
Farmaci, progettazione dei
Farmaci, progettazione dei
di Fulvio Gualtieri
Farmaci, progettazione dei
sommario: 1. Il processo di sviluppo di un farmaco. 2. I principî alla base dell'azione dei farmaci. 3. Il bersaglio dei farmaci. 4. Il meccanismo d'azione dei farmaci. 5. Prodotti biologicamente attivi e farmaci. 6. Saggi farmacologici e biologici ad alta efficienza e capacità. 7. Metodi computazionali nella progettazione dei farmaci. 8. La progettazione dei farmaci. a) L'dentificazione del prototipo. b) L'ottimizzazione del prototipo. □ Bibliografia.
1. Il processo di sviluppo di un farmaco
La scoperta e lo sviluppo di un farmaco sono processi lunghi, ad alto rischio e molto costosi cui contribuiscono principalmente la chimica e la farmacologia preclinica e clinica, ma che poggiano su conoscenze di base derivanti da una varietà di discipline, quali la genetica, la fisiologia, la microbiologia, la patologia, la biologia molecolare e, più in generale, quelle discipline che vanno sotto il nome di scienze della vita. Il cammino che conduce al farmaco inizia generalmente con l'identificazione di una sostanza chimica, di origine naturale o sintetica, dotata di promettenti proprietà farmacologiche; usualmente questa sostanza viene indicata con il termine inglese lead, che in italiano può essere tradotto con 'prototipo'. Dopo che la molecola prototipo è stata identificata, essa in genere subisce un processo di ottimizzazione consistente in variazioni strutturali mirate, eseguite mediante sintesi chimica, tramite le quali si cerca di identificare, nella serie di congeneri che ne deriva, il prodotto con le migliori caratteristiche farmacologiche, farmacocinetiche e di sicurezza. A questo scopo, le molecole studiate sono sottoposte a una varietà di saggi farmacologici in vitro e in vivo su animali di laboratorio. Durante questa fase, che dura tipicamente da tre a cinque anni, viene inoltrata la richiesta di protezione brevettuale. La molecola ottimizzata, se giudicata adatta a essere utilizzata come farmaco, è sottoposta a una sequenza di prove cliniche sull'uomo - indicate come fasi I, II, III - il cui esito positivo permette la presentazione di una richiesta di registrazione presso le autorità sanitarie. Mediamente, le prove cliniche e la registrazione di un farmaco richiedono da 8 a 10 anni a seconda del tipo di azione farmacologica coinvolta, il che significa che dal momento della scoperta del prototipo all'utilizzazione del farmaco passano circa 11-15 anni. Va però osservato che attualmente i tempi per lo sviluppo clinico e l'approvazione sembra si stiano lentamente riducendo. Dopo l'immissione sul mercato, il farmaco entra nella cosiddetta fase IV, che essenzialmente consiste nel monitoraggio della sua azione terapeutica e degli effetti tossici su un numero di pazienti più elevato rispetto a quelli coinvolti nelle fasi precedenti e quindi con risultati statisticamente più significativi. I problemi che emergono in questa fase possono determinare una restrizione d'uso o anche il ritiro del farmaco dal mercato, com'è successo in non pochi casi. Al termine della protezione brevettuale (in genere venti anni), un farmaco può essere prodotto e commercializzato, a ben precise condizioni, da altre società del settore ed entrare così nel gruppo dei farmaci cosiddetti generici.
Le risorse, in uomini e mezzi, necessarie per la realizzazione di un farmaco richiedono ingenti investimenti che, di fatto, rendono impossibile che ciò avvenga al di fuori del sistema industriale. Solo alcune fasi possono aver luogo in sedi diverse, come università, centri di ricerca o strutture ospedaliere. In genere, tuttavia, l'industria titolare del brevetto sovrintende e finanzia le fasi che non possono essere svolte nel suo ambito. La spesa per portare a termine lo sviluppo di un farmaco è cresciuta costantemente negli ultimi decenni - attualmente si aggira tra i 600 e gli 800 milioni di euro, a seconda del tipo di farmaco - per molteplici ragioni; tra esse appare predominante l'esigenza di sicurezza, che induce le autorità sanitarie a richiedere accertamenti sempre più accurati, soprattutto a livello clinico, prima di autorizzare la commercializzazione di una nuova molecola. A questo proposito è necessario ricordare che le autorità sanitarie sono spinte a questo rigore, che potrebbe apparire eccessivo, dai gravi incidenti verificatisi negli ultimi anni (vedi per tutti il caso della talidomide) e dall'allarme che questi hanno suscitato nell'opinione pubblica. Con l'aumento delle spese per lo sviluppo, il ritorno in denaro derivante dalla commercializzazione di una nuova molecola tende a ridursi. Da una parte, per il protrarsi dei tempi di realizzazione il periodo di monopolio coperto dal brevetto si abbrevia e, dall'altra, un prodotto innovativo rimane l'unico del suo genere sul mercato per periodi sempre più brevi; per esempio, il propranololo, che è stato lanciato nel 1968, ha avuto in esclusiva il mercato per dieci anni prima che fosse disponibile il secondo beta bloccante (metoprololo); venti anni dopo, nel 1988, l'antidepressivo fluoxetina dopo solo quattro anni ha dovuto subire la concorrenza della sertralina, mentre nel 1999 l'antinfiammatorio inibitore delle ciclossigenasi 2 (COX-2), celecoxib, ha visto apparire un suo diretto concorrente (rofecoxib) appena quattro mesi dopo la sua immissione sul mercato.
Per ragioni di spazio, verranno inserite nel testo solo alcune delle strutture chimiche delle molecole menzionate; tutte le altre si possono ritrovare in The Merck index (v. O'Neil e altri, 200113).
Considerato che solo il 10° circa dei prodotti studiati raggiunge la registrazione, si può capire quanto sia complesso, costoso e lungo il processo che conduce all'immissione di un farmaco sul mercato. Per fronteggiare questi problemi, l'industria farmaceutica è intervenuta sia con soluzioni strutturali - quali la formazione, per confluenza o per acquisizione, di società sempre più grandi e potenti dal punto di vista finanziario - sia cercando di accelerare al massimo la scoperta di nuovi farmaci. La riduzione dei tempi che portano all'individuazione dei prototipi, l'abbattimento dei tassi di fallimento durante la loro messa a punto e un'accelerazione dei tempi di registrazione (che peraltro dipende essenzialmente dalle autorità sanitarie) sono alcune delle possibili soluzioni. Il sistema industriale farmaceutico ha investito notevolmente nelle tecnologie sviluppate negli ultimi due decenni, che sono essenzialmente la chimica computazionale (v. Leach, 20012; v. chimica computazionale), la bio- e la chemoinformatica (v. Stahura e Bajorath, 2002), i metodi di valutazione farmacologica e biologica ad alta efficienza e capacità (High Throughput Screening, HTS) e la sintesi combinatoriale (v. Gualtieri e altri, 1997) applicati alle conoscenze derivanti dalla genetica (v. Fernandes, 2001; v. genomica), dallo studio delle proteine codificate dai geni (v. Swindells e Overington, 2002; v. proteomica) e dai progressi della biologia molecolare.
Qui di seguito saranno esaminati in maggiore dettaglio i principî e le metodologie che sono alla base della progettazione dei farmaci, dopo aver richiamato alcune nozioni indispensabili sulle relazioni tra struttura chimica e azione biologica e farmacologica e sul meccanismo d'azione dei farmaci (v. Wermuth, 1996).
2. I principî alla base dell'azione dei farmaci
Corpora non agunt nisi fixata. Questo principio, formulato da Paul Ehrlich (v., 1913) agli albori della farmacologia molecolare, può essere così riformulato: non c'è azione senza interazione. Esso è alla base delle scienze del farmaco e permette di descrivere l'azione del farmaco in termini strutturali, sulla base dei legami che una data molecola con una data struttura è in grado di instaurare con la molecola bersaglio. Il principio esalta l'importanza della disposizione spaziale (stereochimica) degli atomi o dei gruppi di atomi che controllano la formazione dei legami tra il farmaco e il suo bersaglio, e rende possibile l'elaborazione di una serie di relazioni, note come 'relazioni struttura-attività' (RSA), tra la struttura chimica di una molecola e la sua azione biologica e farmacologica. Tra i più importanti legami interessati - di norma legami deboli, che permettono la formazione di complessi reversibili e di facile rottura - vi sono i legami di van der Waals, i legami elettrostatici, i legami a ponte di idrogeno, i legami a trasferimento di carica e il cosiddetto legame idrofobico. In alcuni casi, dopo la formazione del complesso reversibile iniziale, la situazione può evolvere verso la formazione di legami covalenti che rendono il complesso più stabile nel tempo. Le conseguenze di ciò a livello terapeutico possono essere molto diverse secondo l'oggetto biologico coinvolto. Nel caso in cui il bersaglio sia, ad esempio, un enzima presente solo in un organismo patogeno e che la sua inibizione conduca alla morte dell'organismo stesso, la formazione di un legame irreversibile è utile e auspicabile. È questo il caso della penicillina e di tutti gli antibiotici beta-lattamici che, formando un legame covalente, bloccano un enzima essenziale per la sopravvivenza dei microrganismi sensibili, impedendo la corretta formazione della loro parete cellulare. Quando però la molecola bersaglio ha funzioni biologiche importanti per l'organismo del paziente, la formazione di legami covalenti può essere una complicazione che induce severi effetti collaterali: è il caso degli antitumorali alchilanti che sono in grado di reagire sia con il DNA delle cellule tumorali sia con quello delle cellule sane, con ovvie conseguenze a livello di tossicità.
