
Analisi chimica strumentale
Analisi chimica strumentale
La chimica analitica si occupa dello studio e dello sviluppo dei metodi mediante i quali si possono individuare le specie chimiche presenti in un campione di materia e determinarne le quantità. Di solito, queste vengono espresse come quantità relative: per ogni specie presente in numero discreto, si determina la parte in peso per cento e, per i costituenti in tracce, la parte in peso per milione (ppm) o per miliardo (ppb) o per trilione (ppt). Con l'espressione 'specie chimiche' si possono intendere sia gli elementi chimici di cui è costituito il campione, sia gli ioni presenti, sia, infine, i composti chimici che come tali esistono nel materiale in esame.
I compiti attuali della chimica analitica sono molteplici, in quanto si tratta di valutare non solo i costituenti principali di un dato sistema, cioè i componenti maggiori, ma anche i costituenti minori o in tracce in miscele particolarmente complesse, la presenza dei quali può avere un'influenza determinante. Mentre fino a qualche tempo fa erano poche le sostanze che potevano essere determinate con sufficiente accuratezza in concentrazioni di poco inferiori a 1 ppm, si è arrivati a eseguire determinazioni di numerose sostanze presenti in ppb e, in casi molto favorevoli, in ppt. Facendo uso di tecniche estremamente raffinate e in condizioni molto particolari, sono state rilevate concentrazioni dell'ordine di decine di atomi per cm3 (si noti che, a pressione atmosferica, il numero di molecole presenti in 1 cm3 di un gas o di un vapore è dell'ordine di 1019).
L'esigenza di poter rilevare elementi o composti in tracce si manifesta in molti campi: per esempio, in quello dell'ambiente allo scopo di valutarne con accuratezza i sintomi del degrado nel tempo; oppure nella preparazione dei semiconduttori impiegati nella costruzione dei componenti elettronici. Il germanio e il silicio, infatti, devono essere preparati allo stato di grandissima purezza prima di inquinarli di proposito con quantità esattamente predeterminate di altri elementi, in modo da conferire loro la semiconduttività desiderata. Occorre, quindi, un accurato controllo della purezza sia delle materie prime impiegate sia dell'ambiente nel corso della preparazione industriale.
Per quanto riguarda gli alimenti, si possono avere inquinamenti da sostanze nocive sia nel corso della produzione (diserbanti, insetticidi, ormoni), sia in quello della conservazione o dei trattamenti successivi ai quali vengono sottoposti.
Altro importante compito della chimica analitica è rappresentato dal tentativo di separare e di determinare sostanze le cui strutture molecolari ‒ e quindi anche le proprietà chimiche ‒ sono assai simili. Per rendersi conto dell'importanza di questa attività è sufficiente ricordare che in un gruppo di composti con struttura molecolare molto simile può accadere di riscontrare che uno di essi è dotato di proprietà biologiche (per es., tossicità) molto più degli altri, e quindi è di fondamentale importanza poter accertare la quantità di quel particolare composto.
Tali compiti sono stati eseguiti utilizzando procedimenti strumentali, e cioè mediante l'impiego di apparecchiature basate su principî fisici o chimico-fisici specifici del sistema in esame o della struttura delle specie che debbono essere determinate. In numerosi casi, le apparecchiature possono fornire una risposta molto selettiva, così da non richiedere una preventiva separazione delle specie da analizzare.
