
Vitamine
Vitamine
di Paolo Cerletti
Vitamine
sommario: 1. Introduzione. 2. Tiamina. 3. Riboflavina. 4. Piridossina. 5. Vitamina B12. 6. Acido folico. 7. Acido nicotinico. 8. Acido pantotenico. 9. Biotina. 10. Vitamina C. 11. Altri fattori idrosolubili. 12. Vitamina A. 13. Vitamina D. 14. Vitamina E. 15. Vitamina K. □ Bibliografia.
1. Introduzione
Le vitamine costituiscono un gruppo eterogeneo di composti a peso molecolare relativamente basso, indispensabili in quantità traccia, come tali o sotto forma di derivati, per processi vitali essenziali; l'organismo umano e gli animali superiori, tuttavia, non sono in grado di sintetizzarle e devono quindi assumerle tramite gli alimenti. Esse sono invece sintetizzate da microrganismi e dai vegetali.
Alcune vitamine per la loro solubilità in acqua sono dette idrosolubili. Queste sono la tiamina, la riboflavina, la Piridossina, la vitamina B12, gli acidi folico, nicotinico e pantotenico, la biotina, la vitamina C. Eccetto quest'ultima, tutte le vitamine idrosolubili vanno a costituire la molecola di coenzimi, cofattori indispensabili a reazioni chiave del metabolismo. Il loro ruolo chimico è generalizzato alla totalità o quasi delle cellule, sia animali sia vegetali, comprese quelle dei microrganismi, indipendentemente dalla capacità che esse hanno di sintetizzarle o meno. Altri fattori di crescita idrosolubili, come l'inositolo, la colina, la carnitina, l'acido lipoico, non sono in genere classificati tra le vitamine, in quanto sono presenti in quantità maggiori ovvero, per taluni, l'uomo non ha dipendenza alimentare.
Le restanti vitamine, A, D, E, K, estraibili con solventi per i lipidi, sono dette liposolubili. Un loro ruolo essenziale in piante e microrganismi non è stabilito. Non appaiono esser parte di coenzimi, ma funzionano secondo altre modalità che richiedono comunque solo quantità traccia.
La possibile biosintesi di specifiche vitamine a opera di popolazioni batteriche simbionti rende le varie specie animali più o meno dipendenti dalla dieta per il loro rifornimento. Intervengono fattori diversi: ad esempio la biosintesi intestinale di vitamina K è inadeguata nei polli per il passaggio troppo rapido della massa alimentare attraverso l'intestino e perché, avvenendo la sintesi nel tratto distale dell'intestino, l'assorbimento è insufficiente. Nei Ruminanti il contributo dei batteri del rumine dipende dal loro grado di sviluppo e di attività. Inoltre la richiesta delle singole vitamine è diversa secondo l'età, il sesso, lo stato fisiologico, l'attività fisica, la razza.
La composizione biochimica particolare di ogni tessuto determina una specifica richiesta e diverse affinità per una data vitamina; da ciò derivano, nel caso di deficienza, effetti distinti per i vari tessuti di uno stesso organismo o tessuti di specie diverse, cioè sintomatologie diverse e caratteristiche. Questa situazione è ben evidente per alcune vitamine idrosolubili. In casi in cui il meccanismo molecolare è stato sufficientemente chiarito, in condizioni di deficienza si possono evidenziare alterazioni di vie metaboliche e modificati equilibri di metaboliti, che scompaiono in seguito alla somministrazione della vitamina senza lasciare una traccia organica, talché si parla di ‛lesioni biochimiche'.
L'osservazione che determinate malattie sono collegate alla dieta e che certi alimenti hanno efficacia terapeutica è vecchia di secoli. Così, ad esempio, fin dal Medioevo si stabili l'utilità della somministrazione di fegato nella cecità crepuscolare; oggi sappiamo che ciò è dovuto al suo contenuto di vitamina A. Nel Settecento si cominciò a usare l'olio di fegato di merluzzo, ricca fonte di vitamina D, per la cura del rachitismo. Tra la fine del XVIII e l'inizio del XIX secolo la Marina britannica introdusse l'uso del succo di agrumi, apportatore di vitamina C, per prevenire lo scorbuto. Osservazioni del genere si moltiplicarono nel corso dell'Ottocento. Nel 1912 G.F. Hopkins, a Oxford, affermò la necessità nella dieta di fattori accessori oltre alle proteine, ai lipidi, ai carboidrati e ai sali minerali, e nello stesso anno C. Funk, a Londra, isolò dal pericarpo del riso un concentrato contenente un'ammina, la vitamina B1, efficace nel curare il beri-beri. Poco dopo E.V. McCollum, negli Stati Uniti, stabilì che i ratti richiedono fattori di crescita idro- e liposolubili la cui mancanza agisce anche sui caratteri della cute, del pelo, ecc.
L'isolamento, la caratterizzazione chimica e la sintesi delle singole vitamine avvenne tra gli anni venti e quaranta e per molte di esse, soprattutto le idrosolubili, si accompagno alla evidenziazione del loro ruolo determinante nel chimismo cellulare. Queste scoperte suscitarono un entusiasmo di cui non godettero altri composti egualmente indispensabili all'organismo e per i quali dipendiamo dall'alimentazione, per esempio gli amminoacidi essenziali. Indubbiamente ai componenti tradizionali della dieta si aggiungeva un gruppo nuovo di sostanze metabolicamente multipotenti, in genere innocue se iperdosate. Inoltre la constatazione, invero sorprendente, delle quantità piccolissime, per talune dell'ordine dei microgrammi giornalieri, richieste nella dieta portò ad attribuire a questi composti virtù pressoché magiche. Sembrò possibile ottenere, una volta conosciuti questi fattori, un sostanziale miglioramento nell'alimentazione umana ovviando a condizioni di parziale deficienza in taluni casi effettive ma in altri ipotetiche. In realtà le prescrizioni poli- e ipervitaminiche che per un lungo periodo furono di moda si rivelarono ingiustificate, salvo casi specifici, ben caratterizzati, di ipovitaminosi. Viceversa il progredire della ricerca sul meccanismo d'azione delle vitamine ne definiva il ruolo centrale nel chimismo cellulare.
Nella sintetica trattazione che faremo di ciascuna vitamina cercheremo di metterne in luce il ruolo biologico e insieme le possibilità che l'uomo ha di provvedere ai fabbisogni relativi.
2. Tiamina
La tiamina è detta anche vitamina B1 o aneurina. È il cloridrato del 3-(4-ammino-2-metilpirimidinil-5-metil)-4-metil-5(β-idrossietil)tiazolo cloruro. Venne isolata in forma cristallina nel 1926 da C. P. Jansen e W. F. Donath. R. R. Williams ne realizzò la sintesi totale nel 1936.
Chimica
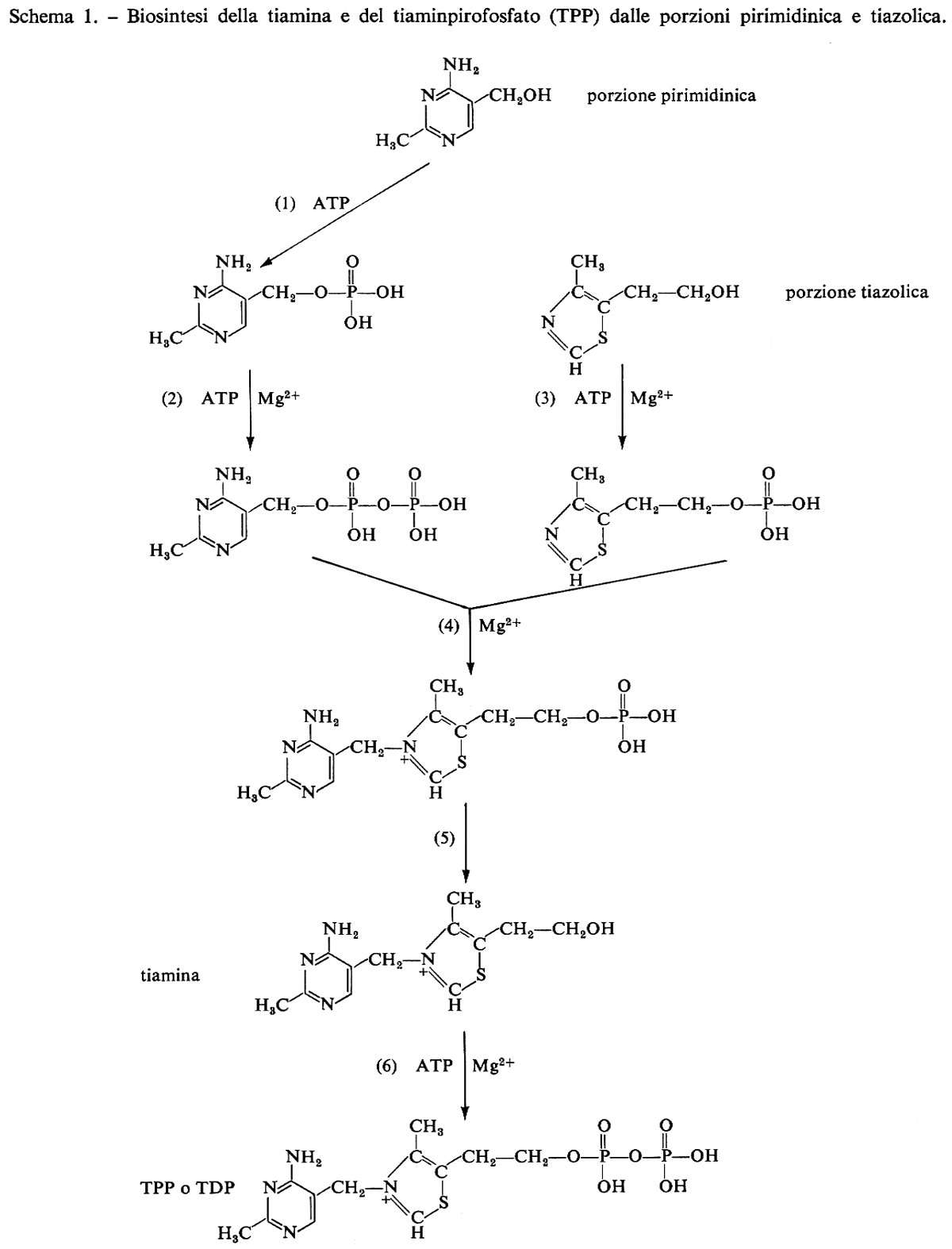
La tiamina consiste di una parte pirimidinica e una tiazolica unite da un ponte −CH2− (v. schema 1). Dà cristalli monoclini, p.f. 250 °C, con odore caratteristico, leggermente amari, stabili all'ossigeno. È insolubile in etere e in altri solventi dei lipidi, poco solubile in alcool. Forma in acqua soluzioni debolmente acide. A pH minore di 5 è relativamente stabile al calore e all'ossidazione; a pH superiore è alterata all'ebollizione o autoclavando; a pH 7 o maggiore si inattiva completamente anche se conservata a temperatura ambiente. Trattata con solfito, si spezza in una parte pirimidinica e una tiazolica. In soluzione fortemente alcalina il ferricianuro la ossida a tiocromo, sostanza la cui forte fluorescenza blu viene utilizzata per determinare la tiamina. Tiamina può essere prodotta sintetizzando separatamente il nucleo pirimidinico e quello tiazolico e poi unendoli, ovvero preparando uno dei due nuclei con un'apposita appendice laterale su cui successivamente viene costruito l'altro. Per la sintesi industriale vengono seguiti entrambi i metodi, ma preferibilmente il primo.
Ruolo biochimico
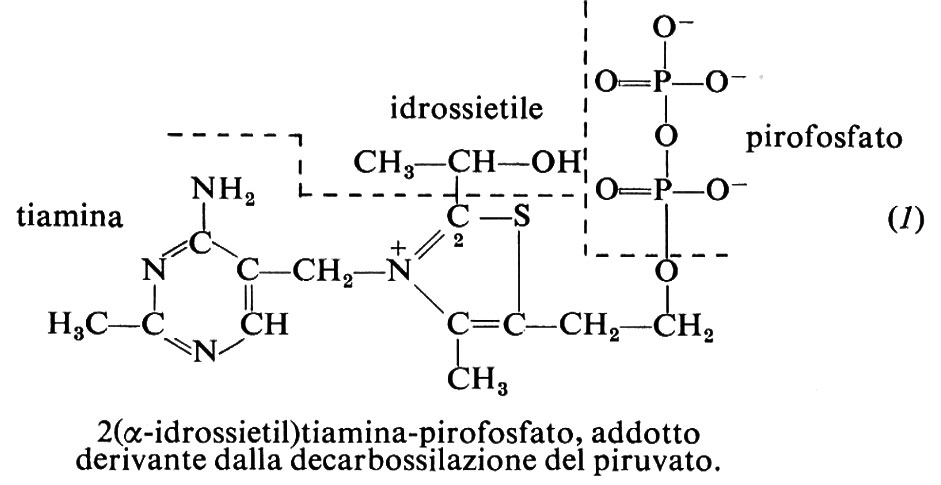
Il pirofosfato di tiamina, TPP, o tiamindifosfato, TDP, è il coenzima in reazioni chiave del metabolismo dei carboidrati, quali la decarbossilazione degli α-chetoacidi e la formazione o degradazione di α-chetoli. In ambedue i casi il meccanismo si basa sulla formazione di un carbanione sul C2 dell'anello tiazolico, il quale addiziona il carbonio carbonilico del composto da trasformare; a questo segue, con gli α-chetoacidi, la decarbossilazione. Nel caso del piruvato, secondo la proteina catalitica, l'addotto (1) si scinde, liberando acetaldeide, per esempio nella fermentazione alcolica, ovvero il gruppo idrossietilico viene trasferito all'acido lipoico (v. cap. 11) e da questo è ossidato nelle reazioni preliminari al ciclo degli acidi tricarbossilici. Nel ciclo stesso con meccanismo analogo avviene la decarbossilazione ossidativa dell'acido α-chetoglutarico a succinil-coenzima A. Nella formazione o degradazione dei chetoli, il frammento bicarbonioso del chetosio donatore, che nell'addotto è glicolaldeide attivata, viene trasferito a un aldosio accettore: così, per es., la transchetolasi forma sedoeptulosio-7-fosfato da xilulosio-5-fosfato e ribosio-5-fosfato o viceversa, reazioni coinvolte nell'utilizzazione dei carboidrati via i pentosi (v. metabolismo dei carboidrati) e nella loro formazione post-fotosintetica nel ‛ciclo di Calvin' (v. fotosintesi).