Una delle conseguenze del principio di interazione è che molecole simili, interagendo in modo analogo, sono in grado di produrre effetti simili. Anche se questo corollario ha trovato un certo numero di eccezioni, esso è alla base dei processi di ottimizzazione e di una delle metodiche principali per la progettazione dei farmaci: la modulazione molecolare di farmaci noti. Al contrario, quando si progettano molecole per sviluppare farmaci innovativi, sia per patologie che necessitano di nuovi interventi terapeutici sia per nuovi tipi di malattie, ci si orienta verso strutture il più possibile differenti da quelle già note.
3. Il bersaglio dei farmaci
Il principale bersaglio dei farmaci attualmente usati sono le proteine e gli acidi nucleici, di cui è fondamentale conoscere le funzioni, la struttura chimica, la stereochimica e le caratteristiche chimico-fisiche per la progettazione di piccole molecole che possano utilmente interagire con loro, modulandone l'azione, e che quindi possano essere utilizzate come farmaci. Per esempio, la conoscenza della struttura a doppia elica del DNA ha permesso di spiegare l'interazione di intere classi di farmaci antitumorali e di conseguenza ha facilitato la progettazione di altri farmaci dello stesso tipo. Attualmente l'attenzione nel campo degli acidi nucleici è focalizzata sull'informazione genetica racchiusa nel DNA cromosomico. In questi ultimi anni il numero delle sequenze geniche note è rapidamente cresciuto e dopo la decodificazione del genoma di organismi inferiori, ma di grande interesse per la progettazione di farmaci, è stata recentemente completata anche quella del genoma umano. È prevedibile che ciò faciliterà la definizione delle funzioni di ciascun gene e quindi l'uso della terapia genica e dei farmaci antisenso (oligonucleotidi modificati sintetici che interferiscono con l'espressione genica), ma soprattutto renderà possibile correlare la funzione dei geni con le proteine derivanti dalla loro espressione nelle cellule e nei tessuti. In altre parole, la definizione del genoma umano, così come quello di altre specie, consentirà la completa conoscenza dei processi biologici cellulari, suggerendo le modalità di intervento per modularli tramite i farmaci.
Le proteine, nella forma di enzimi, recettori, canali ionici, proteine di trasporto e pompe di vario genere, sono sicuramente il bersaglio più comune dei farmaci. Una volta che è stato individuato un bersaglio proteico e ne è stata riconosciuta l'importanza, diventa essenziale conoscerne la struttura chimica e sterica. Ciò è relativamente facile se la proteina è cristallizzabile o ha un peso molecolare inferiore ai 30.000 dalton; in questo caso, infatti, le tecniche di diffrazione ai raggi X e la spettrometria di risonanza magnetica nucleare (NMR) permettono di stabilire la struttura tridimensionale della proteina e di determinare anche la natura dei complessi formati dalla proteina stessa con piccole molecole, siano essi ligandi naturali o di sintesi. È così possibile identificare il sito di interazione, gli amminoacidi che lo compongono e la stereochimica coinvolta, e di conseguenza progettare le piccole molecole in grado di interagire con il sito sulla base della sua struttura. Un caso particolarmente favorevole è quello degli enzimi che, nella maggior parte dei casi, sono in grado di produrre cristalli la cui struttura può essere risolta. In genere, questi stessi cristalli, messi a contatto con una soluzione contenente un ligando, lo inglobano mantenendo la struttura cristallina e rendendo possibile la determinazione delle modalità di legame.
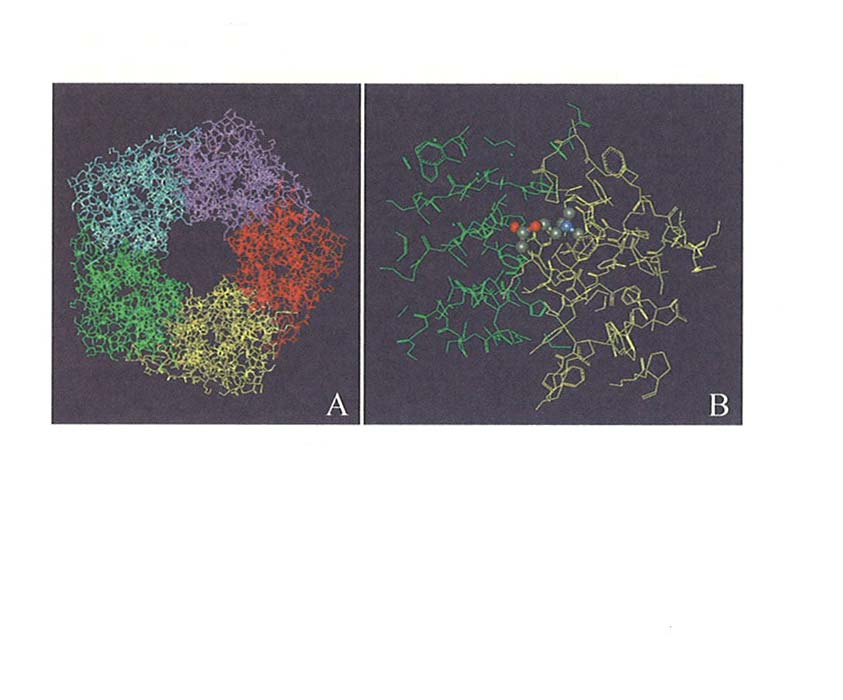
Tuttavia, molte proteine, in particolare quelle di membrana, sono difficili da cristallizzare. In tal caso, se è nota la struttura di un'altra proteina della stessa famiglia si può procedere per omologia (homology modelling) e costruire un modello, che naturalmente sarà meno preciso e attendibile di quello ottenuto direttamente, ma è utilizzabile per la progettazione di piccole molecole in grado di interagire con la proteina stessa e quindi divenire farmaci utili. Questa è, a tutt'oggi, la situazione nel campo delle proteine che costituiscono i recettori di membrana. Nella fig. 3 è riportata la struttura cristallina di una proteina solubile, ricavata dalle cellule gliali del mollusco Lymnaea stagnalis, che ha la proprietà di legare l'acetilcolina. Questa proteina, chiamata AchBP (Acetylcholine Binding Protein) ha un'alta omologia con il recettore colinergico nicotinico ed è stata utilizzata per modellare per omologia la parte extracellulare del recettore stesso.
La recente e attesissima risoluzione a 2,8 Å della struttura della rodopsina bovina (v. Palczewski e altri, 2000), che ha fornito la prima struttura cristallina di un recettore accoppiato alle proteine G (GPCR, G Protein Coupled Receptor), dovrebbe permettere di elaborare modelli di recettori appartenenti a questa famiglia recettoriale molto più attendibili di quanto non sia stato possibile in passato, con gran miglioramento delle prospettive di progettazione dei farmaci relativi.
4. Il meccanismo d'azione dei farmaci
Anche se non mancano esempi illustri di farmaci scoperti e usati con successo prima che fosse chiarito il loro meccanismo d'azione (vedi i sulfamidici), per essere in grado di progettare con sufficiente attendibilità un farmaco è essenziale conoscere, a livello molecolare, i processi fisio-patologici legati alla malattia da trattare. Spesso i meccanismi sono talmente complessi da permettere una certa varietà di linee d'intervento. È questo il caso dell'ipertensione, che può essere controllata da farmaci di diverso tipo, che agiscono su recettori (adrenergici, dell'angiotensina II), su canali ionici (canali del calcio), su sistemi enzimatici (ACE, Angiotensin Converting Enzyme), sui meccanismi di diuresi. È chiaro che in casi del genere, prima di passare alla progettazione del farmaco si deve scegliere il sistema sul quale agire. Questa scelta può essere dettata da ragioni di selettività d'azione, in modo da ridurre gli effetti collaterali non desiderati; per esempio, il blocco della formazione dell'acido tetraidrofolico tramite l'inibizione degli enzimi che sono coinvolti nella sua sintesi è un modo per impedire lo sviluppo di microrganismi patogeni. I sulfamidici inibiscono il funzionamento di un enzima (la pteroatosintetasi) che è presente solo nei Procarioti; pertanto, dato che la loro azione è selettiva verso i microrganismi, provocano meno effetti collaterali rispetto agli inibitori dell'enzima diidrofolatoreduttasi, che è presente sia nei microrganismi sia nei Mammiferi.
