
Sistemi genetici mitocondriali del lievito e dell'uomo
Sistemi genetici mitocondriali del lievito e dell'uomo
II ruolo genetico del DNA mitocondriale consiste nella codificazione di alcuni componenti dell'apparato enzimatico responsabile della fosforilazione ossidativa. Anche se condividono le medesime funzioni genetiche, il genoma mitocondriale dell'uomo e quello di Saccharomyces cerevisiae, di cui verranno in seguito descritte alcune caratteristiche, mostrano di possedere alcune differenze significative dal punto di vista della grandezza, dell'organizzazione e dei meccanismi di espressione. A causa delle loro dimensioni ridotte, questi genomi dipendono in gran parte, per il controllo della propria espressione, da prodotti dei geni nucleari. Inoltre, in seguito a recenti scoperte, è stato possibile associare diverse alterazioni della sequenza del DNA mitocondriale a varie malattie genetiche umane.
Organizzazione, espressione e trasmissione dei geni mitocondriali nel lievito Saccharomyces cerevisiae
La funzione genetica primaria del DNA mitocondriale (mtDNA) consiste nel codificare i componenti dell'apparato enzimatico mitocondriale preposti alla fosforilazione ossidativa: la catena di trasporto degli elettroni e il complesso dell'ATP-sintetasi. La via metabolica della fosforilazione ossidativa produce ATP attraverso la riduzione dell' ossigeno molecolare, per mezzo di equivalenti riducenti forniti dal ciclo degli acidi tricarbossilici e dalla glicolisi citoplasmatica, per generare un gradi ente elettrochimico tra le due superfici dello strato interno della membrana mitocondriale. Per esprimere questi geni mitocondriali i mitocondri si affidano, in gran parte ma non esclusivamente, a prodotti di geni nucleari. Per esempio, nella maggioranza degli eucarioti il genoma mitocondriale codifica gli rRNA e gran parte dei tRNA, mentre gli altri componenti dell'apparato di traduzione mitocondriale sono codificati dal genoma nucleare. Nell'insieme, solo poco più di una dozzina dei polipeptidi che compongono l'apparato di fosforilazione ossidativa sono codificati dall'mtDNA. A eccezione di pochi prodotti di geni mitocondriali coinvolti, in alcune specie, nelle attività di processamento (processing) postrascrizionale, quali la rimozione degli introni (splicing) e la maturazione di tRNA, il controllo dell'espressione dei geni mitocondriali è, principalmente, affidato al genoma nucleare. Ciò che sorprende in questa 'divisione del lavoro' è il numero relativamente alto di geni nucleari, più di 100, che sono coinvolti nell' espressione di appena una dozzina di geni mitocondriali.
Come sarà descritto in seguito, i genomi mitocondriali delle cellule animali sono molto compatti e non contengono introni. Al contrario, quelli di molti funghi e piante non solo presentano delle sequenze introniche, ma negli introni si può rilevare la presenza di ORF (Open Reading Frames, moduli di lettura aperti), corrispondenti a sequenze traducibili ancora non assegnate, che codificano nuovi prodotti coinvolti nello splicing e nella mobilità degli introni. Questi prodotti genici, comunque, così come anche gli introni stessi, non sono strettamente indispensabili per il corretto svolgimento della funzione mitocondriale. Nella prima parte del saggio ci concentreremo sul genoma mitocondriale del lievito Saccharomyces cerevisiae che ha rappresentato, insieme ad altre specie di lieviti, un sistema sperimentale d'elezione per lo studio della genetica e della biologia molecolare del mitocondrio. Infatti, essendo il lievito un organismo anaerobio facoltativo, esso può essere coltivato anche in assenza di attività respiratoria, e ciò permette di ottenere facilmente mutazioni delle funzioni mitocondriali. Inoltre, la disponibilità di metodi molto avanzati di analisi genetica convenzionale e di analisi genetica inversa dei geni nucleari e mitocondriali ha permesso di studiare i geni responsabili di quasi tutti gli aspetti della funzione mitocondriale.

Le dimensioni del genoma mitocondriale di S. cerevisiae (fig. 1) possono variare, a seconda del ceppo considerato, tra 75 e 80 kb, per cui esso risulta circa cinque volte più grande di quello dei mammiferi. Questa maggiore dimensione del genoma mitocondriale del lievito e i suoi polimorfismi sono dovuti, principalmente, alla presenza di 13 introni opzionali e di alcune sequenze di DNA spaziatrici, non codificanti, che non sono presenti nei genomi mitocondriali degli eucarioti superiori. Un introne del genoma mitocondriale di lievito è localizzato nel gene per l'rRNA maggiore (21S), gli altri si trovano nel gene OXI3, che codifica la subunità l della citocromoossidasi, e nel gene COB, che codifica l'apoproteina del citocromo b. Le sequenze spaziatrici ricche di AT contengono elementi che agiscono in cis nei processi trascrizionali e postrascrizionali mitocondriali e una serie di brevi regioni (20 -7- 75 bp) ricche di GC, chiamate gruppi di GC, di cui non si conosce la funzione, sebbene sembra che si tratti di forme semplici di elementi mobili.
Punti di controllo nell'espressione dei geni mitocondriali
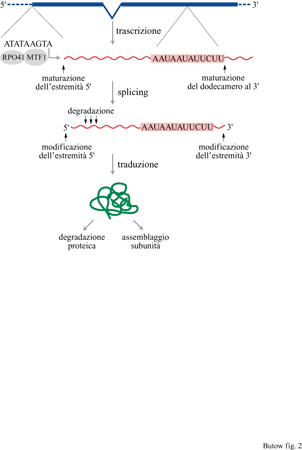
l differenti livelli di controllo dell' espressione genica mitocondriale del lievito sono rappresentati schematicamente nella figura (fig. 2); i controlli postraduzionali, trattati d, L.A. Grivell (1995), che comprendono la stabilità delle proteine e l' assemblaggio dei complessi multiproteici, pw rappresentando aspetti importanti dell'espressione genic, mitocondriale, non saranno discussi in questo saggio. Una delle caratteristiche peculiari dell'espressione de geni mitocondriali nel lievito è l'assoluta mancanza di qual· siasi controllo sull'inizio della trascrizione. A differenza d quanto accade nel nucleo, in cui numerosi elementi che agiscono in cis e in trans sono coinvolti nella regolaziom dell'espressione genica, nel mitocondrio la trascrizione s basa esclusivamente sull'attività di una RNA-polimerasi costituita da due componenti: una subunità catalitica codificata dal gene RP041 e un fattore di specificità codificato dal gene MTF1. La funzione del fattore di specificità consiste nel guidare la subunità catalitica affinché inizi la trascrizione partendo da un promotore semplice, analogamente al fattore (J dei procarioti. L' efficienza di trascrizione che parte da questi promotori non subisce variazioni se non per effetti dovuti al contesto della sequenza. Se ne deduce che i controlli esercitati a livello postrascrizionaIe, traduzionale e postraduzionale rendono conto di tutta la regolazione dell'espressione dei geni mitocondriali.
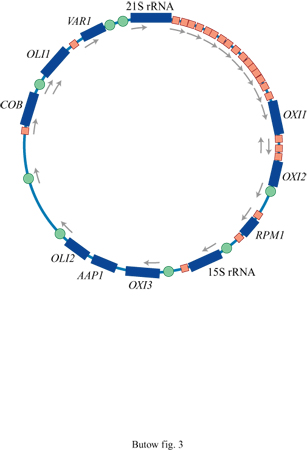
L'elemento centrale del promotore mitocondriale è costituito da una sequenza evolutivamente conservata di nove nucleotidi ATATAAGTA, in cui la trascrizione inizia dal nucleotide A all'estremità 3'. Esistono circa 19 di questi promotori, sparsi lungo il genoma mitocondriale del lievito, che danno origine a ben precisi trascritti mitocondriali (fig. 3). Nel lievito molti dei geni mitocondriali vengono trascritti sotto forma di mRNA policistronici, oppure vengono sottoposti a vari processi postrascrizionali necessari per produrre mRNA e tRNA maturi.
Come sarà descritto in seguito, si ritiene che la proteina HMGl-simile, che nell'uomo è chiamatah-mTFl, giochi un ruolo importante nel controllo della trascrizione dell'mtDNA nelle cellule animali. Sebbene una proteina simile (Abf2p) sia presente anche nei mitocondri del lievito, non esistono prove sufficienti per dimostrare che questo omologo presente nel lievito mostri un analogo coinvolgimento nella traduzione dell'mtDNA. Sia Abf2p sia h-mTFl contengono due domini HMG box, ma la proteina del lievito manca di una coda carbossiterminale, presente invece in h-mTFl, che svolge un ruolo rilevante nell' attività di trascrizione. È molto probabile che entrambe le proteine abbiano una funzione strutturale nell'organizzazione del cromosoma mitocondriale.