La tiamina viene assorbita nel duodeno, è fosforilata nelle cellule della mucosa ed è così trasportata al fegato. I depositi qui e nei vari tessuti sono modesti, per cui è necessario un apporto giornaliero. La vitamina sintetizzata dalla flora intestinale è poco disponibile per l'uomo e contribuisce solo in piccola parte al fabbisogno. La tiaminasi, enzima presente nei microrganismi intestinali e in certi pesci, crostacei e molluschi, inattiva la vitamina separando i due anelli: ne consegue la ‛paralisi di Chastek' in animali, per esempio carnivori da pelliccia, allevati con organi addominali, tra cui gli intestini, residuati da macellazione.
Biosintesi
Diversi microrganismi possono utilizzare invece della tiamina le sue parti pirimidinica e tiazolica o singolarmente o assieme. Negli animali superiori alcuni tessuti sono in grado di formare tiamina, in piccolissime quantità, a partire dai due anelli.
I passi metabolici che portano alla sintesi di tiaminpirofosfato a partire dai nuclei pirimidinico e tiazolico sono indicati nello schema 1.
La porzione pirimidinica della tiamina non viene formata seguendo la via che porta alla biosintesi delle pirimidine degli acidi nucleici, bensì secondo una via comune con le purine, che ha come intermedio il 4-ammidinoimidazoloribonucleotide. Le tappe che conducono da questo composto alla pirimidina non sono ancora tutte note: verosimilmente si ha rottura dell'anello imidazolico tra C4 e CS, associazione di C4 con una unità bicarboniosa e di C2 con un metile, distacco del ribosiofosfato e chiusura dell'anello pirimidinico. La via seguita nella biosintesi della parte tiazolica è ancora molto oscura.
Distribuzione
Tiamina è presente in quasi tutti gli organismi viventi, soprattutto come pirofosfato, ma anche in forma libera e come estere mono- e trifosforico e come mono- e disolfuro. Nelle piante la forma più abbondante è la tiamina libera. Negli animali, uomo incluso, le concentrazioni più elevate si riscontrano (in μg per g di tessuto fresco) nel cuore (2,8-7,9), fegato (2,0-7,6), rene (2,4-5,8) e cervello (1,4-4,4). Contenuti rappresentativi in alimenti sono, in mg/100 g: muscolo di maiale (0,6-0,8), legumi secchi (0,68), cereali integrali (0,37), latte (0,04), lievito alimentare (1,8), germe di grano (2,0). Lo sbiancamento delle farine provoca una cospicua perdita di tiamina. Anche la cottura distrugge dal 30 al 50% della vitamina.
Diffusi nei vegetali sono fattori termostabili che distruggono la tiamina: in genere flavonoidi, o derivati fenolici, chinoni e catecoli. Se presenti nell'alimento agiscono alla temperatura di cottura ma non dopo l'ingestione dell'alimento crudo, in quanto la temperatura corporea è troppo bassa per una loro azione significativa.
Fabbisogno
Dipende dal contenuto calorico e in carboidrati della dieta: quando i grassi hanno una parte importante nella dieta, la richiesta di tiamina diminuisce. L'U. S. National Research Council raccomanda un apporto giornaliero di 0,5 mg/1.000 calorie. La richiesta del bambino, tenuto conto del peso, è analoga a quella dell'adulto e può essere valutata in 0,4 mg/kg. Nel lattante il latte materno è talora insufficiente a fornire questa quantità, dato che il suo contenuto in tiamina può variare considerevolmente (0,09-0,2 mg/1). Il latte vaccino contiene una quantità leggermente maggiore di tiamina (0,35-0,40 mg/1), ma parte di questa viene distrutta nella pastorizzazione. Pertanto i margini di sicurezza sono scarsi.
Ricerche sugli animali hanno indicato che il fabbisogno di tiamina è anche influenzato dallo stato fisico (ipertiroidismo, gravidanza, lattazione, febbre, età, ecc.), temperatura ambiente, microflora intestinale o del rumine, fattori genetici individuali, lavoro muscolare.
La sintomatologia da deficienza si manifesta soprattutto a carico del tessuto nervoso e di quello cardiaco, che dipendono esclusivamente o quasi dalla glicolisi e quindi dal metabolismo del piruvato come sorgente di energia. Si hanno anche disturbi nel tratto gastrointestinale. La sindrome più caratteristica è il beri-beri, in cui la nevrite si associa a insufficienza cardiaca. Si possono avere forme ‛secche', in cui predominano le manifestazioni polinevritiche, e forme ‛umide', specificate dall'insufficienza cardiaca con edema. Nei piccioni la polinevrite dà il caratteristico ‛opistotono'. La pur cospicua sintomatologia scompare, senza lasciare lesioni istologiche, se si somministra la vitamina.
Nella deficienza di tiamina, all'alterazione dei livelli della vitamina nei liquidi biologici - nel sangue si passa dai 6,5-16,5 μmg/100 ml dei soggetti normali a valori sotto i 5 μg/100 ml - si associa un accumulo ematico di acido piruvico e un abbassamento dell'attività transchetolasica eritrocitaria. Tale ‛patologia nutrizionale biochimica' si manifesta quando la quantità della vitamina scende a valori per cui i meccanismi omeostatici vengono trascesi e si instaurano alterazioni nel metabolismo intermedio; per valutare gli stati di deficienza della vitamina si saggiano l'attività della transchetolasi nonché gli effetti delle somministrazioni di TPP.
Farmacologia e tossicologia
Somministrata endovena in dosi che, secondo l'animale, variano da 125 mg/kg nel ratto a 600 mg/kg nella scimmia, la tiamina ha effetti letali che conseguono alla paralisi del centro respiratorio. Dosi inferiori provocano caduta pressoria e broncocostrizione. Una componente importante nell'azione della tiamina è centrale, tuttavia la tiamina blocca la trasmissione dell'impulso nervoso anche a livello dei gangli e, a dosi più alte, a livello della giunzione neuromuscolare. Somministrata per bocca la tiamina non ha effetto tossico sull'uomo. In pazienti che già l'avevano ricevuta per via parenterale, in occasione di una nuova somministrazione per la stessa via si sono osservate reazioni che somigliano allo shock anafilattico.
Analoghi
I requisiti strutturali per l'attività vitaminica sono assai specifici: solo i 2′-etil- e 2′-n-propil- analoghi della tiamina hanno effetti biologici confrontabili con quelli della vitamina. Vi sono tiamine modificate che possono esser trasformate nella vitamina da processi metabolici. Tali il cosiddetto tiamin-disolfuro, che è un derivato della forma a tiolo aperto della tiamina, e l'allitiamina, un artefatto derivante dalla reazione di allina con tiamina. Questo composto e suoi analoghi rappresentano forme particolarmente stabili della vitamina, che sono meglio assorbite. La Piritiamina, in cui l'anello tiazolico è sostituito dalla piridina, e l'ossitiamina, in cui l'anello pirimidinico ha in posizione 2 un idrossile invece di un ammino-gruppo, competono verosimilmente con il TPP per gli enzimi cui si lega, ma non hanno attività catalitica, per cui provocano sintomi di deficienza. Un'altra tiamina modificata, l'amprolio [cloridrato di 1-(4-ammino-2-n-propil-5-pirimidinilmetil)-2-picolinio cloruro] influenza l'assorbimento della tiamina ed è usato come profilattico contro la coccidiosi nei polli.
3. Riboflavina
La riboflavina è detta anche vitamina B2 o lattoflavina. Identificata da Paul György nel 1927, venne isolata e cristallizzata da Theodor Wagner-Jauregg nel 1932. La sintesi fu realizzata da R. Kuhn ed E. Weygand e da P. Karrer nel 1934.
Chimica
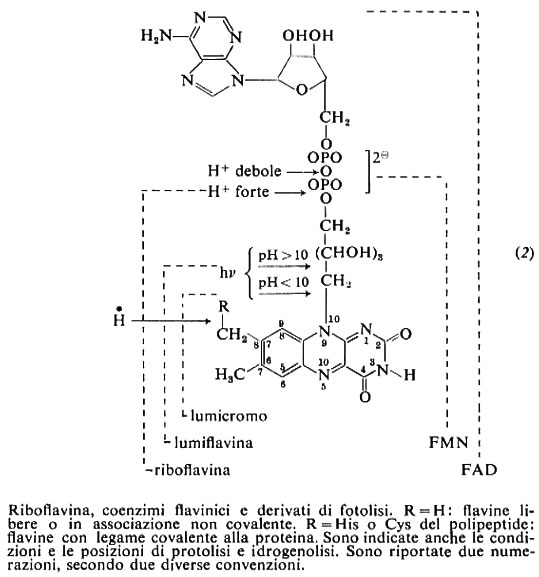
La riboflavina è un derivato dell'isoallossazina con catena laterale ribitilica su N-9 (2). Poco solubile in acqua (12 mg/100 ml a 25 °C), cristallizza da acido acetico 2N; in alcali è solubile, ma instabile. Ha carattere anfotero: Ka=6,3×10-12, Kb=0,5×10-5, pI a pH 6,0. Soluzioni neutre hanno colore giallo-verde e intensa fluorescenza gialla, che è utilizzata per le valutazioni qualitative e quantitative.
Sodio ditionito e altri riducenti la riducono a una diidroflavina pressoché incolore, che si riossida all'aria; Em=−0,185 V a pH 7,0. Illuminata in assenza di ossigeno a pH neutro, subisce un'ossidoriduzione intramolecolare con riduzione dell'anello isoallossazinico e distacco del ribitile. In presenza di O2 la fotolisi dà, in ambiente acido o neutro, lumicromo con distacco del ribitile, in ambiente alcalino, lumiflavina, che al posto del ribitile contiene un metile (2).
La parte isoallossazinica dà chelati radicali con metalli, e complessi, verosimilmente a trasferimento di carica, con adenina, caffeina e altre purine, con indolo, triptofano, serotonina, clortetraciclina e fenoli, ma non con altri derivati benzenici. Nel FAD (v. sotto) il complesso è intramolecolare.
La riboflavina è ottenibile per sintesi seguendo varie vie, alcune di applicazione industriale.
Ruolo biochimico
Forme attive della riboflavina sono i coenzimi flavinadenindinucleotide (FAD) e flavinmononucleotide (FMN) (2). Essi sono presenti ubiquitariamente nelle cellule, con prevalenza del FAD. In alcuni enzimi il legame del FAD alla proteina è covalente tramite il metile in 8 dell'anello isoallossazinico. Il FAD e l'FMN fungono da coenzimi di enzimi ossidoriduttivi. Dato il loro basso potenziale redox (a pH 7,0 EmFAD=−0,219 volt, EmFMN=−0,186 volt), sono intermedi tra l'ossidazione dei piridinnucleotidi ridotti o di substrati e la riduzione dei citocromi o dell'ossigeno molecolare. L'associazione con proteine diverse ne modifica grandemente il potenziale e consente l'accoppiamento con sistemi donatore-accettore diversi. Il FAD e l'FMN intervengono come trasportatori bielettronici nelle ossigenasi, deidrogenasi e ossidasi; d'altra parte la possibilità, subita la riduzione bielettronica, di riossidarsi in due passaggi monoelettronici tramite la formazione di un radicale, consente alla flavina di fungere da intermedio tra un donatore bielettronico (substrato o piridinnucleotide) e un accettore monoelettronico come per esempio i gruppi ferro-zolfo di ferro-solfo-proteine, presenti talora nella flavoproteina stessa, o i citocromi nelle catene redox biologiche. Nelle flavodossine, la classe più semplice di flavoproteine, sia la riduzione che la riossidazione appaiono reazioni monoelettroniche e la flavina oscilla tra la forma completamente ridotta e il semichinone.
Biosintesi
Riboflavina è sintetizzata in piante, lieviti, funghi inferiori e alcuni batteri. Gli animali superiori non sono in grado di sintetizzarla, ma molti di essi ospitano nell'intestino o nel rumine batteri in grado di fornire la vitamina. La isoallossazina origina dalle purine via la 6,7-dimetil-8-ribitillumazina. L'FMN si forma per fosforilazione della vitamina, donatore l'ATP, e da FMN e ATP si ha FAD, con liberazione di pirofosfato (difosfato) inorganico.
Distribuzione
Riboflavina è presente nell'occhio di molti pesci e di alcuni crostacei. Cristalli di riboflavina si riscontrano nel fungo Eremothecium ashbii e nel tapetum oculare del lemuride Galago crassicaudatus agisymbanus. Nei Mammiferi la vitamina libera è presente in quantità significative nella retina, nel siero di latte e nell'urina. In tutti gli altri tessuti prevalgono i coenzimi. Nelle piante è presente in maggior abbondanza nelle foglie. Le migliori sorgenti alimentari di riboflavina sono il latte, l'albume d'uovo, il fegato, il cuore, il rene e le foglie in crescita di vegetali. La relativa termostabilità preserva la riboflavina nel corso della cottura degli alimenti. La vitamina è invece rapidamente distrutta se esposta alla luce.
Fabbisogno
È influenzato dalla costituzione individuale, dall'età, dallo stato di salute, dalla dieta. L'apporto giornaliero raccomandato per un uomo adulto di 70 kg è di 1,7 mg. Per la donna gravida e i bambini la quantità relativa al peso è leggermente maggiore. Nel ratto, nel cane e nella volpe l'ariboflavinosi provoca lesioni oculari, manifestazioni neuropatologiche con degenerazione della mielina nel midollo spinale e paralisi parziale alle gambe, alterazioni istologiche della pelle, granulocitopenia e modificazioni del ciclo riproduttivo. Nei giovani Ruminanti la flora del rumine non è ancora sufficiente a fornire una quantità adeguata di vitamina: se l'apporto alimentare è insufficiente si sviluppano lesioni alla mucosa buccale, salivazione e lacrimazione eccessive, perdita di pelo, diarrea.
Nell'uomo l'ariboflavinosi provoca una sindrome pellagra-simile (pellagra sine pellagra) con stomatite angolare e seborrea nelle pliche cutanee del viso, analoghe a quelle che si manifestano in altre deficienze nutrizionali.
Farmacologia e tossicologia
La bassa solubilità della riboflavina la rende relativamente innocua. La vitamina è escreta prevalentemente tramite le feci. L'escrezione urinaria varia con la quantità assunta con la dieta e il deposito nei tessuti. Alte dosi ritardano il manifestarsi delle lesioni epatiche prodotte da p-dimetilamminoazobenzene. La deficienza spinta di riboflavina rallenta la crescita di tumori negli animali da esperimento.
Analoghi della riboflavina ne antagonizzano l'azione: 1) competendo con il gruppo prostetico dei flavoenzimi, come tali o dopo trasformazione enzimatica in un analogo dell'FMN e del FAD: in questa categoria ricadono la D-araboflavina, la dibromoriboflavina e la duodoriboflavina; 2) inibendo competitivamente la fosforilazione della riboflavina e quindi la formazione del coenzima; così agiscono, per esempio, la lumiflavina e la 2′, 3′, 4′-tridesossiriboflavina.
