
Riconoscimento dell'antigene da parte dei recettori del sistema immunitario
Riconoscimento dell'antigene da parte dei recettori del sistema immunitario
Il riconoscimento specifico degli antigeni estranei all'organismo da parte di strutture complementari sulla superficie dei Iinfociti B e T rappresenta il primo passaggio nella sequenza di eventi che portano all'attivazione del sistema immunitario. Due classi distinte di molecole, entrambe appartenenti alla superfamiglia delle immunoglobuline, sono responsabili del legame dell' antigene: gli anticorpi, che funzionano sia come recettori sui Iinfociti B sia come molecole effettrici rilasciate nel torrente circolatorio dalle plasmacellule, e i recettori delle cellule T (TCR), che si presentano solo nella forma legata alla membrana. Mentre gli anticorpi riconoscono l'antigene nella sua forma nativa, i TCR lo riconoscono solo quando esso si trova sotto forma di peptidi legati a molecole del complesso maggiore di istocompatibilità (MHC). I TCR interagiscono anche con una classe di proteine patogene e immunostimolatrici di origine batterica o virale, note come 'superantigeni'. In questo saggio verranno esposte le conoscenze attuali sulla struttura tridimensionale dei complessi antigene-anticorpo, TCR-peptide/MHC, e TCR-superantigene.
Struttura tridimensionale dei complessi antigene-anticorpo
Le immunoglobuline G (lgG) sono omotetrameri composti da due catene polipeptidiche identiche di circa 250 amminoacidi (catene leggere o L) e da altre due catene, anch'esse identiche, di circa 450 amminoacidi (catene pesanti o H). Ogni catena L contiene due regioni o domini formati da due foglietti β antiparalleli, mentre ogni catena H ne comprende almeno 4. Questi domini a foglietti β sono molto simili dal punto di vista della struttura e sono stati denominati ripiegamento dell'immunoglobulina. l domini amminoterminali variabili (V) delle catene L e H contengono entrambi tre anse che collegano i filamenti β e che presentano una lunghezza e una sequenza molto diverse tra un anticorpo e l'altro. Queste sono le cosiddette regioni ipervariabili o regioni determinanti la complementarità (CDR, Complementarity Determining Regions); esse formano una struttura all'estremità della molecola di immunoglobulina che è complementare all'antigene. Il principale modello dell' interazione antigene-anticorpo è rappresentato dalla struttura tridimensionale delle sei anse CDR la quale riconosce e lega una superficie antigenica specifica, l'epitopo, che è determinata dalla struttura tridimensionale dell'antigene. Le interazioni tra gli anticorpi e le proteine antigeniche, gli antigeni più comuni e diversificati con cui il sistema immunitario viene in contatto, si verificano attraverso ampie aree complementari dal punto di vista sterico ed elettrostatico. Le aree idrofobiche presenti sulla superficie antigenica vengono riconosciute e interagiscono con le aree idrofobiche localizzate sulla superficie anticorpale del sito di legame per l'antigene; gruppi atomici con caratteristiche polari interagiscono, a loro volta, con gruppi atomici dell'anticorpo di carica opposta; donatori e accettori di protoni sono coinvolti nella formazione di legami idrogeno.
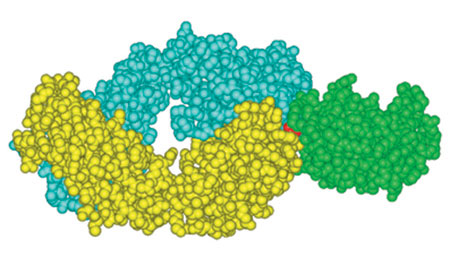
Il modello più studiato di interazione tra antigene e anticorpo è quello dell'anticorpo antilisozima di topo D1.3, una IgG1, K prodotta nella risposta secondaria diretta contro il lisozima dell'albume d'uovo di gallina (HEL, Hen Egg Lysozyme). Inizialmente, le caratteristiche strutturali di questo anticorpo sono state definite (fig. 1) studiando, mediante diffrazione ai raggi X, la struttura cristallina del frammento Fab (Fragment antigen binding, Frammento che lega l'antigene) complessato con HEL, a una risoluzione di 2,8 A (Amit et al., 1986) ed esaminando successivamente, ad alta risoluzione, la struttura cristallina del frammento Fv libero di D1.3, un eterodimero costituito dai soli domini VL e VH, (Bhat et al., 1990) e del complesso Fv-HEL (Bhat et al., 1994). Sedici residui amminoacidici di HEL formano l'epitopo conformazionale discontinuo riconosciuto da D1.3. Diciassette amminoacidi dell'anticorpo vengono a contatto con l'epitopo: 10 residui sono situati sulla catena H e gli altri 7 sulla catena L. Tutti i sei CDR e, in più, due residui provenienti dalla regione strutturale (FR, Framework Region) partecipano al contatto con l'antigene. Rispetto alla catena L, quella H è maggiormente in contatto con l'antigene: in particolare, la sua ansa CDR3 rispetto a qualunque altro CDR; il CDR2 della catena V L è quello che contribuisce in misura minore al legame con l'antigene. Gran parte delle catene laterali (9 su 17) dell' anticorpo che entrano in contatto con l' antigene sono di tipo aromatico e presentano all'antigene, quindi, ampie zone di superfici idrofobiche. Alcune di queste catene laterali (istidina 30 e tirosina 50 del dominio VL, tirosina 101 del dominio VH) contribuiscono anche alla formazione di legami idrogeno con l'antigene attraverso i loro gruppi atomici polari. Quindi, i legami idrogeno e le interazioni idrofobiche di van der Waals descrivono la natura chimica dei contatti tra antigene e anticorpo. La formazione del complesso copre circa 750 Ų della superficie accessibile al solvente di HEL e approssimativamente 700 Ų dell'anticorpo D1.3. La ricombinazione somatica e l'imprecisione presente nei meccanismi di congiungimento dei segmenti VL - h e V H - D - JH delle catene L e H, rispettivamente, generano la diversità di sequenza che interessa la posizione 96 della catena K e il CDR3 della catena H (v. il saggio di C. Rada, nel II volume). Il modello strutturale del complesso D1.3-HEL permette di valutare il contributo di questa diversità al legame con l'antigene. Il residuo di arginina 96 (Arg 96) è relativamente distante dall'antigene (> 4 Å), come lo sono i residui codificati da JK• I residui codificati dal segmento D del CDR3 di VH (arginina 99, acido aspartico 100, tirosina 101 e arginina 102) sono coinvolti in contatti molto specifici con l'epitopo HEL. In contrasto, né i residui codificati dalla regione JH né quelli che potrebbero derivare dall'impreciso congiungimento a livello della giunzione DH - JH contribuiscono direttamente ai contatti presi da D1.3 con il suo antigene.