Replicazione del DNA mitocondriale
Le cellule di lievito aploidi contengono dalle 25 alle 50 molecole di mtDNA, che si replicano continuamente durante il ciclo cellulare. Tuttavia, i dettagli molecolari di tale replicazione non sono ancora stati definiti e, in particolare, non è chiaro quale sia il ruolo degli inneschi di RNA e se vi siano origini di replicazione. Ciò contrasta con il quadro abbastanza ben definito dei meccanismi di replicazione dell'mtDNA nelle cellule animali, i cui dettagli verranno esposti in seguito. A seconda del ceppo di appartenenza, si possono identificare nel genoma mitocondriale di lievito circa otto sequenze evolutivamente conservate che vengono identificate come ORI o REP ed è stato proposto, ma non dimostrato, che esse siano le origini della replicazione del DNA. Le prove sperimentali a supporto dell'ipotesi che gli elementi ORI siano i veri punti di partenza della replicazione del DNA sono costituite, prima di tutto, dal fatto che esse contengono alcuni tratti di sequenza presenti anche nelle origini della replicazione dell'mtDNA dei mammiferi; esse vengono trascritte a partire da un promotore composto da nove nucleotidi conservati, contenuto all'interno di esse, come ci si aspetterebbe in un tipico innesco a RNA. Inoltre esiste una classe di lieviti mutanti petite (ρ̅), cioè mancanti di un tratto di mtDNA, detti petite ipersoppressori, il cui genoma mitocondriale consiste in unità ripetute di elementi ORI. L'aspetto importante caratteristico di questi mutanti è che quando essi vengono incrociati con cellule di tipo ρ+, cioè con un genoma mitocondriale completo, o con altri mutanti p-non soppressori, nelle cellule diploidi risultanti si ritrova sempre il genoma degli ipersoppressori, a discapito di tutti gli altri genomi mitocondriali parentali. Questa osservazione ha originato l'ipotesi, in verità non universalmente accettata, che l'mtDNA ipersoppressore si replichi più rapidamente, fino a soppiantare gli altri mtDNA, a causa dell'alta densità di punti di inizio della replicazione.
Questa controversia sul significato funzionale delle sequenze ORI ha origine, almeno in parte, dall'incertezza esistente sul ruolo di innesco dell'RNA nella replicazione dell'mtDNA di lievito e, più in particolare, dall' osservazione che l'attività dell 'RNA-polimerasi mitocondriale codificata dal gene RP041 non sembra essere necessaria per il mantenimento del mtDNA ρ̅ o per l'effetto di soppressione dei mutanti petite ipersoppressori (Lorimer et al., 1995). Nonostante la diminuzione di circa mille volte della quantità di mtRNA in ceppi con un allele non funzionale di RP041 , rimane comunque possibile che l'iniziatore di RNA sia necessario per la replicazione dell'mtDNA nei ceppi ρ̅, ma che l'enzima ad attività primasica coinvolto non sia stato ancora identificato. Un'ipotesi alternativa è che il meccanismo di replicazione dell'mtDNA di lievito sia un processo dipendente dalla ricombinazione, in cui l'innesco viene fornito dalle estremità 3' libere dei filamenti di DNA che si formano negli intermedi di ricombinazione (Lockson et al., 1995) secondo una strategia adottata da alcuni batteriofagi a DNA. In questo caso, gli elementi ORI potrebbero giocare un ruolo importante nella segregazione dell'mtDNA piuttosto che nella sua replicazione.
Sebbene per decidere tra le varie ipotesi saranno necessari ulteriori esperimenti, l'analisi molecolare dell'mtDNA di lievito mostra che esso si trova sotto forma di molecole più grandi di una singola unità e forse è strutturato come una rete di intermedi di ricombinazione (Bendich, 1996). Inoltre, i ceppi mutanti che hanno perso la capacità di scindere questi intermedi di ricombinazione dell'mtDNA perdono anche la capacità di trasmettere efficientemente il loro mtDNA alle generazioni successive (Lockson et al., 1995). È significativo che nel genoma mitocondriale di lievito la ricombinazione abbia una frequenza estremamente alta, tanto che un valore di ricombinazione dell'1% corrisponde a circa 100 bp! Questo valore così straordinariamente elevato potrebbe essere stato raggiunto nell'evoluzione non tanto per permettere un rapido riarrangiamento dei geni mitocondriali, quanto per generare gli inneschi necessari per la replicazione dell'mtDNA.
Trasmissione del DNA mitocondriale
Si ritiene che l'unità di segregazione dell'mtDNA sia costituita da un complesso DNA-proteina chiamato nucleoide o condriolite, individuabile nelle cellule come una struttura citoplasmatica puntiforme che si colora con coloranti specifici per il DNA. È molto probabile che i nucleoidi contengano più molecole di mtDNA unite tra loro sotto forma di strutture di ricombinazione. l primi studi sulla segregazione dell'mtDNA negli zigoti stabilirono che la trasmissione del DNA non è casuale (Strausberg e Perlman, 1978). Dopo che le cellule aploidi si sono fuse per formare uno zigote, i mitocondri derivati da ciascun genitore si uniscono, mentre la fusione degli mtDNA parentali sembra essere molto limitata e confrnata alla regione del collo dello zigote. Questa conclusione scaturisce dall'osservazione che il genotipo degli mtDNA delle gemme che si formano a entrambe le estremità dello zigote è, in massima parte, quello del progenitore aploide che ha dato origine a quell'estremità. Al contrario, le gemme centrali che si formano nella regione del collo contengono entrambe le forme parentali e le forme ricombinanti degli mtDNA che segregano rapidamente per dare origine a cellule omoplasmatiche, cioè dotate di una popolazione di mtDNA pura. In contrasto con questo ridotto rimescolamento dell'mtDNA ρ+, le proteine della matrice mitocondriale si ridistribuiscono equamente in tutto il reticolo mitocondriale fuso (Azpiroz e Butow, 1993). Nel loro insieme questi risultati suggeriscono l'esistenza di un apparato specifico di segregazione dell'mtDNA.
È noto che numerosi geni sono necessari per la propagazione dell'mtDNA e, sebbene il ruolo dei prodotti di alcuni di questi geni sia scontato come quello della mtDNA-polimerasi, la funzione di molti altri rimane incerta. È stata studiata abbastanza approfonditamente una proteina che lega l'mtDNA e che è necessaria per il mantenimento dell'mtDNA in ceppi ρ+ in particolari condizioni di crescita. Si tratta della proteina Abf2p che contiene un dominio HMG e che, come già menzionato, ha un analogo nell'uomo. Abf2p è necessaria per il mantenimento dell'mtDNA nei ceppi ρ+ in cellule coltivate in presenza di fonti di carbonio fermentabili, mentre non è essenziale se le cellule vengono coltivate su fonti di carbonio non fermentabili. Se ne deduce che Abf2p non è, di per sé, indispensabile per la replicazione o l'espressione dell'mtDNA ma, più probabilmente, il suo ruolo è legato alla capacità di legare e stabilizzare varie forme strutturali dell'mtDNA.
Maturazione dell'RNA
Quasi tutti gli mRNA mitocondriali sono trascritti in grandi molecole contenenti anche dei tRNA e delle regioni spaziatrici intergeniche. Gli mRNA e i tRNA che si trovano in questi trascritti policistronici vanno incontro, oltre allo splicing, a una maturazione che comporta tagli endonucleolitici alle estremità 5' e 3'. La maturazione dell'estremità 3' dei tRNA mitocondriali è un processo che comporta due passaggi e che richiede, in primo luogo, un taglio endonucleolitico operato da un' attività endoribonucleasica codificata nel nucleo e, successivamente, l'aggiunta del trinucleotide CCA all'estremità 3' del tRNA. Quest'ultima reazione è catalizzata da una specifica nucleotidiltransferasi che, fatto piuttosto interessante, è il prodotto di un gene che codifica anche la versione citoplasmatica dell'enzima. Questa strategia di doppia localizzazione di uno stesso prodotto genico nei compartimenti mitocondriale ed extramitocondriale è stata osservata per altri due enzimi che operano modificazioni a carico dei tRNA. La maturazione dell'estremità 5' dei tRNA mitocondriali avviene, invece, a carico di una RNasi P costituita da una molecola di RNA di 490 nucleotidi codificata dall'mtDNA (RMP1) e da una componente proteica codificata dal gene nucleare, RMP2 (Morales et al., 1992).
Gli eventi di maturazione alle estremità 5' e 3' dei trascritti che portano alla produzione degli mRNA maturi non si esauriscono con quanto appena descritto a carico dei tRNA, ma coinvolgono altre attività di maturazione, portate avanti da una serie di elementi che agiscono in cis e in trans. Le estremità 3' degli mRNA mitocondriali di lievito non sono poliadenilate, al contrario di quelle degli mRNA citoplasmatici e dei mitocondri delle cellule animali. Nella regione non tradotta all'estremità 3' dei precursori degli mRNA mitocondriali è presente, però, un dodecamero 5'AAUAAUAUUCUU-3' (v. figura 2), riconosciuto da un'attività endoribonucleasica non ancora caratterizzata che taglia l'RNA precursore esattamente a valle del dodecamero. Mutazioni della sequenza del dodecamero portano a un'alterata maturazione dell'estremità 3' degli mRNA. Benché gli RNA maturati in maniera difettosa siano stabili, le proteine da essi codificate non vengono sintetizzate. Queste osservazioni suggeriscono che il dodecamero abbia un ruolo importante nel rendere traducibili gli mRNA mitocondriali.
l trascritti di quattro degli otto geni che codificano le principali proteine mitocondriali (VAR1, COB, OXI2 e AAP 1) raggiungono la maturazione grazie a eventi che coinvolgono l'estremità 5' . Il trascritto primario dell'A TPasiS; è parte di un grande RNA policistronico che comprende, a monte, l'mRNA di COX1 e, a valle, quello dell'ATPasi6 (v. figura 1). Il trascritto dell'ATPasi8 comprende anche l'ATPasi6, cosicché le due proteine vengono tradotte a partire dalla stessa molecola di RNA. l fattori proteici che sono coinvolti in trans in questi eventi di maturazione all'estremità 5' non sono ancora stati identificati, tranne CBPl, prodotto di un gene nucleare che interviene nel processamento del precursore dell 'mRNA di COB.