Nel caso di miscele complesse, in cui può essere presente un gran numero di composti, si rende spesso necessario il frazionamento, con il conseguente isolamento delle specie che interessano. L'obiettivo viene raggiunto mediante l'utilizzazione di tecniche cromatografiche, che possono essere condotte secondo una varietà di procedimenti. Ovviamente, i progressi compiuti dalla strumentazione nell'analisi chimica riflettono lo sviluppo dell'elettronica, senza il quale sarebbe stata impensabile la progettazione degli attuali strumenti. Tale apporto evidenzia un'altra caratteristica dell'attuale chimica analitica: l'utilizzazione di mezzi strumentali in modo semiautomatico o automatico, al fine di eliminare l'intervento dell'operatore in molte attività e gli errori legati alla manualità, e ottenere il responso finale in forma grafica o digitale. Lo strumento, infatti, può essere collegato con calcolatori che programmano lo svolgimento dell'analisi, elaborano i dati ed eventualmente confrontano la risposta dello strumento con i dati di sostanze note contenuti in una memoria, allo scopo di identificare le sostanze analizzate.
Sommario: 1. Metodi elettroanalitici. 2. Metodi basati sull'emissione e sull'assorbimento di radiazioni elettromagnetiche. 3. Metodi basati sulla distribuzione tra fasi
Metodi elettroanalitici
Si chiamano metodi elettroanalitici tutti i procedimenti di analisi in cui si perviene alla determinazione di una data specie attraverso la misura di una grandezza elettrica, utilizzando idonei dispositivi. I più importanti sono: (a) i metodi potenziometrici, fondati sulla misura della forza elettromotrice di una cella galvanica; (b) i metodi conduttimetrici, nei quali si eseguono misure della resistenza elettrica di soluzioni di elettroliti; (c) i metodi voltammetrici, basati sul rilevamento della variazione dell'intensità di corrente in una cella elettrolitica al variare della differenza di potenziale applicata agli elettrodi; (d) i metodi coulombometrici, con i quali la determinazione della quantità di una sostanza viene ricavata dalla misura del numero di coulomb consumati in un opportuno processo elettrolitico.
Potenziometria
Nella potenziometria si crea una cella galvanica e dalla misura della sua forza elettromotrice si risale alla concentrazione di una specie presente nella soluzione in condizioni di equilibrio. A titolo d'esempio, viene illustrato brevemente il tipo di cella usato per la determinazione della concentrazione degli ioni idrogeno [H+] in una soluzione acquosa. Com'è noto, questa grandezza indica le caratteristiche di acidità (o di basicità) di una soluzione e viene misurata correntemente sia in chimica sia in biologia. La concentrazione viene espressa in grammomolecole per litro. Un parametro correlabile direttamente con la concentrazione degli ioni idrogeno è il pH, definito per semplicità (anche se in modo non del tutto corretto) come l'inverso del logaritmo della concentrazione idrogenionica: pH=−log10[H+]].
