
Prione
Prione
Il termine prione identifica un agente infettivo con dimensioni più piccole dei virus e resistente alle procedure impiegate per rimuovere o inattivare virus, batteri, funghi o altri microrganismi noti. Metodi fisici, come il calore, e chimici, come i trattamenti con fenolo, cloroformio, formaldeide e alcoli, sono inefficaci; una consistente riduzione della carica infettiva si ottiene solo con un prolungato trattamento in autoclave (calore umido a elevata pressione) a 132 °C per almeno un'ora, o con procedure chimiche in grado di idrolizzare le proteine. Non è possibile identificare alcun acido nucleico associato con il p. e pertanto non sono utilizzabili le tecniche di biologia molecolare usate per identificare singole particelle virali in tessuti o fluidi biologici. Il termine prione deriva dall'inglese prion, abbreviazione di proteinaceous infectious particle (particella proteica infettiva), ed è stato coniato per sottolineare l'ipotesi che l'agente infettivo sia composto solo da proteine, senza la presenza di acidi nucleici. I p. sono responsabili delle encefalopatie spongiformi trasmissibili (EST), malattie neurodegenerative dell'uomo a esito fatale (malattia di Creutzfeldt-Jakob o MCJ, sindrome di Gerstmann-Sträussler-Scheinker e insonnia fatale sporadica o familiare) e degli animali (scrapie della pecora e della capra, encefalopatia spongiforme bovina, BSE).
La proteina prionica 'normale' e quella 'patologica'
Nei tessuti infetti da EST è presente una proteina anomala, la proteina prionica patologica o PrPEST, che deriva da un'alterata struttura tridimensionale della proteina prionica cellulare (PrPc). Studi sulla struttura tridimensionale della PrPc hanno dimostrato che questa è altamente conservata tra le diverse specie di mammiferi, e che proteine simili sono presenti anche in rettili, uccelli, anfibi e pesci. Nei mammiferi, la PrPc è legata alle membrane cellulari mediante un'ancora di glicosil-fosfatidil-inositolo (GPI), suggerendo un suo possibile coinvolgimento nella trasduzione del segnale o nell'adesione cellulare. La sua porzione amino-terminale lega il rame e potrebbe quindi far parte dei sistemi di resistenza cellulare allo stress ossidativo. Altri dati sperimentali mostrano che la PrPc può legarsi ad altre proteine della membrana partecipando così a una serie di attività funzionali e di regolazione quali l'adesione cellulare, il differenziamento e la protezione della cellula dai meccanismi di stress. Nonostante questi studi, non si è però ancora giunti a evidenze conclusive su quale sia l'esatto ruolo fisiologico della PrPc. Topi knock-out per la PrPc (topi nei quali il gene della PrP è stato rimosso e che quindi non producono la proteina), per es., hanno uno sviluppo normale e non presentano segni clinici o lesioni patologiche particolari. Questi topi, tuttavia, sono resistenti all'infezione con EST sperimentale e ciò dimostra che la PrPc svolge un ruolo fondamentale nella patogenesi della malattia. Nei soggetti infetti, per ragioni ancora sconosciute, la PrPc assume una conformazione spaziale alterata che consiste principalmente nella trasformazione di alcune strutture α-elica in strutture a foglietti β. Questa modifica strutturale comporta un cambiamento nelle caratteristiche della proteina che diventa resistente alla degradazione cellulare, accumulandosi all'interno della cellula sotto forma di fibrille amiloidee. L'alterata conformazione conferirebbe alla PrPEST le caratteristiche infettanti. La prima molecola di PrPEST deriverebbe dalla conversione spontanea di una molecola endogena di PrPc. La replicazione del p. avverrebbe quindi mediante l'interazione diretta tra una neomolecola di PrPEST e una di PrPc, che a sua volta assumerebbe la conformazione patologica a foglietti β. Questa conversione innescherebbe un meccanismo autocatalitico responsabile della produzione massiva di PrPEST. Quando la PrPEST è inoculata, accidentalmente oppure sperimentalmente, in un ospite sano, questa interagisce con la PrPc dell'ospite attivandone la sua trasformazione in nuova PrPEST e inducendo quindi la malattia nel nuovo ospite. In alternativa all'ipotesi che la PrPEST sia la componente principale, se non la sola, del p., è possibile supporre che l'agente infettivo sia costituito da una particella virale, quindi con un proprio acido nucleico, e che la PrPc funga da proteina recettoriale indispensabile al p. per entrare e quindi replicarsi nella cellula ospite. In questa ipotesi, il legame tra PrPc e virus sarebbe responsabile della trasformazione della PrPc in PrPEST.