Introni mitocondriali
Come già accennato, il genoma mitocondriale di lievito può contenere fino a tredici introni, ognuno dei quali è facoltativo. Tutti appartengono alle due classi di introni capaci di autosplicing: quelli di gruppo l e quelli di gruppo 11. È noto che molti di questi introni, se non tutti, sebbene in vitro siano in grado di compiere autosplicing, richiedono in vivo la presenza di proteine che catalizzano lo splicing. Questi fattori di splicing sono codificati da geni mitocondriali o da geni nucleari e, in alcuni casi (v. oltre), lo splicing di alcuni introni dipende sia da fattori codificati nel mitocondrio sia da fattori codificati nel nucleo. È interessante notare che i fattori di splicing codificati dall'mtDNA, chiamati maturasi, sono codificati da ORF localizzati all'interno degli introni e, spesso, sono necessari per lo splicing degli stessi introni che li codificano. ORF che codificano maturasi note, a eccezione di quelle contenute ne II 'unico introne presente nel gene per l'rRNA 2lS, sono sempre in fase con l' esone a monte, cosicché il prodotto primario della traduzione è una proteina derivante da questa fusione, contenente amminoacidi codificati sia dall'esone sia dall'introne. La maturasi attiva è, probabilmente, una forma processata della proteina codificata dall'introne, liberata dagli ammino acidi codificati dall' esone mediante processamento postraduzionale.
Da esperimenti di incrocio genetico è emerso che alcuni di questi introni di gruppo l e il si comportano come elementi mobili, in grado di spostarsi in maniera replicativa dal proprio sito a quello omologo presente sull'altro allele parentale privo di introne (Lambowitz e Belfort, 1993). Questo fenomeno è, a volte, definito intron homing (ritorno a casa degli introni). La mobilità degli introni di gruppo l si basa su un processo di 'taglio della doppia elica-riparazione' (double strand break-gap repair), simile al meccanismo responsabile del cambiamento del tipo sessuale che si verifica nel lievito, ed è mediato da un'endonucleasi sitospecifica per il DNA a doppio filamento, codificata dall'introne mobile; in alcuni casi l'endonucleasi può agire anche come maturasi. La mobilità degli introni di gruppo II osservata, per esempio, negli introni l e 2 del gene OX/3 (v. figura 1), si basa su un processo di retrotrasposizione, mediato da una trascrittasi inversa codificata anch'essa dall'introne mobile che agisce anche come maturasi per lo splicing dell'introne. l fattori di splicing a tutt'oggi identificati sono numerosi. Alcuni, come la proteina codificata dal gene CBP2, sembrano essere specifici per lo splicing di un singolo introne mitocondriale, in questo caso l'introne 5 (b15) del gene COB. Altri sono necessari per la rimozione di più di un introne mitocondriale, oltre ad avere altre funzioni non connesse con lo splicing. Attualmente, i meccanismi attraverso i quali agiscono molte di queste proteine rimangono incerti, soprattutto nei casi in cui non si hanno prove dirette di un legame della proteina all'RNA intronico. Fa eccezione la proteina Cbp2 che promuove lo splicing dell'introne b15 legandosi fortemente a una struttura terziaria termodinamicamente instabile che rappresenta il nucleo catalitico dell'introne (Weeks e Cech, 1995). Stabilizzando la struttura del nucleo catalitico, Cbp2 facilita la transizione verso una conformazione dell'introne più favorevole allo splicing, aumentando la velocità della reazione di circa mille volte. Come sarà esposto in seguito, questo meccanismo di splicing assistito da proteine di introni in grado di effettuare autosplicing viene affrontato in maniera differente dagli introni di gruppo I.
Un gruppo di fattori di splicing codificati nel nucleo da geni chiamati MRS è stato identificato grazie alla capacità di sopprimere difetti di splicing negli introni di gruppo II o difetti di splicing che insorgono per mutazione di altri geni MRS. È interessante notare che confronti di sequenze delle proteine codificate da MRS3 e MRS4 suggeriscono che questi geni codificano una famiglia di proteine mitocondriali preposta al trasporto di soluti. Sebbene il meccanismo attraverso il quale questi fattori di splicing agiscono rimanga incerto, questi risultati indicano la possibilità che le reazioni di autosplicing dipendano dalle condizioni ambientali all'interno del compartimento mitocondriale. In questo caso, è lecito attendersi che il quadro delle proteine classificabili come fattori di splicing sia lontano dall'essere completo.
È ben noto, infine, che per lo splicing dei pre-mRNA nucleari è necessaria la partecipazione di una classe di proteine appartenenti alla famiglia delle elicasi NTP-dipendenti, generalmente note con il nome di proteine DEAD-box, dal nome di una sequenza amminoacidica evolutivamente conservata (Schmid e Linder, 1992). Queste proteine hanno la capacità di srotolare i doppi filamenti di RNA e intervengono a vari livelli nel processo di splicing, compreso il riconoscimento del sito di splicing e l'assemblaggio e disassemblaggio del complesso dello spliceosoma (particella ribonucleoproteica adibita al compimento dello splicing). Nei mitocondri di lievito esistono due proteine codificate dai geni MSS1l6 e suv3 e appartenenti alla famiglia delle DEAD-box. Entrambe sembrano essere implicate nello splicing poiché mutazioni di uno dei due geni risultano in difetti di splicing. Nei mutanti di suv3, inoltre, si verifica un accumulo di un gran numero di introni di gruppo l rimossi, che non si rileva nelle cellule di tipo selvatico. Come sarà discusso in seguito, la stabilità degli mRNA maturi dipende in maniera critica dalla presenza, all'interno dei mitocondri, di pochi introni di gruppo l rimossi.
Stabilità dell'RNA mitocondriale
Il controllo della stabilità di alcuni mRNA mitocondriali sembra dipendere dai prodotti di specifici geni nucleari. Tre di questi geni nucleari, PET309, CBPl e NCA1, sono stati identificati e associati, rispettivamente, alla stabilità degli mRNA dei geni mitocondriali OX/3, COB e OLll. Nel caso dell'mRNA del gene COB è stata anche individuata una regione, presente all'estremità 5' del suo trascritto primario, che rappresenta un possibile bersaglio per il riconoscimento da parte della proteina Cbp l. Tuttavia, il meccanismo mediante il quale la proteina Cbp l e le altre proteine di questo genere agiscono nella stabilizzazione degli mRNA rimane incerto.
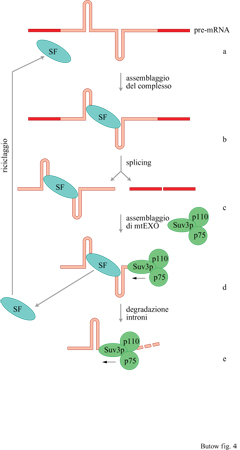
Mentre i precursori degli mRNA nucleari che sfuggono al processo di maturazione vengono rapidamente degradati, i precursori degli mRNA mitocondriali che, a causa di mutazioni di fattori che agiscono in cis o in trans, non riescono a procedere nello splicing di uno o più introni, non vengono riconosciuti per essere degradati e si accumulano all'interno del mitocondrio. Tuttavia, l'apparente mancanza di un meccanismo di degradazione selettiva degli mRNA mitocondriali non funzionali non indica che il controllo della stabilità dell'RNA non abbia alcun ruolo nella regolazione dell'espressione genica mitocondriale. Come già detto, nei mitocondri di cellule di tipo selvatico, gli RNA intronici di gruppo l rimossi vengono degradati, cosicché non vengono trovati allo stadio stazionario. Questo caso di degradazione selettiva rappresenta un esempio di come alcuni geni nucleari, come suv3, si siano evoluti per soddisfare le esigenze dell'espressione genica mitocondriale. Recentemente è stato dimostrato che la proteina cui è attribuita attività RNA-elicasica, codificata dal gene suv3, rappresenta un componente funzionale di mtEXO, una nuova esoribonucleasi 3'-5' mitocondriale (Margossian et al., 1996). Oltre a Suv3p, mtEXO contiene anche due polipeptidi p110 e p75 che, insieme, sono responsabili della degradazione degli RNA intronici di gruppo I rimossi. Come mostrato nel modello rappresentato (fig. 4), l'attività RNAelicasica attribuita a Suv3p dovrebbe consistere nella rimozione dei fattori di splicing, come Cbp2p, dagli RNA intronici rimossi. Ciò dovrebbe ren-dere la struttura dell'RNA intronico più facilmente accessibile alla degradazione da parte di un'attività esonucleasica 3'-5', presumibilmente a opera di una delle subunità del complesso mtEXO.
Per quale motivo le cellule dovrebbero prendersi cura di degradare nei mitocondri gli introni di tipo I rimossi e, sostanzialmente, non degradare altre specie di RNA, in particolare RNA mutanti senza alcuna ovvia funzione? La risposta va cercata, almeno in parte, nelle proprietà chimiche dei ribozimi, quali gli RNA intronici di gruppo I. Questi RNA intronici sono, ad alte concentrazioni, in grado di invertire la reazione di splicing provocando una riapertura degli esoni legati. Questa reazione si può verificare nei mitocondri anche in vivo, se gli RNA intronici di gruppo I rimossi si accumulano in quantità sufficienti come accade, per esempio, nelle cellule con un allele suv3 mutato (Margossian et al., 1996). Quindi, per evitare la possibilità di una dannosa distruzione degli mRNA, le cellule di lievito potrebbero aver sviluppato il complesso dell'esoribonucleasi, allo scopo di mantenere basso il quantitativo di RNA intronici di gruppo I rimossi.