Elettrodi a membrana di vetro
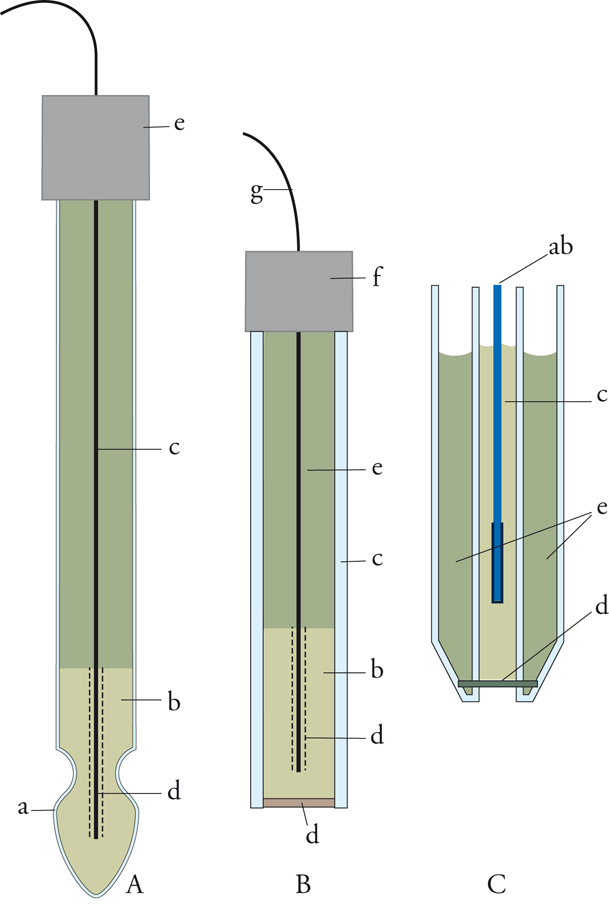
Il classico elettrodo di platino con gorgogliamento d'idrogeno, indicato come elettrodo a idrogeno, è alquanto scomodo nel suo impiego e quindi si preferisce sostituirlo per la misura del pH con l'elettrodo a membrana di vetro, che ha la forma riportata nella fig.1A. Nel suo interno vi è un elettrodo di riferimento argento-cloruro d'argento immerso in una soluzione con acidità costante contenente cloruri. La membrana di vetro, che costituisce il bulbo dell'elettrodo, è in contatto con la soluzione esterna di cui si vuole determinare la [H+]. L'elettrodo descritto assume, rispetto a quello di riferimento, un potenziale che è funzione logaritmica del rapporto tra le concentrazioni in H+ delle soluzioni esterna e interna. Siccome la soluzione interna ha un'acidità fissa, ne consegue che l'elettrodo misura l'acidità della soluzione esterna.
Elettrodi ione-selettivi
Sullo stesso principio dell'elettrodo a membrana ne vengono costruiti altri per determinare la concentrazione di vari ioni, come per esempio rame, argento, cadmio, piombo, e anioni come cloruri, bromuri, tiociana-ti, ecc. Tali elettrodi hanno uno schema simile a quello dell'elettrodo a membrana di vetro: questa è sostituita da una membrana sensibile ottenuta con vari procedimenti. Essa può essere una lamina sottile ricavata da un macrocristallo di un composto poco solubile dello ione considerato, oppure una membrana eterogenea ottenuta incorporando una polvere di un sale poco solubile dello ione in una matrice formata da una materia plastica (fig.1B). In un altro metodo si ricorre all'impregnazione di un diaframma poroso con uno scambiatore di ioni liquido, immiscibile con acqua (fig. 1C). La funzione dello scambiatore ionico è quella di scambiare con altri ioni la specie ionica che si deve determinare.
Elettrodi a enzima
Ulteriori applicazioni della potenziometria diretta sono offerte dagli elettrodi a enzima, nei quali uno o più enzimi sono accoppiati a un opportuno elettrodo selettivo. L'enzima si trova immobilizzato sulla superficie dell'elettrodo: questo dispositivo viene impiegato per misurare un prodotto o un reagente che partecipa alla reazione enzimatica. Quando la sostanza diffonde nello strato di enzima, avviene la reazione enzimatica, che è specifica, e si forma un prodotto o viene consumato un reagente, che l'elettrodo selettivo è in grado di rivelare. Un'estensione dell'elettrodo a enzima è l'elettrodo a batteri, in cui una colonia viva di particolari batteri è immobilizzata sulla superficie di un elettrodo sensibile a un gas, che si forma nel corso di una reazione metabolica. Il gas attraversa lo strato di batteri e raggiunge l'elemento sensibile dell'elettrodo che funge da rivelatore. Un dispositivo di questo tipo può essere impiegato come sensore specifico per vari amminoacidi.