Alcune sostanze ad azione antimalarica, per esempio la mepacrina (Atabrina), pirimidine sostituite e la chinina, inibiscono l'effetto di crescita della riboflavina sui microrganismi; tuttavia l'efficacia non è correlata alla loro somiglianza con il nucleo flavinico. Clortetraciclina, cloramfenicolo, streptomicina, penicillina e clorpromazina si legano al FAD formando un complesso che inibisce la D-amminoacidossidasi, enzima FAD-dipendente. A meccanismi analoghi sembra siano da attribuire le manifestazioni di ariboflavinosi che si osservano in seguito alla somministrazione di antibiotici, le modificazioni elettroencefalografiche conseguenti all'assunzione di clorpromazina e l'azione disaccoppiante sulla fosforilazione ossidativa di questo composto e dell'1,1,3-triciano-2-ammino-1-propene.
4. Piridossina
La piridossina è detta anche vitamina B6. Venne isolata e cristallizzata nel 1938 e l'anno successivo fu ottenuta per sintesi. In mezzo acido è stabile al calore, mentre in soluzione alcalina è relativamente instabile. È fortemente instabile alla luce. È assorbita nell'alto intestino tenue e viene escreta soprattutto come acido piridossico.
Ruolo biochimico
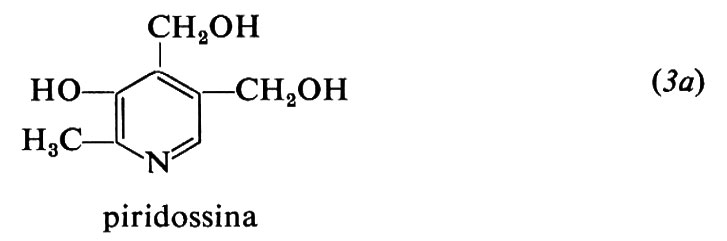
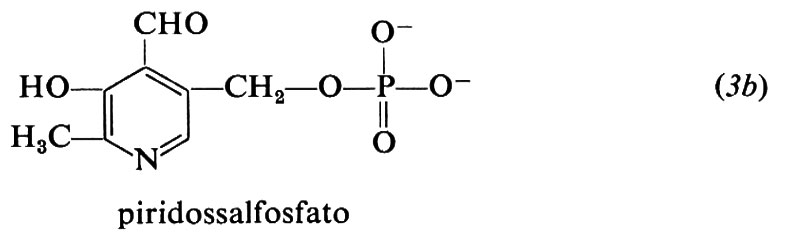
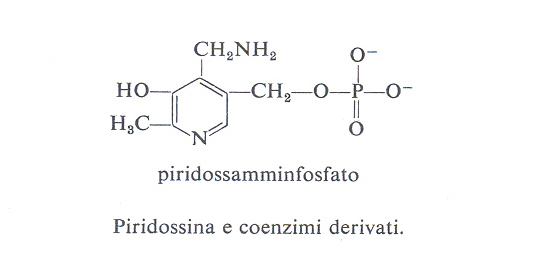
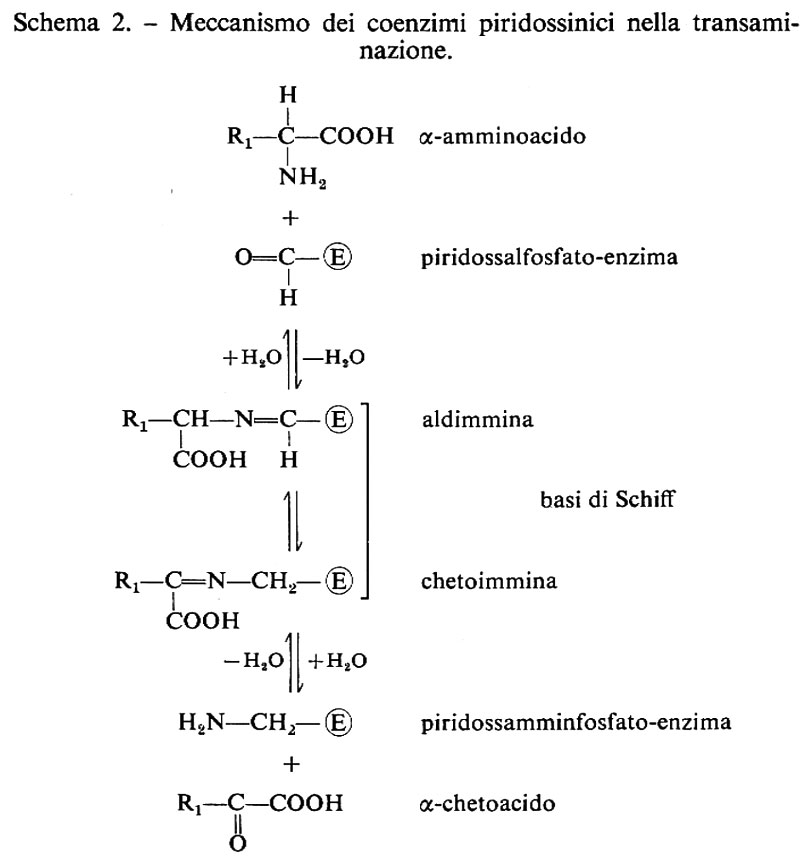
Le forme biologicamente attive della piridossina (3a) sono il piridossalfosfato (PLP) (3b) e il piridossamminfosfato (3c). Esse sono coinvolte come coenzimi in diversi aspetti del metabolismo degli α-amminoacidi, quali la transaminazione, la decarbossilazione, la deidratazione della senna, il distacco dello zolfo della cisteina e reazioni di racemizzazione enzimatica che portano all'interconversione degli L- e D-amminoacidi. L'amminoacido forma una base di Schiff (4) con il coenzima e la struttura elettron-attrattrice dell'enzima labilizza selettivamente i legami a, b o c rendendo così possibili vari tipi di trasformazione.

Nella transaminazione (v. schema 2) il coenzima funziona da trasportatore di ammino-gruppo fra un amminoacido e un chetoacido e viceversa, oscillando tra la forma aldeidica (piridossale) e quella amminica (piridossammina): è questo un esempio tipico di reazione a doppio spostamento, di cui possiede la cinetica a ping-pong. La lisina o altro amminoacido basico presenti nel sito attivo facilitano la conversione aldo-cheto-imminica funzionando da elettron-attrattori. La transaminasi libera lega il PLP non solo via l'azoto pirimidinico, ma anche formando una base di Schiff tramite l'8-amminogruppo di una lisina del sito attivo; l'amminoacido da trasformare si associa al coenzima sostituendosi in questo legame.
Il PLP è anche coinvolto nel metabolismo del triptofano, in particolare è necessario per la trasformazione in acido nicotinico dell'acido xanturenico derivante dal triptofano.
Inoltre è necessario per la trasformazione dell'acido linoleico, un acido grasso essenziale, in arachidonico. La formazione degli sfingolipidi necessari per lo sviluppo della guaina mielinica attorno alle cellule nervose dipende anch'essa dalla vitamina B6.
Il PLP ha uno spettro di assorbimento e una fluorescenza che variano in modo caratteristico all'aggiunta dei diversi substrati: ciò consente di seguire la formazione e la scomparsa di complessi intermedi nella catalisi enzimatica.
Distribuzione e fabbisogno
Alto contenuto di vitamina B6 hanno (in mg/100 g) il lievito (2,0), il germe di grano (1,15), il fegato (0,83). Buon livello anche le carni (0,4-0,7). Basso le verdure e la frutta (0,05-0,2) e il latte (0,04).
Il fabbisogno è in rapporto con l'assunzione alimentare di proteine. La dose consigliata per gli adulti è di 2 mg al giorno; la richiesta è maggiore nel corso dell'allattamento.
In adulti che hanno assunto la desossipiridossina, un antagonista della B6, si è manifestata depressione, nausea, vomito, lesioni delle mucose, neurite periferica e dermatite seborroica insensibile a trattamento con acido nicotinico. Nella deficienza grave si instaurano sintomi a carico del sistema nervoso centrale, soprattutto nei bambini. Una forma di anemia sideroblastica connessa con impedita sintesi dell'eme risponde a terapia con B6. Infatti il PLP interviene nella formazione dell'acido δ-amminolevulinico, richiesto per la sintesi dell'eme. L'isoniazide, un chemioterapico antitubercolare, è un potente antagonista della B6 in quanto si combina con il PLP. L'escrezione urinaria della vitamina aumenta fortemente. Una situazione analoga è provocata dalla penicillammina.
5. Vitamina B12
La struttura base della vitamina B12 è detta cobalamina. G. R. Minot e W. P. Murphy riconobbero nel 1926 che l'anemia perniciosa può essere trattata nutrendo il paziente con forti dosi di fegato, ma il fattore responsabile, la cobalamina, fu purificato e cristallizzato solo nel 1948 da E. L. Smith in Inghilterra ed E. L. Rickes e K. Folkers negli Stati Uniti; la sua struttura fu chiarita nel 1956 da D. C. Hodgkin.
Chimica e biochimica
La vitamina B12 è formata da un anello corrinico con un atomo centrale di cobalto legato all'azoto dei quattro anelli pirrolici (5). Il quinto sostituente del cobalto è fuori del piano dell'anello corrinico, ed è un ribonucleotide nel quale la base, 5,6-dimetilbenzimidazolo, lega ribosio-3-fosfato esterificato da una catena laterale dell'anello pirrolico D. Il sesto sostituente (R), dall'altro lato del piano corrinico, può essere CN-, OH-, −CH3 o il desossiadenosile. Lo ione cianuro non è presente in vivo e viene ad associarsi nel corso della purificazione; la cianocobalamina è la forma commerciale più comune della vitamina. È inattivata da alcali e acidi diluiti, luce, agenti ossidanti e riducenti. Circa il 30% dell'attività vitaminica è perduto nella cottura.
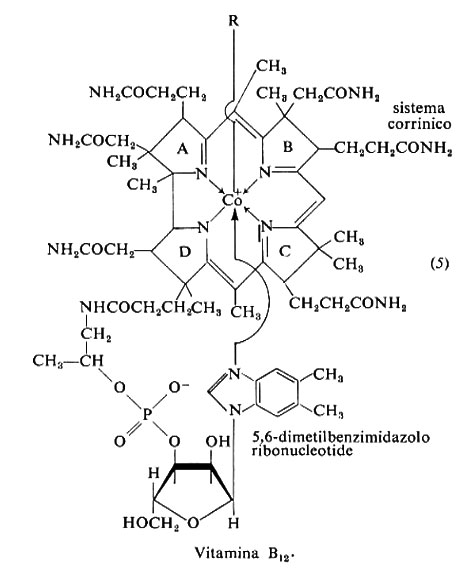
Lo stato di ossidazione del cobalto è +3 nella idrossocobalamina, che è indicata come B12a (Co3+). Due reduttasi flavoproteiche che utilizzano NADH come riducente la riducono successivamente a B12r (Co2+), che contiene cobalto bivalente, e poi a B12s (Co+). Su quest'ultima viene trasferito dall'ATP un gruppo 5-desossiadenosinico e si forma la 5′-desossiadenosil-cobalamina, che nella metilmalonil-CoA-mutasi funge da trasportatore intermedio nello scambio tra un idrogeno di un atomo di carbonio e il gruppo sostituente di un carbonio vicinale. In questo modo il malonil-CoA, che si forma nella degradazione degli acidi grassi a numero dispari di atomi di carbonio e di alcuni amminoacidi (treonina, isoleucina, metionina e valina), origina succinil-CoA e viene avviato alla degradazione terminale dello scheletro carbonioso. Un intervento analogo sembra si abbia nella riduzione di ribonucleotidi a 2-desossiribonucleotidi.
Altro coenzima derivante dalla vitamina B12 è la metilcobalamina, in cui un gruppo metile è il sesto sostituente del cobalto; esso funge da intermedio nel trasferimento di un metile dall'N5-metiltetraidrofolato a un limitato numero di accettori, in particolare la omocisteina, che diventa metionina.
Alcuni batteri anaerobi contengono la pseudovitamina B12, un analogo della cobalamina in cui il dimetilbenzimidazolo è sostituito da adenina. Il coenzima da esso derivato ha il 5-desossiadenosile come legante del cobalto.
Fisiologia, distribuzione e fabbisogno
Animali e piante sono incapaci di produrre cobalamina. Sembra che essa sia sintetizzata esclusivamente dai microrganismi, specie gli anaerobi. Per l'assorbimento della cobalamina lo stomaco secerne una specifica glicoproteina, detta ‛fattore intrinseco', che si associa alla vitamina nel lume intestinale: il complesso viene poi legato a un fattore specifico nelle cellule della mucosa dell'ileo. Esso ne è dissociato da un fattore di rilascio; infine la vitamina, associata a proteine dette trans-cobalamine I e II, subisce un trasporto attivo attraverso la mucosa ileale fino al sangue. La deficienza di fattore intrinseco nell'anemia perniciosa e nella resezione chirurgica delle parti di stomaco che lo secernono (fundus e cardias), come anche l'asportazione di porzioni dell'ileo, compromettono l'assorbimento della cobalamina. Deficienza di vitamina B12 si può avere in malattie intestinali e in sindromi di malassorbimento. La deficienza causa demielinizzazione delle fibre nervose nel midollo spinale e arresta la maturazione degli eritrociti. La migliore terapia consiste nell'iniettare intramuscolo la vitamina a intervalli mensili.
Il contenuto in cobalamina (in μg/100 g) di alcune sorgenti alimentari rappresentative è il seguente: fegato di bue 80, ostriche 18, carne 2,6, uovo 2,0, latte intero 0,4. Il latte pastorizzato e quello evaporato perdono dal 40 al 90% della vitamina.
Dosi giornaliere raccomandate sono 3 μg per gli adulti, 4 μg durante la gravidanza e l'allattamento. Carenze nutrizionali sono rare, in quanto la vitamina è presente praticamente in tutti i tessuti animali, e occorrono diversi anni per esaurire il deposito organico, che nell'uomo si aggira sui 2 mg. La sintesi a opera della flora intestinale avviene nel colon, per cui la vitamina prodotta non è assorbita.
Esistono forme ereditarie - basate, si pensa, su un difetto della transferasi che catalizza la sintesi della desossiadenosil-cobalamina - che si manifestano nel primo anno di vita con acidosi ematica ed eliminazione urinaria di acido metilmalonico. Esse rispondono parzialmente a forti dosi di cobalamina.
6. Acido folico
L'acido folico viene anche indicato come acido pteroilglutammico o folacina (6). Fu sintetizzato nel 1946.