Controlli traduzionali
La regolazione della sintesi delle proteine mitocondriali si attua, in massima parte, con le medesime strategie utilizzate nei diversi tipi di controllo postrascrizionale, ossia mediante fattori gene-specifici che interagiscono con i trascritti mitocondriali. Sono già stati isolati alcuni fattori, codificati nel nucleo, che interagiscono con la regione 5' UTR (Untranslated Region, regione non tradotta) degli mRNA mitocondriali (Dieckmann e Staples, 1994). La tecnica della trasformazione mitocondriale si è rivelata particolarmente utile in questo campo di studi, poiché ha permesso di compiere esperimenti di scambio di 5' UTR, nei quali si è dimostrato che, se tale regione viene fusa con una sequenza codificante differente, essa necessita di fattori di traduzione gene-specifici. Quindi, questi fattori sono probabilmente coinvolti all'inizio della traduzione e nella localizzazione degli mRNA sulla membrana del mitocondrio, dove avviene la traduzione.
Ne è un esempio l'interazione tra i prodotti di tre geni nucleari, PET494, PET54 e PET122, con il 5' UTR dell'mRNA del gene OXI2. Si ritiene che essi formino un complesso che favorisce l'interazione tra l'mRNA e la subunità minore del ribosoma mitocondriale. Mutazioni di questi geni bloccano la traduzione dell'mRNA di OXI2 e possono essere soppresse con la fusione di un diverso 5' UTR alla sequenza codificante di OXI2. Inoltre, sembra che questi fattori siano associati allo strato interno della membrana mitocondriale, e ciò suggerisce che l'assemblaggio della proteina COX3 al complesso della citocromoossidasi dello strato interno della membrana sia un processo cotraduzionale.
Meccanismi di regolazione tra mitocondri e nucleo
Molte discussioni sulle interazioni tra nucleo e mitocondri sono state finora influenzate, a ragione, dal ruolo critico che i geni nucleari svolgono nel controllo dell' espressione genica dei mitocondri. Recentemente è emerso che in S. cerevisiae, così come in altri eucarioti, le cellule sono in grado di rilevare i cambiamenti della funzione mitocondriale e rispondere a essi mediante modificazioni dell'espressione di geni nucleari (Shyjan e Butow, 1993). Questa via di comunicazione tra i mitocondri e il nucleo è stata chiamata regolazione retrograda (retrograde regulation), per indicare una via regolatoria di direzione opposta al ben documentato flusso di informazioni e di materiale che scorre dal nucleo ai mitocondri. La regolazione retrograda agisce in vari modi fungendo da meccanismo omeostatico in grado di regolare numerose attività cellulari in relazione ad alterazioni del metabolismo mitocondriale; essa è osservabile come un aumento dell'espressione di uno specifico gruppo di geni nucleari in cellule con mitocondri non funzionali, quali le petites con difetti della respirazione. Un esempio è rappresentato dalla regolazione dell'espressione del gene CIT2, che codifica un'isoforma perossisomale della citratosintetasi, che interviene nel ciclo del gliossilato. Il ciclo del gliossilato e quello mitocondriale degli acidi tricarbossilici (TCA) condividono alcuni intermedi di reazione, mediante vie di trasporto (shuttle pathway) tra gli organelli. Questo interscambio di metaboliti permette alle cellule di compiere una sintesi netta di carboidrati a partire da unità a due atomi di carbonio, in quanto il ciclo del gliossilato permette quei passaggi del ciclo dei TCA in cui si verifica il rilascio di COz. Sebbene non sia noto se la citratosintetasi perossisomale sia l'enzima limitante nel ciclo del gliossilato, nelle cellule con un metabolismo mitocondriale alterato, per esempio nei casi di riduzione o di blocco del ciclo dei TCA, si assiste a un aumento della trascrizione del gene CIT2, probabilmente per incrementare, nel ciclo dei TCA, il flusso di intermedi verso i mitocondri.
l meccanismi attraverso i quali i mitocondri modulano l'espressione genica nucleare rimangono ancora sconosciuti. Tuttavia è chiaro che la risposta retrograda rappresenta una complessa rete di interazioni tra i mitocondri e gli altri organelli e presto saranno scoperti nuovi geni responsabili del collegamento tra funzioni mitocondriali e altre funzioni cellulari.
Sviluppi futuri
Sebbene il lievito abbia fornito una grande quantità di informazioni sulla genetica dei mitocondri, molto poco si sa sul meccanismo della trasmissione dell'mtDNA. L'ereditarietà mitocondriale nel lievito può essere distinta, per convenienza, in due componenti ereditabili: i mitocondri e l'mtDNA. l primi sono indispensabili per la vita della cellula, anche se priva dell'mtDNA. Attualmente sono stati trovati e studiati geni responsabili della trasmissione della massa mitocondriale. Il fenomeno della trasmissione dell'mtDNA può essere facilmente disaccoppiato dai movimenti della massa mitocondriale e sembra che sia richiesto o, comunque, partecipi alla trasmissione ereditaria dell 'mtDNA un numero sorprendentemente grande di geni, molti dei quali codificano proteine senza un'apparente funzione nel metabolismo dell'mtDNA. La comprensione del ruolo di questi geni nell'eredità dell'mtDNA e della relazione esistente tra la struttura, l'organizzazione e la trasmissione del cromosoma mitocondriale rappresenta la sfida da intraprendere in questo campo.
È noto da lungo tempo che la quantità di mtDNA presente all'interno dell'organulo è relativamente costante, anche nei casi di DNA non funzionale come nelle petite p-. Rimangono ancora sconosciuti i fattori coinvolti nel controllo del numero di copie di mtDNA. Sebbene la biologia degli mtDNA del lievito differisca per alcuni aspetti fondamentali da quella dei vertebrati, che mancano di attività ricombinatoria, è probabile che vengano riscontrate delle somiglianze, in particolare nel modo in cui il DNA è associato all'organulo. È logico supporre, quindi, che le scoperte nel campo della genetica mitocondriale ottenute in uno dei due sistemi possano essere facilmente applicate all'altro.
Il genoma mitocondriale umano
Per la sua organizzazione elegante e compatta, il genoma mitocondriale umano ha da sempre attratto l'attenzione dei ricercatori e dei medici. La molecola di mtDNA si presenta apparentemente priva di interesse dal punto di vista delle dimensioni (circa 16 kb) se confrontata con il massiccio genoma nucleare umano (3 ∙ 10⁶ kb), ma studi di genetica mitocondriale hanno dimostrato il suo coinvolgimento in un numero incredibile di aspetti biologici e medici.
Sebbene le potenti tecniche genetiche applicabili ai lieviti non possano essere sfruttate nell 'uomo e nonostante la mancanza di un sistema che consenta la trasformazione dei mitocondri, gli studi compiuti sul genoma mitocondriale umano hanno prodotto un insieme cospicuo di informazioni riguardanti l'organizzazione, l'espressione e la replicazione dei geni mitocondriali e hanno evidenziato mutazioni che danno origine a specifiche malattie.
Organizzazione ed espressione del DNA mitocondriale umano
Il genoma mitocondriale umano è una molecola circolare di DNA a doppio filamento che misura 16.569 bp (base pairs, coppie di basi). La sequenza completa è stata determinata nel 1981 e ha rappresentato uno dei primi genomi integralmente sequenziati (Anderson et al., 1981). A differenza del lievito, i geni mitocondriali dell'uomo mancano di introni, sono densamente compattati e, quindi, privi di estese regioni intergeniche di DNA non codificante. I filamenti complementari di DNA sono identificati come filamento pesante e filamento leggero, sulla base del differente contenuto di GC che permette una loro separazione tramite centrifugazione all'equilibrio su gradi ente di densità. I geni sono localizzati su entrambi i filamenti ma non si sovrappongono mai e la maggioranza dei prodotti di questi geni è codificata dal filamento pesante.

È stato trovato che tredici ORF codificano alcune subunità dei grandi complessi multiproteici che costituiscono l'apparato della fosforilazione ossidativa presente sullo strato interno della membrana mitocondriale. In particolare, si tratta della subunità I, II e III della citocromoossidasi (complesso IV), della subunità 4 e 6 della Fj, Fo-ATPasi (ATP-sintetasi o complesso V), del citocromo b (un componente del complesso III) e delle subunità 7 della NADHdeidrogenasi (complesso I), tutte sintetizzate unicamente da geni mitocondriali. Ognuno dei prodotti di questi geni mitocondriali viene assemblato con altre subunità codificate da geni nucleari che vengono importate nel mitocondrio per costituire gli oloenzimi. l geni che nell'mtDNA umano codificano proteine sono affiancati da geni più piccoli che codificano i tRNA, il cui set completo viene interamente sintetizzato all'interno dei mitocondri. Inoltre, il genoma mitocondriale umano codifica i due rRNA presenti nelle due subunità ribosomali, necessarie per la traduzione degli mRNA mitocondriali all'interno della matrice mitocondriale. Questo tipo di organizzazione è schematizzato in figura (fig. 5), ed è evolutivamente conservato nelle altre specie di vertebrati esaminate. La trascrizione, la maturazione dell'RNA e la traduzione nel mitocondrio umano somigliano molto alla serie di passaggi sequenziali attraverso cui vengono espressi i geni nucleari, sebbene alcuni dettagli meccanicistici siano tipici del mitocondrio. A eccezione dei tRNA e degli rRNA, tutte le altre macromolecole necessarie per la replicazione dell'mtDNA e per l'espressione dei geni mitocondriali vengono fornite dal nucleo.