L'immagine complessiva dell' interfaccia antigene-anticorpo nel complesso D1.3-HEL è quella di due superfici irregolari, piuttosto piatte ma con protuberanze e depressioni che risultano complementari tra loro (v. figura 1). Nella generazione della complementarità tra le due superfici che interagiscono sono coinvolte piccole variazioni conformazionali nell'anticorpo, in seguito alla genesi del complesso, in accordo con un meccanismo di 'induzione e incastro' nel fenomeno di riconoscimento tra antigene e anticorpo. Questi cambiamenti comprendono aggiustamenti nella conformazione della catena laterale, come pure un piccolo dislocamento dei domini V L e V H rispetto alle loro posizioni nell'Fv libero. Quest'ultimo movimento avvicina all'antigene i residui che prendono i contatti, ottimizzando l'adattamento tra le due proteine. Per anticorpi anti-DNA e antipeptidi sono stati, invece, dimostrati riarrangiamenti significativi di singole anse CDR (Davies e Padlan, 1992; Wilson e Stanfield, 1993). Quando si confronta la struttura del complesso D1.3-HEL con quella di altri complessi antigene-anticorpo, in cui l'antigene è una proteina, si osservano caratteristiche comuni, come aree simili di interazione, un numero simile di residui che entrano in contatto, piccoli aggiustamenti conformazionali a livello di antigene e anticorpo, e la partecipazione di tutti i CDR e di uno o due residui della regione strutturale nei contatti con l'antigene (Davies et al., 1990; Braden e Poljak, 1995). Tuttavia, sono state osservate anche alcune differenze sostanziali, come la presenza di ponti salini nel complesso tra l'anticorpo HyHEL-5 e HEL, assenti nel complesso D1.3-HEL. Un'altra differenza consiste nel fatto che nel complesso D1.3-HEL il maggior numero di contatti avviene tramite la regione CDR3 di VH, mentre nel caso di HyHEL-5 sono le regioni CDRI di VL e CDR2 di VH a entrare maggiormente in contatto con l'antigene.
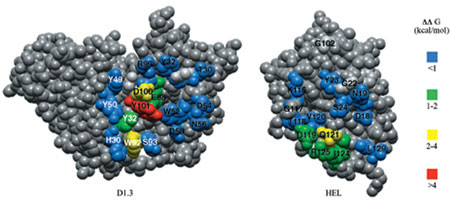
Anche se in grado di fornire informazioni dettagliate sull'architettura molecolare dell' interfaccia antigene-anticorpo, la struttura cristallina del complesso D1.3-HEL non fornisce utili informazioni su quello che è l'effettivo contributo dei singoli residui alla stabilizzazione del complesso. Per rispondere a questa domanda, è stata applicata, a tutti i residui di D1.3 e di HEL in contatto tra loro nella struttura cristallina, la tecnica alanine scanning mutagenesis, una procedura in cui gli amminoacidi coinvolti nei legami vengono mutati in alanina. Successivamente, sono state misurate le affrnità delle proteine mutanti allo scopo di determinare il contributo dei singoli residui alla formazione del complesso (Dall'Acqua et al., 1996). In figura (fig. 2) è mostrata la perdita relativa di energia libera di legame (∆∆G) per singole sostituzioni con alanina in D1.3 e HEL, mappate sulla struttura tridimensionale di ogni proteina. Come si può vedere, l'energia di legame a HEL è dominata da solo 3 dei 13 contatti amminoacidici analizzati mediante mutazione dei singoli residui in alanina (∆∆G > 10,5 kJ/mol): il triptofano 92 nel dominio VL, l'acido aspartico 100 e la tirosina 101 nel dominio VH. Questi amminoacidi formano un'area al centro dell'interfaccia e sono circondati da residui i cui apparenti contributi sono molto meno pronunciati (∆∆G < 6 kJ/mol). Analogamente, riduzioni significative nella capacità di legare D1.3 sono state osservate solo per 4 delle 12 posizioni di contatto di HEL: la glutammina 121, l'isoleucina 124, l'arginina 125 nel dominio VH e l'acido aspartico 119 nel dominio V L. Questi residui formano un' area contigua collocata alla periferia della superficie che entra in contatto con l'antigene (v. figura 2). Di conseguenza, la formazione del complesso D1.3-HEL risulta mediata da poche interazioni produttive che, da sole, dominano le energie di associazione. Si tratta di risultati del tutto simili a quelli ottenuti nel caso del legame dell'ormone della crescita umano al suo recettore (Clackson e Wells, 1995), che dimostrano come i cosiddetti epitopi funzionali, cioè i residui che contribuiscono maggiormente al legame, non siano necessariamente equivalenti ai cosiddetti epitopi strutturali, cioè i residui che prendono contatti diretti nella struttura tridimensionale.
