
Polimeri
Polimeri
di Paolo Corradini e Lido Porri
Polimeri
sommario: 1. Introduzione. 2. Cenni storici. 3. Costituzione, configurazione e conformazione delle macromolecole. 4. Metodi di preparazione delle macromolecole. a) Policondensazione. b) Polimerizzazione radicalica. c) Polimerizzazione ionica. d) Polimerizzazione con catalizzatori a base di metalli di transizione. 5. Metodi fisici di determinazione strutturale dei polimeri. 6. Comportamento nell'uso dei polimeri in relazione alla struttura. a) Possibili stati fisici dei sistemi macromolecolari. b) Proprietà meccaniche dei materiali polimerici. c) I polimeri e l'ambiente. 7. Futuro della chimica macromolecolare. □ Bibliografia.
1. Introduzione
Le macromolecole sono molecole enormemente grandi, aventi cioè un peso molecolare molto più elevato dei più comuni composti organici, ma costituite, come questi ultimi, da strutture covalenti. Per il solo fatto di essere costituite da molecole ad altissimo peso molecolare, le sostanze macromolecolari (dette anche polimeri) presentano peculiari proprietà sia dal punto di vista chimico che fisico. I metodi usati per la loro preparazione e caratterizzazione differiscono inoltre sensibilmente da quelli usati per le sostanze a basso peso molecolare. Per queste ragioni la scienza delle macromolecole costituisce oggi un capitolo a sè nel quadro della chimica moderna. Non esiste un limite netto di divisione tra sostanze a basso e ad alto peso molecolare; si ammette tuttavia che sostanze con peso molecolare al di sopra di 104 circa rientrino nel campo delle macromolecole.
2. Cenni storici
I polimeri sono tra le prime sostanze con cui l'uomo è venuto a contatto fin dai primordi della sua storia; basti pensare che il legno, usato come materiale da costruzione, e alcuni alimenti base (carne, amido) sono essenzialmente costituiti da sostanze polimeriche. La vera natura di queste sostanze è stata chiarita tuttavia solo di recente, intorno agli anni trenta.
I primi studi sui polimeri risalgono alla seconda metà del secolo scorso, a opera soprattutto di studiosi delle sostanze naturali, che si occupavano, tra l'altro, della struttura chimica e del comportamento chimico-fisico della gomma, della cellulosa, delle proteine, e di chimici organici, che nel loro lavoro di sintesi e caratterizzazione di nuovi composti si imbattevano spesso in sostanze polimeriche. T. Graham nel 1861 coniò, per indicare i polimeri naturali, il termine ‛colloide' (cioè ‛simile alla colla') a causa dell'alta viscosità delle loro soluzioni e della lentezza di diffusione nei solventi (v. Graham, 1861). Anche i primi polimeri sintetici, successivamente preparati a partire da sostanze semplici chiamate monomeri, furono classificati come colloidi. Già prima della fine del secolo scorso furono effettuate determinazioni di peso molecolare su vari polimeri; si ottennero valori che oggi appaiono errati per difetto, ma che erano tuttavia notevolmente più alti di quelli dei comuni composti organici. I chimici di allora non presero però in considerazione l'esistenza di grandi molecole e considerarono le sostanze polimeriche come costituite da molecole a basso peso molecolare, in genere cicliche, unite in aggregati da forze secondarie di natura non ben definita. Così C. Harries, il quale nel 1904 aveva stabilito, attraverso 0ssidazione, che la gomma naturale era costituita da unità −CH2−C(CH3)=CH−CH2−, concluse che la molecola della gomma era costituita dal dimero ciclico dell'isoprene
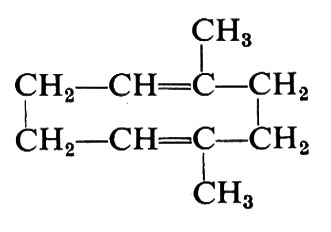
parecchi di questi cicli essendo combinati, mediante valenze secondarie, in aggregati più grandi (v. Harries, 1904). Analoghe formule cicliche costituite da due o più unità monomeriche furono proposte per il polistirene (v. Stobbe e Posnjak, 191 0), per il polibutadiene (v. Lebedev e Skavronskaya, 1911), per la cellulosa (v. Heuser, 1924). Per capire le ragioni per le quali i chimici di allora non presero in considerazione grandi strutture covalenti, che pur erano implicite nei fondamenti della chimica organica, occorre considerare che quelli erano tempi in cui la chimica organica otteneva rapidi e spettacolari successi attraverso sintesi, purificazione e caratterizzazione di composti a basso peso molecolare; l'esistenza di composti ad altissimo peso molecolare veniva in quei tempi semplicemente esclusa. È da tenere presente altresì che nell'ultimo decennio del secolo scorso fu posta particolarmente in evidenza l'associazione secondaria delle molecole; non sorprende quindi che a grandi strutture covalenti si preferissero aggregati di piccole molecole.
H. Staudinger già nel 1920 contestò le teorie correnti sulla natura delle sostanze polimeriche come composti di associazione tenuti insieme da valenze secondarie e propose per i polimeri sintetici dello stirene, della formaldeide e per la gomma naturale le seguenti formule a catena aperta, oggi accettate (v. Staudinger, 1920):

Egli attribuì le proprietà colloidali degli alti polimeri esclusivamente all'elevato peso molecolare delle loro molecole, che propose appunto di chiamare ‛macromolecole'.
Tali concetti non furono subito accettati e la polemica da essi suscitata continuò per tutti gli anni venti. Le teorie degli oppositori finirono però col cadere a una a una di fronte a nuove dimostrazioni sperimentali, quali quelle derivanti dall'indagine con i raggi X di vari polimeri cristallini e dai lavori di sintesi di H. W. Carothers. Questi preparò numerosi polimeri attraverso semplici reazioni organiche, che non lasciavano dubbi circa la struttura lineare delle macromolecole ottenute (v. Carothers, 1929).
Il chiarimento del concetto di macromolecola pose lo sviluppo della chimica macromolecolare su basi scientifiche. Il periodo che va dalla metà degli anni trenta agli inizi degli anni cinquanta vide uno sviluppo rapidissimo di questa scienza, in tutti i suoi aspetti. Furono poste le basi, teoriche e pratiche, per la determinazione dei pesi molecolari medi e fu chiarita la costituzione dei più importanti polimeri. Contemporaneamente veniva chiarito negli aspetti essenziali il meccanismo chimico della polimerizzazione, il che portò a distinguere tra polimerizzazione radicalica, cationica e anionica; venne poi affrontato lo studio della copolimerizzazione, e ne furono chiarite le leggi fondamentali. I problemi connessi con la conformazione delle macromolecole cominciarono a essere affrontati fin dal 1934 (E. Outh e H. F. Mark; W. Kuhn) e furono da allora poste le basi teoriche per affrontare i problemi relativi al comportamento dei polimeri in soluzione e all'elasticità della gomma. Vennero anche affrontati i problemi riguardanti i vari possibili stati fisici delle sostanze polimeriche e furono chiarite negli aspetti essenziali le relazioni tra proprietà e struttura.
Come risultato di questo intenso lavoro la chimica delle macromolecole era ormai, agli inizi degli anni cinquanta, su solide basi. Anche l'industria dei polimeri era in pieno sviluppo e produceva fibre (nailon, poliestere), gomme (gomma butile, gomma stirene-butadiene), varie resine termoplastiche (polietilene o politene, polivinildoruro) e termoindurenti (amminoplasti, resine epossidiche).
I metodi preparativi di cui allora disponeva la chimica macromolecolare non erano tuttavia in grado di fornire, partendo dai più comuni monomeri, come quelli vinilici e diolefinici, macromolecole a struttura ordinata. Dalla maggior parte di questi monomeri si ottenevano infatti polimeri amorfi o a bassissima cristallinità. Si era ben lontani dall'ordine strutturale dei polimeri naturali, tutti altamente cristallini. Un decisivo progresso in questo campo fu raggiunto nel 1954, anno che segna un'altra tappa fondamentale della storia della chimica macromolecolare. K. Ziegler, che da tempo lavorava sugli alluminioalchili (v. composti organometallici), aveva trovato nel 1953 che il prodotto della reazione tra Al(C2H5)3 e TiCl4 era in grado di catalizzare la polimerizzazione dell'etilene ad alto polimero lineare (v. Ziegler, 1964). Nel 1954 G. Natta e la sua scuola ottennero dalle principali α-olefine, con catalizzatori analoghi, una classe di polimeri altamente cristallini, che furono chiamati ‛isotattici', caratterizzati dalla presenza di lunghe sequenze di unità monomeriche aventi la stessa configurazione.
Successivamente Natta e la sua scuola ottennero polimeri stereoregolari di altro tipo, caratterizzati dalla presenza di lunghe sequenze di unità monomeriche aventi alternativamente configurazione opposta. Tali polimeri furono chiamati ‛sindiotattici'. Polimeri altamente regolari furono ottenuti anche dal butadiene e da altre diolefine (v. Natta, 1964).
Questi risultati stimolarono in tutto il mondo ricerche sulla polimerizzazione stereospecifica, che hanno portato alla realizzazione di numerosi polimeri stereoregolari, ottenuti non solo da monomeri idrocarburici lineari, vinilici e diolefinici, ma anche da cicloolefine, aldeidi, epossidi, monomeri acrilici.
L'inizio della polimerizzazione stereospecifica ha aperto un periodo nuovo nella chimica macromolecolare, il cui interesse non è solo scientifico, ma anche pratico: l'industria delle materie plastiche, degli elastomeri e delle fibre ne è stata infatti profondamente influenzata.
3. Costituzione, configurazione e conformazione delle macromolecole
Una macromolecola risulta dal concatenamento di centinaia o migliaia di unità chimiche polifunzionali (generalmente, bifunzionali). Nei polimeri sintetici queste unità chimiche prendono origine da molecole organiche semplici e reattive, dette ‛monomeri', attraverso una ‛reazione di polimerizzazione'. Nel seguito di questo capitolo riserveremo la nostra attenzione alle macromolecole risultanti dal concatenamento di unità chimiche bifunzionali, che danno luogo alle cosiddette ‛macromolecole lineari'. Per esempio, in una reazione di polimerizzazione, le molecole del monomero etilene, CH2=CH2, danno luogo principalmente alla unità chimica bifunzionale −CH2−CH2−.
Una struttura chimica ‛ideale' di una macromolecola di polietilene può essere rappresentata nel modo seguente:
R−(CH2−CH2−)R′
dove x è dell'ordine di qualche migliaio e R e R′, ai terminali della catena lineare di atomi di carbonio, sono due radicali monofunzionali, per esempio −C2H5 e −H.
Dalla discussione che segue apparirà chiaro che tutte le informazioni sulla struttura chimica di materiali polimerici - a cominciare dai più semplici - non possono che avere un carattere statistico, a differenza da quanto avviene per le sostanze chimiche a basso peso molecolare, in cui per definizione sono presenti molecole tutte uguali. Cercheremo perciò in quanto segue di mettere in evidenza questo aspetto peculiare della chimica macromolecolare, attraverso un confronto tra la descrizione strutturale che si può fare di sostanze caratterizzate da un solo tipo di molecole (trattate tradizionalmente in chimica organica) e quella che si può fare di sostanze caratterizzabili solo da grandezze strutturali medie (come quelle trattate nella chimica macromolecolare). Abbiamo ricordato che il decollo della chimica macromolecolare come scienza è stato ritardato per un lungo periodo proprio dal mancato riconoscimento di questa peculiarità.
La ‛costituzione' di un composto organico specifica quali atomi in una molecola sono legati l'uno all'altro, e con che tipo di legami, senza considerare la loro disposizione spaziale. Così la costituzione di un composto semplice quale l'etilene è specificata simbolicamente scrivendo:
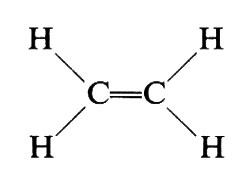
(ogni molecola è composta da due atomi di carbonio e quattro di idrogeno; i due atomi di carbonio sono legati da un doppio legame, ecc.).
Dal punto di vista della costituzione, un polimero può essere considerato una sostanza le cui molecole possono essere descritte da molte unità costitutive (di una o più specie) ripetutamente legate tra loro. Le singole macromolecole possono differire tra loro nel peso molecolare e quindi nel numero di unità costitutive in esse contenute, nonché, quando queste sono di più specie, nella successione delle unità costitutive e nel loro rapporto percentuale. Le informazioni sulla costituzione che i metodi fisici di determinazione strutturale ci possono fornire sui polimeri hanno perciò sempre un carattere eminentemente statistico: esse sono tanto migliori, quanto più includono correlazioni di rango più elevato.
Consideriamo le possibili costituzioni delle macromolecole di un polimero derivato da un monomero vinilico, di formula generale:
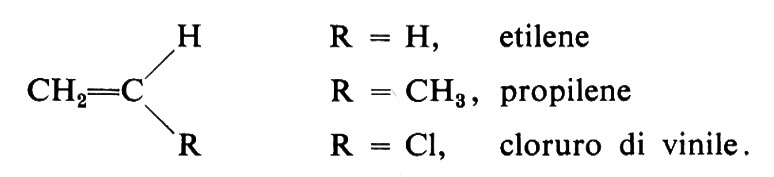
Nell'addizionarsi alla catena del polimero durante la reazione di polimerizzazione, le molecole del monomero possono assumere, in linea di principio, se R ≠ H, due costituzioni non equivalenti rispetto alla direzione di accrescimento:

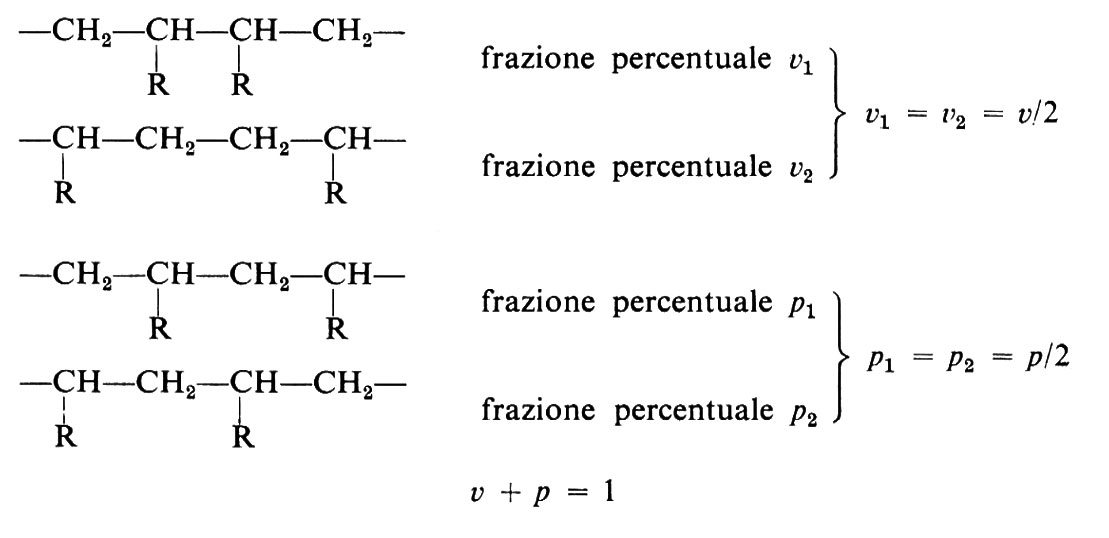
Se unità (a) o (b) si addizionassero a caso, le macromolecole risultanti avrebbero costituzioni irregolari del tipo:
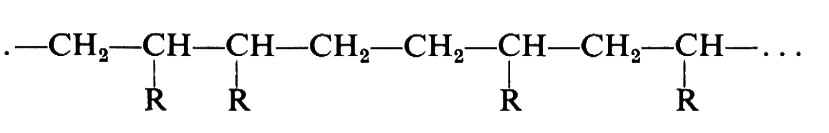
È stato dimostrato tuttavia che la maggior parte delle polimerizzazioni di monomeri vinilici avvengono in modo prevalentemente regolare, per quanto riguarda la successione di unità costitutive, dando luogo a sequenze testa-coda del tipo:
Idealmente, un polimero si dice regolare se le sue molecole sono formate da unità costitutive di una sola specie, in una sola sistemazione sequenziale. Le deviazioni dalla regolarità possono essere analizzate in termini statistici, esaminando la ripartizione percentuale dei vari tipi di coppie, triplette, quadruplette, ecc. di unità costitutive in una catena. Con riferimento alle sole coppie di unità costitutive del tipo sopra descritto, si hanno le possibilità:
Mentre la maggior parte dei polimeri vinilici presenta un solo tipo di costituzione, essenzialmente regolare, nel caso dei polimeri dienici sono possibili due differenti tipi di reazioni di addizione.
Per il più semplice monomero dienico, il butadiene, l'addizione può aver luogo in 1,2 o in 1,4:
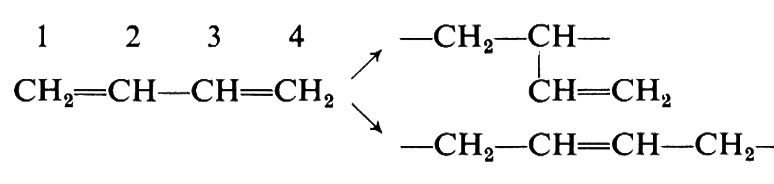
Avremo perciò polimeri regolari a concatenamento 1,2 (testa-coda) oppure a concatenamento 1,4 e polimeri irregolari, in cui unità 1,2 e 1,4 si succedono in differenti proporzioni.
Già in un caso come quello dei polimeri del butadiene l'analisi statistica della costituzione delle macromolecole diventa assai complessa; nel caso di polimeri irregolari del butadiene gli studi finora effettuati non vanno molto oltre la determinazione del rapporto percentuale tra unità 1,2 e unità 1,4 nella catena. Infine, è opportuno notare che, accanto a polimeri caratterizzabili in base a più unità costitutive omogeneamente distribuite lungo la catena, possono essere preparati polimeri in cui più unità costitutive sono segregate in porzioni differenti della stessa macromolecola: tali polimeri vengono chiamati ‛polimeri a blocchi'.
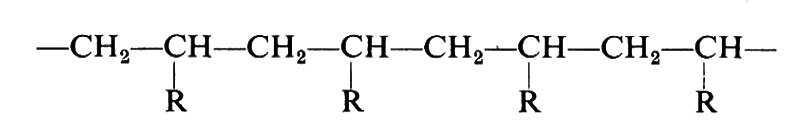
Si vede chiaramente come il problema di determinare la costituzione chimica delle molecole di un polimero non possa che avere una risposta statistica. Il problema è inoltre complicato dal fatto che non sempre si ha a che fare con macromolecole perfettamente lineari. In dipendenza dal metodo di preparazione e dalle condizioni sperimentali possono infatti formarsi macromolecole contenenti ramificazioni laterali corte o lunghe, come indicato nella fig. 1.
Quando un polimero è formato da macromolecole costituite da unità derivanti da due differenti monomeri (per es., A e B), esso viene più propriamente detto ‛copolimero'. Secondo la distribuzione delle unità −A− e −B− nella macromolecola si possono avere vari tipi di copolimeri, come sotto indicato:
1) copolimeri a distribuzione statistica
−A−B−B−B−A−B−A−A−;
2) copolimeri a blocchi
−A−A−A−A−A−A−B−B−B−B−B−B−A−A− A−A−A−;
3) copolimeri alternati
−A−B−A−B−A−B−A−H−A−B−A−B−;
4) copolimeri innestati
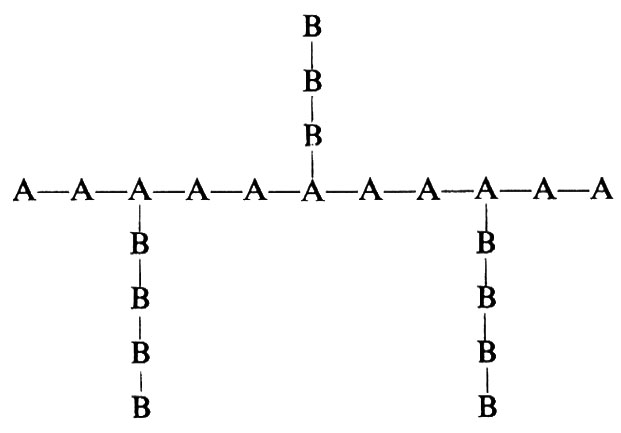
Appare chiaro che il problema di determinare la costituzione di un copolimero è ancora più complesso di quello che si presenta per un polimero derivante da un solo monomero (‛omopolimero').
La necessità d'introdurre grandezze statistiche per caratterizzare un polimero (omo- o copolimero) risulta ancora più evidente quando si passi a valutarne il peso molecolare. A questo riguardo, supponiamo di essere capaci di estrarre dalla massa di un polimero due molecole diverse; indipendentemente dalle differenze nei gruppi terminali e dalle irregolarità di costituzione lungo la catena, sempre presenti, in maggiore o minore misura, in un polimero reale, quasi certamente il grado di polimerizzazione, cioè il numero di unità monomeriche presenti in ciascuna delle due molecole, sarà differente.
Per grado di polimerizzazione di un polimero s'intenderà perciò un'altra importante grandezza statistica utile alla caratterizzazione della costituzione di un polimero: esso rappresenterà un qualche tipo di media (in numero, o in peso, per esempio) dei gradi di polimerizzazione di ogni singola molecola.
Quando le unità costitutive di un polimero lineare hanno tutte lo stesso peso formula, si preferisce far riferimento, anzichè al grado di polimerizzazione, al peso molecolare, che è il prodotto del grado di polimerizzazione per il peso formula dell'unità monomerica.
Con metodi fisici diversi, si ottengono pesi molecolari M, per uno stesso polimero lineare, mediati in modo diverso. Data una certa distribuzione di pesi molecolari M delle singole macromolecole N(M), definita in modo tale che
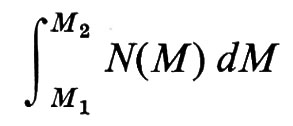
è la frazione in numero di catene con pesi molecolari nell'intervallo
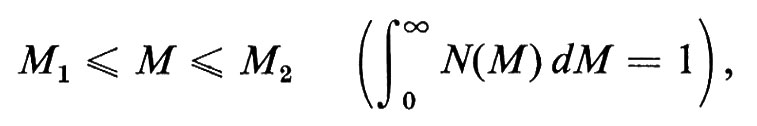
si definisce peso molecolare medio in numero
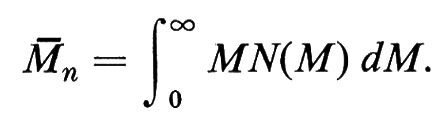
Se definiamo la distribuzione di pesi molecolari M delle singole macromolecole con la funzione W(M), tale che
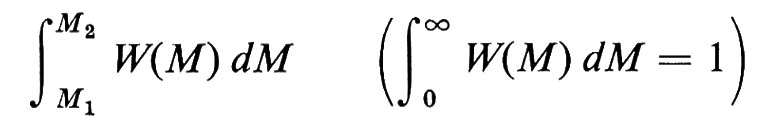
è la frazione ‛in peso' di catene con pesi molecolari nell'intervallo M1 ≤ M ≤ M2, si ha per il peso molecolare medio in peso
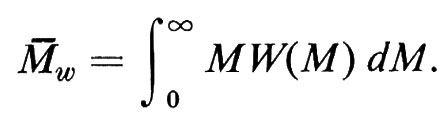
M-n e M-w possono coincidere solo nel caso ideale di un polimero monodisperso, cioè costituito da macromolecole tutte eguali; in tutti gli altri casi sarà Mw > Mn (le macromolecole più grandi influiscono di più sulla media in peso che sulla media in numero); la frazione (M-w − M-n)/M-n può essere presa come misura del grado di ‛polidispersità' del polimero, anche se non è sufficiente a definire la forma di W(M).
Nello studio dei composti organici, il passo successivo alla determinazione della costituzione è la determinazione della configurazione.
La ‛configurazione' di un composto (v. Morawetz, 1965) specifica la disposizione spaziale dei legami in una molecola (di data costituzione) senza riguardo alla molteplicità delle disposizioni spaziali che possono sorgere per rotazione intorno a legami semplici. Per i polimeri, converrà fare riferimento, per semplicità, a polimeri regolari. In un polimero regolare, differenti configurazioni possono prendere origine dall'esistenza di ‛centri di stereoisomeria'.
I più importanti centri di stereoisomeria che si possono trovare lungo la catena di un polimero sono: 1) doppi legami, che possono avere una configurazione cis o trans; 2) centri stereoisomerici tetraedrici, cioè atomi di carbonio lungo la catena legati a due differenti sostituenti R e R′.
Cominciamo a esaminare la stereoisomeria da doppi legami. Data, per esempio, l'unità costituzionale:
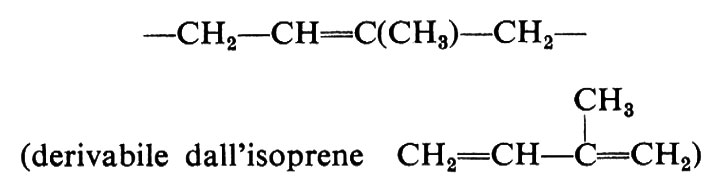
a essa possono corrispondere due diverse configurazioni a seconda che i metileni adiacenti al doppio legame abbiano una disposizione spaziale cis o trans:
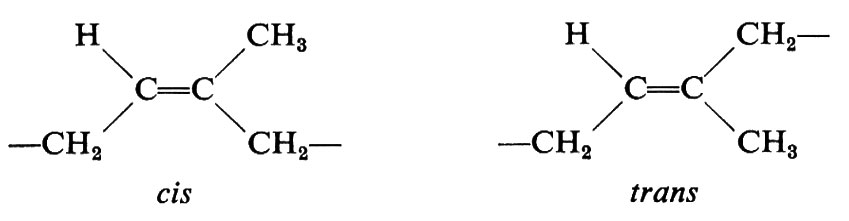
Non è affatto detto, naturalmente, che un polimero dell'isoprene, a concatenamento regolare 1,4 del tipo:
−CH2−CH=C(CH3)−CH2−CH2−CH=C(CH3)− ...
debba necessariamente avere una successione di configurazioni anch'essa regolare.
Se la successione di configurazioni è regolare, il polimero si dice ‛stereoregolare' e il processo di polimerizzazione attraverso il quale si può ottenere ‛stereospecifico'. Processi di polimerizzazione stereospecifica sono stati sviluppati soltanto negli ultimi due decenni, mentre polimeri naturali stereoregolari erano già ben noti in precedenza. Nel caso del polusoprene, a cui si è sopra accennato, le più semplici successioni regolari di configurazioni che si possono concepire corrispondono a doppi legami tutti cis, o tutti trans. In un caso ideale del primo tipo, il polimero si dirà ‛cistattico', nel secondo caso ‛transtattico'.
I polimeri naturali dell'isoprene - la gomma naturale e la guttaperca - sono stereoregolari e corrispondono rispettivamente al polimero cistattico e transtattico dell'isoprene.
Consideriamo ora il caso generico di un polimero vinilico di costituzione regolare:
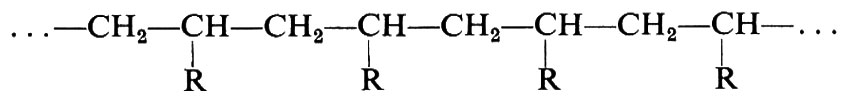
Per discutere le possibili configurazioni di una siffatta catena, conviene rendersi indipendenti dalla molteplicità delle disposizioni spaziali che possono sorgere per rotazione intorno ai legami semplici, facendo riferimento sempre alla conformazione zig-zag planare della catena stessa; rispetto a una tale conformazione, due gruppi R adiacenti possono trovarsi da una stessa parte rispetto al piano dello zig-zag:
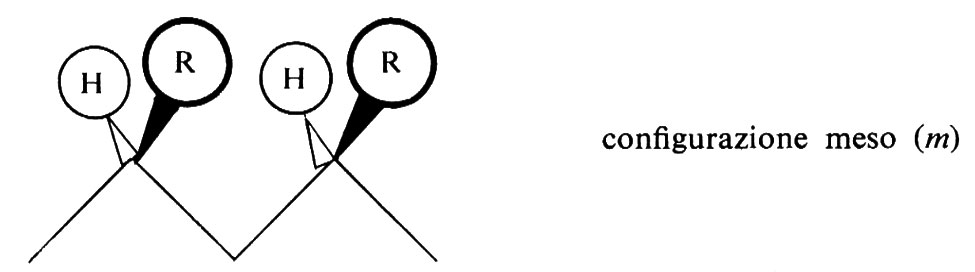
oppure da parti opposte rispetto a esso:
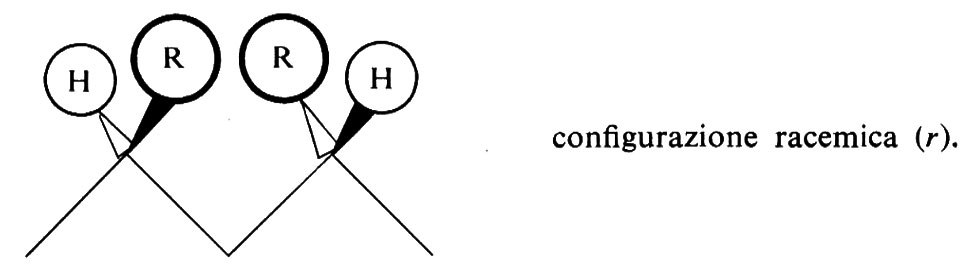
Una configurazione relativa del primo tipo si dice meso (abbreviato m); una configurazione relativa del secondo tipo si dice racemica (abbreviato r).
Le più semplici configurazioni stereoregolari che si possono concepire per un polimero vinilico corrispondono a successioni di configurazioni tutte m (in un caso ideale di questo tipo il polimero si dirà ‛isotattico') oppure a successioni di configurazioni tutte r (in un caso ideale di questo tipo il polimero si dirà ‛sindiotattico'). Un polimero vinilico in cui si succedano casualmente, lungo la catena delle macromolecole di cui è formato, posizionamenti m e posizionamenti r si dirà ‛atattico'.
Il riconoscimento del tipo di ordinamento delle configurazioni ('tassia') nelle macromolecole di un polimero non è confinato alle sole coppie di unità costitutive adiacenti: un polimero col 50% di unità m e il 50% di unità r potrebbe essere al limite costituito da macromolecole, ognuna delle quali formata da due blocchi, uno con unità tutte m e l'altro con unità tutte r. Oltre alla distribuzione delle configurazioni delle coppie (o diadi, meso o racemiche) è pertanto necessario in generale caratterizzare un polimero vinilico anche in funzione della distribuzione delle triadi (mm, isotattica; mr o rm eterotattica; rr, sindiotattica), delle tetradi, delle pentadi, ecc., come vedremo nel cap. 4.
È evidente che le precedenti definizioni di polimero isotattico, sindiotattico e atattico corrispondono a idealizzazioni, cui non potrebbe nemmeno corrispondere un'osservabilità diretta a mezzo di esperimenti (si pensi, per esempio, all'estrema difficoltà di rilevare la presenza di frazioni minori dell' 1% di unità r in catene con una prevalenza di oltre il 99% di posizionamenti m).
Nella pratica si parla perciò di polimero isotattico o sindiotattico quando tutte le sue proprietà tendono al limite dei valori attendibili per un polimero idealmente isotattico o sindiotattico; se del caso, invece, si parlerà di polimeri prevalentemente isotattici o prevalentemente sindiotattici, di polimeri praticamente atattici, ecc.
Le proprietà dei polimeri, sia in soluzione sia nei vari stati fisici di massa in cui essi possono presentarsi, sono legate strettamente alle ‛conformazioni' che le macromolecole possono assumere. I più notevoli progressi realizzati in questi ultimi anni nella comprensione di tali proprietà e le concrete speranze future di ulteriori progressi sono fondati proprio sugli studi sempre più dettagliati che si vengono conducendo sulle conformazioni energeticamente accessibili alle macromolecole di un polimero.
Tali studi vengono condotti da un lato in funzione della costituzione e della configurazione delle macromolecole stesse; dall'altro, a parità di costituzione e di configurazione, in funzione di variabili fisiche o chimiche esterne, quali temperatura, forze applicate, concentrazione di solventi o reagenti.
Riteniamo opportuno perciò accennare, sia pur brevemente, alle conformazioni che possono essere assunte dalle macromolecole, con particolare riguardo al caso più semplice, che corrisponde allo stato cristallino.
Per conformazioni intendiamo le differenti disposizioni spaziali degli atomi in una molecola di data costituzione e configurazione, che si possono realizzare per rotazione intorno a legami semplici. In prima approssimazione, infatti, dell'insieme di coordinate interne che possono essere prese per definire la forma di una molecola, e cioè lunghezze di legame, angoli di valenza e angoli diedri definiti da quaterne di atomi concatenati (v. fig. 2), sono proprio questi ultimi che, in generale, possono assumere un ampio campo di valori.
Gli angoli di valenza, e soprattutto le lunghezze di legame, rimangono invece praticamente costanti nel passare da una conformazione all'altra. L'angolo diedro definito da una quaterna di atomi concatenati, A, B, C, D, e precisamente l'angolo tra i piani ABC e BCD, se BC è un legame semplice, viene anche detto angolo di rotazione interna, relativo al legame BC (e può assumere le designazioni illustrate nella fig. 3). Siccome la rotazione attorno a doppi legami è normalmente impedita, l'assegnamento della conformazione a una macromolecola corrisponde alla specificazione del valore assunto dagli angoli di rotazione interna relativi ai legami semplici in essa presenti.
La conformazione molecolare, per un polimero non cristallino, può essere definita in generale solo in termini statistici, e questo anche nel caso che la costituzione e la configurazione siano regolari. Si dice in tal caso che le macromolecole costituiscono dei gomitoli statistici. Nel caso invece di macromolecole proteiche, aventi la costituzione
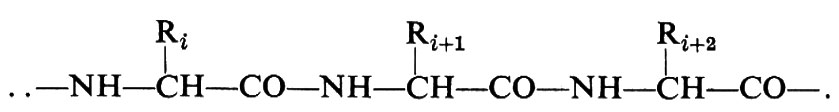
con gli Ri in una ben precisa sequenza, peso molecolare costante e con gli atomi di carbonio tetraedrici della catena aventi tutti la stessa configurazione, le macromolecole possono assumere anche delle precise conformazioni compatte, globulari, in cui generalmente i gruppi R più idrofili risultano all'esterno e i gruppi R più idrofobi sono all'interno del globulo stesso (struttura terziaria delle proteine). Allo stato cristallino, e con l'eccezione delle proteine globulari cristalline, la conformazione della catena di un polimero è definita invece da una successione di unità strutturali equivalenti, che coincidono di norma, ma non necessariamente, con una unità monomerica. Unità strutturali successive occupano posizioni geometricamente (non necessariamente cristallograficamente) equivalenti nei riguardi di un asse di ripetizione cristallografico. Questo principio è stato chiamato da Natta e Corradini (v., 1960) principio di equivalenza.
Per quanto riguarda la previsione di tale conformazione, si è rivelato di grande importanza euristica il principio che la conformazione della catena in un cristallo deve corrispondere da vicino a un minimo dell'energia interna conformazionale (la parte cioè dell'inerzia interna dipendente dalla sola conformazione), che potrebbe essere assunta da una catena isolata, soggetta alle restrizioni imposte dal principio di equivalenza.
Il principio di equivalenza è una conseguenza diretta dell'ordine tridimensionale associato allo stato cristallino; esso implica a sua volta, almeno idealmente, la regolarità della costituzione e della configurazione dei polimeri capaci di cristallizzare. Le eccezioni al postulato di equivalenza possono giustificarsi nel caso che unità costituzionali, configurazionali o conformazionali diverse abbiano un analogo ingombro sterico; si parla allora di isomorfismo costituzionale, configurazionale o conformazionale rispettivamente, così come si parla di isomorfismo tra ioni nella chimica inorganica, quando questi possono vicariarsi tra loro nello stesso cristallo.
I fattori energetici che determinano la conformazione della catena di un polimero cristallino sono essenzialmente due: 1) l'esistenza di un potenziale torsionale, che favorisce un orientamento dei legami semplici per un sistema C−C−C−C in conformazioni gauche (destra o sinistra, v. fig. 4A) oppure trans; mentre per un sistema C−C−C=C favorisce conformazioni skew o ‛anticlinali' (destra o sinistra) oppure cis (v. fig. 4B). Tali conformazioni saranno indicate nel seguito coi simboli G+, G-, T e S+, S-, C rispettivamente; 2) le interazioni tra atomi non direttamente legati della stessa macromolecola, che possono essere repulsive (a piccola distanza) o debolmente attrattive (forze di van der Waals). Queste interazioni possono comportare deviazioni rispetto alle conformazioni corrispondenti ai minimi dell'energia torsionale.
I principi suesposti giustificano completamente le conformazioni assunte dalle macromolecole dei polimeri di cui è stata determinata la struttura allo stato cristallino, e sono stati quantitativizzati di recente in diagrammi che forniscono l'energia conformazionale di una macromolecola di data costituzione e configurazione in funzione degli angoli di rotazione interna. Oltre che a polimeri sintetici, si possono applicare analoghi principi a biopolimeri. Tali applicazioni hanno portato negli anni cinquanta L. Pauling (v. Pauling e Corey, 1951; v. Pauling, 1955) alla scoperta della conformazione elicoidale dei poliamminoacidi (v. fig. 5) e delle proteine (v. proteine), nonché Crick e Watson (v. Watson e Crick, 1953; v. Crick e Watson, 1956; v. Crick, 1963; v. Watson, 1963) alla scoperta della doppia elica del DNA (v. acidi nucleici).
Per i polipropileni stereoregolari si prevede, in base ai principi suesposti, una struttura elicoidale caratterizzata da una successione di conformazioni (TG+)3 o (G-T)3 equivalenti per il polipropilene isotattico (v. fig. 6A). La ripetizione identica si ha dopo tre unità monomeriche, in un passo di elica. Per il polipropilene sindiotattico si sono previste tre conformazioni isoenergetiche, corrispondenti a successioni (TTG+G+)2 o (G-G-TT)2 (equivalenti) oppure TTTT. Le conformazioni (TTG+G+)2 e (G-G-TT)2 (v. fig. 6B) sono quelle normalmente presenti allo stato cristallino; la conformazione TTTT è stata trovata, dopo che ne era stata stabilita la possibilità energetica, in una forma polimorfa ottenibile per stiro a bassa temperatura.
Le conformazioni allo stato cristallino dei quattro polimeri stereoregolari del butadiene sono presentate nella fig. 7.
Un gran numero di ricerche teoriche, infine, sono state fatte in questi ultimi anni per valutare la distribuzione statistica delle conformazioni della catena di un polimero, in soluzione o allo stato fuso, in funzione della costituzione e della configurazione. Per un polimero semplice come il polietilene, che allo stato cristallino presenta una successione di conformazioni trans, si vede che il modello in soluzione è quello di un ‛gomitolo statistico', in cui conformazioni trans dei legami sono intervallate da legami gauche destri e gauche sinistri; stati rotazionali di legami successivi risultano interdipendenti.
I risultati dei calcoli, e la bontà quindi dei parametri energetici usati, possono essere confrontati, per questo e altri polimeri, con misure sperimentali, basate sulla diffusione della luce o sulla viscosità di soluzioni, che aumenta all'aumentare dell'ingombro del gomitolo statistico.
Per gli argomenti trattati in questo capitolo si vedano Birshstein e Ptitsyn (v., 1963), Corradini (v., 1968), Flory (v., 1969), Volkenstein (v., 1963).
4. Metodi di preparazione delle macromolecole
La sintesi di macromolecole avviene attraverso la combinazione (o polimerizzazione) di molecole più piccole dette ‛monomeri'. Un numero vastissimo di sostanze può funzionare da monomero, possiede cioè una funzionalità chimica adatta a fornire unità monomeriche almeno bivalenti. In pratica però sono stati sviluppati solo quei polimeri che impiegano monomeri facilmente accessibili.
Si conoscono vari metodi di polimerizzazione. Essi vengono generalmente classificati in due grandi categorie: polimerizzazione per condensazione e polimerizzazione per addizione.
Nella polimerizzazione per condensazione la formazione del polimero avviene per stadi, e i vari intermedi (dimero, trimero, ecc.) hanno la stessa reattività del monomero. Molto spesso viene liberata una piccola molecola, di solito acqua, a ogni stadio della reazione.
Nella polimerizzazione per addizione gli intermedi sono radicali o ioni oppure sono specie legate, a un'estremità, a un atomo di un metallo di transizione. La crescita della macromolecola avviene esclusivamente per addizione del monomero alla catena crescente ed è molto spesso estremamente rapida. Appartiene a questa seconda categoria la polimerizzazione di tutti i monomeri di tipo vinilico e delle diolefine. Sulla base della natura della specie intermedia e del meccanismo chimico, la polimerizzazione per addizione può essere suddivisa nelle seguenti classi: polimerizzazione radicalica, polimerizzazione ionica, polimerizzazione con catalizzatori a base di metalli di transizione (v. Flory, 1953; v. Lenz, 1967; v. Ham, 1969; v. Kirk e Othmer, 1965).
a) Policondensazione
Questo tipo di polimerizzazione richiede che nelle molecole del monomero siano presenti almeno due gruppi funzionali, capaci di reagire tra loro. Si consideri, per es., un idrossiacido HO−R−COOH. Poiché i gruppi carbossilici, −COOH, sono in grado di reagire con i gruppi idrossilici, −OH, per formare un legame estereo, dall'idrossiacido sopra indicato sarà possibile ottenere un poliestere lineare, secondo l'equazione:
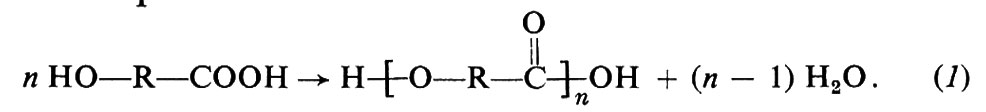
Invece di un solo tipo di monomero contenente due gruppi funzionali diversi, si possono usare due tipi di monomero, ciascuno contenente due eguali gruppi funzionali. Per es., da un glicol, HO−R′−OH, e da un diacido, HOOC−R″−COOH, si otterrà ugualmente una macromolecola lineare:
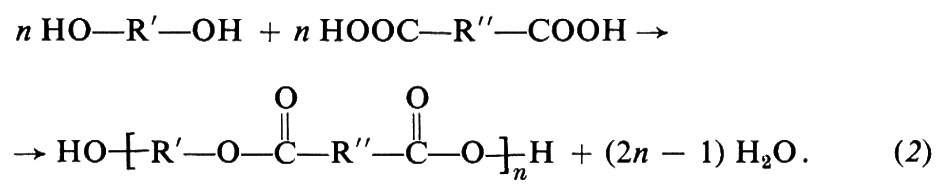
In questi esempi, R, R′, R″ stanno a indicare generici gruppi organici bivalenti, come −(CH2)x−, −C6H4−, −C6H4−CH2−C6H4−, ecc. I polimeri ottenuti secondo le reazioni (1) e (2), in cui le unità monomeriche sono legate tra loro da gruppi esterei,
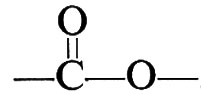
si chiamano comunemente ‛poliesteri'. Altri gruppi mutuamente reattivi, oltre ai gruppi −OH e −COOH sopracitati, sono i gruppi −NH2 e −COOH, che danno luogo a ‛poliammidi'; i gruppi −COOH e −COOH, che danno luogo a ‛polianidridi'; i gruppi −OH e −N=C=O, che danno luogo a ‛poliuretani'.
Quando in una policondensazione una delle molecole reagenti contiene più di due gruppi funzionali, si ha formazione di una struttura tridimensionale. Si ottengono così prodotti infusibili, chiamati comunemente ‛resine termoindurenti'. Così dalla reazione tra glicerolo, CH2OH−CHOH−CH2OH, e acido orto-ftalico,
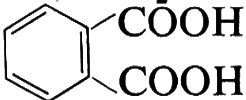
si ottiene un prodotto tridimensionale (resina glicero-ftalica o, in generale, ‛alchidica', da alcool + acido) che può essere schematicamente rappresentata come segue:
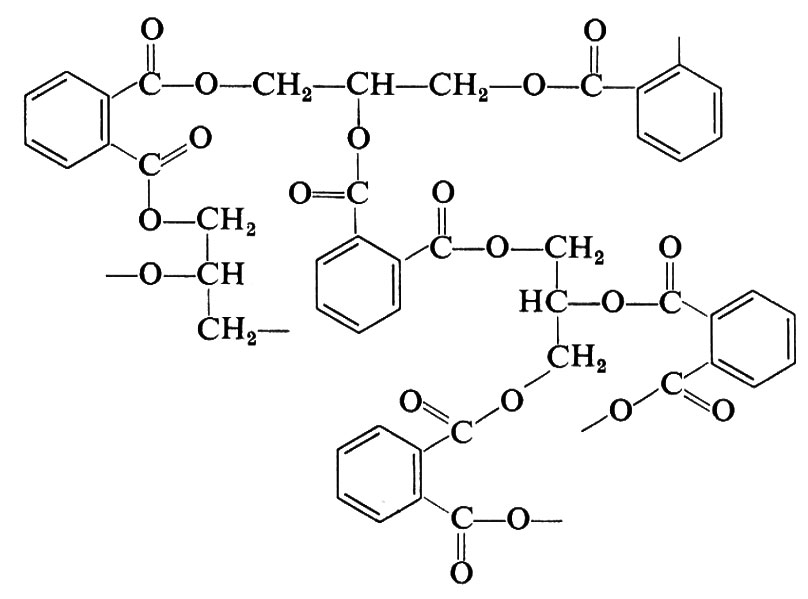
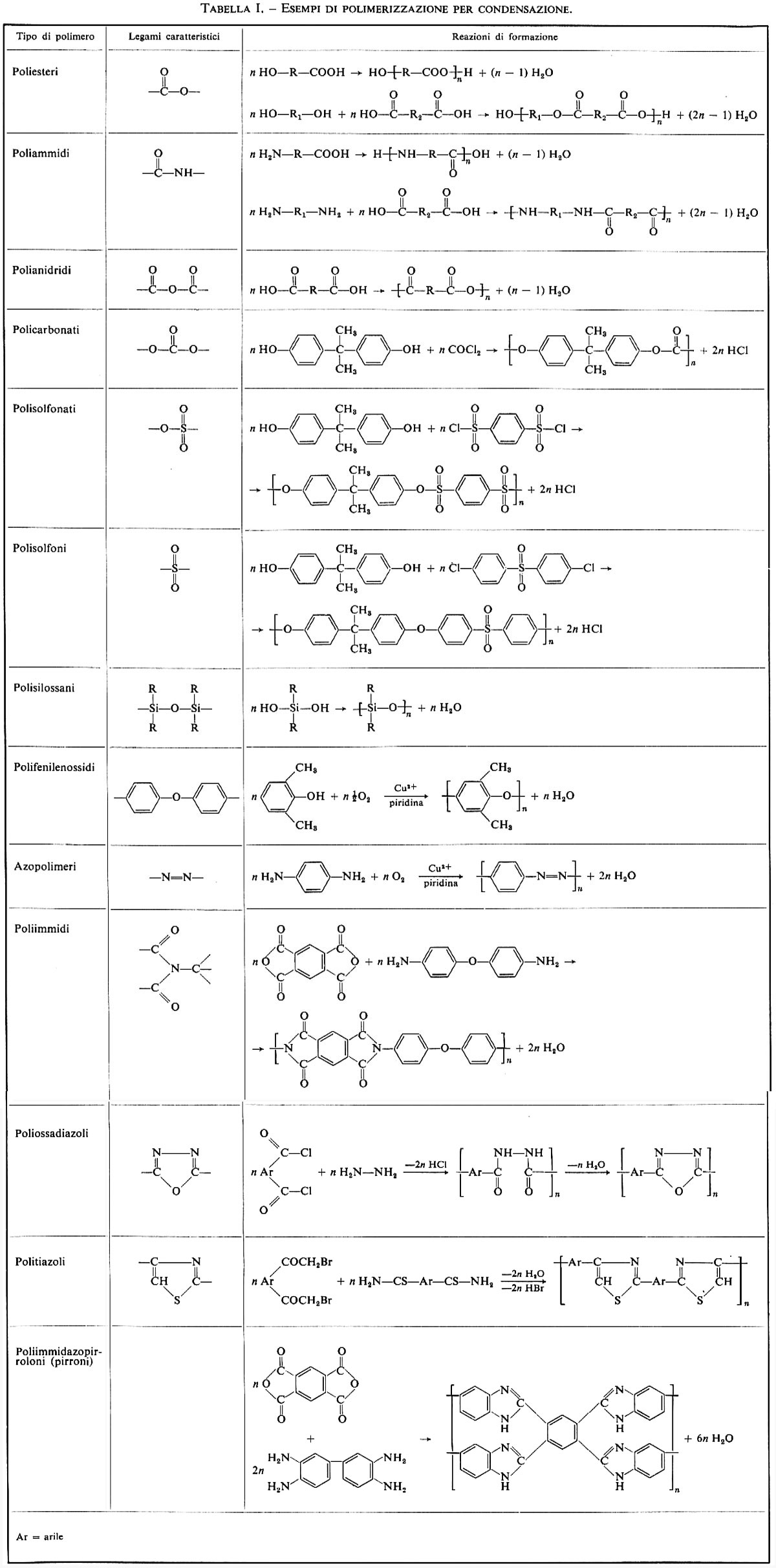
Esempi dei più comuni polimeri ottenuti per policondensazione sono riportati nella tab. I.
Nelle policondensazioni lineari la crescita della macromolecola avviene per stadi. Il monomero reagisce con se stesso per dare un dimero, questo può reagire con un altro monomero per dare un trimero, o con un altro dimero per dare un tetramero. Due catene polimeriche, comunque lunghe, possono reagire tra di loro per dare una nuova catena che è la somma delle due. In un processo di questo tipo la concentrazione del monomero decresce velocemente, fino a essere praticamente nulla poco dopo l'inizio della polimerizzazione. Il peso molecolare medio cresce con il grado di avanzamento della reazione.
L'andamento delle policondensazioni tridimensionali è più complesso. Nella prima fase della reazione si ha formazione di un numero relativamente alto di molecole a peso molecolare non elevato; in questa fase il liquido di reazione diventa gradualmente più viscoso. A un certo punto, detto ‛punto di gelo', si ha il fenomeno della ‛gelazione', cioè la rapida trasformazione del mezzo di reazione da un liquido viscoso, ma solubile, e che può assumere la forma desiderata, in una massa densa, insolubile, che mantiene la sua forma. La gelazione è dovuta al fatto che un numero relativamente grande di molecole a basso peso molecolare si trasforma, per un ulteriore piccolo avanzamento della reazione, praticamente in un'unica macromolecola tridimensionale. Nella pratica industriale, tuttavia, si arriva alla struttura tridimensionale per gradi: si forma dapprima un prepolimero, che può essere essenzialmente lineare o anche ramificato, ma che può assumere la forma che si desidera, e si passa poi alla reazione di ‛reticolazione', cioè alla formazione della struttura tridimensionale.
La polimerizzazione per condensazione è alla base di alcuni importantissimi processi industriali per preparare fibre, film, resine termoplastiche e resine termoindurenti. Per condensazione tra acido adipico ed esametilendiammina si ottiene il tipo di nailon comunemente detto 6-6, per il fatto che deriva da due monomeri entrambi con 6 atomi di carbonio. Da acido tereftalico e glicol etilenico si ha un poliestere che va commercialmente sotto vari nomi, usato sia come fibra che come film. Tra le resine termoplastiche citiamo i policarbonati, i polisolfoni, i polisolfonati.
Si preparano vari tipi di resine termoindurenti, le principali delle quali sono le seguenti: resine fenoliche, che si ottengono da fenolo e formaldeide; resine amminiche, da formaldeide e melamina o urea; resine epossidiche, da bisfenolo A ed epicloridrina; poliuretani, da dusocianati e glicoli o diammine; resine alchidiche, da un acido dibasico (acido ftalico, maleico, adipico) e un alcool poliidrossilico (glicerolo, pentaeritritolo, sorbitolo, ecc.).
Il metodo di polimerizzazione per condensazione è forse il più versatile tra i vari metodi che si conoscono, in quanto permette, attraverso opportuna scelta dei monomeri, di costruire macromolecole aventi la struttura desiderata per ottenere materiali aventi particolari proprietà. A partire dalla fine degli anni cinquanta si è osservato un rinnovato interesse verso questo metodo di polimerizzazione, in relazione soprattutto con la ricerca di sostanze polimeriche aventi elevate proprietà meccaniche anche ad alte temperature, secondo le richieste dell'industria aerospaziale. Sono da ricordare tra i vari polimeri termoresistenti finora preparati le poliimmidi, i poliossadiazoli, i politiazoli, i polifenilenossidi, gli azopolimeri. La tab. I riporta le reazioni base di preparazione di questi tipi di polimeri.
Attraverso policondensazione sono stati ottenuti anche vari polimeri cosiddetti a scala, o a nastro, per il fatto che le loro macromolecole sono costituite da due catene lineari collegate tra di loro da piccole catene trasversali. Citiamo come esempio i poliimmidazopirroloni, detti anche pirroni. I polimeri a scala sono caratterizzati da elevate proprietà meccaniche, anche a temperature elevate.
b) Polimerizzazione radicalica
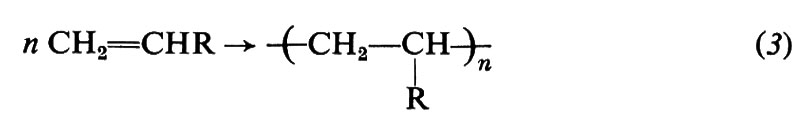
Possono essere polimerizzati per via radicalica vari monomeri di tipo vinilico o vinilidenico, cioè monomeri insaturi di formula generale CH2=CHR o CH2−CRR′. Citiamo tra questi l'etilene (R = H), lo stirene (R = C6H5), l'acrilonitrile (R = CN), gli esteri metilici dell'acido acrilico (R = COOCH3) e metacrilico (R = COOCH3, R′ = CH3), varie diolefine coniugate (per es., butadiene e isoprene), l'allene (propadiene) e alcuni suoi derivati. La reazione di polimerizzazione può essere rappresentata dall'equazione:
Questo metodo di polimerizzazione richiede l'impiego di un ‛iniziatore', cioè di una sostanza o di un sistema capace di generare radicali liberi. S'impiegano oggi vari tipi d'iniziatori, la maggior parte dei quali appartiene alle classi dei perossidi, idroperossidi, azoderivati. Queste sostanze, a una certa temperatura, diversa per ciascun iniziatore e in genere compresa tra 50 e 150 °C, cominciano a decomporsi, con velocità finita, generando radicali. Un altro tipo d'iniziatori, molto usati per polimerizzazioni in fase acquosa, è rappresentato dai cosiddetti sistemi ‛redox', costituiti da due sostanze che reagendo tra di loro generano radicali. I sistemi redox hanno il vantaggio di poter generare radicali anche a temperatura ambiente o inferiore a essa e possono quindi essere usati quando si desideri condurre la polimerizzazione a queste temperature.
Le reazioni relative alla formazione di radicali da parte di tre comuni iniziatori, il perossido di benzoile, l'azobisisobutirronitrile e il sistema sale ferroso-perossido d'idrogeno sono le seguenti:
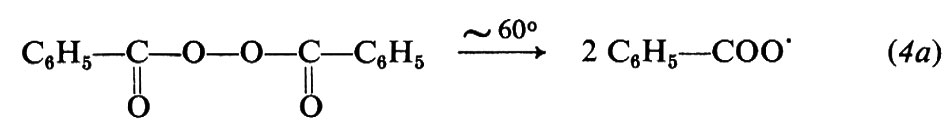
(il radicale C6H5−COO• può ulteriormente decomporsi per dare radicali fenilici: C6H5COO• → C6H5 + CO2)