Accade spesso che le cause di una malattia non siano del tutto chiare e che il processo che determina la patologia da trattare non sia noto a livello molecolare; in tal caso, la progettazione di farmaci assume un aspetto più empirico e viene basata sulla sperimentazione a tappeto o, più razionalmente, su ipotesi di lavoro formulate in base alle informazioni disponibili sulla fisiologia e la patologia in questione. Un esempio istruttivo in proposito è quello della malattia di Alzheimer, le cui origini sono tuttora oggetto di ricerca e discussione. La ricerca di farmaci utili è stata guidata, in un primo tempo, dall'osservazione che il tono colinergico centrale dei pazienti affetti da questa patologia era compromesso; è stata così formulata un'ipotesi di lavoro, nota come 'ipotesi colinergica della malattia di Alzheimer', che attribuisce a un deficit colinergico centrale le manifestazioni della malattia, per cui, riportando la trasmissione colinergica al livello normale, si dovrebbe riuscire a controllarne alcuni sintomi. Su questa base sono stati progettati una notevole varietà di farmaci in grado di aumentare il tono colinergico centrale, ma solo una famiglia di questi, gli inibitori della acetilcolinesterasi, che prevengono la degradazione dell'acetilcolina, ha avuto un certo successo. Sulla base delle nuove informazioni raccolte relativamente alla natura delle placche caratteristiche della malattia e dei processi che conducono alla loro formazione, all'ipotesi colinergica è stata affiancata l'ipotesi amiloide, che attribuisce la responsabilità dei processi neurodegenerativi che si verificano a livello cerebrale alla patologica formazione di una miscela di peptidi costituiti da 40-42 residui amminoacidici, generalmente indicati come proteina amiloide (Aβ), che è un costituente fondamentale delle placche. Attualmente, sulla base di tale ipotesi, si stanno sperimentando farmaci progettati per inibire gli enzimi coinvolti nella formazione della proteina, alcuni dei quali sono stati recentemente isolati e identificati. Parallelamente, si stanno ricercando sostanze in grado di impedire che la proteina amiloide vada incontro a quelle variazioni conformazionali che conducono alla precipitazione di polipeptidi insolubili e quindi alla formazione delle caratteristiche placche.
5. Prodotti biologicamente attivi e farmaci
Il numero delle molecole che hanno una qualche azione sui sistemi biologici è enorme, mentre quelle realmente usate in terapia sono poche migliaia. Ciò è dovuto al fatto che, per ottenere un effetto terapeutico, un prodotto non solo deve avere un'azione biologica, ma deve essere messo nelle condizioni di esercitarla, senza peraltro causare effetti indesiderati. Ciò significa che, in generale, il prodotto deve essere assorbito, trasportato e distribuito nel sito d'azione, resistere al metabolismo e all'eliminazione in misura tale da mantenere una concentrazione sufficiente allo scopo, e infine non essere tossico per l'organismo alle dosi farmacologicamente efficaci, né direttamente né attraverso i suoi metaboliti. Il complesso di questi eventi costituisce la farmacocinetica e viene identificato spesso con l'acronimo ADME (assorbimento, distribuzione, metabolismo, eliminazione; v. Smith e altri, 2001). Le proprietà farmacocinetiche e di tossicità (ADME/Tox) sono determinanti per il destino di un farmaco e contribuiscono a restringere enormemente il numero di prodotti biologicamente attivi utilizzabili come farmaci. In realtà il numero delle molecole attive in via di sviluppo che si debbono abbandonare per problemi di farmacocinetica e tossicità è purtroppo molto elevato e costituisce una delle cause degli altissimi costi richiesti dalla realizzazione di un farmaco. L'esperienza dimostra che le informazioni sulla farmacocinetica e sulla tossicologia di una molecola debbono essere disponibili prima possibile durante il processo di progettazione e di sviluppo, tendenzialmente insieme alle informazioni farmacologiche preliminari (fase preclinica), per minimizzare le perdite in tempo e denaro. A questo scopo sono stati elaborati specifici saggi sperimentali (v. Li, 2001) e procedure computerizzate che forniscono previsioni teoriche sulle proprietà ADME/Tox di una molecola. In generale, è importante che la molecola in via di progettazione abbia caratteristiche il più possibile vicine a quelle della maggioranza dei farmaci (druglikeness).
Per ciò che riguarda l'assorbimento di un farmaco, la 'regola del cinque', elaborata da Christopher Lipinski, stabilisce che molecole che presentano: a) più di cinque gruppi donatori di legami idrogeno; b) un peso molecolare maggiore di 500 dalton; c) un logaritmo del coefficiente di ripartizione calcolato (Clog P) maggiore di cinque; d) un totale di atomi di ossigeno e azoto più alto di dieci, sono potenzialmente male assorbite e quindi inadatte a diventare farmaci. Il passaggio attraverso cellule epiteliali intestinali in monostrato tipo Caco-2 è una procedura utilizzata per valutare sperimentalmente l'assorbimento intestinale, ma sono stati anche elaborati degli algoritmi in grado di valutare tale caratteristica utilizzando il computer (in silico).
La tossicità di un potenziale farmaco è tradizionalmente valutata sugli animali, in genere a uno stadio di sperimentazione del farmaco piuttosto avanzato. Tuttavia, la necessità di avere informazioni attendibili già all'inizio della fase di ottimizzazione ha favorito la ricerca di metodi alternativi, tra i quali sembra particolarmente promettente la proteomica: l'analisi delle proteine presenti nelle cellule di un tessuto prima e dopo l'esposizione alla sostanza da analizzare può dare informazioni preziose sulle conseguenze della sua presenza nelle cellule. La sensibilità del metodo permette l'uso di quantità minime di sostanza, anche nella fase iniziale dello sviluppo del farmaco.
Nell'ambito della farmacocinetica, grande rilievo assumono le trasformazioni metaboliche di un possibile farmaco, anch'esse studiate su animali e poi sull'uomo. Infatti, il metabolismo condiziona il tempo di emivita, e quindi il dosaggio e la via di somministrazione, e può contribuire alla tossicità del farmaco attraverso i prodotti di metabolizzazione. Tuttavia, sempre per evitare costosi fallimenti, appare necessario avere informazioni sulla stabilità metabolica al più presto possibile. A questo scopo sono stati messi a punto saggi in vitro che utilizzano microsomi o epatociti e sono state elaborate procedure computerizzate per prevedere il comportamento metabolico di una molecola. Le informazioni raccolte permettono di modulare la sintesi durante la fase di ottimizzazione, per esempio proteggendo con adatte sostituzioni una posizione che risulta rapidamente ossidata e quindi aumentando la stabilità metabolica della molecola. Il metabolismo gioca un ruolo essenziale anche nella progettazione dei profarmaci, ossia di derivati funzionali di un farmaco che dopo l'assorbimento vengono nuovamente trasformati nel farmaco originale per mezzo di una reazione metabolica. In tal modo si possono superare i problemi di biodisponibilità che spesso condizionano l'uso di un farmaco; ad esempio, la pivampicillina, un profarmaco dell'ampicillina, presenta una biodisponibilità migliore di quella della molecola originaria ed è quindi utile a combattere infezioni batteriche difficili da trattare.
6. Saggi farmacologici e biologici ad alta efficienza e capacità
L'introduzione di metodologie di analisi farmacologica e biologica HTS (High Throughput Screening), ossia estremamente rapide, efficienti e sensibili, ha rappresentato una svolta epocale nella progettazione dei farmaci. Queste metodologie permettono di valutare l'azione biologica e farmacologica di un prodotto, o di una miscela di prodotti, utilizzando quantità minime di sostanza e possono essere quasi completamente automatizzate, consentendo l'analisi di centinaia o migliaia di prodotti in una giornata ed eliminando così quello che per decenni ha rappresentato il fattore limitante nella ricerca di prototipi e nella fase di ottimizzazione dei farmaci. La loro introduzione ha determinato la necessità di disporre di sempre nuove molecole da sottoporre a valutazione farmacologica, che a sua volta ha condotto all'utilizzazione massiccia di un'altra moderna metodologia di cui si parlerà più avanti, la sintesi combinatoriale.
Poiché si può ragionevolmente supporre che nello spazio chimico virtuale, che comprende tutte le molecole progettabili, siano possibili qualcosa come 10100 composti chimici, è del tutto irrealistico pensare che tale spazio possa essere esplorato con librerie di composti ottenute sperimentalmente, per quanto ampie esse possano essere. Per questa ragione sono stati messi a punto degli algoritmi in grado di fare un'analisi biologica virtuale di molecole virtuali comprese in una libreria virtuale (v. Walters e altri, 1998). Questi algoritmi, se efficienti e attendibili, dovrebbero permettere di filtrare (virtual screening) l'enorme spazio chimico virtuale, delimitando il tipo e il numero delle molecole da sintetizzare e provare sperimentalmente.