Basi patogenetiche
Come per altri agenti microbici, esistono vari ceppi di p. che, in mancanza di un acido nucleico specifico, possono essere isolati e caratterizzati solo mediante le caratteristiche cliniche, biochimiche e neuropatologiche che inducono negli animali da laboratorio. Ogni ceppo produce nel topo (o in altri roditori) un caratteristico tempo di incubazione, un pattern elettroforetico della PrPEST e una specifica distribuzione delle lesioni (in particolar modo la spongiosi) nel cervello. Con questa metodica sono stati isolati circa venti ceppi di scrapie, uno o forse due di BSE e almeno tre distinti ceppi di malattia di Creutzfeldt-Jakob. Lo studio di queste patologie si avvale di modelli animali transgenici e non transgenici. I topi transgenici sono ottenuti mediante l'introduzione nel topo di una o più copie del gene della PrPc di una specie diversa. Si hanno così a disposizione modelli murini per lo studio delle EST nella pecora, nel bovino o nell'uomo. Quando il gene inserito contiene mutazioni si ha a disposizione un modello per lo studio delle EST genetiche umane. La tecnica di costruzione più semplice consiste nell'inoculare direttamente negli ovociti murini il gene della PrPc eterologa. Il gene, contornato da sequenze di DNA che ne facilitano l'integrazione nella doppia elica di DNA dell'ospite, si inserisce stabilmente nel genoma del topo. Questa tecnica largamente utilizzata non consente però di controllare né il numero di copie né la posizione di integrazione nel genoma e in questo senso ogni singola linea di animali transgenici ottenuta è unica per le caratteristiche di integrazione e per il livello di espressione genica. Questa tecnica ha permesso di superare la cosiddetta barriera di specie, definita come la scarsa efficienza o l'incapacità, da parte di un determinato ceppo di EST, di propagarsi in una specie diversa da quella in cui è stata isolata. L'introduzione di un gene eterologo per la PrPc consente di riprodurre la malattia in animali normalmente non suscettibili, anche se l'efficienza della transfezione può essere limitata dall'effetto del gene residuo: animali transgenici completamente privi del gene originario sono infatti più sensibili all'infezione con il ceppo di EST eterologo rispetto a quelli in cui sono presenti copie del gene originario. Una tecnica di costruzione più raffinata, che evita i problemi derivanti dall'integrazione in siti casuali e dal livello di espressione, è stata ottenuta inserendo il gene della PrPc endogena (o meglio la sua porzione codificante la proteina) al posto del gene murino, che quindi si ritrova nella posizione fisiologica e sotto il controllo dei fattori di regolazione dedicati. Questo modello ha consentito di dimostrare che le mutazioni del gene della PrP non danno spontaneamente luogo a malattia, ma rendono l'ospite molto più suscettibile all'infezione da prioni. Tra i modelli animali non transgenici quello rappresentato da un roditore selvatico, l'arvicola rossastra (Clethrionomys glareolus) potrebbe fornire un nuovo e potente strumento di studio e di prevenzione in quanto permette di discriminare diversi ceppi di p. delle EST naturali (per es., MCJ sporadica o genetica e scrapie naturale) con una elevata efficienza di infezione e tempi di incubazione paragonabili o addirittura inferiori a quelli dei topi transgenici. Sebbene i modelli animali restino insostituibili in questo ambito di ricerca, le colture cellulari potrebbero rappresentare un conveniente surrogato all'infezione in vivo. Inoltre è stata sviluppata una linea cellulare, derivata