiche per l'intensità della sintomatologia e il tipo di decorso sono le forme acutissime nelle quali l'exitus sopravviene in pochi giorni, le forme parcellari paucisintomatiche, le forme subacute a decorso protratto, ecc. In occasione di episodi epidemici di encefalite letargica sono state osservate forme fruste, oligosintomatiche, caratterizzate ad esempio da febbricola, sonnolenza, diplopia transitoria, singhiozzo. È possibile quindi ipotizzare che, analogamente a quanto accade per altre nevrassiti da virus noti, possano esistere infezioni subcliniche o totalmente asintomatiche. Anche le forme fruste possono, a distanza di mesi o anni, essere seguite da parkinsonismo postencefalitico.
g) Meningite da virus coxsackie
È una sindrome meningea sostenuta da virus di entrambi i gruppi (A: 2, 4, 7, 9, 10 e B: 2, 3, 4, 5, 6) a evoluzione benigna, della durata di pochi giorni, molto raramente associata a manifestazioni dipendenti da localizzazione cerebro-spinale: atassia, turbe sfinteriche, paralisi facciale, nevralgie.
La sintomatologia clinica è uniforme, senza alcun rapporto con il ceppo che l'ha provocata: l'esordio è improvviso, con cefalea, spesso dolore retro-orbitale, nausea, vomito, angina rossa e qualche volta mialgie. La febbre, presente fin dall'inizio o solo più tardivamente, è moderata (38-39 °C) e dura in genere 4-5 giorni. È presente con vario grado d'intensità il complesso sintomatologico della sindrome meningea: rigidità nucale e della colonna toracica, segni di Kernig e di Brudzinsky, ecc.; alla rachicentesi il liquor fuoriesce sotto pressione, è limpido e presenta aumento moderato del contenuto proteico, normale glicorrachia e notevole pleiocitosi linfocitaria con un numero di elementi fino a 500 per mm3. In pochi casi di sindromi paralitiche simili alla poliomielite l'agente eziologico è stato identificato in virus coxsackie A7, A9, e B2, 3, 4, 5.
Nel complesso, la sintomatologia clinica e i reperti liquorali non consentono di distinguere questa forma morbosa da altre meningiti virali, talché la ricerca della sua eziologia esige il contributo indispensabile degli esami di laboratorio: isolamento del virus dal liquor, dimostrazione dell'aumento degli anticorpi neutralizzanti sierici fra inizio della malattia e convalescenza.
All'esame istologico le meningi, in casi di meningite neonatale da virus B, appaiono diffusamente edematose e infiltrate di macrofagi; possono rilevarsi associati piccoli foci di degenerazione disseminati nel SNC, soprattutto nel midollo spinale.
La guarigione senza esiti è l'evoluzione abituale. Nei casi da virus B può essere presente una sindrome mialgica del tipo pleurodinia epidemica che caratterizza la cosiddetta meningite mialgica.
h) Nevrassite da virus del vaiuolo
A esclusione delle sindromi paralitiche localizzate, di tipo periferico, la maggior parte dei sintomi neurologici osservabili nei casi di vaiuolo, la cui durata, come in altre forme di encefalite virale, può protrarsi oltre quella della malattia acuta, è imputabile a localizzazioni encefaliche e midollari precoci del virus, che possono assumere diversi tipi anatomo-clinici. Il tipo atassico è caratterizzato da tremori, disartria, disfagia, ipotonia muscolare, incoordinazione motrice, titubazione, indebolimento dei riflessi: il quadro può giungere fino a simulare una tabe, dalla quale è tuttavia differenziabile sia per mancanza delle crisi dolorose folgoranti, sia per la mancata accentuazione a occhi chiusi dei sintomi motori, sia infine per la regressione della sintomatologia nei mesi o nei primi anni successivi alla malattia. Si sono anche osservate sintomatologie nervose simulanti la sclerosi a placche, caratterizzate da tremori della muscolatura volontaria, disturbi della parola, esagerazione dei riflessi, tutte comunque evolventi favorevolmente entro l'anno successivo a quello della malattia acuta.
i) Encefalite morbillosa
Questa encefalite, che si manifesta in circa lo 0,1% dei casi di morbillo, esordisce fra il 2° e il 6° giorno di malattia con cefalea, vomito, convulsioni, coma; sono evidenti le classiche manifestazioni di irritazione meningea (rigidità nucale, segni di Brudzinsky, di Kernig, ecc.) e nel liquido cefalorachidiano, iperteso, sono rilevabili pleiocitosi linfocitaria e aumento del contenuto proteico senza diminuzione della glicorrachia. Il decorso, diverso da caso a caso, è talora benigno e regredibile, altre volte (nel 15% dei casi) è progressivo fino all'exitus.
Nel 25% dei casi si ha la guarigione con residua sindrome da danno cerebrale, caratterizzata da ritardo mentale, crisi convulsive ricorrenti, modificazioni del comportamento, paralisi o paresi, sordità, ecc. L'interpetrazione eziologica più corrente di questi quadri clinici è quella autoimmunitario-allergica. Esistono anche sindromi nervose regredibili, in rapporto a disturbi della regolazione idrica o elettrolitica, agevolmente controllabili mediante la correzione del parametro alterato.
l) Encefalite postvaccinica
Un posto a parte fra le complicanze della vaccinazione antivaiolosa occupa l'encefalite postvaccinica, che oggi si tende a includere nel gruppo delle encefaliti demielinizzanti postinfettive (postvirali).
Nei soggetti sottoposti alla vaccinazione antivaiolosa, in una percentuale estremamente esigua rispetto alla grandissima massa, senza differenza di sesso, con frequenza maggiore nei mesi primaverili rispetto a quelli invernali, con frequenza inversa all'età dei soggetti, possono insorgere, a variabile distanza di tempo dalla vaccinazione, alcune sindromi nervose di cui la più imponente è quella encefalitica. Il significato di tale distribuzione stagionale deve essere valutato in rapporto all'epoca delle sessioni di vaccinazione eseguite dalle autorità sanitarie. La complicanza sembra essere più frequente nelle regioni nordiche, e in Italia in quelle settentrionali, mentre è sconosciuta ai tropici.
Nella tab. III sono riportate le frequenze della complicanza in relazione all'età e alla data di vaccinazione.

Nella grande maggioranza dei casi la complicanza insorge sopo una prima vaccinazione, solo molto raramente si verifica in occasione di una rivaccinazione.
Non è stato messo in evidenza alcun rapporto fra l'insorgenza della complicanza e l'origine o la qualità della linfa vaccinica: linfe la cui inoculazione è stata seguita dalla comparsa di casi di encefalite in certe regioni si sono rivelate completamente innocue in altre. Egualmente prive di importanza determinante risultano le modalità di inoculazione della linfa vaccinica.
Lo studio istopatologico dei casi mortali ha permesso di osservare lesioni di duplice tipo: vasali, consistenti essenzialmente in dilatazione e congestione con circoscritte emorragie per diapedesi e necrosi perivascolari del tessuto cerebrale; cerebrali, sotto forma di aree circoscritte di necrosi della sostanza nervosa, di origine tossica, senza alcun segno di infiammazione acuta o cronica nè di modificazioni ischemiche del tessuto. La lesione dominante è costituita da una marcata demielinizzazione delle fibre nervose della sostanza bianca, soprattutto intorno ai piccoli vasi venosi, con scarsa degenerazione o necrosi neuronale.
Dal punto di vista clinico la sintomatologia è schematizzabile nei seguenti tipi: paretico sonnolento, meningeo, tetaniforme, mielitico, abortivo. Il suo inizio è brusco, contrassegnato da cefalea, vomito, sonnolenza, accessi convulsivi, e un corteo di manifestazioni meningee di variabile intensità: segno di Brudzinsky, rigidità nucale, iperestesia, segno di Lasègue, ecc. Il reperto liquorale è tuttavia assai scarso, limitato a un lieve aumento delle cellule e dell'albumina, senza modificazione della glicorrachia. In generale i casi a più spiccata impronta meningea presentano un'evoluzione più favorevole.
Assai meno frequente è il rilievo di stati di agitazione o di delirio. I fenomeni motori dell'encefalite postvaccinica consistono in crisi convulsive e paralisi. Le prime si verificano specialmente nelle forme a impronta meningea, sono più frequenti nei primi giorni della malattia e in genere generalizzate, di durata variabilissima, da pochi minuti a qualche ora, e si succedono con frequenza altrettanto variabile; più di rado si osservano attacchi convulsivi circoscritti a determinati gruppi muscolari. Le paralisi possono colpire esclusivamente o prevalentemente la muscolatura oculare (paralisi oculari motorie, ptosi palpebrale, paralisi dell'accomodazione), ma più frequentemente riguardano i vari gruppi muscolari degli arti; sono abitualmente spastiche e tali rimangono anche in seguito, oppure possono insorgere con carattere di flaccidità e così mantenersi o acquisire invece carattere di spasticità. La cefalea è precoce, continua, intensa; v'è iperestesia generale. La febbre è elevata fin dall'inizio della sindrome o diventa tale, nei casi mortali, nell'imminenza dell'exitus.
La durata media della malattia è di 2-3 settimane nei casi favorevoli, di pochi giorni nei casi mortali. Nei primi, i fenomeni convulsivi sono quelli che si dileguano più precocemente, mentre quelli paralitici persistono più a lungo modificandosi e attenuandosi poi gradualmente.
L'encefalite postvaccinica ancor oggi resta di difficile interpretazione eziopatogenetica: l'ipotesi secondo la quale sarebbe determinata da una migrazione del virus vaccinico dotato di un peculiare neurotropismo appare inconciliabile con i risultati negativi dei tentativi di isolare il virus dal tessuto nervoso o dal liquor dei pazienti e con le lesioni di tipo completamente diverso da quelle prodotte sperimentalmente nell'animale mediante inoculazione intracerebrale di virus vaccinico. La negatività dei suddetti tentativi di isolamento è peraltro spiegata dai fautori dell'eziologia vaccinica ammettendo che il virus a livello del tessuto cerebrale esisterebbe mascherato in forma non rilevabile dalle tecniche dell'isolamento, per la presenza di anticorpi neutralizzanti o perchè sotto forma di virioni incompleti.
Una seconda interpretazione suggerisce che responsabile dell'encefalite postvaccinica, così come della postmorbillosa, della postvaricellosa e di quella seguente la vaccinazione antirabbica, sia l'attivazione di virus latenti per opera della stimolazione vaccinale o di altri stimoli.
L'ammissione di un'eziologia virale unica per tutte le forme appartenenti a questo gruppo di encefaliti rende tuttavia difficile la comprensione della loro così variabile letalità: dal 10% dei casi di encefalite postmorbillosa fino talvolta al 55-60% di quelli di encefalite postvaccinica.
Infine, il fatto che l'encefalite postvaccinica insorge in pieno periodo di immunità verso il virus vaccinico ha consentito di prospettare una teoria allergica, che tuttavia non è suffragata da altre prove positive e sembra anzi in contrasto con la constatazione che la complicanza si manifesta di regola nei soggetti vaccinati per la prima volta e non nei rivaccinati, nei quali sarebbe naturalmente più agevole ammettere l'esistenza di una sensibilizzazione.
La terapia, oltre a quella sintomatica comune a tutti i processi encefalitici acuti, è sostanzialmente basata sulla somministrazione di adeguate dosi giornaliere di prednisone (25-30 mg) per 6-7 giorni. Nei casi di ipertensione liquorale saranno consigliabili rachicentesi ripetute.
m) Nevrassite da virus herpes simplex
L'eziologia erpetica di alcuni casi di meningoencefalite è stata dimostrata sia per l'isolamento del virus erpetico dal liquor e dal tessuto cerebrale, sia per la presenza e per l'aumento di anticorpi sierici antierpetici, sia infine per il reperto di inclusioni del tipo corpi di Lipschütz nelle cellule del SNC. La sintomatologia è caratterizzata da inizio improvviso con febbre alta e rapido instaurarsi di segni meningei e di ipertensione endocranica: intensa cefalea, fotofobia, rigidità nucale, asimmetria pupillare, presenza dei segni di Kernig, di Brudzinsky, di Lasègue, paresi e paralisi di determinati gruppi muscolari, parestesie. Nel complesso, sul piano clinico, la sintomatologia non si distingue da quella delle altre nevrassiti virali. Il liquor è limpido, iperproteico e caratterizzato da una modica pleiocitosi.
n) Nevrassite da virus herpes zoster
In questa forma la lesione è elettivamente localizzata nei vari segmenti del neurone sensitivo periferico: a livello del ganglio spinale come necrosi, eventualmente emorragica, con infiltrazione linfocitaria; nel midollo come mielite unilaterale, estesa a un paio di segmenti, interessante le corna posteriori e le radici; nel nervo periferico sotto forma di mononeurite vera estesa a tutto il tratto distale del nervo oltre il ganglio; infine, nelle meningi avvolgenti le radici posteriori e i segmenti spinali come leptomeningite. Rara è la diffusione del processo alle corna anteriori, mentre è possibile l'estensione di un'infiltrazione monocitaria, oltre che alle radici posteriori, al bulbo e ai peduncoli cerebrali.
La riparazione dei fatti necrotici si compie attraverso un processo di iperplasia gliale con eventuale possibile sclerosi secondaria.
L'osservazione di lesioni di tanto rilievo e così diffuse nei casi, relativamente scarsi, pervenuti al tavolo anatomico, ne lascia supporre l'esistenza, sia pure con caratteri notevolmente più lievi, praticamente in tutti i pazienti con herpes zoster, e induce a considerare la malattia come un processo morboso ben diverso e più complesso di una semplice affezione cutanea o nervosa localizzata e periferica.
o) Nevrassiti in corso di varicella
Meno infrequentemente di quanto comunemente si creda è possibile osservare in corso di varicella quadri neurologici di grande interesse, per la cui interpretazione poco convincenti appaiono le due ipotesi da alcuni sostenute: quella dell'affioramento, sotto l'influenza del virus varicelloso, di un virus preesistente nel tessuto nervoso, e quella di Glanzman, che ammette l'esistenza di una reazione anafilattoide tardiva all'antigene varicelloso in soggetti sensibilizzati.
Per quanto resti difficile da spiegare l'insorgenza tardiva della complicanza nervosa se non ammettendo necessariamente una lenta migrazione centripeta del virus lungo le vie nervose, la possibile localizzazione del virus varicelloso al tessuto nervoso appare suggestiva in considerazione del ben noto neurotropismo del virus del gruppo varicella-zoster, e sembra confermata dal rilievo di casi in cui la complicanza nervosa è intervenuta contemporaneamente all'eruzione cutanea varicellosa.
I quadri neurologici di più frequente osservazione sono: le encefalomieliti, in cui si ritrovano misti in variabili proporzioni segni mielitici e segni encefalitici; le encefaliti pure, con compromissione del solo encefalo e integrità di altre parti del nevrasse; le encefaliti caratterizzate da tremore acuto, le encefaliti a varietà cerebellare, le encefaliti a varietà coreo-atetosica, quelle polimorfe non caratteristiche; inoltre le mieliti trasverse ascendenti, le poliomieliti, le sclerosi multiple, le nevriti, le polinevriti, le neuromiositi, le oftalmoplegie soprattutto di tipo nucleare, le nevriti ottiche, le manifestazioni psichiche di difficile classificazione accompagnate in rari casi a perdita della parola o idiozia definitiva.
L'insorgenza più frequente è fra il 4° e il 10° giorno di malattia, ma in un certo numero di casi la sindrome esplode più tardivamente, fino a 90 giorni dopo il periodo eruttivo.
Il liquido cefalorachidiano non mostra alterazioni notevoli oltre il reperto di una moderata linfocitosi; solo nei casi a spiccata sintomatologia meningea si possono osservare quadri liquorali caratterizzati da variabili gradi di pleiocitosi e aumento dell'albumina.
p) Nevrassiti in corso di polmoniti virali
Sono stati descritti nel corso di polmoniti virali primitive casi di manifestazioni neurologiche varie: meningoencefaliti, mieliti, encefaliti, mieloencefaliti, meningite linfocitaria, mielite trasversa, emiplegia, paralisi ascendente, sindrome di Guillain-Barré. In tutti i pazienti esaminati il liquor è apparso sempre chiaro e sotto tensione, con pleiocitosi di grado variabile fino a 350 cellule per mm3, soprattutto linfociti o polinucleati, questi ultimi in qualche caso costituenti il 95% di tutti gli elementi presenti. La letalità è elevata, in media di 12 decessi su 30 casi descritti.
In nessun caso si è riusciti a isolare virus dal tessuto nervoso. È stata avanzata l'ipotesi patogenetica di trombosi del SNC da coagulazione intravasale provocata da sostanze liberate dal parenchima polmonare durante il periodo dell'infiltrazione.
q) Nevrassiti in corso di influenza
Mentre manifestazioni morbose esprimenti lievi segni di compromissione meningea (cefalea tormentosa, lieve rigidità nucale, lieve ipertensione liquorale) possono riscontrarsi non raramente in corso di talune epidemie, è eccezionale la documentazione di eziologia da virus influenzale di casi di nevrassite in corso di influenza, secondo criteri eziologici restrittivi. D'altra parte, anche quando è possibile dimostrare la presenza del virus nel SNC, la sua concentrazione è così bassa che il reperto potrebbe essere interpetrato in rapporto alla semplice diffusione dal torrente circolatorio: si tratterebbe, in altre parole, di una espressione postviremica senza significato eziologico in riferimento alla manifestazione neurologica in atto, che non può essere quindi considerata un elemento probativo.
r) Meningiti da virus ECHO (Enteric Cytopathogenic Human Orphan)
Si manifestano durante epidemie, generalmente estive, nel corso delle quali possono osservarsi anche quadri non nevrassitici di malattie indifferenziate con esantema facoltativo; presentano i caratteri tipici della sindrome meningea, con liquor limpido costantemente contrassegnato da pleiocitosi sia pure di variabile entità (da 10-15 fino a 600-900 cellule mm3, rappresentate per la maggior parte da linfociti); colpiscono prevalentemente ma non esclusivamente i bambini e possono talvolta essere associate a localizzazione nel SNC, come meningoencefaliti o meningoencefalomieliti, sindromi paralitiche, generalmente benigne.
Mediante l'isolamento dalle feci, dalle secrezioni respiratorie e dal liquor è stato possibile accertare che i tipi di virus più frequentemente responsabili delle manifestazioni epidemiche sono 4, 6, 11, 16 e soprattutto 9. Si ammette che i tipi 2, 3, 4, 7, 8, 14, e meno sicuramente quelli 16, 18, 21, 12 e 13, possano provocare casi sporadici.
Oltre la sindrome meningea, di intensità variabile (rigidità nucale, fenomeni di Kerning e di Brudzinsky, atteggiamento a cane di fucile, segno di Lasègue, ecc.), sono presenti: cefalea; febbre fino a 38-39 °C della durata di 3-4 giorni; facoltativamente un esantema maculo-papulo so; sintomi gastrointestinali fra i quali predominano il vomito e la nausea; meno frequentemente diarrea, mialgie, artralgie. Nel 30% dei casi è presente angina rossa con modica iperplasia del tessuto linfatico faringeo. Inoltre i pazienti accusano fotofobia, ed è possibile rilevare iniezione congiuntivale. Non si osservano modificazioni significative della crasi leucocitaria periferica.
Nel complesso si tratta di quadri clinici meningei, per i quali, senza l'ausilio di ricerche virologiche, sarebbe impossibile andare oltre la generica diagnosi di ‛meningite abatterica presumibilmente virale', né tanto meno, una volta accertata l'eziologia da virus ECHO, sospettare un particolare tipo antigenico. La durata della malattia è di pochi giorni, generalmente circa una settimana, e la reversibilità dei quadri meningei puri costituisce la regola.
Accanto a questi quadri possono osservarsi casi mortali di paralisi febbrile pseudopoliomielitica, documentati dall'isolamento del virus dal bulbo. Talora le manifestazioni paralitiche sono associate a segni meningei, si tratta cioè di meningoencefalomieliti. I gruppi muscolari più frequentemente colpiti sono quelli del volto, quelli della deglutizione, della nuca, del dorso, degli arti. La compromissione talora è sfumata, e si estrinseca in paresi, debolezza muscolare, diminuzione transitoria della forza. La reversibilità è comunque di regola e conseguentemente rarissime sono le atrofie.
Più frequentemente responsabili dei fatti paralitici sono i tipi 4, 6, 9, eccezionalmente i tipi 16 e 2.
s) Sintomatologie neurologiche in corso di dengue
Cefalea, insonnia, delirio con allucinazioni, manifestazioni convulsive osservabili specialmente nei bambini, sonnolenza, disorientamento sensoriale, associati a ipertensione liquorale con lieve iperproteinorrachia e pleiocitosi, denotano il frequente cointeressamento del sistema nervoso in corso di dengue, che, in rari casi, può giungere fino all'encefalite vera.
V'è costantemente, specie all'inizio, leucopenia con neutropenia in rapporto soprattutto a una diminuzione degli elementi a nucleo maggiormente segmentato, mentre può osservarsi una graduale ascesa del numero dei linfociti.
Con la caduta della temperatura, non sempre la sintomatologia si estingue altrettanto prontamente: possono soprattutto persistere mialgie ed eventuali disturbi dell'accomodazione, e residua sempre un'intensissima, invincibile astenia. Queste caratteristiche conferiscono alla malattia l'attributo di un'affezione di considerevole interesse sociale, perché colpisce soggetti giovani, in piena capacità lavorativa, inabilitandoli per un considerevole periodo alla loro normale attività produttiva.
t) Sintomatologie neurologiche in corso di febbre da flebotomi
In circa il 15% dei casi di febbre da flebotomi sono presenti sintomi neurologici: cefalea frontale intollerabile, rigidità nucale, segno di Kernig, fotofobia, vomito.
Per le caratteristiche algie muscolari si è considerata la possibilità di una compromissione dei cordoni posteriori del midollo, e per la presenza in alcuni casi di sintomi nervosi marcati con evidente impronta meningea, di localizzazioni nelle meningi: in questi casi l'esame del liquor ha permesso di accertare una discreta ipertensione con lieve aumento dell'albumina e discreta pleiocitosi, fino a un centinaio di cellule per mm.
u) Nevrassite in corso di mononucleosi infettiva
L'interessamento nevrassitico, specie se è ricercato anche nelle sue espressioni minori, risulta abbastanza frequente: la sua incidenza varia infatti dall'1 al 9% dei casi di mononucleosi, e in alcune forme epidemiche della malattia esso conferisce al quadro clinico un'impronta particolare.
Possono essere compromessi i nervi cranici, i nervi periferici, le radici spinali e il midollo spinale, le meningi, il cervello. Si possono osservare quadri clinici di neuriti isolate o di poliradicoloneuriti, di sindrome di Guillain-Barré, di meningite o di meningoencefalite. Il liquor è iperteso con aumento delle proteine e lieve o discreta pleiocitosi prevalentemente linfocitaria.
Sono descritte con variabile frequenza, fino al 40% dei casi di interessamento nevrassitico, manifestazioni oculari che possono essere distinte in due tipi: quelle direttamente interessanti l'apparato oculare, caratterizzate da bruciore, dolore alla rotazione dei bulbi oculari, dolore profondo statico, fotofobia, in rapporto a processi di congiuntivite, edema orbitario, uveite, edema ed emorragie retiniche; e quelle interessanti la funzione visiva e il sistema neurooftalmico, da lesione del SNC, consistenti in paralisi oculari estrinseche, ptosi palpebrali, nistagmo, disordini dei movimenti coniugati, ecc.
v) Nevrassite da virus encefalomiocardite
Il quadro più comune è quello di una malattia acuta del SNC con pleiocitosi liquorale linfocitaria e con variabile corteo di manifestazioni paralitiche. Nell'uomo sono stati osservati segni di miocardite.
Nella cosiddetta malattia di Manila (febbre dei tre giorni), il quadro clinico è caratterizzato da inizio improvviso, con cefalea, febbre alta della durata di 2-3 giorni, angina rossa, sintomatologia meningea di variabile intensità (rigidità nucale, segno di Kernig, iperreflessività profonda, pleiocitosi liquorale variabile da 50 a 500 cellule/mm3 prevalentemente rappresentate da linfociti), sovente corna; il sangue periferico presenta linfocitosi. La guarigione interviene in terza-quarta giornata. La malattia pertanto è clinicamente indistinguibile da quella causata da virus enterici, dai virus del gruppo dengue e dalle altre meningiti linfocitarie virali.
w) Infezioni del SNC da virus lenti: kuru
Il concetto di ‛infezioni da virus lenti' è stato introdotto da Björn Sigurdsson del Keldur Institute di Reykjavik in seguito a osservazioni compiute principalmente in patologia veterinaria, in particolare su alcune malattie delle pecore, conosciute come maedi, visna e scrapie. Tali forme morbose consistono, rispettivamente, in una polmonite lentamente evolvente con profonde modificazioni dell'aspetto, del peso e della consistenza dei polmoni; in una infezione lungamente silente dal punto di vista clinico e rivelantesi successivamente con sintomi neurologici associati a modificazioni di tipo meningitico del liquido cerebro-spinale; in una malattia neurologica caratterizzata da tremore, che successivamente è stata riconosciuta identica allo scrapie.
Originariamente si stabilì che, perché una malattia potesse essere inclusa fra le infezioni da virus lenti, se ne sarebbero dovute dimostrare tre caratteristiche fondamentali: 1) un lunghissimo periodo di latenza (settimane, mesi o anni); 2) una progressione regolare dei sintomi clinici dopo la loro comparsa; 3) l'incidenza in una sola specie animale e la limitazione delle lesioni a un solo organo o sistema. Quest'ultimo criterio ha successivamente subito una revisione, in quanto è stata accertata la possibilità della trasmissione di taluni virus lenti a specie animali diverse da quelle nelle quali la malattia era stata originariamente identificata.