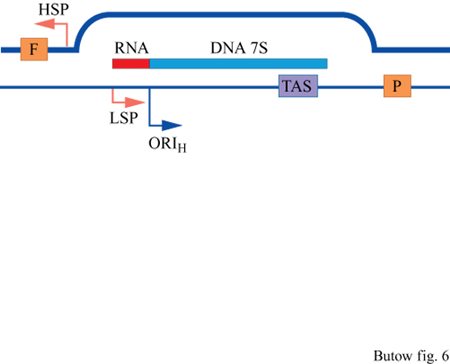
La trascrizione dei geni mitocondriali inizia da promotori bidirezionali raggruppati a un'estremità della regione dell'ansa-D (displacement loop, ansa di dislocazione), l'unico segmento non codificante presente nel genoma mitocondriale umano. Le posizioni dei promotori presenti sul filamento pesante e su quello leggero sono mostrate nella figura (v. figura 5) e, a più alta risoluzione, nella figura successiva (fig. 6), insieme ad altre caratteristiche funzionali fondamentali della regione dell'ansa-D. Entrambi i filamenti sono utilizzati come stampo per la trascrizione e i trascritti primari, analogamente ai batteri, sono policistronici. l singoli rRNA, tRNA e mRNA vengono prodotti in seguito al processamento endonucleolitico di questi trascritti. In particolare, i tRNA maturi sono prodotti dall'azione della RNasi-P mitocondriale. Il codice genetico a triplette impiegato per specificare i residui amminoacidici delle proteine tradotte nel mitocondrio è differente da quello utilizzato per la traduzione dei prodotti dei geni nucleari ed è, perciò, necessario uno specifico set di amminoacil-tRNA-sintetasi per legare gli amminoacidi ai tRNA mitocondriali.
Poiché le regioni codificanti dell'mtDNA umano non sono interrotte da sequenze introniche, non è necessario lo splicing degli RNA per generare trascritti maturi. Gli mRNA mitocondriali sono anche poliadenilati, anche se le code di poli-A sono più corte di quelle della maggior parte degli mRNA di origine nucleare. Il processo di traduzione delle proteine sui ribosomi mitocondriali inizia con un residuo di metionina formilata, analogamente a quanto accade nei batteri.
Molto frequentemente, la trascrizione del filamento pesante si interrompe immediatamente a valle della regione che codifica gli rRNA, grazie all'azione di un fattore di terminazione trascrizionale che si lega a una sequenza specifica presente in questa regione. Tuttavia, la trascrizione dell'intero filamento pesante si verifica con frequenza sufficiente a garantire la produzione degli mRNA e dei tRNA situati a valle di questa posizione. Questo spiega la presenza di un numero maggiore di rRNA maturi rispetto agli mRNA all'interno del mitocondrio. A causa del meccanismo di trascrizione policistronica, tutti gli altri trascritti originati dal filamento pesante vengono prodotti con la stessa velocità, quindi le differenze di concentrazione osservate tra i singoli mRNA e tRNA mitocondriali sono dovute a differente stabilità. Analogamente al lievito, non si verifica nel mitocondrio umano una regolazione della trascrizione dei singoli geni, come tipicamente avviene per la regolazione dei geni nucleari.
L'apparato molecolare necessario per la trascrizione dei geni mitocondriali umani è relativamente semplice, soprattutto se paragonato a quello nucleare che coinvolge complessi macromolecolari costituiti da dozzine di proteine specifiche. La trascrizione in vitro dell'mtDNA richiede, infatti, oltre alla RNA-polimerasi, solo il fattore di trascrizione mtTFI, che si lega specificamente a un elemento della sequenza del DNA posto immediatamente a monte di ciascuno dei due promotori bidirezionali presenti all'interno della regione dell'ansa-D.
La trascrizione e la replicazione del genoma mitocondriale sono intimamente connesse, in quanto l'inizio della replicazione è innescato da piccole molecole di RNA trascritte a partire dal promotore del filamento leggero e successivamente processate da una endonucleasi, la cui identità è ancora controversa. Infatti, è stata purificata dai mitocondri la RNasi-MRP, una ribonucleoproteina costituita da un'apoproteina e da una componente a RNA e codificata da geni nucleari. Essa è in grado di tagliare selettivamente, in vitro, gli RNA trascritti dal promotore del filamento leggero, in corrispondenza del sito di transizione RNA-DNA che si osserva in vivo nei prodotti nascenti della replicazione dell'mtDNA. Anche un'altra proteina, chiamata endonucleasi G, sembrerebbe tuttavia essere in grado di generare gli iniziatori richiesti per la sintesi dell'mtDNA.
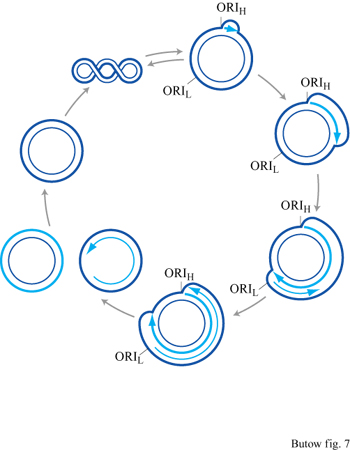
Replicazione del DNA mitocondriale umano La replicazione del DNA inizia da un'origine ben caratterizzata (v. figura 6) e procede con la formazione di un'ansa di dislocazione in cui il filamento pesante nascente rimane appaiato al filamento stampo leggero, spostando il filamento pesante parentale sotto forma di un lungo tratto di DNA a singolo filamento (fig. 7). La replicazione del filamento leggero ha inizio solo dopo che la forca di replicazione del filamento diretto (leading strand) ha percorso circa due terzi del genoma, esponendo così il tratto di DNA che corrisponde all'origine di replicazione del filamento leggero. Si pensa che la tendenza di questa sequenza a formare una struttura a forca permetta l' interazione con una primasi e con altri componenti dell'apparato replicativo, in modo da iniziare la replicazione del DNA usando il filamento pesante come stampo. Quando la replicazione di entrambi i filamenti è completata, le molecole figlie vengono separate da un'attività topoisomerasica.
L'apparato enzimatico preposto alla replicazione dell'mtDNA, analogamente a quello della trascrizione, è meno complesso di quello costituito dal folto gruppo di proteine coinvolte nella replicazione del DNA nucleare. La DNA-polimerasi mitocondriale (detta PoI γ) ha proprietà strutturali e funzionali distinte dalle DNA-polimerasi nucleari presenti nelle cellule umane ed è più simile alla PoI l dei batteri. Analogamente, mtSSB (mitochondrial Single Strand Binding Protein, proteina mitocondriale che lega il DNA a singolo filamento) presenta caratteristiche più simili alla SSB di Escherichia coli che alla più complessa SSB di replicazione presente nei nuclei delle cellule umane (RP-A). È stata caratterizzata anche una topoisomerasi mitocondriale ma rimangono ancora da identificare le altre proteine coinvolte nella replicazione dell'mtDNA.
Una delle caratteristiche più sorprendenti della replicazione dell'mtDNA umano e degli altri vertebrati è rappresentata dall' elevata frequenza di eventi di replicazione abortiva che danno origine a forme stabili di ansa-D dell'mtDNA. Ciò è dovuto alla pausa o all'arresto della forca di replicazione, poche centinaia di nucleotidi a valle dell'origine di replicazione del filamento pesante, in uno o più punti ben definiti del genoma, caratteristici per ogni specie di mammiferi. Sorprendentemente, in alcuni tipi di cellule, più del 90% delle sintesi di mtDNA è dovuto a eventi di replicazione abortiva di questo tipo. È stata identificata una sequenza evolutivamente conservata definita TAS (Termination Associated Sequence, sequenza associata alla terminazione) che è situata in prossimità dei siti in cui la replicazione si interrompe e che, presumibilmente, è coinvolta nella terminazione, o perché rappresenta un sito di legame per proteine che bloccano la progressione dell'apparato di replicazione, o perché interferisce con la topologia della forca di replicazione. Non è stato ancora chiarito il meccanismo molecolare che determina l'arresto o la prosecuzione della replicazione e il significato funzionale di questo fenomeno rimane ancora sconosciuto.
Ogni mitocondrio presente all'interno delle cellule umane possiede numerose copie di mtDNA e ogni cellula umana contiene da centinaia a migliaia di copie di genomi mitocondriali. In seguito alla mitosi, i genomi mitocondriali devono segregare in ciascuna cellula figlia in numero sufficiente per mantenere un'appropriata capacità respiratoria. Tuttavia, la replicazione dell 'mtDNA non è coordinata con nessuno degli stadi specifici del ciclo cellulare che regolano la replicazione e la segregazione dei cromosomi nucleari. Per esempio, la replicazione dell'mtDNA continua anche dopo che la replicazione del DNA nucleare si è interrotta, come nei neuroni e nei miociti di età ormai avanzata e postmitotici, indicando la necessità di sostituire le molecole di mtDNA che vengono degradate nel corso del tempo. Le cellule umane possiedono una limitata capacità di riparazione dei danni subiti dall'mtDNA cosicché il tasso di insorgenza delle mutazioni è maggiore rispetto a quello dei geni nucleari. Analogamente, esperimenti recenti suggeriscono che la ricombinazione tra genomi mitocondriali nelle cellule umane è estremamente rara. Nei vertebrati, l'mtDNA viene ereditato solo per via materna, poiché gli spermatozoi non forniscono mitocondri allo zigote fecondato. Durante la meiosi e le fasi terminali dell'oogenesi solo un numero limitato di genomi mitocondriali viene trasmesso all' oocita maturo e si ritrova nella progenie. Da studi recenti, eseguiti nei topi, è emerso che l'unità minima di segregazione è rappresentata da meno di 20 molecole di mtDNA.