da cellule neuronali tumorali di topo (N2a), che è sensibile all'infezione con p. di scrapie murino con una soglia di sensibilità molto elevata. Tuttavia queste linee cellulari sono resistenti all'infezione con altri ceppi di p., rendendo quindi questa tecnica ancora poco utilizzabile per lo studio delle EST umane o animali. Organo bersaglio del p. è il sistema nervoso centrale, dove si osserva una caratteristica vacuolizzazione (spongiosi), un aumento e a volte un ingrandimento delle cellule microgliali e astrocitarie e spesso anche una più o meno grave perdita dei neuroni. Al contrario di altri microrganismi, il p. non stimola invece l'attivazione di cellule infiammatorie nei tessuti infetti. In sostanza, le lesioni istologiche nel cervello somigliano più a quelle che si osservano nelle malattie neurodegenerative (come, per es., l'Alzheimer o il Parkinson) piuttosto che a quelle riscontrate nelle encefaliti batteriche o virali. Studi con modelli sperimentali di scrapie dimostrano che in seguito a infezione sperimentale per via periferica - endovenosa, intraperitoneale, intramuscolare, sottocutanea oppure orale - l'agente si diffonde nell'organismo per via ematica dando inizio a una prima fase replicativa nelle cellule follicolari dendritiche dei tessuti linfatici (milza, linfonodi, tonsille, placche di Peyer dell'intestino). Dagli organi linfatici attraverso i nervi periferici, mediante il trasporto assonale, il p. raggiunge il midollo spinale e quindi l'encefalo dove continua a replicarsi provocando la formazione di PrPEST e le tipiche lesioni neuropatologiche, quindi l'insorgenza dei sintomi clinici e la morte dell'animale. Alcuni ceppi particolarmente neurovirulenti raggiungono il sistema nervoso centrale dal sito di inoculazione periferico penetrando direttamente nelle terminazioni nervose locali. L'origine endogena della PrPEST e la mancanza di anticorpi anti- PrPEST negli animali infetti hanno per anni indotto a credere che non fosse possibile attuare terapie immunologiche nelle EST. Tuttavia altri studi hanno evidenziato che la somministrazione di anticorpi anti-PrPc rallenta la formazione di PrPEST negli animali infetti con conseguente ritardo della comparsa di segni clinici. Sarebbe quindi possibile 'vaccinarsi' contro i p. se si riuscisse a superare la tolleranza immunologica che ogni organismo mette in atto verso proteine endogene, quale di fatto è la PrPc. Questo ostacolo sembra possa essere superato introducendo, con tecniche di ingegneria genetica, la PrPc in batteri che sarebbero in grado di aggirare la tolleranza immunologica dell'ospite. Ovviamente questi risultati sperimentali sono ancora a uno stadio preliminare e la vaccinazione per prevenire le malattie da p. è ben lontana dal poter essere applicata all'uomo o agli animali d'allevamento. Tra gli approcci terapeutici più tradizionali vi è la sperimentazione di farmaci o molecole attive, basati sul presupposto che la PrPEST rappresenti l'elemento patogenetico principale e quindi indirizzate sia alla destabilizzazione della PrPEST e alla disgregazione degli accumuli di PrPEST sia alla stabilizzazione della PrPc. L'efficacia di queste sostanze (poliammine, porfirine e ftalocianine, peptidi intercalanti, curcumina, piccoli RNA interferenti) è stata valutata nei modelli animali o in vitro. Purtroppo anche i pochi risultati incoraggianti ottenuti sperimentalmente si scontrano con lo scarso indice terapeutico di queste sostanze (elevata tossicità) e con la loro incapacità di superare la barriera ematoencefalica per raggiungere il cervello.