Secondo un'interessante interpetrazione patogenetica, formulata da Adams e Field nel 1968, lo sviluppo di una infezione da virus lento presuppone tre componenti: una particella subvirale autoreplicantesi non patogena, membrane cellulari normali e un materiale di congiunzione (linkage-substance) non autoreplicantesi.
I primi due non possono interreagire senza il terzo, in presenza del quale le particelle subvirali si unirebbero alle membrane cellulari per formare una struttura patologica: questa diffonderebbe sulle superfici cellulari inducendovi la formazione di ulteriori quantità di materiale di congiunzione, cui si unirebbe quindi un maggior numero di particelle subvirali, aumentando così le dimensioni della lesione patologica della membrana. Conseguentemente la malattia potrebbe essere provocata non già dall'inoculazione di particelle subvirali in animali recettivi, bensì da quella del materiale di congiunzione in animali nei quali siano presenti particelle subvirali. Per converso la malattia non si sviluppa in animali semplici portatori di particelle subvirali, in quanto in assenza di materiale di congiunzione non può avvenire l'unione di tali strutture alle membrane cellulari.
Si è tentato di riunire in un unico gruppo denominato CHINA (CHronic Infectious Neuropathic Agents) alcuni virus lenti, quali quelli responsabili del kuru, della malattia di Creutzfeldt-Jakob, di determinate malattie degli animali come lo scrapie (Koprowsky); si potrebbero tentativamente includere anche gli agenti di malattie neurologiche umane a eziologia tuttora sconosciuta, caratterizzate clinicamente dalla lentissima progressione e da quadri anatomopatologici comuni di reazione astrocitaria, degenerazione neuronale e demielinizzazione. La possibilità che la sclerosi multipla sia determinata da un agente virale di questo tipo viene oggi presa in buona considerazione.
Con il termine kuru, che nel dialetto degli indigeni di una zona interna della Nuova Guinea australiana significa letteralmente ‛tremore con paura', viene designata una malattia determinata da un ceppo olandese di virus scrapie.
La malattia, osservata per la prima volta da Mac Artur nel 1953 e accuratamente descritta da Gajdusek e Zigas nel 1957, costituisce il prototipo umano delle ‛infezioni lente'; dal punto di vista epidemiologico è caratteristica la limitazione della sua distribuzione geografica a una ristretta regione montagnosa situata nella parte più occidentale della Nuova Guinea australiana, abitata da popolazioni appartenenti al gruppo linguistico e culturale Fore. Il kuru è diffuso anche ad altri gruppi etnici, ma colpisce soltanto individui di popolazioni strettamente confinanti o apparentate con i Fore; la sua comparsa sembra essere avvenuta negli ultimi 60 anni, per quanto almeno consentano di affermare le non facili comunicazioni con popolazioni il cui livello di civiltà è fermo all'epoca della pietra e tra le quali ancora sopravvive il cannibalismo. La malattia, inizialmente limitata al sesso femminile, si è successivamente estesa ai bambini dei due sessi; in epoca più recente, invece, il suo inizio in età infantile è divenuto un'evenienza piuttosto inconsueta. Il kuru ha fatto registrare la sua massima diffusione nel 1959, probabilmente in rapporto a particolari spostamenti della popolazione colpita che, a carattere nomade, occupa un'area assai vasta, con una densità media di 31 individui per miglio quadrato. La considerazione di tutti questi dati di ordine epidemiologico induce a formulare le seguenti ipotesi: 1) la malattia attecchisce in soggetti geneticamente recettivi; 2) la malattia è trasmessa attraverso l'abitudine al cannibalismo.
Dal punto di vista anatomopatologico, le lesioni proprie del kuru consistono essenzialmente in una degenerazione dei neuroni cerebellari con proliferazione degli astrociti e status spongiosus; manca qualsiasi segno di infiammazione, mentre è presente, come fenomeno secondario, la demielinizzazione. Molti neuroni mostrano neuronofagia e accumulo di lipofuscina e appaiono frequentemente infarciti di vacuoli otticamente vuoti, reperto questo molto simile a quello dello scrapie; i nuclei, raramente doppi, sono frammentati e picnotici. Nel cervelletto si osserva atrofia delle cellule dello strato granuloso. La lesione sembra essere disseminata senza una particolare distribuzione topografica a vaste zone del SNC, mentre il grado di degenerazione è variabile da zona a zona, così come da caso a caso. Non esiste alcun rapporto tra distribuzione topografica e gravità delle lesioni neuronali da un lato, e distribuzione dei vasi sanguigni dall'altro. La proliferazione astrocitaria è intensa e diffusa specie nella materia grigia del midollo spinale: tale rilievo, in considerazione del fatto che gli astrociti sono intimamente associati con la cosiddetta barriera emato-encefalica alle cui variazioni di permeabilità rispondono immediatamente, induce a ritenere valida l'ipotesi che un agente patogeno esistente nel sangue produca una reazione precoce negli astrociti perivascolari. Attivazione della microglia appare invece diffusa sia alla materia bianca sia a quella grigia. I vasi sanguigni sono frequentemente circondati da zone di infiltrati di mononucleati di origine ematica. Sono ancora osservabili status spongiosus di vario grado nella materia grigia, formazioni cistiche in quella bianca, talvolta lesioni degenerative anche a livello del tratto ipotalamo-ipofisario.
Per spiegare l'evoluzione del kuru sono state fornite due principali interpretazioni patogenetiche, secondo le quali la malattia consisterebbe rispettivamente in uno stato degenerativo diffuso avente alcune similarità con la malattia di Creutzfeldt-Jakob, ovvero in un'elettiva degenerazione neuronale del cervelletto associata a una reazione diffusa ma aspecifica con ipertrofia astrocitaria e status spongiosus.
Si ritiene da taluni che l'aspetto ‛diffuso' delle lesioni del kuru sia solo apparente, tale da mascherare l'elettiva localizzazione cerebellare; secondo altri autori, invece, il kuru sarebbe un processo morboso generalizzato nel corso del quale il cervelletto è particolarmente e più gravemente colpito, così da somigliare più alla malattia di CreutzfeldtJakob che non a una malattia cerebellare vera e propria. La trasmissione sperimentale del kuru è stata eseguita con successo da Gajdusek (1967) mediante l'inoculazione in scimpanzé di materiale cerebrale proveniente da individui malati: sette animali, dopo un periodo di incubazione di 18-30 mesi, presentarono una malattia neurologica caratterizzata da atassia, da incoordinazione progressiva e manizione terminale sostenuta da lesioni anatomiche sovrapponibili a quelle osservate nell'uomo. In successivi passaggi nella stessa specie animale il periodo di incubazione si ridusse in modo considerevole, e la comparsa dei sintomi potè essere osservata dopo 11-12 mesi dall'inoculazione del materiale infettante. I risultati dei tentativi sperimentali di provocare la malattia in topi sottoposti a irradiazioni Röntgen sono assai controversi, onde sembra ormai dimostrato che l'agente eziologico del kuru sia un virus, trasmissibile allo scimpanzé, del quale si stanno ora indagando le principali caratteristiche (resistenza termica e dimensioni), che si sospetta debbano essere assai vicine a quelle dei virus dello scrapie e all'agente isolato da casi di malattia di Creutzfeldt-Jakob.
Le caratteristiche cliniche del kuru sono rappresentate dall'inizio insidioso, dall'assenza di febbre e di significativi reperti patologici ematici e liquorali e dalla progressione lentissima della sintomatologia neurologica, che non mostra consistenti remissioni: atassia, tremori, modificazioni della personalità psichica. La morte sopravviene entro un periodo di tempo variabile dai 3 ai 6 mesi dall'inizio della malattia.
y) Possibile eziologia da virus lenti (non ancora identificati) di alcune malattie neurologiche dell'uomo
Esiste un gruppo di malattie del SNC dell'uomo sulla cui eziologia regna tuttora una grande incertezza nonostante le varie ipotesi interpretative di volta in volta formulate. Recenti acquisizioni di ordine clinico-biologico sembrano comunque rappresentare valide prove a favore dell'esistenza di virus responsabili di alcune particolari forme morbose: la panencefalite subacuta sderosante, l'encefalopatia leuco progressiva plurifocale, la sclerosi multipla. Alcuni autori riuniscono in un unico gruppo di malattie a eziologia non accertata ma possibilmente virale la panencefalite nodulare e la panencefalite subacuta sderosante o encefalite a inclusi di Dawson. Quest'ultima è uno stato morboso proprio della giovane età, soprattutto dei bambini in età scolare, il cui quadro clinico, relativamente uniforme, evolve attraverso tre stadi successivi. Il primo stadio è quello dell'inizio, insidioso, caratterizzato soprattutto da deteriorazione del potenziale intellettuale e da disturbi psichici o, in altri casi, da disprassia, convulsioni generalizzate, disordini della vista: nel volgere di settimane o mesi questi disordini si accentuano progressivamente fino a condurre a uno stato di disorientamento che può terminare con una situazione di cecità e decerebrazione. In un secondo stadio la muscolatura degli arti superiori e inferiori è compromessa per l'insorgenza di contratture in flessione e rigidità, interrotte da fasi di mioclonia. Il terzo stadio è caratterizzato da persistente rigidità della muscolatura, in particolare di quella degli arti, e da coma.
L'elettroencefalogramma mostra inizialmente uno stato di disritmia che si modifica, quando compaiono le crisi miocloniche, in onde sincrone di elevata ampiezza e bassa frequenza assumenti significato diagnostico, e comparsa periodica di onde delta trifasiche di notevole ampiezza sulla regione frontale. Il liquido cerebrospinale è sostanzialmente normale, per quanto riguarda sia il contenuto in proteine sia quello in elementi corpuscolati, durante tutto il decorso della malattia: solo negli stadi più avanzati si riscontra una curva di oro colloidale di tipo paretico e un aumento di IgG. Il quadro anatomopatologico è quello di un'encefalomielite virale con compromissione variabile sia della sostanza bianca sia della grigia e compromissione dei neuroni corticali ; sono rilevabili stati di infiammazione cronica perivascolare associata a gliosi fibrillare e demielinizzazione, mentre i neuroni corticali possono presentare, specialmente nei casi a rapido decorso, inclusi del tipo A, secondo alcuni autori indistinguibili da quelli osservabili in casi di encefalite da herpes simplex. La demielinizzazione può derivare dalla distruzione della oligodendroglia, che è frequentemente stipata di inclusioni. Sono anche presenti neuronofagia, noduli microgliali, infiltrazione plasmacellulare e linfocitaria delle meningi, essudati perivascolari, gliosi astrocitaria e fibrillare. Tutta la malattia evolve in maniera non uniformemente progressiva, nel senso che si possono osservare remissioni di variabile durata dei vari sintomi, mentre il destino finale è quello di una progressione inarrestabile.
L'ipotesi della possibile eziologia virale di questa ma- lattia fu inizialmente prospettata da Adams e quindi so- stenuta da Bouteille e collaboratori in base alle loro osservazioni di microscopia elettronica, i cui risultati erano apparsi suggestivi per implicare il virus del morbillo; successivamente sembrò confermata dalla dimostrazione dell'esistenza di alti livelli di anticorpi antimorbillosi nel siero dei malati (Connolly e coll., Sever e Zeman) e di elevate concentrazioni di antigeni morbillosi nei neuroni colpiti (Lennette e coll., Tourtellotte e coll.). Invero, due diversi gruppi di autori riuscirono a riprodurre nella scimmia un quadro morboso paragonabile a quello della leucoencefalite sderosante partendo da materiale umano, ma non poterono identificare nell'agente così trasmesso il virus morbilloso (Pelc e coll., Katz e coll.). Al contrario, Baublis e Payne e Tsu Teh Chen e coll. poterono isolare il virus morbilloso da materiale cerebrale proveniente da malati con elevati livelli di anticorpi antimorbillosi.
Questi dati, la cui interpretazione è tuttora incerta, in- ducono a ritenere possibile che la malattia sia provocata da virus morbilloso rimasto latente dopo un'infezione precoce in soggetti con poteri immunitari depressi. Al pari di quanto è stato ipotizzato a proposito di altre malattie, il reperto di soli anticorpi antimorbillosi, in assenza di isolamento del virus, potrebbe essere spiegato ammettendo che all'atto della replica virale il virione abbia sequestrato antigeni dell'ospite e che questo complesso antigenico ospite-virus provochi l'elaborazione di anticorpi capaci di danneggiare le strutture nervose contenenti gli stessi antigeni sequestrati.
L'encefalopatia leuco progressiva plurifocale fu descritta inizialmente da Astrom, Mancall e Richardson come uno stato morboso associato ad alcune neoplasie ed emopatie, nonché a malattie di varia natura: leucemie, linfosarcoma, carcinomi, granuloma maligno, mieloma multiplo, policitemia vera, ipersplenismo primario, malattia di Whipple, sarcoidosi e tubercolosi, caratterizzate tutte da uno spiccato interessamento del sistema reticoloistiocitano. Il riconoscimento di tale condizione neurologica deve far sospettare la presenza di uno stato neoplastico indiagnosticato. Il decorso è progressivo durante un periodo di 3-4 mesi ed è caratterizzato da disordini della vista, emiparesi, afasia, deterioramente psichico e intellettuale, talora sintomi cerebellari: vertigini, dismetria, atassia. Possono inoltre rilevarsi disfasia, emianestesia, diplopia, paralisi del facciale, disartria e disfagia per interessamento del tronco cerebrale. Il quadro è progressivo e termina con la morte entro qualche settimana o mese. Il reperto anatomopatologico caratteristico è quello di aree confluenti di demielinizzazione prevalenti nelle regioni parieto-occipitali, nel mesencefalo, nel cervelletto e nel midollo spinale, circondate da infiltrati di linfociti e plasmacellule; in tali aree al precoce ingrandimento dei nuclei degli oligodendrociti fa seguito la loro scomparsa seguita dalla comparsa di astrociti giganti nei quali sono presenti numerose mitosi anche atipiche. È possibile rilevare una relativa conservazione degli assoni e la presenza di materiale sudanofilo nelle cellule della microglia. L'esame elettroencefalografico mostra la presenza di onde di scarsa ampiezza in corrispondenza delle zone cerebrali di massima degenerazione. Per quanto riguarda l'eziologia, appare singolare e importante la dimostrazione fornita da Howatson, Nagai e Zu Rhein che in tali casi sono presenti nel SNC virioni simili a quelli del virus del gruppo Papova, e in particolare del polioma SV4O; Zu Rhein, che ha raccolto in letteratura ventisei casi di osservazione di inclusioni intranucleari, ha poi descritto egli stesso in quattro ulteriori casi la presenza in oligodendrociti di virioni intranucleari caratterizzati da una struttura cristallina, dai quali peraltro non è riuscito a coltivare virus. Tuttavia, la frequenza con la quale sono state osservate simili inclusioni e la loro persistenza nei nuclei induce a ritenere che, piuttosto che a parassiti passeggeri, ci si trovi di fronte a patogeni capaci di provocare sia la degenerazione degli oligodendrociti parassitati sia la proliferazione atipica degli astrociti; la demielinizzazione sarebbe conseguente alla lesione degli astrociti. Il significato di tale reperto dovrà comunque essere chiarito dall'isolamento, sistematico e non occasionale, del virus da altri casi della malattia.
La sclerosi multipla (SM) potrebbe a sua volta essere interpretata come un processo morboso provocato da un virus lento. La conoscenza della malattia si deve alle prime osservazioni anatomopatologiche di Cruveilhier e di Carswell e soprattutto alle rigorose descrizioni anatomocliniche di Charcot, le quali hanno consentito di precisarne gli essenziali aspetti morfologici: distruzione delle guaine mieliniche delle fibre nervose e riduzione di numero e alterazioni varie di altri elementi quali cilindrassi, cellule nervose e cellule gliali. Tali lesioni sono disseminate in tutto il SNC in zone adiacenti a piccole venule, con siti di predilezione corrispondenti agli angoli laterali dei ventricoli, ai peduncoli cerebrali, al ponte, al mesencefalo. Talora si osservano anelli concentrici di demielinizzazione, interpretati come zone alterne di propagazione dell'agente patogeno e di resistenza relativa del tessuto, con conservazione dei cilindrassi. Precocemente si nota proliferazione di astrociti, talchè l'intera lesione appare come una cicatrice semitraslucida piuttosto depressa in superficie di sezione; la reazione astrocitaria può essere presente anche a distanza dalla lesione. Nella progressiva evoluzione della lesione, i cilindrassi che attraversano una placca possono degenerare, così come accade anche nell'encefalomielite morbillosa.
La proliferazione astrocitaria, da alcuni ritenuta un fenomeno satellite e secondario alle lesioni degenerative della mielina, è da altri autori interpretata, come del resto già inizialmente lo fu da Charcot, quale lesione primitiva cui solo secondariamente conseguono l'atrofia degenerativa della mielina e l'iperplasia delle pareti vascolari. Si ammette, infatti, la possibilità che le modificazioni primitive della glia interferiscano nei processi di nutrizione delle fibre nervose, producendo a loro volta la degenerazione della mielina. Alcune inclusioni nei vacuoli del citoplasma degli astrociti, di frequente osservazione, sono state interpetrate come centrioli allungati. Accanto alle placche di demielinizzazione che appaiono totalmente scolorate, se ne osservano altre, in cui il processo degenerativo della mielina è incompleto, caratterizzate da una colorazione meno intensa e designate pertanto come ‛ombre mieliniche', espressione forse di una fase iniziale del processo ovvero di un tentativo di rimielinizzazione.
Nel liquido cefalorachidiano è rilevabile un consistente aumento delle gammaglobuline. La microscopia elettronica non ha consentito di osservare strutture virioniche.
Riguardo all'eziopatogenesi, la somiglianza delle lesioni nervose sperimentalmente provocate nell' animale mediante somministrazione di sostanze adiuvanti ha indotto a sostenere la natura autoimmunitaria di un vasto gruppo di malattie demielinizzanti dell'uomo, comprendente vari processi encefalitici, dai postesantematici alla SM, ma tale ipotesi unitaria non appare sostenuta da prove sicure e inoppugnabili (v. neuropatologia: Ricerche immunologiche).
L'ipotesi infettiva si basa sull'analogia di distribuzione delle lesioni fra poliomielite e SM e sulla presunzione che, in ambedue, accanto a casi clinicamente manifesti esista un numero preponderante di casi lievi, subclinici o no, progressivi; il verificarsi di piccoli focolai di casi in determinate località (Hargreaves, Campbell e Steiner) è compatibile con la possibilità che un non identificato agente infettante aggredisca giovani soggetti determinando un processo infettivo lentamente evolvente in un arco di tempo di una o due decadi. In questa ipotesi si inseriscono varie ricerche di ordine sperimentale: Innes e Kurland, in una rassegna sintetica sull'argomento, hanno concluso che di diciotto tentativi di trasmissione della malattia compiuti da vari autori ben undici debbono essere considerati negativi, e che comunque nessuna delle sperimentazioni in esame può dirsi esente da critiche e accettabile come incontrovertibilmente positiva. Già nel 1943 Schaltenbrand, mediante inoculazione nella cisterna di materiale proveniente da schizofrenici e catatonici, riprodusse nelle scimmie una malattia trasmissibile caratterizzata da lesioni cerebrali (non midollari) che tuttavia, pur presentando considerevoli analogie con quelle della SM, non erano sicuramente distinguibili da quelle della leucoencefalite spontanea precedentemente descritta da Scherr negli stessi animali.
È stato solo con il riconoscimento dell'esistenza delle infezioni da ‛virus lenti' che l'ipotesi virale ha ricevuto nuovo credito e le ricerche relative nuovo impulso. Dick tentò inutilmente di riprodurre la malattia in pecore provenienti da greggi dislocati in varie zone. In realtà, negli animali inoculati si osservò la comparsa di un processo morboso la cui alta incidenza ne rese difficile l'identificazione con lo scrapie: è noto infatti che questa malattia non può essere facilmente riprodotta, nemmeno mediante l'inoculazione di materiale biologico sicuramente proveniente da animali che ne sono affetti. Inoltre, il progressivo accorciamento del tempo di incubazione e certi sintomi precoci inconsueti difficilmente potrebbero far accettare l'ipotesi della comparsa di un'encefalite di ‛sortita' in animali portatori di scrapie allo stato latente.
A tale proposito va sottolineato che la differenza immediatamente rilevabile sul piano anatomopatologico tra SM e scrapie, consistente nel fatto che in questa malattia non si osservano le lesioni diffuse che costituiscono invece una inconfondibile caratteristica della prima, è forse più apparente che reale: infatti, come è stato già detto, le tipiche alterazioni della SM rappresentano in effetti un aspetto tardivo della malattia, e d'altro canto molti autori ritengono che l'ipertrofia astrogliale costituisca la lesione precoce e fondamentale piuttosto che un semplice processo cicatriziale, esattamente come si ritiene avvenga per lo scrapie.
L'ammissione che la proliferazione astrogliale possa essere primitiva e la conoscenza sia della possibile associazione di gliomi con la SM, sia dell'esistenza di casi di gliomi a genesi plurifocale hanno indotto a prendere in considerazione l'ipotesi che la proliferazione astrogliale sia in grado di dar luogo a una trasformazione neoplastica; d'altra parte è anche ben noto che la proliferazione astrogliale osservabile nello scrapie presenta una suggestiva somiglianza con una trasformazione neoplastica benigna. Considerando il rapporto esistente tra cellule gliali e nutrizione delle guaine mieliniche, si può supporre che lo stesso tipo di processo morboso possa determinare la comparsa delle lesioni dello scrapie o delle caratteristiche placche della SM a seconda se sia limitato nell'estensione e nella gravità o, rispettivamente, intenso, diffuso e focalizzato.
Con riserbo debbono essere interpretati i risultati delle ricerche sperimentali di Field sulla produzione di scrapie dopo passaggi ciechi nel topo di materiale proveniente da casi di SM: considerando infatti che tali esperimenti furono condotti in laboratori nei quali si lavorava con il virus dello scrapie di cui è nota la possibilità di trasmissione tra topi viventi in gabbie separate, e considerando altresì che il periodo di incubazione della malattia in studio apparve più breve di quello proprio dello scrapie spontaneo tanto da far presumere che l'infezione tra gli animali fosse già in atto al momento dell'inoculazione del materiale umano, non si può certo escludere che si siano verificati scambi e contaminazioni.
Risultati negativi sono stati ottenuti da Thormar e von Magnus in ricerche eseguite mediante l'inoculazione di materiale di SM in colture di tessuti.
D'altra parte, il fatto che non sia stata riscontrata differenza fra il potere inattivante il virus scrapie del siero di soggetti normali e quello di pazienti affetti da SM può far ritenere che tale inattivazione dipenda dalla presenza di un inibitore aspecifico.
Un'ipotesi più complessa è stata formulata da Webb e Hotchin secondo i quali una trasformazione strutturale dei neuroni può essere determinata dall'azione combinata di un virus e di una risposta immunitaria cellulare all'antigene virale. Il riscontro di un alto livello di anticorpi antimorbillosi, l'isolamento di un virus herpes-simile, il riconoscimento di anticorpi specifici in altri casi di SM sono dati oggi difficilmente valutabili, che appaiono comunque piuttosto reperti occasionali privi di significato eziopatogenetico.
8. Considerazioni conclusive
Da quanto precedentemente esposto risultano alcuni punti fondamentali che rappresentano i dati peculiari delle malattie da virus del SNC. In primo luogo l'estrema varietà delle modalità di trasmissione e di penetrazione dell'agente infettante e le scarse conoscenze che a tutt'oggi se ne possiedono: basti pensare che, come s'è visto, in questi ultimi anni è divenuto insostenibile il concetto di neurotropismo obbligato in seguito alla dimostrazione di lesioni extranervose in diverse malattie da virus del SNC e, all'opposto, di lesioni nervose causate da virus abitualmente non neurotropi quali ECHO, coxsackie, herpes simplex, ecc.
Non meno importante è risultato il concetto di ‛recettività specifica di cellula' nei confronti di vari virus nell'ambito del SNC. L'acquisizione che un virus può penetrare ed essere replicato solo in un determinato tipo di cellula nervosa (per es., motoneuroni delle corna anteriori del midollo spinale, cellule di Purkinije del cervelletto, cellule del corno di Ammone, ecc.), lasciando indenni le altre, è risultata di importanza fondamentale per la comprensione sia della sintomatologia clinica delle singole sindromi neurologiche, sia di alcuni caratteristici quadri anatomopatologici; essa inoltre è valsa a fornirci una visione più esatta dell'estrema differenziazione a livello biologico delle strutture nervose, che siamo invece tradizionalmente abituati a considerare come appartenenti a gruppi relativamente omogenei.
Al contrario, lo stato attuale delle tecniche istopatologiche non offre sicuri criteri diagnostici differenziali, così che, se si fa eccezione di alcuni quadri patognomonici quali quelli della rabbia e dell'herpes, le modalità di risposta del SNC alla replicazione dei virus neurotropi e ai fenomeni infiammatori che ne conseguono appaiono generalmente limitate e uniformi. Se i risultati delle indagini morfologiche sono pertanto in gran parte deludenti, ben più ricche di promesse sembrano le ricerche di ordine biochimico, il cui inizio è peraltro ancora recente: si può infatti ragionevolmente sperare che lo studio dei processi chimici e fisicochimici, la cui complessa dinamica è responsabile delle modificazioni tessutali conseguenti all'infezione, possa recare preziosi contributi alla delucidazione della patogenesi delle malattie da virus del SNC.