Struttura tridimensionale dei complessi TCR-peptide/MHC
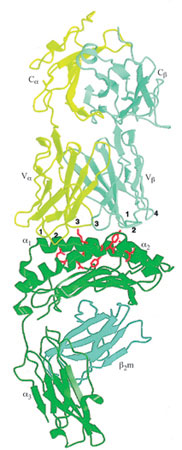
Il riconoscimento dell'antigene da parte delle cellule T è mediato dai TCR (T Cell Receptor, recettore delle cellule T), eterodimeri, legati da un ponte disolfuro, composti da catene α e β (o γ e δ) che presentano regioni variabili (V) e costanti (C) molto simili, nella loro struttura di fondo, a quelle degli anticorpi. L'interazione dei TCR con i relativi ligandi specifici, i complessi formati da peptidi antigenici e molecole MHC del self, è responsabile del destino delle cellule T (Jameson et al., 1995). l timociti, i precursori delle cellule T che esprimono in modo donale i TCR sulla propria superficie, sviluppano il loro repertorio e maturano a cellule T nel timo prima di migrare alla periferia. l timo citi che presentano TCR con moderata affinità (KD = 10 ÷ 100 μM) per i complessi peptide/MHC self vanno incontro a 'selezione positiva' e vengono risparmiati dall'apoptosi (Alam et al., 1996). Al contrario, i timociti nei quali i TCR legano troppo strettamente peptidi/MHC self vengono 'selezionati negativamente' e, come quelli privi di affinità per peptidi MHC/self, muoiono senza raggiungere la periferia, o diventano incapaci di provocare una reazione immunitaria. In questo modo, durante lo sviluppo timico, vengono eliminate le cellule T autoreattive più pericolose. Sorprendentemente, la differenza di affrnità tra ligandi di cellule T che vengono selezionati positivamente e negativamente è relativamente piccola, con valori tra 3 e 10. Quindi, si pensa che i TCR analizzino molti diversi ligandi peptidi/MHCself per evidenziare quelli che forniscono il grado appropriato (né troppo elevato né troppo basso) di stabilizzazione del complesso, necessario per la selezione positiva. Il processo di selezione genera un pool di cellule T mature nella periferia che, per la maggior parte, reagiscono fortemente solo nei confronti di peptidi estranei presentati nel contesto delle molecole MHC self. Studi di cristallografia a raggi X (fig. 3) hanno mostrato che la struttura complessiva dei TCR somiglia a quella dei frammenti Fab degli anticorpi (Bentley et al., 1995; Fields et al., 1995; Garboczi et al., 1996; Garcia et al., 1996). La regione V del TCR è costituita da domini Vα e Vβ, simili a quelli delle immunoglobuline, collegati da piccoli segmenti peptidici ai domini Cα e Cβ della regione C. Il relativo orientamento dei domini Vα e Vβ è simile a quello dei domini VL e VH. Analogamente, i singoli CDR (a eccezione del CDR2 di Va) si trovano in posizioni quasi equivalenti nei TCR e negli anticorpi. Tuttavia, vi sono tre differenze strutturali importanti tra i TCR e gli anticorpi. La prima consiste nel cambiamento di posizione di un filamento β dal foglietto più interno dei domini VL e VH al foglietto più esterno di Vα, che provoca un riposizionamento dell'ansa CDR2 di Vα. La seconda riguarda il dominio Cα, che presenta solo una parziale somiglianza con il tradizionale dominio Ig. Nel dominio Cα, infatti, i tre filamenti β più esterni vengono rimpiazzati da un'ansa, da una breve α-elica, e da un foglietto β, rispettivamente. La terza differenza strutturale tra TCR e Ig riguarda i domini V β e Cβ che sono molto più strettamente in contatto tra loro rispetto ai corrispondenti domini degli anticorpi: l'area di superficie nascosta complessivamente tra Vβ e Cβ è di circa 800 Å2, mentre tra VH e CHI è di 200 ÷ 350 Å2, a seconda dall'angolo formato.
Le nostre conoscenze attuali sulle interazioni TCR-peptide/MHC si basano sulle strutture cristalline di due complessi TCR-peptide/MHC di classe I (Garboczi et al., 1996; Garcia et al., 1998): il complesso tra il TCR 2C e un peptide self (dEV8), proveniente da una proteina mitocondriale coinvolta nella respirazione, legato alla molecola MHC di classe I H-2Kb (v. figura 3); il complesso tra il TCR A6 e un peptide virale (Tax), derivante dalla proteina HTLV-l, legato alla molecola MHC di classe I HLA-A2 (v. oltre figura 6b). Dal confronto di queste due strutture è emersa un'immagine comune del meccanismo di riconoscimento dei complessi peptide!MHC da parte dei TCR.