Malattie da prioni negli animali
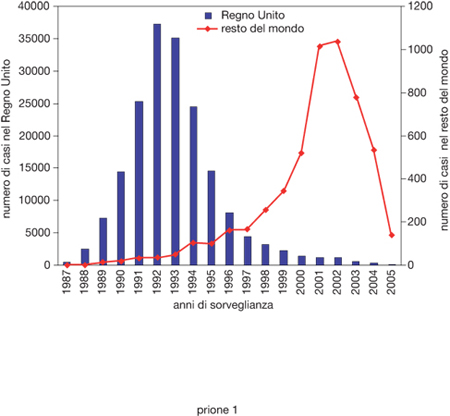
BSE. - L'encefalopatia spongiforme bovina (BSE) si è manifestata per la prima volta nel Regno Unito nel 1986, e successivamente si è diffusa in quasi tutti i Paesi europei, Giappone, Canada e Stati Uniti. La malattia colpisce animali di età compresa tra 22 mesi e 18 anni, con un picco attorno ai 4-5 anni e con un tempo d'incubazione medio compreso tra i 4 e i 6 anni. Dal punto di vista clinico i bovini affetti da BSE presentano modificazioni del comportamento come apprensione e aggressività, iperestesia a stimoli tattili e uditivi; incoordinazione dei quarti posteriori, tremori muscolari e digrignamento dei denti. Le misure di prevenzione e controllo adottate, in particolare il divieto di utilizzare farine di carne per la preparazione di mangimi, hanno ridotto significativamente l'epidemia di BSE nel Regno Unito (fig. 1) e il rischio di infezione ad altre specie animali. Tuttavia, poiché le misure preventive sono state adottate in ritardo dagli altri Paesi europei, si è assistito a un progressivo aumento di casi di BSE in Europa e nel resto del mondo (fig. 1). L'identificazione in Francia di una capra con BSE conferma il timore che anche gli ovicaprini possano essere stati contaminati da mangimi infetti. Se ciò venisse confermato in altri ovicaprini, la BSE sarebbe molto più difficile da eradicare e potrebbe rimanere endemica per molti anni. Al contrario di quanto accade nei bovini con BSE dove il p. è localizzato quasi esclusivamente nel sistema nervoso centrale e periferico, negli ovicaprini il p. della BSE, così come quello della scrapie, è presente in quasi tutti gli organi, compresi i muscoli, e nel sangue. Il consumo di carni ovicaprine potrebbe quindi rappresentare un ulteriore rischio per l'uomo. Una nuova fonte di allarme deriva dall'identificazione di una forma di EST bovina individuata in Italia nel 2004, chiamata encefalopatia spongiosa amiloidotica del bovino (BASE) e caratterizzata dalla presenza di placche amiloidee nel cervello e da una distribuzione delle lesioni neuropatologiche diverse dalla BSE, di cui non si conoscono ancora le caratteristiche di contagiosità e patogenicità per l'uomo. Questi nuovi e preoccupanti eventi epidemiologici hanno stimolato nuove ricerche per la messa a punto di tecniche di inattivazione dei p. che possano essere utilizzate nella produzione di alimenti. A questo scopo è stato dimostrato che il p. è sensibile al trattamento con alte pressioni dell'ordine di centinaia di megaPascal (un megaPascal equivale alla pressione di 10 atmosfere) che, in particolari condizioni, non altera le caratteristiche nutritive e organolettiche dei cibi.
Scrapie. - La scrapie colpisce tutte le razze ovicaprine ed è diffusa in tutto il mondo a eccezione di Paesi come l'Australia, la Nuova Zelanda e la Repubblica Sudafricana, nei quali è stata eradicata. Di solito la scrapie colpisce animali dopo i 2 anni di vita, con un picco di incidenza intorno ai 4 anni. L'esordio clinico della malattia è insidioso e caratterizzato da modificazioni del comportamento e aggressività riconoscibili solo a un attento esame del gregge. Il quadro clinico si aggrava con la comparsa di prurito (da cui scrapie, dall'inglese to scrape, grattare) e atassia, che si manifesta con una errata postura o esecuzione dei movimenti, il tipico andamento trottante. Il prurito è localizzato inizialmente alla regione lombare, diviene poi generalizzato fino alla completa perdita del pelo e alla produzione di escoriazioni e lesioni cutanee profonde. Nella fase terminale della malattia l'animale è in decubito e la morte sopravviene generalmente dopo 4-6 mesi. I meccanismi di trasmissione della scrapie all'interno del gregge non sono completamente chiariti: può trasmettersi dalla pecora all'agnello nel periodo prenatale, al momento del parto o durante lo svezzamento. La via orale e la scarificazione della cute sembrano essere le principali vie d'infezione nella trasmissione orizzontale da pecora a pecora: la placenta proveniente da animali infetti costituirebbe inoltre un rischio per gli animali che se ne nutrono come pure una possibile fonte di contaminazione dei pascoli. Per il controllo della scrapie nella popolazione ovina è stato avviato in tutta la Comunità europea un programma di selezione genetica per ridurre il numero di pecore suscettibili alla maggior parte dei ceppi di scrapie. Quanto queste misure saranno in grado di ridurre la scrapie in Europa e quindi il rischio che possa sorgere una nuova epidemia di EST in animali da allevamento, è difficile da prevedere. Il rischio potrebbe essere quello di eliminare ceppi di scrapie non patogeni per l'uomo o per altri animali e favorire quindi la selezione di altri ceppi che sono in grado di infettare gli ovini geneticamente resistenti e che teoricamente potrebbero essere più aggressivi per altre specie.