7. Metodi computazionali nella progettazione dei farmaci
La possibilità di immagazzinare, richiamare e analizzare un'enorme mole di informazioni rende il computer prezioso, e in alcuni casi indispensabile, nella ricerca biologica (bioinformatica), chimica (chemoinformatica) e di conseguenza in quella farmaceutica. Si è già accennato all'uso del computer e della scienza informatica nell'identificazione delle proteine del proteoma, nella predizione della stabilità metabolica di una molecola, nell'analisi virtuale di librerie di prodotti (v. bioinformatica). Queste applicazioni sfruttano la capacità dei moderni computer, anche di piccole dimensioni, di analizzare con opportuni algoritmi enormi collezioni di dati (data base) per identificare strutture, per dedurne una vasta gamma di proprietà e darne una rappresentazione grafica immediata e facilmente comprensibile. Si tratta di applicazioni estremamente utili ma che non riguardano direttamente la progettazione del farmaco, come accade invece per la modellistica molecolare (molecular modelling; v. Gubernator e Bohm, 1998). Quest'ultima è una tecnica che permette di determinare la geometria di una molecola e le sue possibili conformazioni, calcolando per ognuna di esse l'energia e quindi la probabilità di esistenza, gli orbitali molecolari e la densità elettronica, la lipofilia, il volume, la superficie accessibile al solvente. È possibile simulare il comportamento dinamico della molecola nel vuoto, in un solvente o durante l'interazione con un'altra molecola d'interesse biologico. Se il sito di interazione è noto se ne possono valutare le modalità e prendere in considerazione le variazioni strutturali necessarie per incrementare l'energia di interazione; se il sito di interazione non è noto, possono essere elaborati modelli che aiutino a capire quale esso sia e quindi a progettare molecole più affini basandosi sulle caratteristiche strutturali di altre molecole in grado di interagire positivamente con quel sito.
I programmi di modellistica molecolare sono numerosi, ma sostanzialmente si rifanno a due tipi di approccio: quello quantomeccanico e quello di meccanica molecolare. I programmi di tipo quantomeccanico calcolano le proprietà della molecola risolvendo l'equazione di Schroedinger in maniera approssimata, con i metodi cosiddetti semiempirici, oppure in modo più rigoroso, con i metodi ab initio, che però in genere richiedono più potenza di calcolo. I programmi di meccanica molecolare calcolano l'energia sterica di una molecola utilizzando campi di forza empirici (force field) formati da funzioni di potenziale derivanti dalla meccanica classica, come per esempio la legge di Hook.
Un altro approccio molto utilizzato per lo studio delle dinamiche molecolari è quello della tempra simulata (simulated annealing); in questo caso la molecola in esame viene descritta come una struttura dinamica le cui coordinate atomiche cambiano rispetto al tempo in base all'energia cinetica degli atomi e alle forze esercitate su di loro dagli atomi circostanti e dal campo di forza in cui sono immersi.
Le informazioni raccolte con queste metodiche, in particolare quelle sulla conformazione di minima energia, sono di regola utilizzate per la progettazione dei farmaci, come si vedrà più avanti.
Un altro aspetto della chimica computazionale che trova particolare uso nello studio e nella progettazione dei farmaci è la determinazione delle relazioni quantitative tra struttura chimica e attività biologica (QSAR, Quantitative Structure-Activity Relationships). Il metodo che ha avuto maggior successo tra i molti sperimentati è quello di Hansch (v. Kubinyi, 1993), nel quale queste relazioni sono descritte da un'equazione multiparametrica che mette in relazione l'azione biologica con proprietà chimico-fisiche quali la lipofilia, la distribuzione elettronica e la struttura sterica, definite da opportuni parametri. Quando l'equazione assume significatività statistica, i coefficienti delle variabili forniscono informazioni preziose sulle caratteristiche necessarie a produrre l'attività biologica propria della serie di farmaci in esame, informazioni che possono servire sia a spiegare il meccanismo di azione della classe di farmaci studiata, sia a programmare la sintesi di altre molecole con caratteristiche migliori. L'uso combinato della QSAR e della modellistica molecolare ha condotto alla 3D-QSAR (QSAR tridimensionale), che migliora la qualità e la quantità delle informazioni ottenibili con la tecnica tradizionale. Tra le varie metodologie sviluppate, la più diffusa è l'analisi comparativa dei campi molecolari (CoMFA, Comparative Molecular Field Analysis), la quale si basa sull'assunto che le interazioni molecolari di una sostanza biologicamente attiva con il proprio bersaglio biologico possano essere descritte tramite campi di forza elettronici, sterici e idrofobici, e che le molecole possano quindi essere caratterizzate sulla base del valore di queste interazioni calcolate a una distanza determinata e mediante opportune sonde molecolari. I risultati dell'analisi CoMFA sono visualizzati tramite mappe di contorno dei coefficienti (elettrostatici, sterici, idrofobici), i quali esprimono l'intensità del campo in un particolare punto dello spazio.
8. La progettazione dei farmaci
a) L'identificazione del prototipo
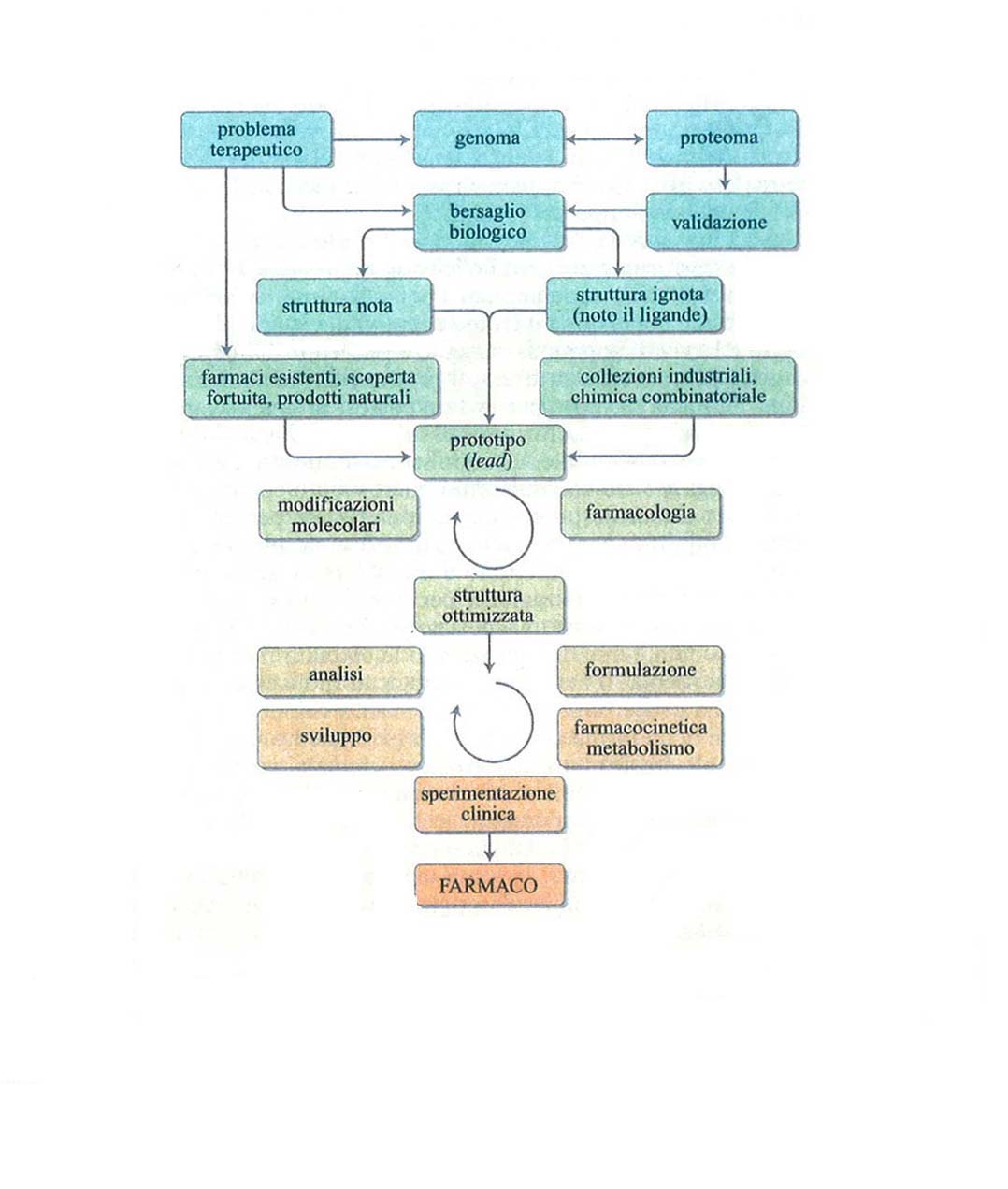
Tenendo presente quanto detto nei capitoli precedenti, è ora possibile scendere nei dettagli del processo di identificazione, progettazione e sviluppo di un farmaco, le cui fasi sono schematicamente riportate nella fig. 4.
Ciò che determina la ricerca di un farmaco è quasi sempre la necessità di fronteggiare un problema terapeutico. Il 'quasi' è opportuno perché talvolta, seppure raramente, la spinta al trattamento farmacologico sorge da esigenze di tipo diverso dall'esistenza di uno stato patologico. È questo il caso degli anticoncezionali steroidei, che costituiscono un gruppo estremamente importante di farmaci sviluppati per prevenire gravidanze indesiderate. La chimica farmaceutica e la farmacologia sono le scienze che si occupano di questa ricerca, l'una identificando, progettando e sintetizzando molecole, l'altra valutandone l'azione farmacologica.