Sul piano clinico spicca una relativa uniformità di qua- dri per ciascun gruppo di affezioni (encefalite, meningite, meningoencefaliti) nei confronti del grandissimo numero di agenti patogeni potenzialmente in causa talchè, molto spesso, solo l'isolamento del virus può consentire di identificare la malattia dal punto di vista eziologico. Fanno eccezione alcune nevrassiti, quali la rabica, la russa estivoprimaverile, la poliomielitica paralitica e la meningoencefalite bifasica, che si presentano invece con quadri clinici più caratteristici.
Secondo i dati più recenti è possibile l'identificazione, mediante appropriate indagini virologiche, del 75% degli agenti eziologici di meningiti virali, ma la natura generalmente benigna delle affezioni e la mancanza di una terapia eziologica specifica riducono grandemente agli occhi del medico e del paziente l'interesse per questo tipo di in- dagine ; lo stesso dicasi per quanto riguarda l'indagine sierologica che, pur se in grado di consentire una diagnosi eziologica nell'89% dei casi, è retrospettiva e quindi di scarsa utilità clinica.
Nel caso delle encefaliti, inoltre, la diagnosi specifica eziologica è complicata notevolmente dal fatto che il virus responsabile di tali forme è raramente reperibile nel liquido cefalorachidiano o che, come per esempio nell'encefalite da herpes simplex, lo si può individuare solo in materiale bioptico prelevato dal lobo temporale: quest'ultima pratica diagnostica è evidentemente alla portata di pochi centri specificamente attrezzati e, per la sua insita traumatizzante complessità, non utilizzabile in pratica.
Assai complessa si presenta invece la collocazione dei ‛virus lenti' come possibili agenti di alcune malattie del sistema nervoso centrale dell'uomo. Se l'agente eziologico del kuru presenta un interesse notevolissimo sul piano biologico e patogenetico, ma assai minore su quello clinico, per le caratteristiche epidemiologiche del tutto particolari della malattia, il riconoscimento del possibile ruolo di agente eziologico svolto da un ‛virus lento' nella sclerosi multipla apre prospettive di imprevedibile portata per comprendere le modalità di diffusione della malattia e per chiarire l'eziopatogenesi di altre sindromi neurologiche di largo interesse clinico.
Va tuttavia rilevato che, per quanto riguarda sia i problemi di ordine biologico relativi all'identificazione e alla caratterizzazione morfologica e antigenica di questi virus, sia quelli di ordine epidemiologico e anatomopatologico, il cammino da percorrere è ancora così lungo che allo stato attuale i ‛virus lenti' rappresentano nella patologia del sistema nervoso centrale un elemento il cui ruolo risulta ancora indefinito.
Infine si apre oggi il capitolo della chemioterapia di queste infezioni. Come in tutte le malattie da virus manca una terapia specifica che possa essere paragonata a quella antibiotica delle infezioni batteriche; tuttavia, tentativi recentemente effettuati da ricercatori americani di impiegare la iododeossiuridina, un antimetabolita che interferisce con la sintesi del DNA, nella terapia dell'encefalite erpetica, sono stati seguiti da risultati di un certo interesse. La limitazione maggiore di questo tipo di terapia è comunque rappresentata dall'interferenza nella sintesi del DNA che l'antimetabolita presenta anche nei confronti delle cellule dell'uomo, con conseguenti e talora assai gravi manifestazioni tossiche generali: leucopenia, trombocitopenia, lesioni della mucosa gastroenterica, epato-tossicità (v. chemioterapia antineoplastica).
bibliografia
Albrecht, P., Pathogenesis of neurotropic arbovirus infections, in ‟Current topics in microbiology and immunology", 1968, XLIII, pp. 44-48.
Anonimo, Current understanding of persistent viral infections and their implications in human disease: summary of a workshop, in ‟Journal of infectious diseases", 1976, CXXXIII, pp. 707-712.
Astrom, K. E., Mancall, E. L., Richardson, E. P. Jr., Progressive multifocal leukoencephalopathy, in ‟Brain", 1958, LXXXI, pp. 93-99.
Brody, J. A., Epidemiology of multiple sclerosis and a possible virus etiology, in ‟Lancet", 1972, II, pp. 173-176.
Brody, J. A., Detels, R., Subacute sclerosing panencephalitis: a zoonosis following aberrant measles, in ‟Lancet", 1970, II, pp. 500-501.
Brownell, B., Campbell, M. J., Greenham, L. W., Peacock, D. B., Experimental transmission of Creutzfeldt-Jacob disease (Letter to the editor), in ‟Lancet", 1975, II, pp. 186-187.
Burnet, F. M., Measles as an index of immunological function, in ‟Lancet", 1968, II, p. 610.
Byington, D. P., Johnson, K. P., Experimental subacute sclerosing panencephalitis in the hamster: correlation of age with chronic inclusion-cell encephalitis, in ‟Journal of infectious diseases", 1972, CXXVI, pp. 18-28.
Connolly, J. H., Allen, I. V., Hurwitz, L. J., Miller, J. H. D., Measlevirus antibody and antigen in subacute sclerosing panencephalitis, in ‟Lancet", 1967, I, pp. 542-543.
Cremer, N. E., Oshiro, L. S., Weil, M. L., Lennette, E. H., Itabashi, H. H., Carney, L., Isolation of rubella virus from brain in chronic progressive panencephalitis, in ‟Journal of general virology", 1975, XXIX, pp. 143-148.
De Ritis, F., Infezioni e malattie da arborivus, in Malattie da virus (a cura di F. De Ritis), Napoli 1969, pp. 343-413.
De Ritis, F., Giusti, G., Encefaliti da virus trasmesse da artropodi, in Trattato italiano di medicina interna (a cura di P. Introzzi), parte IV, Firenze 1963, pp. 2253-2278.
Duffy, P., Wolf, J., Collins, G., De Voe, A. G., Streeten, B., Cowen, D., Possible person-to-person transmission of Creutzfeldt-Jacob disease, in ‟New England journal of medicine", 1972, CCIC, pp. 629-634.
Field, E. J., Invasion of the mouse nervous system by scarpie agent, in ‟British journal of experimental pathology", 1967, XLVIII, pp. 662-666.
Finley, K. H., Post-encephalitis manifestations of viral encephalitides, in Viral encephalitis (a cura di W. S. Field se R. L. Blattner), Springfield, Ill., 1958, pp. 69-94.
Gajdusek, D. C., Gibbs, C. J. Jr., Slow-virus infections of the nervous system and the laboratories of slow, latent and temperate virus infections, in The nervous system (a cura di D. B. Tower), 2 voll., New York 1975, pp. 793-801.
Gajdusek, D. C., Gibbs, C. J. Jr., Alpers, M., Experimental transmission of a kuru-like syndrome to chimpanzees, in ‟Nature", 1966, CCIX, pp. 794-797.
Gajdusek, D. C., Zigas, V., Degenerative disease of the central nervous system in New Guinea: the endemic occurrence of ‛kuru' in the native population, in ‟New England journal of medicine", 1957, CCLVII, pp. 974-979.
Gebhardt, L. P., Hill, D. W., Ovewintering of western equine encephalitis virus, in ‟Proceedings of the Society for Experimental Biology and Medicine", 1960, CIV, pp. 695-698.
Gibbs, C. J. Jr., Gajdusek, D. C., Amyotrophic lateral sclerosis, Parkinson's disease and the amyotrophic lateral sclerosis-parkinsonism dementia complex on Guam: a review and summary on attempts to demonstrate infection as the etiology, in ‟Journal of clincal pathology", 1974, XXV, pp. 131-141.
Gibbs, C. J. Jr., Gajdusek, D. C., Asher, D. M., Alpers, M. P., Beck, E., Daniel, P. M., Matthews, W. B., Creutzfeldt-Jacob disease (spongiform encephalopathy): transmission to the chimpanzee, in ‟Science", 1968, CLXI, pp. 388-392.
Haase, A. T., Stowring, L., Narayan, O., Griffin, D., Price, D., The slow persistent infection caused by visna virus: the role of lisogeny, in ‟Science", 1977, CXCV, pp. 175-178.
Hadlow, W. J., Scrapie and kuru, in ‟Lancet", 1959, II, pp. 289-290.
Herzberg, L., Herzberg, B. N., Gibbs, C. J. Jr., Sullivan, W., Amyx, H., Gajdusek, D. C., Creutzfeld-Jacob disease: hypothesis for high incidence in Libyan Jews in Israel, in ‟Science", 1974, CLXXXVI, pp. 848-857.
Hornabrook, R. W., Kuru, a subacute cerebellar degeneration, in ‟Brain", 1968, XCI, pp. 53-63.
Horta-Barbosa, L., Fuccillo, D., Sever, J. L., Zeman, W., Subacute sclerosing panencephalitis: isolation of measles virus from a brain biopsy, in ‟Nature", 1969, CCXXI, pp. 974-983.
Igarashi, A., Kitano, H., Fukai, K., An infectious and ribonuclease-sensitive fracton from mouse brain infected with Japanese B encephalitis virus, in ‟Biken journal", 1963, VI, pp. 21-23.
Johnson, R. T., The pathogenesis of herpes virus encephalitis. I. Virus pathways to the nervous system of suckling mice demonstrated by fluorescent antibody staining, in ‟Journal of experimental medicine", 1964, CXIX, pp. 343-347.
Johson, R. T., Herndon, R. M., Virologic studies of multiple sclerosis and other chronic and relapsing neurological diseases, in ‟Progress in medical virology", 1974, XVIII, pp. 214-216.
Klatzo, I., Gajdusek, D. C., Zigas, V., Pathology of kuru, in ‟Laboratory investigation", 1959, VIII, pp. 799-803.
Komarov, M., Kalmar, E., A hitherto undescribed disease, turkey meningoencephalitis, in ‟Veterinary record", 1960, LXXII, pp. 257-261.
Koprowski, H., Barbanti-Brodano, G., Katz, M., Interaction between papova-like virus and paramyxovirus in human brain cells: a hypothesis, in ‟Nature", 1970, CCXXV, pp. 1045-1051.
Kreth, H. W., Kackell, Y., Meulen, V., Cellular immunity in SSPE patients, in ‟Medical microbiology and immunology", 1974, CLX, pp. 191-193.
Kuznetsova, R. I., Sukhomlinova, O. I., Churilova, A. A., The character of biphasic meningoencephalitis in the Leningrad oblast, in ‟Journal of microbiology, epidemiology and immunology", 1960, XXI, pp. 262-268.
Lampert, P. W., Gajdusek, D. C., Gibbs, C. J. Jr., Subacute spongiform virus encephalopathies, in ‟American journal of pathology", 1972, LXVIII, pp. 626-632.
Luby, J. P., Sensitivities of neurotropic arbovirus to human interferon, in ‟Journal of infectious diseases", 1975, CXXXII, 4, p. 361.
McCormick, W. F., Schochet, S. S., Sarles, H. E., Caverley, J. R., Progressive multifocal leukoencephalopathy in renal transplant recipients, in ‟Archivs of internal medicine", 1976, CXXXVI, pp. 829-834.
Manuelidis, E. E., Transmission of Creutzfeld-Jacob disease (spongiform encephalopathy): transmission from man to the guinea pig, in ‟Science", 1975, CXC, p. 571.
Mettler, N. E., Identificación de cepas de encefalitis equinas en la República Argentina, in ‟Revista de la Sociedad Argentina de Biologia", 1962, XLVIII, pp. 55-62.
Meulen, V., Katz, M., Muller, D., Subacute sclerosing panencephalitis, a review, in ‟Current topics in microbiology and immunology", 1972, LVII, pp. 1-12.
Morgan, C., Howe, C., Rose, H. M., Structure and development of viruses as observed in the electron microscope. V. Western equine encephalitis virus, in ‟Journal of experimental medicine", 1961, CXIII, pp. 219-233.
Narayan, O., Penney, J. R. Jr., Johnson, R. T., Herndon, R. M., Weiner, L. P., Etiology of progressive multifocal leukoencephalopathy. Identification of papovavirus, in ‟New England journal of medicine", 1973, CCLXXXIX, pp. 1278-1282.
Narayan, O., Silverstein, A. M., Price, D. L., Johnson, R. T., Visna virus infection of North American sheep, in ‟Science", 1974, CLXXXIII, pp. 1202-1209.
Norrby, E., Link, H., Oisson, J. E., Paelius, M., Salmi, A., Vandvik, B., Comparison of antibodies against different viruses in cerebrospinal fluid and serum samples from patients with multiple sclerosis, in ‟Infection and immunity", 1974, X, pp. 688-694.
Oldstone, M. B. A., Perrin, L. H., Welsh, R. M. Jr., Potential pathogenic mechanism of injury in amyotrophic lateral sclerosis, in Recent research trends amyotrophic lateral sclerosis (a cura di J. M. Andrews, R. T. Johnson e M. Brazier), New York 1976, pp. 207-221.
Padgett, B. L., Walker, D. L., Zu Rhein, G. M., Hodach, A. E., Chou, S. M., JC papovavirus in progressive multifocal leukoencephalopathy, in ‟Journal of infectious diseases", 1976, CXXXIII, pp. 686-692.
Payne, F. E., Baublis, J. V., Measles virus and subacute sclerosing panencephalitis, in ‟Perspective virology", 1971, VII, pp. 179-186.
Rawls, W. E., Viral persistence in congenital rubella, in ‟Progress in medical virology", 1974, XVIII, pp. 273-278.
Reeves, W. C., Hammon, W. M., Epidemiology of the arthropod-borne viral encephalitides in California, 1943-1951, in Univesity of California publications in public health, vol. IV, Berkeley 1962, pp. 1-257.
Roos, M., Gajdusek, D. C., Gibbs, C. J. Jr., The clinical characteristics of transmissible Creutzfeldt-Jacob disease, in ‟Brain", 1973, XCVI, pp. 1-15.
Shinner, J. J., St. Louis virus encephalomyelitis, in ‟Archives of pathology", 1963, LXXV, pp. 309-322.
Shope, E. R., Causey, O. R., Theiler, M., The Venezuelan equine encephalomyelitis complex of group A arthropod-borne viruses, including Mucambo and Pixuna from the Amazon region of Brazil, in ‟American journal of tropical medicine and hygiene", 1964, XIII, pp. 723-727.
Sigurdsson, B., Rida, a chronic encephalitis of sheep. With general remarks on infections which develop slowly and some of their special characteristics, in ‟British veterinary journal", 1954, CX, pp. 341-347.
Suckling, A. J., Cerebral acid hydrolases as indicators of the progress of viral encephalitis (proceedings), in ‟Biochemical Society transactions", 1975, V, 4, pp. 1085-1089.
Townsend, J. J., Baringer, J. R., Wolinsky, J. S., Malamud, N., Mednik, N., Mednik, J. P., Panitsch, H. S., Scott, R. A., Oshiro, L. S., Cremer, N. E., Progressive rubelle encephalitis. Late onset after congenital rubella, in ‟New England journal of medicine", 1975, CCXCII, pp. 990-993.
Wear, D. J., Rapp, F., Latent measles virus infection on the hamster central nervous system, in ‟Journal of immunology", 1971, CVII, pp. 1593-1599.
Weil, M. L., Itabashi, H. H., Cremer, N. E., Oshiro, L. S., Lennette, E. H., Carnay, L., Chronic progressive panencephalitis due to rubella virus simulating subacute sclerosing panencephalitis, in ‟New England journal of medicine", 1975, CCXCII, pp. 994-1000.
Weiner, L. P., Narayan, O., Virologic studies of progressive multifocal leukoencephalopathy, in ‟Progress in medical virology", 1974, XVIII, pp. 229-234.
Wiemers, M. A., Immunoglobulin M in Murray Valley encephalitis, in ‟Pathology", 1975, VII, 3, pp. 187-192.
Zhdanov, V. M., Parfanovich, M. I., Integration of measles virus nucleic acid into the cell genome, in ‟Archiv für die Gesamte Virusforschung", 1974, XLV, pp. 225-231.
Ricerche immunologiche sulla demielinizzazione sperimentale
di Charles E. Lumsden
sommario: 1. Introduzione: a) la mielina; b) immunità e sistema nervoso centrale; c) i diversi tipi di demielinizzazione sperimentale. 2. L'encefalomielite allergica sperimentale: a) introduzione; b) notizie storiche. 3. L'EAS nella cavia: a) metodo sperimentale di provocazione; b) quadro clinico e sue varianti; c) le alterazioni istologiche nell'EAS. 4. Patogenesi dell'EAS. 5. Il fattore encefalitogeno: a) la proteina basica: isolamento, caratterizzazione e determinanti strutturali; b) localizzazione e funzione dell'antigene encefalitogeno. □ Bibliografia.
1. Introduzione
a) La mielina
Con il termine ‛mielina' si indica in biologia quella sostanza protoplasmatica membranosa modificata, composta da una mescolanza di lipidi ‛caratteristici' e di circa il 30% di proteine, che forma le guaine relativamente spesse, pluristratificate, delle fibre dei nervi sensitivi e motori, centrali e periferici, di tutte le specie animali, a cominciare dagli Artropodi in su. La speciale composizione biochimica della mielina conferisce alle guaine mieliniche una peculiare combinazione di proprietà elettriche - alta impedenza e bassa capacitanza - e, insieme ai rivestimenti più esterni costituiti dal neurilemma segmentato e dalla nevroglia, permette la rapida conduzione saltatoria caratteristica delle fibre nervose sensitive e motorie, e diversa da quella lenta degli assoni sottili, amielinici. Nella mielina i componenti lipidici sono presenti in combinazioni e rapporti caratteristici, identici tanto nella mielina dei nervi centrali quanto in quella dei nervi periferici; tali lipidi sono però individualmente rappresentati anche in altre membrane cellulari. Al contrario, la componente proteica della mielina, o per lo meno un'importante frazione di essa, è ‛caratteristica' di questa sostanza al punto da rivelarsi immunospecifica non solo nei riguardi della mielina in generale, ma perfino in maniera differenziale specifica per la mielina centrale e periferica. La struttura molecolare di tale proteina basica, caratteristica della mielina del sistema nervoso centrale (SNC) (che costituisce l'antigene dell'EAS - encefalite allergica sperimentale - del quale si tratta largamente in questo articolo), è in gran parte nota; per il momento non si può dire altrettanto per la corrispondente proteina basica che costituisce l'antigene della mielina periferica. Tuttavia esistono buone ragioni per supporre che le differenze molecolari tra la proteina basica della mielina centrale e quella della mielina periferica siano dovute a trasposizioni di amminoacidi relativamente secondarie, in seno ai peptidi che compongono le sequenze immunologicamente determinanti delle due proteine.
Lo stimolante titolo assegnato al presente articolo deve essere inteso dal lettore nel senso che, innanzi tutto, l'argomento della trattazione sarà rappresentato fondamentalmente dall'immunologia del SNC; in maniera particolare verranno discussi quella parte dell'immunologia che riguarda il comportamento normale e patologico del sistema immunitario in esso localizzato (neuroimmunologia) e gli argomenti concernenti la ‛demielinizzazione sperimentale'.
b) Immunità e sistema nervoso centrale
Il SNC possiede caratteristiche speciali che differenziano notevolmente il modo di reagire del suo parenchima da quello degli altri organi per quanto riguarda la risposta immunitaria verso antigeni estranei.
In primo luogo il SNC non è fornito di un apparato linfatico nel senso comunemente inteso, mancando in esso le stazioni linfonodali e la relativa rete linfatica, che tanta importanza hanno nel filtrare e concentrare gli antigeni estranei e nel favorire l'interazione tra questi e le cellule linfocitarie.
In secondo luogo, il SNC è separato dal resto dell'organismo da una barriera particolarmente efficiente, chiamata barriera emato-encefalica (BEE), che rappresenta una struttura caratteristica del cervello e del midollo spinale, i quali vengono così isolati dal circolo ematico generale mediante un sistema complesso di impermeabilità o di permeabilità selettiva verso le più varie molecole semplici o macromolecole. Tale barriera rappresenta fondamentalmente una funzione dell'endotelio dei vasi cerebrali, della sottostante membrana basale e dell'astroglia circostante (v. Lumsden, 1958). Da ciò deriva che vi è un'ampia possibilità teorica che molecole specifiche del SNC rimangano isolate in siti ‛immunologicamente sequestrati' durante la fase precoce dello sviluppo fetale, prima che il sistema linfatico acquisisca la sua piena efficienza.
Inoltre vi sono ulteriori dati che concorrono a far considerare il SNC come un sito ‛immunologicamente privi- legiato' non solo nel limitato periodo dello sviluppo fetale ma anche nella successiva vita autonoma di adulto. Un primo dato a favore di ciò è l'incapacità del cervello di rigettare trapianti omologhi di pelle (v. Medavar, 1948) e trapianti di cellule tumorali omologhe o eterologhe (v. Greene, 1951, 1953 e 1957). Tale tolleranza viene invece abolita se precedentemente si sensibilizza, al livello sistemico, l'animale da esperimento verso gli antigeni in questione. Ciò è ben comprensibile se si considera che, in condizioni di normalità, il sistema immunitario generale dell'organismo partecipa attivamente alla difesa del parenchima del SNC. Infatti è stato dimostrato da Frick e Scheid-Seydel (v., 1958) e da Lippincott e altri (v., 1965) che, in individui con una normale barriera emato-encefalica, le IgG liquorali provengono quasi per intero dal sangue circolante ed è pertanto verosimile che anche a livello del cervello e del midollo spinale avvenga la stessa cosa, data la grande facilità con la quale avvengono gli scambi di macromolecole tra SNC e liquor cerebrospinale (v. Rall e altri, 1962; v. Brightman, 1965).
Benché i dati di Medavar e di Greene non abbiano avuto una conferma definitiva, tuttavia sembra lecito supporre che: 1) molto difficilmente un antigene estraneo che venga impiantato primitivamente nel SNC, proprio a causa della mancanza in esso di vie linfatiche e della presenza dell'ostacolo opposto dalla BEE, sensibilizzi il sistema immunitario generale; 2) è fisiologicamente usuale che questi stessi antigeni vengano aggrediti nell'ambito del SNC quando il sistema immunocompetente generale sia stato sensibilizzato contro di essi in un altro sito dell'organismo. Tutto quanto detto nel punto 1), naturalmente, presuppone l'improbabile eventualità che l'antigene estraneo sia riuscito a localizzarsi nel SNC senza ledere in modo sostanziale la BEE e senza aver dovuto prima far tappa in altre zone dell'organismo, come invece avviene nelle comuni infezioni o infestazioni del SNC.
Sembrerebbe, da quanto è stato detto precedentemente, che più degli altri parenchimi il SNC sia isolato, anatomicamente, dal suo apparato linfatico, e si potrebbe essere indotti nella erronea convinzione che il SNC sia, per quanto riguarda la difesa immunitaria, un sito del tutto passivo, essendo debitore verso l'intero apparato linfatico dell'organismo. Ciò viene subito smentito da alcuni fatti che dimostrano come nelle infezioni croniche si formino e si mantengano nel SNC dei doni di cellule produttrici di anticorpi del tutto autonome (v. Tavolato, 1975). Chiara evidenza di questa affermazione è fornita dai seguenti dati sperimentali: 1) nei processi flogistici cronici del SNC il rapporto tra la concentrazione di IgG liquorali e quella di IgG sieriche è aumentato rispetto al corrispondente rapporto riguardante l'albumina, indice quest'ultimo di integrità della BEE (v. Delpech e Lichtblau, 1972; v. Tibbling e altri, 1977); 2) il rapporto tra catene κ e catene λ è aumentato nel liquor pur restando normale nel siero (v. Link e Zettervall, 1970); 3) la presenza della tipica separazione ‛oligoclonale' delle IgG nell'elettroforesi di molti liquor patologici. Ciò vuol dire che la zona γ, a differenza di quanto avviene nel siero, non si presenta come una banda omogenea, bensì frazionata in bande minori, corrispondenti alla attività anticorpopoietica di selezionati doni plasmacellulari, localizzati nell'ambito del SNC; 4) l'aumento locale delle IgM liquorali nel corso di talune infestazioni croniche (tripanosomiasi), senza che vi sia una evidente lesione della BEE (v. Michaux, 1966); 5) gli studi già citati di Frick e Scheid-Seydel e di Lippincott e altri che dimostrano, mediante iniezioni in vena di IgG marcate con 131I che venivano successivamente ricercate a livello del liquor cerebro-spinale, come nelle infiammazioni croniche del SNC le IgG liquorali abbiano nella maggior parte una origine locale; 6) infine, gli studi di Tourtellotte e altri (v., 1971) e di Tavolato (v., 1975) che hanno dimostrato in modo inequivocabile, mediante l'uso di anticorpi fluorescenti anti-IgG umane, la presenza, nel SNC di malati di sclerosi multipla, di cellule linfoidi ripiene di IgG. Tutto questo dimostra chiaramente la sintesi ‛locale' di immunoglobuline e precisa come tale sintesi sia compiuta, analogamente agli altri organi, dalle normali cellule linfo-plasmocitarie. È verosimile che tali cellule provengano dal circolo generale e che abitualmente un certo numero di esse attraversi la parete dei piccoli vasi venosi a mo' di sentinelle.
Tuttavia è nelle situazioni infiammatorie che le cellule linfo-plasmocitarie sviluppano tutta la loro potenzialità attivandosi nel produrre localmente le immunoglobuline richieste per la difesa dell'organo. Tutto questo avviene nell'intera zona parenchimale interessata dalla noxa patogena, ma tali fenomeni sono particolarmente evidenti al microscopio ottico nei manicotti linfocitari perivenosi o nei valli linfocitari che contornano le placche di demielinizzazione.