Malattie da prioni nell'uomo
La malattia di Creutzfeldt-Jakob (MCJ) è una rara forma morbosa neurologica a esito fatale. Esistono quattro diverse forme di MCJ: la MCJ sporadica, presente in tutto il mondo e della quale non si conoscono né le cause né gli eventuali fattori di rischio; la MCJ iatrogena, causata dalla trasmissione dell'agente infettivo attraverso procedure medico-chirurgiche; la MCJ genetica, legata a mutazioni del gene della proteina prionica (PRNP) e la variante MCJ, causata dall'agente dell'encefalopatia spongiforme bovina (BSE). Notevoli progressi sono stati ottenuti in ambito clinico-epidemiologico e diagnostico anche se mancano esami preclinici per individuare i soggetti infetti e terapie efficaci.
La malattia di Creutzfeldt-Jakob sporadica. - Il progetto di collaborazione europea sulla sorveglianza epidemiologica delle EST umane ha raccolto dati clinico-epidemiologici su oltre 4400 casi di MCJ e sindromi correlate. I risultati di questa collaborazione hanno confermato che la forma più frequente di MCJ è quella sporadica (84% dei casi) con una mortalità annuale, relativamente uniforme nei diversi Paesi europei, pari a 1,39 casi per milione di abitanti. Questo studio ha inoltre evidenziato un aumento della mortalità per MCJ sporadica negli ultimi anni nelle classi di età oltre i 60 anni dovuto probabilmente a un miglioramento della sorveglianza nell'accertamento dei casi (per es., una maggiore attenzione nella diagnostica differenziale della demenza in età avanzata) piuttosto che alla conseguenza dell'esposizione a nuove fonti d'infezione ambientali. Nelle classi di età più giovani, tra le quali è più probabile si sviluppi la variante MCJ, non sono stati invece rilevati aumenti nei tassi di mortalità, suggerendo che è improbabile che negli anni passati si siano persi casi di variante MCJ. In Italia la MCJ sporadica è distribuita in maniera omogenea su tutto il territorio nazionale con un tasso medio di mortalità pari a 1,42 casi per milione di abitanti per anno. Anche in Italia è stato osservato un aumento di mortalità per MCJ sporadica nelle classi di età oltre i 60 anni ma non per i casi al di sotto dei 50 anni. Da un confronto tra i dati di mortalità ISTAT e i dati del registro nazionale della MCJ è stato calcolato che la sottonotifica dei casi di MCJ al registro si è ridotta nel tempo a meno del 5%. La diagnosi in vita della MCJ è basata su criteri clinici e su alcuni esami strumentali, quali l'elettroencefalogramma (EEG) e l'esame della proteina 14-3-3 nel liquido cefalorachidiano. Le anomalie EEG caratteristiche della MCJ sono i complessi periodici trifasici punta-onda (1-2 cicli al secondo) sincroni e bilaterali, che però non sono costanti durante l'intera fase clinica. L'identificazione mediante Western Blot della proteina 14-3-3 è di grande aiuto per confermare il sospetto clinico di MCJ sporadica e ha lo stesso valore diagnostico dell'EEG. La specificità di questo esame in pazienti affetti da disturbi cognitivi è del 93%. Un importante obiettivo della ricerca diagnostica è quello di aumentare la sensibilità delle tecniche di identificazione della PrPEST nei tessuti periferici. Per es., la proteina PrPEST è stata identificata nell'epitelio olfattorio che è facilmente ottenibile mediante la semplice esecuzione in vita della biopsia della mucosa olfattoria. Basandosi principalmente su due criteri (il polimorfismo in posizione 129 della PrP e il pattern elettroforetico della PrPEST che si deposita nel cervello) si distinguono diversi sottotipi clinico-patologici