L'area di superficie totale nascosta a livello dell' interfaccia è simile nei due complessi TCR-peptide!MHC, e occupa approssimativamente 1800 Å2; il peptide risulta coperto molto più profondamente da parte dell'MHC che non dal TCR. La catena α del TCR copre i residui amminoterminali del peptide, mentre la catena β stabilisce contatti con i residui carbossiterminali (tab. I). I CDR l α e l β sono adagiati tra le α-eliche della catena pesante dell'MHC di classe I e, contemporaneamente, sono in contatto con il peptide e la molecola MHC. I CDR 2α e 2β interagiscono esclusivamente con l'MHC e sono posti direttamente sopra le eliche α2 e αl, rispettivamente. I CDR 3α e 3β sono quindi posizionati in modo tale da 'ispezionare' quanto è contenuto nel solco dove si lega il peptide. Sembra che questa disposizione tragga pieno vantaggio dalla diversità di sequenza dei TCR. Il CDRI e il CDR2 sono codificati dai geni della linea germinale Vα e Vβ. Al contrario, il CDR3α e il CDR3β sono il risultato di eventi di ricombinazione a livello delle giunzioni Vα-Jα e Vβ-Dβ-Jβ, e mostrano quindi una variabilità molto più elevata. Pertanto, essi sono posizionati in modo tale da interagire con il peptide legato, la cui diversità è naturalmente molto più grande di quella delle molecole MHC che lo ospitano. Comunque, è opportuno notare che anche i CDRI entrano in contatto con il peptide. Similmente, anche i CDR3 contattano l'MHC (v. tabella l). In tal modo, il riconoscimento del complesso peptide/MHC risulta mediato da combinazioni particolari di CDR, unici per ogni sito di legame del TCR. Le altre due regioni di variabilità del TCR, le regioni ipervariabili 4α (VH4α e VH4β), partecipano solo in misura minore alla formazione del complesso. Nel complesso TCR A6-Tax/HLA-A2, i CDR lβ e 2β interagiscono in misura minima con il peptide/MHC; il CDR3β, che è solitamente lungo, presenta la superficie di contatto più ampia di tutti i CDR. Nel complesso TCR 2CdEV8/H-2Kb, tuttavia, il CDR3β è posizionato sopra una tasca in gran parte vuota e stabilisce solo contatti limitati con il complesso peptide/MHC. Sulla base del numero di contatti e dell' area di superficie nascosta, sembra che, in entrambi i complessi, il Va abbia un ruolo dominante nell'orientare il TCR verso il peptide/MHC.
La generalità del meccanismo di legame visto nei complessi TCR 2C-dEV8/H-2Kb e TCR A6-Tax/HLA-A2 si basa su diverse considerazioni (Garboczi et al., 1996). Sembra che il legame diagonale sia la conseguenza di caratteristiche strutturali condivise sia dalle molecole MHC di classe l sia da quelle di classe II. La superficie all'apice di queste proteine non è piatta, ma presenta due protuberanze vicino alle posizioni amminoterminali delle regioni ad αelica che formano il solco nel quale viene accomodato il peptide. Un orientamento diagonale è necessario affinché la superficie di contatto, discretamente piatta, del TCR si abbassi sull'MHC in modo da poter venire a contatto diretto con il peptide in tutta, o quasi, la sua lunghezza. Ampie traslazioni lungo il solco dove si lega il peptide o rotazioni dall'orientamento diagonale distruggerebbero l'adattamento tra il TCR e il complesso peptide/MHC.
Nonostante questa complementarità di forma complessiva, sembra che l'interfaccia tra il TCR e il peptide/MHC sia impacchettata, in modo significativo, molto meno strettamente rispetto a quella di altri complessi proteina-proteina, compreso il complesso antigene-anticorpo (Garcia et al., 1998). Nel caso del complesso TCR 2C-dEV8/H-2Kb, lo scarso adattamento è, per la maggior parte, dovuto al numero limitato di contatti tra il TCR e il peptide legato, piuttosto che ai contatti del TCR con l'MHC. In particolare, vi è un'ampia cavità vuota al centro dell'interfaccia tra le anse CDR3 delle catene α e β del TCR. Questa complementarità relativamente scarsa è in rapporto con le deboli affinità dei TCR per i complessi peptide/MHC (10-5 ÷10-6 M rispetto all'affrnità di 10-8 ÷10-10 M tipiche delle interazioni antigene-anticorpo). La bassa affinità tra TCR e ligandi implica l'esistenza di speciali meccanismi per l'attivazione dei linfociti T, quali serial triggering (attivazione in serie) o kinetic proofreading (correzione cinetica). Si rimanda ai classici lavori di S. Valitutti e collaboratori (1995) e di J.D. Rabinowitz e collaboratori (1996) per maggiori dettagli. In breve, la debole affinità dipende da un'alta velocità di dissociazione dei legami tra TCR e ligando. Ciò consente a un singolo complesso peptide/MHC di impegnare e attivare più di un TCR, durante il periodo in cui illinfocita T viene a contatto con la cellula presentante l'antigene. Questi modelli spiegano anche come ligandi con affinità molto elevata per il TCR siano effettivamente meno efficaci nello scatenare l'attivazione delle cellule T. A questo proposito, non vi sono prove di ipermutazioni somatiche a livello dei geni del TCR, come evidenziato dall' analisi della sequenza di un vasto numero di catene α e β. Questo suggerisce che, in contrasto con quanto osservato per gli anticorpi, non vi è alcun vantaggio selettivo nel generare TCR con alta affinità tramite l'aumento della complementarità a livello dell'interfaccia di contatto.

