
Proteoma
Proteoma
I progetti di sequenziamento dei genomi hanno posto le basi per lo studio dei fenomeni biologici con metodologie globali, in genere descritte dal suffisso -omica. Lo studio della globalità delle proteine, nella loro attualità funzionale, viene comunemente indicato con la denominazione di proteomica. Non sarebbe possibile affrontare questo tipo di studi senza la disponibilità delle banche dati genomiche e dei mezzi informatici che ne consentono la consultazione e, soprattutto, senza lo sviluppo di tecnologie e metodologie sofisticate di separazione e di identificazione delle proteine. Si riconosce al ricercatore australiano M. Wilkins il merito di aver coniato nel 1994 il vocabolo proteoma, un acronimo di proteine e genoma, volendo con tale vocabolo intendere il complemento proteico espresso da un genoma, cioè l'intero complesso dei prodotti dell'espressione di un genoma. Il p. ha un significato dinamico, a differenza del concetto statico di genoma, poiché in corrispondenza di stimoli, interni o esterni alla cellula, i prodotti dell'espressione del genoma possono notevolmente variare; pertanto si può affermare che a un genoma corrisponda una molteplicità di p. il cui limite superiore, almeno per ora, non è definibile. È importante sottolineare come la regolazione dell'espressione genica, nella sua eccezionale capacità di modulazione, rappresenti un continuo che si estende per almeno nove ordini di grandezza; ne discende che una singola proteina in un p. può essere differentemente rappresentata da una a un miliardo di volte. La proteomica, pertanto, può essere definita come lo studio, nella loro complessità, dei proteomi. Da questa definizione di carattere del tutto generale discendono varie altre definizioni di proteomica che, opportunamente specificate o aggettivate, si riferiscono agli obiettivi specifici che l'approccio proteomico intende affrontare. Questi obiettivi si possono riferire, in definitiva, agli aspetti meramente strutturali (proteomica strutturale) e a quelli funzionali (proteomica funzionale).
Metodi in proteomica
La proteomica si basa essenzialmente su due differenti passaggi analitici consecutivi costituiti dalla separazione delle proteine che costituiscono il p. e dalla loro successiva identificazione individuale. I primi studi, ora definibili di proteomica, si possono far risalire alla metà degli anni Settanta del 20° sec., quando, utilizzando la tecnica dell'elettroforesi bidimensionale, fu possibile ottenere una mappa dell'insieme delle proteine presenti in cellule sia procariotiche sia eucariotiche. Tuttavia, non erano in quel momento disponibili sistemi altrettanto efficienti e sensibili per l'identificazione delle proteine stesse né, d'altra parte, erano disponibili banche dati di sequenza di una certa consistenza. Solo dopo tre lustri furono messi a punto metodi di sequenziamento a livello subnanomolare delle bande proteiche mediante l'impiego del sequenziatore in fase gassosa. In questo modo però era possibile identificare soltanto le proteine più abbondanti e, mediante confronto, solamente quelle la cui sequenza completa era già stata determinata, eventualmente dedotta dalla sequenza del gene codificante, e resa disponibile, prima in cataloghi e in seguito in banche dati. La nascita della moderna proteomica, pertanto, si può far risalire alla possibilità d'accesso a queste informazioni conseguenti al sequenziamento dei genomi. Successivamente l'evoluzione, della spettrometria di massa ha radicalmente cambiato i tempi e i metodi d'analisi dei proteomi. L'elevatissima sensibilità e l'ampio intervallo dinamico di analisi di questo tipo di strumentazione risponde, pienamente, alle esigenze della proteomica, così come sono state delineate nel paragrafo introduttivo. Si può sostenere che l'impiego dello spettrometro di massa nella fase di 'identificazione' ha, in seguito, consentito di superare una serie di limiti dell'elettroforesi bidimensionale e ha suggerito importanti soluzioni metodologiche non più dipendenti da questo sistema di separazione. Di seguito sono descritti i principali metodi di separazione e d'identificazione utilizzati in proteomica.
Elettroforesi bidimensionale (2DE). - L'elettroforesi bidimensionale è una tecnica di separazione ortogonale che sfrutta essenzialmente le due principali caratteristiche delle proteine, ossia la carica netta valutata in termini di punto isoelettrico (pI) e la grandezza molecolare valutata in termini di peso molecolare (Mw). Nella 2DE si utilizza nella prima dimensione la separazione in funzione del punto isoelettrico e, quindi, si sottopongono le proteine così frazionate all'ulteriore processo elettroforetico ortogonale in presenza di sodio dodecilsolfato (SDS), che separa le proteine in funzione del peso molecolare. Le proteine vengono evidenziate come macchie per trattamento del gel con sistemi di colorazione (blu di Comassie, argento o oro colloidale, coloranti fluorescenti) e descrivono una complessa mappa elettroforetica in funzione delle due coordinate, Mw e pI. Naturalmente la riproducibilità di tale mappa dipende da una molteplicità di fattori e ha costituito, per molti anni, uno dei punti deboli di questa tecnica.