della MCJ sporadica. Nell'uomo il gene della PrP (PRNP) ha diversi siti polimorfici ma quello al codone 129 (codificante metionina o valina) è l'unico che abbia una chiara influenza sull'età d'insorgenza della malattia e sulla sopravvivenza. Anche la struttura tridimensionale che assume la PrPEST in corso di malattia influisce sulle caratteristiche cliniche della MCJ. Nell'uomo la PrPEST può assumere almeno due diverse conformazioni, tipo 1 e tipo 2, che si distinguono all'esame elettroforetico della proteina. In elettroforesi la PrPEST, come d'altronde il suo precursore fisiologico, migra in tre bande distinte che corrispondono alla forma non glicosilata e a quelle mono- e diglicosilata. Nel tipo 1 la banda non glicosilata (che è quella dove si notano meglio piccole differenze di migrazione) è leggermente più grande (21 KDa) di quella del tipo 2 (19 KDa) che migra più velocemente. La PrPEST tipo 2 è ulteriormente divisa in due isoforme in base al grado di glicosilazione della proteina, nel tipo 2A la banda tipica è quella monoglicosilata mentre nel tipo 2B è quella diglicosilata. La PrPEST tipo 2B si trova solo nella variante MCJ e nell'insonnia fatale familiare. I pazienti con MCJ sporadica sopravvivono mediamente circa cinque mesi dall'esordio della malattia, ma ci sono pazienti che presentano una rapidissima evoluzione (meno di un mese) e altri che sopravvivono oltre due anni. I fattori che influenzano la sopravvivenza sono il sesso, l'età all'esordio della malattia, il genotipo del paziente al codone 129 del gene PRNP e il tipo di PrPEST. La sopravvivenza è inferiore nei maschi, nei soggetti di età più avanzata, nei portatori di omozigosi per metionina al codone 129 e nei pazienti con il tipo 1 di PrPEST. Identificare le variabili che influenzano la sopravvivenza nelle diverse forme di MCJ sporadica costituisce la base per una strategia di valutazione dell'effetto di potenziali regimi terapeutici nei soggetti trattati rispetto alla storia naturale di queste malattie.
La malattia di Creutzfeldt-Jakob iatrogena. - La MCJ iatrogena si riscontra essenzialmente nel Regno Unito e in Francia, soprattutto in pazienti che hanno ricevuto l'ormone della crescita ipofisario di tipo estrattivo. In Italia, dal 1993 al 2004, si sono registrati solo tre casi di MCJ iatrogena dovuta a impianto di dura mater umana durante interventi neurochirurgici.
Le forme genetiche. - La MCJ genetica, insieme con la sindrome di Gerstmann-Sträussler-Scheinker e l'insonnia fatale familiare, costituisce circa il 10-15% dei casi, ma queste forme, al contrario della sporadica, sono distribuite in maniera molto disomogenea nei diversi Paesi europei: in Slovacchia e in Italia il tasso di mortalità per MCJ genetica è rispettivamente di quasi 7 e 2 volte più alto della media europea. Anche in Italia le forme genetiche sono distribuite in maniera disomogenea con una maggiore prevalenza di casi in Calabria e Campania. La durata della malattia nei pazienti con MCJ genetica è generalmente più breve (la media è 4 mesi) che nei pazienti con MCJ sporadica e il sesso e l'età d'insorgenza della malattia ne influenzano la sopravvivenza in maniera simile alla MCJ sporadica, eccetto che per i soggetti con MCJ genetica associata alla mutazione V210I, dove gli omozigoti per metionina hanno una sopravvivenza media di circa 2 mesi superiore agli eterozigoti. Pazienti con insonnia fatale familiare hanno una sopravvivenza media di circa un anno mentre i pazienti con sindrome di Gerstmann-Sträussler-Scheinker di oltre 3 anni.