La difesa immunitaria del SNC non è tuttavia prerogativa dei soli anticorpi circolanti, quelli cioè che sono prodotti e secreti dai B-linfociti (linfociti borsa-dipendenti), ma è strettamente collegata anche all'attività sinergica dei T-linfociti (linfociti timo-dipendenti). Tali linfociti non secernono nel sangue o nei liquidi biologici gli anticorpi che essi stessi producono, ma li conservano adesi alla propria membrana cellulare che, in tal modo, diviene uno strumento attivo di risposta immunitaria. La risposta dei T-linfociti è essenziale nel rigetto dei trapianti e nella difesa contro le infezioni virali. È noto infatti che l'infezione morbillosa, che non produce particolari problemi in pazienti con grave immunodeficienza di tipo B-linfocitaria, è spesso fatale nei pazienti con deficit dei T-linfociti.
Lo studio delle differenti funzioni dei B- e T-linfociti e dei loro sinergismi è di fondamentale importanza per comprendere i fenomeni immunitari patogenetici dell'EAS (v. sotto, cap. 4), e quelli che avvengono in altre malattie neurologiche con coinvolgimento immunopatologico. Nella panencefalite sderosante subacuta, ad esempio, la risposta di blastizzazione linfocitaria verso il virus morbilloso è assente o diminuita (v. Valdimarsson e altri, 1974). Nella sclerosi multipla vi è, nel liquor cerebrospinale, una riduzione dei T-linfociti attivi rispetto ai corrispettivi linfociti ematici e rispetto ai valori riscontrati nel liquor e nel siero di individui normali (v. Kam-Hansen, 1978).
Anche per i T-linfociti è verosimile supporre quanto già detto per i B: in condizioni di normalità, un certo numero di essi attraversa la parete dei vasi venosi del parenchima del SNC e si attiva solamente in presenza di uno stimolo antigenico sufficientemente energico e prolungato, quando, contro tale antigene, abbia già ricevuto la competenza a livello del sistema immunitario generale.
Tuttavia questi aspetti caratteristici del tessuto nervo- so non sono limitati al SNC, come risulta chiaro dalle ricerche sulla neurite allergica sperimentale - il corrispettivo nervoso periferico della malattia trattata in questo saggio. Il sistema nervoso periferico (SNP) condivide, con qualche differenza, queste peculiarità. L'intera portata di questi dati da un punto di vista strutturale e funzionale, ossia la natura della barriera emato-nervosa, se esiste, e il rapporto fra nervi periferici e apparato linfatico, è stata oggetto fino a ora di scarsa considerazione critica.
c) I diversi tipi di demielinizzazione sperimentale
Tenendo presenti questi tre importanti dati di fatto possiamo ora tornare all'argomento della nostra trattazione. È preferibile parlare di ‛demielinizzazione' invece che di ‛demielinazione' quando si voglia sottolineare l'evoluzione continua del processo di distruzione attiva della mielina; il termine di ‛demielinazione', meno preciso, potrebbe essere confinato più opportunamente a una condizione stabilizzata nella quale la perdita della mielina rappresenti la conseguenza di un processo già esauritosi precedentemente.
Ma nella definizione di ‛demielinizzazione', oltre al concetto di continuità, deve soprattutto essere compreso quello di un certo grado di distruzione ‛selettiva' della mielina. L'involucro di mielina è un indicatore sensibile dello stato di salute della fibra nervosa sottostante; per merito soprattutto del suo alto contenuto in lipidi è possibile dimostrarne facilmente qualsiasi alterazione mediante il semplice esame microscopico (perfino su sezioni non colorate) e per mezzo di svariati coloranti dei grassi capaci di rivelare gli esteri di colesterolo e i trigliceridi liberati dai lipidi della mielina. Per questa ragione la ‛demielinizzazione' rappresenta il segno più ovvio della degenerazione secondaria dell'assone ossia della degenerazione di tipo walleriano - un processo essenzialmente identico nella fibra nervosa centrale e in quella periferica. Si è fatta e si fa ancora un'assurda confusione fra la demielinizzazione vera, selettiva, che si verifica nel corso di una malattia primaria demielinizzante, e la pseudodemielinizzazione che accompagna la patologia gangho-assonale. Le ragioni di questa confusione, e la complessità dei processi pseudodemielinizzanti nel corso di malattie naturali, sono state l'oggetto di un rapporto molto ampio dell'autore (v. Lumsden, Pathogenetic mechanisms..., 1970) e quindi non possono essere nemmeno riassunte in un saggio di questa brevità. Tanto difficile era distinguere in pratica fra demielinazione ‛primaria' e ‛secondaria' che, ancora nel 1944, E. W. Hurst, un esperto di notevole levatura, doveva concludere abbastanza vagamente che ‟la demielinazione sembra rappresentare la risposta della sostanza bianca a tutti gli stimoli che non risultino immediatamente letali per i tessuti". Sulla base delle stesse considerazioni c'erano ancora, a quel tempo, molti neuropatologi di fama che mettevano in dubbio l'entità della sclerosi multipla. A questo proposito è bene perciò considerare due fatti storici d'importanza vitale: 1) tutto questo accadeva prima che venisse chiaramente dimostrato, per mezzo della coltura di tessuti, che la guaina mielinica è prodotta non dall'assone ma interamente dalla cellula satellite (oligodendrocita centralmente, cellula di Schwann in periferia); 2) benché il concetto di ‛encefalomielite allergica sperimentale' (EAS) fosse probabilmente già nell'aria (v. sotto), sarebbero dovuti trascorrere ancora anni prima che si cominciasse anche solo a considerarla alla stregua di un autentico modello di un processo ‛primariamente' demielinizzante paragonabile alla sclerosi multipla. La selettività, tanto dell'EAS quanto della sclerosi multipla, per l'involucro mielinico, è un fatto tutt'altro che unanimamente riconosciuto. Prenderemo in seguito in considerazione questo problema in relazione con l'EAS, come è stato fatto recentemente altrove nei riguardi della sclerosi multipla (v. Lumsden, The neuropathology..., 1970).
2. L'encefalomielite allergica sperimentale
a) Introduzione
Si è molto discusso a riguardo delle similitudini e delle differenze intercorrenti tra EAS e sclerosi multipla. In passato è parso a taluni di poter intravvedere nella prima entità un modello sperimentale di studio della demielinizzazione che è caratteristica della seconda. Tuttavia è opinione oramai concorde che non vi è nulla che autorizzi a sovrapporre le due patologie. Prova fondamentale ne è la mancanza di anticorpi anti-proteina basica della mielina nei sieri di pazienti affetti da sclerosi multipla, al contrario di quanto avviene nei sieri di animali affetti da EAS che sono ricchi in tali anticorpi (v. Biggins e altri, 1978; v. Ulrich e Lardi, 1978).
Tuttavia l'EAS dimostra di possedere alcune caratteristiche eccezionali fra i modelli sperimentali di malattie autoimmunitarie (e occorre dire che qualunque affermazione circa l'esistenza di un unico meccanismo in tutte le malattie autoimmuni è ancora del tutto ingiustificata). Innanzi tutto l'antigene responsabile è conosciuto, e dimostra, per quel che se ne sa, le seguenti caratteristiche, uniche fra gli autoantigeni.
1. Possiede un peso molecolare insolitamente basso - 16.200 dalton - nella condizione che corrisponde probabilmente a quella naturale nella quale si trovano i tessuti, benché, in vitro, frazioni di esso con un peso pari a un decimo o anche meno di quello citato mantengano un'attività encefalitogena completa. Invece la tireoglobulina, uno dei pochi altri autoantigeni caratterizzati chimicamente, ha un peso molecolare di 669.000 dalton.
2. Nonostante le piccole dimensioni della molecola attiva, il fattore chimico dimostra di essere strettamente caratteristico dell'oligodendroglia-mielina, particolare importante ai fini della patogenesi, perché in base a esso sembra possibile escludere in maniera assoluta la possibilità di risposte autoimmunitarie crociate fra l'oligodendrogliamielina e altre cellule e componenti tessutali. Ne risulta che il processo patologico che si verifica nell'EAS, paragonato ai processi autoimmuni dei tessuti connettivi e delle strutture emovascolari, dimostra un carattere eccezionalmente selettivo (se indotto dall'antigene purificato), il tessuto bersaglio primario essendo rappresentato unicamente dall'oligodendroglia e dalla mielina. Tale caratteristica selettività si ritrova anche nella sclerosi multipla e rappresenta infatti, come già detto, l'argomento più forte a favore di quanti sostengono la fondamentale identità tra sclerosi multipla ed EAS.
3. È assai notevole l'esiguità della dose di antigene sufficiente a produrre la malattia in forma grave: in un animale di 500 gr bastano 0, 1 microgrammi di proteina basica pura, o appena 180 micromoli di peptide peptico encefalitogeno (v. sotto). Partendo da queste considerazioni è stato calcolato dall'autore (v. Lumsden, The clinical pathology..., 19722, p. 594) che nell'uomo un solo milligrammo di sostanza bianca del cervello o del midollo contiene abbastanza molecole encefalitogene della variante umana da causare l'EAS.
4. Il basso peso molecolare e la struttura semplice e li- neare dell'antigene, con pochi legami intramolecolari, gli impartiscono una resistenza al calore e agli agenti chimici abbastanza eccezionale per questa categoria di sostanze. Le implicazioni relative a quanto sopra esposto, riferite al rischio di autosensibilizzazione nell'uomo, vengono prese in considerazione solo ora.
b) Notizie storiche
È opportuno ricordare che l'argomento di queste ricerche emerse da un problema squisitamente pratico - un problema che in effetti attrasse l'attenzione dello stesso autore nel corso della sua esperienza in Oriente durante la seconda guerra mondiale - vale a dire la natura degli ‛incidenti' neuroparalitici in corso di trattamento antirabbico. Le teorie più vecchie, secondo le quali tali incidenti erano dovuti a infezione da ‛virus fisso' del vaccino, divennero scarsamente sostenibili quando fu ripetutamente provato che diverse specie di neurovaccini sterili, preparati da animali non infetti, risultavano egualmente capaci di provocare paralisi. Fra i molti studi dedicati alla natura di tali accidenti neuroparalitici vi erano quelli, oggi citati come classici, di Rivers e altri (v., 1933), Rivers e Schwentker (v., 1935) e Ferraro e Jervis (v., 1940), che descrissero l'insorgenza di encefalomielite sperimentale in circa il 70% delle scimmie iniettate ripetutamente con tessuto nervoso, e indicarono la demielinizzazione perivenulare come un aspetto caratteristico delle lesioni. Una conclusione importante, che sarebbe stato possibile trarre da queste osservazioni, ma che fu trascurata, è che negli esperimenti iniziali la ripetizione delle iniezioni di cervello da solo (vale a dire senza adiuvante) accrebbe di circa il 70% il rischio naturale degli accidenti paralitici (che nell'uomo era dell'ordine di un caso su 2.500 con i primi vaccini di tipo Semple). Questa costituisce certamente l'obiezione più significativa che si possa opporre all'uso di vaccini di cervello sterile nel trattamento della sclerosi multipla dell'uomo, che ebbe una certa voga pochi anni fa (v. Lumsden, The clinical pathology..., 19722). Col tempo, il riconoscimento della validità del fenomeno dell'EAS si riflettè sui metodi di vaccinazione antirabbica, e i vaccini contenenti tessuto cerebrale furono quasi del tutto (ma non completamente) rimpiazzati da quelli coltivati su embrione di anatra e inattivati con β-propiolattone. Tuttavia è interessante notare che nemmeno questo procedimento è riuscito a eliminare completamente il rischio di accidenti neuroparalitici nell'uomo (v. Cohen, 1969); conviene sottolineare questo punto, perché esso potrebbe indicare una possibile direzione di ricerca sul ruolo dei virus come potenziali scatenatori di processi di autosensibilizzazione a carico del sistema nervoso umano.
Inoltre si trascura spesso il fatto che, quasi contemporaneamente ai classici esperimenti di Rivers sul cervello e sul midollo, Burky (v., 1934) descrisse lesioni simili in un organo di senso, l'occhio. La sua ‛endoftalmite sperimentale' veniva provocata in maniera analoga usando tessuto omologo di lente - ma con l'interessante differenza che per la prima volta insieme all'antigene veniva iniettato un adiuvante, in questo caso tossina stafilococcica. È difficile stabilire quanto reale fosse l'effetto di tale adiuvante senza ripetere gli esperimenti, e l'autore non è al corrente di alcun lavoro di questo genere; ma nel campo dell'EAS l'introduzione dell'adiuvante di Freund ebbe effetti drammatici, facendo aumentare la produzione dell'EAS a tal punto che praticamente nei primi esperimenti tutti gli animali stimolati con tessuto cerebrale o midollare emulsionato in adiuvante completo di Freund svilupparono rapidamente lo stesso tipo di lesioni descritte da Rivers e dai suoi colleghi. L'adozione di questa modifica alla tecnica originale di Rivers culminò in una pioggia di rapporti sulla possibilità di riprodurre regolarmente con successo la malattia in una vasta serie di specie di Mammiferi. I lavori originali più importanti sono elencati, in ordine di apparizione, nella tab. I. Storicamente, questo fiorire di rapporti sulla produzione rapida dell'EAS rese possibile l'enorme cumulo di ricerche realizzate in seguito, dirette verso l'identificazione chimica dell'agente responsabile e verso la natura del processo patogenetico - i due argomenti che qui verranno trattati in dettaglio.
Ma bisogna anche osservare che in un certo senso tale facilità di ricerca ebbe l'effetto di stornare l'attenzione dal possibile ruolo, in campo umano, di adiuvanti più ‛naturali' e di più ‛naturali' interventi di agenti privi di adiuvanti che potrebbero essere coinvolti nell'insorgenza di malattie quali la sclerosi multipla e le polineuriti.
La tab. I è stata preparata in parte per ragioni storiche, in parte per dare un'impressione, anche a prima vista, dell'EAS in differenti specie animali. Non sarebbe possibile, se non in una lunga monografia, rendere giustizia alle osservazioni originali contenute nei lavori citati, specialmente per ciò che riguarda le interessanti differenze di dettaglio messe in luce da un esame dell'EAS dal punto di vista della patologia comparata. Torneremo in seguito su alcune delle differenze di specie emerse nella composizione chimica del ‛fattore allergico' o proteina basica. Ma un'analisi di questo genere serve a rammentare come, fra tutte le caratteristiche biologiche delle specie animali, la più proteiforme sia proprio la risposta immunologica (un classico esempio è rappresentato dalla reazione delle diverse specie al bacillo tubercolare). I dettagli rilevati dai lavori citati nella tab. I non permettono di affermare che la malattia sia strettamente identica in tutte le specie. Le differenze esistono: per esempio, secondo l'esperienza dell'autore, la malattia comporta una mortalità alta nelle cavie, trascurabile nelle capre; esistono grandi differenze fra ratti di ceppi diversi (quelli di razza Wistar sono quasi totalmente resistenti, i Lewis sono altamente suscettibili), mentre le cavie di qualunque ceppo sembrano suscettibili in misura praticamente eguale; la dieta determina una differenza significativa nel topo ma non in altre specie; reazioni crociate tra le varianti del sistema nervoso centrale e periferico (EAS e NAS) si verificano in alcune specie ma non in altre; secondo alcuni risultati il ruolo dei linfociti e quello degli anticorpi antimielina circolanti sarebbero diversi nella cavia e nel coniglio e finalmente, mentre nei Mammiferi non è mai stato messo in evidenza alcun segno di reazioni crociate fra sistema nervoso e altri organi, i risultati delle ricerche condotte da kanzenhofer e altri (v., 1958) nel pollo indicano l'esistenza di una doppia patologia nel SNC e nel testicolo. Nonostante tali differenze (forse dovute a differenze di specie nei determinanti antigenici del fattore encefalitogeno, o forse legate a differenze più generali della risposta immune) esiste in tutte le specie una generale somiglianza che fa dell'EAS una definita entità patologica. Forse la conclusione finale che si può trarre dalla patologia comparata dell'EAS è che essa si può sviluppare in numerose specie (o forse nella totalità) dei Mammiferi.
3. L'EAS nella cavia
a) Metodo sperimentale di provocazione
La provocazione dell'EAS ‛senza adiuvanti' non è stata finora adeguatamente sperimentata. Il metodo adottato quasi uniformemente in tutti i laboratori consiste nell'uso di tessuto cerebrale o spinale, omologo o eterologo - oppure dell'antigene estratto chimicamente da essi - emulsionato con adiuvante completo di Freund. Quest'ultimo è composto da 85 parti di Arlacel A (Honeywill Atlas Ltd.) e da 15 parti di Bayol F (Esso Petroleum Co.) e contiene 5mg/ml di Mycobacterium tuberculosis ceppo H. 37 ucciso al calore. Lo stimolo consiste in una sola iniezione di adiuvante di Freund emulsionato nella siringa con la quantità stabilita del materiale liofilizzato (omogenato semplice, o estratto) previamente disciolto o sospeso in soluzione fisiologica o acqua bidistillata.
Sebbene siano state usate con successo le vie sottocutanea e intraperitoneale, e perfino la sottopleurica, esse risultano senza dubbio meno efficaci della via intradermica. Così, per esempio, nelle cavie di ceppo Hartley, virtualmente tutte suscettibili alla malattia, mentre si ottiene regolarmente una risposta del 90-100% con uno stimolo altamente efficace per via intradermica, con la stessa dose per via sottocutanea si riduce l'efficacia della risposta al 50-60%. La via orale, sperimentata esaurientemente dall'autore parecchi anni fa (osservazioni non pubblicate), si è dimostrata del tutto inefficace.
In parecchie specie è stato dimostrato con una varietà di antigeni che questi vengono trasportati ai linfonodi regionali assai meglio se iniettati per via intradermica che non per altre vie, e la speciale efficacia dello stimolo intradermico nell'EAS è senza dubbio dovuta semplicemente a questo. Per la stessa ragione appare logico scegliere un punto vicino a un linfonodo. I punti preferiti sono due: 1) la parte alta della pelle del petto, circa 1-2 cm sotto e appena medialmente al linfonodo ascellare anteriore (noi usiamo abitualmente il lato destro); 2) alcuni autori eseguono le iniezioni nel cuscinetto plantare; ma a parte l'obiezione estetica, questo procedimento fa zoppicare l'animale e può mascherare una reale debolezza neurologica.
Insieme alle variazioni legate al punto di stimolazione, bisogna tener presente che, anche lavorando con un antigene solubile il più puro possibile e pesato con estrema accuratezza, si realizza sempre un test biologico e non un test chimico. L'iniezione stimolante non è strettamente dosabile perché, per quanto accuratamente si determini la dose iniettata nel derma (in genere 0,1 ml, talvolta 0,2 ml della totale massa emulsionata), la dose effettiva è presumibilmente rappresentata dal numero di macrofagi infarciti di materiale stimolante che giunge al linfonodo, e questo varia indubbiamente da animale ad animale. Perciò anche quando si voglia effettuare la determinazione di routine del potere encefalitogeno di estratti chimici o residui, l'autore considera indispensabile usare un minimo di 5 animali per gruppo - non 3 o 4, come a volte si legge. Infine anche la tecnica di iniezione dell'operatore rappresenta un fattore di fondamentale importanza in un tentativo di stimolo ‛quantitativo' di questo genere.
b) Quadro clinico e sue varianti
Dopo l'iniezione stimolante gli animali rimangono apparentemente in buone condizioni fino all'insorgere dell'EAS; sebbene si possa verificare tra il 7° e il 14° giorno una sensibile perdita di peso, non ci sono altri segni riferibili a un disordine di carattere generale; la loro eventuale presenza, anzi, deve far pensare a una malattia concomitante indipendente dall'EAS. In tutti gli animali stimolati, in corrispondenza del punto d'iniezione si sviluppa lentamente un granuloma piccolo e duro, che fra il 12° e il 21° giorno tende a ulcerare, spesso all'improvviso, senza suppurazione: a questo stadio l'esame microscopico mette in evidenza necrosi, emorragie e infiltrazione leucocitaria, mentre in precedenza il punto iniettato era occupato da una vasta massa di olio e di macrofagi carichi di lipidi - cioè da un piccolo oleogranuloma. La lesione acquista così i caratteri di una reazione di Arthus.
I segni clinici neurologici della malattia compaiono nella cavia talora improvvisamente, talora in maniera insidiosa, con una evidente debolezza del treno posteriore (che si può mettere bene in evidenza inseguendo l'animale nella gabbia con la mano e fingendo di afferrarlo, oppure distendendolo sul dorso e osservando i suoi tentativi di raddrizzarsi). Tale debolezza può rimanere di grado lieve e va ricercata regolarmente ogni giorno, potendosi a volte verificare la guarigione completa. Ma più spesso la paresi è progressiva e conduce alla paralisi completa del treno posteriore, con incontinenza urinaria e ristagno fecale, segni d'interessamento grave delle vie lunghe del midollo. Gradi inferiori di compromissione neurologica sfuggono facilmente all'osservazione, come risulta evidente dal successivo esame istologico del SNC. Con un'osservazione sistematica è facile notare la debolezza e instabilità del dorso, l'interessamento localizzato del treno anteriore, il torcicollo e le deviazioni posturali di tipo cerebellare; a volte lo sguardo amaurotico e il non ritrarsi da un oggetto tenuto vicino agli occhi tradiscono l'interessamento delle vie ottiche. Purtroppo è impossibile praticare un adeguato esame neurologico e, in particolare, mettere in evidenza l'interessamento delle vie sensitive.
A causa della difficoltà d'interpretare con accuratezza le condizioni neurologiche nella cavia noi preferiamo registrare i reperti clinici separatamente da quelli istologici. È evidente che così facendo si possono verificare discrepanze grossolane. Così, dopo stimolazioni deboli (con estratti chimici o residui privati parzialmente del fattore encefalitogeno - v. sotto) era facile scoprire che a volte non venivano registrati segni clinici in interi gruppi di animali (5 o 6), mentre 4 o 5 di essi potevano mostrare indubbi segni istologici di EAS di grado leggero (v. sotto). Probabilmente questa fu la ragione che indusse Alvord e Kies (1959) a proporre di adottare un ‟indice per il saggio quantitativo dell'attività encefalitogena dell'antigene". Tuttavia l'autore, insieme ad altri ricercatori, pensa che l'idea non sia applicabile. Appare ovvio, a ben riflettere, che una simile mescolanza di rilievi clinici e istologici risulti scarsamente valida dal momento che, in alcuni casi, per esempio nelle lesioni midollari, i segni istologici e quelli neurologici si riferiscono alla stessa lesione; mentre l'assenza di un reperto clinico non esclude la presenza di gravissime alterazioni istologiche nel punto d'elezione - le regioni periventricolari del cervello - che sono clinicamente silenti nella cavia. Inoltre è difficile comprendere la logica d'includere la perdita di peso e la lipemia nell'elenco dei criteri di valutazione, dal momento che la loro importanza nei riguardi del disordine neurologico non è affatto chiara. La perdita di peso rappresenta senza dubbio l'effetto della tossiemia da vaccino, che comporta perdita d'appetito e diminuzione dell'assunzione di cibo, ed è probabilmente provocata dagli altri costituenti proteici della sostanza stimolante (soprattutto tubercoloproteina) piuttosto che dall'antigene encefalitogeno. La lipemia rappresenta probabilmente l'effetto di uno stress che comporta la mobilizzazione di acidi grassi liberi (in seguito a stimolo neuroendocrino o del sistema nervoso autonomo), ma non è manifestazione dell'EAS più di quanto le alterazioni dei lipidi del siero in corso di sclerosi multipla non siano un effetto diretto della malattia (v. Lumsden, The clinical pathology..., 19722). La valutazione quantitativa dell'EAS secondo parametri clinici che riflettono solo debolmente e senza accuratezza le lesioni istologiche non è realizzabile, né teoricamente valida.
D'altra parte, così come avviene in patologia umana nel caso della sclerosi multipla, il tentativo di mettere in relazione la gravità della paralisi con la gravità del processo patologico che ne è alla base, comporta scarso vantaggio pratico e minima validità teorica quando le lesioni siano multiple e localizzate a caso senza una degenerazione sistemica delle vie. Nei casi in cui la malattia si presenta sempre in forma leggera - come nella capra o nella neurite allergica sperimentale (NAS) della cavia - i segni neurologici possono non essere mai rilevabili, mentre si possono ritrovare piccoli infiltrati linfocitari nelle pareti delle venule diffusi nell'intero sistema nervoso, centrale o periferico a seconda del caso. Inoltre, usando mezzi più sensibili per esempio la ricerca di anticorpi anti-mielina o di linfociti sensibilizzati contro la mielina in tessuti coltivati - gli animali apparentemente resistenti facenti parte di un gruppo di cavie stimolate per l'EAS possono reagire esattamente come gli altri. Nel caso di una specie come la cavia che reagisce così uniformemente a dosi di stimolo efficace ampiamente variabili, si finisce per concludere che quando anche solo uno o due animali sui cinque o sei trattati in maniera identica mostrano una chiara risposta istologica, anche gli altri dello stesso gruppo sono affetti dalla stessa malattia immunologica. Considerazioni di questo peso servono opportunamente a rammentare che la valutazione quantitativa dello stimolo encefalitogeno è un problema più difficile di quanto non appaia da alcuni lavori pubblicati. A questo proposito, inoltre, è opportuno ricordare che occasionalmente le cavie rispondono a uno stimolo efficace con chiari segni clinici di sofferenza piramidale, eppure istologicamente non mostrano alcuna traccia dei caratteristici infiltrati linfocitari in sezioni seriate del SNC, benché presentino in genere una notevole congestione venosa nella zona di drenaggio del sistema venoso terminale. Questo tipo di ‛risposta encefalopatica', in contrasto con la più comune risposta encefalitica, è assai più frequente nel coniglio. Poiché almeno alcune di queste cavie encefalopatiche danno risultati fortemente positivi ai test per la ricerca degli anticorpi anti-mielina nel siero, è possibile che in questi casi l'effetto clinico sia dovuto interamente all'anticorpo, senza intervento locale di linfociti sensibilizzati - un punto a cui abbiamo già fatto riferimento e che assume importanza nella genesi delle varianti encefalitiche ed encefalopatiche dell'encefalomielite acuta disseminata postesantematica dell'uomo (v. Lumsden, The clinical pathology..., 19722).