In alcune specie animali il residuo pteroico è legato a più molecole di acido glutammico associate con legame peptidico tramite il γ-carbossile. Alcuni organismi sono in grado di sintetizzare acido folico una volta che dispongano di acido p-amminobenzoico.
L'acido folico tramite due successive riduzioni è trasformato nel coenzima acido 5,6,7,8-tetraidrofolico (FH4). Questo trasporta, legati a N-5 e/o N-10, gruppi idrossimetile (−CH2OH), formile (−CHO) o metile (−CH3) in numerose reazioni enzimatiche connesse con il metabolismo di amminoacidi, purine e pirimidine, nelle quali tali gruppi sono trasferiti da uno a un altro metabolita o sono interconvertiti.
Il sintomo biochimico più cospicuo della deficienza di acido folico consiste in difficoltà nella sintesi delle purine e della timina, una pirimidina. Nei Mammiferi la deficienza si manifesta con riduzione della crescita e svariate forme di anemia.
L'acido folico è ampiamente distribuito nelle piante, in particolare nelle foglie. E molto diffuso negli alimenti, soprattutto nella forma poliglutammica. I contenuti più alti sono nel fegato, nei fagioli e nei vegetali verdi, soprattutto gli spinaci. Buone fonti anche le patate e il pane integrale. Poveri ne sono la frutta, molte carni, latte e uova.
La forma poliglutammica, una volta ingerita, è degradata al composto monoglutammico, che è assorbito a livello intestinale ed è depositato nel fegato. Si ha anche sintesi di folato a opera della flora intestinale.
La vitamina è instabile al calore in mezzo acido. Se ne perde assai nei vegetali durante la conservazione a temperatura ambiente e in tutti gli alimenti durante la cottura. Il latte essiccato, per esempio, è privo di acido folico.
L'assunzione giornaliera consigliata è di 0,4 mg per gli adulti, 0,8 mg in gravidanza e 0,6 mg in allattamento. Stati di malattia e alcolismo aumentano la richiesta di acido folico.
Forme di anemia sensibili a terapia con acido folico sono presenti nelle regioni tropicali, ovvero si osservano in donne gravide o in neonati di madri deficienti della vitamina. L'acido folico allevia lo stato anemico nell'anemia perniciosa, ma non agisce sui sintomi gastrointestinali e neurologici.
La deficienza di acido folico rallenta la crescita, causa disturbi ematici e gastrointestinali, glossite e altri sintomi. Deficienza si può instaurare nelle sindromi di malassorbimento, malnutrizione proteica, morbo di Hodgkin, con alcuni farmaci e in determinati carcinomi.
Alcuni farmaci, quali l'amminopterina e l'ametopterina (metotressato), per la loro somiglianza strutturale con l'acido folico inibiscono competitivamente la riduzione del diidrofolato a tetraidrofolato a opera della diidrofolatoriduttasi. Essi sono detti agenti antifolici e, bloccando la biosintesi delle basi, specie la timina, rallentano la biosintesi del DNA: l'effetto si nota in varie forme tumorali di cui viene rallentato lo sviluppo, in particolare in leucociti leucemici in trasformazione maligna e nel coriocarcinoma (v. chemioterapia antineoplastica).
7. Acido nicotinico
L'acido nicotinico (o niacina) (7a) e la nicotinammide (o niacinammide) (7b) sono anche chiamati vitamina PP (Pellagra Preventive). La pellagra fu riconosciuta come una malattia alimentare fin dagli anni 1915-1920, ma il fattore la cui carenza è responsabile della malattia fu identificato con l'acido nicotinico (composto già noto da tempo) solo nel 1937 da C. A. Elvehjem e D.W. Woolley, in seguito alla caratterizzazione, a opera di O. Warbury e W. Christian, nel 1934, della nicotinammide come componente dei due coenzimi piridinici.
Ruolo biochimico
I coenzimi contenenti nicotinammide (7b) sono il nicotinammide-adenin-dinucleotide (NAD+), una volta detto difosfo-piridin-nucleotide (DPN), e il NAD-fosfato (NADP+) o trifosfo-piridin-nucleotide (TPN). Nella loro ossidoriduzione, reversibile, sono coinvolti due atomi di idrogeno. Di essi uno è trasferito sotto forma di ione idruro ed è legato in posizione 4 all'anello nicotinammidico, dando le forme ridotte indicate con NADH e NADPH; l'altro diviene, o deriva da, un idrogenione: il potenziale dei coenzimi piridinici è quindi pH-dipendente. I piridinnucleotidi hanno basso potenziale redox; secondo il valore di potenziale del substrato, l'idrogeno è acquisito o ceduto dal coenzima. I nucleotidi piridinici hanno in soluzione due tipi di conformazione, aperta - o estesa - e chiusa, in cui gli anelli nicotinamrnidico e adeninico, planari, sono paralleli l'uno all'altro. Questa conformazione predomina in soluzione; il coenzima assume la forma aperta quando si lega alla proteina.
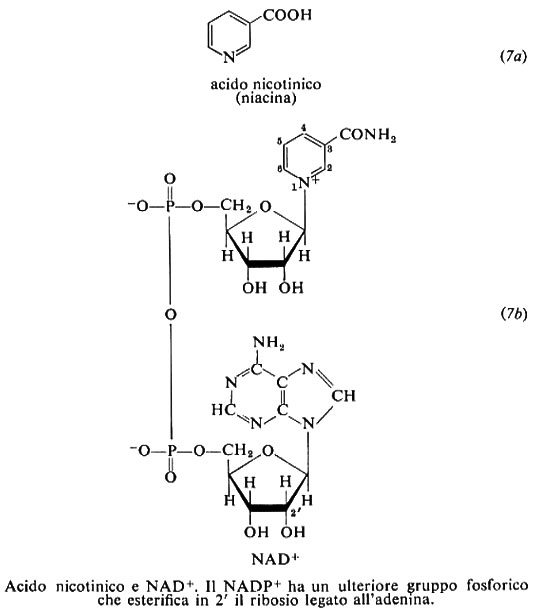
Le numerose ossidoriduttasi di cui i piridin-nucleotidi fungono da coenzimi sono indicate come deidrogenasi piridiniche. In genere esse sono specifiche per l'uno o l'altro dei due piridin-nucleotidi: il NAD prevale nelle deidrogenasi respiratorie, ed è presente in prevalenza nei mitocondri; il NADP interviene in vie biosintetiche. La maggior parte delle deidrogenasi piridiniche contiene, strettamente associati, ioni metallici divalenti. Il coenzima ridotto può agire da modulatore negativo per l'enzima, come per esempio il NADH per la isocitratodeidrogenasi mitocondriale. La forma ridotta del coenzima ha un caratteristico assorbimento a 340 nm, che viene utilizzato per seguire lo svolgersi delle reazioni.
Le piante e molti animali formano acido nicotinico da diversi precursori, specie il triptofano via l'acido 3-idrossiantranilico. Si valuta che per l'uomo 60 mg di triptofano equivalgano a 1 mg di niacina.
Distribuzione e fabbisogno
Materiali alimentari assai ricchi in niacina sono il fegato, il lievito da panettiere, le arachidi; buone quantità si trovano anche nella carne e nel pesce. Verdura e frutta ne sono invece assai povere. Il latte e le uova contengono poca niacina ma molto triptofano.
Il fabbisogno è valutato in 6,6 mg per ogni 1.000 kcal ingerite, con un minimo di 13 mg giornalieri.
Non si conoscono effetti tossici. L'acido nicotinico è stato utilizzato in dosi massive (3 g al dì, o più) per abbassare il livello ematico del colesterolo.
La deficienza di niacina causa la pellagra, i cui sintomi caratteristici sono dermatite, diarrea, demenza, i ‛3 D'.
La niacina è nettamente più stabile di altre vitamine, per esempio la tiamina e la riboflavina.
8. Acido pantotenico
L'acido pantotenico è stato sintetizzato per la prima volta nel 1940. È più stabile in soluzione che in forma anidra; è facilmente decomposto da acidi, alcali e calore secco; è stabile al calore umido e in soluzione neutra. E prodotto dalle piante e dai microrganismi, ma i Vertebrati lo richiedono nella dieta. L'assunzione giornaliera adeguata è valutata sui 5-10 mg.
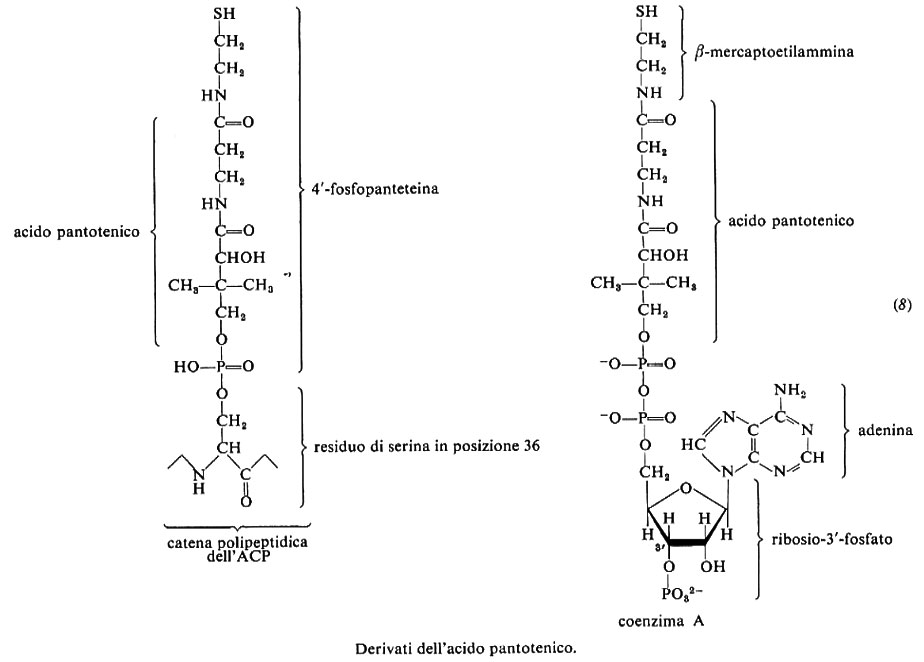
Come indicato dalla formula (8), l'acido pantotenico è parte del coenzima A e della 4′-fosfopanteteina che esterifica la senna 36 nella proteina trasportatrice di acili (ACP), uno dei componenti della sintetasi degli acidi grassi. Ambedue le molecole di cui l'acido pantotenico fa parte funzionano da trasportatrici di acili: il coenzima A nelle più svariate reazioni metaboliche, la fosfopanteteina nella biosintesi degli acidi grassi. L'acile forma un tioestere con l'−SH della β-mercaptoetilammina cui l'acido pantotenico è legato.
L'acido pantotenico è presente in materiali vegetali e animali; particolarmente ricchi ne sono le uova, il rene, il fegato, il salmone e il lievito. Altri alimenti che lo contengono in notevoli quantità sono i cavolfiori, i broccoli, le carni, le patate, i pomodori. Nella cottura delle carni se ne perde circa un terzo, e circa la metà nella molitura del grano.
È così diffuso negli alimenti che malattie da deficienza non sono osservate in individui a dieta normale. Non ha effetti tossici.
9. Biotina
La biotina fu isolata da F. Kögl nel 1935; V. Du Vigneaud e altri ne stabilirono la struttura. La sintesi fu realizzata nel 1943. Contiene un anello imidazolico e uno tiofenico condensati (9). È stabile al calore, ma è suscettibile agli ossidanti, agli alcali e agli acidi forti.

Si lega all'ε-amminogruppo di una lisina della proteina enzimatica a dare la biotinillisina o biocitina (9), che funziona da trasportatore di CO2 nell'azione di alcune carbossilasi. In particolare è presente in una subunità della acetil-CoA-carbossilasi, che catalizza la formazione di malonil-CoA, tappa essenziale nella biosintesi degli acidi grassi.
È presente nel fegato, nel latte, nelle carni, nel tuorlo d'uovo, in quasi tutti i vegetali e in molti frutti.
È prodotta da batteri intestinali; è quindi difficile provocare una deficienza di biotina semplicemente omettendola dalla dieta. Tuttavia l'albume d'uovo crudo contiene una proteina, l'avidina, che lega assai fortemente la biotina e ne impedisce l'assorbimento intestinale: pertanto in persone che consumino grosse quantità di uova crude si può instaurare una deficienza di biotina. L'avidina viene denaturata nella cottura. Animali a dieta di albume d'uovo crudo presentano una tossicosi caratteristica, da cui la biotina protegge.
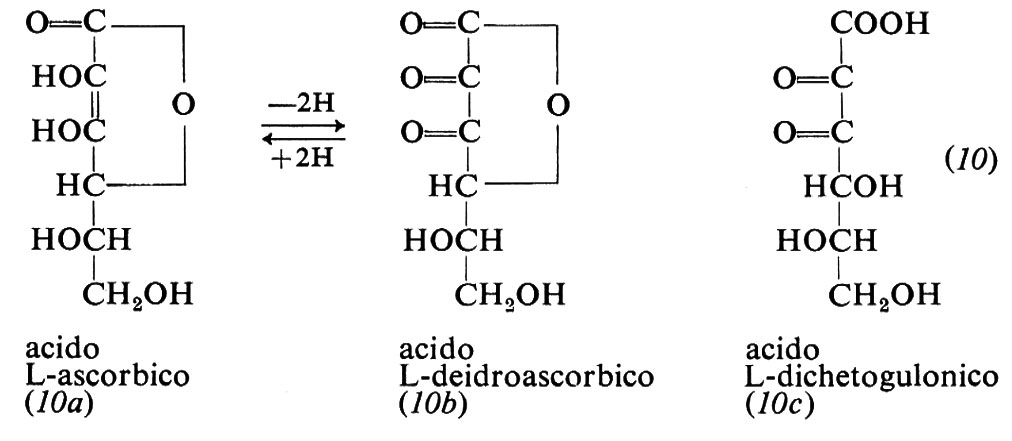
10. Vitamina C
La vitamina C, detta anche acido ascorbico, è il γ-lattone della forma enolica dell'acido α-chetogulonico (10a); essa fu isolata e cristallizzata tra il 1928 e il 1932 da A. Szent-Györgyi in Ungheria e da C. G. King e W. A. Waugh negli Stati Uniti.
Chimica
Il carattere acido della vitamina C deriva dalla dissociabilità di un protone dal gruppo idrossilico in C-3; la sua struttura dienolica la rende facilmente e reversibilmente ossidabile ad acido deidroascorbico (10b), che in ambiente alcalino subisce l'idrolisi dell'anello lattonico trasformandosi in acido dichetogulonico (10c). L'acido ascorbico è dotato di un forte potere riducente, cui si può attribuire gran parte delle sue peculiari proprietà biologiche.