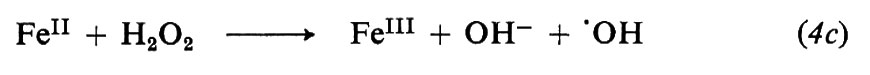
Il radicale generato dall'iniziatore (detto anche radicale primario) reagisce prontamente col monomero con formazione di un nuovo radicale (‛iniziazione'), al quale poi si addizionano, in un processo rapidissimo, nuove molecole di monomero (‛propagazione'). Le macromolecole crescenti sono quindi radicali liberi, contengono cioè un elettrone spaiato. La terminazione della crescita avviene quando due macromolecole crescenti reagiscono tra di loro, con scomparsa dei due radicali. La terminazione può avvenire per accoppiamento (equazione 8a), che porta alla formazione di un'unica macromolecola da due radicali crescenti, o per disproporzionamento (equazione 8b), che porta alla formazione di due macromolecole. Le varie fasi che portano alla formazione di una macromolecola sono rappresentate dalle equazioni seguenti:
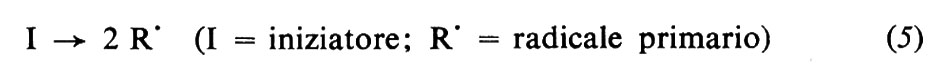

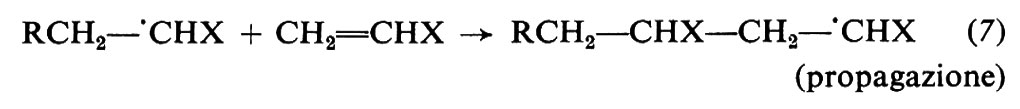

Già pochi secondi dopo l'inizio della polimerizzazione si ha l'instaurazione di uno stato stazionario, in cui la concentrazione dei radicali crescenti rimane costante nel tempo. Ciò è dovuto al fatto che i radicali che scompaiono per incontro con un altro radicale secondo l'equazione (8) sono sostituiti da nuovi radicali che si generano attraverso la continua decomposizione nel tempo dell'iniziatore, secondo l'equazione (5).
La crescita di una macromolecola per via radicalica è una tipica reazione a catena e avviene in un periodo di tempo che è dell'ordine di una frazione di secondo. Ciò significa che se si arresta la polimerizzazione dopo aver trasformato, per es., il 5% del monomero, le macromolecole formate risultano tutte a peso molecolare elevato. A differenza, cioè, di quanto avviene nella policondensazione, il peso molecolare del polimero non aumenta praticamente all'aumentare della trasformazione.
Si conosce oggi come regolare il peso molecolare medio dei polimeri ottenuti per via radicalica. Il metodo si basa sull'aggiunta, nell'ambiente di polimerizzazione, di sostanze che sono in grado di reagire col radicale crescente per dare una catena ‛morta' (cioè non più in grado di crescere) e un nuovo radicale, che dà inizio a una nuova catena polimerica. Queste sostanze, dette ‛regolatori', sono in genere composti che hanno un atomo di idrogeno o altri atomi molto reattivi verso i radicali. La reazione che avviene è la seguente:
~CH2−•CHX + RY → ~CH2−HXY + R• (Y = H, CI, Br, ecc.). (9)
Sono composti molto attivi i mercaptani, usati nella regolazione del peso molecolare del polibutadiene, il bromoformio, il tetracloruro di carbonio.
Reazioni come la (9), dette reazioni di trasferimento perché non portano alla scomparsa di un radicale, ma solo al suo trasferimento dalla macromolecola crescente a un'altra sostanza, possono avvenire anche con il solvente, col monomero e altresi con catene polimeriche già formate. In quest'ultimo caso si ha la crescita di una nuova catena su di un'altra già formata, il che porta al formarsi di una ramificazione:
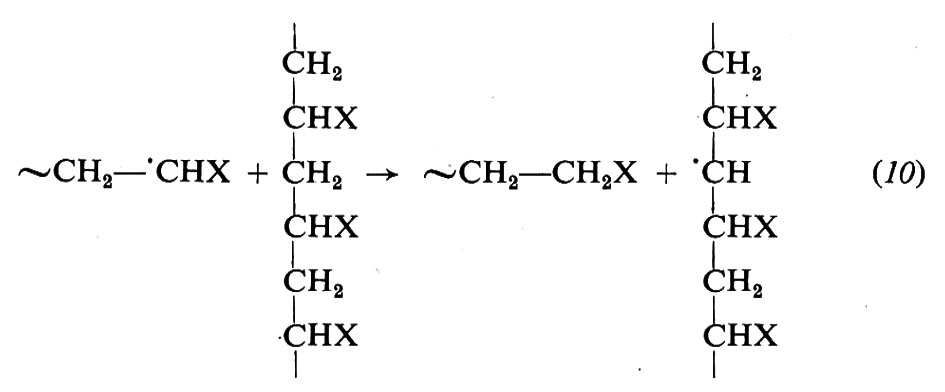
Si conoscono sostanze in grado di reagire con i radicali crescenti per dare luogo a nuovi radicali che non sono più capaci di reagire col monomero. Ciò porta all'arresto della catena crescente. Queste sostanze, le più comuni delle quali appartengono alla classe dei fenoli, o dei nitro- o nitrosoderivati aromatici, vengono chiamate ‛ritardanti', in quanto, provocando una diminuzione nel numero dei radicali crescenti, abbassano la velocità di reazione. Quando il ritardante ha una reattività verso i radicali di gran lunga più elevata del monomero, si preferisce chiamarlo ‛inibitore', in quanto finché esso è presente non si ha polimerizzazione. Gli inibitori hanno interesse pratico poiché vengono di solito aggiunti al monomero puro allo scopo di permetterne il trasporto, la conservazione in magazzino, ecc. Il monomero viene separato dall'inibitore attraverso distillazione, prima di essere sottoposto a polimerizzazione.
La polimerizzazione radicalica fornisce, da monomeri vinilici, polimeri di costituzione sostanzialmente regolare, ma a stereoregolarità molto bassa. Quest'ultima risulta normalmente di tipo sindiotattico e di solito aumenta al diminuire della temperatura di polimerizzazione. Dal butadiene la polimerizzazione radicalica fornisce a 50-60 °C polimeri amorfi contenenti il 65% circa di unità 1,4-trans; la percentuale di tali unità aumenta polimerizzando a più bassa temperatura.
La polimerizzazione radicalica può essere condotta in soluzione, ma nella pratica industriale si opera preferibilmente senza solvente (polimerizzazione in blocco), oppure in sospensione o in emulsione. Nella polimerizzazione in blocco è piuttosto scarso il controllo termico della reazione, e appunto per questo in siffatte polimerizzazioni ci si arresta di solito a basse percentuali. Nella polimerizzazione in sospensione il monomero è disperso nell'acqua, in sferette di 0,1-1 mm, con l'aiuto di un agente disperdente (alcool polivinilico, acido poliacrilico, metilcellulosa, gelatina, fosfati, ecc.). L'iniziatore è disperso nel monomero; la cinetica di polimerizzazione è sostanzialmente identica a quella che si ha in soluzione. Questa tecnica permette una buona dissipazione del calore di reazione; inoltre i polimeri vengono ottenuti sotto forma di piccolissime sferette, che vengono facilmente separate e filtrate. Nella polimerizzazione in emulsione le particelle di monomero disperse nella fase acquosa hanno dimensioni molto più piccole, 10-510-6 mm; inoltre l'iniziatore (di solito un sistema redox) è disciolto nella fase acquosa. La cinetica è diversa da quella che si ha in soluzione; inoltre, si ottengono pesi molecolari molto più alti.
La polimerizzazione radicalica ha assunto un notevole interesse industriale. I principali monomeri che sono polimerizzati industrialmente per tale via sono: etilene (150-300 °C; 1.000-2.000 atm), stirene, acrilonitrile, acrilato e metacrilato di metile, tetrafluoroetilene, acetato di vinile, cloruro di vinile, butadiene-stirene (gomma SBR). (Su questo argomento v. Bevington, 1961).
c) Polimerizzazione ionica
La polimerizzazione ionica è una tipica polimerizzazione a catena, come la radicalica, e procede attraverso uno schema simile, cioè iniziazione, propagazione e terminazione. Si differenzia dalla radicalica per il fatto che la specie attiva, cioè la macromolecola crescente, porta una carica positiva (polimerizzazione cationica) o negativa (polimerizzazione anionica).
Polimerizzazione cationica. - Nella polimerizzazione cationica di un monomero M la reazione di propagazione può essere rappresentata dall'equazione:
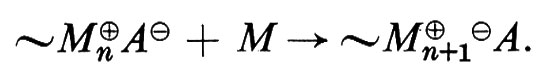
Per questo tipo di polimerizzazione sono necessari dei catalizzatori; questi sono sostanze o sistemi capaci di cedere un protone o un catione al monomero. Si possono distinguere tre classi di catalizzatori: a) acidi protonici, come H2SO4, HClO4; b) acidi di Lewis, per es., AlCl3, BF3, TiCl4, SnCl4; c) altri generatori di cationi, come t-C4H9ClO4, I2, (C6H5)3CCl. Gli acidi di Lewis richiedono di solito (ma non sempre) la presenza di piccole quantità di promotori, detti anche cocatalizzatori; questi sono sostanze (per es., H2O, HCl, CCl3COOH, alogenuri alchilici) capaci di generare protoni o cationi per reazione con l'acido di Lewis. Per es.:
AlCl3 + HCI →H+ AlCl4-; C2H5CI + AlCl3 → C2H5+ AlCl4-. (12)
Vari tipi di monomeri possono essere polimerizzati per via cationica ad alti polimeri lineari: 1) alcuni monomeri idrocarburici insaturi (stirene e derivati, isobutene; quest'ultimo monomero è stato finora polimerizzato solo per via cationica); 2) alchilvinileteri e alchilvinilammine; 3) aldeidi e alcuni altri monomeri carbonilici; 4) eteri ciclici; 5) esteri ciclici e ammidi cicliche. Alcuni di questi monomeri possono essere polimerizzati in modo stereospecifico.
1. Per la polimerizzazione dei monomeri idrocarburici insaturi gli acidi di Lewis sono i catalizzatori tipici. Riportiamo, come esempio di polimerizzazione cationica di questi monomeri, lo schema di polimerizzazione dell'isobutene con BF3 in presenza di tracce di H2O come catalizzatore:
iniziazione:

propagazione:
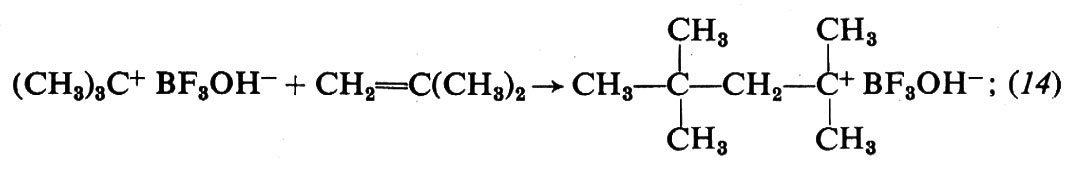
terminazione:
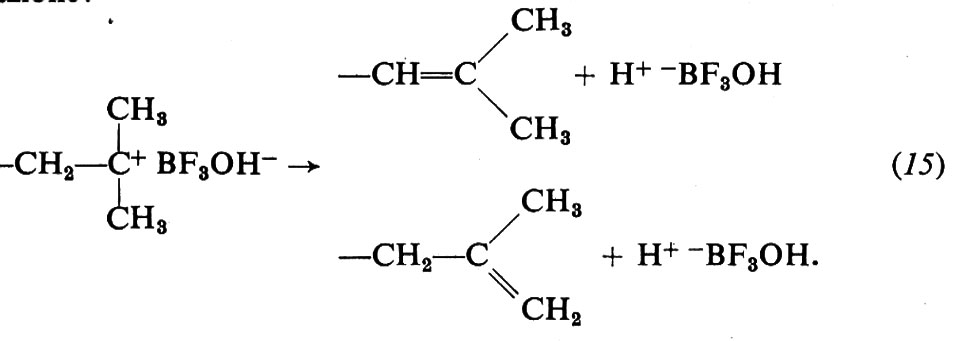
Durante la polimerizzazione lo ione negativo rimane vicino al catione costituito dalla macromolecola crescente e influenza la velocità di crescita di quest'ultima. Il grado di associazione tra i due ioni dipende dalla loro natura, oltre che dal tipo di solvente. Solventi a elevata costante dielettrica aumentano la velocità di polimerizzazione e il peso molecolare dei polimeri.
La polimerizzazione dei monomeri idrocarburici viene di solito effettuata a temperatura molto bassa, ed è estremamente rapida; l'isobutene, per es., viene polimerizzato a −100 °C circa e dà in qualche secondo pesi molecolari superiori al milione.
Lo stirene viene polimerizzato in modo non stereospecifico, ma alcuni suoi derivati, come l'orto-metossistirene, forniscono con vari catalizzatori [AlCl2C2H5, Al(C2H5)2Cl, TiCl2(O n-C4H9)2], a −78 °C, polimeri stereoregolari isotattici. La polimerizzazione cationica di diolefine, come butadiene e isoprene, ha scarso interesse perché fornisce prodotti reticolati insolubili.
Ha interesse industriale la copolimerizzazione dell'isobutene con isoprene (3% circa), che fornisce un copolimero da cui si ottiene la cosiddetta gomma butile (v. Kennedy, 1968).
2. Gli eteri vinilici, CH2=CH−O−R, polimerizzano attraverso il doppio legame, dando unità

(v. Gaylord, 1963; v. Lal, 1968; v. Ketley, 1967). Sono stati ottenuti, oltre a polialchilvinileteri atattici, anche polialchilvinileteri cristallini o cristallizzabili, che presentano stereoregolarità di tipo isotattico. È da ricordare che i polimeri cristallini degli alchilvinileteri rappresentano il primo caso di polimerizzazione stereospecifica di monomeri vinilici, anteriore alla scoperta di Natta, anche se all'epoca non fu riconosciuto che la cristallinità era da attribuire alla stereoregolarità isotattica delle macromolecole (v. Schildknecht e altri, 1948).
Polimerizzazioni stereospecifiche degli alchilvinileteri sono state effettuate con BF3.O(C2H5)2, ma si conoscono vari catalizzatori più stereospecifici di quest'ultimo, per es. Al(C2H5)2Cl, AlC2H5Cl2, Al(OC2H5)Cl2, TiCl2(OC2H5)2, TiF3, TiF4+Al(OR)3, Al(OR)3~ + H2SO4. Vari aspetti della polimerizzazione di questi monomeri non sono stati ancora chiariti, e tra questi il meccanismo del controllo sterico nella polimerizzazione.
3. Le aldeidi,

, polimerizzano (v. Pregaglia e Binaghi, 1967; v. Vogl, 1968) attraverso il doppio legame carbonilico, dando unità monomeriche
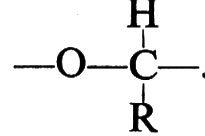
La formaldeide e l'acetaldeide possono essere polimerizzate sia con acidi protonici, sia con acidi di Lewis. L'acetaldeide deve essere polimerizzata a temperatura di −40 °C e fornisce, con questo metodo, polimeri atattici.
L'acetaldeide clorurata (CH2Cl−CHO, CHCl2−CHO, CCl3−CHO) e gli omologhi superiori dell'acetaldeide possono essere polimerizzati con acidi di Lewis (BF3, AlBr3, AlC2H5Cl2, SnBr4) a bassa temperatura (−78 °C circa); a differenza dell'acetaldeide, questi monomeri danno polimeri isotattici.
Ha interesse industriale il polimero della formaldeide, che viene usato principalmente come materia plastica.
4. Possono essere polimerizzati per via cationica eteri ciclici a 3, 4 o 5 membri (v. Ledwith e Fitzsimmonds, 1968; v. Tsuruta, 1967). I catalizzatori cationici sono, per questo tipo di monomeri, molto più attivi di quelli anionici, che del resto fanno polimerizzare solo gli epossidi (eteri ciclici a tre membri). Si ammette che la polimerizzazione cationica degli eteri ciclici avvenga attraverso la formazione di ioni ossonio. Per esempio, nel caso del tetraidrofurano (THF):
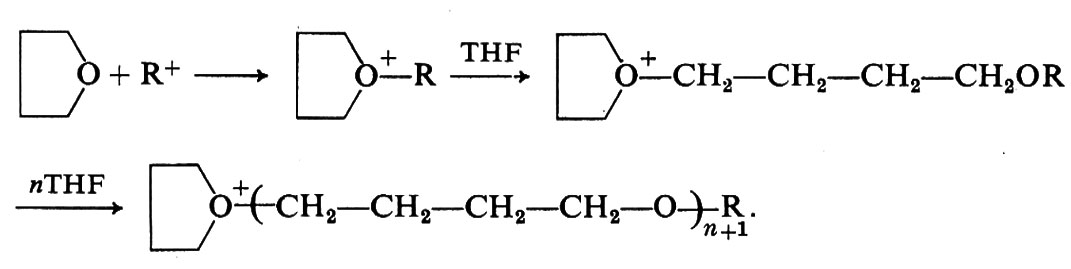
La reazione di terminazione, in questi processi, non è ancora chiarita.
L'ossido di etilene, il più semplice etere ciclico, può essere polimerizzato con i più comuni acidi di Lewis (BF3) con o senza cocatalizzatore, dà tuttavia pesi molecolari molto bassi, mentre pesi molecolari più elevati si ottengono con SnCl4. Il catalizzatore più attivo, tuttavia, è quello che si ottiene dalla reazione tra Zn(C2H5)2 e H2O, in rapporto 1: 1; questo sistema, che è omogeneo ed è ritenuto da vari autori di tipo cationico, fornisce, a partire da ossido di etilene, polimeri a peso molecolare altissimo. Si ritiene che l'effettivo catalizzatore sia una specie che contiene legami Zn−O−Zn, verosimilmente C2H5Zn−O−ZnC2H5.
Anche il sistema Al(C2H5)3-H2O (1 : 1) è un catalizzatore molto attivo, sebbene meno di quello a base di zinco.
Nei polimeri dell'ossido di propilene,
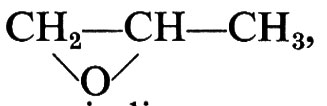
esistono atomi di carbonio asimmetrici e quindi possono prevedersi polimeri atattici, isotattici o sindiotattici. Vari catalizzatori cationici danno dal monomero racemo polimeri atattici, da considerarsi copolimeri dei due enantiomeri che costituiscono il monomero. I sistemi Zn(C2H5)2-H2O e Al(C2H5)3-H2O danno invece, partendo dal monomero racemo, polimeri cristallini isotattici. Ciò indica che ogni macromolecola è ottenuta prevalentemente da uno solo dei due enantiomeri (processo stereoselettivo).
Nella polimerizzazione del tetraidrofurano, gli acidi di Lewis non sono molto efficienti. Si usano il sistema Al(C2H5)3-H2O e altri particolari sistemi, come FeCl3+ +(C6H5)8CCl, FeCl3 + TiCl4, FeCl3 + AlCl3, sali di ossonio (per es. (C2H5)3O+PF6-).
5. I monomeri più studiati, tra gli esteri e le ammidi cicliche, sono i lattoni e i lattami. Nel caso dei lattami, il monomero di gran lunga più studiato è il caprolattame, che serve per la preparazione del nailon 6. Industrialmente questo monomero, tuttavia, viene polimerizzato per via anionica. La polimerizzazione cationica non ha grande importanza. (Su questo argomento v. Plesh, 1963).
Polimerizzazione anionica. - Nella polimerizzazione anionica la reazione di propagazione può essere rappresentata dall'equazione:

Mediante questo tipo di polimerizzazione possono essere polimerizzate varie classi di monomeri e cioè alcuni monomeri idrocarburici (stirene, diolefine coniugate), monomeri acrilici, aldeidi, epossidi, esteri ciclici, ammidi cicliche. Vengono usati vari tipi di catalizzatori, che si differenziano tra loro per quanto riguarda il meccanismo di iniziazione e cioè: a) metalli alcalini; b) derivati alchilici o ammidi di metalli alcalini, di Mg, Cd, Zn, Al; c) alcossiderivati di metalli vari, ammine.
I monomeri idrocarburici polimerizzano con i catalizzatori di tipo a) e b) (v. Braun, 1967). Con i metallo-alchili o con le metallo-ammidi l'iniziazione consiste nell'addizione di uno ione negativo al monomero. Per es., nella polimerizzazione dello stirene con litio-butile in solventi eterei:

La propagazione procede per addizione del monomero all'anione:

Con i metalli alcalini l'iniziazione è più complessa: avviene con trasferimento di un elettrone dal metallo al monomero e formazione di un anione radicale (19); quest'ultimo si trasforma rapidamente, per accoppiamento con un altro anione-radicale, in un dianione (20), che è attivo alle due estremità:
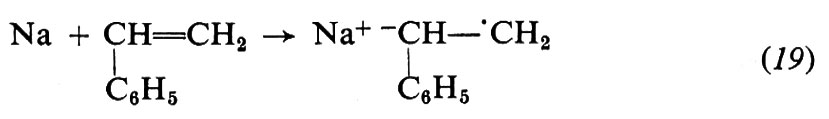
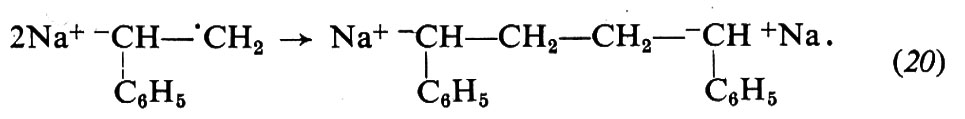
Invece dei metalli alcalini, che essendo insolubili reagiscono solo in superficie, si preferisce usare come catalizzatore il complesso che si forma per reazione in tetraidrofurano tra Na o Li e naftalene:
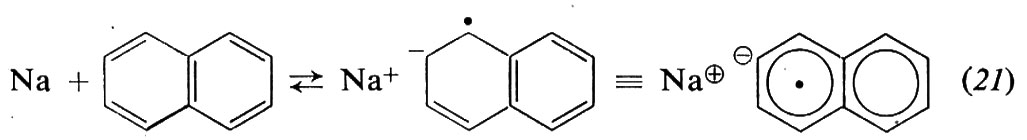
L'anione-radicale sodio-naftalene (naftaluro di sodio) formato in (21) reagisce col monomero dando lo stesso anione-radicale (22) che si ottiene in (19):
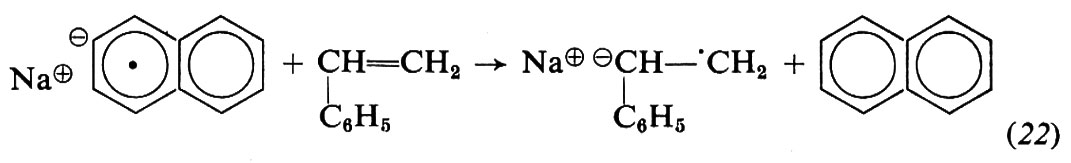
Nella polimerizzazione anionica di monomeri idrocarburici in solventi eterei non si ha di solito terminazione; ciò significa che, in assenza di impurezze che possano distruggere la coppia ionica (alcoli, acqua, ecc.), le macromolecole crescono fino a che praticamente tutto il monomero è consumato; riprendono poi a crescere all'aggiunta di nuovo monomero, col risultato che il peso molecolare delle macromolecole è proporzionale al monomero consumato. Polimeri aventi questa proprietà vengono chiamati ‛polimeri viventi' (v. Szwarc, 1956).
Questo tipo di polimerizzazione si presta alla preparazione di copolimeri a blocchi, cioè del tipo −A−A−A− A−A−B−B−B−B−A−A−A−A−A−, dove A e B indicano le unità dei due differenti monomeri. Basta a questo scopo aggiungere alla soluzione del catalizzatore una certa quantità del monomero A, quindi, quando A è tutto consumato, una certa quantità del monomero B, poi di nuovo A e così via.
In solventi diversi dagli eteri non si hanno di solito polimeri viventi. Si osservano infatti fenomeni di trasferimento col monomero e altri tipi di terminazione non del tutto chiariti.
Un certo interesse ha il catalizzatore eterogeneo costituito dall'associazione di sodioallile, isopropilato di sodio e cloruro di sodio. Questo sistema, che si ritiene agisca con meccanismo anionico, va sotto il nome di ‛catalizzatore Alfin' ed è in grado di polimerizzare il butadiene, l'isoprene e lo stirene ad altissimi pesi molecolari (v. Morton, 1946, 1952 e 1957). Tale catalizzatore ha acquistato un potenziale interesse industriale per la preparazione di polibutadiene, dopo che è stato trovato il modo di regolare il peso molecolare del polimero mediante diidroo tetraidronaftalene.
La polimerizzazione anionica degli acrilati e metacrilati non ha interesse pratico (industrialmente questi monomeri vengono polimerizzati per via radicalica), ma ha interesse scientifico, perché, in particolari condizioni, può essere stereospecifica. Nel caso degli acrilati, danno polimeri stereoregolari (isotattici) solo quei monomeri che contengono gruppi alchilici ramificati (per es. isopropilo terz-butilacrilato). Dal metilmetacrilato, invece, sono stati ottenuti sia polimeri iso- che sindiotattici. In queste polimerizzazioni si opera di solito a bassa temperatura, usando come catalizzatori composti organometallici del litio o sodio, magnesioalchili e cadmioalchili. I vari fattori che determinano la stereospecificità non sono stati peraltro del tutto chiariti.
Le aldeidi possono essere polimerizzate da vari catalizzatori, quali derivati alchilici, idruri, alcolati di metalli del 1°, 2°, 3° gruppo del sistema periodico (v. Pregaglia e Binaghi, 1967; v. Vogl, 1968). La specie attiva risulta la stessa qualunque sia il composto di partenza. La macromolecola cresce per inserimento del monomero sul legame metallo-ossigeno, verosimilmente attraverso un intermedio a quattro centri. Per es., usando LiC4H9 o LiOC4H9:
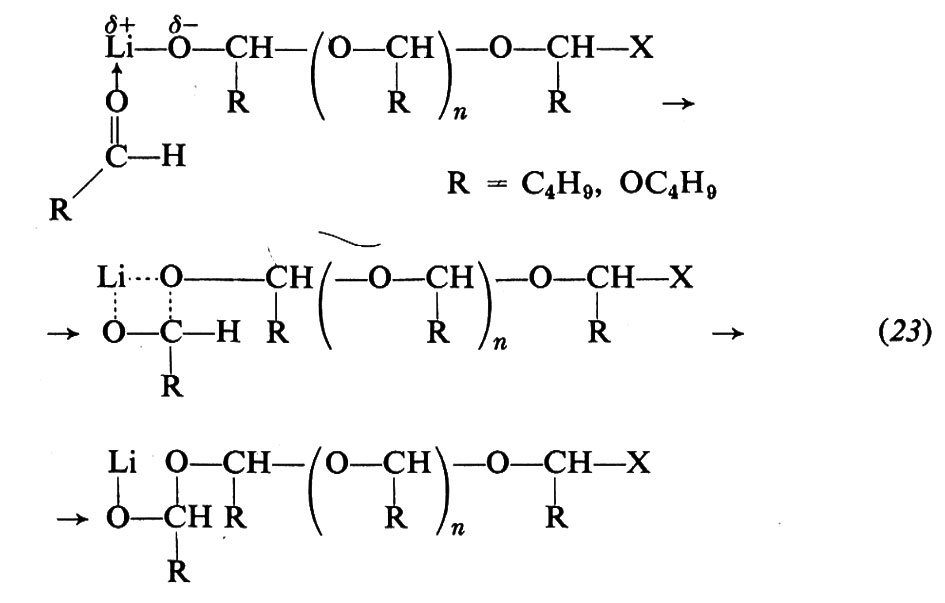
La formaldeide può essere polimerizzata anche con basi più deboli, quali ammine, fosfine, stibine. La polimerizzazione anionica delle aldeidi, essendo altamente stereospecifica, ha permesso di ottenere polimeri isotattici dall'acetaldeide e omologhi superiori. Le polimerizzazioni vengono condotte a bassa temperatura (−70 ÷ 80 °C).
Tra gli eteri ciclici solo gli epossidi possono essere polimerizzati per via anionica; si ottengono però pesi molecolari molto più bassi che con iniziatori cationici (v. Ledwith e Fitzsimmonds, 1968; v. Tsuruta, 1967).
La specie attiva nella polimerizzazione iniziata, per esempio, da NaOH è la seguente:
Na+ -O−CH2−CH2−(O−CH2−CH2−)O−CH2−CH2OH.
Tra le ammidi cicliche, di notevole interesse industriale è il caprolattame, che, come si è detto sopra, è il monomero impiegato per la preparazione del nailon 6. Si usano come catalizzatori basi forti, quali metalli alcalini, metalloammidi e metalloalchili.
Scarso interesse ha la polimerizzazione anionica di esteri ciclici.
d) Polimerizzazione con catalizzatori a base di metalli di transizione
L'impiego di questi catalizzatori ebbe inizio praticamente nel 1953; dal nome dei due scienziati che dettero il maggiore contributo in questo campo, essi vengono comunemente indicati come catalizzatori Ziegler-Natta (v. Natta, 1964; v. Ziegler, 1964).
Essi costituiscono una classe molto vasta e comprendono sia sistemi omogenei che eterogenei. Vengono preparati in genere per reazione tra un metalloalchile e un composto di un metallo di transizione. Si conoscono anche catalizzatori preparati solo da composti di metalli di transizione, che però sono molto meno importanti in pratica. Come metalloalchile viene di solito usato un composto di alluminio, come AlR3, AIR2Cl, AlRCl2 (R = etile o altro gruplo alchilico), ma possono essere usati anche litioalchili, zincoalchili, gallioalchili; come composto di metallo di transizione può essere usato un alogenuro (per es. TiCl4, TiCl3, VOCl3, VCl3, ecc.), un alcolato, un sale di un acido organico, un acetilacetonato (acac) e svariati altri tipi di complessi. Si vede subito come possono essere effettuate numerosissime combinazioni tra un metalloalchile e un composto di un metallo di transizione. In realtà, come vedremo nel seguito, solo un numero limitato di queste combinazioni ha interesse pratico.
La vera natura dei catalizzatori a base di metalli di transizione non è ancora del tutto chiarita; nei casi sopra indicati possono formarsi complessi bimetallici, contenenti cioè sia l'alluminio sia il metallo di transizione, ma in nessun caso si conosce con precisione la struttura della specie cataliticamente attiva.
Per quanto riguarda il loro meccanismo, tali catalizzatori vengono considerati di tipo ionico e più precisamente anionico, ma tra questi sistemi e quelli anionici classici esistono differenze notevoli che appariranno chiare in seguito e che ne giustificano una trattazione a parte.
I catalizzatori a base di metalli di transizione sono in grado di polimerizzare le seguenti classi di monomeri idrocarburici: olefine, diolefine coniugate, alleni, cicloolefine. L'interesse di tali catalizzatori consiste nel fatto che essi sono in grado di polimerizzare in modo altamente stereospecifico queste classi di monomeri (v. Boor, 1970; v. Raff e Doak, 1965; v. Reich e Schindler, 1966; v. Jordan, 1967).
Olefine. 1. Etilene La più semplice olefina, l'etilene, può essere polimerizzata con un numero molto grande di catalizzatori, sia omogenei che eterogenei, per es. AlR3-TiCl4, AlR3-TiCl3, Al(C2H5)3-VCl4, Al(C2H5)3Cl-V(acac)3, Al(C2H5)3-VCl3, Al(C2H5)3Cl-VCl3. È sufficiente far gorgogliare etilene, a temperatura ambiente, in una soluzione o sospensione di uno di questi catalizzatori in un solvente idrocarburico (per es. n-eptano), per ottenere una rapida polimerizzazione. In pratica, si opera sotto una leggera pressione, che rende ancora più veloce la polimerizzazione. Le condizioni sperimentali sono quindi notevolmente più blande di quelle impiegate nella polimerizzazione radicalica, che richiede temperature di 150-300 °C e pressioni di 1.000-2.000 atm. Il polimero che si ottiene è inoltre più regolare di quello preparato per via radicalica, che è caratterizzato dalla presenza di corte ramificazioni laterali (0,5-2 per 100 atomi di carbonio). Il polimero ottenuto dall'etilene con catalizzatori a base di metalli di transizione (detto anche ‛politene a bassa pressione') è più cristallino e quindi più denso, e ha una temperatura di fusione più elevata, 138 °C contro 110 circa, del polimero radicalico (detto anche ‛politene ad alta pressione'). I due tipi di politene vengono entrambi prodotti industrialmente, perché le loro caratteristiche diverse si prestano a impieghi diversi. Attualmente, per la preparazione industriale del politene, si vanno affermando alcuni tipi di catalizzatori ottenuti da AlR3 e TiCl4 supportato su MgCl2 e altre sostanze. Questi catalizzatori danno politene con altissima resa (vari chilogrammi di polimero per grammo di catalizzatore), cosicché non è necessaria alcuna purificazione del polimero per allontanare la parte minerale.
Tra i catalizzatori usati per la polimerizzazione dell'etilene sono da ricordare due sistemi eterogenei, a base, rispettivamente, di ossido di cromo e ossido di molibdeno supportati su allumina. Anche questi catalizzatori danno politene altamente lineare.
2. α-Olefine. Sono state polimerizzate per la prima volta nel 1954 (v. Natta e altri, 1954 e 1955). In generale, tutte le α-olefine di tipo vinilico, cioè di formula CH2=CHR (R = gruppo alchilico lineare o ramificato, alchilaromatico o aromatico), possono essere polimerizzate sia a polimero atattico sia a polimero isotattico. Il propilene (propene) è, fino a oggi, l'unica α-olefina di cui sia stato caratterizzato anche un polimero sindiotattico (v. Natta e altri, 1960).
I più importanti sistemi catalitici per la polimerizzazione di α-olefine alifatiche (R = gruppo alchilico) sono ottenuti da alogenuri di titanio o vanadio e derivati alchilici di litio, berillio o alluminio. Tutti questi sistemi sono eterogenei. La polimerizzazione viene condotta tra temperatura ambiente e 90-100 °C, a bassa pressione di olefina. Si ottiene in genere un prodotto di polimerizzazione che contiene sia macromolecole atattiche che isotattiche. Le temperature di fusione di alcuni polimeri isotattici di α-olefine alifatiche sono riportate nella tab. Il. Tra questi polimeri quello di gran lunga più importante dal punto di vista pratico è il polipropilene isotattico. I sistemi più stereospecifici per la preparazione di polipropilene isotattico sono quelli ottenuti da TiCl3 violetto (si conosce anche una modificazione bruna, detta TiCl3 β) e AlR3, AlR2Cl, o BeCl2. Alcuni di questi sistemi danno prodotti di polimerizzazione contenenti fino al 95% di polimero altamente isotattico. Nella pratica industriale s'impiegano catalizzatori Al(C2H5)2Cl-TiCl3 violetto, a 70 °C. Il peso molecolare del polimero può variare, secondo il catalizzatore usato e le condizioni sperimentali, da 10.000 circa fino a più di un milione.