Molto è stato scritto intorno alle variazioni del tempo d'insorgenza della malattia, sottintendendo una notevole variabilità e usando i termini ‛induzione rapida' e ‛risposta accelerata' in modo ingannevole. L'autore ritiene, in base a un'esperienza che gli deriva dall'aver stimolato più di 5.000 cavie con antigeni vari, che vanno da omogenati crudi di SNC a peptidi encefalitogeni, e con dosi diverse, che tale variabilità sia stata esagerata. La curva del rapporto risposta-dose presenta un andamento chiaramente sigmoide. Almeno per quanto riguarda la cavia, l'inibizione dell'attività dell'antigene per un presunto effetto ‛protettivo' esercitato da dosi alte, descritta originariamente da Alvord e Kies (1959),non è confermata dai nostri risultati personali. Questi provengono dai test di pseudotitolazione regolarmente impiegati nel nostro laboratorio per controllare i procedimenti di estrazione del fattore encefalitogeno. A questo scopo stimoliamo gruppi di animali con dosi di 0,1, 1 e 10 microgrammi quando si tratta di saggiare una sostanza attivamente encefalitogena, oppure con dosi di 100 microgrammi o 1 mg quando invece si voglia escludere ogni attività encefalitogena residua. Quantunque Alvord (v., 1970) abbia criticato questo metodo di ‛titolazione' di routine ritenendolo unicamente ‟pseudo quantitativo", è difficile pensare a un test migliore per questo scopo particolare; oltre tutto nei riguardi del problema in discussione esso presenta il vantaggio di permettere l'uso di dosi da 10 a 100 volte maggiori di quella pienamente efficace. Ma nemmeno le dosi maggiori mostrano di possedere effetti protettivi costanti. Un certo grado di variabilità nel tempo d'insorgenza è invece chiaramente evidente quando s'impiegano dosi pienamente efficaci di uno stimolo altamente encefalitogeno. L'avverarsi di lesioni gravi nel 100% degli animali del gruppo stimolato significa che tali lesioni diventano clinicamente evidenti assai presto - ossia che fra l'inizio della malattia sperimentale e l'evidenziarsi dei segni clinici che rendono necessaria la soppressione dell'animale intercorre solo un breve periodo di latenza. In più occasioni abbiamo adottato il sistema di sopprimere ogni giorno, a partire da quello della stimolazione, un gruppo rappresentativo di 2 o 3 animali. In tal modo, quando s'impieghino dosi inferiori a quelle pienamente efficaci, si può dimostrare che le lesioni istologiche compaiono dopo un periodo di tempo abbastanza costante. In altre parole, variando la dose della sostanza stimolante si modifica piuttosto la gravità della malattia clinica che non il tempo necessario allo sviluppo degli infiltrati linfocitari nel SNC; è discutibile se sia possibile allo sperimentatore operare una qualunque vera ‛accelerazione' del processo encefalitico. Un problema diverso è ovviamente rappresentato da un ritardo nella risposta allo stimolo: agli inizi delle ricerche sull'EAS, quando gli stimoli venivano somministrati per via sottocutanea, l'insorgenza della paralisi avveniva con un ritardo di almeno una settimana rispetto a quella che segue la stimolazione intradermica adottata successivamente.
Analogamente è stato esagerato l'effetto degli agenti en- hancing (intensificanti), se con tale termine s'intende qualcosa di diverso dall'effetto primario dello stesso adiuvante consistente nell'impegnare fin dall'inizio nella risposta immunitaria un numero di cellule linfoidi maggiore di quello che verrebbe altrimenti reclutato. L'effetto enhancing di una sostanza come l'eparina (v. Levine e Wenk, 1966) consiste soprattutto nel modificare la permeabilità della barriera emato-encefalica, o nell'accelerarne la distruzione, oppure nell'aumentare il numero dei punti in cui questa avviene. Si può pensare che, di conseguenza, tanto gli anticorpi circolanti quanto i linfociti sensibilizzati possano raggiungere più rapidamente la mielina del parenchima - benché il vantaggio non superi 1 o 2 giorni e possa avverarsi solo durante il periodo della normale risposta immune del processo encefalitico. Le sostanze enhaneing, al pari del danno termico (v. Levine, 1970), permettono la fuoriuscita di fibrinogeno e di emazie, che attraggono a loro volta i leucociti polimorfonucleati, trasformando il primitivo infiltrato dell'EAS (composto da linfociti e monociti) in una forma acuta o emorragica. Tali variazioni d'insorgenza acquistano interesse specialmente nei riguardi della genesi di malattie naturali quali la leucoencefalite acuta emorragica, l'encefalomielite acuta necrotizzante, la mielite di Landry, la malattia di Devic, ecc., e possono risultare importanti anche in relazione al ruolo scatenante o potenziante dei ‛fattori precipitanti' nelle ricadute della sclerosi multipla (McAlpine, 1972). Ma in sostanza esse, invece di contraddirla, riconfermano la tesi secondo la quale i caratteri temporali dell'EAS obbediscono alla legge biologica fondamentale che regola la risposta immune primaria. È stato già accennato alla confusione semantica che può insorgere fra l'effetto di enhancement e il vero e proprio effetto ‛adiuvante'. In questo senso risulta particolarmente illustrativo il ruolo del vaccino antipertosse nella produzione dell'EAS iperacuta. È questa la forma di EAS per ora descritta solo nel ratto - che si verifica quando al posto dell'adiuvante di Freund viene usato un vaccino antipertosse concentrato (circa 10 miliardi di microrganismi). Gli autori di queste ricerche (v. Levine e Wenk, 1964, 1965 e Central nervous..., 1966; v. Levine e altri, 1966) affermano che nella risultante EAS: 1) il periodo di latenza è diminuito; 2) è aumentata la gravità della reazione; 3) è alterata la qualità della reazione del SNC, perché predominano edema, emorragia, necrosi, essudazione di fibrina e reazione leucocitaria polimorfonucleare. Tali differenze rispetto alla classica forma di EAS rappresentano in realtà gli effetti apparenti della sensibilizzazione verso il fattore pertosse, sovrapposti alla risposta EAS e attirati localmente dalla sua reazione specifica. Questo è dimostrato da un esperimento di trasporto passivo eseguito più tardi dagli stessi autori (v. Levine e Wenk, 1967). In esso un gruppo di animali normali viene iniettato con cellule linfatiche provenienti da animali con EAS classica (adiuvante di Freund) mentre un altro gruppo simile riceve linfociti di animali con EAS iperacuta (adiuvante pertosse): tutti gli animali sviluppano solo la forma ordinaria della malattia, mai l'iperacuta. D'altra parte, se i due gruppi di animali ricettori vengono trattati con vaccino antipertosse e poi trasfusi con cellule linfatiche - un gruppo con cellule provenienti da animali con EAS ‛normale', un altro con cellule di animali con EAS iperacuta - ambedue i gruppi sviluppano solamente la forma iperacuta della malattia. Questi risultati dimostrano pertanto il verificarsi di due processi allergici diversi, sovrapposti uno all'altro, il primo rappresentato dalla sensibilizzazione verso l'antigene nervoso favorita dall'adiuvante, l'altro dalla sensibilizzazione verso l'antigene della pertosse; quest'ultima tuttavia si manifesta attraverso un'accentuazione delle lesioni venulari cerebrali conseguenti all'aumentata permeabilità dei vasi colpiti dall'EAS. Dai protocolli degli autori appare inoltre chiaro che la sensibilizzazione verso la pertosse modifica le manifestazioni cliniche e istologiche dell'EAS, ma non il suo corso temporale.
In conclusione, secondo l'autore, nell'EAS sono possibili solo due modificazioni importanti della risposta temporale. Una consiste nella piccola differenza già descritta legata al luogo dell'iniezione; la seconda nel trapasso alla cronicità nei rari casi di guarigione parziale - un argomento che verrà ripreso in seguito.
c) Le alterazioni istologiche nell'EAS
Nel corso di questo paragrafo analizzeremo la natura delle alterazioni dei tessuti nell'EAS.
Patologia generale. - Nel corso delle prime ricerche (v. Lumsden, Experimental ‛allergic'... I, 1949) fu possibile notare l'insorgenza, durante la prima settimana dopo lo stimolo, di una leggera febbre remittente e di una modica leucocitosi contemporanee allo sviluppo della linfoadenite localizzata. L'aumento di peso della milza palesava, inoltre, una reazione linfoide generalizzata contemporanea a quella, predominante, a livello del linfonodo regionale (ibid.).
Il progredire della lesione nel luogo dell'iniezione cutanea, dall'oleogranuloma alla risposta di tipo Arthus, è stato già menzionato. Vedremo in seguito che i linfociti sensibilizzati verso la mielina compaiono in circolazione 7 giorni dopo lo stimolo, mentre 10-12 gioni dopo la stimolazione possono essere messi a volte in evidenza gli anticorpi antimielina circolanti.
È interessante rilevare che il timo non mostra di partecipare alla reazione. A volte può essere messa in evidenza nei polmoni una lieve disseminazione embolica del materiale iniettato, anche quando si sia usato il solo adiuvante di Freund. A questa evenienza è stato attribuito il nome di ‛malattia da adiuvante', a torto secondo noi, dal momento che si tratta di una complicazione concomitante e non di una malattia ‛per sé'. All'infuori di queste lesioni lontane, e della possibilità (rara in uno stabulario ben condotto) d'infezioni intercorrenti, alle quali alcune specie sono notoriamente suscettibili e che lo sperimentatore ben conosce, non si verificano nel corso dell'EAS complicazioni viscerali diffuse. È stata già menzionata la possibilità nel pollo di una reazione crociata con il testicolo.
Neuropatologia. - Nonostante l'occasionale ‛interessamento sistemico' sopra descritto, nell'EAS, come nella sclerosi multipla, le alterazioni fondamentali istologicamente caratteristiche sono limitate al SNC.
La lesione tipica della malattia classica è rappresentata da una infiltrazione o da un ammasso di linfociti, cellule plasmocitoidi e monociti del sangue a livello della parete delle piccole vene del parenchima, della tela coroidea, del plesso coroideo e - in misura più modesta - delle leptomeningi di tutto il sistema nervoso centrale. I particolari reperti della forma ‛iperacuta' sono stati descritti in precedenza.
È interessante analizzare gli infiltrati di linfociti e di monociti delle pareti venose. Anche con la semplice colorazione ematossilina-eosina fu possibile evidenziare precocemente (v. Lumsden, Experimental ‛allergic'... I, 1949) la predominanza di grandi cellule linfatiche indifferenziate (o ‛trasformate'). Il microscopio elettronico permette di accertare che le cellule ematiche giacciono fra quelle endoteliali delle pareti delle venule, e si raccolgono nello spazio perivenulare di Virchow-Robin occludendolo. Esse si insinuano inoltre fra i prolungamenti degli astrociti attaccati alle pareti delle venule, ma raramente si estendono molto al di là. L'esame ultrastrutturale dimostra una mescolanza di grandi linfociti di tipo ‛trasformato' e di macrofagi. Le plasmacellule pienamente mature sono in realtà assai rare, ma in un buon preparato alla pironina si può notare che molti dei ‛linfociti' mostrano una chiara pironinofilia citoplasmatica e, al microscopio elettronico, presentano la caratteristica disposizione dei poliribosomi raccolti nel reticolo endoplasmatico.
Benché molti dei macrofagi si colorino con l'argento come la microglia (v. Field, 1961), le ricerche di Kosunen e altri (v., 1963) con la timidina triziata hanno dimostrato chiaramente che essi sono per più del 70% di origine extracerebrale (ossia ematica), e che proliferano attivamente nelle lesioni. In realtà, negli ammassi cellulari delle pareti venose non è difficile notare, anche in sezioni colorate solo con ematossilina-eosina, delle mitosi a carico di queste cellule.
La densità di tali infiltrati di ‛cellule rotonde' miste nelle pareti venulari è altamente variabile. Nelle lesioni più lievi tutto l'infiltrato può limitarsi a un singolo strato di cellule - la cui interpretazione richiede una notevole esperienza di preparati istologici di cervello di roditore - ma in altri casi io stesso ho contato da 20 a 30 strati di cellule, che formano in pratica un granuloma parietale. Mentre in base agli studi già riferiti non è più lecito dubitare che tali cellule siano di provenienza ematica, ciò non significa che esse non proliferino localmente (come accade per lo meno ai fagociti) e continuino a maturare e a secernere localmente immunoglobuline (linfociti).
Fino a ora non abbiamo ancora menzionato la mielina a proposito di tali lesioni istologiche. Già prima dell'uso del microscopio elettronico era stata definitivamente dimostrata l'alterazione della mielina situata immediatamente intorno alle venule (v. Kabat e altri, 1949; v. Lumsden, 1949). Nelle ricerche iniziali sull'EAS si attribuì grande importanza alla differenza in estensione o in gravità della demielinizzazione nelle diverse specie animali, arrivandosi ad affermare da parte di alcuni che nella cavia l'EAS non costituiva affatto un processo demielinizzante. Per un breve periodo fu inoltre attribuita grande importanza al fatto che gli infiltrati cellulari non fossero confinati alla regione callosa e alle vie bianche lunghe, ma si riscontrassero, a volte con la stessa frequenza, anche nella corteccia cerebrale, nei nuclei grigi della base e perfino nelle meningi. Nei piccoli roditori, per la scarsa quantità di sostanza bianca centrale ammassata nel cervello, il grado di distruzione della mielina può essere in realtà assai elevato, pari alla demielinizzazione totale della sostanza bianca centrale. Tuttavia analogie e differenze basate sul raffronto dell'anatomia macroscopica del cervello umano e di quello dei piccoli roditori rivestono un'importanza secondaria. La constatazione importante era e rimane che le lesioni si verificano là dove è presente mielina, tanto attorno a una vena profonda quanto immediatamente sotto una superficiale; che, in generale, esse sono maggiori nella sostanza bianca centrale sia del cervello sia del cervelletto; che lo stesso avviene nel midollo spinale. I piccoli roditori presentano una corteccia cerebrale assai spessa e con strati profondi ricchi di mielina; lo stesso si riscontra anche a livello dei nuclei grigi della base. La distribuzione degli infiltrati perivenulari in queste zone di sostanza ‛grigia', insieme all'aspetto delle lesioni della sostanza bianca periventricolare (con i suoi oligodendrociti), conferma l'ipotesi che proprio la mielina di tutte le zone colpite costituisce il bersaglio dell'assalto immumologico. In questo consiste una ‛malattia demielinizzante', non nel fatto di essere anatomicamente limitata alla regione del centro ovale. Mentre l'alterazione mielinica è confermata dalla microscopia elettronica, è chiaro che anche nelle forme ‛ordinarie' (ossia non solo nella forma iperacuta) il danno a carico dei cilindrassi vicini può essere considerevole anzi senza di esso probabilmente le lesioni risulterebbero asintomatiche. È difficile distinguere il danno primario dagli effetti secondari; l'ammissione conclusiva del carattere ‛demielinizzante' dell'EAS deve piuttosto derivare dai dati dell'istopatologia (al microscopio elettronico), dalle prove immunologiche sull'intervento di fattori specifici antimielina, dall'azione diretta di tali fattori sulla mielina nei modelli sperimentali semplificati rappresentati dalle colture di tessuto nervoso centrale. Per tornare al nostro riferimento a Toynbee, le conclusioni diventano ‛razionali' solo quando derivano da esperimenti che impiegano una serie di tecniche sufficientemente vasta. È per questo che in seguito prenderemo in considerazione anche i dati immunologici.
La migrazione focale delle cellule linfoidi ematiche attraverso le pareti delle venule, che rappresenta la lesione istologica della forma ordinaria di EAS, si accompagna anche a un aumento localizzato della permeabilità vascolare. L'affermazione è dimostrata dal fatto che il tripanblau, iniettato endovena a una cavia appena prima della soppressione, colora elettivamente quelle zone della sostanza bianca nelle quali in seguito gli infiltrati saranno dimostrabili istologicamente (osservazioni personali; v. Barlow, 1956); in maniera analoga si comportano le proteine marcate con iodio radioattivo (v. Vulpé e altri, 1960; v. Cutler e Barlow, 1967). Secondo Oldstone e Dixon (v., 1968) ciò può accadere fin dal primo giorno di malattia clinica, anche prima della comparsa delle cellule infiltranti; gli stessi autori hanno recentemente dimostrato con tecniche di immunoistochimica che in queste stesse zone le gammaglobuline diffondono all'interno dei tessuti parenchimali, localizzandosi nei tratti mielinici. Oldstone e Dixon attribuiscono anzi speciale importanza al fatto che la fuoriuscita di gammaglobuline preceda il formarsi degli infiltrati, scorgendo in ciò l'indicazione di un ruolo patogenetico esplicato da fattori umorali identificabili come anticorpi antimielina - una conclusione con la quale l'autore si trova d'accordo (v. sotto).
Infine, sempre in campo di citologia delle lesioni dell'EAS, è necessario spendere qualche parola sugli astrociti. L'attenzione quasi esclusiva che nel corso degli ultimi venti anni è stata rivolta alla determinazione quantitativa degli eventi riscontrabili in corso di EAS - in termini di un processo ‛acuto' - e l'uso che di tali misurazioni si è fatto per identificare la natura chimica del fattore encefalitogeno hanno portato a trascurare notevolmente le lesioni ‛croniche'. Eppure Ferraro e Cazzullo (v., 1948) dimostrarono chiaramente nella scimmia l'esistenza di tali lesioni. Nelle forme clinicamente ‛croniche' delle loro scimmie gli infiltrati cellulari erano presenti ancora 105 giorni dopo lo stimolo e si accompagnavano a una considerevole proliferazione dell'astroglia. Nelle cavie che guariscono dopo aver presentato i sintomi della malattia, e in quelle che non ne hanno presentato alcuno, pur dopo uno stimolo encefalitogeno risultato efficace in altri animali dello stesso gruppo, gli infiltrati cellulari persistono sino a 60 giorni o più, e in seguito scompaiono completamente lasciando una cicatrice di astroglia facilmente visibile in sezioni colorate con ematossilina-eosina (osservazione personale). Tuttavia non esistono ancora prove definitive che nelle scimmie come nelle cavie si verifichi alcun processo ciclico ricorrente o ‛autopropagante' di danno mielinico, in qualche modo paragonahile a quello tipico della sclerosi multipla (v. Lumsden, The neuropathology..., 1970).
Esistono ancora aspetti curiosi e non spiegati per quanto riguarda i luoghi di predilezione delle lesioni dell'EAS. Nella cavia, una zona di predilezione è chiaramente identificabile nell'intera tela coroidea o zona subependimale al di sopra del tetto e dei lati del sistema ventricolare, seguita immediatamente dal midollo spinale. Inoltre si possono trovare anche lesioni delle vie ottiche. Talvolta nell'EAS le cellule linfatiche infiltrano a tal punto la zona subependimale da formare una cospicua massa pseudogranulomatosa o pseudotumorale. Inversamente in queste regioni possono reperirsi infiltrati piccoli, di tipo focale, chiaramente limitati a minuscoli manicotti cellulari intorno alle vene subependimali più grandi, a parete sottile (e tuttavia non necessariamente limitati alle estremità mediali delle vene, talché la localizzazione non sembra aver nulla a che vedere con la circolazione del liquido ventricolare).
Come si è già detto, infiltrati focali perivenulari di linfociti sono frequenti nel midollo spinale (e secondo i nostri reperti sembra verosimile che la maggior parte delle paralisi cliniche grossolane sia dovuta a essi). Essi possono essere estesi o limitati, e lesioni midollari distanziate e così distribuite possono rappresentare le sole lesioni istologiche reperibili in un animale, sì che è possibile non notarle a meno di non sottoporre sempre il midollo a un esame microscopico sistematico. (Noi prendiamo di norma i 2/3 o i 3/4 superiori del midollo, lo tagliamo in sezioni longitudinali di circa 2 cm di lunghezza e riuniamo parecchie di queste sezioni in un blocco; in questo modo, sezionando ciascun blocco a livelli diversi, si riduce il rischio di farsi sfuggire le lesioni sparse).
Le alterazioni del cervello, del peduncolo e del midollo, se distanziate, sono spesso circoscritte alle zone sottopiali dove le vene superficiali s'immettono nel parenchima (da cui senza dubbio l'ipotesi sostenuta da alcuni che la lesione sia una meningite o una coriomeningite piuttosto che un'encefalomielite); tuttavia bisogna riconoscere che raramente si osserva un'intensa infiltrazione leptomeningea al di sopra del vertice. Al di là di ciò sarebbe assurdo cercar di distinguere nel cervello dei roditori fra ‛leucoencefalite' e ‛polioencefalite' da EAS. Nonostante le affermazioni di Waksman (v., 1959), sono del tutto insolite nell'EAS lesioni a carico delle radici spinali e dei gangli delle radici - formazioni comunemente incluse nelle sezioni di midollo. Una lesione descritta, ma raramente ricercata, e l'uveite (v. Bullington e Waksman, 1958), che insieme all'iridociclite comporta lesioni tipiche a carico della papilla ottica e dello strato di fibre nervose; lesioni simili si verificano a livello del chiasma ottico. Secondo Bullington e Waksman la stimolazione eseguita con tessuto di nervo ottico in adiuvante di Freund al posto di tessuto cerebrale spinale provocherebbe un'incidenza assai maggiore di lesioni encefalitiche intraoculari. Se questo é vero, se ne potrebbe dedurre l'esistenza di una certa specificità regionale dell'antigene mielinico - una conclusione che acquisterebbe senso alla luce di quello che diremo in seguito circa la sottostruttura dell'antigene encefalitogeno.
4. Patogenesi dell'EAS
Finora abbiamo considerato, esclusivamente in termini descrittivi, l'EAS alla stregua del modello per eccellenza di una demielinizzazione sperimentale prodotta con metodi immunologici. Rivolgeremo adesso la nostra attenzione alla patogenesi, tenendo a mente che per gli scopi di quest'opera è necessario limitarsi ai principi fondamentali e ai fatti che aprono nuove strade alla ricerca futura nel vasto campo della biologia del sistema nervoso.
Anche se molti dettagli sulla genesi dell'EAS sono ancora tutt'altro che chiariti, vedremo che questo, per essere l'EAS a priori una malattia artificiale o sperimentale, ci offre un enorme vantaggio. In linea generale infatti conosciamo con sicurezza due punti essenziali: 1) ci è nota la causa della malattia - un componente chimico del tessuto del SNC; 2) sappiamo che la sua patogenesi coinvolge il sistema immunitario, e cioè che l'EAS rappresenta la risposta immunologica a un costituente antigenico del tessuto nervoso, identificato come un costituente naturale, un autoantigene. Occorre ora arricchire quest'impalcatura essenziale della nostra conoscenza.
Daremo per il momento per conosciuta la natura chimica del fattore encefalitogeno, di cui tratteremo più avanti. Sulla base di ciò che si è detto, e in parte di ciò che si dirà, possiamo così riassumere i ‛principi della patogenesi dell'EAS'.
1. Il ‛vaccino' necessario per provocare sperimentalmente la malattia deve sempre contenere l'antigene EAS sotto una delle seguenti forme: a) come tessuto crudo di SNC contenente mielina; b) come mielina separata all'ultracentrifuga; c) come proteina basica mielinica purificata; d) come uno dei determinanti attivi in senso EAS della proteina basica; e) forse come uno dei due ultimi componenti citati legato ad altri costituenti chimici del SNC.
2. Tutti gli agenti suddescritti debbono venire in contatto col tessuto linfatico al di fuori del cervello e del midollo. Questo principio possiede la forza di una ‛legge'.
Ma nell'ambito dei limiti definiti da questa legge emergono numerose le domande sussidiarie. Per esempio, quale tipo di cellula linfoide - B-linfocita o T-linfocita - è interessata? Qual è il ruolo dei monociti (macrofagi)? Qual è la funzione precisa dell'adiuvante di Freund e che cosa lo sostituisce quando l'EAS si verifica senza il suo intervento? Qual è il reale aggressore - l'anticorpo o il linfocita sensibilizzato, o tutti e due in maniera sinergica? Quali anticorpi e/o cellule sensibilizzate vengono prodotti al di fuori di quelli specifici antimielina? Quale significativa ‛immunità' ne può risultare? Conosciamo risposte parziali solo per alcune di queste domande.
Alla luce dell'immunogenesi dell'EAS possiamo iniziare con l'esporre alcuni fatti di cui sinora non si è discusso.
Per quanto riguarda la sostanza iniettata, sappiamo che nel punto d'iniezione essa viene elaborata poco o niente; infatti l'antigene somministrato da solo rimane intatto anche per tre settimane prima che l'aggiunta di adiuvante, iniettato nello stesso punto, provochi l'insorgere dell'EAS 10-15 giorni più tardi, secondo il procedimento normale (v. Lipton e Freund, 1953; v. Alvord e altri, 1964). Se ne deduce che l'elaborazione immunologica dell'antigene avviene altrove e non nella zona d'iniezione. Che nell'EAS l'intera risposta immune avvenga nel sistema linfatico è dimostrato sperimentalmente dalla necessità di asportare entro tre giorni la zona iniettata, se si vuole ‛prevenire' la malattia. Che la maggior parte della risposta sia limitata al linfonodo locale regionale è dimostrato dalla possibilità di prevenire la malattia con l'asportazione di esso entro circa 10 giorni dallo stimolo (v. Good, 1957). Peraltro il risultato dipende strettamente dalla localizzazione del punto di iniezione. Così, negli esperimenti di Levine e Wenk (v., 1962) dopo inoculazione nel cuscinetto plantare del ratto non fu possibile prevenire l'EAS rimuovendo solamente i linfonodi poplitei e inguinali (l'escissione dovette essere estesa ai linfonodi intra-addominali); ma quando, sempre nel ratto, lo stimolo veniva effettuato a livello del fianco, ne risultava evidentemente una disseminazione più circoscritta e l'EAS poteva essere prevenuta dall'asportazione dei linfonodi ascellari e inguinali effettuata in qualunque momento dal giorno dello stimolo fino a 7 giorni dopo o più.
