
Sangue
Sangue
Per sopravvivere, tutti gli organismi hanno bisogno sia di un continuo rifornimento di sostanze nutritive e di energia, indispensabile per lo svolgimento dei processi metabolici, sia di espellere i prodotti di rifiuto di tali processi. Negli organismi più semplici, come per esempio i Poriferi, i Celenterati e i Platelminti, non è presente un fluido circolante, mentre nei gruppi più evoluti composti con ruolo nutritivo, gas respiratori, prodotti del metabolismo e sostanze di scarto vengono veicolati tramite un apposito sistema circolatorio, utilizzando come mezzo di trasporto un complesso eterogeneo di consistenza liquida. Nei Vertebrati questo vettore viene definito 'sangue', negli invertebrati 'emolinfa'.
Dato che l'ossigeno è relativamente poco solubile nel plasma sanguigno, negli animali a più complessa organizzazione il suo trasporto dalla superficie alle varie parti del corpo necessita di speciali molecole proteiche trasportatrici, i pigmenti respiratori, che, legando reversibilmente l'ossigeno, sono in grado di aumentare notevolmente la capacità del fluido di trasportarlo. Nei Molluschi e negli Artropodi il trasportatore di ossigeno è una proteina di colore azzurro, l'emocianina, contenente rame; il ferro si trova invece nell'emeritrina, riscontrabile nei Brachiopodi e nei Priapulidi. Il principale pigmento respiratorio è comunque l'emoglobina: onnipresente nei Vertebrati, si ritrova anche in diversi invertebrati, nei Protozoi, nelle piante e perfino in qualche batterio.
Dal momento che le curve di assunzione e cessione dell'ossigeno molecolare sono funzione della pressione parziale dell'ossigeno e della temperatura alla quale hanno luogo gli scambi gassosi, tra gli animali è riscontrabile una grande variabilità funzionale dell'emoglobina. I Mammiferi, per esempio, che durante la vita embrionale sono soggetti a penuria di ossigeno, possiedono un'emoglobina, chiamata appunto 'emoglobina fetale', capace di assumere ossigeno a tensioni più basse di quella dell'adulto. I Mammiferi di piccola taglia hanno, rispetto a quelli di dimensioni maggiori, un metabolismo più attivo e quindi un'emoglobina con minore affinità per l'ossigeno, che viene così ceduto più facilmente ai tessuti. Dato che un innalzamento della temperatura indebolisce il legame tra l'emoglobina e l'ossigeno, rendendone più facile la dissociazione, l'emoglobina dei Pesci ha un comportamento diverso da quella degli omeotermi di fronte alla temperatura, che nell'acqua è sempre inferiore a quella degli animali a sangue caldo; inoltre il pigmento respiratorio nei Pesci deve avere una maggiore affinità per l'ossigeno, la cui tensione parziale è decisamente minore nell'acqua che nell'aria. Un altro parametro che influenza la curva di dissociazione è l'acidità del plasma sanguigno, poiché a un aumento della concentrazione di anidride carbonica o di altri acidi corrisponde una maggiore liberazione di ossigeno.
Nei Vertebrati il numero e la grandezza degli eritrociti variano molto da gruppo a gruppo; essi diventano più numerosi e più piccoli mano a mano che si sale la scala sistematica; a parità di volume di sangue, quanto più gli eritrociti sono numerosi e piccoli, tanto più aumenta la loro superficie respiratoria. Gli unici Vertebrati a essere privi sia di emoglobina sia di eritrociti sono alcuni Pesci antartici che presentano il sangue totalmente incolore. Anche il numero dei globuli bianchi varia a seconda del gruppo animale; essi sono comunque assai meno numerosi dei globuli rossi: nei Pesci possono raggiungere e superare il 10% delle cellule sanguigne, mentre negli Amnioti non superano mai l'1%. I linfociti, il cui nome deriva dal fatto che nei Mammiferi abbondano nella linfa, sono presenti in tutte le classi di Vertebrati, specialmente in quelle meno evolute.
sommario
1. Emopoiesi. 2. Anatomia. 3. Fisiologia. 4. Patologia. □ Bibliografia.
Emopoiesi
L'emopoiesi è il processo di produzione delle cellule del sangue. Nelle prime fasi dello sviluppo embrionale, prima ancora della formazione del fegato, all'emopoiesi provvedono particolari cellule mesenchimali delle pareti vascolari. In un secondo tempo vi partecipa anche il fegato, la cui attività emopoietica va poi decrescendo dopo il secondo mese di vita intrauterina, quando interviene gradatamente il midollo osseo. Durante la vita extrauterina, all'emopoiesi è preposto fondamentalmente il midollo rosso delle ossa; in misura minore vi provvedono il tessuto linfoide e, secondo alcuni, il tessuto reticoloendoteliale, distribuito in tutto l'organismo. L'emopoiesi midollare è responsabile della formazione degli elementi della serie rossa, delle piastrine, dei granulociti, dei linfociti, dei monociti e dei precursori.
Le ipotesi sull'origine delle cellule ematiche sono state dibattute per lungo tempo tra i sostenitori dell'esistenza di un unico elemento staminale capostipite (teoria unitarista) e i fautori di teorie dualistiche (o trialistiche) che ne ammettevano più di uno. Esiste ampio consenso nel ritenere un'unica cellula (cellula staminale pluripotente, in inglese stem cell) l'elemento progenitore comune di tutte le altre cellule del sangue. Un elemento primordiale è stato storicamente definito da Adolfo Ferrata come 'emoistioblasto', cellula mesenchimale indifferenziata totipotente. Attualmente il problema dell'origine delle cellule ematiche viene affrontato su basi non solo morfologiche ma anche funzionali (in modo particolare correlandolo al ruolo svolto da influenze esterne sulla capacità delle cellule staminali di proliferare o differenziarsi). Le cellule staminali pluripotenti, originariamente poco rappresentate, vanno incontro a uno sviluppo selettivo che porta alla comparsa di progenitori committed (commissionati od orientati) verso la produzione di precursori differenziati dei vari tipi di cellule ematiche.
Le cellule staminali situate nel midollo osseo, dove hanno luogo gli eventi dell'emopoiesi, possono autoreplicarsi o entrare in una fase di sviluppo che le porterà a differenziarsi completamente. La prima tappa di biforcazione separa i precursori delle cellule linfatiche da quelli delle altre linee cellulari. Nella fase preliminare di differenziazione, la morfologia della cellula appare sostanzialmente invariata, mentre durante le fasi successive i caratteri di riconoscibilità sono gradualmente meglio delineati. La cellula staminale mieloide, secondo ramo della biforcazione originaria della cellula staminale, dà luogo a diverse linee cellulari, che portano alla formazione di eritrociti, granulociti, monociti e piastrine. Ricerche recenti hanno dimostrato che esistono fattori di accrescimento proteici (veri e propri ormoni ematopoietici) in grado di regolare la produzione di linee specifiche di cellule ematiche. Le sostanze capaci di stimolare la replicazione e di far maturare le cellule del sangue (essenzialmente i globuli bianchi) sono dette Colony stimulating factors (CSF). L'ormone proteico che induce la proliferazione e la maturazione dei precursori della serie rossa, ossia l'eritropoietina, è prodotto e liberato da alcune cellule del rene in risposta a variazioni del contenuto di ossigeno nel sangue. Il passaggio in circolo delle cellule, a maturazione avvenuta, è regolato da influenze vitaminiche, enzimatiche e ormoniche.
Il processo di formazione degli elementi cellulari della serie rossa del sangue viene denominato 'eritropoiesi'. In condizioni normali tali elementi derivano dal proeritroblasto (generato a sua volta dall'emocitoblasto), caratterizzato da un grande nucleo, nucleoli e citoplasma intensamente basofilo. Esso si trasforma successivamente in eritroblasto basofilo (privo di nucleoli), in eritroblasto policromatofilo (il cui citoplasma è in parte basofilo e in parte acidofilo), in eritroblasto ortocromatico (citoplasma acidofilo, nucleo piccolo) e finalmente in eritrocito o normocito (privo di nucleo). Giunti a questo stadio evolutivo, i globuli rossi abbandonano il midollo e passano in circolo.
La formazione delle piastrine (piastrinogenesi o trombocitopoiesi) prende avvio dal megacarioblasto, cellula capostipite la quale, attraverso diverse fasi, passa allo stadio di megacariocito e infine a quello di piastrina, unico elemento della serie che in condizioni normali entra in circolo. Nella leucopoiesi, il processo di formazione dei leucociti, si distinguono, in relazione alla morfologia delle cellule, una granulocitopoiesi, una linfocitopoiesi e una monocitopoiesi. La granulocitopoiesi si suddivide, in base al colore delle granulazioni citoplasmatiche, in tre sezioni (neutrofila, eosinofila, basofila) che passano attraverso i medesimi stadi maturativi. La presenza, esclusiva oppure prevalente, di note di immaturità (essenzialmente nucleo nucleolato, citoplasma basofilo con granulazioni aspecifiche, debole positività delle reazioni citochimiche come quelle per i lipidi e per le perossidasi) o di maturità (nucleo anucleolato, citoplasma acidofilo per scomparsa dell'acido ribonucleico, intensa positività delle reazioni citochimiche) permette di suddividere gli elementi in immaturi e maturi, distinzione importante perché la comparsa in circolo dei primi caratterizza i processi leucemici o pseudoleucemici, mentre quella dei secondi, anche se abnorme, non è di per sé segno di emopatia, potendo rappresentare soltanto una vivace risposta midollare a particolari stimoli (infezioni, gravidanza, ecc.). Cellule immature sono il mieloblasto, capostipite della serie, e il promielocito; cellule mature tutte le altre.