Ruolo biochimico
L'acido ascorbico è un fattore necessario per i processi idrossilativi catalizzati da alcune monossigenasi dipendenti da rame e da diossigenasi dipendendenti da ferro e α-chetoglutarato. Essi sono i seguenti.
1. Formazione della noradrenalina dalla DOPA, catalizzata dalla dopammina-β-monossigenasi, presente nel cervello e nella midollare delle surrenali; la catalisi è accoppiata all'ossidazione dell'acido ascorbico.
2. Idrossilazione della prolina e della lisina nella formazione del collageno. E una reazione post-traslazionale caratterizzata da una diossigenasi ferro-α-chetoglutarato-dipendente. L'acido ascorbico è l'attivatore quasi specifico di queste diossigenasi. Si pensa che esso sia necessario per mantenere nello stato ferroso il ferro legato all'enzima. L'idrossilazione della prolina è necessaria perché tre polipeptidi si associno nella tipica conformazione a tripla elica e la molecola del collageno venga secreta dai fibroblasti. L'acido ascorbico attiva anche la γ-butirrobetainaidrossilasi, un'altra diossigenasi ferro-α-chetoglutarato-dipendente, che catalizza la biosintesi della carnitina.
3. Idrossilazione in 7α del colesterolo. Questa, che è una reazione chiave nella trasformazione del colesterolo in acidi biliari, risulta notevolmente diminuita nel fegato di cavie sottoposte a deficienza cronica latente di acido ascorbico: sembra che l'acido agisca stimolando la riduttasi del citocromo P450, che è responsabile dell'idrossilazione.
4. Numerosi processi di idrossilazione legati al sistema microsomiale (citocromo P450) o, nelle corticosurrenali, ai mitocondri, e che sembra siano condizionati o stimolati dall'acido ascorbico. Essi verosimilmente controllano la biosintesi degli ormoni tiroidei. L'azione di stimolo dell'ACTH sulla steroidogenesi viene interpretata come conseguenza diretta della deplezione dell'acido ascorbico delle surrenali indotta dall'ACTH. Si pensa che l'acido ascorbico agisca inibendo il sistema citocromo P450 delle surrenali o il sistema ‛adrenossina, adrenossina-riduttasi'.
L'acido ascorbico può venire alchilato e così compete con l'alchilazione di siti nucleofili cellulari, quali quelli presenti negli acidi nucleici e nelle proteine. In tal modo in forti dosi sembra possa esercitare un'azione protettiva contro l'azione di agenti tossici o cancerogeni.
Biosintesi e fisiologia
La stretta affinità chimica fra vitamina C e glucidi è evidente; infatti quasi tutti gli animali la possono ricavare dal glucosio via acido glucuronico. L'uomo, i Primati e la cavia hanno perso questa capacità a causa di una mutazione che ha portato alla delezione di un tratto di DNA che codifica la sintesi di una ossidasi necessaria per la conversione del γ-lattone dell'acido α-chetogulonico in acido ascorbico. Questi animali devono pertanto introdurre obbligatoriamente la vitamina C con la dieta. Gli animali incapaci di sintetizzare acido ascorbico sono in grado di assorbirlo tramite un sistema di trasporto facilitato, Na+-dipendente, di cui è dotato l'orletto a spazzola delle cellule epiteliali del loro intestino tenue; gli altri animali lo assorbono con un processo di semplice diffusione.
La L-gulonolattoneossidasi compare a livello degli Anfibi. Ciò significa che la necessità di acido ascorbico è associata all'emersione degli animali dall'acqua. Il loro adattamento alla vita terrestre, e la conseguente esposizione a una molto più elevata tensione di ossigeno, impone più efficaci sistemi di difesa contro la tossicità dell'ossigeno e precisamente dei suoi prodotti di parziale riduzione (O−2, -OH, O²2-). In effetti l'acido ascorbico ha concentrazioni particolarmente elevate nel liquido che bagna le pareti degli alveoli polmonari. Il tessuto cerebrale che, per l'assenza o l'insufficienza dei sistemi enzimatici (glutationeperossidasi, catalasi, perossidasi, citocromo P450 e P420) adibiti alla protezione contro radicali liberi dell'ossigeno, è particolarmente esposto alla loro azione deleteria, trova nelle concentrazioni fisiologiche di acido ascorbico (1,5×10-3 M) il più importante sistema di difesa controi processi perossidativi.
Le manifestazioni di deficienza di acido ascorbico, come lo scorbuto, sono a carico dei tessuti di sostegno di origine mesenchimale (ossa, cartilagine, tessuto connettivo), proprio per un difetto di formazione della sostanza cementante intercellulare, che ha fra i componenti principali il collageno.
Dosi elevate di acido ascorbico possono modificare il metabolismo di alcuni farmaci di natura fenolica, come la salicilammide o l'isoprenalina, in quanto la vitamina compete con essi per la coniugazione con solfati. Ne consegue un aumento di farmaco allo stato libero, e quindi attivo nel sangue. Forti dosi parrebbero anche aumentare la tolleranza e forse l'attività della L-dopa, che è un farmaco usato nella terapia del morbo di Parkinson. Alcuni anoressigeni, cioè sostanze che servono per ridurre l'appetito, hanno un effetto depletante sulla vitamina C tessutale.
Distribuzione e fabbisogno
L'acido ascorbico abbonda negli agrumi, in verdure con foglie verdi e nei pomodori. Perdite cospicue si hanno quando il materiale che le contiene è esposto per periodi lunghi all'aria. La distruzione è limitata durante una cottura rapida in piccola quantità di acqua entro recipienti ben chiusi. L'aggiunta di bicarbonato di sodio nel corso della cottura è assai dannosa.
Il Food and Nutrition Board americano consiglia la somministrazione quotidiana di 60 mg per un soggetto di 70 kg. Tuttavia, dato che la tossicità dell'acido ascorbico è molto bassa e che le malattie infettive in genere e virali in particolare inducono una deplezione di acido ascorbico nei globuli bianchi e nelle ghiandole surrenali, dove la sua concentrazione soverchia di gran lunga quella esistente in ogni altro tessuto (400-500 mg/100 g), emerge l'opportunità di dosaggi molto più elevati (fino a qualche grammo pro die), specie nella profilassi di queste malattie.
L'uomo catabolizza l'acido ascorbico fino ad acido ossalico. Ciò può essere causa di inconvenienti quando si somministrino dosi molto elevate di vitamina C. Megadosi possono essere dannose per il feto, nel senso che il neonato bruscamente sottratto a un ambiente ricco di acido ascorbico può andare incontro a scorbuto (condizionamento).
11. Altri fattori idrosolubili
Esistono alcuni altri fattori nutritivi essenziali richiesti nella dieta solo da alcune specie o solo in circostanze particolari. Essi sono i seguenti.
Mio-inositolo (11). Noto da tempo come composto chimico, viene considerato una vitamina dal 1940. Non sembra che serva da vitamina per l'uomo. È un polialcool ciclico necessario nella dieta del topo per una crescita normale del pelo e per prevenire nel ratto lo stato di ‛occhi occhialuti'. Il ratto sintetizza facilmente l'inositolo dal glucosio, ma evidentemente non in quantità sufficienti. L'inositolo non è costituente di alcun coenzima, ma è presente in quantità relativamente abbondanti nei tessuti animali come componente degli inositol-fosfogliceridi. È presente nella frutta, nel grano, nei vegetali, nelle noci, nei legumi, nel fegato e nel cuore. Le piante lo contengono come estere esafosforico, cioè come acido fitico. Questo interferisce nell'assorbimento di calcio e ferro, cui si lega formando complessi insolubili.
Colina (12). È un componente dei colin-fosfogliceridi o fosfatidilcoline. Come tale interviene nel trasporto di grassi dal fegato, evitando il formarsi di fegato grasso. Nell'estremità presinaptica dell'assone nervoso viene acetilata dalla colina-acetiltransferasi per dare acetilcolina, che funge da neurotrasmettitore nelle sinapsi colinergiche e alle giunzioni neuromuscolari. Insieme a betaina e metionina è una sorgente di metile labile. Le tre sostanze si vicariano parzialmente l'una con l'altra. L'uomo è in grado di sintetizzare la colina dalla senna in presenza di metionina, che fornisce il metile, e delle vitamine B12 e folacina come coenzimi. Negli animali superiori e nell'uomo, tuttavia, la sintesi è insufficiente a supplire alle esigenze corporee, per cui la colina è considerata un nutriente essenziale.
La fonte alimentare più abbondante di colina è il tuorlo d'uovo. Altre fonti sono il fegato, il cervello, i reni, il cuore e le Leguminose. Frutta, latte e verdura ne contengono scarse quantità. Il contenuto medio nella dieta è stato stimato in 400-900 mg giornalieri. Deficienza di colina causa infiltrazione grassa nel fegato ed emorragie renali.

Carnitina (13). È un fattore di crescita necessario per il coleottero Tenebrio molitor, ma non per i Mammiferi, che sono in grado di sintetizzarla. Abbonda particolarmente nel muscolo.
La carnitina accetta il gruppo acile di acidi grassi a catena lunga dagli acilcoenzimi A extramitocondriali e forma acil-carnitina. Questa trasporta gli acili attraverso la membrana mitocondriale al coenzima A intramitocondriale, passaggio preliminare alla loro ossidazione enzimatica.
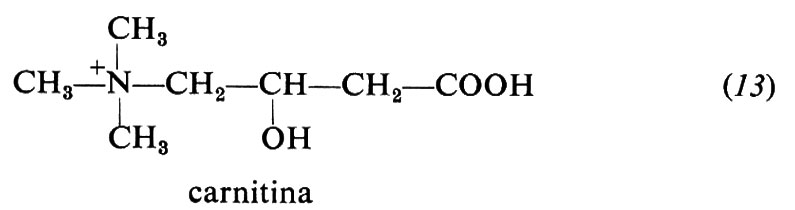
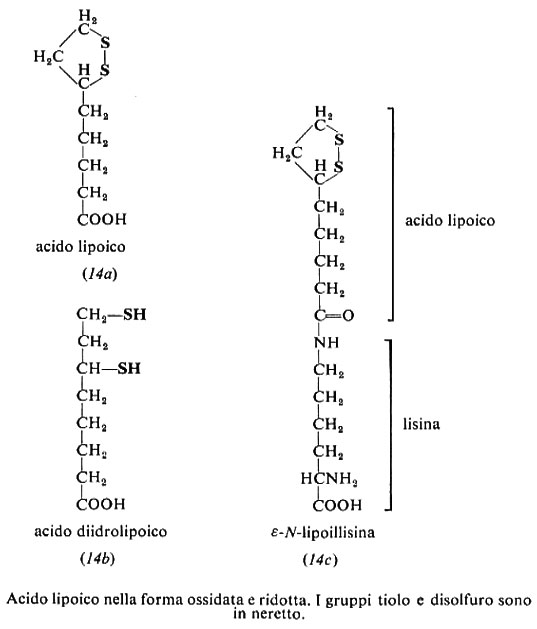
Acido lipoico, acido tiottico (14a). L'acido lipoico è stato isolato e cristallizzato nel 1953 da L. J. Reed e I. C. Gunzalus negli Stati Uniti. L'uomo è in grado di sintetizzarlo e non lo richiede nella dieta. È presente nelle cellule, in equilibrio con la forma ridotta, acido dudrolipoico (14b).
L'acido lipoico funziona da coenzima nella decarbossilazione ossidativa del piruvato e di altri α-chetoacidi. Nel caso del piruvato l'idrossietile proveniente dalla decarbossilazione (v. cap. 2) viene trasferito all'acido lipoico e lo riduce cedendogli due elettron-equivalenti: l'acetile neoformato si lega al ditiolo come tioestere. Successivamente l'acetile è passato al coenzima A; l'acido dudrolipoico residuo è riossidato a dare la forma disolfurica. Nella dudrolipoil-transacetilasi, che catalizza il trasporto, l'acido lipoico è legato all'ε-amminogruppo di una lisina: la lipoillisina risultante (14e) è nota anche come lipoammide.
12. Vitamina A
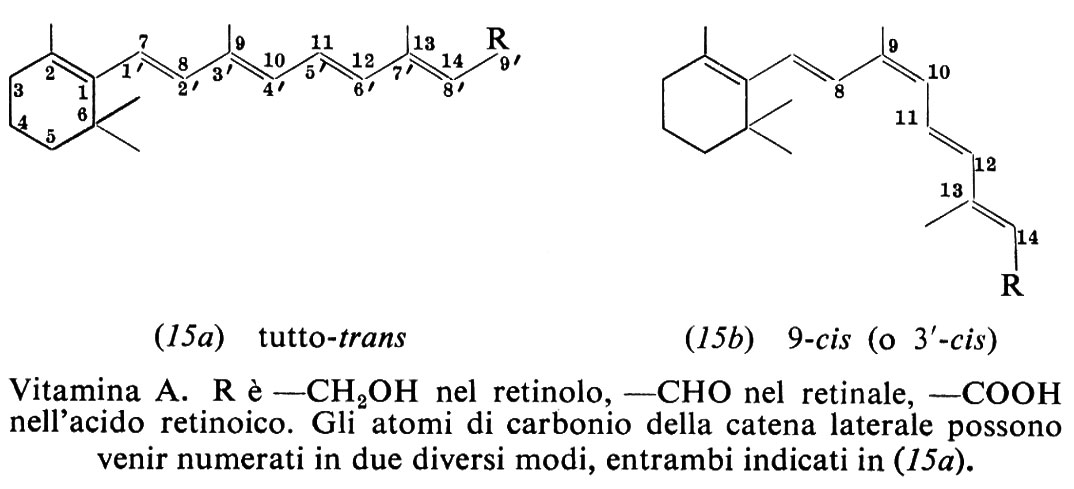
La vitamina A è tutto-trans-retinolo. La forma più comune è la vitamina A1 o retinolo 1 (15a), presente nei tessuti dei Mammiferi e dei pesci marini. Nei pesci di acqua dolce prevale la vitamina A2 o retinolo 2, che ha un ulteriore doppio legame tra gli atomi di carbonio 3 e 4 dell'anello.
L'efficacia di fattori liposolubili del rosso d'uovo nei riguardi della xeroftalmia e della cecità crepuscolare fu riconosciuta rispettivamente da E. V. McCollum e N. Simmonds nel 1917 e L. S. Fridericia ed E. Holm nel 1925. Il lipide attivo fu denominato ‛vitamina A' da J. C. Drummond nel 1920. H. N. Holmes e R. E. Corbet riuscirono a cristallizzarlo, dal fegato di pesce, nel 1937. La sintesi totale fu descritta da O. Isler e altri nel 1947. Il ruolo di provitamina del β-carotene apparve chiaro dopo che P. Karrer e collaboratori stabilirono la sua struttura nel 1930 e quella del retinolo nel 1931-1933. Nel 1935 O. Wald e collaboratori isolarono il cromoforo della retina illuminata e R. A. Morton dimostrò nel 1944 che esso era retinale, cioè l'aldeide del retinolo.