I dati appena esposti, insieme a quelli tratti dagli esperimenti sul trasporto passivo e dai test di ‛tossicità' dei linfociti (v. sotto), ci permettono di misurare il tempo impiegato dalla maturazione dei linfociti - o forse dallo stabilirsi di un clone - necessario per organizzare la risposta immunoaggressiva. Poiché il trasporto passivo risulta massima- mente efficace al settimo giorno dallo stimolo (è inefficace prima e declina rapidamente dopo), e poiché cellule linfoidi prevelate dal linfonodo al 7°-8° giorno risultano tossiche in colture di mielina (v. sotto) mentre l'asportazione del linfonodo risulta efficace nel prevenire la malattia fino a 10 giorni dopo lo stimolo, possiamo affermare con considerevole sicurezza che il periodo di massima circolazione dei linfociti mielinotossici o mielinopatici nel sangue si verifica fra il 70 e il 100 giorno. Quest'informazione potrebbe assumere un grande valore pratico per le ricerche future sulla terapia dell'EAS (v. Lumsden, The clinical pathology..., 19722).
Nel frattempo, senza voler decidere se l'anticorpo umorale antimielina (v. anche in seguito) intervenga o meno in alcuni animali al posto dei linfociti, o magari in maniera a questi sinergica, possiamo affermare che i linfociti circolanti rappresentano i mediatori efficaci delle lesioni dell'EAS nel SNC sulla base dei seguenti gruppi di osservazioni.
1. Si è riusciti fino a ora a ottenere il ‛trasferimento passivo' in tre specie di animali: ratti Lewis (v. Paterson, 1960 e 1968; v. Levine e Wenk, 1967); cavie inbred (v. Stone, 1961; v. Falk e Kies, 1968); conigli (v. Aström e Waksman, 1962). Torneremo in seguito sulla scelta del ceppo nel caso dei ratti. Considerazioni di istocompatibilità impongono naturalmente, per tutte le specie, la scelta di animali inbred. Incidentalmente, mentre gli autori suddetti impiegano tutti cellule linfonodali, Levine e Sowinski (v., 1968) ottennero dei successi anche con sangue intero.
Esperimenti come quelli sopra citati hanno fornito importanti informazioni sull'insorgenza della trasferibilità cellulare nell'EAS, sulla qualità delle linee cellulari interessate e sul loro destino nel recettore (la loro capacità di centrare il bersaglio della mielina del SNC è un fenomeno biologico estremamente drammatico, che riesce a impressionare il più incallito degli istopatologi). Abbiamo già visto come l'impiego di queste tecniche ha reso possibile distinguere, nel caso dell'EAS iperacuta, la risposta neurospecifica dell'EAS dall'ipersensibilizzazione da B. pertussis. Inoltre esse hanno fornito forse la migliore prova biologica in nostro possesso della specificità della reazione linfatica dell'EAS nei riguardi della proteina basica della mielina, dal momento che tale proteina purificata, aggiunta alla sospensione di cellule da trasfondere, esplica un'azione inibitrice specifica (v. Levine e altri, 1970). D'altra parte, poiché la ricerca in questo campo è stata orientata fondamentalmente verso lo studio dei fenomeni acuti, niente ci è noto ancora circa la durata dell'ipersensibilizzazione antimielinica dei linfociti. Infine tali esperimenti ovviamente non spiegano ‛come' i linfociti sensibilizzati danneggino la mielina. Per studiare quest'ultimo problema è necessario far ricorso alle tecniche di coltura della mielina.
2. Il secondo gruppo di osservazioni concerne gli effetti degli agenti immunosoppressivi e citotossici; si tratta in questo caso di informazioni di natura empirica, dal momento che la farmacologia di questi agenti, nei riguardi dei fenomeni immunitari, è ancora incerta. La tab. II elenca alcuni dei modi impiegati per sopprimere e prevenire l'EAS nell'animale da esperimento, in tali termini puramente empirici.
Strettamente parlando, le osservazioni empiriche elencate nella tab. II non permettono di chiarire se la mediazione nell'EAS sia cellulare o umorale - perché anche la produzione di anticorpi dipende dalla stessa specie di cellule linfoidi - ma i risultati terapeutici positivi, annotati nella seconda colonna, certamente sostengono la tesi generica di un meccanismo immunogenetico dell'EAS.
È difficile decidere se, per analogia, tali risultati siano in grado di provvedere una falsariga per una possibile terapia della sclerosi multipla e delle altre malattie neuro-immunologiche dell'uomo citate all'inizio di questo capitolo. La maggior parte delle sostanze ‛immunosoppressive' elencate nella tavola sono dei medicamenti semplicemente citotossici e perciò non in grado di distinguere fra i doni linfocitari specificamente sensibilizzati e gli elementi linfocitari ed emopoietici normali. Per questo, come avviene nelle altre malattie nelle quali vengono usate, gli effetti collaterali tossici possono facilmente superare i benefici temporanei. E, al momento, d'obbligo mettere in guardia contro l'uso di farmaci immunosoppressori nella sclerosi multipla. In questa malattia, infatti, il complesso meccanismo delle relazioni tra B- e T-linfociti non è ancora completamente chiarito, e vi è pertanto il rischio di introdurre con tali terapie un ulteriore fattore di aggravamento in un equilibrio di per sé molto precario.
Noi abbiamo dimostrato per primi (v. Lumsden, Experimental ‛allergic'... I, 1949) che un animale guarito dall'EAS diventa resistente a stimoli successivi e la resistenza si prolunga per almeno un anno dallo stimolo primario, che rappresenta un lungo periodo di tempo nella vita di una cavia. Occorre però notare che anche il granuloma iniziale persiste per parecchi mesi. Sappiamo oggi (v. Lumsden, 1964; v. Alvord, 1970) che esistono diversi tipi di resistenza, e che questa può essere indotta sia con l'adiuvante sia con pretrattamenti encefalitogeni. Anche la somministrazione della proteina basica, effettuata 7-10 giorni dopo lo stimolo primario, si dimostra capace di desensibilizzare le cellule sensibilizzate che distruggono l'antigene, inducendo in tal modo lo stabilirsi di una tolleranza, analogamente a quanto avviene col pretrattamento con proteina basica (v. Alvord, 1970). In campo immunologico si cerca di spiegare questa specie di proiezione in vari modi - parlando di ‛tolleranza', ‛deviazione dell'antigene', ‛rivalità di antigeni' - termini che in definitiva servono solo a mascherare l'ignoranza di ciò che accade nella realtà. Poiché gli stessi adiuvanti - e nel caso della ‛protezione' contro l'EAS il termine include anche la tubercolina e il vaccino BCG - sono in grado di proteggere da un successivo stimolo completo (contenente cioè la proteina basica), l'effetto non è necessariamente specifico in senso immunochimico. Per questo non c'è ragione di pensare che l'effetto dei pretrattamenti con tessuto di SNC si esplichi necessariamente attraverso un meccanismo diverso da quello degli adiuvanti: in altre parole ambedue i tipi di pretrattamento potrebbero semplicemente limitarsi a stimolare i linfonodi, riducendo così il numero delle cellule non impegnate in grado di reagire allo stimolo completo specifico per la mielina attraverso una risposta primaria (con formazione di doni specifici). La resistenza transitoria indotta dalla somministrazione endovenosa della proteina basica durante la fase d'induzione attiva dell'EAS è un semplice effetto di ‛eccesso di antigene' e non offre grandi speranze di protezione a lungo termine contro sensibilizzazioni ricorrenti, quali quelle che potremmo supporre si verifichino nella sclerosi multipla.
Esistono tuttavia degli indizi secondo i quali gli anticorpi circolanti sarebbero in grado di conferire un'immunità per lo meno temporanea nei riguardi dell'EAS (v. Paterson e Harwin, 1963). Più recentemente è stato affermato che è possibile ottenere immunità mediante vaccinazione preventiva con proteina basica non encefalitogena (v. Roboz Finstein e altri, 1968): è probabile che tale effetto sia dovuto alla presenza di un anticorpo circolante diretto verso l'antigene non encefalitogeno ma in grado di reagire in maniera crociata con l'antigene encefalitogeno. Parleremo in seguito delle ricerche in corso sui differenti determinanti antigenici della proteina basica, e della possibilità che reazioni immuni interessanti i determinanti non encefalitogeni siano in grado di proteggere contro quelli encefalitogeni della molecola della proteina basica. Sarebbe interessante, se vi fosse conferma di una patogenesi autoaggressiva nella sclerosi multipla, esplorare le possibilità di dissociare nell'EAS l'immunità protettiva dalla risposta immune da autosensibilizzazione. A tale scopo sarà necessario studiare gli effetti protettivi (ammesso che esistano) dei sieri degli animali trattati in tal modo, come si è fatto negli esperimenti di Paterson e Harwin.
3. Tornando all'argomento fondamentale di questa relazione, vale a dire alla natura immunologica dell'EAS, esamineremo adesso i fatti a favore del carattere aggressivo della risposta immune primaria nei riguardi della mielina. Per questo scopo è giocoforza ricorrere a esperimenti condotti su mielina vivente in coltura di tessuti. I metodi e i principi relativi sono stati descritti altrove (v. Lumsden, 1968).
I primi risultati concernenti gli anticorpi umorali antimielina nell'EAS (v. Bornstein e Appel, 1961) erano basati sullo studio del siero di 12 conigli trattati con uno stimolo encefalitogeno standard. La riproducibilità dei risultati risultò pressoché perfetta: infatti 178 colture saggiate con sieri provenienti da conigli affetti da ‛EAS acuta' ed ‛EAS latente' mostrarono tutte segni di demielinizzazione, mentre non se ne riscontrò alcuna traccia in 94 colture saggiate con sieri di conigli normali. L'effetto positivo si palesava per mezzo di rigonfiamenti fusiformi delle guaine mieliniche, a volte sporgenti in modo grottesco verso le aree circostanti, arrivando perfino al distacco completo. La frammentazione non costituiva la sola manifestazione del danno mielinico: secondo la descrizione degli autori la mielina in qualche zona ‟sembrava semplicemente disciogliersi". La risposta positiva iniziava col rigonfiamento delle cellule dell'oligodendroglia, che in poche ore diventavano enormi, e i cui nuclei, lisati o raggrinziti, si addossavano alla membrana cellulare mentre il citoplasma si riempiva di particelle ripiene di liquido, dotate di movimenti browniani. Abbiamo illustrato in dettaglio altrove (v. Aparicio e altri, 1968), per mezzo di microfotografie elettroniche, la sequenza temporale dell'effetto demielinizzante del siero, e abbiamo dimostrato come l'attacco primario sembri effettuarsi a livello delle membrane plasmatiche degli oligodendrociti e delle zone corrispondenti alla ‛linea intraperiodica' della mielina lamellare, sotto forma di una rapida imbibizione osmotica di siero fino a formare delle vescicole intramieliniche. Ricerche biochimiche successive effettuate nel nostro laboratorio (v. Dickinson è altri, 1970) hanno messo in luce che la proteina basica encefalitogena è localizzata elettivamente nelle linee intraperiodiche della mielina lamellare. Queste due osservazioni prese insieme forniscono perciò una prova circostanziale di notevole valore a favore della teoria secondo la quale l'anticorpo umorale antimielina nell'EAS - è diretto specificamente contro la proteina basica encefalitogena localizzata nelle linee mieliniche intraperiodiche, che rappresentano il sito della lesione primaria. La successiva frammentazione dell'intera guaina mielinica, la mielinofagia da parte dei macrofagi, la reazione astrogliale, sono tutte conseguenze neuropatologiche non specifiche. La chemiotassi dei monociti ematici - tanto abbondanti negli infiltrati istologici dell'EAS - va perciò forse considerata un semplice effetto non specifico della demielinizzazione già iniziata dall'anticorpo circolante.
Tuttavia nell'EAS i linfociti si ritrovano sempre in abbondanza nelle lesioni istologiche del SNC e, come abbiamo visto, è possibile trasferire la malattia mediante trasfusioni di cellule linfatiche sospese in soluzione fisiologica e presumibilmente quasi del tutto prive di anticorpo antimielina. (Per inciso, è proprio vero che, come si ammette comunemente, il trasferimento dei linfociti sensibilizzati capaci di danneggiare la mielina s'identifichi necessariamente col trasferimento della malattia? Varrebbe forse la pena di studiare più da vicino le possibili differenze fra il grado di demielinizzazione dell'EAS ‛naturale' e quello dell'EAS ‛trasferita'). Inoltre, usando antigene altamente purificato, è possibile mettere in evidenza durante gli stadi precoci della malattia alterazioni istologiche in assenza o in presenza di quantità non misurabili di anticorpi liberi circolanti (v. Lumsden, 1965; v. Seil e altri, 1968). Yonezawa e Ishihara (citati da Alvord, 1970) sembra abbiano reperito anticorpi circolanti in cavie stimolate con adiuvante di Freund e proteina basica mielinica: se ne potrebbe dedurre che la presenza di anticorpi circolanti potrebbe essere una semplice questione di variabilità quantitativa. Poiché è possibile che durante la fase d'induzione della malattia l'anticorpo mielinotossico si fissi assai rapidamente ai tessuti, è possibile ipotizzare un suo ruolo patogenico nell'EAS. Tuttavia (v. sotto, e in base agli esperimenti di trasferimento) il ruolo attivo ‛demielinizzante' dei T-linfociti sensibilizzati non può essere messo in dubbio. L'autore ritiene che la possibilità di un'azione sinergica sia più che reale.
La natura immunologica del fattore umorale demielinizzante nell'EAS richiede una breve descrizione. Esso è legato al complemento, non è specie-specifico, ma strettamente organo-specifico, al punto che danneggia solo colture di tessuto nervoso centrale e non agisce sulla mielina dei gangli sensori periferici. Inversamente, sieri di animali affetti da neurite allergica sperimentale demielinizzano solo colture di mielina periferica e sono inefficaci su colture di tessuto nervoso centrale, il che rappresenta uno degli argomenti più forti in favore della natura immunologica dei due fattori, poiché un enzima mielinolitico non potrebbe possedere un tale grado di specificità tessutale. È stato dimostrato inoltre che il fattore antimielinico dell'EAS è una gammaglobulina di tipo 7S; la sua specificità tessutale per la mielina è confermata dal fatto che se si allontana il siero dalla coltura dopo completa demielinizzazione, gli assoni non danneggiati mielinizzano di nuovo nello spazio di 7-10 giorni in seguito a maturazione degli oligodendrociti indifferenziati non danneggiati.
Da ultimo, secondo dati personali (Lumsden e Howard, non pubbl.) è ormai provato che nell'EAS come nella sclerosi multipla si può ritrovare un fattore umorale di natura anticorpale ad azione antisinaptica (per dati più approfonditi relativi alla sclerosi multipla v. Lumsden, The clinical pathology..., 19722), e sembra provato che l'endecapeptide sintetico contenente triptofano, come suggerito da Westall e collaboratori (v. sotto), sia in grado d'indurre la formazione di questo fattore bloccante polisinaptico (Carnegie, Bornstein e Crain, comunicazione personale; v. Bornstein e Crain, 1965 e 1971).
Le stesse tecniche sono state usate recentemente per saggiare in vitro la mielinotossicità dei T-linfociti dell'EAS. Alcuni dati riguardanti la tecnica e i risultati conseguiti sono stati riferiti altrove (v. Lumsden, 1971 e i contributi del 1972). I linfociti vengono raccolti dal linfonodo regionale di drenaggio del punto stimolato, lavati, sospesi in liquido di coltura, e applicati direttamente alla coltura di mielina come uno spesso strato di cellule che viene lasciato in contatto per alcune ore onde permettere alle cellule sensibilizzate di aderire alla mielina. Ovviamente i linfociti che aderiscono sono quelli ‛trasformati', come è confermato dall'esame del loro aspetto ultrastrutturale. Gli effetti sugli elementi bersaglio si scorgono adeguatamente solo al microscopio elettronico, essenziale per la lettura di questi test, trattandosi di alterazioni ristrette al microambiente dei linfociti profondamente insinuati. I linfociti ‛positivi' lisano la mielina delle fibre nervose adiacenti in archi distinti o segmenti in corrispondenza della zona di contatto. A parte l'estensione assai più ristretta, la lesione fino a questo momento sembra identica a quella provocata dai sieri demielinizzanti, ivi compreso il rigonfiamento dell'oligodendroglia. Uno sviluppo ulteriore tuttavia è rappresentato dal fatto che le lamelle esterne di mielina disaggregata, invece di restare distaccate e libere come accade nelle colture trattate con siero, tendono ad arrotolarsi completamente intorno ai linfociti sensibilizzati, come se fossero attirate o ‛assorbite' dalle membrane plasmatiche di queste cellule, mentre dalla superficie dei linfociti si dipartono dei microvilli che investono quelle zone della mielina lamellare distaccata che ancora contengono componenti della linea intraperiodica. Questi aspetti forniscono un'interessante indicazione di come i linfociti sensibilizzati (a differenza di quelli normali di controllo che ‛non' posseggono questa proprietà) siano in grado di fagocitare particelle di mielina contenenti la proteina basica encefalitogena intatta. Nonostante che l'immunità ritardata, vale a dire quella dipendente dai T-linfociti, nell'EAS e nelle malattie demielinizzanti non sia stata ancora indagata a fondo e che numerosi dati contrastanti ancora persistano, si può dire con certezza che proprio da questi studi ci verrà detto quali siano le fondamentali somiglianze e le differenze tra EAS e malattie demielinizzanti spontanee. Infatti, laddove appare ormai accertato un ruolo attivo dei T-linfociti nella demielinizzazione sperimentale, ciò non è provato in quella delle malattie spontanee, anzi pare che vi siano dati a favore di una depressione dei T-linfociti a livello del SNC (v. Valdimarsson e altri, 1974; v. Kam-Hansen, 1978).
5. Il fattore encefalitogeno
Abbiamo ricordato in precedenza, nel breve paragrafo storico, come lo studio sperimentale dell'EAS (e per estensione quello della sclerosi multipla) ebbe inizio con la ricerca del fattore encefalitogeno nel vaccino antirabbico. Le ricerche iniziali portarono alla scoperta: 1) che una proteina altamente stabile di basso peso molecolare può essere regolarmente isolata dal tessuto del SNC per semplice estrazione salma o acida; 2) che la sua capacità encefalitogena é in realtà limitata a poche particolari sequenze di amminoacidi situate nella molecola di una proteina basica caratteristica relativamente libera all'interno della linea intraperiodica della mielina lamellare - o per lo meno ivi trattenuta da legami crociati molto labili; 3) che la sua struttura peptidica interna varia un poco da specie a specie in una maniera che spiega ampiamente le differenze biologiche prima riferite fra l'EAS delle diverse specie animali.
Cercheremo ora di sostanziare queste tre conclusioni ed esamineremo perché, di tutti i potenziali autoantigeni del SNC, la proteina basica mielinica debba esercitare come autoantigene un'azione tanto più efficace.
Un filone di ricerca nell'ambito dell'approccio biochimico al problema dell'EAS, lo stesso che ha largamente dominato il lavoro di ricerca in questo campo negli Stati Uniti, consiste nell'isolamento e nella caratterizzazione chimica di un antigene puro - vale a dire omogeneo - mediante tecniche estrattive applicate a omogenati di tessuti di SNC. I risultati sono stati estremamente positivi. Un filone complementare, seguito nel laboratorio dell'autore, consiste nell'identificare la sorgente precisa dell'antigene e la sua funzione naturale nell'ambito del tessuto cerebrale. Sembra logico inoltre prendere in considerazione allo stesso tempo la ricerca chimica sull'EAS.
a) La proteina basica: isolamento, caratterizzazione e determinanti strutturali
Nei primi lavori sul fattore encefalitogeno furono messi in luce la sua notevole resistenza al trattamento col calore e a quello chimico con formaldeide, acetone, cloroformio, alcool, cloroformio-alcool (v. Lumsden, Experimental... II, 1949). Più tardi l'attenzione venne concentrata sull'estrazione salma (con KCl e NaCl) delle proteine solubili a pH neutro e acido (v. Roboz Finstein e Henderson, 1959; v. Kies e Alvord, 1959). L'estrazione acquosa comporta un breve trattamento preliminare sgrassante del materiale - in genere midollo spinale di bue - mediante acetone; dopo dialisi per rimuovere il sale, la soluzione può essere concentrata per liofilizzazione ottenendo una polvere leggera, bianca, altamente solubile, che può essere facilmente ridisciolta in acqua distillata o soluzione fisiologica e mescolata con adiuvante di Freund a costituire l'iniezione ‛stimolante'. Successivamente si è visto che diversi metodi di estrazione acida semplice - per esempio in tampone di glicina a pH 2 oppure in HCl 0,0lN - sono egualmente efficaci (v. Kies e altri, 1961; v. Lumsden e altri, 1964 e 1966), e alcune varianti di essi costituiscono oggi i metodi usati correntemente per una produzione rapida dell'antigene solubile dell'EAS (v. Eylar e altri, 1969). Quando, con l'elettroforesi, fu identificata la natura altamente basica della proteina solubile che si sposta verso l'estremo catodico del campo, si comprese la ragione dell'alta resa dei sistemi di estrazione fortemente acidi. Una larga serie di ricerche mediante tecniche di cromatografia, gradiente di densità e resine a scambio di ioni permise l'ulteriore purificazione e caratterizzazione dell'agente (v. Robertson e altri, 1962; v. Kibler, Fox e Sbapira, 1964; v. Nakao e Roboz Einstein, 1965; v. Kies, 1965; v. Lumsden e altri, 1966; v. Carnegie e Lumsden, 1966 e 1967; v. Kibler e Shapira, 1968). Negli studi preliminari sugli amminoacidi i pesi molecolari risultarono aggirarsi su 3.000-15.000 secondo coloro che usavano allontanare i grassi con acetone (v. Lumsden e altri, 1964; v. Nakao e Roboz Einstein, 1965; v. Carnegie e Lumsden, 1967; v. Kibler e Shapira, 1968), su 20.000-50.000 secondo quei ricercatori che impiegavano cloroformio-metanolo (v. Kies e altri, 1961; v. Caspary e Field, 1965). Il disaccordo circa il peso molecolare non era però dovuto solamente a differenze legate al solvente usato nello sgrassamento iniziale: esso derivava in gran parte dall'ignoranza delle caratteristiche fisico-chimiche della proteina basica e conseguentemente delle condizioni opportune per la filtrazione molecolare e le tecniche all'ultracentrifuga. In seguito la proteina basica purificata fu identificata in una molecola provvista di alta carica elettrica e molto asimmetrica, variamente raggomitolata (v. Eylar e Hashim, 1969). Il riconoscimento di tale struttura permise di adottare uno standard comparabile, in riferimento al quale il peso molecolare fu stabilito aggirarsi intorno ai 17.000 dalton.
L'analisi recente (v. Eylar e Thompson, 1969) di una preparazione omogenea proveniente da midollo spinale di bue ha considerevolmente aumentato la nostra conoscenza circa le insolite proprietà della proteina. Essa contiene 38 grammomolecole per mole di amminoacidi basici - arginina, lisina e istidina - e solo 7 grammomolecole per mole di amminoacidi acidi - acidi glutammico e aspartico -: da qui il suo carattere fortemente basico. Differisce dagli istoni (le proteine basiche dei nuclei cellulari) per un contenuto assai maggiore in istidina, glicina e senna e minore in alanina. Sono presenti solo un residuo di triptofano e due di metionina. La posizione N-terminale è bloccata. Il peso molecolare, calcolato in base all'analisi degli amminoacidi, risulta aggirarsi sui 15.000-18.200 dalton. Studi sulla viscosità intrinseca indicano una molecola lineare molto aggrovigliata. Il peso molecolare è risultato di 16.400, se calcolato mediante equilibrio di sedimentazione; di 16.200, se calcolato mediante viscosità di sedimentazione; di 16.200, se calcolato mediante assorbimento all'ultravioletto basato sul contenuto in triptofano-tirosina.
Discuteremo in seguito se questa proteina basica di 16.000 daltons possa identificarsi o meno con la forma naturale reperibile nella mielina. Esamineremo invece subito i dati comprovanti come le frazioni molecolari della proteina basica posseggano esse stesse attività encefalitogena - siano capaci cioè di provocare la malattia al pari della molecola intera.