Voltammetria, polarografia
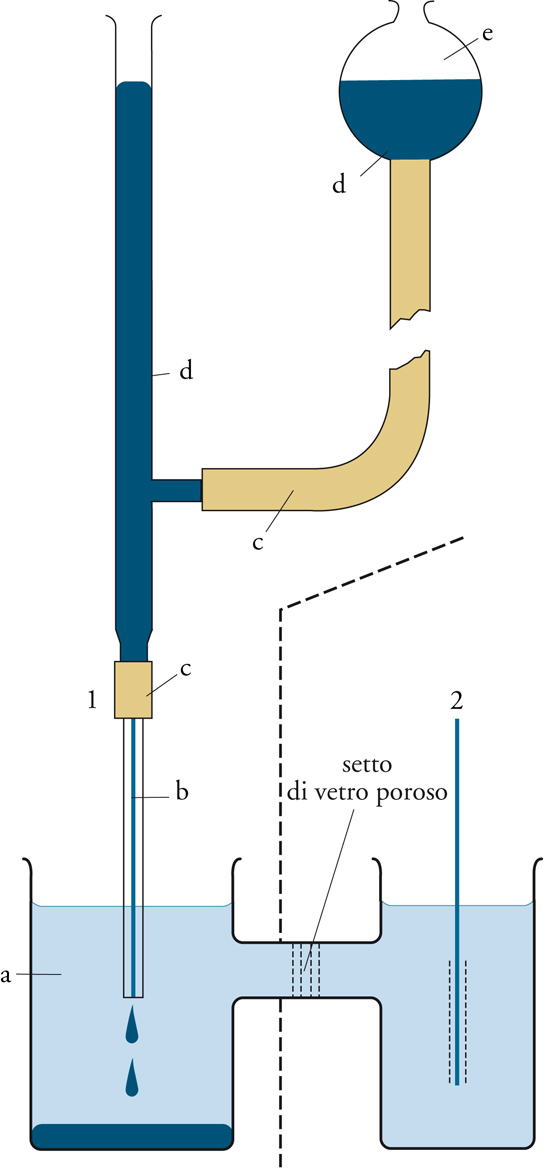
Se una soluzione contenente sostanze ossidabili oppure riducibili viene sottoposta a elettrolisi, il soluto può venire ridotto al catodo, oppure ossidato all'anodo. Operando in opportune condizioni, l'intensità della corrente di elettrolisi che viene misurata è correlabile con la concentrazione del soluto e può quindi consentirne la determinazione. L'applicazione di questo principio ha dato origine alle tecniche voltammetriche di analisi. Tra queste, riveste particolare importanza la polarografia. Nella cella elettrolitica in cui si trova la soluzione da analizzare sono posti due elettrodi: uno di riferimento a potenziale costante e uno indicatore. Nella polarografia, quest'ultimo è il cosiddetto elettrodo a goccia di mercurio, che consiste in un tubo capillare di vetro collegato, tramite un tubo flessibile, con un serbatoio contenente mercurio (fig. 2). Il mercurio riempie il capillare e sgocciola dalla sua estremità inferiore, mentre il serbatoio contribuisce a mantenere costante il livello superiore, assicurando una velocità di efflusso costante. La superficie della goccia varia continuamente nel corso della sua vita, ma è sempre molto esigua, e l'elettrodo si comporta come un microelettrodo, facilmente polarizzabile.
Tecniche di ridissoluzione (stripping) anodica
Un metodo elettroanalitico che può permettere la determinazione di concentrazioni dell'ordine di 10−10 moli/l di ioni metallici consiste nel concentrare su una superficie molto esigua di un catodo, per esempio di grafite, un metallo per riduzione da una soluzione molto diluita di un suo ione. Questo risultato si ottiene eseguendo l'elettrolisi della soluzione in esame per un tempo sufficientemente lungo. Dopo di ciò, la polarità dell'elettrodo viene invertita, in modo da farlo funzionare come anodo. Variando il potenziale applicato all'elettrodo, si raggiunge un valore per il quale il metallo si ossida e passa in soluzione, dando luogo a un picco di corrente dalla cui altezza si misura la concentrazione. Il metodo non richiede che tutto il metallo presente nella soluzione da analizzare venga depositato sull'elettrodo. Infatti, è possibile ricavare la concentrazione per confronto con i risultati ottenuti operando in condizioni identiche con soluzioni a concentrazione nota del metallo da analizzare.