Tuttavia, l'introduzione dei gradienti immobilizzati di pH nella prima dimensione ha consentito non solo di ottenere una riproducibilità notevolmente maggiore, ma anche di coprire intervalli di pH più ampi (pH 3 - 12) di quelli ottenibili con gli anfoliti in soluzione, di utilizzare una maggiore quantità di proteine senza disturbare il gradiente di pH, di ottenere separazioni in intervalli estremamente ristretti di pH (fino a un'unità di pH) e, conseguentemente, unrisoluzione nella prima dimensione. Questo e altri accorgimenti sperimentali, insieme con sempre più potenti sistemi di scansione e di ricostruzione di immagini, hanno consentito di costruire importanti banche dati proteomiche, come, per es., quella reperibile in ExPASy (http://www.expasy.ch/ch2d/2d-index.html), le quali riportano le separazioni bidimensionali di proteine di svariati organismi, tessuti e linee cellulari.
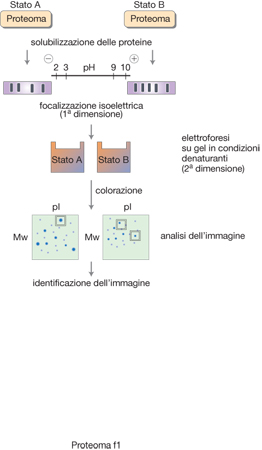
È da sottolineare che la ricostruzione computerizzata delle immagini consente anche una valutazione quantitativa delle bande proteiche e quindi la determinazione dei livelli di espressione in particolari condizioni fisio-patologiche. In fig. 1 è schematicamente riassunta la metodologia impiegata. Mediante l'impiego di reattivi-rivelatori con diversa fluorescenza, è stato introdotto un protocollo sperimentale innovativo che prende il nome di DIGE (DIfference Gel Electrophoresis) e prevede la separazione su un unico gel di due diversi p. che sono stati fatti reagire con due diversi marcatori fluorescenti. In questo modo vengono individuate direttamente e immediatamente le macchie da sottoporre all'analisi per l'identificazione delle proteine e che caratterizzano uno dei due p. (Patton 2002).
Appare evidente che il sistema di separazione così ottimizzato richiede un sistema d'identificazione rapida, sensibile e affidabile delle macchie d'interesse. All'inizio dell'era proteomica ci si affidava alla composizione in aminoacidi o alla determinazione della sequenza N-terminale e alla successiva comparazione dei dati di sequenza ottenuti con quelli presenti nelle banche dati. Tuttavia questi metodi presentavano serie limitazioni in termini di affidabilità, di rapidità di esecuzione, di sensibilità. A partire dagli anni Novanta del 20° sec., grazie anche all'introduzione di nuovi accorgimenti strumentali, la spettrometria di massa si è imposta come strumentazione elettiva per l'identificazione delle proteine.