Una scoperta importante nel caso del complesso TCR 2CdEV8/H-2Kb (Garcia et al., 1998) riguarda i riarrangiamenti strutturali, relativamente estesi, che si osservano a livello del sito di combinazione del TCR quando esso si associa al ligando. Come mostrato in figura (fig. 4) questi riarrangiamenti coinvolgono inizialmente le anse CDRI e CDR3 del dominio Vα che vengono a essere dislocate a 4 ÷ 6 Å rispetto alla posizione che assumono nella struttura del TCR A6 non legato. Alcuni di questi cambiamenti sono necessari per evitare scontri sterici con il complesso peptide/MHC, mentre altri servono probabilmente per migliorare le interazioni produttive con illigando. Questa flessibilità strutturale, o 'plasticità', permetterebbe a un singolo TCR di adottare conformazioni multiple, consentendogli quindi di interagire con differenti peptidi presentati nel contesto della stessa molecola MHC. Il che, a sua volta, potrebbe contribuire a spiegare la ben nota capacità di molte cellule T di andare incontro a reazioni crociate con i complessi peptide/ MHC sia estranei sia propri all'organismo, con il pericolo di scatenare reazioni autoimmunitarie. Tuttavia, è importante enfatizzare che i cambiamenti conformazionali osservati nel TCR A6 sono localizzati nel sito di combinazione e non vengono trasmessi alle regioni C. Quindi, come nel caso del complesso TCR A6-Tax/HLA-A2 (Garboczi et al., 1996), è improbabile che cambiamenti nella conformazione del TCR, in seguito al legame con illigando, siano responsabili dell'avvio dei segnali trasmessi dalle cellule T. Piuttosto, sembra che meccanismi basati sull'induzione dell'oligomerizzazione del TCR da parte delligando (Reich et al., 1997) siano responsabili dell'attivazione delle cellule T da parte del complesso peptide/ MHC. Aggiustamenti conformazionali dell'entità evidenziata nel complesso TCR 2CdEV8/H-2Kb sono stati osservati anche nei complessi antigene-anticorpo in cui l'antigene è un peptide o una molecola di DNA (Davies e Padlan, 1992; Wilson e Stanfield, 1993), ma non con antigeni proteici (v. sopra).
Struttura tridimensionale dei complessi TCR-superantigene
Alcune molecole, per esempio certe tossine batteriche, sono capaci di legarsi a più di un TCR. Per questa proprietà vengono chiamate superantigeni. Il gruppo di superanti geni meglio caratterizzati dal punto vista strutturale e immunologico è rappresentato dalle enterotossine dello Staphylococcus aureus, che causano sia la sindrome da shock tossico sia l'avvelenamento da cibo (Marrack e Kappler, 1990). Alcuni ceppi di S. aureus producono anche la tossinal della sindrome da shock tossico (TSST-l, Toxic Shock Syndrome Toxin-l), coinvolta nella maggior parte dei casi di shock tossico mestruale. Inoltre, i virus del tumore della mammella di topo (MMTV, Mouse Mammary Tumor Viruses) codificano superantigeni endogeni che permettono a questi retrovirus di sfruttare il sistema immunitario dell'ospite per espandersi e trasmettersi (Kotzin et al., 1993). È stato anche proposto che alcuni superantigeni, derivanti da batteri, micoplasmi o virus, siano in grado di avviare malattie auto immuni attraverso l'attivazione di cellule T specifiche per antigeni self. Per esempio, l'analisi di cellule T infiltrate in isolotti pancreatici, derivanti da pazienti affetti da diabete mellito insulinodipendente (lDDM, lnsulin Dependent Diabetes Mellitis) ha permesso di evidenziare l'espressione preferenziale del segmento genico Vβ7, ma nessuna selezione per particolari segmenti Vα o segmenti di giunzione Vβ-Dβ-Jβ (Conrad et al., 1994). Questa osservazione ha indotto a pensare che nella patogenesi del diabete mellito insulinodipendente sia coinvolto un superantigene associato con gli isolotti pancreatici.

Di recente, B. Conrad e collaboratori (1997) hanno isolato, dal supernatante di isolotti di pazienti affetti da lDDM, un nuovo retrovirus endogeno umano e hanno mostrato che il gene dell'involucro virale codifica un superantigene, dipendente dalle molecole MHC di classe II, specifico per il Vβ7. B.A. Fields e collaboratori (1996) hanno determinato la struttura tridimensionale del complesso che si forma in seguito all'interazione tra una catena β del TCR murino (Vβ8.2-Jβ2.1-Cβ1) e un superantigene rappresentato dall'enterotossina C₃ di stafilococco (SEC₃). Il complesso viene formato tramite contatti tra il dominio Vβ e i due domini, piccolo e grande, dell'enterotossina (fig. 5). L'area dell'interfaccia in cui non può penetrare il solvente è di 1300 Ų, simile quindi a quella osservata a livello delle interfacce nei complessi antigene-anticorpo e TCR-peptide/MHC. l residui amminoacidici di Vβ che entrano in contatto con l' enterotossina sono i seguenti: asparagina 28 e 30 del CDRl; tirosina 50, glicina 51, alanina 52, glicina 53, serina 54 e treonina 55 del CDR2; lisina 57 e 66 dell'FR3; prolina 70 e glutammina 72 della VH4. Le regioni CDRl, CDR2, FR3 e VH4 sono responsabili, rispettivamente, del 30, 47, 13 e 10% dei contatti totali con il superantigene.