Ruolo biochimico
L'intervento di vitamina A e/o dei suoi derivati è stato stabilito a tre livelli distinti: visione, funzioni sistemiche, funzioni riproduttive.
Funzione visiva. Circa metà della proteina di membrana dei dischi, vescicole appiattite recettrici della luce parallele alla superficie retinica presenti nel segmento esterno delle cellule a bastoncello, è costituita da rodopsina. Essa contiene, legato come base di Schiff all'ε-amminogruppo di una lisina, il retinale. Dei suoi vari isomeri solo l'11-cis, a catena laterale piegata, forma il cromoforo biologicamente attivo. Grazie alla sua natura polienica con sei legami singoli e doppi alternati, esso è in grado di assorbire luce in una larga fascia di lunghezze d'onda nel visibile con un massimo a 498 nm, ottimale rispetto alle radiazioni solari che pervengono sulla superficie terrestre, e con un'efficienza altissima, in quanto il coefficiente di estinzione molare a 498 nm è 40.000.
L'assorbimento di un fotone, in una reazione puramente fotochimica che si svolge anche alla temperatura dell'azoto liquido, costituisce il primo evento nel processo visivo e trasforma l'isomero 11-cis in quello tutto-trans, alterando profondamente la geometria molecolare, in quanto la catena laterale diventa rettilinea; segue una serie di modificazioni conformazionali nella molecola della rodopsina, che terminano con la sua dissociazione in opsina e tutto-trans-retinale.
Quest'ultimo sembra formi una base di Schiff con la fosfatidiletanolammina. La luce, agendo su questo composto con una reazione fotochimica, riforma l'isomero 11-cis, che si riassocia con l'opsina rigenerando la rodopsina.
L'11-cis-retinale è il cromoforo anche dei recettori presenti nelle cellule a cono, deputate alla percezione dei colori. I diversi spettri di assorbimento dei tre tipi di recettori sono dovuti all'azione della proteina cui il retinale è legato: la mancanza di una o più di queste proteine provoca quindi cecità ai colori.
L'11-cis-retinale si forma in due tappe enzimatiche per ossidazione della vitamina A e successiva isomerizzazione dell'aldeide.
Tutti i sistemi visivi conosciuti, in Molluschi, Artropodi, Vertebrati, pur essendo i tipi di occhio notevolmente diversi tra loro e verosimilmente frutto di un'evoluzione indipendente, utilizzano 11-cis-retinale come cromoforo per le molecole fotorecettrici: esempio dimostrativo di evoluzione convergente, connessa alle prestazioni efficientissime della molecola.
In Halobacterium halobium la luce genera un ciclo fotochimico agendo sulla conversione a tutto-trans del 13-cis-retinale contenuto nella batteriorodopsina.
Meno dell'1% della vitamina A corporea è impegnata nei processi visivi. Il resto interviene in funzioni riproduttive e sistemiche.
Funzioni riproduttive. La deficienza di retinolo lede uno stadio della spermatogenesi antecedente la divisione meiotica degli spermatozoi a spermatidi. A differenza delle lesioni causate da deficienza di vitamina E, che sono irreversibili, quelle derivanti da mancanza di vitamina A vengono eliminate somministrandola nuovamente. Inoltre è efficace anche l'acido retinoico, che non ha effetto sulla funzione visiva; ciò evidenzia la diversità dei processi coinvolti.
Funzioni sistemiche. Nella deficienza sperimentale di vitamina A negli animali si ha un'iperplasia del periostio e una squilibrata attività di osteoblasti e osteoclasti, con prevalere dei primi. Tali effetti sono particolarmente evidenti a carico delle vertebre e delle ossa facciali.
La vitamina A è anche importante per la funzione dei tessuti epiteliali di origine ecto- o endodermica. Questi ultimi in assenza della vitamina subiscono cospicue modificazioni morfologiche. Il meccanismo di queste azioni non è chiaro; si hanno però una serie di informazioni su processi che vengono alterati nella deficienza. Si è osservata un'influenza della vitamina, dell'acido retinoico e altri derivati, sulla sintesi, e apparentemente anche sul metabolismo, dell'RNA. Effetti favorenti sono stati riscontrati anche sulla sintesi di glicopeptidi e glicoproteine contenenti fucosio e mannosio. Sembra che il retinilfosfato funga da trasportatore di mannosio.
Biosintesi e metabolismo
La biosintesi della vitamina A inizia dall'acido mevalonico (v. biosintesi), che subisce una serie di successive fosforilazioni e condensazioni e, via il geranil-geranil-pirofosfato, dà il fitoene e altri caroteni acidici più insaturi, ultimo il licopene, dal quale per chiusura dell'anello si formano i caroteni mono- e diciclici, rispettivamente γ- e δ-, α- e β-carotene. Tutte le reazioni sono stereospecifiche.
La vitamina A naturale è in genere esterificata da un acido grasso, di solito l'acido palmitico. Gli esteri assunti con la dieta liberano retinolo nell'intestino tenue. Questo passa nelle cellule della mucosa, dove è nuovamente esterificato. I carotenoidi in parte sono assorbiti come tali e contribuiscono al colore giallastro del siero di sangue. La parte più cospicua, però, a opera di una ossigenasi che manca nei microrganismi ed è presente nei Mammiferi, viene scissa nelle cellule della mucosa intestinale in due molecole di retinale che, sempre nell'intestino o nel fegato, è ridotto a retinolo e come tale è esterificato. Gli esteri retinilici delle varie provenienze complessati ai chilomicroni della linfa vengono trasportati al fegato, dove costituiscono la forma di deposito della vitamina A. Da qui il retinolo è trasportato ai vari organi legato in complesso 1 : 1 a una proteina specifica - proteina legante il retinolo, RBP - di peso molecolare 21.000, che a sua volta è associata a una prealbumina. Sembra che i sintomi di tossicità si presentino quando la quantità della vitamina eccede quella della RBP e la vitamina perviene alle membrane verosimilmente sotto forma di esteri retinilici.
Acido retinoico può essere prodotto per sintesi, ma si forma anche biologicamente, per esempio nel ratto. Pur essendo attivo nei saggi di crescita, non si deposita nel fegato come la vitamina, né ripristina la rodopsina o la visione, né mantiene le normali funzioni riproduttive.
Distribuzione e fabbisogno
L'attività dei preparati vitaminici è misurata in retinol-equivalenti (R.E.). Un R.E. è pari a 1 μg di retinolo, 6 μg di β-carotene, 12 μg di altri caroteni con funzione di provitamine. L'assunzione giornaliera raccomandata è di 1.000 R.E. per l'uomo e di 800 R.E. per la donna. Le dosi sono maggiori nel corso della gravidanza e nella lattazione, come anche nell'infanzia in rapporto al peso.
Sintomi di tossicità sono stati osservati con assunzioni che superino i 15.000 R.E. al giorno per diversi giorni o i 6.000 per mesi. Se scoperti in tempo, essi scompaiono in un periodo relativamente breve eliminando la vitamina dalla dieta. L'eccesso di carotenoidi alimentari può causare depositi giallastri, peraltro innocui, nella pelle e negli occhi.
Sorgenti alimentari di vitamina A sono fegato, rene, burro, margarine arricchite, tuorlo d'uovo, latte intero, crema di latte e formaggi. I caroteni sono presenti in vegetali gialli, o con abbondante fogliame verde scuro, per esempio carote, patate dolci, ecc., e in frutta gialla, come albicocche, pesche, meloni ecc. L'olio di fegato di merluzzo è una sorgente di vitamina a dosi terapeutiche.
La vitamina A è facilmente distrutta da agenti ossidanti e dalla luce ultravioletta, soprattutto a temperature elevate.
La deficienza di vitamina A può essere primaria, ovvero secondaria a disfunzioni corporee che interferiscano con l'assorbimento o il deposito della vitamina, per esempio la colite ulcerosa, la cirrosi epatica, l'ostruzione dei dotti biliari, o con la trasformazione del carotene in vitamina A, per esempio il diabete e l'ipotiroidismo, ovvero che causino rapida perdita della vitamina, per esempio la polmonite, l'ipertiroidismo, la scarlattina e alcune infezioni respiratorie nell'infanzia. Le sindromi più caratteristiche corrispondenti sono la cecità crepuscolare, lesioni oculari (xeroftalmia e cheratomalacia) e modificazioni cutanee (ipercheratosi follicolare e xeroderma). La somministrazione di vitamina A può farle recedere in misura che dipende dalla gravità delle lesioni.
13. Vitamina D
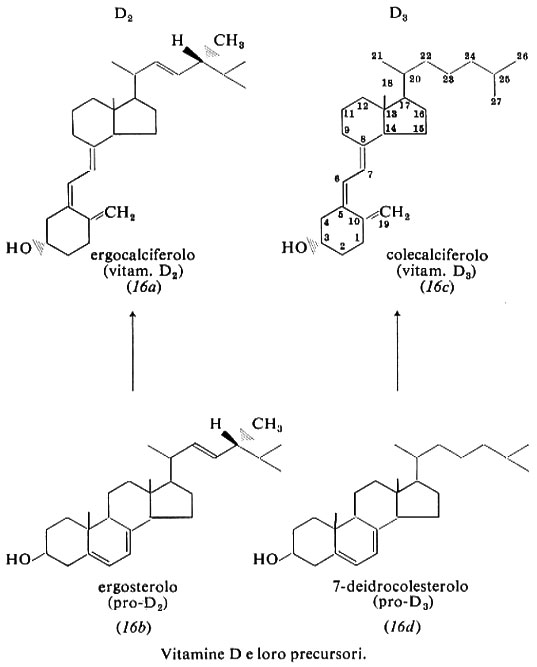
Vitamina D è il nome collettivo per un gruppo di composti: hanno significato biologico la vitamina D2, ergocalciferolo, derivata per fotolisi dallo sterolo vegetale ergosterolo, e la vitamina D3, colecalciferolo, derivata per irradiazione dello sterolo animale 7-deidrocolesterolo (16).
Nel 1922 E. V. McCollum, dell'Università John Hopkins, dimostrò che gli effetti antirachitici dell'olio di fegato di merluzzo erano dovuti a una vitamina che egli denominò ‛vitamina liposolubile D'. Quasi contemporaneamente fu accertato che irradiando con luce solare o ultravioletta i malati (K. Huldshinsky, 1919) o la loro dieta (H. Steenbock e A. F. Hess, 1924) si produceva una sostanza liposolubile antirachitica. Essa fu isolata come vitamina D2 da F. A. Askew e altri nel 1931 e, nel 1932, da Windaus e altri, che negli anni successivi ne determinarono la struttura. Nel 1936 Windaus e altri sintetizzarono il 7-deidrocolesterolo e dai suoi prodotti di irradiazione isolarono la vitamina D3. Il 22,23-diidroergocalciferolo, o vitamina D4 completamente sintetica, venne preparato da A. Windaus e da O. Trautmann nel 1937. I metaboliti della vitamina D furono scoperti nei tardi anni sessanta da H. F. De Luca e altri.
Chimica
Gli aspetti importanti della chimica della vitamina D dipendono largamente dalla sua struttura di cis-triene. Essa è responsabile del caratteristico assorbimento U.V. con massimo a 265 nm e minimo a 228 nm e rapporto di assorbanza di 1,8 nei prodotti puri. Inoltre ne deriva instabilità all'ossidazione, cui il composto sottostà facilmente, e instabilità agli acidi. La vitamina è invece stabile in solventi organici e in oli non acidi, in presenza di antiossidanti, per esempio α-tocoferolo, atmosfera di azoto e assenza di luce.
Produzione e distribuzione
La pelle è in grado di biosintetizzare attivamente 7-deidrocolesterolo. Luce U.V. di lunghezza d'onda intorno ai 300 nm che penetri a livello dell'epidermide, dove si trova il composto, lo trasforma, verosimilmente per fotolisi dell'anello B, in previtamina D3, che è in equilibrio temperatura-dipendente con la vitamina D3. Un'irradiazione eccessiva non provoca i sintomi tossici dell'ipervitaminosi. La contaminazione dell'aria e la copertura della pelle con i vestiti riducono la produzione cutanea a quantità insufficienti e pertanto diventa necessaria la vitamina presente negli alimenti.
La vitamina è contenuta soprattutto nell'olio di fegato di pesce, da 3.000 a 12.000 μg ogni 100 g di tessuto, secondo la specie; notevole anche il contenuto nell'uovo (66 μg/100 g), nel burro (2,3 μg/100 g) e nel latte arricchito (1,1 μg/100 g), mentre il latte comune ne contiene 0,1 μg/100 g. La vitamina scarseggia nel mondo vegetale; fa eccezione Solanum glaucophyllum, del Sudamerica, che contiene nelle foglie una forma coniugata di 1,25-(OH)2-D3 e di 1α-OH-D3.
La quantità giornaliera raccomandata è di 10 μg, pari a 400 U.I.; fino a 25-50 possono essere ingenti senza danni, salvo in casi di malattia.
Assorbimento, metabolismo e ruolo fisiologico
La vitamina D, liposolubile, è assorbita nell'intestino tenue, verosimilmente nella parte distale, ed è trasferita ai chilomicroni, che passano nel dotto toracico e poi nel torrente sanguigno. È noto che la steatortea e altre condizioni, come la sprue e il morbo celiaco, che interferiscono con l'assorbimento dei grassi, provocano anche deficienza di vitamina D.
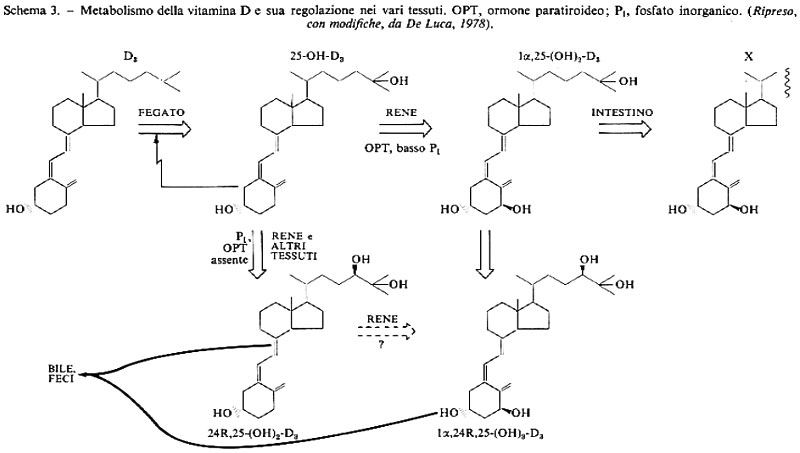
La vitamina somministrata ad animali che ne sono deficienti si accumula nel fegato, ma il sito primario di deposito nella somministrazione cronica è invece il tessuto adiposo. La forma di deposito è lo sterolo della vitamina libera. Nel fegato la vitamina è trasformata in 25-OH-D3 da un'idrossilasi microsomiale che viene inibita da un accumulo del prodotto. Il 25-OH-derivato è 2,5 volte più attivo della vitamina D3. Esso è trasportato ai reni, dove un'idrossilasi specifica localizzata nei mitocondri lo trasforma in 1,25-(OH)2-D3. Nel mitocondrio integro l'idrossilazione richiede il procedere della fosforilazione ossidativa. Il composto formatosi (o un suo metabolita) è la forma metabolicamente attiva della vitamina; dato che viene formato esclusivamente nel rene e ha come bersagli l'intestino e il tessuto osseo, esso può essere considerato un ormone.