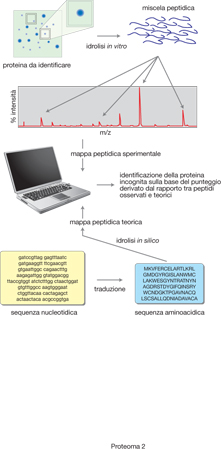
Spettrometria di massa per l'identificazione delle proteine. - L'identificazione di una proteina mediante spettrometria di massa avviene attraverso l'analisi dei peptidi generati utilizzando proteasi specifiche (tripsina, Asp-N proteinasi, Glu-C proteinasi). In questo caso, tuttavia, i peptidi sono caratterizzati non più dai loro parametri chimico-fisici come polielettroliti, ma più semplicemente dalla loro massa attraverso la determinazione, effettuata dallo spettrometro di massa, dei loro pesi molecolari. Il principio dell'identificazione delle proteine mediante l'utilizzo della spettrometria di massa è abbastanza semplice e si basa sull'osservazione che proteine con una diversa sequenza aminoacidica, in seguito all'azione di una proteasi specifica, generano un insieme discreto di peptidi, definiti dalla loro massa, che è unico per quella proteina. In particolare, la banda proteica di un gel bidimensionale, la cui colorazione è condotta in condizioni da non interferire con la successiva analisi, viene direttamente trattata con proteasi (analisi in-gel) generando una miscela di peptidi. La miscela peptidica viene direttamente esaminata con lo spettrometro di massa, ottimizzando la procedura analitica per ottenere il maggior numero di segnali che consentano di valutare con la massima accuratezza possibile, anche in relazione alla strumentazione disponibile, i valori delle masse degli ioni molecolari. Questi valori sono paragonati con le masse teoriche dei peptidi ottenuti, simulando una digestione proteica dello stesso tipo di quella utilizzata dallo sperimentatore su tutte le sequenze proteiche presenti nelle banche dati per mezzo di opportuni programmi che sono facilmente disponibili in rete: Mascot (http://www.matrixscience.com), Matrix Science Ltd. Londra; Protein Prospector (http://prospector.ucsf.edu) University of California; ProFound (http://prowl.rockefeller.edu) Rockefeller University; PepMAPPER (http://wolf.bms.umist.ac.uk/mapper) UMIST, Manchester. Il programma fornisce quindi il risultato come punteggio di probabilità statistica, punteggio che risulterà tanto più elevato quanto maggiori e accurati saranno i dati sperimentali. La metodologia d'identificazione è rappresentata schematicamente nella fig. 2.
Per questo tipo di analisi è utilizzato come sistema di ionizzazione il MALDI (Matrix Assisted Laser Desorption Ionization), in cui si sfrutta la radiazione laser per indurre la produzione di ioni molecolari protonati degli analiti opportunamente incorporati in matrici di natura organica. Di solito le sorgenti MALDI sono accoppiate con analizzatori a tempo di volo (TOF, Time of Flight), che misurano il rapporto massa/carica degli ioni generati nella sorgente sulla base del tempo che questi impiegano nel percorrere uno spazio definito in assenza di campi elettrici e magnetici. Con questi apparecchi, purché dotati di particolari accorgimenti per evitare dispersioni di energia degli ioni, si riesce a ottenere una misura delle masse dei peptidi con un'accuratezza alla seconda cifra decimale. È evidente che a una maggiore accuratezza della misura corrisponde un numero minore di peptidi con masse isobariche, con vantaggi nei tempi di esecuzione del programma e di affidabilità del risultato.
L'approccio descritto è valido soprattutto per lo studio di p. non particolarmente complessi e può essere completamente automatizzato, dall'individuazione della banda proteica da analizzare fino all'elaborazione del dato informatico. Una limitazione intrinseca di tale metodologia è dovuta al fatto che, per effetto di permutazioni nella sequenza aminoacidica, si possono ottenere più specie molecolari isobariche, ossia che a un valore di massa anche notevolmente accurato possano corrispondere una molteplicità di peptidi. Tuttavia, i peptidi isobari sono comunque dotati di differenti successioni degli aminoacidi che li costituiscono. È allora evidente che la ricerca in banche dati, combinando i valori delle masse della mappa peptidica con le informazioni di sequenza, è da considerarsi certamente più affidabile. Infatti in questo caso il programma di ricerca fornirà tutte le sequenze proteiche che contengono la sequenza del peptide analizzato. È anche evidente che a una maggiore quantità di dati sperimentali, sequenze sufficientemente lunghe (>5 residui) o più peptidi sequenziati, corrisponderà una risposta generalmente non ambigua.

Ancora una volta la spettrometria di massa ha consentito di fornire risposte più sensibili (a livello di femtomoli) e più rapide (qualche minuto di analisi) rispetto ai metodi di sequenziamento degli anni Ottanta. Utilizzando spettrometri di massa dotati di un secondo analizzatore, è possibile ottenere uno spettro di massa dei frammenti dei peptidi separati con il primo analizzatore e, quindi, dallo spettro di frammentazione risalire alla sequenza aminoacidica. La successione degli eventi analitici può essere considerata la seguente: a) il primo analizzatore dello spettrometro di massa separa i peptidi, ottenuti per proteolisi della banda proteica di interesse, nella loro forma ionizzata; b) il secondo analizzatore separa i frammenti ottenuti da ciascun peptide ionizzato la cui frammentazione viene indotta in un'opportuna camera di collisione che, fisicamente, lo precede; c) dallo spettro di massa dei frammenti peptidici si deduce la sequenza del peptide. Gli spettri di frammentazione si possono far risalire a frammentazioni statistiche del legame ammidico con la conseguente generazione di due serie di ioni che ritengono la carica o sulla parte N-terminale (ioni di tipo b) o su quella C-terminale (ioni di tipo y). Nella fig. 3 è rappresentata l'interpretazione di uno spettro di frammentazione e l'identificazione della corrispondente proteina.