Il polipropilene sindiotattico è stato finora ottenuto da vari catalizzatori a base di vanadio, tutti omogenei; un sistema tipico è il seguente: VCl4-Al(C2H5)2Cl-anisolo in rapporto 1 : 20: 1. Questi sistemi sono stereospecifici solo se adoperati al di sotto di −50 °C circa; la stereospecificità aumenta al diminuire della temperatura.
I monomeri vinilaromatici (R = gruppo aromatico) sono stati polimerizzati a polimeri isotattici sia col sistema AlR3-TiCl4 (Al/Ti = 2,3 ÷ 2,5) sia con quello AlR3-TiCl3 violetto. Il polistirene che si ottiene con questi sistemi è altamente stereoregolare; il peso molecolare può essere molto elevato, superiore a un milione circa.
Un capitolo interessante della polimerizzazione stereospecifica è quello che riguarda la polimerizzazione di monomeri asimmetrici CH2=CH−R* (R* gruppo alchilico contenente un atomo di carbonio asimmetrico, per es., −CH2~*CH(CH3)−C2H5). La polimerizzazione stereospecifica di questi monomeri fornisce macromolecole isotattiche ciascuna delle quali risulta costituita prevalentemente da uno dei due enantiomeri che costituiscono il monomero (polimerizzazione stereoselettiva). Inoltre, è stato osservato che polimerizzando un monomero CH2= CH−R* con catalizzatori asimmetrici, per es. col sistema otticamente attivo Al [CH2−*CH(CH3)C2H5]3-TiCl4, polimerizza prevalentemente uno dei due enantiomeri del monomero (polimerizzazione stereoelettiva) (v. Pino, 1965).
3. Copolimeri olefinici. Mediante catalizzatori ZieglerNatta possono essere preparati vari copolimeri di olefine. Due tipi di copolimeri presentano particolare interesse: a) i copolimeri etilene-propilene (v. Pasquon e altri, 1967; v. Natta e altri, 1969); b) i copolimeri stereoregolari alternati etilene-butene-2-cis ed etilene-cicloolefine (v. Natta e altri, 1961; v. Dall'Asta e Mazzanti, 1963).
a) Presentano una notevole importanza pratica i copolimeri aventi dal 25 al 30% in moli di propilene, che, essendo amorfi a temperatura ambiente, hanno carattere di elastomeri. Vengono preparati industrialmente con catalizzatori Al(C2H5)2Cl-V(acac)3 oppure Al(C2H5)2Cl-VCl4, a temperatura inferiore a 0 °C. Possono essere vulcanizzati con perossidi; vengono oggi molto utilizzati come materiale di rivestimento per cavi. Viene preparato industrialmente anche un terpolimero contenente, oltre a etilene e propilene nel rapporto sopra indicato, una diolefina, che può essere esadiene-1,4 CH2=CH−CH2−CH=CH-−CH3 o etilidennorbornene,
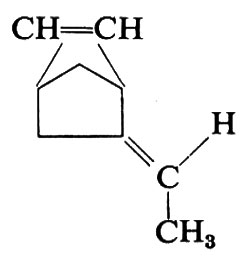
La diolefina, che è presente di solito in piccola percentuale, 4-5% rispetto alle moli totali, fornisce unità contenenti un doppio legame, permettendo così di usare per la vulcanizzazione i metodi classici che si usano per la gomma naturale, il copolimero butadiene-stirene e altri elastomeri insaturi.

Sia i copolimeri etilene-propilene, sia i terpolimeri, a causa della mancanza di insaturazione in catena, presentano elevata resistenza all'ossidazione.
b) Per ragioni steriche è difficile, mediante catalizzatori Ziegler-Natta, ottenere polimeri di olefine interne quali butene-2 o pentene-2. Queste olefine, tuttavia, possono essere copolimerizzate con etilene, usando come catalizzatori Al(C2H5)2Cl-V(acac)3 in toluene, o Al(C6H13)3-VCl4 in n-eptano.
Con quest'ultimo sistema è possibile ottenere copolimeri cristallini alternati etilene-butene-2-cis, purché si operi con largo eccesso di butene-2-cis rispetto all'etilene. Le macromolecole hanno configurazione (che è stata chiamata entrodusotattica), dalla quale si deduce che il butene entra con apertura in cis del doppio legame:
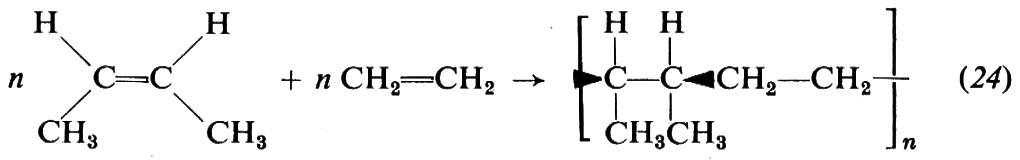
Queste macromolecole possono essere riguardate come polimeri stereoregolari del propilene a concatenamento testa-testa.
È da notare che dal butene-2-trans è stato ottenuto un copolimero alternato, ma non cristallino, perché non stereoregolare.
Anche il ciclopentene, il cicloeptene e altre cicloolefine superiori danno, con entrambi i sistemi sopra indicati, copolimeri alternati con etilene. I copolimeri ottenuti da cicloolefine con numero dispari di atomi di carbonio risultano cristallini ai raggi X. Anche questi copolimeri hanno struttura eritro-diisotattica:
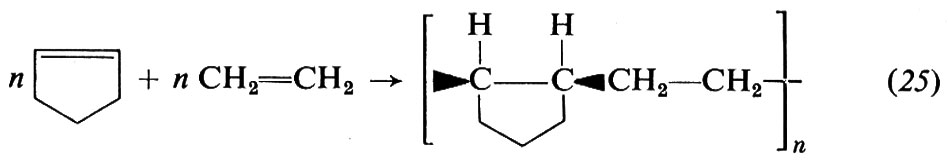
4. Meccanismo di polimerizzazione. Per quanto molti aspetti della polimerizzazione delle olefine mediante catalizzatori Ziegler-Natta siano ancora da chiarire, le caratteristiche principali di questo tipo di polimerizzazione sono ormai note. Si ammette che la macromolecola crescente sia legata, mediante un legame di tipo σ, al metallo di transizione del complesso catalitico. L'inserimento del monomero avverrebbe attraverso i seguenti stadi: a) coordinazione del monomero al metallo di transizione; b) inserimento del monomero nella catena crescente, attraverso un intermedio a quattro centri. Per es., nella polimerizzazione dell'etilene si avrebbe (cat = catalizzatore):
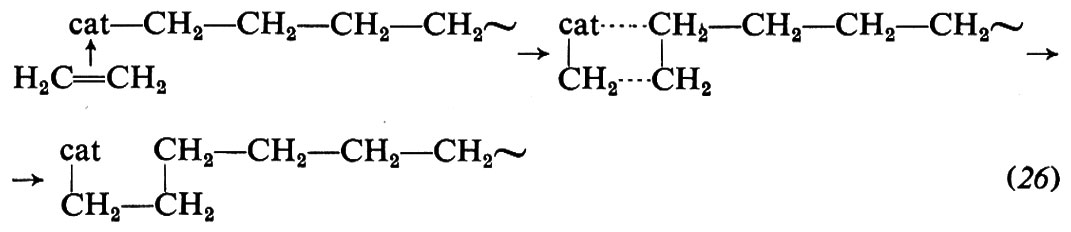
Questo meccanismo spiega l'apertura in cis del doppio legame del monomero entrante, sperimentalmente dimostrata.
Circa le cause che determinano la stereospecificità, la quale costituisce l'aspetto rilevante di questi catalizzatori, sono state avanzate molte ipotesi, ma il fenomeno non è chiarito. Si ritiene comunque che la stereospecificità dipenda dalla particolare struttura sterica (non nota) del complesso catalitico, la quale imporrebbe al monomero di coordinarsi sempre in uno solo dei due possibili modi sotto indicati, durante la crescita di una macromolecola:
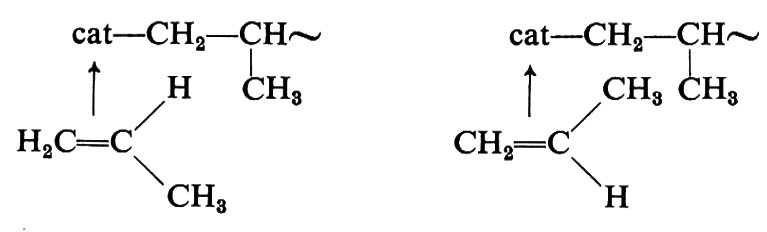
Poiché la coordinazione del monomero al metallo di transizione del complesso catalitico è uno stadio essenziale del processo di formazione della macromolecola, la polimerizzazione dell'etilene e delle a-olefine con catalizzatori Ziegler-Natta viene comunemente indicata come di tipo ‛anionico coordinato'.
Nella polimerizzazione stereoselettiva e in quella stereoelettiva la crescita di ciascuna macromolecola avviene prevalentemente con uno dei due enantiomeri, il che significa che uno dei due enantiomeri si coordina al centro catalitico a preferenza dell'altro, durante la crescita di una macromolecola. La coordinazione preferenziale di un enantiomero dipende evidentemente dalla asimmetria del centro catalitico, ma il fenomeno non è ancora del tutto chiarito.
Diolefine coniugate. - I catalizzatori a base di metalli di transizione hanno permesso di ottenere dalle diolefine coniugate vari tipi di polimeri stereoregolari (v. Natta e Porri, 1969; v. Marconi, 1967). Questi monomeri, come è stato visto in precedenza, possono essere polimerizzati sia con iniziatori radicalici sia con iniziatori ionici, ma tali metodi non assicurano, se non in casi particolari, un controllo sterico della polimerizzazione. Con iniziatori radicalici, per es., si ottengono dal butadiene polimeri costituiti in prevalenza da unità 1,4-trans (circa 70%), che sono amorfi a temperatura ambiente e presentano una debole cristallinità solo se esaminati a bassa temperatura e sotto stiro. Polibutadieni leggermente più stereoregolari, che presentano a temperatura ambiente una debole cristallinità dovuta a sequenze 1,4-trans, sono stati ottenuti con i catalizzatori Alfin. Il litiobutile in etere fornisce polibutadieni costituiti dal 97-98% di unità 1,2, ma questi polimeri risultano atattici. Il cloroprene è l'unica diolefina da cui, prima del 1954, fosse stato ottenuto un polimero altamente stereoregolare; con iniziatori radicalici infatti, operando a bassa temperatura (−30 °C circa), erano stati ottenuti polimeri costituiti dal 95% di unità 1,4-trans, cristallini a temperatura ambiente.
La scoperta dei catalizzatori stereospecifici a base di metalli di transizione ha cambiato radicalmente la situazione nel campo della polimerizzazione delle diolefine coniugate. Dal butadiene sono stati ottenuti quattro polimeri stereoregolari, e cioè un polimero costituito solo da unità 1,4trans, uno costituito solo da unità 1,4-cis e due polimeri costituiti solo da unità 1,2, a struttura iso- e sindiotattica rispettivamente (v. fig. 7). I catalizzatori usati e le principali caratteristiche fisiche dei polimeri ottenuti sono elencati nella tab. III.
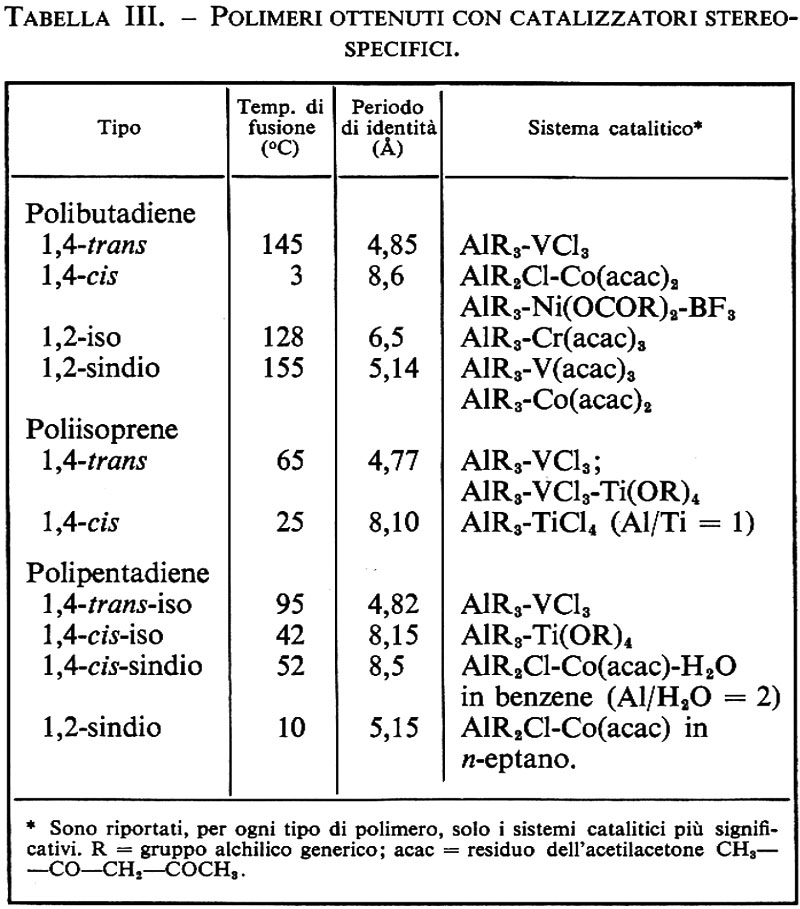
Il polibutadiene 1,4-cis ha notevole interesse pratico come elastomero ed è ora prodotto su scala industriale.
Dall'isoprene sono stati ottenuti due polimeri regolari, uno a struttura 1,4-cis, praticamente identico alla gomma naturale, e uno a struttura 1,4-trans, identico alla guttaperca naturale. Dall'isoprene è stato ottenuto anche un polimero costituito per oltre il 90% da unità 3,4, che però è atattico.
Recentemente, sia dal butadiene sia dall'isoprene sono stati ottenuti, con catalizzatori vari a base di metalli di transizione, polimeri costituiti da due tipi di unità monomeriche, in rapporto praticamente 50 : 50. Questi polimeri sono stati chiamati ‛polidieni equibinari' (v. Dawans e Teyssiè, 1971). La realizzazione di vari polimeri stereoregolari costituiti da unità di un solo tipo, così come quella dei polimeri equibinari, indica le vaste possibilità offerte dai catalizzatori a base di metalli di transizione nella polimerizzazione del butadiene e dell'isoprene.
Vari polimeri stereoregolari sono stati ottenuti dal pentadiene-1,3 e precisamente un polimero a struttura 1,4-trans isotattica, due polimeri a struttura 1,4-cis, iso- e sindiotattica rispettivamente, un polipentadiene 1 ,2-sindiotattico. Polimeri stereoregolari, e quindi cristallini o cristallizzabili, sono stati ottenuti anche da altre diolefine coniugate (2,3-dimetilbutadiene, 4-metilpentadiene-1,3, esadiene-2,4).
Non tutti gli aspetti del meccanismo della polimerizzazione delle diolefine mediante catalizzatori a base di metalli di transizione sono stati chiariti. Sui seguenti punti tuttavia esiste concordanza di vedute tra i vari studiosi: 1) la macromolecola crescente è legata al metallo di transizione del complesso catalitico attraverso un legame che è verosimilmente di tipo π-allilico o σ-π; 2) la crescita della macromolecola avviene attraverso i seguenti stadi: a) coordinazione del monomero al metallo di transizione; b) incorporazione del monomero coordinato sul legame tra il metallo di transizione e l'ultima unità polimerizzata, attraverso un intermedio a più centri.
Il modo di coordinarsi del monomero è verosimilmente uno dei fattori che determinano il tipo di unità monomerica. Vari fatti concorrono a far ritenere che un'unità 1,4-cis deriva da una coordinazione del monomero al metallo di transizione con entrambi i doppi legami, nella conformazione cis
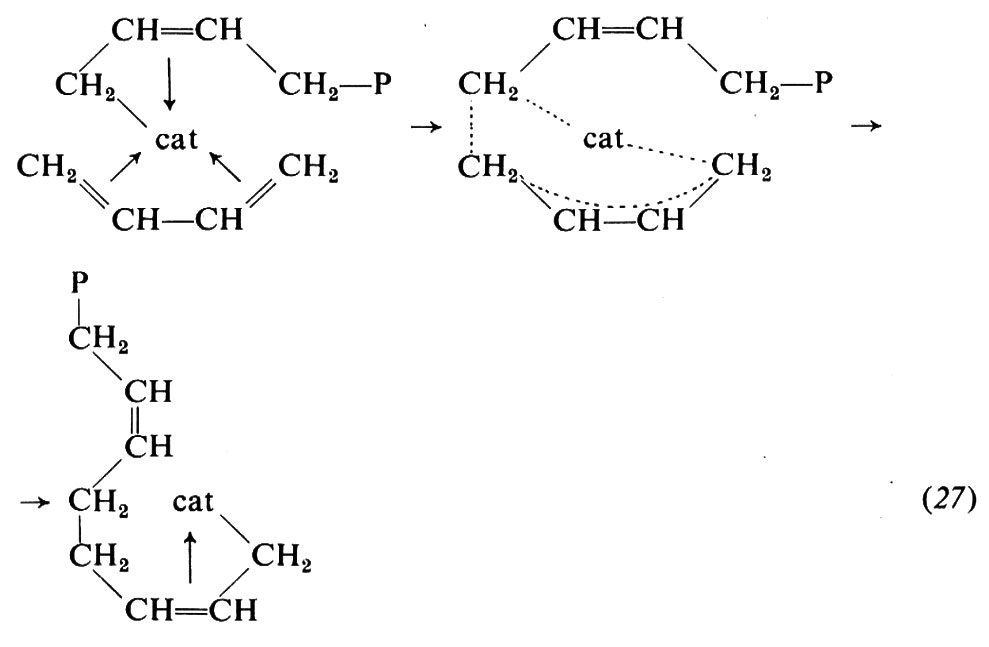
mentre un'unità 1,2 deriva verosimilmente da una coordinazione del monomero con un solo doppio legame:

Circa i fattori che determinano la configurazione sterica dell'unità monomerica, la situazione rimane tuttora oscura. Modelli plausibili, che possono aiutare a comprendere il fenomeno, sono stati presentati per la polimerizzazione del pentadiene a polimero 1,4-cis iso- e sindiotattico.
Cicloolefine. - Da questi monomeri possono essere ottenuti due tipi di unità monomeriche (v. Natta e Dall'Asta, 1969), una derivante da addizione al doppio legame (29a), l'altra derivante da apertura dell'anello (29b):
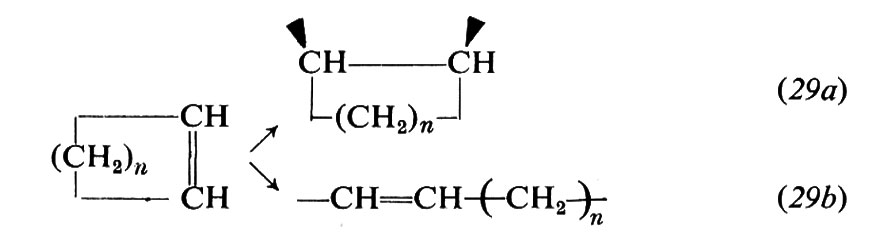
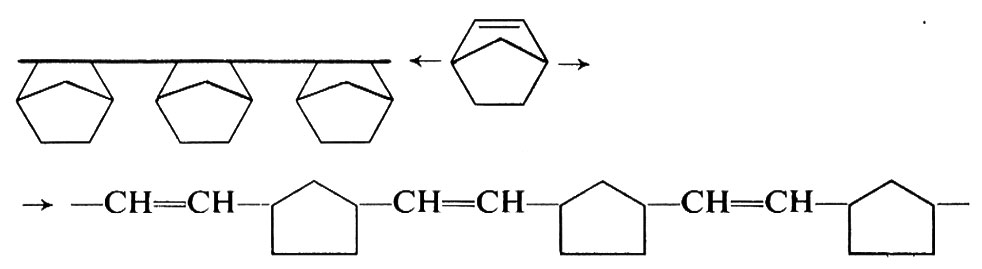
Dalle cicloolefine che hanno un anello fortemente tensionato (ciclobutene, norbornene e derivati) sono stati ottenuti, con catalizzatori a base di metalli di transizione, sia omopolimeri costituiti praticamente solo da unità sature (29a), sia omopolimeri costituiti praticamente solo da unità insature (29b). Dal norbornene, per es., sono stati ottenuti i due tipi di polimeri sotto indicati, secondo il catalizzatore usato:
Dal ciclopentene e dagli omologhi superiori (a eccezione del cicloesene) sono stati ottenuti solo omopolimeri costituiti da unità insature (29b). Il fatto che da queste cicloolefine non siano stati ottenuti, mediante catalizzatori Ziegler-Natta, omopolimeri costituiti solo da unità cicliche (29a) dipende verosimilmente da ragioni di carattere sterico. È stato già detto (v. sopra, 3, copolimeri olefinici) che questi monomeri danno tuttavia copolimeri con etilene in cui le unità derivanti dalla cicloolefina sono di tipo ciclico.
I catalizzatori tipici per la polimerizzazione del ciclopentene e degli omologhi superiori mediante apertura dell'anello sono a base di tungsteno o molibdeno. I più comuni catalizzatori sono ottenuti per reazione tra Al(C2H5)2Cl o AIC2H5Cl2 con WCl6, in presenza di attivatori vari.
È possibile polimerizzare, mediante apertura dell'anello, anche cicloolefine contenenti più di un doppio legame. Così dal cicloottadiene vengono ottenuti polimeri aventi la struttura di un polibutadiene-1,4:
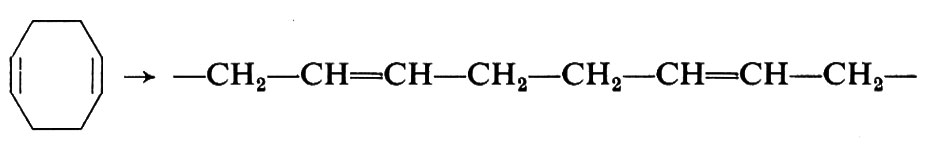
Anche cicloolefine aventi sostituenti possono essere polimerizzate; ciò offre la possibilità di preparare macromolecole equivalenti a copolimeri perfettamente alternati, difficili a preparare per altra via. Così il 5,6-dimetilcicloottadiene fornisce polimeri che possono essere considerati copolimeri perfettamente alternati del butadiene con il 2,3-dimetilbutadiene:
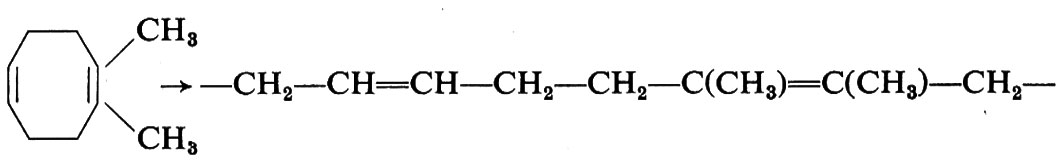
Analogamente il 5,6-dimetilcicloottene fornisce prodotti equivalenti a copolimeri alternati tra un'unità butadienica e due unità propileniche concatenate testa-testa. Appena alcuni anni fa la possibilità di preparare copolimeri di questo tipo sarebbe apparsa utopistica.
Meccanismo di polimerizzazione. - La polimerizzazione di cicloolefine a polimeri costituiti da unità cicliche (29a) si ammette avvenga con meccanismo analogo a quello della polimerizzazione delle α-olefine. La polimerizzazione mediante apertura dell'anello, cioè a polimeri costituiti da unità insature (29b), avviene invece con meccanismo del tutto diverso, e precisamente attraverso reazione di metatesi (v. Scott e altri, 1969; v. i contributi di Calderon, 1972). È questa una reazione generale delle olefine, aperte o cicliche, che può essere rappresentata dall'equazione:
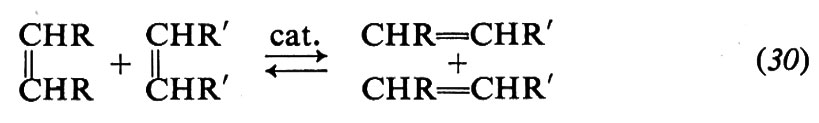
Nella polimerizzazione di cicloolefine a polimeri di tipo insaturo la rottura del ciclo avviene quindi al doppio legame.
In un primo tempo si era ritenuto che la polimerizzazione procedesse secondo lo schema bis-olefinico sotto indicato, attraverso rottura del doppio legame e formazione di macrocicli:
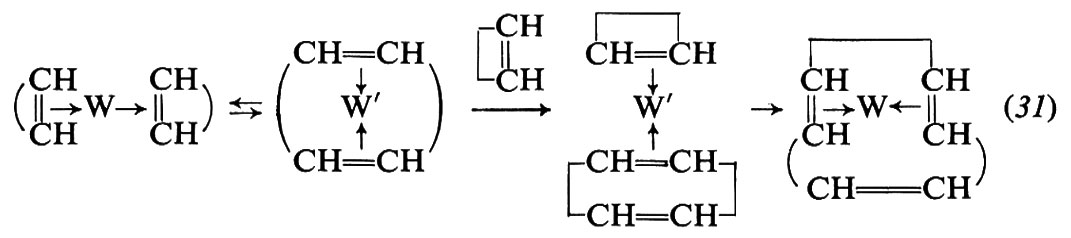
Successivamente è stato proposto un nuovo meccanismo, secondo il quale il sito attivo nella polimerizzazione è un legame metallo-carbene, M=CH−CH2 (M = metallo di transizione del catalizzatore). L'incorporazione del monomero avverrebbe attraverso la formazione di un intermedio metallociclobutano, che poi, scindendosi, darebbe luogo a un nuovo doppio legame e a un nuovo legame metallocarbene, che inizia di nuovo il ciclo catalitico (v. Hérisson e Chauvin, 1971):

Questo meccanismo interpreta meglio del precedente i risultati sperimentali ed è oggi largamente accettato. Non è ancora chiarito, tuttavia, come si arrivi alla formazione del legame metallo-carbene nel complesso catalitico.
5. Metodi fisici di determinazione strutturale dei polimeri
Molte delle proprietà dei polimeri lineari sono dipendenti dal peso molecolare delle macromolecole da cui essi sono costituiti. Tale peso molecolare è generalmente compreso tra 103 e 107; le singole macromolecole differiscono nel loro peso molecolare per la natura del processo di polimerizzazione da cui hanno preso origine. Quando si parla perciò di peso molecolare di un polimero si tratta sempre, come abbiamo già fatto osservare, di un certo tipo di media statistica, a meno che la determinazione non sia stata eseguita su di un polimero frazionato in componenti di peso molecolare in prima approssimazione uniforme. Siccome le misure sperimentali, che si basano sostanzialmente su proprietà fisiche di soluzioni polimeriche, portano in generale a tipi di media diversi, una verifica del fatto che un frazionamento sia stato bene eseguito può essere effettuata constatando che da diverse tecniche si ottengono identici valori del peso molecolare stesso.
Un elenco delle tecniche che consentono la determinazione del peso molecolare è riportato nella tab. IV.