La tabella (tab. I) fornisce una sintesi delle caratteristiche specifiche della genetica mitocondriale dell'uomo, in parallelo con quella nucleare. Queste peculiarità dei geni mitocondriali umani hanno ovvie implicazioni nella comprensione delle malattie dell'uomo che si originano da mutazioni dei geni mitocondriali e che saranno discusse in seguito. Gli studi eseguiti sul genoma mitocondriale umano e di altri vertebrati hanno contribuito anche allo sviluppo della conoscenza in altre aree della biologia. Per esempio, le somiglianze osservate tra alcuni aspetti della replicazione e della trascrizione dell'mtDNA umano e i corrispondenti processi nei batteri hanno fornito prove a sostegno dell'ipotesi endosimbiontica circa le origini evolutive degli organuli subcellulari. L'eredità materna dell'mtDNA implica che i marcatori genetici, per esempio i polimorfismi nucleotidici nell'mtDNA, vengono trasmessi attraverso numerose generazioni, senza essere diluiti dagli alle li patemi, fornendo così un potente mezzo per evidenziare le relazioni genetiche esistenti tra popolazioni geograficamente separate. Questo metodo di analisi ha permesso di formulare alcune ipotesi circa le migrazioni delle popolazioni umane preistoriche; inoltre la caratterizzazione dei genotipi mitocondriali è generalmente accettata come utile tecnica di studio delle relazioni genetiche ed evolutive.
l differenti tipi cellulari dei vertebrati mostrano di possedere capacità di respirazione mitocondriale marcatamente variabili, corrispondenti alla variazione del volume relativo dei mitocondri e dell'attività specifica (relativa alla massa cellulare) degli enzimi mitocondriali. Alcune forme specializzate di miociti striati costituiscono i due estremi di un ampio spettro di capacità respiratoria tra le cellule dei mammiferi: i mitocondri occupano meno dell'1% del volume cellulare nelle fibre del muscolo scheletrico glicolitico del coniglio, ma possono arrivare fino al 50% nei cardiomiociti di piccoli mammiferi. Durante lo sviluppo, i tessuti fetali dei mammiferi si servono principalmente della glicolisi per soddisfare le richieste energetiche all'interno dell'ambiente ipossico della circolazione placentare. Tuttavia, dopo la nascita, uomini e animali neonati dipendono in modo più rilevante dalla fosforilazione ossidativa mitocondriale per sopperire al maggior fabbisogno energetico e, di conseguenza, viene stimolata la bio genesi mitocondriale. Anche in animali adulti, i cambiamenti delle esigenze fisiologiche possono promuovere rilevanti variazioni nella concentrazione cellulare dei mitocondri, nonché nell'espressione degli enzimi mitocondriali. Queste risposte legate allo sviluppo, o all'adattamento, sono mediate da variazioni nell'espressione di geni nucleari che codificano le proteine mitocondriali, ma richiedono anche cambiamenti nell'espressione di geni mitocondriali.
In alcuni tessuti, i fenomeni adattativi e di sviluppo che comportano un aumento del volume relativo dei mitocondri sono accompagnati da un incremento nei livelli di mtDNA, in assenza di replicazione del DNA nucleare; ciò suggerisce che l'amplificazione dei geni mitocondriali abbia un ruolo chiave nel mantenere la stechiometria dei prodotti dei geni mitocondriali e nucleari, quando la bio genesi mitocondriale è alterata. Ci sono, tuttavia, anche esempi in cui i cambiamenti dell' espressione dei geni mitocondriali non sono associati a una variazione della quantità di mtDNA, suggerendo l'esistenza di meccanismi di controllo che alterano la stabilità di specifici mRNA mitocondriali o l'efficienza con cui essi vengono tradotti. Le basi molecolari di queste regolazioni non sono state ancora determinate.
Anomalie fenotipiche causate da mutazioni nel DNA mitocondriale
Sono state descritte numerose specifiche anomalie a carico del genoma mitocondriale umano che comprendono delezioni, duplicazioni dell'mtDNA e mutazioni punti formi nei geni che codificano le proteine, i tRNA o gli rRNA. Tutti questi difetti genetici sono potenzialmente in grado di causare malattie nell'uomo, e in determinate circostanze è provato che ciò si verifica (Wallace, 1992; Luft, 1994). Un'alta frequenza di forme mutate di mtDNA viene osservata anche in individui anziani e nei tessuti di pazienti con diverse patologie comuni, per le quali non è chiaro il reale contributo fisiopatologico dei difetti dei geni mitocondriali. Molto raramente, vi sono pazienti con malattie basate sulla perdita degli interi genomi mitocondriali.
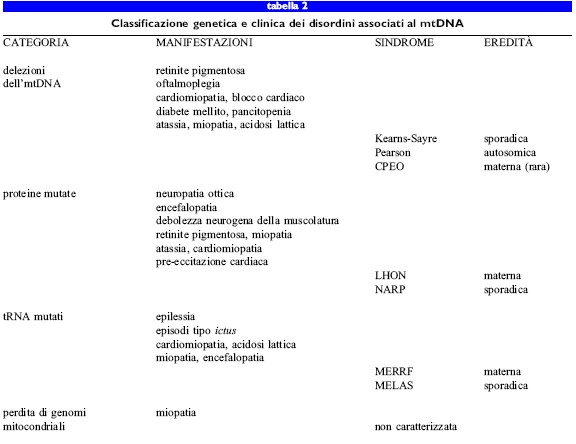

Nelle tabelle sono riportate una classificazione genetica (tab. 2) dei difetti a carico dei geni mitocondriali che causano malattie nell'uomo e sono elencati (tab. 3) gli aspetti principali dei difetti genici associati a questi disordini. È piuttosto frequente che le anomalie fenotipiche riflettano disfunzioni dei muscoli striati e dei neuroni, mentre altre situazioni meno comuni includono difetti nell'emopoiesi o nella funzionalità delle cellule secretorie endocrine.
Lo stato patologico può essere evidente fin dalla nascita ed essere associato a devastanti anomalie del metabolismo sistemico, che portano alla morte dell'individuo durante l'infanzia, oppure le manifestazioni possono risultare assenti o poco evidenti fino a quando, in tarda età, i pazienti sviluppano sintomi più gravi. Molti casi sono sporadici, apparentemente senza evidenti anomalie dell'mtDNA nei genitori o nei parenti prossimi; altri casi, invece, sono familiari. L'eredità materna è il tratto distintivo di alcune malattie genetiche mitocondriali, sebbene altre situazioni presentino un tipo di ereditarietà coincidente con una trasmissione autosomica. I recenti progressi compiuti nella genetica del mitocondrio ci hanno permesso di comprendere la complessa relazione esistente tra genotipo e fenotipo in questi disordini patologici, benché molte questioni importanti rimangano ancora prive di risposta. La gravità delle anomalie fenotipiche associate a difetti del genoma mitocondriale dipende dalla specifica natura delle mutazioni dell'mtDNA, dalla quantità di mtDNA difettoso rispetto all'mtDNA normale, dal contesto genetico nucleare e dalle necessità metaboliche di fosforilazione ossidativa delle cellule malate.
Ogni cellula umana presenta un gran numero di copie del genoma mitocondriale e, spesso, forme mutanti di mtDNA coesistono con le molecole di mtDNA normali (eteroplasmia). Anche se è teoricamente possibile che una proteina o un RNA mutato, prodotti da un ristretto numero di genomi mitocondriali difettivi, possano esercitare un effetto deleterio sulle funzioni di una cellula, in realtà le anomalie fenotipiche riflettono quasi sempre un accumulo di una grande percentuale di genomi difettivi. I pazienti con un'evidente patologia mitocondriale generalmente mostrano, nei tessuti colpiti, dal 75 al 100% di forme mutate di mtDNA. Studi sperimentali hanno dimostrato che cellule in coltura inizialmente prive di mtDNA e ripopolate con rapporti diversi di mtDNA mutato e normale (King e Attardi, 1989) mostrano un difetto respiratorio solo quando il genoma mitocondriale mutato è in eccesso rispetto alla forma normale. Questo effetto-soglia è un determinante importante del fenotipo cellulare in presenza di mutazioni dell'mtDNA.
Nei casi sporadici di malattie mitocondriali, la fase di sviluppo in cui la mutazione dell'mtDNA insorge determina la distribuzione dei genomi mutati nei vari tessuti, in base alla discendenza cellulare. Sia nei casi sporadici sia in quelli familiari, alcune forme difettive dell'mtDNA sembrano avere un vantaggio nella replicazione rispetto al genoma normale e, col tempo, si accumulano in un gran numero di copie. Durante la citocinesi di cellule in crescita proliferativa, le cellule figlie possono ricevere le copie di DNA normale e mutato in rapporto variabile (segregazione replicativa); questo conduce a un'eterogeneità nel numero di copie di mtDNA difettivo, anche tra cellule di uno stesso tessuto o organo.