La maturazione della serie linfocitaria (linfocitopoiesi) deriva da un unico precursore staminale indifferenziato che, in funzione del microambiente nel quale risiede o migra, segue due diverse vie: formazione dei linfociti B (nel midollo osseo) e dei linfociti T (nel timo). Gli studi di immunologia hanno chiarito come la popolazione linfocitaria matura, circolante nel sangue periferico, sia caratterizzata da varie sottopopolazioni in grado di svolgere specifiche funzioni. Quindi, nel caso dei linfociti, a una sostanziale uniformità morfologica si affianca una specifica diversità funzionale. Per la monocitopoiesi si ritiene che il capostipite si trasformi direttamente nell'elemento maturo osservabile in circolo, o monocito. Questa trasformazione è graduale, ma priva di caratteristiche morfologiche che consentano di stabilire con sicurezza lo stadio di promonocito.
Anatomia
Plasma
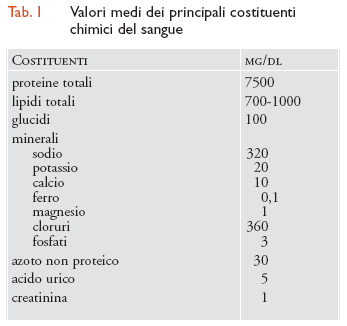
Il plasma è un fluido omogeneo di colore giallo chiaro, costituito per il 90% da acqua e per il 10% da sostanze solide. La sua densità specifica è compresa tra 1,052 e 1,064. I costituenti del plasma sono numerosissimi e rispecchiano parte delle molteplici funzioni che il sangue assolve; in tab. 1 sono riportati i più importanti, tra i quali ricordiamo le proteine, i lipidi, i glucidi o zuccheri, alcuni minerali come il sodio, il potassio, il calcio, ecc. Le proteine sono i costituenti principali del plasma in termini sia quantitativi sia qualitativi. Sono normalmente circa 7500 mg/dl e, a seconda del loro peso molecolare e della loro conformazione, si dividono in albumine e globuline. Le albumine hanno peso molecolare 69.000 e vengono prodotte dal fegato, le globuline hanno peso molecolare maggiore. Le funzioni delle proteine sono numerosissime: mantengono la pressione oncotica opponendosi alla fuoriuscita dai capillari dei liquidi circolanti (soprattutto le albumine), sono vettrici di sostanze o assolvono precise funzioni, per esempio di difesa (anticorpi) o nei processi coagulativi (fattori della coagulazione).
Le immunoglobuline (Ig) o anticorpi sono una classe molto eterogenea di proteine deputate alla difesa dell'organismo; sono sintetizzate dai linfociti in risposta a uno stimolo antigenico estraneo a esso. La concentrazione sierica delle IgG, gli anticorpi che si sviluppano come risposta secondaria agli antigeni e hanno la proprietà di attraversare la placenta proteggendo il feto e il neonato, varia tra 800 e 1600 mg/dl nell'adulto. La concentrazione sierica normale delle IgM, gli anticorpi prodotti per primi nella risposta immune, tanto che la loro presenza è spesso segno di infezione recente, è di 80-300 mg/dl. Le IgA, che si possono dividere in due categorie ‒ quelle circolanti e presenti nelle secrezioni interne (umore acqueo, liquor, ecc.) e quelle presenti nelle secrezioni esterne (saliva, bile, ecc.) ‒, sono capaci di bloccare l'adesione alle mucose di virus e batteri, soprattutto delle vie respiratorie e dell'apparato gastrointestinale, e sono presenti nel siero a una concentrazione di 200-300 mg/dl. Le IgD, immunoglobuline sulle quali poco è noto, sono presenti a basse concentrazioni nel siero (0,3-40 mg/dl). Le IgE, gli anticorpi della risposta immediata, qualche volta anche pericolosa per la vita (per es., nell'anafilassi), sono normalmente contenute a basse concentrazioni nel siero (0,01-0,07 mg/dl) e tipicamente aumentano nei soggetti affetti da asma, febbre da fieno, ecc.
Altre proteine plasmatiche che rivestono un ruolo importante sono i fattori della coagulazione. Nei processi di coagulazione, infatti, queste proteine hanno la capacità di attivarsi con un meccanismo 'a cascata', modificando la propria struttura per arrivare a formare il coagulo. Tappa finale di questa attivazione è la trasformazione del fibrinogeno in fibrina. Il fibrinogeno è una grossa proteina prodotta dal fegato, normalmente presente nel plasma a una concentrazione di 200-400 mg/dl. L'attivazione dei vari fenomeni che portano alla formazione del coagulo può schematicamente avvenire in due modi: attraverso una via detta intrinseca o una via detta estrinseca. La via intrinseca è iniziata dal contatto con superfici estranee, come quelle che si scoprono quando viene leso un vaso arterioso o venoso; la via estrinseca è attivata dalla liberazione di sostanze endogene (fattori tessutali). Una volta innescato il meccanismo di attivazione dei vari fattori della coagulazione, questo porta alla formazione di trombina, un enzima che ha un'altissima affinità per il suo substrato, il fibrinogeno, del quale causa il distacco di due piccole porzioni e che trasforma così in fibrina. Le varie unità di fibrina si uniscono fra di loro polimerizzando e precipitando, venendo così a formare il coagulo.
Un metodo molto usato per lo studio delle proteine plasmatiche è l'elettroforesi. Se una corrente elettrica continua viene fatta passare nel mezzo che le contiene, le proteine migreranno dal polo negativo verso il polo positivo. A seconda del tipo di proteina, la migrazione avverrà più o meno velocemente, distribuendosi nelle frazioni del tracciato elettroforetico. La prima frazione è data dalle albumine, che costituiscono circa il 55-56% delle proteine totali e che nel tracciato sono riconoscibili per la cuspide più alta; seguono poi le α1-globuline (3-3,5%), le α2-globuline (8-8,5%), le β-globuline, che sono normalmente circa il 12-15%, e le γ-globuline. Queste ultime sono rappresentate perlopiù dagli anticorpi e hanno una concentrazione tra il 16 e il 18%.
I lipidi o grassi, presenti in concentrazione variabile tra 700 e 1000 mg/dl, comprendono i trigliceridi, il colesterolo e piccole quantità di acidi grassi liberi. Sono veicolati nel sangue da proteine, a formare complessi lipoproteici. Sottoponendo il plasma a ultracentrifugazione è possibile suddividere questi complessi, a seconda della densità, in: a) chilomicroni, a più bassa densità, composti per il 90% da trigliceridi con l'1% di proteine; b) lipoproteine a bassissima densità (VLDL, Very low density lipoproteins), che trasportano i trigliceridi sintetizzati dal fegato; c) lipoproteine a bassa densità (LDL, Low density lipoproteins), deputate al trasporto di colesterolo; d) lipoproteine ad alta densità (HDL, High density lipoproteins), che trasportano un quarto del colesterolo totale e sono ricche in protidi e fosfolipidi; sembra che il colesterolo contenuto in quest'ultima frazione abbia un ruolo protettivo nei confronti dell'aterosclerosi. Quando queste lipoproteine vengono poste a migrare in un campo elettroforetico, si separano in tre frazioni: le α-lipoproteine, corrispondenti alle HDL, le pre-β-lipoproteine, corrispondenti alle VLDL, e le β-lipoproteine, riconoscibili nelle LDL. I chilomicroni non migrano dalla linea di partenza.