Per molto tempo e fino a parte del secolo passato, le origini molecolari di una malattia erano il più delle volte ignote e quindi mancavano informazioni sul bersaglio del farmaco destinato a curarla. Di conseguenza, per individuare il prototipo da sviluppare la chimica farmaceutica procedeva in modo largamente empirico, attraverso tentativi ed errori, utilizzando una delle metodologie che saranno descritte in seguito; del resto, ciò avviene ancora oggi nei casi in cui non sia nota l'origine della malattia. Sempre più spesso però, la biologia molecolare, lo studio dei geni e delle proteine da questi espresse in una cellula, fanno sì che la natura e la struttura del bersaglio del farmaco siano note in modo più o meno dettagliato. Ciò permette al chimico farmaceutico di intervenire in modo certamente più razionale (v. Fernandes, 2001) e di giungere all'identificazione del prototipo in tempi generalmente più rapidi. Parallelamente, la sperimentazione farmacologica può essere più mirata e informativa.
In ogni caso, però, la ricerca di un farmaco inizia con la ricerca di una molecola prototipo (v. Gualtieri e altri, 1997), vale a dire una molecola che abbia una promettente azione sulla malattia che si vuole curare (o sul suo modello sperimentale) senza possedere, però, tutte le caratteristiche per poter diventare farmaco. Queste caratteristiche sono numerose e quasi tutte essenziali. Una, ovviamente discriminante, è quella della potenza: il prodotto deve essere attivo a dosi molto basse per poter essere somministrato in quantità tali da minimizzare gli effetti collaterali indesiderati. Tipicamente, un prototipo ha un'attività nell'ordine micromolare (10-6 moli/l), mentre per un prodotto ottimizzato ci si aspetta un'attività nell'ordine nanomolare (10-9 moli/l). Anche la selettività di azione, spesso insufficiente nei prototipi, è una caratteristica importante per minimizzare gli effetti secondari, soprattutto per i farmaci che agiscono sui sistemi recettoriali. L'importanza cruciale della farmacocinetica è già stata trattata: spesso un prototipo ha una farmacocinetica insufficiente e una stabilità metabolica inadeguata, che devono essere migliorate prima di poterlo utilizzare come farmaco. Altre proprietà importanti per un farmaco sono la stabilità chimica e la facilità di sintesi, che spesso debbono essere opportunamente modulate. Infine, in molti casi il prototipo non ha le caratteristiche di brevettabilità che sono indispensabili per iniziare lo sviluppo di un farmaco. Comunque, i progressi della chimica farmaceutica, della sintesi organica e della sperimentazione farmacologica permettono di modulare e ottimizzare qualsiasi prototipo portandolo alle soglie dello sviluppo. Il problema fondamentale diventa quindi quello dell'individuazione del prototipo stesso.
Una molecola prototipo può essere individuata attraverso varie strategie, alcune molto ben sperimentate e tradizionali, altre che si stanno affermando solo ora e sulla cui efficacia sarà possibile esprimersi solo tra qualche tempo.
Una via sempre attuale per individuare un prototipo, o addirittura il farmaco, è quella della scoperta casuale. Gli esempi in proposito sono numerosi e illustri, a partire dalla penicillina G, passando al clordiazepossido per arrivare al sildenafil. In questi casi la fortuna è andata di pari passo con l'attenzione e la preparazione dei ricercatori, microbiologi, chimici o farmacologi che fossero. È chiaro, comunque, che la ricerca moderna non può essere affidata solo alla sorte e quindi sono necessarie ben altre strategie.
Una di queste - concettualmente poco gratificante, ma che può dare risultati molto importanti dal punto di vista sia terapeutico che commerciale - è la manipolazione di farmaci già esistenti. Basandosi sul principio già menzionato che molecole simili generalmente producono azioni farmacologiche simili, si può utilizzare come prototipo un farmaco già sperimentato. È chiaro che per ottenere la brevettabilità del prodotto, tali manipolazioni debbono introdurre delle novità a livello sia strutturale che terapeutico. Questa strada, che di norma non produce farmaci con caratteristiche originali e che quindi tende a non essere seguita quando si vogliono identificare farmaci innovativi, è però quella percorsa da industrie farmaceutiche che vogliono entrare nella quota di mercato di un farmaco innovativo. Tale strategia ha comunque ricadute importanti dal punto di vista terapeutico, perché i prodotti di seconda generazione presentano in genere proprietà farmacologiche e farmacocinetiche migliori del farmaco originale. Un esempio di questo approccio è rappresentato dai farmaci antiulcera ranitidina e famotidina, comparsi dopo la cimetidina, il primo a essere immesso sul mercato.
Accade spesso che, durante le manipolazioni molecolari di farmaci noti, si identifichino prodotti con azioni biologiche importanti e diverse da quelle del farmaco di partenza, come è esemplificato dalla scoperta dell'antidepressivo imipramina durante la manipolazione molecolare dell'antipsicotico clorpromazina. Ciò sembrerebbe dimostrare i limiti del concetto che strutture simili siano in grado di dare effetti farmacologici simili, ma spesso studi più approfonditi, soprattutto a livello conformazionale, evidenziano che, in tali casi, le strutture in esame sono simili solo superficialmente.
In qualche modo legata alla strategia precedente è l'utilizzazione come prototipi di metaboliti attivi di farmaci già in uso. È il caso del cicloguanile, che è stato introdotto in terapia dopo la constatazione che la cloroguanide, per divenire attiva, doveva essere ciclizzata nell'organismo.
Molti farmaci hanno effetti collaterali, che spesso si evidenziano nella fase IV, attribuibili a un meccanismo di azione diverso da quello responsabile dell'azione principale. Se tale meccanismo può essere utile per trattare un altro problema terapeutico, il farmaco può diventare il prototipo per l'identificazione di una nuova molecola che agisca esclusivamente con questo meccanismo. La nuova molecola deve essere il più possibile priva dell'azione originale, che rappresenterebbe un effetto secondario indesiderato. L'intero processo, da ottenersi con la manipolazione molecolare di cui parleremo in dettaglio in seguito, consiste quindi nell'amplificazione e nella separazione dei meccanismi d'azione del prototipo. La classe dei sulfamidici è stata, da questo punto di vista, estremamente produttiva. Gli effetti diuretici e ipoglicemizzanti di alcuni sulfamidici, apparsi evidenti quando il loro uso è diventato massiccio, hanno stimolato la sintesi di nuove molecole correlate che presentassero esclusivamente l'azione diuretica o ipoglicemizzante. Si sono così individuate due nuove classi di farmaci ipoglicemizzanti e diuretici - clorpropamide e clorotiazide - prive dell'azione antimicrobica, ora indesiderata. Tuttavia, la separazione completa delle attività non è facile da ottenere e talvolta l'attività farmacologica originale resta come effetto secondario nelle nuove molecole; ad esempio, nell'anabolizzante stanazololo - così come in molti prodotti analoghi ottenuti per manipolazione molecolare del testosterone, che è allo stesso tempo anabolizzante e androgeno - permane una seppur lieve azione androgena responsabile di effetti collaterali indesiderati.
Il complesso delle sostanze naturali, d'origine vegetale o animale, è da sempre una fonte preziosa di prototipi e di farmaci. Dalla chinina, al tassolo, alla ziconotide, sono numerosi i farmaci ricavati da fonti naturali. Alcuni, oltre ad avere grande efficacia terapeutica, hanno poi costituito i prototipi per successive modificazioni molecolari come è successo per la penicillina G che è stata il punto di partenza per tutte le penicilline semisintetiche.
Un metodo praticato diffusamente nella ricerca di prototipi è quello della sperimentazione a tappeto, che può consistere nel provare gli effetti di una molecola su una vasta gamma di modelli farmacologici per individuare eventuali attività terapeutiche (extensive screening), oppure nella sperimentazione di un numero molto grande di molecole su un unico modello farmacologico per individuare il prototipo da sviluppare (random screening). Entrambi gli approcci, dopo esser stati praticati per un certo tempo, erano stati accantonati perché molto dispendiosi in termini di tempo e di investimenti; tuttavia, i metodi di binding e, successivamente, lo sviluppo di saggi farmacologici e biologici HTS, ad alta efficienza e capacità, li hanno resi nuovamente praticabili e altamente produttivi. È ora una pratica diffusa nelle industrie farmaceutiche possedere una vasta collezione di sostanze che possono essere saggiate rapidamente su un dato modello farmacologico, per esempio su di un nuovo sistema recettoriale. Usualmente, questa procedura conduce all'identificazione di uno o più prototipi che possono essere opportunamente sviluppati con i metodi della modulazione molecolare. Un esempio di questo approccio è quello che ha portato alla scoperta dell'inibitore dell'acetilcolinesterasi donepezil, usato nel trattamento della malattia di Alzheimer, a partire da una molecola con debole azione inibitoria, identificata nell'analisi a tappeto della collezione di sostanze chimiche della ditta produttrice.