Vari tessuti contengono un sistema idrossilasico capace di dare 24 (R), 25-(OH)2-D3, che ha attività circa pari al 25-OH-composto, e 1,24 (R), 25-(OH)3-D3, che è invece assai meno attivo dell'1,25 (OH)2-D3: potrebbe trattarsi di un meccanismo di inattivazione di questo potente ormone. Vi è anche la possibilità che 25-OH-D3 venga trasformato in vivo in 25,26-(OH)2-D3, che ha scarsa attività metabolica e di cui non si conosce la funzione.
La produzione di 1,25-(OH)2-D3 è controllata dal livello del calcio nella dieta, e quindi nel siero, in quanto è stimolata dall'ormone paratiroideo che viene secreto in risposta a un'ipocalcemia.
L'1,25-(OH)2-D3 stimola l'assorbimento intestinale di calcio e di fosfati e anche la loro mobilizzazione dall'osso, e aumenta il riassorbimento renale del calcio. L'ormone paratiroideo contribuisce ad aumentare la mobilizzazione del calcio dall'osso e il suo riassorbimento renale, ma, con una reazione indipendente dall'1,25-(OH)2-D3, provoca una diuresi dei fosfati. In tal modo il livello serico dei fosfati resta immodificato, mentre quello del calcio cresce, il che sopprime la secrezione di ormone paratiroideo e la produzione di 1,25-(OH)2-D3. In caso di deplezione di fosfati aumenta la produzione di 1,25-(OH)2-D3, ma non vi è secrezione di ormone paratiroideo, per cui la liberazione di calcio dall'osso è minore, mentre aumenta il trasporto intestinale di calcio e di fosfati. Il rene è in grado di riassorbire tutto il fosfato ultrafiltrato e come risultato il livello dei fosfati nel siero aumenta considerevolmente, mentre quello del calcio varia di poco. L'insieme di questi processi è riassunto nello schema 3.
Negli Uccelli la 25-OH-D3-1-idrossilasi viene fortemente stimolata dagli ormoni che si producono al momento della deposizione delle uova: questo tipo di controllo a opera di ormoni sessuali apre nuove prospettive sulla patogenesi dell'osteoporosi post-menopausa di cui soffrono molte donne. Anche l'insulina e il diabete influenzano la menzionata idrossilasi, il che può essere importante per le malattie ossee associate al diabete.
I controlli sopra accennati agiscono sul metabolismo del calcio con un diverso impatto temporale: la secrezione di ormone paratiroideo segue nell'ambito dei secondi lo stimolo ipocalcemico e l'ormone ha una vita in circolo di alcuni minuti; la produzione di 1,25-(OH)2-D3 o l'aumento di idrossilasi richiedono ore e l'ormone ha efficacia sui suoi bersagli per molte ore e anche giorni.
Nel citoplasma delle cellule della mucosa intestinale esiste una proteina 3,2S, recettrice dell'1,25-(OH)2-D3: legato a essa, questo viene trasferito al nucleo, dove stimola la sintesi di RNA messaggero e di altri RNA che intervengono nella biogenesi di proteine deputate al trasporto intestinale del calcio.
Patologia
Malattia classica da deficienza di vitamina D è il rachitismo, che colpisce i giovani e si manifesta con insufficiente calcificazione dell'osso neoformato e della cartilagine epifisaria. Nell'adulto la deficienza si manifesta con osteomalacia. Un sintomo comune è la debolezza muscolare. A ipovitaminosi D consegue anche tetania ipocalcemica. Pazienti ipoparatiroidei non sono in grado di percepire l'ipocalcemia né conseguentemente di sintetizzare 1,25(OH)2-D3: essi hanno valori bassi di calcio e alti di fosfati nel siero. Nell'osteodistrofia renale la produzione di 1,25-(OH)2-D3 è diminuita o annullata, con conseguente sintomatologia, dalla diminuzione della massa del rene o da accumulo di fosfati nelle cellule deputate alla biosintesi. I metaboliti della vitamina D curano l'osteoporosi indotta da glucocorticoidi.
Una forma di rachitismo con eredità autosomica recessiva si manifesta anche in presenza di assunzioni normali di vitamina D: scompare, però, con somministrazioni elevate. Un'altra forma, dominante, correlata al cromosoma X, resiste anche a forti dosi.
Quantità eccessive di vitamina possono essere tossiche in quanto provocano demineralizzazione dell'osso, ipercalcemia e calcificazione in diversi tessuti: i sintomi sono nausea, perdita di appetito, vomito, poliuria, cute scagliosa e infine la morte. Contro questi effetti sono attivi i glucocorticoidi, specie il prednisone.
14. Vitamina E
La vitamina E è l'α-tocoferolo, 5,7,8-trimetiltocolo (17). Nel 1922 H. M. Evans e altri, dell'Università della California, descrissero l'esistenza di un fattore alimentare necessario per la riproduzione normale; Evans isolò l'α e il β-tocoferolo (5,8-dimetiltocolo) dal germe di grano nel 1936. Successivamente furono isolati altri tocoferoli. La sintesi di (2RS, 4′R, 8′R)-α-tocoferolo venne descritta da P. Karrer e altri e da S. E. Smith e altri nel 1938.
Chimica
I tocoferoli sono derivati dal croman-6-olo. L'α-tocoferolo naturale ha la configurazione 2R, 4′R, 8′R. Le proprietà chimiche della molecola dipendono: a) dall'idrossile fenolico libero che può essere acilato o eterificato; b) dal fatto che il tocoferolo è essenzialmente il monoetere di un idrochinone che viene facilmente ossidato; c) dalla presenza - non però nell'α-tocoferolo - di una posizione libera sull'anello aromatico, disponibile per nitrosazione, clorometilazione, idrossimetilazione, ecc.
I tocoferoli sono oli viscosi di color giallo chiaro, p.f. 2,5-3,5 °C per l'α-tocoferolo, solubili in solventi dei lipidi, insolubili in acqua. Sono lentamente ossidati dall'ossigeno atmosferico. In assenza di aria sono stabili fino a circa 200 °C. Si possono preparare per sintesi totale a partire da alcoli terpenici insaturi, essi stessi ottenibili per via sintetica.
La vitamina E può esser saggiata per via chimica tramite la riduzione dello ione ferrico a ferroso in presenza di α, α′-dipiridile, o mediante reazione con acido nitroso. Risultati più qualificanti si hanno però con la cromatografia gas-liquido o su strato sottile. 1 mg di d-α-tocoferolo è uguale a 1,49 U.I.; 1 mg di dl-α-tocoferolo è uguale a 1 U.I.
Ruolo biochimico
Risiede principalmente nella protezione di membrane biologiche, in particolare quelle dei mitocondri, microsomi e lisosomi. La catena laterale fitilica dà luogo a interazioni specifiche con le catene degli acidi grassi polunsaturi, specie i derivati dell'acido arachidonico; la vitamina in parte si lega anche alla proteina strutturale della membrana. Funziona da antiossidante neutralizzando i radicali liberi e così impedisce la perossidazione degli acidi grassi polunsaturi che porterebbe a un aumento di permeabilità della membrana. Verosimilmente inibisce anche la degradazione dei fosfolipidi di membrana a opera delle fosfolipasi legate alla membrana. La maggior efficacia della vitamina E rispetto agli antiossidanti sintetici è dovuta al fatto che essa è legata attivamente alle membrane lipoproteiche, mentre questi non lo sono.

La liberazione di enzimi idrolitici dai lisosomi è stata proposta come una delle cause dei fenomeni distrofici che si instaurano a carico del muscolo nella deficienza di vitamina E.
Esiste un effetto sinergico fra vitamina E e glutatione ridotto, ovvero glutationeperossidasi, ovvero selenio. Questo perché l'enzima citato, che è una selenio-proteina, ossidando il glutatione rimuove il perossido d'idrogeno e in tal modo interrompe la catena perossidativa. L'H2O2, generato dalla superossidodismutasi, ove permanga, reagisce con lo ione superossido, formato in condizioni anaerobiche dalla xantinossidasi, e dà origine al radicale libero idrossile che attacca i lipidi di membrana ossidandoli. La complementarità tra enzima e vitamina è avvalorata dal fatto che questa, liposolubile, è legata alle membrane, mentre la glutationeperossidasi è presente nella fase acquosa del plasma o della cellula.
La vitamina E appare anche coinvolta nell'assimilazione e nel metabolismo della vitamina A e del ferro e, in modo meno chiaro, di altri fattori nutritivi.
Biosintesi e fisiologia
I tocoferoli sono costituenti di tutte le piante verdi e di alcune forme vegetali inferiori.
Nelle piante verdi l'α-tocoferolo è localizzato nei cloroplasti, mentre le parti non fotosintetizzanti e non in crescita attiva, come semi e frutti, contengono in gran parte tocoferoli non α. La biosintesi dell'α-tocoferolo inizia con la comparsa delle prime foglie contenenti clorofilla e si accompagna in genere a diminuzione dei tocoferoli non α, dai quali l'α-composto può derivare per metilazione. Esso si forma però anche direttamente da precursori intermedi. Per l'anello aromatico viene seguita la via dell'acido scichimico (v. biosintesi).
Per l'assorbimento intestinale della vitamina E sono essenziali le micelle di lipidi e sali biliari, che non si formano ove vi sia deficienza di selenio. Le micelle trasportano la vitamina, assieme ad altri composti liposolubili, attraverso la fase acquosa del lume intestinale adiacente alla superficie a spazzola dei microvilli. Essa penetra nella cellula epiteliale tramite la membrana plasmatica apicale e, passando attraverso la membrana laterale o basale e la lamina basale, entra nel fluido della lamina propria e di qui nei capillari linfatici, dove è trasportata nei chilomicroni. Gli acidi grassi polunsaturi inibiscono l'assorbimento della vitamina E in quanto competono per i siti di assorbimento e di trasporto.
Tessuti come il fegato e il sangue l'assorbono e la rilasciano più rapidamente di altri organi. Nel plasma la vitamina E si associa alle lipoproteine e in tal modo viene trasportata, ma in parte è anche legata ai grassi neutri. La porzione intracellulare si trova prevalentemente nelle frazioni mitocondriali e microsomiali.
Dosi elevate di vitamina E non sono tossiche e sembra riducano la tossicità di alte dosi di vitamina A.
Analoghi
La racemizzazione in posizione 4′ e 8′ non modifica l'attività biologica, ma il dl composto in 2′ mantiene solo il 75% dell'attività. Gli analoghi β-, γ- e ζ2-tocoferolo (rispettivamente 5,8-, 7,8- e 5,7dimetii-tocolo) hanno attività vitaminiche rapportabili a circa il 30, 15 e 50% dell'α-tocoferolo. Lo ζ1-tocoferolo (5, 7, 8-trimetil-toco-3′, 7′, 11′-trienolo) o α-tocotrienolo, con catena laterale insatura, ha circa il 20% dell'attività vitaminica dell'α-tocoferolo.
Distribuzione e fabbisogno
Sorgenti alimentari principali della vitamina sono gli oli vegetali; essa abbonda anche nell'olio di fegato di merluzzo. È da tener presente però che grosse perdite possono aversi nella cottura e soprattutto nel corso della conservazione di cibi cotti.
Vitamina E è richiesta in phyla superiori di Invertebrati, nelle forme larvali di taluni Insetti, in molte specie di Uccelli e nei Mammiferi. La quantità necessaria è in rapporto con la perossidabilità relativa degli acidi grassi presenti nella dieta e nei tessuti e con la presenza di amminoacidi solforati, di selenio e di antiossidanti. Alcune sintomatologie da deficienza, evidenziabili soprattutto negli animali, richiedono solo vitamina E, altre anche selenio, altre vitamina E e antiossidanti, altre infine vitamina E e cisteina. Queste correlazioni sono interpretabili in base alle considerazioni prima fatte sul ruolo e le interazioni della vitamina a livello biochimico.
L'uomo adulto ha un livello serico di vitamina pari in media a 1 mg/100 ml. Nella dieta sono richiesti 10-15 mg giornalieri, ovvero 30 U.I./kg di dieta consumata. La semplice deficienza di vitamina non produce una sindrome grave e palese, a meno che non sia associata ad altri danni o stress patologici.
Il trasferimento transplacentare della vitamina al feto è scarso: pertanto nel neonato essa ha concentrazioni piasmatiche di circa un terzo rispetto all'adulto. Nei neonati pre-termine e nei bambini a diete inadatte essa può scendere a livelli ancora più bassi, che producono forme di anemia emolitica.
15. Vitamina K
Vitamina K è il nome generico per il 2-metil-1, 4-naftochinone, indicato come ‛vitamina K3' o menadione (18a), e per i suoi derivati che abbiano attività antiemorragica in animali a dieta deficiente. Il 3-fitil-derivato è detto ‛vitamina K1' o fillochinone (18b); vitamine K2(n) o menachinoni sono una serie di composti che hanno in posizione 3 catene laterali polusopreniche di diversa lunghezza (18c). H. Dam propose la vitamina K come fattore liposolubile antiemorragico nel 1935, la isolò in forma grezza l'anno successivo e nel 1939 la purificò, in collaborazione con P. Karrer, dall'erba medica, sotto forma di un olio giallo. Karrer ne riconobbe la natura chinonica; la sintesi del fitilderivato venne effettuata, sempre nel 1 939, quasi contemporaneamente dal gruppo di E. R. Doisy e da altri gruppi. Doisy isolò altresì dalla farina di pesce un composto cristallino che denominò ‛vitamina K2'. La caratterizzazione completa della struttura dei vari composti indicati come vitamina K2 avvenne negli anni cinquanta a opera di O. Isler e collaboratori.
Caratteristiche
Il fillochinone è un olio, a temperatura ambiente, mentre i menachinoni sono facilmente cristallizzati da solvente organico e hanno p.f. tra 35 e 60 °C secondo la lunghezza della catena poliisoprenica. Le forme ridotte degli 1,4-naftochinoni si ossidano spontaneamente.