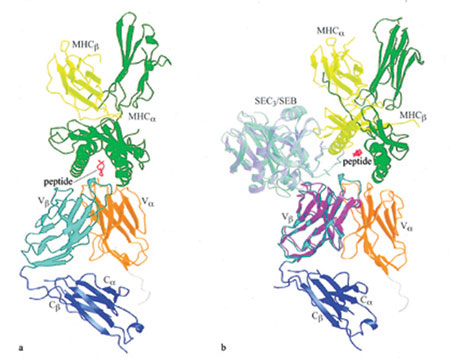
Come mostrato nella figura 5, non vi sono contatti diretti tra il CDR3 della catena Vβ e SEC₃. Tale osservazione fornisce una spiegazione semplice della stimolazione da parte dei superantigeni batterici e virali delle cellule T che presentano particolari elementi Vβ, senza però tener conto della lunghezza o della sequenza amminoacidica del CDR3 di Vβ (Marrack e Kappler, 1990). Tuttavia, ciò non esclude la possibilità che, in alcuni casi, il CDR3 di Vβ riesca a modulare la re attività del superantigene. Per esempio, quando il dominio Vβ del TCR A6 (Garboczi et al., 1996) viene sovrapposto al dominio l4.3.d di Vβ nella struttura VβC13-SEC3, si osserva che i residui dal 96 al 100 del CDR3 di Vβ A6, localizzati all'estremità di questa lunga ansa sporgente, stabiliscono contatti estesi con i residui dal 99 al 104 dell' enterotossina. lnterazioni simili possono spiegare l'influenza dei residui del CDR3 di Vβ sulla reattività delle cellule T verso il superantigene retrovirale murino Mtv-9 (Ciurli et al., 1998). l residui dell' enterotossina stafilococcica che prendono contatto con il Vβ sono i seguenti: asparagina 60, tirosina 90, valina 91 (nel dominio piccolo) e glicina 19, treonina 20, asparagina 23, tirosina 26, fenilalanina 176 e glutammina 210 (nel dominio grande). Questi residui sono localizzati nella fenditura situata tra il dominio piccolo e quello grande che, come appare nella struttura cristallina (v. figura 5), forma l'interfaccia con la catena β. L'allineamento delle sequenze di alcuni superantigeni batterici ha evidenziato che tre di essi (SEA, SED e SEE) non sono in grado di attivare cellule T che presentano il Vβ8.2; essi differiscono da SEB e SEC a livello di quasi tutte le posizioni di contatto con il Vβ. D'altra parte, alcuni superantigeni capaci di legare la catena β 14.3.d, con un'affrnità simile a quella dell'enterotossina stafilococcica (KD = 6,2 JLM), mantengono numerosi residui che svolgono un ruolo chiave nei contatti, in particolare asparagina 60, tirosina 90 e glutammina 210. L'analisi della struttura cristallina di questi complessi permette di comprendere come l'enterotossina stafilococcica possa stimolare cellule T che esprimono domini Vβ appartenenti a diverse famiglie: tutti i ponti idrogeno tra SEC₃ e Vβ si formano, infatti, tra le catene laterali di SEC₃ e gli atomi della struttura portante di Vβ, Un simile meccanismo di legame, in cui la struttura portante, piuttosto che le catene laterali degli amminoacidi, ha un ruolo rilevante per la formazione dei legami idrogeno, è stato osservato nei complessi peptide/ MHC (Fremont et al., 1992). In accordo con questo meccanismo, le strutture tridimensionali di altri domini Vβ rivelano che, nei Vβ che reagiscono con l' enterotossina e nel V₁₃14.3.d, le posizioni degli atomi della struttura portante che prendono contatto con il superantigene sono molto simili. Al contrario, essi differiscono in modo significativo nei Vβ che non legano il superantigene. Per esempio, il SEC₃ attiva le cellule T che presentano il Vβ8 di topo e il Vβ12 umano, ma non il Vβ2.3 murino (Kotzin et al., 1993). Quando il Vβ12.3 (Garboczi et al., 1996) è sovrapposto al Vβ8.2 di topo, la differenza media nella posizione del carbonio α per i residui che stabiliscono contatti con SEC₃ è di soli 0,9 Å. Al contrario, quando il Vβ2.3 murino che non lega il superantigene (Housset et al., 1997) è sovrapposto al Vβ8.2 murino, la differenza media è di 3 Å. Essa è dovuta principalmente allo spostamento di un filamento nel Vβ2.3 rispetto ad altri domini Vβ di struttura nota: infatti, nel Vβ2.3 il filamento c" è legato con ponti idrogeno al filamento d del foglietto β adiacente (più esterno), mentre in altri Vβ il filamento c'è associato con il filamento c' all'interno dello stesso foglietto (più interno). In seguito alla formazione del complesso, non si verificano altri grandi cambiamenti conformazionali né a livello della catena β 14.3.d né a livello del superantigene, sebbene sia possibile osservare alcuni cambiamenti nella conformazione delle catene laterali dei residui all'interfaccia; queste modificazioni strutturali delle catene laterali potrebbero indicare un meccanismo di 'induzione e incastro' per il riconoscimento TCR-superantigene, come descritto da K.C. Garcia e collaboratori (1998) per il complesso TCR 2C-peptide/MHC. La stretta associazione tra i domini Vβ e C₁₃, riscontrata nella struttura sia delle catene β libere sia negli eterodimeri associati Ciβ di TCR, viene mantenuta anche nel complesso formato da TCR ed enterotossina stafilococcica. Analogamente, non si osservano grandi cambiamenti conformazionali in seguito alla formazione del complesso tra HLA-DRI e SEB (Jardetzky et al., 1994). Quindi, escludendo possibili