Il frazionamento di un polimero (v. Cantow, 1967), le cui macromolecole abbiano analoga costituzione e configurazione, può essere effettuato per precipitazione frazionata. L'importanza della configurazione di un polimero sulla sua frazionabilità per pesi molecolari è stata mostrata in modo chiaro dalla scuola di Natta: polimeri del propilene a diversa distribuzione di configurazioni hanno solubilità completamente diverse in solventi idrocarburici, anche a parità di peso molecolare (v. fig. 8).
Talora è usabile il metodo della precipitazione frazionata, che, consiste nell'aggiunta di un precipitante a una soluzione diluita di polimero, il quale precipita gradualmente in frazioni di diverso peso molecolare. In generale, precipitano per prime le frazioni ad alto peso molecolare; solo aggiungendo grandi quantità di precipitante si separano le frazioni a basso peso molecolare. Un accurato frazionamento di un polimero per questa via è lungo e tedioso; si ricorre oggi con successo a tecniche più rapide, quali la cromatografia di permeazione su gel (gel permeation chromatography). In tale tecnica, la Separazione di macromolecole aventi differente peso molecolare avviene attraverso un insieme di colonne impaccate con un materiale poroso. Quando una soluzione di polimero è iniettata nella corrente del solvente eluente, le macromolecole a peso molecolare più alto, capaci in minor misura di permeare i pori della fase stazionaria, vengono eluite per prime. La concentrazione della soluzione in uscita dalle colonne può essere analizzata, per es., con un rifrattometro differenziale.
Un tipico risultato del frazionamento di un polimero è mostrato nella fig. 9. Appare evidente l'ampiezza di distribuzione dei pesi molecolari: per il polimero mostrato, questa si riflette in un valore di M-n = ~1,05•105 e di M-w = ~2•105. Il rapporto M-w/ M-n fornisce un indice della polidispersità di un polimero: per il caso di monodispersità si deve avere = 1; in tutti gli altri casi, come abbiamo già notato (v. cap. 3), M-w > M-n.
È opportuno accennare rapidamente alle tecniche usate per la determinazione dei pesi molecolari (v. Allen, 1959; v. Bonnar e altri, 1958; v. Ke, 1964).
L'uso delle proprietà colligative - ebullioscopia, crioscopia, osmometria - fornisce il peso molecolare medio in numero (M-n). La proprietà misurata - innalzamento del punto di ebollizione, abbassamento del punto di congelamento, pressione osmotica di una soluzione polimerica - è tanto più piccola quanto più alto è M-n. Ciò rende impossibile praticamente effettuare misure crioscopiche o ebullioscopiche quando M-n > ~ 3•104. Il metodo osmometrico permette invece la determinazione dei pesi molecolari in un più ampio campo. Se il peso molecolare di un polimero è 1,5•106 e la sua concentrazione in soluzione è 1•10-2 g/ml, la sua pressione osmotica in una soluzione benzenica, a 25 °C, è ancora equivalente a una colonna di liquido alta 2 mm. La precisione di lettura, effettuata con un catetometro, può essere migliore dell' 1%.
Il peso molecolare medio in peso (M-w) è il risultato, invece, di misure di diffusione della luce, dovuta alle fluttuazioni di concentrazione in una soluzione polimerica. Viene di solito misurata l'intensità di luce, diffusa a un angolo di 90° rispetto alla direzione di incidenza: essa è tanto più grande, a parità di concentrazione in peso di polimero, quanto più grande è M-w.
I metodi sopraindicati sono tecnicamente assai delicati e quindi poco convenienti per lavori di routine; il metodo più usato è quello viscosimetrico, che è semplice e rapido, anche se richiede una previa taratura. È stato visto che per tutti i polimeri vale la relazione:
[η] = KMa.
Il loro peso molecolare è pertanto funzione di una grandezza detta viscosità ‛intrinseca':
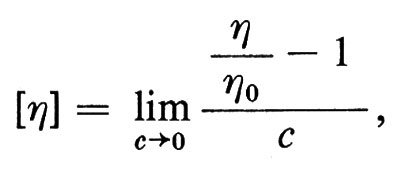
dove c è la concentrazione (g/dl), η è la viscosità della soluzione e η0 la viscosità del solvente.
K e a sono due costanti, tipiche del polimero in esame, da determinare per taratura in base a misure del tipo precedentemente descritto su polimeri frazionati, a peso molecolare noto.
È da osservare che per polimeri diversi provenienti dallo stesso monomero, disciolti in uno stesso solvente e misurando [η] alla stessa temperatura, i valori di K e di a dipendono in una certa misura sia dalla costituzione (compresa la distribuzione di pesi molecolari), sia dalla configurazione del polimero stesso. Il peso molecolare ricavato applicando la relazione è il cosiddetto ‛peso molecolare medio viscosimetrico'; esso risulta generalmente compreso tra M-n e M-w.
È stato dimostrato che il valore di a è maggiore di 0,5 (fino a raggiungere il valore massimo di 1,0) per soluzioni di macromolecole lineari in buoni solventi; in un solvente o miscela di solventi in cui il polimero sia vicino al punto di precipitazione a = 0,5.
Flory ha dimostrato che, sotto queste condizioni, vale la relazione:
[η]0 = KM1/2 = K′(-h-20)1/2
dove -h-02 è la distanza quadratica media tra i due punti terminali della macromolecola, nelle cosiddette condizioni imperturbate, calcolabile come se la macromolecola fosse allo stato gassoso, cioè come se ‛la sua conformazione non risentisse nè d'interazioni a lunga distanza tra singoli segmenti della macromolecola stessa, nè d'interazioni tra molecole di solvente e di polimero.
Come è stato già mostrato nel cap. 2, una viva corrente di ricerca si occupa attualmente proprio della determinazione dei valori di -h-20 e della loro dipendenza dalla temperatura, partendo da primi principi, in funzione della costituzione e configurazione dei sistemi polimerici esaminati.
L'uso di metodi fisici di determinazione strutturale per lo studio della costituzione e della stereoregolarità dei polimeri ha avuto un enorme sviluppo in questi ultimi anni, raggiungendo livelli di sofisticazione sempre più alti.
Riteniamo opportuno accennare brevemente alle tecniche dell'infrarosso (v. Zbinden, 1964), della risonanza magnetica nucleare (v. Bovey, 1969) e dei raggi X (v. Kakudo e Kasai, 1972), illustrandole con qualche esempio.
Abbiamo già visto che il butadiene nella polimerizzazione può dar luogo a due unità costitutive diverse, corrispondenti al concatenamento 1,4 o 1,2; le unità a concatenamento 1,4 possono avere una configurazione del doppio legame cis o trans. Gli spettri infrarossi di soluzioni in solfuro di carbonio ad altissimo tenore rispettivamente in unità a concatenamento 1,2(A), in unità a concatenamento 1,4 trans (B), in unità a concatenamento 1,4 cis (C) sono riportati nella fig. 10, nella regione spettrale tra 9 e 16 μm: bande caratteristiche compaiono rispettivamente a 10,89 μm, a 10,35 μm e a 13,50 μm. Una volta determinati i coefficienti di assorbimento di tali bande, è possibile pervenire al rapporto delle varie composizioni per un polimero qualunque.
L'uso della risonanza magnetica nucleare (RMN) si è rivelato in tempi recentissimi uno dei metodi più adatti per pervenire allo studio della distribuzione di configurazioni lungo la catena di un polimero.
Riteniamo interessante riferirci al caso del polimetilmetacrilato:
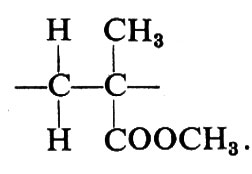
Consideriamo i due protoni del gruppo metilenico nelle due possibili configurazioni locali:
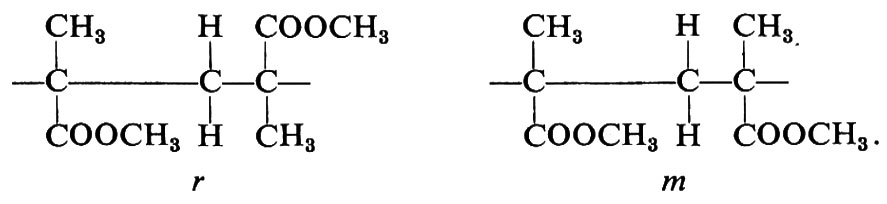
Nel primo caso i due protoni sono in una situazione sterica identica e daranno luogo a un unico segnale (se non si tiene conto delle possibili distribuzioni di configurazioni delle unità monomeriche adiacenti); nel secondo caso i due protoni risultano in una situazione sterica diversa e dovranno dar luogo a spostamenti chimici diversi, generando un tipico quartetto (v. fig. 11).
L'uso di campi magnetici sempre più alti ha consentito di ottenere spettri a sempre più elevata risoluzione, permettendo di gettar luce sulla distribuzione di configurazioni in polimeri non completamente tattici. Consideriamo per questo lo spettro RMN di un polimetilmetacrilato prevalentemente sindiotattico, nella regione di risonanza del gruppo metilenico: il suo spettro a 60 MHz è mostrato nella fig. 11. Già per tale campo si vede che per le configurazioni r non si ha una sola riga, ma questa accenna a risolversi in più linee. A 220 MHz (v. fig. 12) la risonanza dovuta a diadi r si risolve chiaramente in tre linee, dovute alle tetradi che si possono associare a una configurazione diadica r: esse sono state attribuite, anche basandosi sulla loro intensità relativa, alle tetradi mrm (la meno intensa), rrm (equivalente a mrr) e rrr (la più intensa).
Un analogo ragionamento si può fare per la risonanza dei protoni dei gruppi CH3 legati direttamente alla catena: già a 60 MHz si osservano nettamente tre risonanze, che debbono essere ragionevolmente attribuite alle tre possibili triadi mm (la meno intensa), mr (equivalente a rm) e rr (la più intensa). Nello spettro a 220 MHz vi sono chiari indizi di un'ulteriore suddivisione di tali risonanze in quelle delle corrispondenti pentadi. Siccome l'intensità di ciascuna risonanza è proporzionale alla concentrazione della singola sequenza configurazionale che l'ha originata, è evidente come il metodo RMN consenta - specie se usato in condizioni di alta risoluzione - di fornire un quadro statistico pressoché completo della distribuzione di configurazioni.
Nel caso in esame, l'analisi degli spettri ha consentito di stabilire al di là di ogni dubbio che la distribuzione di configurazioni in un polimero radicalico (come quello da noi esaminato, preparato a bassa temperatura) è di tipo bernoulliano.
Riteniamo infine utile dare al lettore un'idea delle informazioni conformazionali che possono essere ricavate su di un polimero cristallino con studi röntgenografici.
L'immagine di diffrazione fornita da una fibra orientata consente di determinare il periodo d'identità (C) lungo l'asse della catena e il numero di unità monomeriche (M) che si ripetono nel periodo d'identità. Nel caso di polimeri con strutture elicoidali complesse, la distribuzione d'intensità consente di determinare anche il numero di passi dell'elica necessari alla ripetizione identica dopo M unità monomeriche (N).
Le grandezze M, N, C dipendono a loro volta dall'insieme di coordinate interne che caratterizzano l'unità strutturale e, in particolare, dall'insieme di angoli di rotazione interna che caratterizzano la conformazione macromolecolare.
La determinazione del modo d'impacchettamento delle catene di un polimero è resa invece più difficile, rispetto ad analoghe determinazioni su composti a basso peso molecolare, per l'impossibilità di ottenere cristalli singoli di dimensioni sufficienti. Numerose determinazioni strutturali sono state tuttavia effettuate, in particolare dalla scuola di Natta e Corradini, in questi ultimi anni; è stato possibile anche, in alcuni casi semplici, arrivare alla previsione del modo d'impacchettamento delle macromolecole stesse.
Un'informazione diretta e tecnologicamente interessante, che può essere fornita dal solo spettro di polveri di un polimero, è la percentuale di cristallinità: a una variazione di cristallinità corrisponde la variazione di numerose altre proprietà fisiche di un polimero, quali la densità, il carico di rottura, il modulo di elasticità.
6. Comportamento nell'uso dei polimeri in relazione alla struttura
a) Possibili stati fisici dei sistemi macromolecolari
I materiali polimerici trovano le loro più importanti applicazioni pratiche perché sono plastici durante la lavorazione (in modo da poter essere foggiati nella forma desiderata), mentre, nell'uso, devono assumere una forma sufficientemente rigida e duratura.
L'uomo ha utilizzato la plasticità di alcuni materiali fin dagli albori della civiltà: egli imparò presto che un impasto di acqua e argilla può essere modellato, anche sotto la pressione delle dita, nella forma di vasi e stoviglie e può mantenere la forma acquistata per il tempo sufficiente a essere posto in un forno, dove perde la plasticità per trasformarsi in un solido rigido.
Le sostanze organiche di natura macromolecolare, di cui noi utilizziamo la plasticità in opportune condizioni di lavorazione e la rigidità nell'uso, sono dette ‛materie plastiche'. Anche se alcune materie plastiche, come la ceralacca, erano ben note all'uomo fin dall'antichità, soltanto in tempi recenti egli è pervenuto alla scoperta, alla produzione e all'utilizzazione di una varietà enorme di tali materiali, il cui consumo aumenta di giorno in giorno; nel breve volgere di dieci anni, per esempio, si è passati da consumi di meno di 1 kg pro capite l'anno a oltre 10 kg pro capite l'anno.
Gli stati fisici delle materie plastiche sono classificati come: 1) stato amorfo-vetroso; 2) stato termoindurito; 3) stato amorfo-viscoso; 4) stato semicristallino.
Le materie plastiche semicristalline possono essere tirate in fili per dar luogo a fibre.
Ad alcuni materiali polimerici (le gomme) corrisponde invece uno stato detto elastico-gommoso (5).
In questo capitolo esamineremo questi cinque stati.
Lo stato amorfo-vetroso è analogo, dal punto di vista fisico, allo stato vetroso che è tipico dei vetri inorganici: la temperatura di transizione vetrosa denota, in entrambi i casi, la transizione da un liquido vetroso, caratterizzato da rigidità, a un liquido viscoso. Anche la lavorazione di un polimero usato nello stato amorfo-vetroso (per es., il polistirene, che è fragile e trasparente) è analoga a parte la temperatura a cui viene condotta la lavorazione, molto più bassa a quella di un vetro inorganico. Dal punto di vista strutturale vi sono però notevoli differenze. In un vetro inorganico, per es. silice vetrosa, noi abbiamo un insieme di legami chimici che reticolano gli atomi in tre dimensioni. La struttura è caratterizzata dal fatto che ciascun atomo di silicio è coordinato tetraedricamente a quattro atomi di ossigeno, e ciascun atomo di ossigeno è legato a due atomi di silicio. A causa della natura parzialmente ionica dei legami, il vetro di silice diventa un liquido viscoso al di sopra della temperatura di transizione, in quanto la mobilità a livello molecolare è assicurata da un continuo reversibile rompersi e riformarsi di legami. La rottura dei legami covalenti può avvenire, naturalmente, anche in un polimero, a temperature sufficientemente elevate; tuttavia una tale rottura di legami sarebbe irreversibile, né può essere invocata per spiegare la fluidità di un polimero al di sopra della temperatura di transizione vetrosa. Al di sotto della temperatura di transizione vetrosa, le molecole polimeriche divengono incapaci di scorrere l'una sull'altra, per l'insufficiente mobilità dovuta proprio alla loro natura macromolecolare, nè possono cristallizzare, per le irregolarità nella loro costituzione o configurazione.
Lo stato termoindurito dei polimeri somiglia viceversa, dal punto di vista strutturale, a quello di un vetro, anche se ne differisce in molti aspetti fisici. Esso è proprio di quelle materie plastiche che induriscono e perdono la loro plasticità per riscaldamento prolungato, e sono dette perciò ‛resine termoindurenti'. L'indurimento e la perdita di plasticità sono causati dalla formazione di una struttura molecolare non lineare, ma reticolata, che impedisce alle macromolecole di muoversi l'una rispetto all'altra. È ovvio che uno dei monomeri, tra quelli che vengono polimerizzati per dare una materia plastica termoindurente, deve essere almeno trifunzionale, a differenza dei monomeri che danno materie plastiche capaci di essere modellate a caldo un numero indefinito di volte e che prendono perciò il nome di ‛resine termoplastiche'.
La prima materia plastica termoindurente fu scoperta da L. H. Baekeland nel 1909. Facendo reagire fenolo e formaldeide si forma un prodotto resinoso, che diventa plastico per riscaldamento; in queste condizioni può essere compresso in oggetti di varia forma. Prolungando il riscaldamento dello stampo, il materiale indurisce e mantiene permanentemente la forma che gli è stata data. Questo materiale fu chiamato ‛bachelite' (dal nome dello scopritore). Nella bachelite i legami chimici sono sostanzialmente covalenti, come è mostrato nella fig. 13.
I polimeri termoindurenti si differenziano dai vetri perché non hanno una temperatura di transizione vetrosa; riscaldati ad alte temperature, vengono irreversibilmente degradati a prodotti a basso peso molecolare.
I ‛polimeri semicristallini' vengono facilmente lavorati allo stato fuso e posseggono una serie di proprietà fisiche interessanti: essi costituiscono perciò il settore in più rapida espansione dell'intera chimica macromolecolare.
Il ‛grado di cristallinità' di un polimero è quella frazione del polimero stesso che dà luogo, all'esame ai raggi X, a linee nette e non ad aloni slargati (v. fig. 14). La cristallinità è associata all'esistenza, nella massa del polimero, di strutture ordinate in tre dimensioni.
Le condizioni perché un polimero possa essere cristallino sono state accennate nel cap. 2; le macromolecole debbono poter assumere una conformazione regolare lungo un asse e si associano poi, parallele l'una all'altra, per dar luogo a cristalli. La conformazione regolare non si mantiene nel fuso e non può realizzarsi per un polimero non stereoregolare. Per esempio, il polipropilene atattico è amorfo a temperatura ambiente e non può essere mai cristallizzato; al di sotto di −20 °C è nello stato amorfo-vetroso; al di sopra è nello stato amorfo-viscoso, che è analogo a quello posseduto dai polimeri semicristallini al di sopra del punto di fusione. Invece, il polipropilene isotattico fonde a + 176 °C e costituisce quindi un'importante materia plastica, capace anche di dare eccellenti fibre.
Un certo grado di ordine delle macromolecole di un polimero lineare si mantiene anche allo stato fuso; nel caso del polietilene, l'analisi della distribuzione radiale delle distanze interatomiche mostra che nel polimero fuso si mantiene un certo grado di correlazione tra gli angoli di rotazione interna di ogni singola catena; orientamenti correlati di macromolecole vicine sussistono solo, però, in un intorno molto piccolo dei punti di contatto. I lavori teorici e sperimentali di numerosi ricercatori sembrano indicare la giustezza di un modello siffatto.
Nello stato amorfo-viscoso le macromolecole possono muoversi l'una rispetto all'altra. Tuttavia, a differenza da quanto accade per i composti a basso peso molecolare, tali movimenti non riguardano, in ogni stadio elementare, l'intera molecola, che è costituita da una catena molto lunga: sembra che l'unità cinetica per ogni movimento elementare sia costituita da segmenti che comprendono 20-40 atomi della catena con energie di attivazione dello stesso ordine di grandezza di quelle occorrenti per lo scorrimento viscoso. Non è pertanto illogico supporre che le forze di frizione interne in un polimero fuso ritardino la velocità di deformazione tanto di più, quanto più alto è il peso molecolare medio in peso Mw (v. fig. 15).
Comunque, il comportamento reologico di un polimero è molto complesso e risulta dipendente anche dalla distribuzione dei pesi molecolari, dalla presenza o assenza di ramificazioni nella catena e, ovviamente, dalla natura chimica del polimero stesso. La reologia del fuso è estremamente importante per le tecniche di lavorazione. Per esempio, le tecniche di formatura sotto vuoto richiedono un fuso polimerico che sia viscoso anche per piccole ‛velocità di deformazione'. Al contrario, la formatura per iniezione sotto pressione richiede fusi assai meno viscosi a più alte velocità di deformazione: lo stesso prodotto polimerico non può essere perciò accettabile che per l'una o per l'altra tecnica.
Per un polimero che sia incapace di cristallizzare per ragioni strutturali intrinseche (basti pensare a un polimero vinilico atattico) o per ragioni cinetiche (per es., alcuni polimeri isotattici di stireni sostituiti), a una certa temperatura, detta ‛temperatura di transizione vetrosa' Tg si ha una transizione dallo stato amorfo-viscoso allo stato amorfo-vetroso.
Termodinamicamente, la transizione vetrosa corrisponde a una transizione del 2° ordine. Mentre in una transizione del 1° ordine (per es. da liquido a solido cristallino, per un composto a basso peso molecolare) funzioni termodinamiche come V, H, S manifestano una discontinuità, in una transizione del 2° ordine esse rimangono continue (v. fig. 16), mentre le loro derivate parziali rispetto alla temperatura o alla pressione sono discontinue.
Dal punto di vista strutturale la capacità di scorrimento segmentale delle macromolecole l'una nei riguardi dell'altra viene meno quando si passa dallo stato viscoso a quello vetroso; al di sotto di Tg le posizioni relative delle macromolecole risultano congelate.
La temperatura Tg dipende fortemente dalla struttura: può essere più bassa di −100 °C per certi polimeri e più alta di + 100 °C per altri. Al di sotto di Tg, i polimeri diventano fragili in maggiore o minor misura; la resistenza all'urto è pure una sensibile funzione della struttura. E infatti, pur essendo le macromolecole congelate nelle loro posizioni, sotto a Tg sono possibili ancora rotazioni di gruppi laterali o rotazioni cooperative attorno ai legami della catena principale che coinvolgono un piccolo numero di atomi, ma nessun movimento globale di traslazione di un segmento di macromolecola. Questi movimenti locali possono assorbire l'energia di un urto, evitando la rottura.
Un esempio può essere costituito dal policarbonato di struttura
che esiste nello stato amorfo-vetroso al di sotto di 150 °C. La sua eccezionale resistenza all'urto anche a basse temperature è da ritenersi associata alla possibilità di rotazione cooperativa del gruppo
Formula
, mentre tutto il resto della molecola resta congelato nella posizione che aveva in precedenza.
Per quanto riguarda lo stato semicristallino, c'è da premettere che in un materiale solido sono cristalline quelle porzioni in cui è presente ordine a lunga distanza, caratterizzato in generale dalla ripetizione in tre dimensioni del contenuto della cella elementare. Quando un polimero cristallizza, le macromolecole assumono conformazioni periodiche lungo un asse e si impacchettano in modo parallelo tra loro. A differenza delle sostanze a basso peso molecolare, ciascuna macromolecola fa parte di più celle elementari, e uno degli assi della cella elementare coincide perciò con l'asse di ripetizione periodica della conformazione di una singola macromolecola. Affinché una traslazione sia un'operazione di simmetria possibile per la ripetizione di atomi della stessa molecola, occorre che questa sia, almeno idealmente, di lunghezza infinita e di costituzione e configurazione perfettamente regolari. Salvo casi particolari, infatti, la presenza di quantità più o meno grandi d'irregolarità costituzionali e/o configurazionali diminuisce e talvolta annulla la capacità di cristallizzare di un polimero, cambiandone in modo drastico le proprietà fisiche.
Negli ultimi anni sono stati condotti molti studi sulla morfologia di cristalli singoli di polimeri e sulla morfologia di aggregati policristallini, formati sotto varie condizioni sperimentali. Un curioso risultato di tali ricerche è che la dimensione più corta di un cristallo di polimero è nella direzione dell'asse della catena, ed è dell'ordine del migliaio di nanometri (v. fig. 17). Siccome la lunghezza di una macromolecola singola è molto maggiore della dimensione dei cristalli lungo l'asse della catena, ne segue che ciascuna macromolecola deve ripiegarsi su se stessa parecchie volte sulla superficie esterna del cristallo. Pertanto, la superficie esterna di un cristallo singolo di polimero non ha lo stesso grado di ordine riscontrabile all'interno del cristallo stesso; si sta sempre più facendo strada l'ipotesi che l'alone amorfo, sempre presente nello spettro X di un polimero cristallino, possa derivare in tutto (per i polimeri più cristallini) o in parte dalle regioni superficiali più disordinate dei cristalli. Per i polimeri più cristallini, al modello bifasico, che comprendeva regioni amorfe e regioni cristalline, si va sostituendo un modello costituito sostanzialmente solo da cristalli molto piatti, associati tra loro in modo intricato. Ciascun cristallo di polimero, proprio per la sua forma di piattina, risulta avere una notevole flessibilità senza per questo rompersi. Infatti alla piegatura si oppongono solo deboli forze di van der Waals, perpendicolarmente alla direzione degli assi delle catene, mentre sulla superficie la struttura è tenuta insieme ancora da legami covalenti.
È stato inoltre ipotizzato che una cospicua frazione percentuale di macromolecole possa far parte di più cristalli, nella massa di un materiale polimerico, dando ulteriore coerenza al materiale stesso (v. fig. 18), e sono state addotte convincenti prove sperimentali al riguardo. Le macromolecole che fanno parte di più cristalli conferiscono ai materiali polimerici orientati ('fibre') un'alta tenacità nella direzione di allungamento, come vedremo più avanti (v. fig. 19).
Come in un metallo, sono presenti in un polimero cristallino difetti puntuali e dislocazioni. Studi teorici e sperimentali indicano che tali difetti possono essere imputati, oltre che a difetti costituzionali e/o configurazionali eventualmente tollerati dal reticolo, a terminali di catena non segregati nella parte amorfa, piegature di catena e attorcigliamenti (v. fig. 20). Questi ultimi possono essere responsabili del trasporto di materia che può avvenire nella massa di un polimero cristallizzato. Sembra che i fenomeni che avvengono durante la deformazione di un polimero sotto sforzo possano essere spiegati in modo coerente, in maniera analoga a quanto avviene nei metalli.
Il tipo più comune di aggregato di cristalliti è lo sferulita (v. fig. 21). Osservati al microscopio, gli sferuliti mostrano una simmetria radiale rispetto a un centro. La crescita degli sferuliti si arresta quando essi vengono a contatto tra di loro, e ciò conduce a una distorsione rispetto alla simmetria sferica.
Le osservazioni microscopiche e röntgenografiche hanno dimostrato che le fibrille che s'irradiano dal centro dello sferulita sono costituite da nastri spiralizzati di cristalli. Ne risulta che il raggio dello sferulita è perpendicolare all'asse della catena, e parallelo a uno degli altri assi del cristallo (v. fig. 22).
b) Proprietà meccaniche dei materiali polimerici
I materiali polimerici amorfi, al di sotto della temperatura di transizione vetrosa, si comportano come materiali fragili. Non è possibile infatti misurare nessuna deformazione plastica e la duttilità, misurata dall'allungamento a rottura, è molto bassa. In maniera analoga a quanto si riscontra per i metalli (pur tenendo conto della diversa entità delle deformazioni e dei carichi), il diagramma sforzo-allungamento di un materiale polimerico semicristallino mostra una regione di allungamento reversibile e una regione di deformazione plastica irreversibile, finché non viene raggiunto l'allungamento a rottura (v. fig. 23). Per materiali polimerici già deformati plasticamente e orientati (fibre), la regione di deformazione plastica sparisce nel diagramma e i carichi, riferiti al peso di un campione già orientato di data lunghezza, diventano comparabili con quelli misurabili per un metallo.
Nei materiali metallici le proprietà meccaniche sono governate dalla generazione e dal movimento di dislocazioni attraverso il reticolo cristallino. Nei materiali polimerici, i primi stadi della deformazione comportano lo scorrimento viscoso delle parti non cristalline e un riassestamento delle lamelle che si irradiano dal centro degli sferuliti. Questi ultimi diventano ellissoidali e perdono la simmetria sferica. I legami interlamellari, forniti dalle macromolecole che fanno parte di più di un cristallo, si oppongono sempre di più a queste deformazioni, per cui da un certo momento in poi cominciano a prevalere gli scorrimenti su piani cristallografici associati al movimento di dislocazioni. Ne risulta che un materiale polimerico semicristallino può essere allungato di parecchie centinaia per cento senza rompersi. In un campione allungato completamente, abbiamo già notato come i cristalli di polimero diventino tutti orientati nella direzione di stiro.