Gli spettrometri di massa che consentono analisi di questo tipo si sono evoluti in termini di complessità (e quindi di costi e dimensioni) e di sensibilità. Dagli strumenti dotati di due analizzatori magnetici (tandem MS) della metà degli anni Ottanta si è passati a quelli dotati di più analizzatori di tipo quadrupolo (Q), di trappola ionica (IT o FT-ICR), di quadrupolo e TOF ortogonale (Q-TOF) e di due analizzatori a tempo di volo (TOF-TOF). Occorre notare che solo nel caso di quest'ultimo strumento, peraltro da poco disponibile commercialmente, viene utilizzata la sorgente MALDI, laddove nel caso degli strumenti con analizzatori quadrupolari si usa la sorgente a ionizzazione per elettronebulizzazione o Electro-Spray Ionization (ESI). In questo tipo di sorgente gli analiti in soluzione, opportunamente purificati da sali con un passaggio cromatografico, vengono introdotti attraverso un capillare nella sorgente stessa, che, per effetto combinato del vuoto spinto e di un'opportuna differenza di potenziale, genera dal campione nebulizzato ioni a carica multipla. Questo tipo di sorgente è stata successivamente ottimizzata con l'introduzione del sistema nanospray, che, facendo uso di un microcapillare con flussi di qualche nl/min, consente un tempo di residenza in sorgente dell'analita dell'ordine dei minuti con un notevole incremento del rapporto segnale/rumore. Gli spettrometri ES-nanospray con i doppi analizzatori, in particolare quadrupolo e a tempo di volo ortogonale, collegati a un cromatografo capillare, hanno rivoluzionato la pur breve storia della proteomica. È da sottolineare che, una volta riconosciuta l'enorme potenzialità di questo sistema, molti ricercatori hanno valutato l'opportunità di non utilizzare più l'elettroforesi bidimensionale come sistema di separazione e hanno suggerito e messo a punto nuove metodologie di separazione che meglio si integrano con questo potente sistema di analisi.
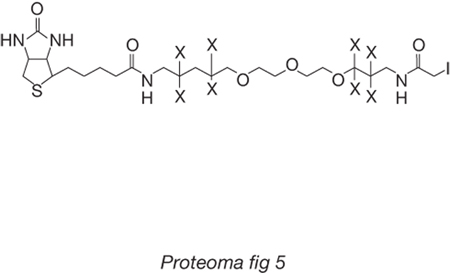
La proteomica di seconda generazione. - La proteomica di seconda generazione nasce dall'osservazione che, invece di separare le proteine e quindi ottenere poi da ciascuna banda separata i peptidi da analizzare, il sistema possa essere reso più efficiente sottoponendo a proteolisi (per es., con tripsina) l'intero p. di un sistema cellulare o di una frazione subcellulare e, dopo separazione cromatografica, sequenziare direttamente i peptidi con uno spettrometro di massa del tipo descritto. In questo modo viene apparentemente esasperato il problema analitico, giacché la complessità della miscela da separare aumenta di almeno un ordine di grandezza. Tuttavia, è possibile sfruttare il potere risolutivo della cromatografia capillare (in qualche caso è stata impiegata anche l'elettroforesi capillare), che risulta molto più elevato di quello dell'elettroforesi bidimensionale. L'approccio metodologico che ha avuto il maggiore successo consiste nell'impiego di un sistema cromatografico capillare multidimensionale che combina la separazione a scambio ionico con quella su fase inversa, e di una potente piattaforma informatica per la ricostruzione della sequenza dei peptidi dagli spettri di frammentazione e per l'interrogazione delle banche dati. La limitazione maggiore di tale metodologia è legata al fatto che, in questo modo, sono assai difficili studi di carattere differenziale, nel caso in cui si è principalmente interessati alle variazioni di livelli dell'espressione. Una metodologia innovativa, intesa anche a rispondere alle problematiche quantitative della proteomica differenziale, è stata proposta da R. Aebersold e dai suoi collaboratori (Gygi, Rist, Gerber et al. 1999). Tale metodologia si basa sull'utilizzo di una molecola che è stata denominata Isotope Coded Affinity Tag, conosciuta con l'acronimo ICAT (fig. 5).