Parte corpuscolata
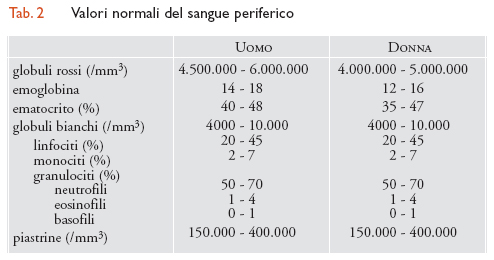
La parte corpuscolata del sangue è costituita da tre tipi cellulari: i globuli rossi (detti anche 'eritrociti' o 'emazie'), i globuli bianchi e le piastrine, i cui valori normali sono indicati in tab. 2. Queste cellule hanno origine nel midollo osseo dove, da una cellula primordiale totipotente (cellula staminale), per successiva differenziazione, originano le cellule progenitrici di ciascuna linea. I globuli rossi sono privi di nucleo, di forma biconcava, del diametro di circa 7-7,5 mm. Il loro numero normale è variabile nell'uomo adulto tra 4.500.000 e 6.000.000/mm3 e nella donna tra i 4.000.000 e i 5.000.000/mm3. I globuli rossi contengono emoglobina, una proteina complessa, deputata al trasporto di ossigeno. Con le normali colorazioni gli eritrociti appaiono come elementi rotondeggianti di colore rosa, con una zona più chiara al centro. La struttura dei globuli rossi, i quali presentano notevoli capacità di deformabilità, è adattissima alle funzioni che essi espletano. Infatti l'alto rapporto superficie/volume facilita il passaggio dei gas e la deformabilità permette alle emazie di passare senza danno nei capillari più piccoli. I globuli rossi, come le altre cellule del sangue, vengono ormai perlopiù contati mediante contaglobuli automatici che forniscono anche i valori relativi ad altri parametri importantissimi: l'emoglobina e l'ematocrito.
I globuli bianchi sono cellule nucleate che si trovano nel sangue in condizioni normali in numero tra 4000 e 10.000/mm3. Si differenziano in granulociti, linfociti e monociti. I granulociti sono così chiamati perché nel citoplasma contengono delle granulazioni; vengono inoltre chiamati 'polimorfonucleati' perché il nucleo si può presentare di forma variabile. A seconda del tipo di granuli che presentano, essi si distinguono in neutrofili, basofili ed eosinofili. I granulociti neutrofili rappresentano la maggior parte dei globuli bianchi, arrivando in condizioni normali a costituire circa il 50-70% di tutti i leucociti. Il nucleo è multilobato e il citoplasma presenta fini granulazioni specifiche: questi granuli contengono numerosi enzimi, quali fosfatasi alcalina, perossidasi, esterasi, ecc.: sono sostanze che intervengono nella funzione primaria dei granulociti neutrofili, quella di difesa. È importante ricordare che queste cellule sono capaci di movimenti attivi: oltre alla quota circolante esiste anche una quota di neutrofili, detta 'marginata', sulle pareti vascolari.
In seguito a uno stimolo infettivo i neutrofili convergono verso il focolaio di infezione per attaccare, fagocitandoli, gli agenti responsabili dell'infezione, che vengono distrutti con l'ausilio degli enzimi contenuti nei granuli citoplasmatici. È quindi comprensibile come, in caso di infezione in atto, si osservino una grande quantità di neutrofili nella sede del processo e, in genere, un aumento della quota circolante. I granulociti eosinofili sono cellule che presentano un nucleo meno segmentato e caratteristici granuli citoplasmatici, che con le normali colorazioni assumono una tonalità rosso-arancio; anche questi granuli contengono enzimi, che però differiscono da quelli dei neutrofili. Gli eosinofili sono inoltre dotati di attività fagocitaria che si esplica in genere nei confronti dei complessi antigene-anticorpo; aumentano in soggetti sensibilizzati per un determinato antigene e che presentano una reazione allergica verso di esso. Sono inoltre coinvolti in maniera determinante nella difesa da infezioni parassitarie. I granulociti basofili, presenti in numero ridottissimo nel sangue, sono cellule costituite da un nucleo bilobato o trilobato e granulazioni citoplasmatiche grossolane di colore viola scuro. I basofili possiedono movimenti lenti, sono in grado di fagocitare, ma non sembra questa la loro principale funzione. In seguito a uno stimolo liberano istamina, un enzima responsabile dei fenomeni allergici, compresi quelli di una certa gravità.
I linfociti sono cellule mononucleate di diametro intorno ai 10 mm, rotondeggianti, con un nucleo ovalare che occupa gran parte della cellula; qualche volta sono di dimensioni maggiori, con piccoli granuli citoplasmatici; nel sangue periferico rappresentano il 20-45% dei leucociti. Sono differenziabili, tramite tecniche particolari, in varie classi che assolvono funzioni diverse. Queste cellule, prodotte nel midollo osseo, migrano verso gli organi dove avvengono le prime modificazioni funzionali. Alcuni linfociti migrano nel timo dove acquistano capacità particolari, come quelle di regolazione dell'immunità e di cellule effettrici dell'immunità cellulare: sono i cosiddetti 'linfociti T'. Dal timo i linfociti T passano negli organi linfatici (linfoghiandole, milza, tessuti linfoidi dell'apparato gastroenterico). I linfociti B, invece, sono così chiamati perché negli Uccelli acquisiscono le loro caratteristiche in un organo detto 'borsa di Fabrizio'; nell'uomo non è ancora chiarito quali possano essere gli organi equivalenti, ma si pensa che essi siano rappresentati dal midollo osseo e da alcuni organi linfatici quali, per esempio, le tonsille e l'appendice. I linfociti B producono gli anticorpi attraverso vari processi maturativi che li conducono alla loro forma terminale, le plasmacellule; hanno la capacità di rispondere a un singolo antigene in maniera specifica e di proliferare in seguito a stimolazione di questo; possono sopravvivere a lungo e sono dotati di memoria; ciò fa sì che possano rispondere prontamente a nuova stimolazione antigenica, ricircolando fra i tessuti e il sangue periferico.
I monociti sono cellule piuttosto grandi, del diametro di 15-20 mm; normalmente si trovano nel sangue periferico, in quantità variabile tra il 2 e il 7% di tutti i globuli bianchi. Il monocita circolante è caratterizzato da un nucleo a forma di ferro di cavallo e da un citoplasma di colore bluastro, talvolta con fini granulazioni. È una cellula di passaggio, in quanto transita nel sangue per poco tempo per poi raggiungere i tessuti, trasformandosi in macrofago, cellula funzionalmente attiva, che varia nella forma e nelle funzioni a seconda del tessuto in cui si trova (polmone, fegato, midollo osseo, ecc.) e viene considerata lo 'spazzino' dell'organismo, in quanto è capace di inglobare qualsiasi detrito, sia organico sia inorganico. I monociti macrofagi svolgono un ruolo importantissimo nei meccanismi di difesa, sia mediante un'attività fagocitaria diretta, sia per la loro capacità di riconoscere l'antigene sia per la possibilità di interferire nella produzione di anticorpi.
Le piastrine sono piccoli elementi privi di nucleo, di forma ellittica o rotondeggiante, che si trovano nel sangue periferico in numero di 150.000-400.000/mm3. Rivestono un ruolo primario nei processi dell'emostasi, mediante la loro complessa struttura. La membrana è dotata di fosfolipidi e proteine necessari per i processi di coagulazione; all'interno si trovano filamenti e microtubuli, che contribuiscono alle modificazioni di forma che precedono l'aggregazione delle piastrine, e numerosi granuli contenenti alcuni fattori della coagulazione, enzimi, calcio, serotonina. Queste sostanze, liberate nel corso di processi di aggregazione, favoriscono quest'ultima scatenando un meccanismo irreversibile che porta alla formazione di un agglomerato di piastrine, detto 'trombo'.
Gruppi sanguigni
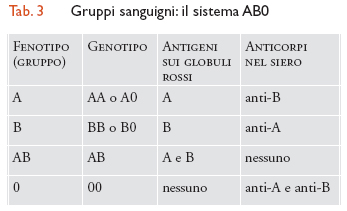
Ogni individuo possiede caratteristiche proprie determinate geneticamente, derivate cioè dall'unione di alcuni caratteri materni con alcuni caratteri paterni. Il gruppo sanguigno è costituito dall'insieme di antigeni presenti sulla superficie dei globuli rossi e deriva dal corredo materno e paterno. Sono noti numerosi antigeni che corrispondono ad altrettanti gruppi sanguigni. I sistemi più conosciuti e quelli di importanza pratica maggiore sono i sistemi AB0 e Rh. Gli antigeni AB0 si ereditano come semplici caratteri e determinano i gruppi sanguigni di tipo A, B, AB e 0 (tab. 3): ogni individuo possiede nel proprio corredo genetico due geni corrispondenti a due di questi antigeni, uno di derivazione paterna e uno di derivazione materna, che si combinano tra loro a formare le varie possibilità. Inoltre nel siero di ogni persona si ritrova l'anticorpo corrispondente all'antigene mancante nelle emazie: per esempio, un individuo di gruppo A avrà nel siero un anticorpo anti-B, uno di gruppo B un anticorpo anti-A. Il sistema Rh è più complesso di quello AB0 e comprende tre coppie di geni strettamente collegati, Cc, Dd, Ee, variamente combinati fra di loro. Sono soggetti Rh positivi tutti coloro che portano sulle emazie l'antigene D e Rh negativi tutti coloro che mancano di questo antigene. I soggetti Rh negativi presentano nel siero un anticorpo anti-D. In caso di trasfusione AB0 o Rh incompatibile, gli anticorpi relativi al sistema AB0 e Rh scatenano reazioni trasfusionali anche molto gravi.