La tendenza delle mutazioni di geni mitocondriali a causare encefalomiopatie è basata su almeno due considerazioni. I neuroni e molti tipi di miociti richiedono una cospicua respirazione mitocondriale per soddisfare il forte fabbisogno energetico da parte della cellula. Per questo motivo possono presentarsi, in queste cellule, anomalie funzionali prima che in altri tipi cellulari, pur essendo esse portatrici dello stesso difetto genetico. In secondo luogo, questi tipi cellulari 'a lunga vita' escono dal ciclo cellulare nel periodo neonatale e non vengono sostituiti da cellule staminali proliferanti. Se, col tempo, le forme mutanti di mtDNA aumentano rispetto alle forme selvatiche e le cellule con un'alta proporzione di genomi mutanti non vengono sostituite da cellule normali, ne consegue che la probabilità di una disfunzione cellulare è maggiore rispetto a quanto avviene nelle popolazioni di cellule proliferanti. A conferma di questo concetto, va osservato che le manifestazioni cliniche delle mutazioni dei geni mitocondriali diventano frequentemente più gravi durante l'invecchiamento.
Le mutazioni dell'mtDNA, che causano anomalie qualitative o quantitative delle proteine della catena di trasporto degli elettroni, danneggiano la respirazione mitocondriale e compromettono l'apporto di energia alla cellula. Pertanto, ci si potrebbe aspettare che le manifestazioni fenotipiche e cliniche di diversi difetti dei geni mitocondriali siano simili tra loro, variando per gravità ma non per tipologia. In realtà, tali aspettative si dimostrano errate: le malattie mitocondriali umane si presentano con tipologie sovrapposte, ma distinte, di anomalie fenotipiche associate con differenti categorie di difetti genetici.
Delezioni del DNA mitocondriale umano
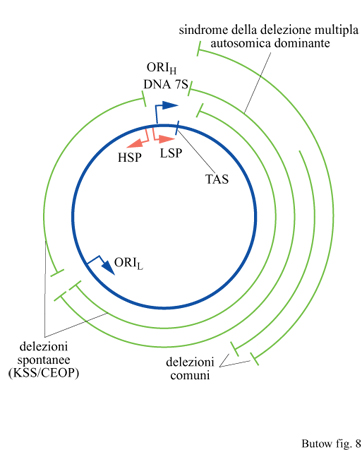
Delezioni del genoma mitocondriale portano all'eliminazione di diversi geni mitocondriali, che il più delle volte includono una o più regioni codificanti proteine e vari geni di diversi tRNA (fig. 8). In quasi tutti i pazienti con malattie clinicamente manifeste predomina una singola e ben precisa forma deleta, che comprende molti, se non tutti, i genomi mitocondriali presenti nelle cellule dei tessuti malati, cosicché il rapporto quantitativo tra le forme delete e quelle normali può variare tra pazienti consanguinei, e anche tra le diverse cellule di un singolo paziente. Sono state osservate delezioni che coinvolgono quasi tutti i segmenti del genoma mitocondriale. La regione dell'ansa-D che si estende dal promotore del filamento leggero fino alla sequenza di terminazione (che contiene l'origine di replicazione del filamento pesante) e l'origine di replicazione del filamento leggero sembrano rappresentare gli unici elementi assolutamente necessari per la replicazione e la conservazione dell'mtDNA.
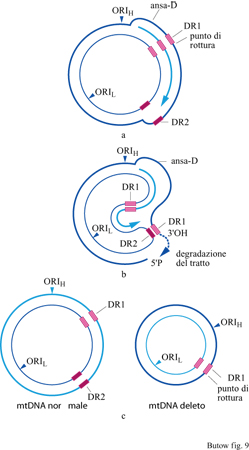
Le regioni più comunemente delete nell'mtDNA sono affiancate da sequenze ripetute dirette (Schon et al., 1989), suggerendo un possibile meccanismo attraverso il quale queste mutazioni possono insorgere. Il modello di replicazione 'a filamento scivolato' (slipped strand) si basa sulla formazione di appaiamenti nucleotidici tra le sequenze ripetute dirette nella regione dislocata del filamento pesante e le regioni complementari del filamento leggero parentale (fig. 9). Il tratto del filamento pesante escluso viene degradato e la replicazione continua, ottenendo come risultato finale che una delle due molecole di mtDNA prodotte presenta una delezione. È piuttosto frequente che forme delete dell'mtDNA siano accompagnate da molecole di DNA in cui sono presenti ampie regioni duplicate.
La sindrome di Kearns-Sayre è una malattia umana caratterizzata da retinite pigmento sa, oftalmoplegia esterna cronica e anomalie cardiache, tra cui frequentemente il 'blocco di conduzione'. Alcuni pazienti manifestano miopatie limitate ai muscoli extraoculare o cardiaco, mentre altri presentano disfunzioni cerebellari. Gli individui affetti da questa malattia presentano un'alta percentuale di forme delete di mtDNA nelle biopsie muscolari, o nei leucociti da sangue periferico. La malattia si manifesta normalmente nel primo ventennio di vita e il più delle volte è sporadica. Nei casi familiari, solo raramente vi è indicazione di ereditarietà materna; potrebbe, quindi, esservi un difetto di un gene nucleare che determina una predisposizione alle delezioni dell'mtDNA in molti familiari (Zeviani et al., 1989).
Una patologia meno comune, la sindrome pancreaticamidollare di Pearson, si sviluppa in seguito all'accumulo di forme delete di mtDNA. Questa malattia è molto grave, generalmente letale durante l'infanzia, poiché causa pancitopenia, acido si lattica e insufficienza pancreatica endocrina. l casi riportati sono sporadici e il limitato quadro di disfunzioni cellulari suggerisce che è colpita una specifica linea di discendenza cellulare, in seguito a un evento mutazionale avvenuto durante lo sviluppo embrionale.
Sostituzioni nucleotidiche e mutazioni di genomi codificanti tRNA
Le sostituzioni nucleotidiche in regioni dell'mtDNA che codificano proteine causano una serie di disturbi clinici simili, ma ben distinti tra loro. La sindrome nota come LHON (Leber's Hereditary Optical Neuropathy, neuropatia ottica ereditaria di Leber) causa cecità e una serie variabile di disordini associati che includono pre-eccitazione cardiaca e complessi sintomi neurologici. I pazienti affetti da LHON presentano mutazioni puntiformi nei geni mitocondriali che codificano il citocromo b o le subunità della NADH-deidrogenasi; i casi familiari si trasmettono per eredità materna. Un quadro differente caratterizza la sindrome NARP (Neurogenic muscle weakness, Ataxia and Retinitis Pigmentosa, debolezza muscolare neurogenica, atassia e retinite pigmentosa), che è associata a una mutazione del gene che codifica la subunità 6 dell'ATPasi mitocondriale.
Alterazioni di geni che codificano tRNA mitocondriali comportano una serie di patologie chiamate MERRF (Myoclonic Epilepsy with Ragged Red Fibers, epilessia mioclonica con lacerazione delle fibre rosse) o MELAS (Mitochondrial Encephalopathy, Lactic Acidosis and Stroke-like episodes, encefalopatia mitocondriale, acidosi lattica ed episodi tipo ictus). Il fenotipo MERRF è associato a mutazioni nel tRNA Lys, mentre il fenotipo MELAS deriva da un difetto nel tRNA Leu. Una delle mutazioni nel tRNA Leu comporta anche una perdita del sito di legame per il fattore di terminazione della trascrizione che controlla il rapporto dei tassi di trascrizione dell'rRNA mitocondriale e dei geni codificanti proteine, presenti nel filamento pesante, contribuendo così al fenotipo malato. Altre mutazioni dei tRNA mitocondriali sono associate a encefalomiopatie e sia i pazienti affetti da MERRF sia quelli affetti da MELAS mostrano uno spettro di anomalie cardiache, oltre alle manifestazioni cliniche classiche che caratterizzano queste sindromi. In alcuni casi, mutazioni dei tRNA mitocondriali sembrano portare a una cardiomiopatia ipertrofica che somiglia al fenotipo derivato da mutazioni nella catena pesante della miosina e di altre proteine del sarcomero. È stato dimostrato che mutazioni dei tRNA mitocondriali causano un difetto nell'amminoacilazione e una terminazione prematura della traduzione. È difficile spiegare, tuttavia, solo sulla base di una generale diminuzione della sintesi proteica mitocondriale, la manifestazione di differenti anomalie fenotipiche in caso di mutazioni di differenti tRNA; sembrano esserci complesse e differenti variazioni nell'espressione dei geni mitocondriali in seguito a specifiche mutazioni dei tRNA. Per tutti i fenotipi malati discussi finora si hanno prove convincenti che un difetto genetico rilevante riguarda l'mtDNA. Queste patologie mitocondriali ben definite sono relativamente rare; sono state descritte, forse, solo poche centinaia di pazienti nella letteratura di tutto il mondo. La conoscenza del problema da parte dei medici potrebbe portare a un aumento del numero conosciuto di pazienti con malattie mitocondriali e potrebbero essere scoperte altre sindromi, anche se la loro incidenza si rivelerebbe comunque molto bassa. Molti studiosi, tuttavia, hanno presentato dati che suggeriscono un ruolo ancora più importante delle anomalie dell'mtDNA nella biologia e nelle malattie dell'uomo.