Coulombometria
La coulombometria è una tecnica elettroanalitica che si basa sull'impiego delle leggi di Faraday riguardanti il processo di elettrolisi. La quantità di una determinata specie chimica presente in una cella elettrolitica e riducibile al catodo, oppure ossidabile all'anodo, può essere dedotta dal consumo di elettricità nel corso dell'elettrolisi, a condizione che la quantità di elettricità che passa nella cella sia stata esclusivamente utilizzata per il processo di riduzione (o di ossidazione) preso in esame. Siccome, adottando opportuni accorgimenti, questa condizione può essere in molti casi realizzata, ne conseguono le molteplici possibilità di applicazione del metodo.
Metodi basati sull'emissione e sull'assorbimento di radiazioni elettromagnetiche
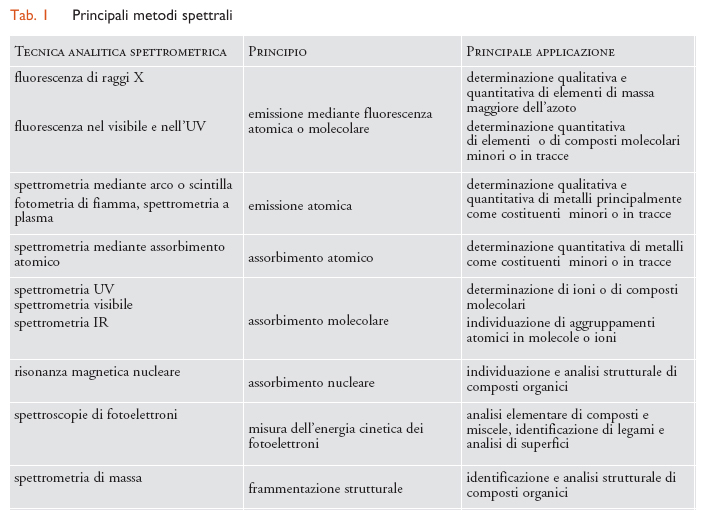
I metodi di analisi basati sull'emissione e sull'assorbimento di radiazioni, genericamente indicati come metodi spettrali, sono quelli di più diffuso impiego in chimica analitica e sono ricchi di informazioni sia sotto l'aspetto qualitativo sia sotto quello quantitativo. Le tecniche spettrali hanno come fondamento l'interazione delle radiazioni con sistemi atomici o molecolari; le radiazioni vengono emesse o assorbite quando hanno luogo transizioni tra livelli energetici stazionari. Data l'ampiezza dello spettro elettromagnetico, si possono distinguere numerose tecniche spettrali, ciascuna delle quali copre un particolare intervallo di lunghezze d'onda. Le apparecchiature impiegate presentano caratteristiche costruttive assai diverse, conseguenti sia alle caratteristiche specifiche dell'intervallo spettrale interessato sia alle prestazioni richieste. Uno schema dei metodi spettrali più importanti è riportato nella tab. 1.