La variante della malattia di Creutzfeldt-Jakob. - La variante MCJ si discosta dalla forma sporadica di MCJ per la durata della malattia superiore ai 6 mesi e le caratteristiche cliniche di esordio di tipo psichiatrico (depressione, ansietà, apatia, illusioni). La sintomatologia evolve nei mesi successivi con un'atassia della marcia, disturbi sensoriali di tipo dolorifico, che non si osservano nella forma sporadica, movimenti involontari, progressivo deterioramento intellettivo e mutismo acinetico con un quadro neurologico franco sostanzialmente non dissimile dalla forma sporadica della malattia. Fondamentale per la diagnosi clinica di variante MCJ è l'esecuzione della risonanza magnetica del cranio che mostra un'iperintensità bilaterale di segnale a livello del pulvinar e il tracciato elettroencefalografico che non evidenzia il caratteristico periodismo della forma sporadica. La presenza della proteina liquorale 14-3-3 è incostante. Quando la sintomatologia clinica e gli esami strumentali non permettono di distinguere la variante MCJ da altre forme di MCJ, si ricorre alla biopsia tonsillare per identificare - mediante tecniche immunochimiche - la presenza di PrPEST che è presente nei tessuti linforeticolari dei soli pazienti affetti da variante MCJ. L'analisi del gene PRNP non ha identificato alcuna mutazione mentre tutti i casi sono risultati omozigoti per metionina al codone polimorfico 129 del gene della PrP. La maggior parte dei casi di variante MCJ sono stati descritti in pazienti giovani (tra i 15 e i 30 anni) senza distinzione di sesso, anche se la malattia può colpire a qualsiasi età (il paziente più anziano aveva 74 anni). Anche nel caso della variante MCJ la diagnosi è confermata solo dall'esame neuropatologico che dimostra, oltre alla spongiosi, numerose e diffuse placche amiloidee circondate da vacuoli (placche floride), non riscontrabili in altre forme di MCJ, e una forte positività immunocitochimica alla PrPEST. In aggiunta alla neuropatologia, l'analisi molecolare è in grado di differenziare il pattern di glicosilazione della PrPEST della variante MCJ (tipo 2B) da quello delle forme sporadiche (tipo 1 o 2A). Fino a ottobre 2006 sono stati descritti 199 casi di variante MCJ (fig. 2). Il maggiore numero è riscontrato nel Regno Unito con 164 casi; altri casi di variante MCJ sono stati identificati in Francia (21), Irlanda (4), Olanda (2), Stati Uniti (2), Italia, Portogallo, Giappone, Arabia Saudita e Canada (un caso ciascuno). Due dei quattro casi irlandesi e quelli nordamericani hanno a lungo soggiornato nel Regno Unito durante il periodo considerato maggiormente a rischio d'infezione da BSE, il caso giapponese ha trascorso solo un mese nel Regno Unito, mentre gli altri casi, incluso quello italiano, si sono infettati nel proprio Paese d'origine. Questo dato è importante per una corretta valutazione del rischio BSE nei diversi Paesi europei e indica, sebbene in modo approssimativo, che il livello d'infettività circolante in Italia negli anni Novanta, prima cioè dell'introduzione di provvedimenti di sanità pubblica mirati a eliminare tessuti bovini ad alto rischio per il consumo umano, è stato circa 100 volte inferiore che nel Regno Unito e 10 volte inferiore che in Francia. Il numero di casi di variante MCJ nel Regno Unito ha avuto un picco nel 2000 e da allora è in costante diminuzione (fig. 2). Questo dato epidemiologico è però in contrasto con un primo studio di prevalenza nel Regno Unito basato sull'analisi retrospettiva di circa 13.000 campioni di appendici e tonsille. Questo studio ha individuato 3 appendici positive per la proteina prionica patologica (PrPEST) fornendo una stima di prevalenza di 237 persone infette per milione di abitanti o, considerando che l'83% dei campioni testati era nella fascia di età dai 10 ai 30 anni, di 3808 persone che, in questa fascia di età, potrebbero incubare la variante MCJ. Non è ben chiaro come interpretare questa discordanza di dati: è possibile ipotizzare che in alcuni soggetti l'infettività rimanga localizzata al di fuori del sistema nervoso centrale senza pertanto causare alcun segno clinico o, in alternativa, che questi soggetti stiano incubando la malattia e che in un prossimo futuro ci sarà un nuovo incremento di variante MCJ. L'aver trovato l'appendice positiva alla PrPEST in due pazienti con variante MCJ nei quali era stata eseguita appendicectomia 8 e 24 mesi prima dell'esordio clinico della malattia, avvalora quest'ultima ipotesi. In entrambe le ipotesi, questi soggetti potrebbero trasmettere la variante MCJ attraverso il sangue o prodotti farmaceutici derivati dal plasma, quali, per es., il fattore viii, immunoglobuline o altro. Recentemente due pazienti hanno sviluppato la variante MCJ 6-8 anni dopo aver