effetti di cooperazione, questi risultati suggeriscono che la formazione del complesso ternario tra un recettore T, un superantigene e una molecola MHC non coinvolge movimenti dei domini principali o riarrangiamenti nello scheletro polipeptidico delle molecole che interagiscono. In modo simile, fatta eccezione per il riarrangiamento di alcune anse CDR, non sono state osservate grosse variazioni conformazionali del TCR nelle strutture cristalline dei complessi TCR-peptide/MHC di classe I studiate, come riportato in precedenza, da Garboczi e collaboratori (1996) e da Garcia e collaboratori (1998). Sono stati proposti numerosi modelli di interazione delle cellule T con i superantigeni (Kotzin et al., 1993). Questi modelli differiscono l'uno dall'altro per l'entità della reazione tra il TCR e l'MHC nel complesso TCR-superantigene-MHC. Le strutture cristalline disponibili permettono di discriminare chiaramente tra questi modelli, per lo meno per le enterotossine di stafilococco. Abbiamo costruito un modello del complesso TCR-superantigene-MHC (fig. 6) attraverso un metodo di sovrapposizione delle seguenti strutture cristalline: il complesso VβC13-SEC3; il complesso SEBpeptide/HLA-DRl (Jardetzky et al., 1994); l'eterodimero αβ TCR A6 (Garboczi et al., 1996). Questo modello presenta numerose caratteristiche importanti: il superantigene è posizionato in modo ideale per collegare la cellula che presenta l' antigene (APC, Antigen Presenting Cell) alla cellula T. Il dominio Va non prende contatti diretti con il superantigene, in accordo con la capacità della sola catena β di legare i superantigeni (Malchiodi et al., 1995). La molecola MHC è orientata con la sua catena α sopra la catena β del TCR e con la sua catena β sopra la catena α del TCR (v. figura 6a). Tuttavia, in contrasto con il complesso TCRpeptide/MHC convenzionale, non vi sono contatti diretti tra la catena β del TCR e le catena α o β dell'MHC. D'altra parte, il dominio βl dell'MHC interagisce con il dominio Va del TCR. Questi dati possono essere considerati come la prova di un'interazione funzionale tra la catena β di classe 11 e quella α del TCR nel riconoscere i superantigeni batterici, come pure del fatto che la capacità di alcune regioni Va di interagire in modo più favorevole di altre con differenti alleli di classe II è responsabile dell'espressione preferenziale di questi Va tra cellule T re attive ai superantigeni. Comunque, è importante sottolineare che l'entità dell'interazione tra il Va del TCR e il dominio βl dell'MHC dipende, in larga misura, dal relativo orientamento dei domini Va e Vβ nell'eterodimero del TCR e che un confronto tra diversi TCR ha evidenziato una variabilità significativa nella geometria dell'associazione VaN 13 (Garcia et al., 1998). Nel nostro modello la molecola MHC è solo parzialmente impegnata dal TCR, con il superantigene che si inserisce come un cuneo tra la catena β del TCR e la catena α dell'MHC (v. figura 6b). Come conseguenza, il peptide antigenico viene effettivamente rimosso dal sito di combinazione con il TCR. In questo modo, il superantigene è in grado di aggirare il normale meccanismo per l'attivazione delle cellule T attraverso specifici complessi peptide/MHC. Il risultato è l'attivazione policlonale dell' intera popolazione di cellule T che esprimono particolari elementi Vβ, che prescinde, in gran parte, dalla specificità del complesso peptide-MHC dei TCR corrispondenti.
Bibliografia citata
ALAM, S.M., TRAVERS, P.I., WUNG, J.L., NASHOLDS, W., REDPATH, S., JAMESON, S.C., GASCOIGNE, N.R. (1996) T cell receptor affinity and thymocyte positive selection. Nature, 381, 616-620.
AMIT, A.G., MARIUZZA, R.A., PHILLIPS, S.E.V., POLJAK, R.J. (1986) Three-dimensional structure of an antigen-antibody complex at 2.8 Å resolution. Science, 233, 747-753.
BENTLEY, G.A., BOULOT, G., KARJALAINEN, K., MARIUZZA, R.A. (1995) Crystal structure of the beta chain of a T cell antigen receptor. Science, 267, 1984-1987.
BHAT, T.N., BENTLEY, G.A., BOULOT, G., GREENE, M.I., TELLO, D., DALL'ACQUA, W., SOUCHON, H., SCHWARZ, F.P., MARIUZZA, R.A., POLJAK, R.I. (1994) Bound water molecules and conformational stabilization help mediate an antigen-antibody association. Proc. Nat!. Acad. Sci. USA, 91, 1089-1093.
BHAT, T.N., BENTLEY, G.A., FISCHMANN, T.O., BOULOT, G., POLJAK, R.J. et al. (1990) Small rearrangements in structures of Fv and Fab fragments of antibody D1.3 on antigen binding. Nature, 347, 483-485.
BRADEN, B.C., POLJAK, R.I. (1995) Structural features ofthe reactions between antibodies and protein antigens. F ASEB J, 9, 9-16.
CIURLI, C., POSNETT, D.N., SEKALY, R.-P., DENIS, F. (1998) HighIy biased CDR3 usage in restricted sets of beta chain variable regions during viraI superantigen 9 response. J. Exp. Med., 187, 253-258.