Fluorescenza di raggi X
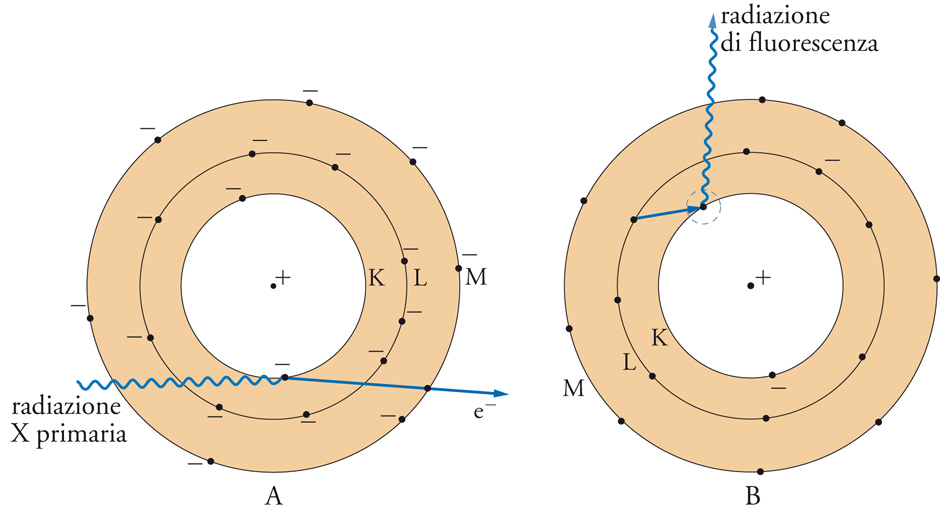
Se un fascio di raggi X di lunghezza d'onda opportuna viene inviato su una sostanza allo stato solido o liquido, una frazione del fascio incidente sarà assorbita dagli atomi presenti nel campione, con l'espulsione di elettroni appartenenti ai cosiddetti gusci interni (K, L). Come indicato nella fig. 3, la lacuna che si crea viene immediatamente colmata dalla transizione di uno degli elettroni dei gusci più esterni. In questo processo, si ha emissione di energia sotto forma di raggi X secondari (radiazione di fluorescenza). Siccome le transizioni possibili sono relativamente poche, il fascio emesso può essere scomposto in una serie non numerosa di radiazioni con lunghezza d'onda caratteristica: ciascuna di esse ha lunghezza d'onda maggiore della radiazione eccitatrice. È noto che le frequenze delle radiazioni X emesse da ogni elemento obbediscono alla legge di Moseley e sono quindi correlabili con il numero atomico dell'elemento considerato. Lo spettro di emissione di raggi X di un elemento consente di individuarlo con una certa facilità. Se il materiale in esame è costituito da più elementi, sia liberi sia combinati tra loro, l'individuazione delle frequenze nello spettro di raggi X emesso permette di risalire agli elementi presenti. La misura dell'intensità delle radiazioni emesse, confrontata con quella di un elemento di riferimento, aggiunto al campione in quantità nota, permette di ricavare indicazioni sulla loro concentrazione.
Emissione atomica
È noto che gli atomi isolati possono trovarsi sia allo stato fondamentale sia in stati elettronici eccitati. Le transizioni da uno stato a un altro, che danno luogo a liberazione di energia, causano l'emissione di radiazioni con lunghezze d'onda ben definite, cui corrispondono righe diverse nello spettro di emissione. Il numero di righe presenti nello spettro cresce fortemente con l'aumentare del numero atomico. Ogni elemento emette uno spettro caratteristico nella regione dall'ultravioletto al visibile; pertanto, se un campione di una data sostanza viene portato allo stato di vapore atomico ed eccitato, lo spettro di emissione consente di individuare la maggior parte degli elementi presenti. L'intensità delle righe emesse è proporzionale alla quantità di atomi presenti, se si opera in condizioni di eccitazione perfettamente riproducibili.
Assorbimento atomico
Se un vapore formato prevalentemente da atomi di un elemento allo stato fondamentale viene irradiato con un fascio monocromatico, la cui energia corrisponda alla differenza di energia richiesta per l'eccitazione degli atomi, la radiazione viene assorbita. L'entità dell'assorbimento è proporzionale al numero di atomi presenti nello stato fondamentale. Se I0 è l'intensità del fascio monocromatico incidente e I l'intensità dopo che esso ha attraversato uno strato di vapore atomico di spessore S, è valida la relazione A=log(I0/I)=KCS, dove C è la concentrazione di atomi per unità di volume e K una costante di proporzionalità che può essere esplicitata. La grandezza A viene chiamata assorbanza. L'intensità della radiazione incidente viene modulata per distinguerla dalla radiazione emessa dagli atomi eccitati sempre presenti nel vapore atomico. Il rivelatore, che è un tubo fotomoltiplicatore, raccoglie la radiazione e la converte in una corrente. Solo la corrente modulata viene amplificata.