Fisiologia
Le funzioni che vengono assolte dal sangue, sia dalla componente cellulare sia da quella plasmatica, sono numerosissime. Ricordiamo qui le più importanti: trasporto, difesa ed emostasi.
Trasporto. Il sangue trasporta una moltitudine di componenti (sostanze nutritive come le proteine, i lipidi, gli zuccheri, ecc.) che vengono assorbiti nel tratto gastroenterico e trasferiti ai vari organi, principalmente al fegato, dove vengono metabolizzati e da dove si ridistribuiscono a tutto l'organismo. È inoltre specializzato nel trasporto di un elemento indispensabile per la vita: l'ossigeno. Questo si lega all'emoglobina contenuta nei globuli rossi nel passaggio del sangue attraverso i polmoni. Dai polmoni, una volta ossigenato, il sangue attraverso il circolo arterioso si distribuisce a tutti i tessuti cedendo ossigeno e acquistando anidride carbonica. Tale scambio avviene a livello dei più piccoli capillari periferici: da questi, tramite il circolo venoso, il sangue ritorna ai polmoni per rifornirsi nuovamente di ossigeno. È mediante questa serie di processi che tutti i tessuti, e quindi tutte le cellule del corpo, acquistano l'ossigeno necessario ai fenomeni di combustione essenziali per la vita.
Difesa. La difesa dell'organismo è un meccanismo complesso, assolto da diversi tipi di cellule, la maggior parte delle quali sono circolanti nel sangue. È infatti il risultato della cooperazione tra le varie classi linfocitarie, il sistema monocito-macrofagico, i granulociti. I meccanismi di difesa si distinguono in umorali e cellulari. L'immunità umorale è costituita dalla produzione di sostanze attive contro gli organismi estranei (virus, batteri, funghi, ecc.), prime fra queste gli anticorpi prodotti dalle plasmacellule in risposta a un preciso stimolo antigenico. La presenza di un antigene già noto all'organismo determina la rapida proliferazione di un clone linfocitario specifico e la conseguente produzione di anticorpi diretti verso quell'antigene. Gli anticorpi possono neutralizzare direttamente l'antigene oppure facilitare la sua eliminazione da parte di cellule, quali, per esempio, i macrofagi. Nell'immunità cellulare sono le cellule ad agire direttamente contro gli antigeni estranei. I linfociti T rappresentano la risposta specifica verso alcuni antigeni da parte dell'organismo, non solo intervenendo direttamente nei processi di difesa ma anche producendo fattori che attivano il sistema macrofagico. La produzione di anticorpi è, inoltre, il risultato della cooperazione tra linfociti B e T. Infine i granulociti neutrofili hanno un ruolo importante nella difesa contro le infezioni: fanno parte dell'immunità aspecifica e, come già detto, sono capaci di accorrere verso il focolaio infettivo e di fagocitare l'agente causa dell'infezione.
Emostasi. L'emostasi comprende una serie di complesse reazioni biochimiche volte a impedire fuoriuscite del sangue dai vasi. Quando si crea una lesione in una vena o in un'arteria, intervengono tre tipi di processi: a) un processo immediato, che consiste in una rapida costrizione del vaso leso e nell'adesione delle piastrine alla lesione; b) un processo intermedio, in cui le piastrine si modificano di forma e si aggregano irreversibilmente a costituire il trombo piastrinico; contemporaneamente vengono attivati quei processi di coagulazione che portano alla formazione del coagulo di fibrina; c) i processi tardivi, che consistono nella lisi del coagulo da parte di alcune proteine plasmatiche addette ai fenomeni di fibrinolisi e nella riparazione del vaso. È comprensibile come questa serie di meccanismi sia molto efficiente per lesioni di vene o arterie di piccole dimensioni o comunque per lesioni di non grave entità. Nelle rotture di grossi vasi, sia spontanee sia traumatiche, i meccanismi fisiologici dell'emostasi non sono sufficienti e l'emorragia deve essere arrestata chirurgicamente.
Patologia
Le malattie del sangue, o emopatie, comprendono una serie di affezioni congenite o acquisite che riguardano le cellule del sangue (globuli rossi, globuli bianchi e piastrine) o i loro precursori midollari. Vengono compresi nelle emopatie anche i disordini della coagulazione, di tipo sia emorragico sia trombotico. I globuli rossi possono essere in numero inferiore alla norma (anemia) o superiore (poliglobulie).
Poliglobulie
Le poliglobulie sono caratterizzate da un aumento della massa eritrocitaria circolante. Possono essere secondarie a condizioni di diminuzione dell'ossigenazione tessutale, a malattie neoplastiche o renali, oppure primitive. La sintomatologia è legata all'aumento del volume e della viscosità del sangue ed è contraddistinta da cefalea, vertigini, parestesie, rischio di trombosi arteriose o venose, incluso l'infarto del miocardio; caratteristico è il colore rosso acceso della cute e delle mucose. Le cause più frequenti di poliglobulia secondaria sono le malattie dell'apparato respiratorio (per es., insufficienza respiratoria cronica, alcune cardiopatie, permanenza prolungata a basse pressioni di ossigeno, come si può verificare in alta montagna). Inoltre, una poliglobulia può accompagnare alcune malattie renali per un aumento della produzione, che avviene in questa sede, di eritropoietina, l'ormone che stimola la produzione dei globuli rossi. Esiste anche una forma di poliglobulia primitiva, la 'policitemia vera' o 'malattia di Vaquez', in cui l'incremento dei globuli rossi può accompagnarsi a quello di globuli bianchi e piastrine. Si tratta di una malattia del midollo osseo, nel quale i precursori delle cellule circolanti proliferano senza causa apparente. Quando la massa eritrocitaria aumenta in maniera notevole si impone un trattamento che può essere effettuato con periodici salassi o impiego di farmaci che riducono la proliferazione cellulare.
Leucemie acute
Delle malattie dei globuli bianchi quelle più note e più studiate sono senz'altro le leucemie. La leucemia è una malattia sistemica dovuta alla proliferazione midollare incontrollata di un tipo cellulare (clone) che ha subito un blocco maturativo e che, oltre a ritrovarsi nel sangue periferico, può invadere praticamente tutti gli organi. È possibile classificare le leucemie in diversi modi, in riferimento alle loro caratteristiche biologiche, immunologiche o cliniche. Una classificazione grossolana le divide in due grandi gruppi, molto differenti tra loro per andamento clinico e prognosi: forme acute e forme croniche.
Le leucemie acute sono di due tipi: linfoidi e mieloidi. Le prime, che dal punto di vista immunologico possono essere distinte in B e T e in diverse fasi maturative, sono più frequenti nei bambini (circa l'80% dei casi), mentre le seconde prevalgono negli adulti e negli anziani (più del 50% dei casi in pazienti di età superiore ai 60 anni). Per le leucemie mieloidi acute l'incidenza è di circa 2 nuovi casi l'anno su 100.000 soggetti; per le linfoidi acute l'incidenza è simile, il che corrisponde a circa 600 nuovi casi l'anno in Italia nei bambini di età inferiore ai 15 anni e a circa 200 casi per i soggetti di età superiore ai 15 anni. L'eziologia delle leucemie acute è tuttora sconosciuta: è stato comunque osservato l'aumento dell'incidenza nei soggetti esposti a radiazioni ionizzanti, o a seguito di incidenti nucleari o per motivi professionali, e nei pazienti sottoposti a terapie con farmaci antineoplastici, soprattutto gli alchilanti. Anche fattori ambientali, in particolare il benzene e derivati, possono aumentare l'incidenza delle leucemie. Deve essere inoltre tenuto presente che alcune condizioni genetiche, come per esempio la sindrome di Down, si accompagnano a una maggiore incidenza di leucemie.
Nel processo della leucemogenesi sono state acquisite nuove conoscenze, anche se molti aspetti sono ancora da chiarire. A livello cromosomico sono state identificate numerose alterazioni, sia nelle forme linfoidi sia in quelle mieloidi; è importante sottolineare che alcune di queste alterazioni sono specifiche di varietà come la traslocazione 15:17 nella leucemia mieloide promielocitica, o la traslocazione 8:14 in quella linfoide a immunofenotipo B maturo, e che il loro riconoscimento può essere molto utile sotto il profilo diagnostico e terapeutico. Numerose alterazioni cromosomiche interessano i siti dove sono stati identificati alcuni oncogeni che sono senz'altro coinvolti nel processo di leucemogenesi. Il coinvolgimento di geni specifici per determinate leucemie (per es., nella leucemia promielocitica, una varietà di leucemia mieloide acuta, è coinvolto il gene del recettore dell'acido retinoico) può consentire nuove strategie terapeutiche mirate nei confronti del clone neoplastico, con possibilità di straordinari risultati come quelli che si ottengono con l'impiego dell'acido retinoico (ATRA, All-trans-retinoic acid) nella leucemia promielocitica.