La prima prova in questo senso venne fornita dalle ricerche effettuate nel laboratorio dello stesso autore (v. Robertson e altri, 1962; v. Lumsden e altri, 1964 e 1966). Secondo la nostra tecnica estrattiva di routine, l'omogenato di tessuto midollare, estratto con sali in ambiente acido, veniva dializzato a pH 9 per liberarlo dai sali e dai piccoli istoni. Risultò presto evidente che questo passaggio provocava nel residuo non dializzabile una resa di materiale encefalitogeno inferiore a quella prevedibile secondo calcoli grossolani, e che quanto più la dialisi veniva prolungata tanto maggiore era la perdita di attività nel residuo. L'osservazione indusse a recuperare tutti i dializzati e a estrarne laboriosamente i solidi per liofilizzazione. I saggi sul dializzato liofilizzato permisero di accertare la presenza in esso di materiale encefalitogeno attivo: era evidente che attraverso la membrana di dialisi si verificava un certo passaggio di materiale attivo. Test paralleli impieganti vari indicatori misero in evidenza una porosità della membrana equivalente a un peso molecolare di circa 3.000-4.000 dalton, cifra suffragata da determinazioni di pressione osmotica. Studi successivi su idrolizzati enzimatici della proteina basica originale fornirono anch'essi peptidi, perfino più piccoli (v. sotto), forniti di attività encefalitogena. Se ne dedusse perciò che il reperto originale di peptidi attivi in un idrolisato alcalino fosse la conseguenza di un certo grado di idrolisi enzimatica avvenuta durante l'estrazione originale del tessuto con acetone, che può estrarre la proteinasi acida attiva, mentre l'uso del cloroformio-metanolo elimina la ‛contaminazione'. Tuttavia l'esame accurato delle tavole di estrazione nel nostro lavoro più approfondito (v. Lumsden e altri, 1966) dimostra come in parecchi casi questa spiegazione non appaia verosimile, mentre sembrerebbe che, oltre all'azione della proteinasi, il semplice trattamento alcalino sia sufficiente a rompere i legami deboli nell'ambito della proteina basica originale. Qualunque sia la spiegazione, è facile concludere che, se i determinanti encefalitogeni della mielina sono realmente assai resistenti ai trattamenti chimici, nelle condizioni naturali la molecola intera della proteina non è invece molto resistente.
Per quel che riguarda gli effetti dell'idrolisi enzimatica sistematica effettuata per determinare le sequenze di amminoacidi nella proteina basica, le analisi digestive iniziali furono effettuate con tripsina, chimotripsina e pepsina (v. Carnegie e altri, 1967; v. Chao e Roboz Einstein, 1968; v. Eylar e Hashim, 1968; v. Hashim e Eylar, 1969). I trattamenti fornivano regolarmente polipeptidi attivi in senso encefalitogeno, nell'ambito dei quali non tardarono a emergere differenze strutturali legate alla specie animale. Così Kibler e altri (v., 1969) isolarono un polipeptide di 45 residui, fortemente basico, molto simile in corrispondenza dell'estremità amminica alla proteina basica del cervello umano, ma del tutto differente all'estremità carbossilica (v. Carnegie, 1971). Kibler e collaboratori tuttavia erano inizialmente interessati alle differenze di struttura fra i polipeptidi encefalitogeni di bue e quelli di coniglio, che a loro volta presentavano zone d'identità e zone di differenza strutturale. Nell'ambito di un polipeptide più grande proveniente dalla loro preparazione di coniglio esistevano chiaramente due zone attive distinte. Nella proteina umana (peso molecolare 18.400) Carnegie (v., 1971) isolò non meno di tre sequenze peptidiche ancora fornite di attività encefalitogena.
Per Carnegie il principale determinante encefalitogeno della proteina basica umana è un piccolo peptide di soli 15 amminoacidi disposti secondo la sequenza: Ser-Arg-Phe-Ser-Trp-Gly-Ala-Glu-Gly-Gln-Arg-Pro-Gly-Phe-Gly. Lo stesso autore afferma che 180 micromoli di questo quindecapeptide sono sufficienti a provocare l'EAS nella cavia.

La struttura completa della proteina basica encefalitogena umana secondo Carnegie è riprodotta nella fig. 19. Il quindecapeptide fortemente attivo descritto sopra è situato in posizione 111-125 (sottolineato). Nella sua rassegna Alvord (v., 1970) include uno schema modificato della proteina A. 1 di bue proposto da Eylar (v., 1970) nel quale si vede che la stessa serie di amminoacidi si ripete in posizione 111-125 con l'eccezione del residuo 121 dove la lisina sostituisce l'arginina della proteina umana.
L'analisi delle differenze di specie nell'ambito del fattore encefalitogeno rappresenta dunque il progresso più drammatico verificatosi dal tempo delle osservazioni empiriche iniziali sugli accidenti paralitici dei vaccini antirabbici.
Recentemente questi studi hanno ricevuto un ulteriore impulso in avanti: Westall e altri (v., 1971), usando peptidi sintetici, hanno approfondito ulteriormente l'identificazione del sito intramolecolare dell'attività encefalitogena. Vale la pena di citare in dettaglio il loro lavoro. Usando la sintesi peptidica in fase solida di Merrifield questi autori hanno prodotto parecchi peptidi analoghi tra loro, ognuno dei quali differisce soltanto per un residuo dal peptide naturale dell'EAS (derivato dalla proteina basica A. 1 del midollo spinale di bue). Nella fig. 20 sono rappresentate le sequenze amminoacidiche di tali peptidi, numerati da S1 a S10, paragonate con quelle dei peptidi naturali dell'EAS, T. 27 ed E. Gli amminoacidi sostituiti sono indicati in corsivo.
Westall e collaboratori concludono che la regione del triptofano è la più importante dal punto di vista encefalitogeno per tre ragioni: 1) i peptidi derivati e quelli sintetici posseggono su base molare attività eguale, che si avvicina a quella della proteina basica di origine; 2) l'interazione selettiva del residuo del triptofano col bromuro di 2-idrossi-5-nitrobenzene inattiva la sequenza encefalitogena; 3) ogni volta che è conservata la specificità della sequenza Trp-Gln-Lys (oppure Arg) nelle proteine mieliniche naturali (come accade nell'uomo, nella scimmia, nel cane, nel coniglio, nella cavia, nel topo e nel cavallo) è presente l'attività encefalitogena, mentre là dove tale sequenza è alterata, come nel pollo e nella tartaruga, l'attività scompare.
b) Localizzazione e funzione dell'antigene encefalitogeno
L'identificazione della struttura molecolare della proteina basica rappresenta un enorme progresso tecnico e si rivelerà senza dubbio d'importanza fondamentale in immunobiologia, rendendo disponibile uno dei più piccoli antigeni naturali isolati finora. Essa permette inoltre di trarre analogie che potrebbero assumere un valore pratico nella desensibilizzazione o nell'immunizzazione contro l'EAS e nello studio di eventuali analogie con la sclerosi multipla. Eppure queste ricerche non aiutano a risolvere l'inquietante problema della precisa localizzazione e funzione del peptide basico e della sua esclusiva localizzazione nella mielina, poiché, come abbiamo visto, la malattia da esso evocata è strettamente un processo antimielinico.
Il concetto che il fattore encefalitogeno fosse localizzato ‛nella' mielina (ossia non solamente che la risposta immune al fattore fosse diretta ‛contro' la mielina) si è appoggiato per due decadi unicamente su metodi molto grossolani, consistenti nel paragonare l'attività encefalitogena di regioni del cervello anatomicamente ricche di mielina con quella di zone anatomicamente povere, o su paragoni simili fra tessuti appartenenti a periodi di pre- e post-mielinizzazione del SNC in sviluppo. Prove più solide dovettero attendere l'applicazione dei metodi di ultracentrifugazione alla separazione della mielina (v. Laatsch e altri, 1962). I test biologici quantitativi per l'attività encefalitogena della mielina così preparata dimostrarono che essa possiede un'attività specifica molto più alta di quella dell'intero cervello (nella cavia).
La maggior parte di queste tecniche di ultracentrifugazione (v. Autilio e altri, 1964; v. Gerstl e altri, 1965; v. Eng e altri, 1968) non era diretta a studiare il potere encefalitogeno della mielina ma a determinarne accuratamente la composizione nelle diverse specie, soprattutto nei riguardi dei costituenti lipidici, e a chiarire la presenza o meno in essa di enzimi. Tali ricerche mettevano scarsamente in discussione il presupposto, basato su reperti istologici, che la mielina fosse un componente tessutale relativamente stabile, benché, in ultima analisi, finissero per confermarlo almeno per quanto riguarda la componente lipidica.
Ma il problema iniziale negli studi di caratterizzazione chimica della ‛mielina pura', consiste nel definire la mielina stessa. A parte ogni definizione elettro-fisiologica (praticamente ancora impossibile), è lecito tentare una definizione in termini di un'ultrastruttura fissa? Nel nostro laboratorio questo rappresentò, cinque anni fa, il punto di partenza della ricerca in questa particolare area della neurochimica. Adottammo allora due postulati: 1) che al microscopio elettronico una mielina pura ottenuta per ultracentrifugazione deve rispettare alcuni criteri ultrastrutturali basati su quelli del tessuto intatto di provenienza, pur rimanendo allo stesso tempo praticamente priva di materiale non mielinico; 2) che la sua composizione chimica sia riproducibile.
Una tale mielina, separata all'ultracentrifuga dai componenti non mielinici (nuclei, mitocondri, ribosomi), forma costantemente delle vescicole chiuse del diametro di 1-20 micron. Le osservazioni di Autilio e collaboratori, inoltre, dimostrano chiaramente che tali vescicole posseggono densità variabile, dal momento che nelle separazioni al gradiente di sucrosio tendono a disporsi a livelli differenti secondo una serie di bande ben definite, piuttosto che formare una singola banda omogenea. Studiando tali bande, noi accertammo che, mentre la composizione in lipidi rimaneva virtualmente costante, esisteva una considerevole differenza nel contenuto proteico, che variava dal 20% nella mielina più leggera al 32% in quella più pesante. Quale di queste doveva essere considerata la mielina ‛normale'? O non si doveva piuttosto concludere che allo stato naturale esistono più tipi di mielina? All'esame morfologico le variazioni della distribuzione del volume delle vescicole e del contenuto dei costituenti non mielinici nelle differenti mieline non sembrava spiegare completamente i diversi strati o bande. Inoltre, a parità di età e di sesso, fra una preparazione di midollo spinale di bue e un'altra esisteva un'ampia variazione nella quantità di proteina mielinica, contenuta nei campioni finali ottenuti all'ultracentrifuga e scelti per l'analisi chimica. L'unica tendenza costante venuta in seguito alla luce era che il numero e la larghezza (o altezza) rispettivi delle bande variava - anche solo entro 6 ore - a seconda del tempo trascorso fra l'uccisione dell'animale e l'omogeneizzazione e il raffreddamento a 4 °C del materiale escisso. Se l'intervallo non superava i 20 minuti l'aspetto era quello di una sola banda centrata in corrispondenza di 1,084 g/cm-3 di densità. Tale prodotto era rappresentato da una mielina di aspetto morfologico normale e ben conservato al microscopio elettronico, con una disposizione lamellare ripetuta di 110-120 Å. Quando il tempo intercorrente tra la morte e il raffreddamento dell'omogenato si prolungava anche solo di due ore comparivano bande addizionali, la principale delle quali era centrata in corrispondenza della densità di flottazione di 1,063 g/cm-3 e mostrava, insieme a mielina morfologicamente normale, una larga componente di strutture lamellari ‛collabite' di una periodicità pari a 100-104 Å. Nel caso di queste strutture lamellari ‛collabite' la normale ‛linea intraperiodica', meno densa, risultava cancellata e rimpiazzata da una condensazione della sostanza lamellare interposta tra linee dense principali adiacenti, come illustrato nel nostro lavoro originale (v. Dickinson e altri, 1970). Quanto più leggere sono le bande di mielina nella densità di flottazione, tanto maggiore è il numero di strisce di lamelle collabite nelle pareti delle vescicole. Inoltre, test biologici delle differenti bande di flottazione dimostrarono che la mielina più pesante possedeva un potere encefalitogeno significativamente più elevato, e nel caso di una mielina disposta su quattro bande, le due più alte, col minore contenuto proteico, erano praticamente prive di attività encefalitogena. Perfino nella più leggera delle due bande pesanti si verificava una considerevole caduta di attività rispetto alla banda più pesante. L'incubazione di mielina inizialmente normale ottenuta all'ultracentrifuga (priva di proteinasi) in un tampone di NaCl-bicarbonato pH 7,4 a 37° per periodi fino a 7 gg. non provocava alterazioni morfologiche o chimiche. Ma l'incubazione in tampone lattato pH 3,5 o in HCl 0,1M provocava la comparsa quasi istantanea delle strutture mieliniche collabite già descritte, e nello stesso tempo compariva proteina nel mezzo d'incubazione. Tale proteina risultò identica alla proteina basica all'elettroforesi su acrilamide, alla cromatografia su colonna e all'analisi degli amminoacidi, e i test biologici ne misero in evidenza l'alto potere encefalitogeno nella cavia alla dose di 1 microgrammo.
I risultati fondamentali di questi ultimi studi sono riassunti nella fig. 21. Essi permettono chiaramente di concludere che la proteina basica encefalitogena è strettamente localizzata alle ‛linee intraperiodiche' lamellari, delle quali forma il principale e forse l'unico costituente. La proteina rappresenta, secondo i nostri dati quantitativi, circa un terzo del contenuto proteico totale dell'intera guaina mielinica (essa stessa circa il 30-32% della mielina in peso secco), per cui circa il 10% in peso secco della mielina è costituito da proteina basica encefalitogena. Si libera facilmente dalla mielina isolata a basso pH, senza l'intervento a questo stadio di alcuna attività enzimatica. Così, benché dal punto di vista degli altri suoi componenti la mielina sia chimicamente una sostanza altamente stabile, essa possiede una curiosa instabilità fisica differenziale nei riguardi della proteina basica che forma la linea intraperiodica. (Questa conclusione attira l'attenzione sull'importante ruolo esplicato dalla membrana limitante più esterna della guaina mielinica vera e propria, costituita dalla membrana plasmatica degli oligodendrociti; le alterazioni precoci post mortem di questa membrana, che permettono alla mielina di rivelare la sua instabilità differenziale nei riguardi della proteina basica della linea intraperiodica, potrebbero essere dovute all'attività delle proteinasi acide cellulari).
La sequenza di amminoacidi immunologicamente attiva (il ‛sito attivo' della molecola della proteina basica) è stata identificata in precedenza in questo lavoro. È stata anche dimostrata la localizzazione della proteina basica nelle linee intraperiodiche delle lamelle di mielina e la sua selettiva liberazione senza bisogno di alcuna degradazione enzimatica della mielina. I test di tossicità linfocitaria in vitro hanno dimostrato come nell'EAS i linfociti specificamente sensibilizzati per la mielina assumano frammenti di mielina lamellare che ancora contengono materiale di linea intraperiodica - cioè che ancora contengono l'antigene attivo della mielina.
(Si ringrazia il prof. Angelo Massaro per l'aggiornamento dell'articolo e della bibliografia).
bibliografia
Allegranza, A., Cazzullo, C. L., Encefalomielite sperimentale ‛allergica' prodotta in cavie mediante iniezioni di cervello umano più adiuvanti, in ‟Bollettino della Società Italiana di Patologia", 1950, III, pp. 664-676.
Allegranza, A., Cazzullo, C. L., Il quadro anatomopatologico della encefalomielite diffusa sperimentale della cavia prodotta da emulsioni di cervello omologo ed eterologo più adiuvanti, in ‟Bollettino della Società Italiana di Patologia", 1950, I, pp. 3-8.
Alvord, E. C., Studies on the etiology and pathogenesis of experimental meningoencephalomyelitis in the guinea pig, in ‟Journal of immunology", 1949, LXI, pp. 355-367.
Alvord, E. C., Acute disseminated encephalomyelitis in ‛allergic' neuroencephalopathies, in Handbook of clinical neurology (a cura di P. J. Vinken e G. W. Bruyn), vol. IX, Amsterdam 1970, pp. 500-570.
Alvord, E. C., Correll, J. W., Ladd, A. T., Kellner, A., The effect of pyribenzamine in beeswax in experimental meningoencephalomyelitis in the guinea pig, in ‟Journal of immunology", 1948, LX, pp. 345-348.
Alvod, E. C., Shaw, C. M., Fahlberg, W. J., Kies, M. W., An analysis of various types of inhibition of experimental ‛allergic' encephalomyelitis in the guinea pig, in ‟Zeitschrift für Immunitats-und Allergieforschung", 1964, CXXVI, pp. 217-227.
Alvord, E. C., Shaw, C. M., Hruby, S., Kies, M. W., Encephalitogen-induced inhibition of experimental allergic encephalomyelitis: prevention, suppression and therapy, in ‟Annals of the New York Academy of Sciences", 1965, CXXII, pp. 333-345.
Anderson, W. R., Vogel, F. S., Encephalomyelitis induced in pigeons with homologous neural tissue and adjuvants, in ‟Journal of neuropathology and experimental neurology", 1961, XX, pp. 548-553.
Aparicio, S. G., Lumsden, Ch., E., Jennings, M., Electron microscopy of glial reactions observed in CNS cultures, in ‟Brain research", 1968, VII, pp. 268-285.
Arnason, B. G., Janković, B. D., Waksman, B. H., Wennersten, C., Role of the thymus in immune reaction in rats. II. Suppressive effect of thymectomy at birth on reactions of delayed (cellular) hypersensitivity and the circulating small lymphocyte, in ‟Journal of experimental medicine", 1962, CXVI, pp. 177-186.
Aström, K. E., Waksman, B. H., The passive transfer of experimental allergic encephalomyelitis and neuritis with living lymphoid cells, in ‟Journal of pathology and bacteriology", 1962, LXXXIII, pp. 89-106.
Autilio, L. A., Norton, W. T., Terry, R. D., The preparation and some properties of purified myelin from the central nervous system, in ‟Journal of neurochemistry", 1964, XI, pp. 17-27.
Barlow, C. F., A study of abnormal blood-brain permeability in experimental allergic encephalomyelitis, in ‟Journal of neuropathology and experimental neurology", 1956, XV, pp. 196-207.
Biggins, J. A., Taylor, A., Caspary, E. A., Circulating antibody to myelin basic protein in relapsing-remitting multiple sclerosis, in ‟Journal of neurology neurosurgery and psychiatry", 1978, XLI, pp. 1131-1134.
Bornstein, M. B., Appel, S. H., The application of tissue culture to the study of experimental ‛allergic' encephalomyelitis, in ‟Journal of neuropathology and experimental neurology", 1961, XX, pp. 141-157.
Bornstein, M. B., Crain, S. M., Functional studies of cultured brain tissues as related to ‛demyelinative' disordes, in ‟Science", 1965, CXLVIII, pp. 1242-1244.
Bornstein, M. B., Crain, S. M., Lack of correlation between changes in bioelectric functions and myelin in cultured CNS tissues chronically exposed to sera from animals with EAE, in ‟Journal of neuropathology and experimental neurology", 1971, XXX, pp. 129 ss.
Brandriss, M. W., Smith, J. W., Friedman, R. M., Suppression of experimental allergic encephalomyelitis by antimetabolites, in ‟Annals of the New York Academy of Sciences", 1965, CXXII, pp. 356-368.
Brightman, M. W., The distribution within the brain of ferritin injected into cerebrospinal fluid compartments. II. Parenchimal distribution, in ‟American journal of anatomy", 1965, CXVII, pp. 193-220.
Bullington, S. J., Waksman, B. H., Uveitis in rabbits with experimental allergic encephalomyelitis, in ‟Archives of ophthalmology", 1958, LIX, pp. 435-445.
Burky, E. L., Experimental endophthalmitis phaco-anaphylactica in rabbits, in ‟Achives of ophthalmology" (New Series), 1934, XII, pp. 536-546.
Calne, D. B., Liebowitz, S., Suppression of experimental allergic encephalomyelitis by cytotoxic drugs, in ‟Nature", 1963, CXCVII, pp. 1309-1310.
Carnegie, P. R., Properties, structure and possible neuroreceptor role of the encephalitogenic protein of human brain, in ‟Nature", 1971, CCXXIX, pp. 25-28.
Carnegie, P. R., Bencina, B., Lamoureux, G., Isolation of bsic proteins and polypeptides from central nervous tissue, in ‟Biochemical journal", 1967, CV, pp. 559-568.
Carnegie, P. R., Lumsden, Ch. E., Encephalitogenic peptides from spinal cord, in ‟Nature", 1966, CCIX, pp. 1354-1355.
Carnegie, P. R., Lumsden, Ch. E., Fractionation of encephalitogenic polypeptides from bovine spinal cord by gel filtration in phenolacetic acid-water, in ‟Immunology", 1967, XII, pp. 133-145.
Caspary, E. A., Field, E. J., An encephalitogenic protein of human origin: some chemical and biological properties, in ‟Annals of the New York Academy of Sciences", 1965, CXXII, pp. 182-188.
Cazzullo, C. L., Allegranza, A., Contributo allo studio delle encefalomieliti sperimentali ‛allergiche' nella cavia, in ‟Sistema nervoso", 1950, II, pp. 258-272.
Cazzullo, C. L., Ferraro, A., Ricerche ed osservazioni semeiologiche sulla encefalomielite allergica sperimentale acuta e cronica nella scimmia, in ‟Rivista di patologia nervosa e mentale", 1950, LXXI, pp. 533-620.
Chao, L. P., Roboz Einstein, E., Isolation and characterization of an active fragment from enzymatic degradation of an encephalitogenic protein, in ‟Journal of biological chemistry", 1968, CCXLIII, pp. 6050-6055.
Clausen, J., Möller, J., Allergic encephalomyelitis induced by brain antigen after deficiency in polyunsaturated fatty acids during myelination, in ‟Acta neurologica scandinavica", 1967, XLIII, p. 375.
Cohen, A., Textbook of medical virology, Oxford 1969.
Condie, R. M., Kelly, J. T., Campbell, B., Good, R. A., Prevention of experimental encephalomyelitis by prior injections of homologous spinal cord, in ‟Federation proceedings", 1957, XVI, p. 24.
Condie, R. M., Kelly, J. T., Thomas, L., Good, R. A., Prevention of experimental allergic encephalomyelitis by removal of the regional lymph node, in ‟Anatomical record", 1957, CXXVII, p. 405.
Condie, R. M., Nicholas, T., Effect of total body irradiation on the development of experimental allergic encephalomyelitis, in ‟Federation proceedings", 1962, XXI, p. 43.
Cutler, R. W. P., Barlow, C. F., Alteration in brain vascular permeability in allergic encephalomyelitis, in ‟Journal of neuropathology and experimental neurology", 1967, XXVI, pp. 558-571.
Delpech, B., Lichtblau, E., Étude quantitative des immunoglobulines et de l'albumine du liquide céphalo-rachidien, in ‟Clinica chimica acta", 1972, XXXVII, pp. 15-23.
Dickinson, J. P., Jones, K. M., Aparicio, S. G., Lumsden, Ch. E., Localization of encephalitogenic basis protein in the intraperiod line of lamellar myelin, in ‟Nature", 1970, CCXXVII, pp. 1133-1134.
Eng, L. F., Chao, F. C., Gerstl, B., Pratt, D. T., Tavaststjerna, M. G., The maturation of human white matter. Fractionation of the myelin membrane proteins, in ‟Biochemistry", 1968, VII, pp. 4455-4465.
Eylar, E. H., Basic Al protein: relationship to experimental allergic encephalomyelitis, in ‟Research publications. Association for Research in Nervous and Mental Diseases", 1970.
Eylar, E. H., Hashim, G. A., Allergic encephalomyelitis: the structure of the encephalitogenic determinant, in ‟Proceedings of the National Academy of Sciences", 1968, LXI, pp. 644-650.
Eylar, E. H., Hashim, G. A., Allergic encephalomyelitis; cleavage of the C-tryptophyl bond in the encephalitogenic basis protein from bovine myelin, in ‟Archives of biochemistry and biophysics", 1969, CXXXI, pp. 215-222.
Eylar, E. H., Salk, J., Beveridge, G. C., Brown, L.V., Experimental encephalitogenic basic protein from bovine myelin, in ‟Archives of biochemistry", 1969, CXXXII, pp. 34-48.
Eylar, E. H., Thompson, M., Allergic encephalomyelitis: the physiochemical properties of the basic protein encephalitogen from bovine spinal cord, in ‟Archives of biochemistry and biophysics", 1969, CXXIX, pp. 468-479.
Falk, G. A., Kies, M. W., Passive transfer of experimental allergic encephalomyelitis (EAE) and delayed hypersensitivity to myelin basic protein (BP), in ‟Federation proceedings", 1968, XXVII, p. 544.
Falk, G. A., Kies, M. W., Alvord, E. C., Passive transfer of experimental allergic encephalomyelitis: mechanisms of suppression, in ‟Journal of immunology", 1969, CIII, pp. 1248-1253.
Ferraro, A., Cazzullo, C. L., Chronic experimental allergic encephalomyelitis in monkeys, in ‟Journal of neuropathology and experimental neurology", 1948, VII, pp. 235-260.
Ferraro, A., Jervis, G. A., Experimental disseminated encephalopathy in the monkey, in ‟Archives of neurology and psychiatry (Chicago)", 1940, XLIII, pp. 195-209.
Ferraro, A., Roizin, L., Experimental allergic encephalomyelitis during and following cortisone acetate treatment, in ‟Journal of neuropathology and experimental neurology", 1953, XII, pp. 373-386.
Field, E. J., The early lesions of experimental ‛allergic' encephalomyelitis, in ‟Experimental neurology", 1961, IV, pp. 233-240.
Field, E. J., Effect of imuran (Axathioprine) on susceptibility of guinea pigs to allergic encephalomyelitis, in ‟Archives internationales de pharmacodynamie et de thérapie", 1967, CLXV, pp. 138-141.
Fischel, E. E., Kabat, E. A., Stoerk, H. C., Skolnick, M., Bezer, A. E., Suppression by cortisone of granuloma formation and antibody in guinea pigs receiving egg albumen with Freund's albumin, in ‟Journal of allergy", 1954, XXV, pp. 195-200.