CLACKSON, T., WELLS, J.A. (1995) A hot spot ofbinding energy in a hormone-receptor interface. Science, 267, 383-386.
CONRAD, B., WEIDMANN, E., TRUCCO, G., RUDERT, W.A., BEHBOO, R., RICORDI, C., RODRIQUEZ-RILO, H., FINEGOLD, D., TRUCCO, M. (1994) Evidence for superantigen involvement in insulin dependent diabetes mellitus aetiology. Nature, 371, 351-355.
CONRAD, B., WEISSMAHR, R.N., BONI, J., ARCARI, R., SCHUPBACH, J., MACH, B. (1997) A human endogenous retroviral superantigen as candidate autoimmune gene in type I diabetes. Cell, 90, 303-313.
DALL' ACQUA, W., GOLDMAN, E.R., EISENSTEIN, E., MARIUZZA, R.A. (1996) A mutational analysis of the binding of two different proteins to the same antibody. Biochemistry, 35, 9667-9676.
DAVIES, D.R., PADLAN, E.A. (1992) Twisting into shape. Curr. Biol., 2, 254-256.
DAVIES, D.R., PADLAN, E.A., SHERIFF, S. (1990) Antigen-antibody complexes. Annu. Rev. Biochem., 59, 439-473.
FIELDS, B.A., OBER, B., MALCHIODI, E.L., LEBEDEVA, M.L, BRADEN, B.C., YSERN, X., KIM, J.K., SHAO, X., WARD, E.S., MARIUZZA, R.A. (1995) Crystal structure of the V alpha domain of a T cell antigen receptor. Science, 270, 1821-1824.
FIELDS, B.A. MALCHIODI, E.L., LI, H., YSERN, X., STAUFFACHER, C.V., SCHLIEVERT, P.H., KARJALAINEN, K., MARIUZZA, R.A. (1996) Crystal structure beta-chain of a T cell receptor complexed with a superantigen. Nature, 384, 188-192.
FREMONT, D.H. MATSUMURA, M., STURA, E.A., PETERSON, P.A., WILSON, I.A. (1992) Crystal structures of two viraI peptides in complex withmurine MHCclass I H-2Kb. Science, 257, 919-927.
GARBOCZI, D.N., GHOSH, P., DTZ, D., FAN, Q.R., BIDDISON, W.E., WILEY, D.C. (1996) Structure ofthe complex between human T cell receptor, viraI peptide and HLA-A2. Nature, 384, 134-141.
GARCÍA, K.C., DEGANO, M., PEASE, L.R., HUANG, M., PETERSON, P.A., TEYTON, L., WILSON, I.A. et al. (1998) Structural basis of plasticity in T cell receptor recognition of a self peptideMHC antigen. Science, 279, 1166-1172.
GARCÍA, K.C., DEGANO, M., STANFIELD, R.L., BRUNMARK, A., SACKSON, M.R., PETERSON, P.A., TEYTON, D., WILSON, I.A. (1996) An alphabeta T cell receptor structure at 2.5 Å and its orientation in the TCR-MHC complex. Science, 274, 209-219.
HOUSSET, D. MAZZA, G., GREGOIRE, C., PIRAS, C., MALISSEN, B., FONTECILLA-CAMPS, J.C. (1997) The three-dimensional structure of a T -cell antigen receptor V alpha V beta heterodimer reveals a novel arrangement of the V beta domain. EMBO J., 16, 4205-4216.
JAMESON, S.C., HOGQUlST, K.A., BEVAN, M.J. (1995) Positive selection ofthymocytes. Annu. Rev. lmmunol., 13, 93-125.
JARDETZKY, T.S., BROWN, J.H., GORGA, J.C., STERN, L.J., DRBAN, R.G., CHI, Y.L, STAUFFACHER, C., STROMINGER, J.L., WILEY, D.C., (1994) Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature, 368, 711-718.
KOTZIN, B.L., LEUNG, D.Y.M., KAPPLER, J., MARRACK, P. (1993) Superantigens and their potential role in human disease. Adv. lmmunol., 54, 99-166.
MALCHIODI, E.L, EISENSTEIN, E., FIELDS, B.A., OHLENDORF, D.H., SCHLIEVERT, P.M., KARJALAINEN, K., MARIUZZA, R.A. (1995) Superantigen binding to a T cell receptor beta chain of known three-dimensional structure. J. Exp. Med., 182, 1833-1845.
MARRACK, P., KAPPLER, J. (1990) The staphylococcal enterotoxins and their relatives. Science, 248, 705-711.
RABINOWITZ, J.D., BEESON, C., LYONS, D.S., DAVIS, M.M., Mc CONNELL, H.M. (1996) Kinetic discrimination in T cell activation. Proc. Natl. Acad. Sci. USA 93, 1401-1405.
REICH, Z., BONIFACE, J.J., LYONS, D.S., BOROCHOV, N., WACHTEL, E.I., DAVIS, M.M. (1997) Ligand-specific oligomerization of T cell receptor molecules. Nature, 387, 617-620.
VALITUTTI, S., MULLER, S., CELLA, M., PADOVAN, E., LANZAVECCHIA, A. (1995) Serial triggering ofmany T cell receptors by a few peptide-MHC complexes. Nature, 375, 148-151.
WILSON, LA., STANFIELD, R.L. (1993) Antigen-antibody interaction. Curr. Opin. Struct. Biol., 3, 113-118.