La catena poliisoprenica rappresenta il sostituente più efficace in posizione 3. Il metile in 2 è molto importante per l'attività vitaminica, tuttavia anche il 2-fitil-1, 4-naftochinone esplica considerevole attività. Animali (o batteri intestinali) utilizzano una vasta gamma di composti, come il 2-metil-4-ammino-1-naftolo e il 2-metil-1-naftolo.
Le radiazioni U.V. decompongono la vitamina; pertanto le manipolazioni che la coinvolgono devono esser fatte al riparo dalla luce.
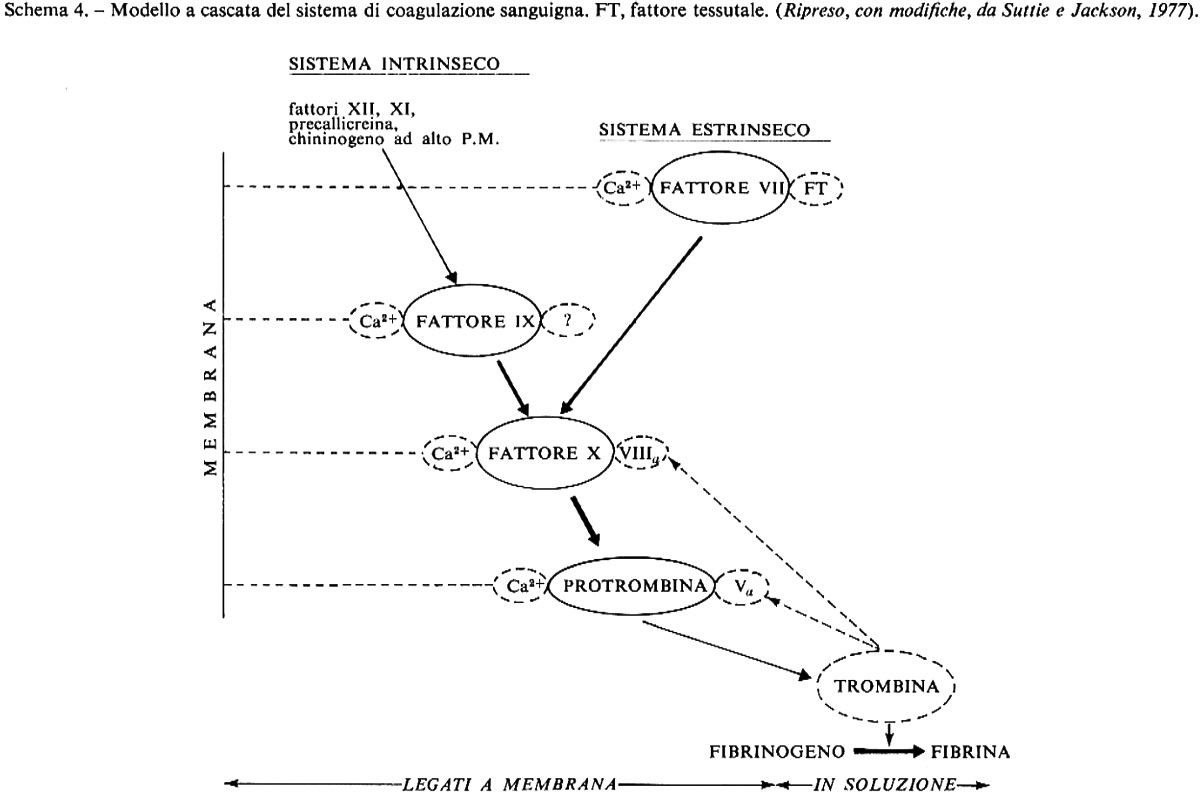
Ruolo biochimico
La vitamina K interviene nelle trasformazioni delle proteine che presiedono alla coagulazione del sangue. L'attivazione della protrombina e la trasformazione del fibrinogeno in fibrina con formazione del coagulo avverrebbero, stando alle attuali interpretazioni, secondo lo schema 4. Il processo può essere iniziato da fattori presenti nel sangue (sistema intrinseco) o rilasciati dal tessuto ferito (sistema estrinseco). A ogni stadio, esclusi i due iniziali, una proteinasi, in associazione a fattori accessori, trasforma la proteina indicata nell'ellisse in una proteasi attiva che catalizza lo stadio successivo. Tutte le molecole racchiuse a tratto pieno contengono nella regione ammino-terminale del polipeptide - del polipeptide leggero nel caso del fattore X che ne contiene 2 - acido γ-carbossiglutammico, che interviene nel legame calcio-dipendente alla matrice fosfolipidica. La carbossilasi che catalizza la formazione di questo particolare amminoacido dal glutammato, oltre al CO2 richiede vitamina K ridotta e O2. Per il fattore VII è dubbio se la vitamina intervenga a livello del processo di carbossilazione o in altra reazione.
Sono state fatte varie ipotesi circa il ruolo molecolare della vitamina nella reazione di carbossilazione: essa potrebbe funzionare da attivatore, o da trasportatore del CO2 nella membrana, analoga in questo alla biotina, fattore idrosolubile, ovvero riossidandosi potrebbe labilizzare il legame di un idrogeno al carbonio γ del glutammile, e in tal modo facilitare l'attacco a opera del CO2, ovvero infine, ed è l'eventualità meno probabile, potrebbe agire da attivatore per un enzima coinvolto nella reazione.
Il preparato microsomiale capace di effettuare la γ-carbossilazione è in grado anche di ossidare la vitamina K a epossido e di ridurre l'epossido. Quest'ultima reazione è bloccata dal warfarin e da altri anticoagulanti cumarinici.
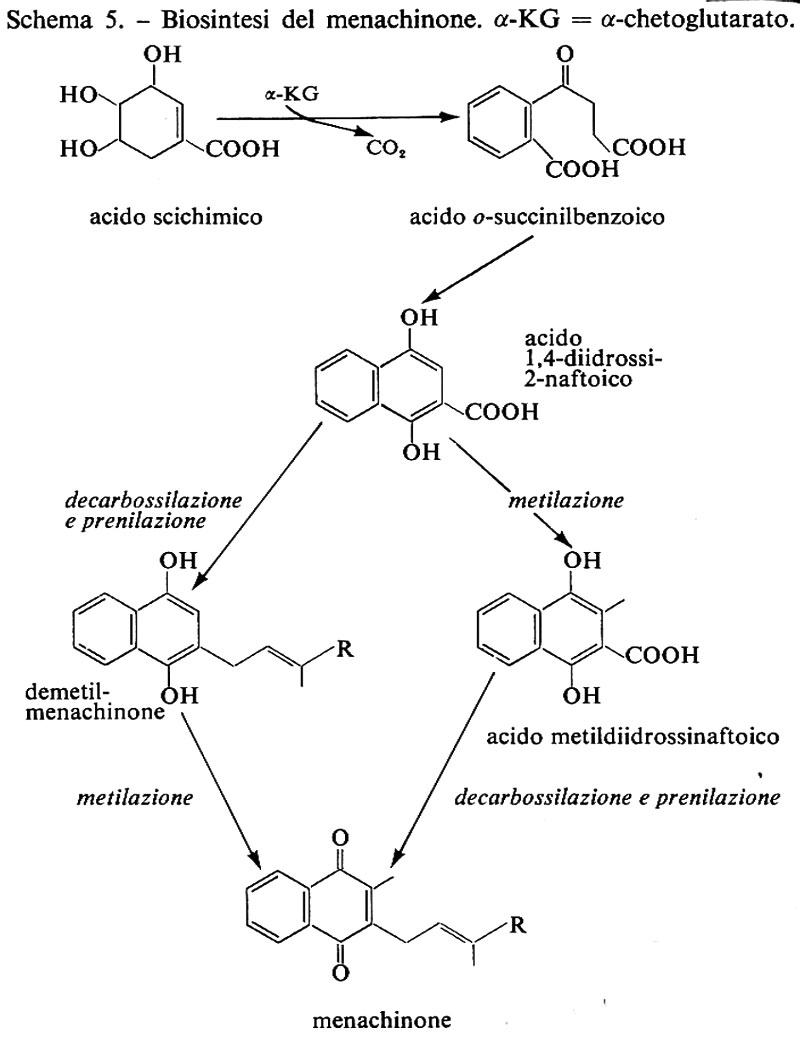
Si è perciò ipotizzato che la carbossilazione possa essere collegata alla formazione dell'epossido e che la vitamina fornisca gli equivalenti riducenti necessari per far procedere la reazione. Questa formulazione e le proposte alternative sul meccanismo d'azione degli anticoagulanti cumarinici attendono però ancora una conferma sperimentale.
Presenza, vitamina K-dipendente, di acido γ-carbossiglutammico è stata descritta anche in due altre proteine plasmatiche, la proteina C nei bovini e la proteina S nell'uomo; non si sa però se esse intervengano nel processo di coagulazione.
La vitamina K interviene anche in processi non correlati con la coagulazione. In alcuni batteri è parte della catena di trasporto degli elettroni: in Mycobacterium phlei, per esempio, occupa il posto generalmente tenuto dall'ubichinone tra flavine e citocromi.
Biosintesi
Gli animali non sono in grado di sintetizzare l'anello del 2-metil-1, 4-naftochinone. La sintesi è stata studiata nei Batteri. Il carbonio per l'anello aromatico è fornito dall'acido scichimico: i restanti carboni dell'anello naftalenico derivano dall'α-chetoglutarato; la sintesi sembra procedere tramite acido succinilbenzoico, che passerebbe poi ad acido 1,4-diidrossi-2-naftoico. La via di questo composto a menachinone varia secondo l'organismo, in quanto la metilazione in 2 può precedere o seguire la decarbossilazione e la prenilazione (v. schema 5). Gli animali sono in grado di alchilare il menadione a menachinone, soprattutto menachinone-4.
Metabolismo
La vitamina K è assorbita dall'intestino nel sistema linfatico. Come per altre sostanze liposolubili, l'assorbimento massimo si ha in presenza di bile e di succo pancreatico. Forme diverse della vitamina vengono assorbite a livelli diversi dell'intestino: così il fillochinone è assorbito nell'intestino tenue prossimale con un processo saturabile ed energia-dipendente, mentre menadione e menachinone-9 sono assorbiti con un processo passivo a livello del tenue distale e del colon.
Il fillochinone si deposita specificamente nel fegato; il menadione è distribuito in tutti i tessuti, dove viene rapidamente coniugato a glucuronide e a solfato e come tale è escreto, mentre solo in piccola parte è trasformato in menachinone-4 biologicamente attivo. Il fillochinone e i menachinoni hanno un metabolismo più lento del menadione.
La quantità più cospicua di vitamina K nella cellula epatica è nella frazione microsomiale, ma un livello elevato si riscontra anche nei mitocondri. Nelle urine la vitamina è escreta sotto forma di glucuronidi, mentre nella bile e nelle feci prevale un composto meno polare, che verosimilmente è una forma esterificata degli agliconi. È probabile l'esistenza di altri prodotti di escrezione non identificati.
Il metabolita quantitativamente più importante della vitamina K è il 2,3-epossido, detto comunemente ‛ossido della vitamina K'. In condizioni di terapia con warfarin rappresenta la forma più cospicua di vitamina nel plasma.
Se per lo studio dell'attività biologica si evita la somministrazione orale, che potrebbe portare a modificazioni a livello intestinale del composto usato, e si usa, per esempio, l'iniezione intracardiaca, il menadione e i menachinoni a breve catena appaiono avere scarsa o nulla attività e risultano più attivi i composti ad alto peso molecolare, più di tutti il menachinone-9.
Il composto più diffuso nei vegetali è il fillochinone, mentre i Batteri contengono una serie di menachinoni.
Distribuzione e fabbisogno
Vitamina K è presente nei vegetali a foglie verdi, nei pomodori, nella crusca di grano, nei semi e nell'olio di soia, nel formaggio, nel tuorlo d'uovo e nel fegato; quantità minori sono contenute nelle ossa, nei muscoli scheletrici e nel sangue. È scarsa negli altri organi. La flora del basso intestino sintetizza vitamina K2: solo parte di questa è però disponibile per essere assorbita.
La richiesta giornaliera di vitamina per l'uomo è di 0,5-1,0 μg/kg. Oltre all'apporto con la dieta sono importanti la presenza di bile nell'intestino e condizioni di normalità nella superficie assorbente dell'intestino e nel fegato. Qualora tali requisiti non siano soddisfatti si può instaurare malassorbimento e deficienza.
L'uso principale di vitamina K in zootecnia viene fatto nelle diete per il pollame, in quanto l'impiego di antibiotici nella dieta ne riduce la sintesi intestinale. Per agevolare l'assorbimento si usano derivati idrosolubili.
Antagonisti
Vari gruppi di sostanze agiscono come antagonisti della vitamina K. Innanzitutto i derivati cumarinici, il primo dei quali, 3,3′-metilen-bis(4-idrossicumarina), fu caratterizzato nel 1941 e denominato dicumarolo. Accanto a esso il warfarin e l'etilbiscumacetato vengono usati come anticoagulanti in clinica e anche come rodenticidi (v. pesticidi). Negli anni sessanta-settanta si sono però manifestate popolazioni di topi resistenti al warfarin, il che ha portato alla sintesi di composti molto più efficaci. Una seconda classe di antagonisti sono gli 1 ,3-indandioni, che per la loro potenziale tossicità epatica non sono più usati in clinica. Infine vi sono i 2-alo-3-fitil-1,4-naftochinoni, che agiscono da veri inibitori competitivi bloccando il sito di azione della vitamina.
bibliografia
Barker-David, M., Bender, A. (a cura di), Vitamins in medicine, London 19804.
De Luca, H. F. (a cura di), The fat-soluble vitamins, New York 1978.
György, P., Pearsons, W. N. (a cura di), The vitamins, voll. VI-VII, New York 1967.
Lewin, S., Vitamin C: its molecular biology and medical potential, London-New York 1976.
Machlin, L. J., Vitamin E: a comprehensive treatise, New York-Basel 1980.
Norman, A. W., Vitamin D: the calcium homeostatic steroid hormone, New York 1979.
Rivlin, R. S. (a cura di), Riboflavin, New York 1975.
Sebrell, W. H. Jr., Harris, R. S. (a cura di), The vitamins, voll. I-V, New York 1967-1972.
Suttie, J. W., Jackson, C. M., Prothrombin structure, activation and biosynthesis, in ‟Physiological reviews", 1977, LVII, pp. 1-70.
Tryfiates, G. P. (a cura di), Vitamin B6: metabolism and role in growth, Westport, Conn., 1980.
Zagalak, R., Friedrich, W. (a cura di), Vitamin B12, Berlin-New York 1979.