Le leucemie acute possono presentarsi con una sintomatologia molto grave, dovuta sia a insufficiente produzione di globuli rossi, piastrine e granulociti da parte del midollo, con la comparsa rispettivamente di anemia, sindrome emorragica e infezioni, sia a localizzazione della malattia leucemica, con possibile interessamento di tutti gli organi e apparati (linfonodi, milza, fegato, sistema nervoso centrale, cute, ecc.); sia infine alla liberazione di sostanze tossiche da parte delle cellule leucemiche, con febbre più o meno elevata e astenia profonda. La sintomatologia può essere molto grave ‒ tale da richiedere l'immediato ricovero del malato per la terapia del caso ‒ o di lievissima entità, o addirittura assente. Sempre più spesso la diagnosi viene effettuata in pazienti del tutto asintomatici, sulla base dei risultati degli esami di laboratorio, soprattutto quello emocromocitometrico, eseguiti in controlli di routine. In corso di leucemia acuta nel sangue periferico, oltre alla presenza di anemia, piastrinopenia o granulocitopenia più o meno grave, vi può essere un aumento del numero dei globuli bianchi, fino a valori anche molto elevati (superiori a 100.000/mm3), per la comparsa in circolo di cellule leucemiche. Per l'accertamento diagnostico, che nei casi con presenza di elementi leucemici nel sangue periferico è molto facile, si rende sempre necessaria la valutazione del midollo osseo, che viene effettuata mediante aspirato midollare; esso può essere eseguito a livello del manubrio sternale e/o delle ossa iliache.
Diagnosticata e caratterizzata la varietà di leucemia acuta, deve essere stabilita la strategia terapeutica, che, nella fase di attacco, deve portare alla scomparsa della maggioranza delle cellule leucemiche presenti in tutto l'organismo, compreso il midollo osseo. Se il risultato è ottimale si ottiene la cosiddetta 'remissione completa', che consiste nella scomparsa di tutti i sintomi clinici soggettivi e obiettivi legati alla malattia e nella normalizzazione dei valori dei globuli rossi, dei globuli bianchi, delle piastrine e del midollo osseo. Si parla di remissione completa e non di guarigione, in quanto il risultato ottenuto corrisponde alla spiccata riduzione (oltre il 99%) della massa leucemica, ma non alla sua scomparsa; infatti, mentre all'esame microscopico non sono più riconoscibili le cellule leucemiche, con l'impiego di tecniche più sensibili (fenotipo immunologico, citogenetica, biologia molecolare) è sempre possibile dimostrarne la persistenza come malattia minima residua.
Durante la terapia di attacco, per la mancata attività selettiva dei farmaci nei confronti delle cellule leucemiche, si ha un aggravamento dell'anemia, della piastrinopenia e soprattutto della granulocitopenia dovuto all'ulteriore riduzione a livello midollare dei precursori ematopoietici normali (aplasia midollare terapeutica). È in questa fase che, soprattutto nelle leucemie mieloidi acute, si devono attuare tutti i presidi diretti a prevenire e a combattere le complicanze del trattamento antileucemico, rappresentate essenzialmente da emorragie e da infezioni gravi, talvolta mortali. La probabilità di conseguire la remissione completa dipende dalla varietà di leucemia e dall'età del paziente: si ottiene, infatti, in oltre il 95% dei bambini e in più dell'80% degli adulti con leucemia linfatica acuta, mentre nelle leucemie mieloidi acute si ha in circa l'80% dei bambini e nel 65-70% degli adulti. Nei pazienti di età superiore ai 60-65 anni la probabilità di remissione è inferiore ‒ circa il 50% ‒ in entrambe le varietà di leucemia acuta. Molto recentemente nella leucemia promielocitica la prognosi è radicalmente cambiata rispetto a qualche anno fa, quando questa era ritenuta la forma più grave di tutte le leucemie acute. In tale variante di leucemia, indipendentemente dall'età, da quando viene impiegato in associazione alla chemioterapia l'acido retinoico si ottiene con la terapia di attacco una percentuale di remissione completa molto elevata (circa il 90% dei casi).
Dopo la remissione, tutti i pazienti affetti da leucemia acuta devono essere curati con la cosiddetta 'terapia postremissionale'; tale trattamento ‒ come quello d'attacco ‒ utilizza più farmaci (polichemioterapia) dotati di meccanismi d'azione diversi. La polichemioterapia è diversa nelle forme linfoidi rispetto a quelle mieloidi: più protratta (per almeno due anni) nelle leucemie linfoidi, più breve (sei mesi ca.) e più intensa nelle leucemie mieloidi. Per quanto riguarda le forme linfoidi, va tenuto presente che, per la frequente comparsa di localizzazioni a livello del sistema nervoso (meningosi leucemica), deve essere effettuato un trattamento specifico per la loro prevenzione, trattamento che prevede la somministrazione di farmaci per via intratecale, associata a radioterapia craniale o ad alte dosi di farmaci per via endovenosa che oltrepassano la barriera emato-encefalica. Circa il 60% dei bambini con leucemia linfoide acuta e il 25-30% degli adulti ha la probabilità di diventare un lungo sopravvivente senza recidiva, e quindi di guarire. Nelle leucemie mieloidi acute la probabilità di guarigione è minore (ca. il 35-40% dei bambini e il 20-25% degli adulti). Fa sempre eccezione la leucemia promielocitica, in cui, con una terapia postremissionale intensa e di breve durata, la probabilità di guarigione è superiore al 50% dei casi sia nei bambini sia negli adulti.
Nonostante la terapia postremissionale, nella maggior parte dei pazienti ‒ con percentuali diverse nelle varie forme di leucemia acuta e nelle diverse età ‒ si verifica la ricomparsa della malattia, cioè la 'recidiva leucemica'. La prima recidiva deve essere curata in modo intensivo allo scopo di ottenere una nuova remissione che, se persistente, permetterà ancora al paziente di guarire. In questa fase di malattia più avanzata trova indicazione elettiva, se le condizioni generali del paziente lo permettono, un trattamento intensivo ad alte dosi, seguito da infusione di cellule staminali (procedura trapiantologica).
Leucemie croniche
Le leucemie croniche differiscono sostanzialmente dalle acute per modalità di presentazione della malattia, andamento, terapia e prognosi. In generale l'esordio è piuttosto lento, insidioso, tanto che spesso la diagnosi viene fatta occasionalmente in seguito a esami di controllo. La sintomatologia può essere caratterizzata da astenia, febbricola o infezioni recidivanti, dimagrimento, sensazione di ingombro addominale dovuta alla frequente presenza di una milza notevolmente aumentata di volume, comparsa di tumefazioni linfonodali. Possono inoltre coesistere i sintomi legati all'anemia e alla piastrinopenia, quando queste siano presenti. Le cellule leucemiche sono ad abito più maturo che nelle leucemie acute e nella stragrande maggioranza l'età di insorgenza è quella adulta avanzata. Le forme più comuni sono rappresentate dalla leucemia mieloide cronica, dalla leucemia linfatica cronica e dalla leucemia a tricoleucociti.
Leucemia mieloide cronica. - Questa forma, rara nei bambini e più frequente nell'adulto e nell'anziano, con un'età media di circa 50 anni, ha un'incidenza di un nuovo caso ogni 100.000 soggetti all'anno, per cui in Italia vi sono circa 600 nuovi casi all'anno. È dovuta alla trasformazione neoplastica della cellula staminale ed è caratterizzata dalla presenza a livello cromosomico di una specifica alterazione, presente in oltre il 95% dei casi, il cromosoma Philadelphia (Ph1). Si tratta della prima specifica alterazione cromosomica dimostrata in una malattia neoplastica, consistente in una traslocazione reciproca tra il cromosoma 9 e il cromosoma 22, con la formazione a livello molecolare di un gene ibrido, dovuto alla fusione dell'oncogene ABL localizzato nel cromosoma 9, con la regione BCR, localizzata nelle braccia lunghe del cromosoma 22.
La diagnosi della malattia in circa il 30% dei casi è casuale; in pazienti del tutto asintomatici viene rilevato un aumento del numero dei globuli bianchi, talvolta anche superiore ai 100.000/mm3. L'alterazione numerica dei globuli bianchi si accompagna a un aumento percentuale e assoluto degli elementi granulocitari, con comparsa in circolo di elementi nelle diverse fasi di maturazione. Dal punto di vista dell'obiettività clinica si può rilevare un aumento di volume del fegato e della milza, talvolta molto spiccato. Fondamentale per la diagnosi, inoltre, è lo studio del midollo osseo, non tanto per la netta prevalenza degli elementi della serie granulocitaria a livello citomorfologico, quanto per la dimostrazione del cromosoma Ph1, anomalia citogenetica caratteristica. Fino a pochi anni fa, prima dell'avvento di terapie innovative quali il trapianto di cellule staminali e le terapie mirate a livello molecolare, il decorso della malattia era in genere cronico, con una sopravvivenza media di 4-5 anni. Quasi sempre la morte avviene per la trasformazione della malattia in una forma acuta (crisi blastica), caratterizzata da una sintomatologia molto più grave e da referti di laboratorio e midollari sovrapponibili a quelli delle leucemie acute.