Flad, H. D., Paterson, P. Y., Miescher, P. A., In vitro effects of phytohemagglutinin and brain antigen on cell transfer of experimental autoimmune encephalomyelitis in Lewis rats, in ‟Journal of immunology", 1967, C, pp. 647-653.
Fog, T., Bardram, M., Experimental disseminated encephalomyelitis with iridocyclitis in pigs, in ‟Nordisk medicin", 1953, XLIX, pp. 851-855.
Freund, J., Lipton, M. M., Morrison, L. R., Demyelination in the guinea pig in chronic allergic encephalomyelitis, in ‟Archives of pathology", 1950, L, pp. 108-121.
Freund, J., Stern, E. R., Pisani, T. M., Isoallergic encephalomyelitis and radiculitis in guinea pigs after one injection of brain and myelobacteria in water-in-oil-emulsion, in ‟Journal of immunology", 1947, LVII, pp. 179-194.
Frick, E., Scheid-Seydel, L., Untersuchungen mit J131-markiertem Albumin über Austauschvorgänge zwischen Plasma und Liquor cerebro-spinalis, in ‟Klinische Wochenschrift", 1958, XXXVI, pp. 66-69.
Gerstl, B., Tavaststjerna, M. G., Hayman, R. B., Eng, L. F., Smith, J. K., Alterations in myelin fatty acids and plasmalogens in multiple sclerosis, in ‟Annals of the New York Academy of Sciences", 1965, CXXII, pp. 405-415.
Good, R. A., Experimental allergic disseminated encephalitis, in Etiologic factors in mental retardation. 23rd Ross pediatric research conference, Columbus 1957.
Good, R. A., Campbell, B., Good, T. A., Prophylactic and therapeutic effect of para-aminobenzoic acid and sodium salicylate on experimental allergic encephalomyelitis, in ‟Proceedings of the Society for Experimental Biology and Medicine", 1949, LXXII, pp. 341-347.
Greene, H. S. N., Transplantation of tumors to brains of heterologous species, in ‟Cancer research", 1951, XI, pp. 529-534.
Greene, H. S. N., Transplantation of human brain tumors to brains of laboratory animals, in ‟Cancer research", 1953, XIII, pp. 422-426.
Greene, H. S. N., Heterotransplantation of tumors, in ‟Annals of the New York Academy of Sciences", 1957, LXIX, pp. 818-829.
Halpern, B. N., Bertrand, I., Lhermitte, F., L'encéphalomyélite allergique expérimentale, in ‟Presse médicale", 1950, LVIII, pp. 684-687.
Hashim, G. A., Eylar, E. H., Allergic encephalomyelitis: isolation and characterization of encephalitogenic peptides from the basic protein of bovine spinal cod, in ‟Archives of biochemistry and biophysics", 1969, CXXIX, pp. 635-655.
Hausmanova, I., Saper, J., Wplyw ACTH na doswiadczalne schorzenia ukladu nerwowego zwierzat laboratoryjnych, in ‟Neurologia, neurochirurgia in psychiatria polska", 1956, VI, pp. 747-759.
Hausmanova, I., Saper, J., Experimental encephalomyelitis, in Proceedings of the third international congress of neuropathology, Bruxelles, Amsterdam 1957, p. 44.
Hoyer, L. W., Good, R. A., Condie, R. M., Experimental allergic encephalomyelitis: the effect of 6-mercaptopurine, in ‟Journal of experimental medicine", 1962, CXVI, pp. 311-327.
Hurst, E. W., A review of some recent observations on demyelination, in ‟Brain", 1944, LXVII, p. 103.
Innes, J. R. M., Experimental ‛allergic' encephalitis: attempts to produce the disease in sheep and goats, in ‟Journal of comparative pathology and therapeutics", 1951, LXI, p. 241-250.
Janković, B. D., Draskoci, Paunović, D., Popesković, Suppression of experimental allergic encephalomyelitis in rats and chickens treated with reserpine, in ‟Nature", 1964, CCIV, pp. 1101-1102.
Janković, B. D., Isvaneski, M., Experimental allergic encephalomyelitis in thymectomized, bursectomized and normal chickens, in ‟International archives of allergy and applied immunology", 1963, XXIII, pp. 188-206.
Jervis, G. A., Burkhardt, R. L., Koprowski, H., Demyelinating encephalomyelitis in the dog associated with antirabies vaccination, in ‟American journal of hygiene", 1949, L, pp. 14-26.
Jervis, G. A., Koprowski, H., Experimental allergic encephalomyelitis, in ‟Journal of neuropathology and experimental neurology", 1948, VII, pp. 309-320.
Kabat, E. A., Wolf, A., Bezer, A. E., Rapid production of acute disseminated encephalomyelitis in rhesus monkeys by injection of brain tissue with adjuvants, in ‟Science", 1946, CIV, pp. 362-363.
Kabat, E. A., Wolf, A., Bezer, A. E., The rapid production of acute disseminated encephalomyelitis in rhesus monkeys by injection of heterologous and homologous brain tissue with adjuvants, in ‟Journal of experimental medicine", 1947, LXXXV, pp. 117-130.
Kabat, E. A., Wolf, A., Bezer, A. E., Experimental studies on acute disseminated encephalomyelitis in the rhesus monkey, in ‟Annals of allergy", 1948, VI, pp. 109-130.
Kabat, E. A., Wolf, A. Bezer, A. E., Studies on acute disseminated encephalomyelitis produced experimentally in rhesus monkeys. IV. Disseminated encephalomyelitis produced in monkeys with their own brain tissue, in ‟Journal of experimental medicine", 1949, LXXXIX, pp. 395-398.
Kabat, E. A., Wolf, A., Bezer, A. E., Studies on acute disseminated encephalomyelitis produced experimentally in monkeys. VII. The effect of cortisone, in ‟Journal of immunology", 1952, LXVIII, pp. 265-275.
Kam Hansen, S., Reduced active T cell subpopulation in multiple sclerosis cerebrospinal fluid, in NATO Advanced Study Institute. Humoral immunity in neurological disease. Abstacts, New York 1978, p. 32.
Kesting, G., Petite, E., Zur Pathohistologie und pathogenese der experimentellen ‛allergischen' Encephalomyelitis des Affen, in ‟Deutsche Zeitschrift für nevenheilkunde", 1957, CLXXVI, pp. 387-426.
Kibler, R. F., Fox, R. H., Shapira, R., Isolation of a highly purified encephalitogenic protein from bovine cord, in ‟Nature", 1964, CCIV, pp. 1273-1275.
Kibler, R. F., Shapira, R., Isolation and properties of an encephalitogenic protein from bovine, rabbit and human central nevous system tissue, in ‟Journal of biological chemistry", 1968, CCXLIII, pp. 281-286.
Kibler, R. F., Shapira, R., Partial amino acid sequence of an encephalitogenic protein, in ‟Federation proceedings", 1968, XXVII, p. 464.
Kibler, R. F., Shapira, R., McKneally, S., Jenkins, J., Selden, P., Chou, F., Encephalitogenic protein: structure, in ‟Science", 1969, CLXIV, pp. 577-580.
Kies, M. W., Chemical studies on an encephalitogenic protein from guinea pig brain, in ‟Annals of the New York Academy of Sciences", 1965, CXXII, pp. 161-169.
Kies, M. W., Alvord, E. C., Prevention of allergic encephalomyelitis by prio injection of adjuvants, in ‟Nature", 1958, CLXXII, p. 1106.
Kies, M. W., Alvod, E. C., Encephalitogenic activity in guinea pigs of watersoluble protein fractions of nervous tissue, in ‛Allergic' encephalomyelitis (a cura di M. W. Kies e E. C. Alvord), Springfield, Ill., 1959, pp. 293-299.
Kies, M. W., Murphy, J. B., Alvord, E. C., Studies on the encephalitogenic factor in guinea pig central nervous system, in Chemical pathology of the nervous system (a cua di J. Folch-Pi), Oxford 1961, pp. 197-204.
Kolb, L. C., Karlson, A. G., Sayre, G. P., Prevention of experimental allergic encephalomyelitis by various agents, in ‟Transactions of the American Neurological Association", 1952, LXXVII, pp. 117-121.
Kopeloff, L. M., Kopeloff, N., Neurologic manifestations in laboratory animals produced by organ (adjuvant) emulsions, in ‟Journal of immunology", 1947, LVII, pp. 229-237.
Kosunen, T. U., Waksman, B. H., Samuelsson, I., Radioautographic study of cellular mechanisms in delayed hypersensitivity. II. Experimental allergic encephalomyelitis in the rat, in ‟Journal of neuropathology and experimental neurology", 1963, XXII, pp. 367-380.
Laatsch, R. H., Kies, M. W., Gordon, S., Alvord, E. C., The encephalomyelitic activity of myelin isolated by ultracentrifugation, in ‟Journal of experimental medicine", 1962, CXV, pp. 777-788.
Lee, J. M., Immunologic aspects of experimental ‛allergic' encephalomyelitis in susceptible and resistant mice, 1964, CXXVI, pp. 14-24.
Levine, S., Allergic encephalomyelitis: cellular transformation and vascular blockade, in ‟Journal of neuropathology and experimental neurology", 1970, XXIX, pp. 6-20.
Levine, S., Hoenig, E. M., Kies, M. W., Allergic encephalomyelitis: immunologically specific inhibition of cellular passive transfer by encephalitogenic basis proteins, in ‟Clinical and experimental immunology", 1970, VI, pp. 503-517.
Levine, S., Sowinski, R., Passive transfer of allergic adrenalitis and encephalomyelitis with whole blood, in ‟Proceedings of the Society for Experimental Biology and Medicine", 1968, CXXIX, p. 221.
Levine, S., Wenk, E. J., Lymphatic dissemination of encephalitogenic emulsion and its relation to prevention of allergic encephalomyelitis by lymphadenectomy, in ‟Proceedings of the Society for Experimental Biology and Medicine", 1962, CXI, pp. 385-387.
Levine, S., Wenk, E. J., Allergic encephalomyelitis: a hyperacute form, in ‟Science", 1964, CXLVI, pp. 1681-1682.
Levine, S., Wenk, E. J., A hyperacute form of allergic encephalomyelitis, in ‟American journal of pathology", 1965, XLVII, pp. 61-88.
Levine, S., Wenk, E. J., Central nervous system haemorrhages induced by heparin in rats with allergic encephalomyelitis, in ‟Thrombosis et diathesis haemorhagica", 1966, XVI, pp. 296-301.
Levine, S., Wenk, E. J., Exacerbation and transformation of allergic encephalomyelitis by pertussis vaccine, in ‟Proceedings of the Society for Experimental Biology and Medicine", 1966, CXXII, pp. 115-118.
Levine, S., Wenk, E. J., Rapid passive transfer of allergic encephalomyelitis, in ‟Journal of immunology", 1967, XCIX, pp. 1277-1285.
Levine, S., Wenk, E. J., Devlin, H. B., Pieroni, R. E., Levine, L., Hyperacute allergic encephalomyelitis: adjuvant effecet of pertussis vaccine and extracts, in ‟Journal of immunology", 1966, XCVII, pp. 363-368.
Link, H., Zettervall, O., Multiple sclerosis: disturbed Kappa/Lambda light chain ratio of immunoglobulin G in cerebrospinal fluid, in ‟Clinical and experimental immunology", 1970, VI, pp. 435-438.
Lippincott, S. W., Korman, S., Lax, L. C., Corcoran, C., Transfer rates of gammaglobulin between cerebrospinal fluid and blood plasma, in ‟Journal of nuclear medicine", 1965, VI, pp. 632-644.
Lipton, M. M., Freund, J., Encephalomyelitis in the rat following intracutaneous injection of central nervous system tissue with adjuvant, in ‟Proceedings of the Society for Experimental Biology and Medicine", 1952, LXXXI, pp. 260-261.
Lipton, M. M., Freund, J., The efficiency of the intracutaneous route of injection and the susceptibility of the Hartley strain of guinea pigs in experimental allergic encephalitis, in ‟Journal of immunology", 1953, LXX, pp. 326-329.
Lisak, R. P., Falk, G. A., Heinze, R. G., Kies, M. W., Effect of methotrexate in specific prevention of experimental allergic encephalomyelitis (EAE), in ‟Federation proceedings", 1968, XXVII, p. 473.
Lumsden, Ch. E., Experimental ‛allergic' encephalomyelitis, I., in ‟Brain", 1949, LXXII, pp. 198-226.
Lumsden, Ch. E., Experimental ‛allergic' encephalomyelitis, II. On the nature of the encephalitogenic agent, in ‟Brain", 1949, LXXII, pp. 517-537.
Lumsden, Ch. E., Histological and histochemical aspects of normal neuroglia cells, in Biology of neuroglia (a cura di W. F. Windle), Springfield, Ill., 1958, pp. 141-161.
Lumsden, Ch. E., The problem of prevention and control of autoallergic inflammation in the nervous system, in ‟Zeitschrift für Immunitäts-und Allergieforschung", 1964, CXXVI, pp. 209-216.
Lumsden, Ch. E., The clinical immunology of multiple sclerosis, in Multiple sclerosis: a re-appraisal (a cura di D. McAlpine, Ch. E. Lumsden ed E. D. Acheson), Edinburgh 1965, pp. 372-379.
Lumsden, Ch. E., Nervous tissue in culture, in The structure and function of nervous tissue (a cura di G. H. Bourne), vol. I, London 1968, pp. 67-140.
Lumsden, CH. E., Pathogenetic mechanisms in the leucoencephalopathies, in anoxic-ischaemic processes, in disordes of the blood and in intoxications, in handbook of clinical neurology (a cura di P. J. Vinken e G. W. Bruyn), vol. IX, Amsterdam 1970, pp. 572-663.
Lumsden, Ch. E., The neuropathology of multiple sclerosis, in handbook of clinical neurology (a cura di P. J. Vinken e G. W. Bruyn), vol. IX, Amsterdam 1970, pp. 217-309.
Lumsden, Ch. E., The immunogenesis of the multiple sclerosis plaque, in ‟Brain research", 1971, XXVIII, pp. 365-390.
Lumsden, Ch. E., The clinical pathology of multiple sclerosis, in Multiple sclerosis: a re-appraisal (a cura di D. Mc Alpine, Ch. E. Lumsden ed E. D. Acheson), Edinburgh 19722, pp. 309-619.
Lumsden, Ch. E., The immunogenesis of the demyelinating plaque in multiple sclerosis, in ‟Revue médicale de Liège", 1972.
Lumsden, Ch. E., Robertson, D. M., Blight, R., New and unexpected findings on the chemical nature of the ECM-promoting factor(s) in bovine spinal cord, in Symposium on pathogenesis of demyelinating encephalomyelitis, Hamburg 1962, Stuttgart 1964, pp. 168-183.
Lumsden, Ch. E., Robertson, D. M., Blight, R., New and unexpected findings on the chemical nature of the ECM-promoting factor in bovine spinal cord, in ‟Zeitschrift für Immunitäts- und Allergieforschung", 1964, CXXVI, pp. 168-182.
Lumsden, Ch. E., Robertson, D. M., Blight, R., Chemical studies on experimental allergic encephalomyelitis. Peptide as the common denominator, in all encephalitogenic ‛antigens', in ‟Journal of neurochemistry", 1966, XIII, pp. 127-162.
McAlpine, D., Lumsden, Ch. E., Acheson, E. D. (a cura di), Multiple sclerosis: a re-appraisal, Edinburgh 19722.
Medavar, P. B., Immunity of homologous grafted skin. III., in ‟British journal of experimental pathology", 1948, XXIX, pp. 58-69.
Michaux, J. L., Les immunoglobulines des Bantous à l'état normal et pathologique, in ‟Annales de la Société Belge de Médecine Tropicale", 1966, XLVI, pp. 483-679.
Morgan, I. M., Allergic encephalomyelitis in monkeys in response to injectoin of normal monkey nervous tissue, in ‟Journal of experimental medicine", 1946, LXXXV, pp. 131-140.
Morrison, L. R., Disseminated encephalomyelitis experimentally poduced by the use of homologous antigen, in ‟Archivs of neurology and psychiatry", 1947, LVIII, pp. 391-416.
Mueller, P. S., Kies, M. W., Alvord, E. C., Shaw, C. M., Prevention of experimental allergic encephalomyelitis (EAE) by vitamin C deprivation, in ‟Journal of experimental medicine", 1962, CXV, pp. 329-338.
Nakao, A., Roboz Einstein, E., Chemical and immunochemical studies with a dialyzable encephalitogenic compund from bovine spinal cord, in ‟Annals of the New York Academy of Science", 1965, CXXII, pp. 171-181.
Oldstone, M. B. A., Dixon, F. J., Immuno histochemical study of allergic encephalomyelitis, in ‟American journal of pathology", 1968, LII, pp. 251-263.
Olitsky, P. K., Yager, R. H., Acute disseminated encephalomyelitis produced in albino mice, in ‟Proceedings of the Society for Experimental Biology and Medicine", 1949, LXX, pp. 600-601.
Olitsky, P. K., Yager, R. H., Experimental disseminated encephalomyelitis in white mice, in ‟Journal of experimental medicine", 1949, XC, pp. 213-224.
Palffy, G., Endröczi, E., Über die experimentelle allergische Encephalomyelitis beim Hund, in ‟Deutsche Zeitschrift für Nervenheilkunde", 1958, CLXXVII, pp. 405-416.
Paterson, P. Y., Transfer of allergic encephalomyelitis in rats by means of lymph node cells, in ‟Journal of experimental medicine", 1960, III, pp. 119-136.
Paterson, P. Y., Immunological mechanisms in nervous system inflammation, in ‟Research publications. Association for Research in Mental and Nervous Diseases", 1968, XLIV, pp. 21-29.
Paterson, P. Y., Brand, E. D., Production of experimental encephalomyelitis in a new host, the cat, in ‟Federation proceedings", 1957, XVI, pp. 428-429.
Paterson, P. Y., Hanson, M. A., Studies of cyclophosphamide suppresion of experimental allergic encephalomyelitis in Wistar rats, in ‟Journal of immunology", 1969, CIII, pp. 795-803.
Paterson, P. Y., Hanson, M. A., Gerner, E. W., Cyclophosphamide inhibition of experimental allergic encephalomyelitis in Wistar rats, in ‟Proceedings of the Society for Experimental Biology and Medicine", 1967, CXXIV, pp. 928-932.
Paterson, P. Y., Harwin, S. M., Suppression of allergic encephalomyelitis in rats by means of antibrain serum, in ‟Journal of experimental medicine", 1963, CXVII, pp. 755-774.
Pattison, I. H., Jebbett, J. N., Clinical and histopathological observations on Cuprizone toxicity and scrapie in mice, in ‟Research in veterinary science", 1971, XII, pp. 378-380.,
Pattison, I. H., Jebbett, J.N., Histopathological similarities between scrapie and Cuprizone toxicity in mice, in ‟Nature", 1971, CCXXX, pp. 115-117.
Pousines, Y., Brahic, J., L'encéphalomyélite expérimentale du cobaye consécutive à l'administration répétée par voie générale d'antigéne et d'adjuvants. Données histologiques, in ‟Compte rendu des séances de la Société de Biologie", 1950, CXLIV, pp. 1198-1199.
Poursines, Y., Tamalet, J., Résultats neurologiques chez le chien de l'administration prolongée d'antigèene et d'adjuvants suivant la technique de Kabat-Morgan. Lésions diffuses reactionelles gliales et dégéneratives neuronales, in ‟Compte rendu des séances de la Société de Biologie", 1954, CXLVIII, pp. 125-127.
Rall, D. P., Oppelt, W. W., Patlak, C. S., Extracellular space of brain as determined by diffusion of inulin from the ventricular system, in ‟Life science", 1962, I, pp. 43-48.
Ranzenhofer, E. R., Lipton, M. M., Steigman, A. J., Effect of homologous spinal cord in Freund's adjuvants on Cockerel comb, testicular and body growth, in ‟Proceedings of the Society for Experimental Biology and Medicine", 1958, XCIX, p. 280.
Rivers, T. M., Schwentker, F. F., Encephalomyelitis accompanied by myelin destruction experimentally produced in monkeys, in ‟Journal of experimental medicine", 1935, LXI, pp. 689-702.
Rives, T. M., Sprunt, D. H., Berry, G. P., Observations on attempts to produce acute disseminated encephalomyelitis in monkeys, in ‟Jounal of experimental medicine", 1933, LVIII, pp. 39-53.
Robertson, D. M., Lumsden, Ch., Blight, R., Dialysable peptide as the causative factor in experimental ‛allergic' encephalomyelitis, in ‟Nature", 1962, CXCVI, p. 1005.
Roboz Einstein, E., Csejtey, L., Davis, W. J., Protective action of the encephalitogen and other basic proteins in experimental allergic encephalomyelitis, in ‟Immunochemistry", 1968, V, pp. 567-575.
Roboz Einstein, E., Henderson, N., Preparation and properties of water-soluble proteins from bovine cord with ‛allergic' encephalomyelitic activity, in ‛Allergic' encephalomyelitis (a cura di M. W. Kies ed E. C. Alvord), Springfield, Ill., 1959, pp. 281-292.
Schneider, H.A., Genetic and nutritional aspects of experimental ‛allergic' encephalomyelitis, in ‛Allergic' encephalomyelitis (a cura di M. W. Kies ed E. C. Alvord), Springfield, Ill., 1959, pp. 130-136.
Seil, F. J., Falk, G. A., Kies, M. W., Alvord, E. C., The in vitro demyelinating activity of sera from guinea pigs sensitized with whole CNS and with purified encephalitogen, in ‟Experimental neurology", 1968, XXII, pp. 545-555.
Shaw, C. M., The effect of phytohemagglutinin on experimental allergic encephalitis in guinea pigs, in ‟Federation poceedings", 1968, XXVII, p. 473; in ‟Journal of immunology", 1969, CII, pp. 63-68.
Stone, S. H., Transfer of allergic encephalomyelitis by lymph node cells in inbred guinea pigs, in ‟Science", 1961, CXXXIV, pp. 619-620.
Stone, S. H., Lerner, E. M., Goode, J. H., Influence of homologous versus heterologous antigen on age, strain and sex dependency in autoimmune encephalomyelitis, in ‟Federation proceedings", 1967, XXVI, p. 531.
Svet-Moldavsky, G. J., Svet-Moldavskaya, I. A., Raffkina, L. I., Various types of acquired resistance to experimental ‛allergic' encephalomyelitis, in ‟Nature", 1959, CLXXXIV, pp. 1552-1553.
Tavolato, B., Le immunoglobuline liquorali: aspetti fisiopatologici, clinici e metodologici, in Atti del XIX Congresso nazionale della SIN, 1975, pp. 33-53.
Thomas, A. K., Patterson, P. Y., Smithwick, B., Acute disseminated encephalomyelitis following immunization with homologous brain extracts. I. Studies in the role of a circulating antibody in the production of the condition in dogs, in ‟Journal of experimental medicine", 1950, XCII, pp. 133-152.
Tibbling, G., Link, H., Ohman, S., Principles of albumin and IgG analyses in neurological disorders. I. Establishment of reference values, in ‟Scandinavian journal of clinical laboratory investigations", 1977, XXXVII, pp. 385-390.
Tourtellotte, W. W., Kokmen, E., Tavolato, B., Kruger, P. S., Fluorescent cell tracing in multiple sclerosis brain tissue, in ‟Transactions of the American Neurological Association", 1971, XCVI, pp. 318-320.
Toynbee, A. J., A study of history, vol. I, London 1955.
Uchmura, I., Shiraki, H., A contribution to the classification and the pathogenesis of demyelinating encephalomyelitis with special reference to the central nervous system lesions caused by preventive inoculation against rabies, in ‟Journal of neuropathology and experimental neurology", 1957, XVI, pp. 139-203.
Ulrich, J., Lardi, H., Multiple sclerosis: demyelination and myelination inhibition of organotypic tissue cultures of the spinal cord by sera of patients with multiple sclerosis and other neurological diseaes, in ‟Journal of neurology", 1978, CCXVIII, pp. 7-16.
Valdimarsson, H., Agnarsdottir, G., Lachmann, P. J., Cellular immunity in subacute sclerosing panencephalitis, in ‟Proceedings of the Royal Society of Medicine", 1974, LXVII, pp. 1125-1129.
Vitale, B., Allegretti, N., Matôsić, M., Influence of X-irradiation on experimental allergic encephalomyelitis in rats, in ‟Radiation research", 1966, XXVIII, pp. 727-734.
Vulpé, M., Hawkins, A., Rozdilsky, B., Permeability of cerebral blood vessels in experimental allergic encephalomyelitis studied by radioactive iodinated bovine albumin, in ‟Neurology", 1960, X, pp. 171-177.
Waksman, B. H., Allergic encephalomyelitis in rats and rabbits pretreated with nervous tissue, in ‟Journal of neuropathology and experimenta neurology", 1959, XVIII, pp. 397-417.
Waksman, B. H., Experimental allergic encephalomyelitis and the ‛autoallergic' diseases, in ‟International archives of allergy and applied immunology", 1959, XIV, (suppl.), pp. 1-87.
Waksman, B. H., Arbouys, S., Arnson, B. G., The use of specific ‛lymphocyte' antisera to inhibit hypersensitive reactions of the ‛delayed' type, in ‟Journal of experimental medicine", 1961, CXIV, pp. 997-1022.
Waksman, B. H., Morrison, L. R., Tuberculin type sensitivity to spinal cord antigen in rabbit with isoallergic encephalomyelitis, in ‟Journal of immunology", 1951, LXVI, pp. 421-444.
Westall, F. C., Robinson, A. B., Caccam, J., Jackson, J., Eylar, E. H., Essential chemical requirements for induction of allergic encephalomyelitis, in ‟Nature", 1971, CCXXIX, pp. 22-24.