ricevuto una trasfusione di globuli rossi provenienti da donatori che svilupparono segni clinici di malattia tre anni e mezzo dopo la donazione. Nel 2004 un altro paziente, morto per cause non neurologiche, risultò positivo alla PrPEST nella milza: cinque anni prima era stato trasfuso con globuli rossi donati da un soggetto che aveva sviluppato la variante MCJ 18 mesi dopo la donazione. Al contrario di tutti i pazienti con variante MCJ che sono omozigoti per metionina nell'aminoacido 129 della proteina prionica, quest'ultimo paziente era eterozigote, facendo ipotizzare che la presenza di omozigosi per metionina non sia indispensabile per essere suscettibili all'infezione. Da questi ultimi dati è pertanto ragionevole ipotizzare che il sangue di pazienti con variante MCJ sia infetto diversi anni prima dell'esordio clinico di malattia. Tenendo conto che nel Regno Unito circa il 10% di pazienti con variante MCJ hanno donato sangue prima di sviluppare segni clinici di malattia e che in Francia 3 pazienti su 21 erano donatori di sangue, è possibile che la trasmissione interumana della variante MCJ attraverso il sangue influenzi in un prossimo futuro l'entità dell'epidemia nel Regno Unito e in altri Paesi europei. L'incertezza sul numero di persone che potrebbero incubare la variante MCJ nel Regno Unito e in Europa, la totale mancanza di esami diagnostici preclinici per identificare i soggetti infetti o per lo screening del sangue, hanno imposto il divieto di utilizzare il plasma di donatori britannici per la preparazione di plasma-derivati. Questa disposizione è stata estesa, in maniera precauzionale, anche a tutti i donatori che hanno soggiornato per più di sei mesi nel Regno Unito durante il periodo di massima esposizione della popolazione alla BSE (1980-1996). Queste misure possono sembrare particolarmente restrittive, ma la BSE ha insegnato che per combattere queste malattie l'unica arma efficace è quella di attuare strategie preventive di lunghissimo periodo.
Terapia. - Non vi sono al momento terapie efficaci per la MCJ e sindromi correlate. Sono in corso due trials clinici, uno negli Stati Uniti e uno nel Regno Unito, con la quinacrina, un farmaco noto da tempo come antimalarico in grado di inibire la formazione di PrPEST in coltura cellulare di neuroblastoma infetto con scrapie. Gli studi sperimentali in modelli animali non hanno però confermato l'efficacia del farmaco e, sebbene non siano ancora disponibili i dati conclusivi di questi trials clinici, i risultati del trattamento su singoli pazienti non sono incoraggianti. Nel Regno Unito è stata provata in alcuni pazienti la somministrazione intraventricolare di pentosan polisolfato, un farmaco che in modelli sperimentali animali ha dato risultati promettenti, che sembrerebbe aver stabilizzato il decorso clinico in un paziente affetto da variante MCJ.
bibliografia
P. Brown, R. Meyer, F. Cardone et al., Ultra-high-pressure inactivation of prion infectivity in processed meat: a practical method to prevent human infection, in Proceedings of the National academy of science of the United States of America, 2003, 100, 10, pp. 6093-97.
G. Zanusso, S. Ferrari, F. Cardone et al., Detection of pathologic prion protein in the olfactory epithelium in sporadic Creutzfeldt-Jakob disease, in The New England journal of medicine, 2003, 348, 8, pp. 711-19.
C. Casalone, G. Zanusso, P. Acutis et al., Identification of a second bovine amyloidotic spongiform encephalopathy: molecular similarities with sporadic Creutzfeldt-Jakob disease, in Proceedings of the National academy of science of the United States of America, 2004, 101, 9, pp. 3065-70.
F.L. Heppner, A. Aguzzi, Recent developments in prion immunotherapy, in Current opinion in immunology, 2004, 16, 5, pp. 594-98.
A.H. Peden, M.W. Head, D.L. Ritchie et al., Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient, in Lancet, 2004, 364, pp. 527-29.
M. Pocchiari, M. Puopolo, E.A. Croes et al., Predictors of survival in sporadic Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies, in Brain: a journal of neurology, 2004, 127, pp. 2348-59.
C. Casalone, C. Corona, M.I. Crescio et al., Pathological prion protein in the tongues of sheep infected with naturally occurring scrapie, in Journal of virology, 2005, 79, pp. 5847-49.
J. Collinge, Molecular neurology of prion disease, in Journal of neurology, neurosurgery and psychiatry, 2005, 76, 7, pp. 906-19.
A. Ladogana, M. Puopolo, E.A. Croes et al., Mortality from Creutzfeldt-Jakob disease and related disorders in Europe, Australia, and Canada, in Neurology, 2005, 64, 9, pp. 1586-91.
A. Ladogana, M. Puopolo, A. Poleggi et al., High incidence of genetic human transmissible spongiform encephalopathies in Italy, in Neurology, 2005, 64, 9, pp. 1592-97.