Il trattamento di elezione della leucemia mieloide cronica è basato sull'impiego del farmaco imatinib e, in casi selezionati, sul trapianto di cellule staminali emopoietiche o di midollo allogenico. Le indicazioni all'impiego dell'interferone si sono notevolmente ridotte con l'avvento dell'imatinib. Il farmaco imatinib è un inibitore di uno specifico enzima, chiamato tirosina-chinasi, il cui ruolo è quello di trasferire fosfati alle proteine che danno segnali di crescita alle cellule umane; questo enzima risulta particolamente sregolato e abbondante in alcuni tumori umani. Il più studiato tra questi enzimi si chiama 'tirosina-chinasi BCR-ABL', una proteina prodotta dalla fusione di due geni, BCR e ABL. Negli individui normali questi geni si trovano su due cromosomi differenti (22 e 9), ma nei soggetti affetti da leucemia mieloide cronica una mutazione genica fa sì che i due geni si ritrovino insieme sul cromosoma Ph1, una versione troncata del cromosoma 22. L'imatinib inibisce la produzione della tirosina-chinasi BCR-ABL, bloccando la crescita incontrollata delle cellule leucemiche positive per il cromosoma Ph1.
Da un punto di vista clinico, questo si traduce nella disponibilità di un farmaco, somministrabile per via orale, che ha ottenuto risposte ematologiche complete (cioè scomparsa di tutte le cellule leucemiche) nel 95% dei pazienti con leucemia mieloide cronica e risposte citogenetiche complete (cioè scomparsa di tutti i cromosomi Ph1) in più del 50% dei casi. Queste risposte sono spesso di durata molto prolungata e, fattore importante, si ottengono con sintomi collaterali molto ridotti (soprattutto nausea ed edema), che soltanto in una minima percentuale dei pazienti (meno del 3%) causano l'interruzione della terapia. I risultati ottenuti dall'imatinib sono molto meno favorevoli nella fase accelerata e blastica della leucemia mieloide cronica, e ciò indica che mutazioni ulteriori del gene BCR-ABL in cellule che proliferano molto rapidamente possono dare origine a fenomeni di resistenza al farmaco. In questa fase di malattia, l'impiego combinato dell'imatinib con un trattamento polichemioterapico simile a quello impiegato nelle leucemie acute mira a ottenere il ripristino della fase cronica, che consente di eseguire successivamente il trapianto di cellule staminali emopoietiche o di midollo.
Leucemia linfatica cronica (LLC). - È la più frequente emopatia neoplastica dell'adulto nel mondo occidentale, con un'incidenza di 243 nuovi casi l'anno per 100.000 individui, per cui in Italia vi sono circa 1500 nuovi casi all'anno. È rara al di sotto dei 40 anni; la massima incidenza si ha nei soggetti ultrasessantenni. La diagnosi viene effettuata in più del 50% dei casi nel corso di esami di laboratorio routinari. Il rilievo di un aumento del numero dei globuli bianchi dovuto a una linfocitosi assoluta in persone del tutto asintomatiche porta ad approfondire l'accertamento diagnostico. Nella leucemia linfatica cronica, come nelle leucemie acute, tutti gli organi e apparati possono essere coinvolti dal processo leucemico, anche se in genere questo aspetto si verifica nelle fasi più avanzate. Gli esami di laboratorio dimostrano costantemente una linfocitosi assoluta, che può superare 100.000 linfociti/mm3. Nelle forme più gravi può essere presente un'insufficienza midollare con anemia e/o piastrinopenia; in questi casi a livello del midollo osseo è evidente una sostituzione del normale tessuto emopoietico da parte dei linfociti leucemici, che devono essere studiati dal punto di vista immunologico per dimostrarne il carattere monoclonale.
Il decorso è spesso lento, con una durata media di sopravvivenza che supera comunque i cinque anni. Vi sono pazienti che per l'assenza di sintomi non richiedono alcun trattamento anche per anni e muoiono per cause diverse dalla malattia; altri presentano un andamento più aggressivo e devono essere trattati precocemente, talvolta già all'esordio. La maggioranza dei pazienti, soprattutto se di età superiore ai 60-65 anni, viene curata con terapie molto blande, effettuabili a domicilio, praticamente prive di tossicità, che consentono di controllare il processo patologico per molti anni con una buona qualità di vita. Più difficile è la scelta terapeutica nei pazienti giovani, con forme di leucemia linfatica cronica ad andamento più aggressivo, per cercare di modificare il decorso irreversibilmente fatale della malattia. In questi pazienti vengono utilizzati farmaci nuovi, quali gli analoghi delle purine, che hanno una forte tossicità, con possibile comparsa di complicanze emorragiche e soprattutto infettive, anche mortali, analoghe a quelle che si osservano durante la terapia aggressiva che viene impiegata nelle leucemie acute. Nella leucemia linfatica cronica vengono attualmente utilizzate con risultati estremamente interessanti, pur se del tutto preliminari, le terapie intensive includenti il trapianto autologo o allogenico di cellule staminali emopoietiche.
Leucemia a tricoleucociti (hairy cell leukemia). - È una malattia in cui il clone leucemico è costituito da linfociti con caratteristiche sfrangiature citoplasmatiche, somiglianti a capelli. All'esordio i pazienti presentano anemia, piastrinopenia e leucopenia, con aumento di volume, anche notevole, del fegato e della milza. La prognosi di questa patologia è notevolmente migliorata con l'impiego nella terapia dell'interferone e di farmaci di sintesi, i quali permettono lunghe sopravvivenze.
Da ciò che si è detto è comprensibile come un aumento del numero dei globuli bianchi non sia sinonimo di leucemia e viceversa. Una leucocitosi può essere la norma in caso di infezione o anche in corso di eventi fisiologici quali, per esempio, la gravidanza. Può presentare qualche difficoltà di diagnosi differenziale, rispetto a una forma leucemica vera e propria, la comparsa di una cosiddetta 'reazione leucemoide', del tutto benigna e autolimitantesi nel tempo. La reazione leucemoide è un evento reattivo contraddistinto dalla presenza in circolo di un elevato numero di leucociti (anche 30-50.000/mm3), talvolta con forme immature, tanto da poter essere scambiato per un quadro tipico di leucemia mieloide cronica. La patologia di base (un'infezione oppure una neoplasia) e l'assenza dell'aumento della milza e del cromosoma Ph1 orienteranno verso la diagnosi corretta.
Linfomi
Nell'ambito della patologia neoplastica a carico delle cellule di derivazione linfoide, un capitolo importante è rappresentato dai linfomi, che presentano un'eterogeneità clinica e istologica notevoli e hanno prevalente carattere di malignità. Secondo una classificazione accettata a livello internazionale, vengono distinti in linfoma di Hodgkin e linfomi non-Hodgkin. Questi ultimi non rappresentano una singola entità clinica e a loro volta vengono classificati in base a criteri particolari (malignità istologica, caratteristiche microscopiche, proprietà immunologiche). Rispetto alle leucemie, malattie sistemiche nelle quali è presente un interessamento del sistema linfatico, i linfomi non determinano abitualmente la comparsa nel sangue di particolari alterazioni della morfologia cellulare, quanto piuttosto infiltrano i linfonodi superficiali e profondi, la milza e in alcuni casi organi quali fegato, polmoni, tubo digerente, cervello, ecc. Esistono peraltro taluni rapporti fra leucemie e linfomi, in quanto si possono talora identificare forme di transizione e forme simili, sia dal punto di vista clinico sia per quanto riguarda la derivazione cellulare.
Linfoma di Hodgkin. - Questo linfoma, detto anche 'linfogranulomatosi maligna', è una forma di neoplasia del sistema linfatico. Colpisce a tutte le età, ma soprattutto fra i 20 e i 40 anni e dopo i 70 anni. Predilige il sesso maschile e nella maggior parte dei casi ha un andamento cronico, anche fino a 10 anni. L'Epstein-Barr virus, il virus della sindrome da immunodeficienza acquisita (HIV) e l'esposizione alle sostanze tossiche sono indicati come fattori di predisposizione a tale malattia. Essa si caratterizza per la presenza nel contesto della massa linfonodale di alcuni elementi francamente neoplastici, peculiari di questa forma (cellule di Reed-Sternberg e cellule di Hodgkin) accanto a normali cellule di verosimile natura reattiva (polimorfonucleati, eosinofili, monociti macrofagi, ecc.).
Secondo la classificazione REAL (Revised european-american classification for lymphoid neoplasms), in seguito modificata dalla WHO (World Health Organization), formulata nel 2000, si distinguono due tipi di Hodgkin: il classico ‒ a sua volta distinto in quattro entità istopatologiche, varietà sclero-nodulare, a cellularità mista, a deplezione linfocitaria, ricco di linfociti ‒ e il predominante nodulare. Quest'ultimo si differenzia per l'espressione di alcuni antigeni monoclonali diversi rispetto alla forma classica (CD20+, CD30−), per lo stadio iniziale generalmente più favorevole, per la predominanza del sesso maschile in età giovanile e per localizzazione primaria a livello dei linfonodi cervicali e inguinali. Le varietà che contraddistinguono la forma classica differiscono tra di loro per il tipo di cellule presenti nella componente reattiva, mentre sono sempre riconoscibili le cellule di Reed-Sternberg e quelle di Hodgkin, in proporzioni variabili a seconda dell'istotipo. Tali cellule si identificano immunologicamente per la presenza sulla loro superficie dell'antigene CD30. Questa classificazione è basata sugli aspetti istopatologici del linfonodo colpito e sul fenotipo immunologico della cellula alterata.