L'uso di tecniche molto sensibili basate sulla reazione a catena della polimerasi (PCR) permette di identificare forme delete o mutate di mtDNA nelle cellule di molti soggetti umani di età superiore a 70 ÷ 80 anni, mentre è raro riscontrare genomi mitocondriali difettivi in individui apparentemente sani di età inferiore ai 40 anni. I genomi mitocondriali anormali presenti negli esseri umani sani, ma anziani, costituiscono una piccola frazione delle molecole di mtDNA, generalmente meno dell'1% del totale. I pazienti di qualsiasi età con cardiopatia ischemica in seguito a un'ostruzione aterosclerotica delle arterie coronarie, o i pazienti con cardiomiopatie provocate da causa ignota, presentano delezioni nell 'mtDNA in campioni bioptici cardiaci; il rapporto tra genomi mitocondriali mutati e normali è basso (inferiore al 5%), come nel caso degli ottuagenari sani. Un simile incremento delle forme mutate del genoma mitocondriale è stato osservato nel cervello di pazienti affetti da morbo di Parkinson o da corea di Huntington.
Queste osservazioni indicano che forme delete di mtDNA insorgono negli esseri umani come conseguenza dell'avanzare dell'età, e che la frequenza di eventi replicativi anormali aumenta in alcuni casi di malattie degenerative. In queste condizioni, la quantità di molecole di mtDNA mutate rispetto alla forma normale è al di sotto del valore soglia che è stato associato a chiare anomalie nella fosforilazione ossidativa, in cellule umane in coltura o nei tessuti di pazienti con malattie mitocondriali ben definite. Tale osservazione suggerisce che l'aumento della frequenza delle mutazioni mitocondriali negli anziani, o nei pazienti con malattie degenerative, ha poco o nessun effetto sul processo di invecchiamento o sulla fisiopatologia delle comuni malattie cardiache e neurologiche. Potrebbe essere prematuro, tuttavia, escludere completamente un potenziale contributo delle mutazioni dell'mtDNA all'invecchiamento o alle malattie degenerative umane.
La maggior parte degli studi ha preso in esame una singola forma mutata di mtDNA, ma è possibile che in un'unica cellula possano insorgere molteplici anomalie del genoma mitocondriale e che queste, nel loro insieme, possano avvicinarsi a un valore soglia che determina un fenotipo anormale. È anche possibile che il carico di genomi mitocondriali mutati, insignificante per le cellule sane, possa avere un impatto negativo sul metabolismo di cellule già danneggiate da altri processi patologici.
Frontiere della genetica mitocondriale umana Come è stato illustrato, ricerche recenti hanno permesso di chiarire gli aspetti fondamentali dell' espressione e della replicazione dei geni mitocondriali in cellule umane e hanno anche permesso di identificare mutazioni specifiche nell'mtDNA, stabilendo la base molecolare per un gruppo di malattie umane precedentemente considerato eterogeneo. Tuttavia, molte questioni riguardanti la genetica del mitocondrio, di importanza sia scientifica sia medica, devono ancora essere chiarite.
L'assenza di tecnologie per la propagazione di genomi mitocondriali ricombinanti nelle cellule di mammifero ha rappresentato un grande ostacolo al progresso in questo campo. Le tecniche di trasformazione mitocondriale che hanno avuto successo nel lievito non si sono rivelate utili per gli studi su cellule o tessuti di mammiferi. Tecniche innovative in grado di raggiungere tale scopo potrebbero recare enormi vantaggi.
Le nostre conoscenze sulle relazioni tra genotipo mitocondriale e fenotipo si basano quasi interamente su studi descrittivi eseguiti su materiale bioptico e su linee cellulari ottenute da pazienti con malattie genetiche mitocondriali. Sono necessari nuovi approcci per lo sviluppo di modelli animali recanti geni mitocondriali difettivi, al fine di accrescere le nostre conoscenze relative alla curiosa e insospettata diversità delle anomalie fenotipiche determinate da specifiche mutazioni del genoma mitocondriale. l modelli animali dovrebbero rappresentare il miglior approccio per risolvere la controversia sul significato fisiologico delle anomalie del genoma mitocondriale riscontrate nelle malattie degenerative umane e in quelle associate all'invecchiamento.
Sarà altrettanto importante identificare la serie completa dei prodotti dei geni nucleari richiesta per la replicazione, l'espressione e la segregazione dei geni mitocondriali. Quest'area di ricerca si potrà sviluppare molto rapidamente utilizzando le tecniche genetiche applicabili agli eucarioti unicellulari, ma le caratteristiche distintive della genetica del mitocondrio dei mammiferi dovranno essere affrontate attraverso studi diretti sulle proteine dei mammiferi e dell'uomo. La regolazione di questi geni nucleari è, probabilmente, di importanza fondamentale per la modulazione della biogenesi mitocondriale durante lo sviluppo o in risposta a stimoli fisiologici. Inoltre, difetti dei geni nucleari sembrano causare o modificare i fenotipi associati con delezioni o con mutazioni puntiformi dell'mtDNA.
Infine, gli studi futuri dovranno tendere a sviluppare nuovi approcci per la terapia di pazienti afflitti dalle rare ma gravi malattie che si manifestano in seguito a difetti nell'mtDNA. La sostituzione diretta del genoma mitocondriale mutante con quello normale è concettualmente molto attraente, ma le tecnologie attuali non permettono di realizzarla, nemmeno in esperimenti dimostrativi compiuti su cellule in coltura. Le tecniche per l'espressione stabile di transgeni nucleari continuano a migliorare, sebbene i metodi attuali siano inadeguati per essere applicati, nell'uomo, alle cellule del sistema nervoso centrale e dei muscoli striati, le più rilevanti per le manifestazioni cliniche delle malattie genetiche mitocondriali. Tuttavia, progressi nella comprensione della replicazione e dell' espressione dei geni mitocondriali potranno suggerire nuove strategie con cui i prodotti dei transgeni nucleari, diretti ai mitocondri mediante il ben noto meccanismo di importazione delle proteine, possano modificare il fenotipo determinato dalle mutazioni genetiche mitocondriali. Tali strategie potranno, infine, fornire una base per nuovi approcci biotecnologici alla terapia delle malattie genetiche del mitocondrio.
Bibliografia citata
ANDERSON, S., et al. (1981) Sequence and organization of the human mitochondrial genome. Nature, 290, 457-465.
AZPIROZ, R., BUTOW, R.A. (1993) Patterns of mitochondrial sorting in yeast zygotes. Mol. Biol. of the CelI, 4, 21-36.
BENDICH, A.J. (1996) Structural analysis of mitochondrial DNA molecules from fungi and plants using moving pictures and pulsed-field gel electrophoresis. J. Mol. Biol., 255, 564-588.
DIECKMANN, C.L., STAPLES, R.R. (1994) Regulation of mitochondrial gene expression in Saccharomyces cerevisiae. Intl. Rev. Cyt., 152, 145-181.
GRIVELL, L.A. (1995) Nucleo-mitochondrial interactions in mitochondrial gene expression. Crit. Rev. Biochem. Molec. Biol., 30, 121-164.
KING, M.P., ATTARDI, G. (1989) Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science, 246, 500-503.
LAMBOWITZ, A.M., BELFORT, M. (1993) Introns as mobile genetic elements. Annu. Rev. Biochem., 62, 587-622.
LOCKSON, D., ZWEIFEL, S.G., FREEMANCOOK, L.L., LORIMER, H.E., BREWER, B.J., FANGMAN, W.L. (1995) A role for recombination junctions in the segregation of mitochondrial DNA in yeast. CelI, 81, 947-955.
LORIMER, H.E., BREWER, B.J., FANGMAN, W.L. (1995) A test of the transcription model for biased inheritance of yeast mitochondrial DNA. Mol. CelI. Biol., 15, 4803-4809.
LUFT, R. (1994) The development of mitochondrial medicine. Proc. Natl. Acad. Sci. USA, 91, 8731-8738.
MARGOSSIAN, S.P., LI, H., ZASSENHAUS, H.P., BUTOW, R.A. (1996) The DExH box protein Suv3p is a component of a yeast mitochondrial 3' - to - 5' exoribonuclease that suppresses group I intron toxicity. CelI, 84, 199-209.
MORALES, M.J., DANG, Y.L., LOU, Y.K., SULO, P., MARTIN, N.C. (1992) A 105-kDz protein is required for yeast mitochondrial RNase P activity. Proc. Natl. Acad. Sci. USA, 89, 9875-9879.
SCHMID, S.R., LINDER, P. (1992) D-E-A-D protein family of putative RNA helicases. Mol. Micro., 6, 283-292.
SCHON, E.A., RIZZUTO, R., MORAES, C.T., NAKASE, H., ZEVIANI, M., DI MAURO, S. (1989) A direct repeat is a hotspot for largescale deletion of human mitochondrial DNA. Science, 244, 346-349.
SHYJAN, A.W., BUTOW, R.A. (1993) Intracellular dialogue. Curr. Biol., 3, 398-400.
STRAUSBERG, R.L., PERLMAN, P.P. (1978) The effect of zygotic bud position on the transmission of mitochondrial genes in Saccharomyces cerevisiae. Mol. Gen. Genet., 163, 131-144.
WALLACE, D.C. (1992) Diseases of the mitochondrial DNA. Annu. Rev. Biochem., 61, 1175-1212.
WEEKS, K.M., CECH, T.R. (1995) Assembly of a ribonucleoprotein catalyst by tertiary structure capture. Science, 271, 345-348.
ZEVIANI, M., SERVIDEI, S., GELLERA, C., BERTINI, E., DIMAURO, S., DIDONATO, S. (1989) An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D100p region. Nature, 339, 309-311.