Il reattivo viene utilizzato nella sua forma 'leggera', ossia contenente il braccio molecolare senza atomi di deuterio, e in quella 'pesante', contenente il braccio molecolare con 8 atomi di deuterio, rispettivamente per marcare il proteoma dello stato 1 e quello dello stato 2. I due campioni vengono riuniti e sottoposti a un'estesa proteolisi. I peptidi ottenuti vengono sottoposti a un passaggio di cromatografia di affinità utilizzando un supporto derivatizzato con avidina (una proteina che forma complessi dotati di notevoli costanti di stabilità con la biotina), che consente di trattenere soltanto i peptidi marcati dall'ICAT, semplificando in questo modo notevolmente la complessità della miscela. A ciascun peptide corrisponderanno due valori di peso molecolare che differiranno di 8 Dalton con un rapporto di intensità che può essere direttamente relazionato a quello delle proteine da cui provengono, presenti nelle due diverse condizioni cellulari. Una successiva analisi MS-MS consente di determinare la sequenza dei peptidi di interesse e di identificare le proteine la cui espressione è stata variata per effetto delle diverse condizioni delle popolazioni cellulari dello stato 1 e dello stato 2. È importante evidenziare che il metodo dell'ICAT non permette l'identificazione delle proteine in cui non siano presenti residui di cisteina, in alcuni organismi fino al 20% del proteoma. Per ovviare a questo inconveniente è possibile utilizzare ICAT con gruppi reattivi che consentono la marcatura isotopica di altri gruppi funzionali presenti nelle proteine.
Successivamente è stato introdotto un nuovo reattivo che sfrutta in maniera molto brillante l'uso combinato di isotopi stabili per fare in modo che il derivato con il reattivo, che prende il nome di iTRAQ (Ross, Huang, Marchese et al. 2004), dia luogo a derivati isobari quando analizzati in MS1 e, per effetto della frammentazione, dia luogo in MS2 a due, o eventualmente a quattro, diversi frammenti isotopici che con la loro intensità danno conto della popolazione della proteina marcata in due, o addirittura in quattro, differenti proteomi. Questo nuovo reattivo isotopico presenta molti vantaggi rispetto all'ICAT: a) reagisce con i gruppi aminici per cui è da considerarsi un reattivo universale; b) evita il passaggio di purificazione cromatografico su cromatografia di affinità, che avviene, generalmente, con una scarsa resa; c) impiega isotopi che non comportano un notevole 'effetto isotopico' come il deuterio, che provoca talvolta la separazione cromatografica delle specie marcate; d) non comporta perdita di sensibilità in MS1 in quanto tutte le specie marcate hanno la stessa massa; e) sfrutta a pieno la potenzialità degli spettrometri di massa tandem.
Prospettive. - La proteomica, anche se ha sfruttato un bagaglio di conoscenze e di metodologie già note, ha appena iniziato il suo percorso scientifico. Certamente l'ambizioso progetto della mappa del p. umano (http://www.hupo.org) rappresenterà la prossima grande sfida scientifica della proteomica. Occorre inoltre sottolineare che, comunque, proprio per la natura dinamica del p., quello umano non dovrà risultare in una catalogazione delle differenti forme di espressione dei circa 40.000 geni dell'uomo, ma piuttosto dovrà proporsi di rilevare le sottili relazioni esistenti tra le condizioni fisiopatologiche dell'individuo e la realizzazione del proprio programma dell'espressione genica. A tale scopo risulterà cruciale la messa a punto di metodi per consentire un'ancora più rapida analisi di miscele complesse di proteine, comprese quelle di membrana, e di specie transienti e scarsamente rappresentate.
bibliografia
S.P. Gygi, B. Rist, S.A. Gerber et al., Quantitative analysis of complex protein mixtures using isotope-coded affinity tags, in Nature biotechnology, 1999, 17, pp. 994-99.
S. Fields, Proteomics in genomeland, in Science, 2001, 291, pp. 1221-24.
W.F. Patton, Detection technologies in proteome analysis, in Journal of chromatography B. Analytical technologies in the biomediacal and life sciences, 2002, 771, pp. 3-31.
P.L. Ross, Y.N. Huang, J.N. Marchese et al., Multiplexed protein quantitation in saccharomyces cerevisiae using amine-reactive isobaric tagging reagents, in Molecular and cellular proteomics, 2004, 3, pp.1154-69.
S.E. Ong, M. Mann, Mass spectrometry-based proteomics turns quantitative, in Nature chemical biology, 2005, 1, pp. 252-62.