La malattia si manifesta generalmente con tumefazione di un gruppo di linfoghiandole, la quale tende a regredire spontaneamente per essere seguita, a distanza di tempo variabile, dalla tumefazione di altri gruppi linfonodali. La sintomatologia comprende febbre (ondulante o ricorrente), prurito, aumento di volume della milza e talora anche del fegato, aumento del numero dei globuli bianchi, soprattutto eosinofili. Nel decorso della patologia remissioni spontanee si alternano a periodi di recrudescenza. La diagnosi si basa sulla sintomatologia e sui referti bioptici. Per quanto concerne la terapia, chemio- e radioterapica, è importante considerare una divisione in quattro stadi in base al numero e alla sede dei linfonodi colpiti e alla presenza di interessamento extralinfonodale. L'origine di questa neoplasia è linfoide e un'attenta stadiazione del paziente alla diagnosi e la successiva scelta terapeutica permettono la guarigione nel 70-80% dei casi.
Linfomi non-Hodgkin. - Costituiscono un gruppo eterogeneo di neoplasie che hanno origine dai linfociti B e T e sono state distinte dal linfoma di Hodgkin per le caratteristiche istologiche e per il diverso comportamento clinico-biologico. Da un punto di vista clinico è presente un coinvolgimento più o meno massiccio dell'apparato linfonodale, della milza, del fegato e di strutture extralinfatiche, con un andamento clinico assai differente tra le varie forme di linfoma. Questa eterogeneità dipende dai vari aspetti istologici, dalla diversa sede di localizzazione, dall'età del paziente e dalla tendenza del linfoma a rimanere localizzato o a disseminarsi nell'organismo. La diagnosi va confermata sull'esame istologico della biopsia linfonodale. Una caratterizzazione istologica e immunofenotipica sul materiale prelevato o su materiale fresco permette di valutare il tipo e la natura del linfoma, e di conseguenza di effettuare una scelta terapeutica.
Una valutazione strumentale, che prevede l'uso di una tomografia assiale computerizzata (TAC) 'total body' e la tomografia a emissione di positroni (PET), permette di eseguire una corretta stadiazione del paziente. La biopsia del midollo osseo conclude la stadiazione ed è necessaria per rilevare l'eventuale infiltrazione del midollo da parte del linfoma. Un esame del liquido cefalorachidiano si effettua se il tipo istologico si rivela aggressivo e se la localizzazione è a livello orofaringeo o del sistema nervoso centrale. Il livello della latticodeidrogenasi (LDH) e la sierologia per HIV sono sempre richiesti. I quadri istopatologici sono molto vari e costituiscono la base dei criteri di classificazione, la più recente delle quali è la REAL /WHO. Questo sistema tiene conto dell'istologia, dell'immunologia e delle alterazioni citogenetiche, quali le traslocazioni cromosomiche, di ogni tipo di linfoma.
Il linfoma a piccoli linfociti/LLC rappresenta la variante linfoma della leucemia linfatica cronica; costituisce il 4-5% di tutti i linfomi, che sono molto eterogenei nella presentazione clinica e nella possibilità di risposta alle diverse terapie. Può coinvolgere i linfonodi o le sedi extranodali (milza, midollo) e l'infiltrazione periferica deve essere minima, altrimenti la diagnosi è di sola LLC. Il linfoma follicolare rappresenta il 20-25% di tutti i linfomi, spesso diffusi e costituiti da cellule dette 'centrociti'. Il grado di questo linfoma si basa sulla proporzione di cellule di piccola e di grossa taglia (centroblasti). Le cellule esprimono di solito il fenotipo dei linfociti B. È presente la traslocazione dei cromosomi 14 e 18 che causa un riarrangiamento detto 'bcl-2', rilevabile nel 70-80% dei pazienti. La localizzazione è generalmente diffusa già dall'esordio con infiltrazione midollare. La malattia può evolvere in gradi di aggressività maggiore.
Il linfoma mantellare costituisce il 5-8% di tutti i linfomi; è predominante nel sesso maschile dopo i 50 anni, con localizzazioni già diffuse alla diagnosi e invasione midollare, periferica e gastrointestinale. È generalmente non aggressivo all'esordio, ma successivamente può evolvere in tal senso nei casi di recidiva. Si associa alla traslocazione dei cromosomi 11 e 14 e al riarrangiamento definito 'bcl-1' che attiva una proteina del ciclo regolatorio cellulare. I linfomi della zona marginale comprendono quei linfomi identificati come associati al tessuto linfoide delle mucose, detti anche MALT (Mucosa associated lymphoid tissue), linfomi splenici e linfomi nodali a cellule B monocitoidi. Essi sono estremamente eterogenei nel comportamento, e possono evolvere in alto grado alla recidiva.
I linfomi ad alto grado B diffusi a grosse cellule comprendono linfomi differenti per tipo istologico, morfologia cellulare e presentazione clinica. È tipica la presentazione nel mediastino, accompagnata nelle giovani donne da elevata fibrosi. Il linfoma di Burkitt è molto comune nei bambini e si può presentare come tumore addominale, ovarico, toracico o testicolare. È altamente proliferativo. I linfomi linfoblastici possono essere di forma B o di forma T. Si presentano generalmente già con quadri leucemici. La forma T si localizza più facilmente a livello del mediastino. I linfomi cutanei T comprendono la micosi fungoide e la sindrome di Sezary. Si presentano come infiltrazione del derma sotto forma di noduli o placche o sotto forma di eritrodermia diffusa. L'infiltrazione dei linfonodi e degli organi ha andamento lento, mentre nella Sezary si ha un quadro leucemico nel sangue periferico. I linfomi T periferici, il 10-15% di tutti i linfomi, sono più diffusi in alcuni Paesi come il Giappone. La localizzazione iniziale è nodale e sistematica e i sintomi, quali febbre, sudorazione e calo ponderale, sono quasi sempre evidenti. Il linfoma anaplastico T si può presentare in due forme, sistemica o esclusivamente cutanea; quest'ultima ha andamento variabile, in genere benigno con possibilità di regressione spontanea.
I vari sottotipi si dividono in quattro categorie: cronici, nodali o extranodali con presentazione indolente o aggressiva, linfomi/leucemie acute. Nei diversi sottotipi, quando si ha una recidiva si possono avere delle caratteristiche di aggressività maggiore, anche per un cambiamento delle caratteristiche istologiche. Insieme al tipo istologico sono stati elaborati diversi sistemi che valutano la prognosi del paziente: il più usato è l'IPI (International prognostic index) che si basa sull'età del paziente, lo stadio del linfoma, le condizioni generali del paziente, il valore dell'LDH sierico e il numero di sedi extranodali. È possibile così identificare quattro classi di rischio, che si correlano con la risposta al trattamento e con la sopravvivenza. La terapia viene impostata sulla base del tipo istologico secondo la classificazione REAL dei linfomi e prevede l'impiego di diverse associazioni di farmaci antiblastici, anticorpi monoclonali (anti-CD20) e radioterapia locale. L'uso di chemioterapie ad alte dosi associate a trapianto di cellule staminali viene riservato ai pazienti con linfomi aggressivi o in recidiva.
Mielomi
Il mieloma multiplo è un'altra patologia neoplastica dovuta all'accumulo di cellule di derivazione linfoide immunologicamente differenziate, le plasmacellule. Queste hanno la capacità di sintetizzare una proteina, riconoscibile come immunoglobulina o parte di essa, che ha caratteristiche di omogeneità (è sempre uguale), in quanto deriva da un unico clone di cellule alterate. Questa proteina si può ritrovare nel siero e/o nelle urine. Le cellule del mieloma hanno la particolarità di distruggere la matrice dell'osso creando delle fratture patologiche. È una patologia in aumento, più frequente nel sesso maschile in età avanzata (età media di 60 anni). Tra i vari fattori che possono essere predisponenti per tale malattia si ricordano possibili fattori ereditari (sono noti diversi casi di mieloma familiare), presenza di anomalie cromosomiche, riscontrate nel 40-50% dei casi all'esordio, con possibile attivazione di alcuni oncogeni che possono essere coinvolti nella genesi della malattia.