Naturalmente, più consistente è il numero e più diversa la struttura chimica delle molecole che si sottopongono all'analisi farmacologica, più alta è la probabilità di identificare un numero adeguato di prototipi attivi. Siccome le collezioni delle ditte farmaceutiche hanno un'ampiezza limitata e allo stesso tempo la loro eterogeneità chimica non è quella desiderata, si è posto il problema di avere accesso a numeri molto elevati di molecole più diverse possibile in modo da aumentare le probabilità di individuare nuovi prototipi. La risposta a questa necessità è stata la sintesi combinatoriale (v. Gualtieri e altri, 1997; v. Antonenko e altri, 2000), una strategia mediante la quale si fa reagire una serie di miscele di reagenti ottenendo una miscela di prodotti, indicata come libreria (library), raccolta in un unico contenitore. Il numero di molecole diverse presenti in ciascuna libreria è derivabile dal calcolo combinatorio. Ad esempio, facendo reagire dieci diversi acidi carbossilici con dieci alcoli diversi si otterrà, teoricamente, una libreria di cento esteri diversi. Se esiste una metodica per la valutazione dell'attività biologica di questa miscela e una metodica per individuare il prodotto, o i prodotti, che rendono eventualmente attiva la miscela, si possono individuare in tempi molto rapidi nuovi prototipi. Infatti, con il metodo classico si procederebbe alla sintesi di ogni singolo estere facendo reagire un solo acido e un solo alcool, quindi il prodotto verrebbe purificato e caratterizzato per essere infine sottoposto alle prove farmacologiche. È chiaro che i tempi necessari per individuare un eventuale prototipo sarebbero molto più lunghi. Naturalmente, per rendere praticabile la procedura si sono dovuti risolvere molti e delicati problemi: sono state adattate e sviluppate metodiche sintetiche, come la sintesi in fase solida; è stato necessario mettere a punto tecniche di dosaggio farmacologico sensibili, rapide e affidabili (HTS), escogitare metodi per individuare in modo rapido e sicuro i componenti attivi di una libreria, aumentare l'automazione delle procedure sintetiche e analitiche in modo da poter valutare farmacologicamente il più ampio numero possibile di molecole. L'industria farmaceutica ha creduto in questa metodica e ha investito ingenti risorse nel suo sviluppo che è stato esponenziale negli ultimi dieci anni. Al momento attuale, peraltro, l'interesse iniziale per le librerie contenenti alti numeri di molecole (fino a molte migliaia) si è molto ridotto e, per individuare nuovi prototipi, vengono progettate librerie dell'ordine di centinaia o poche migliaia di prodotti, o anche poche decine quando la chimica combinatoriale (che in questo caso è definita anche sintesi parallela) è utilizzata per l'ottimizzazione. Sono oggi in via di sviluppo numerose molecole identificate e ottimizzate con questa metodica.
Le metodiche di progettazione dei farmaci fino a ora illustrate si possono applicare sempre, ma sono particolarmente utili quando manca qualsiasi informazione sull'oggetto biologico che costituisce il bersaglio del farmaco. Fortunatamente, però, i progressi delle scienze di base hanno fatto sì che sempre più frequentemente le informazioni sui geni (genomica) e sulle molecole proteiche (proteomica) di una cellula o di un tessuto abbiano reso nota la causa della patologia e il bersaglio del farmaco da progettare, e che spesso sia nota nei dettagli anche la struttura del bersaglio stesso. Questo rende possibile quella che viene in genere chiamata sintesi razionale di un farmaco, cioè una progettazione a partire dalla conoscenza, più o meno approfondita, dell'oggetto con cui il farmaco deve interagire.
Le procedure che costituiscono la sintesi razionale sono diverse, a seconda che la disposizione nello spazio degli atomi dell'oggetto biologico con cui il farmaco deve interagire sia conosciuta in dettaglio oppure no. Il primo caso è quello in cui la cristallografia a raggi X, la risonanza magnetica o tecniche equivalenti permettono di localizzare nello spazio, con accettabile precisione, gli atomi dell'oggetto biologico, il più delle volte una proteina. Sarà allora possibile, come già detto in precedenza, conoscere nei dettagli il sito di interazione, studiare con precisione le modalità di legame di una molecola e quindi apportare tutte quelle modifiche che risultano necessarie a un'efficiente interazione (ottimizzazione). In questa metodica, che è generalmente indicata come progettazione basata sulla struttura (SBDD, Structure Based Drug Design), sono di importanza cruciale i metodi della chimica computazionale e della modellistica molecolare già visti. Naturalmente il procedimento assume di regola un carattere ciclico del quale può far parte la già citata co-cristallizzazione di ligando e proteina. Utilizzando questa procedura - che è la più frequente quando debbono essere progettati farmaci che interagiscono con enzimi (costituiti spesso da proteine cristallizzabili) - sono stati identificati ad esempio gli inibitori dell'enzima aspartatoproteasi del virus HIV, saquinavir e indinavir, i quali hanno rappresentato un progresso importante nella cura dell'AIDS.
Nel caso in cui sia conosciuto l'oggetto dell'interazione ma non la sua struttura fine, se sono noti il ligando naturale e altre molecole di sintesi che si legano al sito di azione con affinità variabile, la sintesi razionale può partire dalla struttura delle molecole che si sono dimostrate affini per l'oggetto biologico (LBDD, Ligand Based Drug Design). Questa situazione è frequente nel caso dei recettori, in particolare i recettori di membrana, la cui struttura è risultata fino a ora molto difficile da risolvere. Partendo dalla struttura delle molecole che si legano all'oggetto biologico si possono seguire due vie: manipolare chimicamente tali molecole sulla base di ipotesi di lavoro e verificare l'effetto della modifica sull'affinità, oppure costruire, sulla base di descrittori molecolari e di programmi di modellistica molecolare, un modello del farmacoforo da verificare con la sintesi di nuove molecole suggerite dal modello stesso. Questi due metodi - che sono naturalmente del tutto complementari e spesso, anche se meno di quanto sarebbe auspicabile, sono utilizzati in combinazione - hanno avuto larghissimo impiego, e la maggior parte dei farmaci che agiscono sui recettori attualmente in uso sono stati realizzati in questo modo: basterà citare il caso del sumatriptano, un ligando selettivo per un sottotipo del recettore triptaminergico e che è attualmente usato per il trattamento dell'emicrania, la cui sua struttura appare chiaramente correlata a quella del mediatore fisiologico 5-idrossitriptamina (5HT).
Un caso particolare ma molto importante di sintesi razionale è quello in cui il ligando naturale o sintetico è di natura peptidica. I prodotti di questi tipo non si prestano a divenire farmaci, a causa della sfavorevole farmacocinetica dovuta alla presenza dei legami peptidici (scarsa biodisponibilità e ridotta stabilità metabolica). In situazioni come queste è altamente desiderabile individuare delle molecole che abbiano la stessa azione farmacologica ma struttura non peptidica: questi prodotti vengono indicati come peptidomimetici. La progettazione di un peptidomimetico parte usualmente dall'esame della disposizione spaziale dei gruppi caratterizzanti gli amminoacidi ritenuti essenziali per l'interazione. In genere, si tende a mantenere tali gruppi nella stessa disposizione, sostituendo però i legami peptidici con altre strutture chimiche aventi caratteristiche più accettabili dal punto di vista farmacocinetico. Un peptidomimetico individuato a posteriori è la morfina, che si lega ai recettori oppioidi come i due agonisti naturali met-encefalina e leu-encefalina, mentre il losartan, un antagonista del recettore dell'angiotensina II usato come antipertensivo, è un esempio di peptidomimetico ottenuto per sintesi razionale.
Naturalmente la ricerca di nuovi prototipi è spasmodica e sempre nuove tecniche vengono utilizzate per questa fondamentale tappa del percorso che porta alla nascita di un farmaco. Una del tecniche più recenti, nota come ricerca delle relazioni struttura-attività per mezzo di NMR (v. Roberts, 2000), utilizza la risonanza magnetica nucleare ed è particolarmente adatta a individuare ligandi per proteine delle quali non siano noti la struttura del sito di azione o il ligando naturale, come è il caso della grande maggioranza delle proteine individuate con la genomica e la proteomica. Si è già accennato al fatto che l'uso della tecnica NMR può dare informazioni sull'interazione di una piccola molecola con una proteina; quando si sono individuate due molecole che si legano in zone vicine della proteina, anche se con affinità molto bassa, si procede a legare opportunamente i due frammenti con un adatto connettore. In genere la nuova molecola avrà un'affinità molto più alta e potrà essere utilmente usata come prototipo. In alcuni casi la giunzione di molecole con affinità micromolare, ma anche millimolare, ha prodotto ligandi con affinità nanomolare.
b) L'ottimizzazione del prototipo
Una volta individuato un prototipo è necessario migliorarne le caratteristiche farmacologiche e farmacocinetiche che non si ritengono adeguate. Si cerca di raggiungere questo scopo attraverso successive modificazioni molecolari, rese razionali dal fatto che, come già detto, molecole con struttura simile danno in genere lo stesso tipo di risposta biologica. I chimici farmaceutici, utilizzando l'esperienza di circa un secolo di modulazioni molecolari, hanno messo a punto alcune metodologie che permettono di ridurre il numero dei fallimenti derivanti da modificazioni molecolari non accettate dal sistema biologico in oggetto. Ai giorni nostri, l'aiuto principale per evitare questi errori viene dalla modellistica molecolare, che naturalmente è utilizzabile al meglio quando è nota la struttura del sito di azione. Bisogna però tener ben presente che l'ottimizzazione dell'interazione con il bersaglio biologico è solo un aspetto del processo di ottimizzazione, il quale deve considerare anche le conseguenze farmacocinetiche della modulazione.