La rottura avviene allora anche per lo spezzamento di legami covalenti.
Lo stato gommoso-elastico. - La fig. 24 mostra il diagramma sforzo-allungamento di un campione gommoso-elastico di polimero. Mentre i solidi hookeiani mostrano un estensione reversibile fino a valori dell' 1% o meno, si possono avere, per materiali polimerici elastici, estensioni fino al 1.500% completamente reversibili.
I requisiti perché un materiale si comporti alla stregua di gomma sono principalmente tre: il primo è che sia costituito da molecole a lunga catena; il secondo è che le sue catene siano flessibili, a causa della facilità di rotazione attorno a legami C−C (questo requisito è soddisfatto in generale da quei materiali polimerici in cui sono presenti solo deboli forze di van der Waals tra macromolecole diverse e che hanno Tg e, se capaci di cristallizzare, temperatura di fusione Tm al di sotto della temperatura ambiente); il terzo è che le macromolecole del polimero vengano qua e là reticolate tra loro con legami chimici in modo che passino dallo stato viscoso-amorfo allo stato gommoso, in cui sotto tensione non possono più dar luogo a scorrimento viscoso (v. fig. 25). La reticolazione tridimensionale richiede la precedente presenza nelle macromolecole di punti chimici reattivi nei riguardi degli agenti reticolanti (per es., doppi legami, che reagiscono con lo zolfo per formare ponti). Il processo si chiama ‛processo di vulcanizzazione'.
Un polimero ben noto che soddisfa i requisiti surriportati è il poliisoprene 1,4-cis; esso costituisce in natura la gomma naturale, ma oggi viene in gran quantità sintetizzato con processi di polimerizzazione stereospecifica.
Altri polimeri di cui s'intravedono importanti sviluppi futuri sono i copolimeri etilene-propilene-diene e il polipentenamero trans.
Nel caso di un materiale gommoso-elastico, l'elasticità non è associata a cambiamenti dell'energia interna, ma a cambiamenti di entropia. Per la facile rotazione intorno ai legami semplici e in presenza di molte diverse conformazioni praticamente isoenergetiche, un segmento di catena può essere esteso sotto sforzo da una situazione di alta entropia in cui risulta raggomitolato a una situazione di minor entropia in cui risulta più esteso; la rimozione dello sforzo provoca il ritorno alla situazione di partenza proprio per la presenza delle reticolazioni indotte dalla vulcanizzazione, che, senza irrigidire completamente le catene, ne impediscono lo scorrimento viscoso.
Nella relazione tra sforzo e allungamento:
F = (δE/δL))T,P − T(δS/δL)T,P,
dove F è la forza applicata, L è la lunghezza del campione, E è l'energia interna, S è l'entropia, P e T sono la pressione e la temperatura, rispettivamente, si può notare come il primo termine sia zero per le gomme polimeriche ideali, mentre sia zero il secondo termine per i solidi hookeiani ideali.
È da notare infine che una certa elasticità è presente nei solidi polimerici fusi, il cui comportamento è pertanto definito come visco-elastico. A causa dell'alto peso molecolare, le catene si intrecciano tra loro; gli intrecciamenti agiscono come labili reticolazioni.
c) I polimeri e l'ambiente
Può essere interessante discutere infine dell'influenza dell'ambiente sui polimeri.
La temperatura è una variabile molto importante per decidere sull'uso di un polimero per un certo scopo. Al di sotto di Tg tutti i polimeri sono in maggiore o minor misura fragili; il polipropilene isotattico può essere usato per fare oggetti che occasionalmente possono andare a temperature sotto lo zero, solo se opportunamente modificato chimicamente in modo da abbassare la Tg delle sue parti amorfe. Occorre inoltre ricordare che la transizione dallo stato semicristallino a quello viscoso-amorfo inizia all'incirca 30 °C al di sotto del punto di fusione Tm. Cosi il propilene isotattico è capace di resistere all'azione dell'acqua bollente (Tm = 176 °C), mentre il polietilene a bassa densità non lo è (Tm 115 °C).
Sono stati di recente sviluppati copolimeri a blocchi di polipropilene isotattico (in prevalenza) e polietilene. Tali polimeri hanno praticamente la Tg del polietilene, e quindi resistono bene agli urti, alle basse temperature, mentre posseggono le caratteristiche di resistenza alle alte temperature del polipropilene. È chiaro da questo esempio come si possa trovare la maniera di regolare le proprietà di un polimero a seconda dell'uso a cui si intende destinarlo.
Notevoli sforzi sono stati fatti di recente per ottenere polimeri dotati di resistenza alle alte temperature; e infatti uno dei più grossi svantaggi delle materie plastiche in confronto ai metalli e ai materiali ceramici è la inutilizzabilità (in generale) alle alte temperature. Un irrigidimento della catena principale e delle catene laterali o un aumento delle forze intermolecolari sono tutti fattori che innalzano il punto di fusione del polimero. Essendo infatti Tm = ΔHf/ΔSf, dove Δf e ΔSf sono rispettivamente l'entalpia e l'entropia di fusione, si vede che un irrigidimento della catena porta a una diminuzione di ΔSf, mentre un aumento delle forze intermolecolari (legami a idrogeno, interazioni dipolo-dipoli) porta a un aumento di ΔHf. L'introduzione di anelli aromatici nello scheletro di un polimero ha permesso di ottenere polimeri che si comportano in modo eccellente per lunghi tempi a temperature superiori ai 300 °C.
Un altro problema delle materie plastiche è la loro infiammabilità. Questa può essere ridotta per aggiunta di opportuni ritardanti o con cambiamenti della struttura chimica. I polimeri che contengono cloro e/o fluoro sono molto meno infiammabili dei corrispondenti idrocarburi. Il teflon - un polimero che contiene solo carbonio e fluoro - è virtualmente ininfiammabile.
Inoltre, i polimeri subiscono una maggiore o minore degradazione quando restano esposti agli agenti atmosferici (aria, luce). Il fenomeno trova il suo parallelo nella corrosione dei metalli. Per esempio, le gomme possono lentamente perdere la loro elasticità, mentre i materiali plastici possono aumentare la loro fragilità. Tutti questi fenomeni sono dovuti a cambiamenti strutturali che avvengono nei materiali, e sono principalmente causati da reazioni con l'ossigeno, che hanno luogo anche a temperatura ambiente (autossidazione). Si tratta di reazioni a catena, la cui velocità è aumentata dall'esposizione alla luce o da tracce di vari materiali catalitici. Esse possono essere ritardate, anche per anni, dall'aggiunta di piccole quantità di sostanze organiche ‛sacrificiali', chiamate ‛antiossidanti', capaci d'interrompere le catene cinetiche delle reazioni di ossidazione.
Anche sforzi prolungati hanno un'influenza negativa sulle proprietà meccaniche delle materie plastiche, al di là di certi valori dello sforzo. Come esempio, si riporta il comportamento di un campione di polietilene sotto sforzo e nel tempo, che illustra il fenomeno chiamato creep (v. fig. 26).
Infine, sempre più importante sta diventando il problema inverso, cioè quello dell'influenza dei polimeri sull'ambiente. Le produzioni petrolchimiche sono di per sè produzioni che portano a un notevole inquinamento dell'ambiente. La distruzione delle grandi quantità di oggetti di materia plastica fuori uso è pure un problema, perché con l'aumento continuo della produzione, destinata prevalentemente a beni poco durevoli, si finirà con l'avere un accumularsi di oggetti in plastica fuori uso, di lentissima o nulla degradabilità molecolare. Anche la combustione di tali oggetti presenta gravi problemi d'inquinamento, soprattutto per i polimeri che contengono cloro o fluoro. Ricerche sono in corso, in varie parti del mondo, intese ad arrivare alla produzione di polimeri di minore potenziale inquinante, sia in relazione alle possibili tecnologie che si sviluppano per la loro distruzione, sia in quanto biodegradabili (v. inquinamento ambientale). Per quanto riguarda quest'ultimo punto, gli studi finora condotti evidenziano la possibilità di rendere biodegradabili dei materiali polimerici quando in essi siano presenti gruppi che, assorbendo nell'ultravioletto (doppi legami, gruppi carbonilici, gruppi aromatici, ecc.), facilitano l'attacco fotochimico del polimero da parte dell'ossigeno atmosferico. Il polimero così ossidato diventa biodegradabile nella maggior parte dei casi (fotobiodegradazione).
7. Futuro della chimica macromolecolare
Nelle pagine precedenti è stato esaminato lo stato attuale della chimica macromolecolare nei suoi vari aspetti. Possiamo chiederci ora quali saranno gli sviluppi futuri di questa scienza.
I prossimi decenni vedranno certamente un ampliarsi delle nostre conoscenze sui metodi di polimerizzazione oggi usati. La polimerizzazione radicalica può ormai essere considerata un metodo ‛classico' e per questa non sembrano da prevedersi progressi straordinari, ma piuttosto una lenta acquisizione di nuovi dati. Nel campo della polimerizzazione ionica ci si attende di arrivare a una migliore comprensione dei processi di trasferimento e di terminazione che si verificano nella polimerizzazione di vari monomeri, sia idrocarburici che contenenti eteroatomi (aldeidi, eteri ciclici, eteri vinilici). Ciò potrebbe condurre all'individuazione di nuovi polimeri viventi, il che permetterebbe, tra l'altro, la preparazione di nuovi polimeri a blocchi.
Un campo che continuerà ancora per lungo tempo ad attirare l'attenzione dei ricercatori è quello della polimerizzazione con catalizzatori a base di metalli di transizione. Il lavoro fatto negli ultimi due decenni ha condotto a notevolissimi risultati pratici, ma moltissimo rimane da fare sul piano interpretativo, soprattutto per quanto riguarda i fattori che determinano la stereospecificità. I progressi in questo campo verranno sia dallo studio dei processi di polimerizzazione sia dallo studio della chimica organometallica.
Una maggiore conoscenza dei fattori che determinano la stereospecificità si tradurrà anche in miglioramenti di carattere pratico.
Notevole interesse attirerà ancora la polimerizzazione per condensazione. È questo il più versatile tra i metodi di polimerizzazione e appunto per questo è stato oggetto di rinnovata attenzione in questi ultimi anni, soprattutto per la preparazione di polimeri aventi buone caratteristiche meccaniche anche ad alta temperatura. Molti problemi rimangono ancora da risolvere, sul piano della tecnica preparativa, che oggi limitano l'impiego di questo metodo.
Le maggiori conoscenze nel campo dei metodi di polimerizzazione condurranno alla preparazione di polimeri di nuovo tipo. Già in questi ultimi anni sono apparsi numerosi nuovi tipi di polimeri che per ora rimangono curiosità di laboratorio, ma che danno un'idea delle possibilità di sviluppo in questo settore.
Nel campo dei polimeri del butadiene o dell'isoprene, per es., sono stati preparati, con catalizzatori a base di metalli di transizione, polimeri cosiddetti ‛equibinari', perchè costituiti da due tipi di unità monomeriche, in rapporto praticamente 1 : 1 (polibutadiene 1,4-cis-1,4-trans; polusoprene 1,2-1,4-cis). Inoltre, sempre nel campo dei polimeri dienici, sono stati ottenuti, con catalizzatori vari, copolimeri alternati butadiene-acrilonitrile in cui le unità butadieniche sono esclusivamente 1,4-trans. Sia il meccanismo di sintesi, sia le possibili applicazioni di questi polimeri, che sono solo un esempio fra tutti quelli possibili, sono ancora da esplorare.
Una classe di polimeri che ha recentemente attratto l'attenzione dei chimici è quella dei polimeri organometallici, cioè polimeri contenenti atomi di metalli, in particolare di metalli di transizione, nella catena principale o in una catena laterale. Da quel poco che finora si conosce si prevedono notevoli usi potenziali per questi polimeri nel campo dei materiali resistenti ad alta temperatura, dei semiconduttori, dei fotoconduttori e anche della catalisi.
L'esigenza di ottenere macromolecole aventi una struttura preordinata adatta a usi particolari spinge oggi i chimici ad approfondire un metodo di sintesi che è attualmente solo agli inizi, ma che si mostra di notevole interesse potenziale. Il metodo consiste nella preparazione di segmenti più o meno lunghi di macromolecole lineari contenenti gruppi reattivi alle due estremità (gruppi idrossilici, amminici, carbossilici, isocianici, ecc.). Potranno verosimilmente essere ottenuti, anche sul piano industriale, blocchi di polietilene, di polistirene, di poliisobutene, di poliacrilonitrile, ecc. Questi segmenti di macromolecole (detti anche ‛prepolimeri' o building blocks) possono essere uniti tra loro, per dare luogo a macromolecole più lunghe. Appare chiaro che il numero di combinazioni possibili è enorme, potendosi ottenere attraverso questo metodo copolimeri a blocchi contenenti segmenti di due o tre tipi, ciascuno di lunghezza appropriata. Si offre la possibilità di ottenere prodotti aventi proprietà che variano in modo continuo entro un largo intervallo, al variare della struttura.
Si può quindi concludere che, come risultato del lavoro di ricerca oggi in corso, si arriverà abbastanza presto a disporre di un numero elevato di nuovi polimeri, capaci di soddisfare le più svariate esigenze pratiche.
Al di fuori del campo della chimica macromolecolare preparativa, sensibili progressi sono da prevedersi nel campo della caratterizzazione dei polimeri. Si avranno metodi più rapidi per la determinazione dei pesi molecolari medi, per il frazionamento e per la determinazione della distribuzione dei pesi molecolari. Inoltre, attraverso i metodi di risonanza magnetica nucleare sarà possibile arrivare a una caratterizzazione molto migliore dei polimeri, soprattutto per quanto riguarda la regolarità configurazionale.
Un esame delle prospettive future della chimica macromolecolare non può fare a meno di prendere in considerazione i campi di applicazione dei materiali polimerici, cioè quello delle fibre, degli elastomeri e delle materie plastiche.
Le fibre tessili artificiali presentano alcune eccellenti proprietà, quali la resistenza all'usura, alla rottura, agli agenti chimici e biologici; esse possono inoltre essere usate in miscela con le fibre naturali. Tuttavia, per nessuna delle fibre tessili artificiali finora ottenute si può immaginare un impiego così universale come per il cotone e la lana. La realizzazione di fibre tessili aventi proprietà vicine il più possibile al cotone o alla lana è certamente uno degli obiettivi della chimica macromolecolare per il prossimo futuro. Già ora si cerca di preparare fibre aventi carattere più idrofilo di quelle esistenti ed è assai probabile che in un futuro più o meno vicino, con lo scopo di accostarsi alle fibre naturali, si giunga a produrre industrialmente fibre da α-amminoacidi.
Progressi più rapidi sono da prevedersi nel campo delle fibre industriali. In questo settore il chimico ha già preparato materiali che sono molto superiori ai prodotti naturali per l'alto modulo di elasticità, l'alta temperatura di fusione, la resistenza all'usura. Il futuro, tuttavia, vedrà l'avvento di nuovi materiali con proprietà molto superiori.
Sviluppi notevoli sono da prevedersi anche nel campo delle fibre resistenti ad alta temperatura e delle fibre incombustibili. Già ora si preparano, in laboratorio, fibre che mantengono le loro proprietà fino a 500-600 °C. Queste sono preparate da polimeri contenenti sistemi aromatici condensati, che impartiscono elevata rigidità alle macromolecole. Appare ragionevole supporre che in un prossimo futuro si potranno ottenere fibre resistenti a temperature molto più elevate.
Nel campo degli elastomeri disponiamo oggi di una serie di gomme di uso generale, così come disponiamo di gomme speciali, adatte cioè a particolari impieghi (ad alta o a bassa temperatura; resistenti a solventi, all'ossidazione, non infiammabili). È da prevedere che in futuro saranno ottenute nuove gomme speciali, che presentino maggiore resistenza all'ossidazione o ai solventi di quelle attuali, alcune adatte per l'impiego a temperature molto basse (per es. −100 °C), altre a temperature molto alte (per es. 350 ÷ 450 °C). Già ora sono in preparazione in laboratorio gomme da polimeri completamente fluorurati, e gomme che contengono atomi metallici nella catena principale.
Nel campo delle gomme per pneumatici è da prevedere che si accentuerà in futuro la tendenza attuale a impiegare covulcanizzati di vari polimeri per ottenere un prodotto finale che assommi le migliori caratteristiche dei componenti. È infatti praticamente impossibile che un unico polimero racchiuda in sè, al massimo grado, tutte quelle caratteristiche che appaiono altamente desiderabili in un elastomero.
Un campo per il quale è da prevedere un notevole sviluppo negli anni futuri è quello delle gomme termoplastiche. Queste gomme non necessitano della vulcanizzazione convenzionale con zolfo, la quale, come è noto, ha la funzione di creare, a intervalli, legami covalenti tra le molecole. Nelle gomme termoplastiche tali legami sono realizzati in vari modi, quali la formazione di legami ionici termolabili, o l'impiego di opportuni copolimeri a blocchi. Questi ultimi sono generalmente costituiti da lunghi blocchi flessibili (per es. di polibutadiene o polusoprene), che assicurano l'elasticità, e da blocchi capaci di formare zone rigide o cristalline (per es. di poli stirene o polietilene), le quali hanno la funzione di tenere unite tra loro le catene. Il vantaggio delle gomme termoplastiche, oltre a quello sopradetto, per cui non necessitano del laborioso processo di vulcanizzazione con zolfo, consiste nel fatto che il materiale può essere reimpiegato. Siamo appena agli inizi in questo campo, che probabilmente avrà largo sviluppo nei prossimi decenni.
Per quanto riguarda le materie plastiche, è facile prevedere che esse entreranno sempre più nel mercato ora tenuto dai metalli, dal vetro, dalla ceramica e dal legno. Il chimico macromolecolare ha ormai i mezzi per ottenere prodotti resistenti come l'acciaio, ma molto più leggeri e capaci di mantenere eccellenti proprietà meccaniche anche ad alte temperature (350-500 °C).
Con le materie plastiche si possono ottenere materiali opachi o trasparenti, facilmente colorabili e facilmente lavorabili: anche per queste ragioni essi sono preferiti in molti casi ai materiali classici (metalli, legno, ceramica) per fabbricare oggetti della vita di tutti i giorni. Oggi si produce anche il cuoio artificiale, che presenta proprietà non dissimili da quelle del cuoio naturale, ma che comunque è destinato a migliorare nel futuro. Sono già in produzione sperimentale alcuni tipi di carta sintetica, che si ritiene possano sostituire per alcuni usi il prodotto di origine naturale.
Appare chiaro che le macromolecole, nelle varie applicazioni, stanno assumendo un'importanza sempre maggiore nella vita del nostro tempo.
bibliografia
Allen, P.W., Techniques of polymer characterization, London 1959.
Bevington, J. C., Radical polymerization, London 1961.
Birshstein, T. M., Ptitsyn, O.B., Conformation of macromolecules, New York 1963.
Bonnar, R. U., Dimbat, M., Stross, F. H., Number average molecular weights, New York 1958.
Boor, J., Review on recent literature on Ziegler-Natta catalysts, in ‟Industrial and engineering chemistry, product research and development", 1970, IX, pp. 437-456.
Bovey, F. A., Polymer conformation and configuration, New York 1969.
Braun, D., Stereospecific polymerization of vinyl-tipe monomers and dienes by alkali-metal-based catalysts, in The stereochemistry of macromolecules (a cura di A. D. Ketley), vol. II, New York 1967, pp. 1-35.
Calderon, N., The olefin metathesis reaction, in ‟Accounts of chemical research", 1972, V, pp. 127-132.
Calderon, N., Ring-opening polymerization of cycloolefins, in ‟Journal of macromolecular science. Part C. Reviews in macromolecular chemistry", 1972, CVII, pp. 105-159.
Cantow, M. J. R., Polymer fractionation, New York 1967.
Carothers, H.W., Studies on polymerization and ring formation, in ‟Journal of the American Chemical Society", 1929, LI, pp. 2548-2559.
Corradini, P., Chain conformation and crystallinity, in The stereochemistry of macromolecules (a cura di A. D. Ketley), vol. III, New York 1968, pp. 1-60.
Crick, F. C. H., Über den genetischen Code, in ‟Angewandte Chemie", 1963, LXXV, pp. 425-429.
Crick, F. C. H., Watson, J. D., Structure of small viruses, in ‟Nature", 1956, CLXXVII, pp. 473-475.
Dall'Asta, G., Mazzanti, G., Reaktivität verschiedener Cycloolefine bei der anionisch-koordinierten Copolymerisation mit Äthylen, in ‟Die makromolekulare Chemie", 163, LXI, pp. 178-197.
Dawans, F., Teyssié, P., π-Allyl-type polymerization, in ‟Industrial and engineering chemistry, product and research development", 1971, X, pp. 261-269.
Flory, J. P., Principles of polymer chemistry, New York 1953.
Flory, J. P., Statistical mechanics of chain molecules, New York 1969.
Furukawa, J., Saegusa, T., Polymerization of aldehydes and oxides, New York 1963.
Gaylord, N., Polyethers, New York 1963.
Graham, T., Anwendung der Diffusion der Flüssigkeiten zur nalyse, in ‟Philosophical transactions", 1961, CLI, pp. 183-224; in ‟Annalen der Chemie und Pharmazie", 1962, CXXI, pp. 1-77.
Ham, J. E., Vinyl polymerization, New York 1969.
Harries, C., Über den Abbau des Parakautschuks vermittelst Ozon, in ‟Berichte der deutschen chemischen Gesellschaft", 1904, XXXVII, pp. 2708-2711.
Hérisson, J. L., Chauvin, Y., Catalysis of olefin transformations by tungsten complexes. II. Telomerization of cyclic olefins in the presence of acyclic olefins, in ‟Die makromoleculare Chemie", 1971, CXLI, pp. 161-176.
Heuser, H., Textbook of cellulose chemistry, New York 1924.
Jordan, D. O., Ziegler-Natta polymerization: catalysts, monomers and polymerization procedures, in The stereochemistry of macromolecules (a cura di A.D. Ketley), vol. I, New York 1967, pp. 1-45.
Kakudo, M., Kasai, N., X-ray diffraction by polymers, Amsterdam 1972.
Ke, B., Newer methods of polymer characterization, New York 1964.
Kennedy, J. P., Polyisobutene and butyl rubber, in Polymer chemistry of synthetic elastomers (a cura di J. P. Kennedy ed E. Tornqvist), parte I, New York 1968, pp. 291-329.
Ketley, A. D., Stereospecific polymerization of vinyl ethers, in The stereochemistry of macromolecules (a cura di . D. Ketley), vol. II, New York 1967, pp. 38-109.
Kirk, R. E., Othmer, D. F. (a cura di), Encyclopaedia of polymer science and technology, New York 1965.
Lal, J., Poly (vinyl) ethers, in Polymer chemistryu of synthetic elastomers (a cura di J. P. Kennedy ed E. Tornqvist), parte I, New York 1968, pp. 331-376.
Lebedev, S. V., Skavronskaya, N. A., Polymerization of diethylene hydrocarbons. Polymerization of divinyl (orig. russo), in ‟Journal of the Russian Physical and Chemical Society" (‟Zhurnal Russkago fiziko-khimicheskago obshchestva pri Imperatorskago St-Peterburgskago universitetê"), 1911, XLIII, pp. 1124-1131.
Ledwith, A., Fitzsimmonds, C., Elastomers from cyclic ethers, in Polymer chemistry of synthetic elastomers (a cura di J. P. Kennedy ed., E. Tornqvist), parte I, New York 1968, pp. 377-417.
Lenz, R. W., Organic chemistry of synthetic high polymers, New York 1967.
Marconi, W., The polymerization of dienes by Ziegler-Natta catalysts, in The stereochemistry of macromolecules (a cura di A. D. Ketley), vol. I, New York 1967, pp. 239-307.
Mark, H., Whitbyu, G. S. (a cura di), Collected papers of Wallace Hume Carothers on high polymeric substances, New York 1940.
Morawetz, H., Macromolecules in solution, New York 1965.
Morton, A.A., The reaction of organosodium compounds with butadiene, in ‟Journal of the American Chemical Society", 1946, LXVIII, pp. 93-96.
Morton, A. A., Effect of associated salts on the polymerization of butadiene by organosodium reagents, in ‟Industrial and engineering chemistry", 1952, XLIV, pp. 2876-2882.
Morton, A. A., The alfin reagent, in Advances in catalysis (a cura di D. D. Eley, W.G. Frankenburg, V. I. Komrewsky e P.B. Weisz), vol. IX, New York 1957, pp. 743-753.
Natta, G., Von der stereospezifischen Polymerisation zur asymmetrischen autokatalytischen Synthese von Makromolekülen, in ‟Angewandte Chemie", 1964, LXXVI, pp. 553-566.
Natta, G., Corradini, P., General considerations on the structure of crystalline polyhydrocarbons, in ‟Nuovo cimento", 1960, XV, pp. 9-39.
Natta, G., Dall'Asta, G., Elastomers from cyclic olefins, in Polymer chemistry of synthetic elastomers (a cura di J. P. Kennedy ed E. Tornqvist), parte II, New York 1969, pp. 703-726.
Natta, G., Dall'Asta, G., Mazzanti, G., Pasquon, I., Valvassori, A., Zambelli, ., Ethylene-butene-2 alternating copolymers, in ‟Journal of the American Chemical Society", 1961, LXXXIII, pp. 3343-3344.
Natta, G., Pasquon, I., Corradini, P., Peraldo, M., Pegoraro, M., Zambelli, A., Alti polimeri lineari del propilene aventi struttura sindiotattica, in ‟Atti dell'Accademia Nazionale dei Lincei. Rendiconti", 1960, XXVIII, 8, pp. 539-544.
Natta, G., Pino, P., Corradini, P., Danusso, F., Mazzanti, G., Moraglio, G., Crystalline high polymers of α-olefins, in ‟Journal of the American Chemical Society", 1955, LXXVII, pp. 1708-1710.
Natta, G., Pino, P., Mazzanti, G., Alti polimeri lineari e cristallini di α-olefine, Brevetti italiani 535712 e 526101 (1954).
Natta, G., Porri, L., Diene elastomers, in Polymer chemistry of synthetic elastomers (a cura di J. P. Kennedy ed E. Tornqvist), parte II, New York 1969, pp. 597-678.
Natta, G., Valvassori, A., Sartori, G., Ethylene-propylene rubbers, in Polymer chemistry of synthetic elastomers (a cura di J. P. Kennedy ed E. Tornqvist), parte II, New York 1969, pp. 679-702.
Pasquon, I., Valvassori, A., Sartori, G., The copolymerization of olefins by Ziegler-Natta catalysts, in The stereochemistry of macromolecules (a cura di A. D. Ketley), vol. I, New York 1967, pp. 177-238.
Pauling, L., Moderne Strukturchemie, in ‟Angewandte Chemie", 1955, LXVII, pp. 241-244.
Pauling, L., Corey, R. B., Atomic coordinates and structure factors for two helical configurations of polypeptide chains, in ‟Proceedings of the National Academy of Sciences", 1951, XXXVII, pp. 235-240.
Pino, P., Optically active addition polymers, in ‟Advances in polymer science", 1965, IV, pp. 393-456.
Plesh, P. H. (a cura di), The chemistry of cationic polymerization, New York 1963.
Pregaglia, G. F., Binaghi, M., Ionic polymerization of aldehydes, ketones and ketenes, in The stereochemistry of macromolecules (a cura di A. D. Ketley), vol. II, New York 1967, pp. 111-175.
Raff, R. A. V., Doak, K. W., Crystalline olefin polymers, New York 1965.
Reich, L., Schindler, A., Polymerization by organometallic compounds, New York 1966.
Schildknecht, C. .E, Gross, S. T., Davidson, H. R., Lambert, J. M., Zoss, A. O., Polyvinyl isobutyl ethers, in ‟Industrial and engineering chemistry", 1948, XL, pp. 2104-2115.
Scott, K. W., Calderon, N., Offstead, E. A., Judy, W. A., Ward, J. P., Ring-opening polymerization of cycloolefins. Some mechanicistic aspects, in ‟Advances in chemistry series", 1969, XCI, pp. 399-418.
Staudinger, H., Über Polymerisation, in ‟Berichte der deutschen chemischen Gesellschat", 1920, LIII, pp. 1073-1085.
Stobbe, H., Posnjak, G., Der wahre Zustand des Metastyrols und die Polymerisation des Styrols durch Licht und durch Wärme, in ‟Liebig's Annalen der Chemie", 1910, CCCLXXI, pp. 259-286.
Szwarc, M., Living polymers, in ‟Nature", 1956, CLXXVIII, pp. 1168-1169.
Tsuruta, T., Stereospecific polymerization of epoxides, in The stereochemistry of macromolecules (a cura di A. D. Ketley), vol. II, New York 1967, pp. 177-217.
Vogl, O., Elastomeric polyacetals, in Polymer chemistry of synthetic elastomers (a cura di J. P. Kennedy ed E. Tornqvist), parte I, New York 1968, pp. 419-453.
Volkenstein, M. V., Configurational statistics of polymeric chains, New York 1963.
Watson, J. D., Die Beteiligung der Ribonucleinsäure an der Proteinsynthese, in ‟Angewandte Chemie", 1963, LXXV, pp. 439-449.
Watson, J. D., Crick, F. C. H., Genetical implications of the structure of deoxyribose nucleic acid, in ‟Nature", 1953, CLXXI, pp. 964-967.
Zbinden, R., Infrared spectroscopy of high polymers, New York 1964.
Ziegler, K., Folgen und Werdegang einer Erfindung, in ‟Angewandte Chemie", 1964, LXXVI, pp. 545-553.