La forma classica del mieloma si manifesta come malattia ossea: il dolore osseo è caratteristico di questa malattia e può essere il sintomo di esordio. Colpisce prevalentemente la colonna vertebrale, il bacino e le coste, ma qualsiasi osso può essere interessato. Il riassorbimento può determinare un quadro diffuso di osteoporosi: questa diviene responsabile dei crolli vertebrali, con possibili conseguenze neurologiche legate allo schiacciamento del midollo spinale e delle fratture patologiche a livello delle ossa lunghe. In alcuni casi vi può essere uno stato di ipercalcemia legato all'importante riassorbimento di osso con liberazione di eccessive quantità di calcio. Si può invece soffrire di una malattia prevalentemente renale, legata alla produzione della proteina anomala e alla conseguente eliminazione urinaria di questa. Il mieloma può causare insufficienza renale, dovuta principalmente a un danno tubulare per la precipitazione di frammenti di immunoglobuline. Infezioni e anemia possono essere legate all'infiltrazione midollare; le infezioni sono anche dovute al disturbo immunitario che la produzione anomala della immunoglobulina monoclonale crea e a un deficit delle funzioni degli altri globuli bianchi. L'anemia è spesso causata anche da un'infiltrazione del rene con una minore produzione di eritropoietina. Un'aumentata produzione di immunoglobulina può essere responsabile anche di una sindrome di iperviscosità con disturbi coagulativi, visivi e neurologici.
Quadri alternativi alla forma classica di mieloma possono essere: a) mieloma micromolecolare, nel quale le plasmacellule producono solo parti di immunoglobuline dette 'catene leggere' (sono le forme con frequente insufficienza renale); b) non secernente, in cui non vi è produzione di una paraproteina; c) plasmocitoma solitario, localizzato esclusivamente in un osso o a livello extramidollare; d) leucemia plasmacellulare, con plasmacellule presenti in numero elevato anche nel sangue periferico; e) indolente, asintomatico e con assenza di lesioni ossee.
La diagnosi si effettua mediante l'elettroforesi delle proteine plasmatiche che dimostra la produzione della componente monoclonale anomala, confermata dall'immunofissazione, che consente di tipizzare la composizione di questa proteina anomala. L'esame morfologico del midollo osseo deve sempre dimostrare un'infiltrazione da parte delle plasmacellule superiore al 30% delle cellule midollari. L'esecuzione di radiografie e TAC dello scheletro è utile per riconoscere la presenza di lesioni ossee. La valutazione della componente urinaria della paraproteina (proteinuria di Bence-Jones) conclude il quadro diagnostico. Alcune alterazioni citogenetiche appaiono ricorrenti, come le alterazioni dei cromosomi 13 e 14, che sembrano avere un impatto prognostico sulla sopravvivenza dei pazienti. La cura consiste nella possibile somministrazione sequenziale di chemioterapia convenzionale, alla quale risponde circa il 60-80% dei pazienti. Negli anziani la chemioterapia tradizionale è basata su uno schema che prevede la combinazione di un farmaco chiamato melphalan (o la ciclofosfamide) insieme al cortisone.
Se il paziente è giovane si usano generalmente protocolli di chemioterapia a più farmaci seguiti da un trapianto allogenico di midollo o cellule staminali emopoietiche, se esiste la possibilità di un donatore familiare compatibile o, in alternativa, autologo. Ottenuta una risposta si possono seguire due vie: un periodo di osservazione con l'astensione dalla terapia o una terapia di mantenimento. È risultato efficace in questo senso l'interferone α ricombinante. Tra i nuovi farmaci attivi nel trattamento del mieloma multiplo è da menzionare la talidomide, che agisce in più direzioni: inibendo la crescita e la sopravvivenza delle cellule mielomatose; modulando e impendendo l'adesione delle cellule del mieloma sullo stroma midollare; interferendo direttamente con il DNA; stimolando il compartimento immunitario T e NK (Natural killer) ha inoltre un effetto antiangiogenetico diretto. Questo farmaco, usato in associazione al desametasone o ad altri farmaci antiblastici, è in grado di indurre un'elevata percentuale di risposte nei pazienti anziani.
Altro farmaco di recente impiego è il bortezomib, un composto con azione inibente proteosomica (componenti primari della degradazione proteica cellulare): inibisce la proliferazione e induce l'apoptosi delle cellule mielomatose. Le risposte ottenute in pazienti già trattati con altri farmaci sono nell'ordine del 35% e sembra che l'associazione con il desametasone abbia un effetto sinergico. Gli effetti collaterali più noti sono disturbi gastrointestinali, febbre, citopenia e neuropatia periferica. Per il trattamento del dolore osseo si usano farmaci a scopo analgesico e, dove possibile, una buona terapia del dolore. Una volta al mese si può inoltre infondere il pamidronato o molecole nuove come lo zoledronato per ridurre il riassorbimento di calcio dalle ossa.
Malattie emorragiche
Le malattie emorragiche possono coinvolgere uno qualsiasi dei meccanismi che fanno parte dei processi di emostasi; esse possono quindi essere causate da alterazioni dei vasi, delle piastrine o di uno dei fattori della coagulazione. La malattia di Rendu-Osler costituisce una rara patologia ereditaria nella quale tanto i capillari quanto le arteriole presentano un'alterazione di struttura che li porta a dilatarsi; la sintomatologia emorragica, caratterizzata in genere da emorragie della bocca (gengivorragie) e del naso (epistassi), è dovuta alla rottura di piccoli vasi. Alterazioni acquisite dei vasi possono riscontrarsi in corso di infezioni, nelle persone anziane e in seguito a prolungata assunzione di corticosteroidi.
Le alterazioni delle piastrine, di tipo sia quantitativo sia qualitativo, sono caratterizzate da sintomatologia emorragica a carico della cute (petecchie, ecchimosi) o delle mucose (epistassi, gengivorragia, ematuria, ecc.). La diminuzione del numero delle piastrine rappresenta l'evenienza più frequente. Si tratta di patologie nella grande maggioranza acquisite, che possono insorgere in tutte le età, primitive o secondarie all'assunzione di farmaci, a infezioni spesso di natura virale, a epatopatie o a malattie neoplastiche. Le forme primitive riconoscono un'origine autoimmune (porpora trombocitopenica autoimmune), sono causate cioè da anticorpi diretti contro le piastrine stesse che così vengono precocemente distrutte da organi come la milza e il fegato; possono essere acute, di breve durata, o croniche; in genere è acuta la piastrinopenia del bambino, spesso secondaria a un'infezione e rapidamente rispondente alla terapia, mentre nell'adulto sono prevalenti le forme croniche. Normalmente solo un grave decremento delle piastrine è sintomatico (〈30.000-20.000/mm3), mentre piastrinopenie di lieve entità danno raramente sintomatologia emorragica spontanea. Attualmente si tende quindi a trattare solo le forme gravi; la terapia di prima scelta è con cortisonici ed eventualmente, nelle forme croniche, si interviene con la splenectomia.
Più rare sono le alterazioni qualitative delle piastrine (piastrinopatie); si tratta di forme acquisite o congenite, dove l'alterazione di un costituente delle piastrine ne determina il malfunzionamento. Tra queste vi è la tromboastenia di Glanzmann, una malattia grave e rara nella quale l'alterazione è a carico di alcune glicoproteine di membrana e che può causare un'importante sindrome emorragica; l'unica terapia efficace in caso di emorragia è la trasfusione di concentrati piastrinici. Le piastrinopatie acquisite possono riscontrarsi in corso di malattie mieloproliferative, come la leucemia mieloide cronica, oppure epatiche; più frequenti sono le piastrinopatie secondarie all'assunzione di aspirina o altri farmaci antinfiammatori: in questi casi l'alterazione è reversibile. La carenza totale o parziale di uno o più fattori della coagulazione rappresenta un'altra causa di sintomatologia emorragica; essa può essere acquisita, come spesso succede nelle epatopatie gravi, o congenita, come nell'emofilia.
Bibliografia
Catovsky, Matutes 1992: Catovsky, Daniel - Matutes, Estella, The classification of acute leukemia, "Leukemia", 6 (suppl. 2), 1992, pp. 1-6.
Lee 1999: Wintrobe's clinical hematology, 10. ed., edited by Richard G. Lee e altri, Baltimore-London, Williams & Wilkins, 1999, 2 v.
Padoa 1996: Padoa, Emanuele, Manuale di anatomia comparata dei Vertebrati, 15. ed., Milano, Feltrinelli, 1996 (1. ed.: 1963).
Pinkel 1992: Pinkel, Daniel, Therapy of acute lymphoid leukemia in adults, "Leukemia", 6 (suppl. 2), 1992, pp. 127-131.
Schmidt-Nielsen 1983: Schmidt-Nielsen, Knut, Animal physiology. Adaptation and environment, 3. ed., Cambridge-New York, Cambridge University Press, 1983 (trad. it.: Fisiologia animale. Adattamento e ambiente, Padova, Piccin, 1988).
Williams 1990: Hematology, 4. ed., edited by William J. Williams e altri, New York-London, McGraw-Hill, 1990.
Zittoun 1992: Zittoun, Robert A., Chemotherapy of acute myelogenous leukemia, "Leukemia", 6 (suppl. 2), 1992, pp. 36-38.