Un altro approccio largamente usato è la sostituzione isosterica. Il concetto di isosteria ha origini chimico-fisiche, ma è stato utilizzato soprattutto in chimica farmaceutica, evolvendosi fino alla definizione, molto ampia, della bioisosteria (v. Patani e Lavoie, 1996). In breve, una modificazione isosterica è quella che conduce a una variazione molecolare tale che la nuova molecola possa essere sempre riconosciuta dal bersaglio biologico di quella originale. Sono stati così individuati raggruppamenti vicendevolmente sostituibili sulla base delle caratteristiche steriche ed elettroniche, il che permette di prevedere con una certa sicurezza le conseguenze di manipolazioni molecolari fatte secondo questo principio. Tra i moltissimi disponibili si possono citare due esempi di sostituzioni isosteriche effettuate con successo. Il primo riguarda la sostituzione del gruppo dimetilamminico del fenazone con il suo isostere isopropile per dare l'isopropilfenazone allo scopo di impedire la formazione nell'intestino della corrispondente nitrosoammina, cui sembrano da attribuirsi casi di cancro al colon in pazienti trattati con il fenazone. Il secondo esempio è la sostituzione isosterica del gruppo amminico della carbutamide con un metile, per dare la tolbutamide, che ha permesso di eliminare l'azione antibatterica, non più desiderata, in un prodotto usato come antidiabetico; la successiva sostituzione del metile con l'isosterico atomo di cloro ha condotto alla clorpropamide, che ha infine permesso di aumentare il tempo di emivita di quest'ultima molecola, impedendone la rapida metabolizzazione (trasformazione del metile nel carbossile, che rende inattivo il prodotto).
La semplificazione molecolare è un metodo di modulazione della struttura che viene utilizzato soprattutto quando si debba operare su prodotti di origine naturale. In genere, tali prodotti - che sono chimicamente complessi, e quindi difficili da sintetizzare - presentano tossicità rilevante e biodisponibilità non ideale. Inoltre, l'esperienza indica che spesso solo una parte della struttura molecolare è necessaria per l'azione biologica. Per questo motivo, uno dei primi interventi su una molecola naturale è quello che tende a spogliarla della parte superflua della sua struttura molecolare per identificarne quella essenziale per l'azione biologica (farmacoforo). Questo procedimento è stato utilizzato in quasi tutti i prodotti di origine naturale con risultati contrastanti. In qualche caso la semplificazione molecolare conduce a prodotti attivi ma che perdono di selettività, cosa facilmente intuibile se si pensa che più una molecola è complessa, meno indiscriminatamente si legherà ad altri bersagli biologici. In altri casi i prodotti mantengono l'azione biologica, ma anche la tossicità originaria per superare la quale si era iniziata la manipolazione. È il caso della morfina, alla cui semplificazione strutturale hanno lavorato generazioni di chimici; il lavoro ha condotto a potenti analgesici, quali levorfanolo, metazocina e alfaprodina, che però hanno mantenuto la capacità di indurre tossicodipendenza.
Il procedimento inverso, quello tendente a complicare la struttura del prototipo, è applicato in modo più esteso del precedente. Si può infatti utilizzare in numerose situazioni per ottenere scopi diversi. In genere si basa sul concetto, speculare al precedente, che la complicazione molecolare può condurre a un aumento della specificità di azione ma anche all'inversione del tipo di azione: per esempio da agonista ad antagonista. La complicazione molecolare è anche utilizzata per modulare la farmacocinetica, come appare chiaramente dall'esempio fatto in precedenza riguardante la clorpropamide.
Uno dei modi concettualmente più semplici per aumentare la complessità molecolare, tra i molti disponibili, è quello della omologazione, termine con cui normalmente si intende l'aumento di un atomo di carbonio, ma che può indicare anche un generico incremento del numero di atomi di carbonio di una catena laterale o di un ciclo. Proprio per la specificità dell'interazione tra farmaci e molecole biologiche questa semplice operazione può aver effetti importanti, introducendo vincoli sterici o forze supplementari di interazione con il bersaglio biologico. Un esempio significativo di questo procedimento è la sostituzione di uno dei due idrogeni della noradrenalina (l'agonista naturale in grado di attivare sia i recettori α che β) con un gruppo isopropilico per dare l'isoproterenolo, un agonista selettivo verso il sottogruppo β dei recettori adrenergici. Evidentemente il sito di riconoscimento del sottogruppo α non possiede lo spazio sufficiente ad accomodare l'aumento di volume del sostituente sull'azoto, al contrario del sottogruppo β. Un secondo esempio è rappresentato dalla sostituzione con due gruppi stericamente ingombranti e lipofili dei due idrogeni del gruppo metilico dell'acetilcolina, che trasforma la molecola da agonista ad antagonista; è stato provato che la molecola che ne risulta, l'adifenina, interagisce con un sito accessorio vicino al sito di riconoscimento dell'acetilcolina tramite interazioni lipofile dovute ai due gruppi fenilici che ne aumentano di almeno due ordini di grandezza l'affinità per il recettore.
Un altro procedimento molto usato è quello che tende a modificare la libertà conformazionale del prototipo tramite la chiusura o, al contrario, l'apertura di strutture cicliche. La prima è utile per studiare le relazioni tra conformazione e attività biologica; infatti solo una tra le numerose conformazioni possibili di una molecola attiva interagisce con il bersaglio biologico e pertanto avere informazioni su di essa è cosa della maggiore importanza per la progettazione di altri farmaci attivi sullo stesso sito. L'analisi conformazionale non è decisiva in proposito, in quanto non è detto che la conformazione più stabile sia quella attiva, poiché l'energia messa in gioco nel processo di legame può bastare a compensare la maggiore energia di un'altra conformazione meno stabile. Per affrontare questo problema, i chimici farmaceutici usano spesso il sistema di congelare, per via sterica o con l'introduzione di nuovi legami, una conformazione ritenuta importante, andando poi a verificare le variazioni nell'azione farmacologica (v. Gualtieri e altri, 1996); se l'attività è mantenuta, molto probabilmente si è identificata la conformazione attiva, altrimenti è necessario verificare che nella manipolazione molecolare non siano state fatte modifiche tali da cambiare le caratteristiche steriche e chimico-fisiche della molecola e quindi tali da pregiudicarne l'interazione per ragioni che sono indipendenti dalla conformazione. Un classico esempio di questo tipo di metodologia è quello che ha condotto a identificare la conformazione antiperiplanare dell'acetilcolina come quella attiva nell'interazione con il recettore colinergico. L'introduzione, formale, di un metilene nella struttura dell'acetilcolina produce due ciclopropilderivati isomerici, ognuno dei quali può essere considerato la forma congelata delle due conformazioni più probabili dell'acetilcolina. L'isomero cis corrisponde al conformero sinclinale presente allo stato solido e predetto dalla chimica computazionale; l'isomero trans corrisponde al conformero antiperiplanare che è di poco meno stabile, perché dotato di energia più elevata. Il fatto che solo l'isomero trans sia attivo come colinergico indica che, almeno in questo caso, la conformazione più stabile non è quella attiva.
L'ibridazione molecolare è un'altra metodologia che tende a riunire in un'unica molecola le caratteristiche strutturali e farmacologiche di due farmaci. Può essere utilizzata per coordinare dal punto di vista farmacocinetico l'azione di due farmaci, ovviando al fatto che questi abbiano farmacocinetiche differenti, non compatibili con la semplice associazione. In questo caso la nuova molecola, una volta giunta sul luogo di azione, è destinata a scindersi ripristinando i due principî attivi. Un farmaco realizzato usando l'ibridazione molecolare è la sultamicillina, un ibrido ottenuto dall'ampicillina (un antibiotico) e dal sulbactam (un inibitore delle beta-lattamasi) che protegge l'ampicillina dalla degradazione. I due prodotti hanno farmacocinetiche differenti, per cui non giungono a distribuirsi nel sito di azione in modo coordinato; se però vengono trasformati in un'unica molecola tramite un legame labile che può essere scisso per via metabolica, come avviene appunto nella sultamicillina, la cinetica sarà ovviamente identica e le due molecole, una volta ripristinate, si troveranno sul sito di azione contemporaneamente.
L'ibridazione molecolare può essere utilizzata anche per produrre una nuova molecola non destinata ad alcuna scissione e con proprietà farmacologiche proprie. L'ibridazione è considerata un successo se la nuova molecola presenta, in modo bilanciato, le proprietà farmacologiche di entrambe le molecole ibridate.
Il raddoppiamento molecolare è un altro metodo che viene spesso utilizzato nella manipolazione di un prototipo. Questo metodo si basa sull'assunzione che possano venire coinvolti nell'interazione due siti di riconoscimento del bersaglio, anche se, alla luce della struttura spaziale della maggioranza delle molecole biologiche che costituiscono il bersaglio dei farmaci, questa assunzione è spesso errata e sembra più realistico ritenere che i successi ottenuti siano da ascrivere alla casuale intercettazione di siti con caratteristiche di interazione simili a quelle del sito di riconoscimento, pur non coincidendo con esso. Per esempio, la tacrina è stato il primo inibitore delle acetilcolinesterasi utilizzato nel trattamento della malattia di Alzheimer, anche se ora, a causa della sua tossicità, è stato sostituito da farmaci dello stesso tipo ma più sicuri. Nel tentativo di utilizzarla come prototipo per ottenere prodotti più potenti e meno tossici, la struttura base della molecola è stata raddoppiata con connettori di varia lunghezza. Si è trovato che quando il braccio che unisce le due metà è di lunghezza opportuna, la capacità di inibire la acetilcolinesterasi aumenta in modo impressionante. Utilizzando la tecnica della co-cristallizzazione, descritta in precedenza, si è potuto costatare che la molecola raddoppiata interagisce da una parte con il sito di riconoscimento dell'enzima e dall'altra con un sito accessorio del tutto distinto che si trova a una certa distanza.
Una delle metodiche usate più frequentemente nell'ottimizzazione di un prototipo è quella della modulazione delle proprietà chimico-fisiche, soprattutto la distribuzione elettronica, la lipofilia e l'ingombro sterico. Modificazioni molecolari di questo tipo sono in grado di modulare sia l'interazione con il bersaglio biologico, sia la farmacocinetica e la tossicità. La modulazione è in genere ottenuta con l'introduzione di atomi, o gruppi di atomi, di volume opportuno, che modificano la distribuzione elettronica o le caratteristiche lipofile dalla molecola del prototipo. La scelta dei gruppi è guidata dal loro ingombro sterico, dall'effetto elettronico e da quello lipofilo, indicati da costanti tabulate, come ad esempio Es (parametro sterico), π (costante idrofobica) o σ (costante elettronica). La strategia da usare è suggerita da regole pratiche, come quella delle ramificazioni di Topliss (Topliss tree) o il diagramma bidimensionale di Craig. Per esempio, la trasformazione dell'atropina in un sale ammonico quaternario come l'ipratropium fa sì che il farmaco sia attivo solo a livello periferico, in quanto molecole con carica permanente non riescono a penetrare nel cervello a causa della barriera ematoencefalica, mentre la sostituzione con il cloro di uno o entrambi gli anelli benzenici è in grado di modulare decisamente l'effetto ansiolitico delle benzodiazepine.
La modulazione chirale di un prototipo è anch'essa di grande importanza, poiché la struttura tridimensionale è senza dubbio uno dei fattori principali nell'interazione di un farmaco con il suo obiettivo. Tra l'altro, le autorità sanitarie hanno posto negli ultimi anni molta attenzione a questo problema, in particolare alla purezza ottica dei farmaci contenenti centri stereogenici e quindi in grado di presentare isomeri diastereomerici ed enantiomerici. Di norma, sono presi in considerazione per la registrazione solo enantiomeri puri, a meno che non sia dimostrato che il secondo enantiomero non presenti rilevanti effetti farmacologici o tossici, oppure che la forma enantiomerica pura sia instabile nelle condizioni d'uso, casi nei quali è possibile veder registrato il racemo. Infatti, molecole isomeriche posseggono, normalmente, differenti azioni farmacologiche, non solo, com'è intuibile, nel caso di isomeri con differenti caratteristiche chimico-fisiche, ma anche in quello di enantiomeri con proprietà chimico-fisiche identiche. Questo è dovuto alla natura diastereomerica dei complessi che si formano con i bersagli biologici, che in larghissima parte sono essi stessi chirali. La stereoisomeria è quindi un aspetto di primaria importanza nella progettazione di un farmaco: essa può costituire un problema per lo sviluppo, dal punto di vista sintetico e dei costi, e pertanto uno dei primi approcci al prototipo sarà quello di verificare l'importanza della chiralità sull'azione farmacologica e se, caso del resto abbastanza raro, tale importanza risultasse minima, sarà conveniente cercare di sviluppare una molecola achirale modificando opportunamente il prototipo; ma se, come è logico aspettarsi, l'introduzione di uno o più centri chirali in una molecola fa aumentare il suo potere discriminante nell'interazione e quindi la sua specificità d'azione, può essere estremamente conveniente trasformare un prototipo achirale in uno chirale, anche se si debbono affrontare difficoltà a livello sintetico, separativo e di sviluppo.
Infine, non si può trascurare un metodo di modulazione molecolare molto semplice dal punto di vista teorico ma che può alcune volte risolvere problemi enormi nello sviluppo di un farmaco: si tratta della derivatizzazione molecolare, che consiste essenzialmente nel fare opportuni derivati di funzioni reattive presenti sulla molecola del prototipo, o su quella ottimizzata che presenti ancora problemi, ad esempio solubilità o stabilità chimica e metabolica inadeguate. I casi di problemi risolti con questa semplice operazione sono innumerevoli. Il dapsone si trasforma in un farmaco a lunga durata d'azione per semplice acetilazione dei due gruppi amminici (acedapsone); l'esterificazione con acido succinico dell'oxazepam, che è poco solubile in acqua, per dare l'oxazepam emisuccinato, permette di solubilizzare il prodotto in acqua; l'esterificazione dell'adrenalina con acido pivalico per dare la dipivefrina aumenta la lipofilia e rende il prodotto utilizzabile nel glaucoma; per finire con il caso classico della penicillina G, il cui sale con la dibenziletilendiammina ha permesso di prolungare adeguatamente il suo tempo di emivita troppo breve.
Naturalmente, la ricerca di nuove metodologie per la scoperta e l'ottimizzazione di prototipi e lo sviluppo di nuovi farmaci è in continua evoluzione e sarebbe illusorio pensare che questa breve trattazione copra esaurientemente la materia. Inoltre, prima di concludere va ricordato che, come indicato nella fig. 4, una volta ottimizzata la molecola e iniziato il suo sviluppo ci sono problemi come la sintesi industriale, la messa a punto di saggi analitici sensibili e riproducibili, la ricerca di un'adatta formulazione e di un'accettabile via di somministrazione, che sono altrettanto impegnativi e importanti per l'esito finale: mettere a disposizione del paziente un farmaco efficace, potente e sicuro.
bibliografia
Antonenko, V. V. e altri, Combinatorial chemistry, in New trends in synthetic medicinal chemistry (a cura di F. Gualtieri), Weinheim: Wiley-VCH, 2000, pp. 39-80.
Ehrlich, P., Address in pathology on chemotherapeutics, scientific principles, methods and results, in "The lancet", 1913, n. 2, pp. 445-451.
Fernandes, P. B., Back to basic: from genes to medicines, in "Medicinal chemistry research", 2001, X, pp. 421-430.
Gualtieri, F., Romanelli, M. N., Teodori, E., The frozen analog approach in medicinal chemistry, in Progress in medicinal chemistry (a cura di M. I. Choudhary), Amsterdam: Harwood Academic Publishers, 1996, vol. I, pp. 271-318.
Gualtieri, F., Romanelli, M. N., Teodori, E., Disegno e sintesi di farmaci e mezzi di indagine farmacologica, Bologna: CLUEB, 1997.
Gubernator, K., Bohm, H. J., Structure-based ligand design, Weinheim: Wiley-VCH, 1998.
Kubinyi, H., Hansch analysis and related approaches, Weinheim: Wiley-VCH, 1993.
Leach, A. R., Molecular modelling: principles and applications, Harlow: Pearson Education Ltd., 20012.
Li, A. P., Screening for human ADME/Tox drug properties in drug discovery, in "Drug discovery today", 2001, VI, pp. 357-366.
O'Neil, M. J. e altri, The Merck index. An encyclopedia of chemicals, drugs & biologicals, Whitehouse Station, N. J.: Merck & Co., 200113.
Palczewski, K. e altri, Crystal structure of rhodopsin: a G protein-coupled receptor, in "Science", 2000, CCLXXXIX, pp. 739-745.
Patani, G. A., Lavoie, E. J., Bioisosterism: a rational approach in drug design, in "Chemical reviews", 1996, XCVI, pp. 3147-3176.
Roberts, G. C. K., Applications of NMR in drug discovery, in "Drug discovery today", 2000, V, pp. 230-240.
Smith, D. A., Van De Waterbeemd, H., Walker, D. K., Pharmacokinetics and metabolism in drug design, Weinheim: Wiley-VCH, 2001.
Stahura, F. L., Bajorath, J., Bio- and chemo-informatics beyond data management: crucial challenges and future opportunities, in "Drug discovery today", 2002, VII, pp. S41-S47.
Swindells, M. B., Overington, J. P., Prioritizing the proteome: identifying pharmaceutically relevant targets, in "Drug discovery today", 2002, VII, pp. 516-521.
Walters, W. P., Stahl, M. T., Murcko, M. A., Virtual screening. An overview, in "Drug discovery today", 1998, III, pp. 160-177.
Wermuth, C. G., The practice of medicinal chemistry, San Diego, Cal.: Academic Press, 1996 (tr. it.: Le applicazioni della chimica farmaceutica, Napoli: EdiSES, 2000).