Per produrre il vapore atomico si immette la soluzione del campione, nebulizzata, in una fiamma capace di portare il campione a una temperatura sufficientemente alta da dissociare in atomi i composti presenti. Si opera in atmosfera di argo, per evitare la formazione di ossidi difficilmente dissociabili. La radiazione monocromatica incidente proviene da una lampada spettrale capace di emettere lo spettro dell'elemento sottoposto ad analisi. Un monocromatore isola la riga spettrale desiderata. La spettrometria di assorbimento atomico è particolarmente adatta per la determinazione dei metalli. I limiti di rivelabilità sono particolarmente bassi con la maggior parte dei metalli, perché nel vapore atomico presente nella fiamma o nel fornetto di grafite gli atomi si trovano prevalentemente nello stato fondamentale e sono quindi in grado di assorbire la radiazione incidente.
Assorbimento nel visibile e nell'ultravioletto
Le sostanze capaci di assorbire radiazioni nel visibile o nell'ultravioletto sono molto numerose. Alcune di esse si diseccitano dando luogo alla fluorescenza, mentre la maggior parte dissipa l'energia assorbita sotto forma di calore. Le misure di assorbimento vengono eseguite con uno spettrofotometro capace di operare nella regione spettrale compresa tra circa 195 e 900 nm. Gli strumenti di questo tipo, generalmente, sono costituiti da una sorgente, un monocromatore, un rivelatore e un dispositivo di registrazione e lettura, collegato con un amplificatore. La sorgente emette una radiazione policromatica. Il monocromatore isola le radiazioni comprese in un piccolo intervallo Δλ di lunghezze d'onda (spesso 0,2 nm) intorno a un certo valore λ. L'intervallo Δλ nell'intorno di λ viene detto banda passante. Negli strumenti a doppio raggio, il pennello di radiazioni isolato dal monocromatore viene fatto passare alternativamente attraverso una vaschetta contenente il solvente e una vaschetta con la soluzione in esame.
I due segnali vengono raccolti dal rivelatore, il quale li trasforma in segnali elettrici che vengono elaborati e presentati come trasmittanza T=(I/I0), dove I è l'intensità della radiazione trasmessa dalla soluzione e I0 quella trasmessa dal solvente ‒ oppure come assorbanza A. Quest'ultima grandezza è correlata alla concentrazione dalla relazione di Lambert-Beer A=εbc, dove c è la concentrazione, solitamente espressa in grammomolecole per litro, e b lo spessore dello strato di soluzione, espresso in cm, ε viene detta assorbività molare e rappresenta il reciproco dello spessore di soluzione contenente una grammomolecola/l, che dà luogo a un'assorbanza unitaria. La relazione è applicabile generalmente in un intervallo di concentrazioni di circa 2 ordini di grandezza (per es., di 0,1÷60 ppm).
Gli spettrofotometri IR a trasformate di Fourier (detti più brevemente spettrofotometri FT-IR) adottano uno schema costruttivo completamente diverso. Il fascio policromatico che ha attraversato il campione raggiunge un interferometro del tipo Michelson e dà luogo a un interferogramma che viene raccolto dal rivelatore e poi elaborato da un calcolatore. L'interferogramma contiene tutte le informazioni relative alle radiazioni di diversa lunghezza d'onda prodotte dalla sorgente e assorbite in varia misura dal campione. Anche il fascio policromatico emesso dalla sorgente dà un interferogramma non modificato dall'assorbimento del campione. Il calcolatore elabora i dati facendo uso delle trasformate di Fourier e li presenta registrati come spettro di assorbimento.
Risonanza magnetica nucleare (NMR)
Alcuni nuclidi, e tra di essi l'1H e il 13C, hanno nuclei che possiedono un momento angolare di spin. Se un insieme di nuclei di 1H (ossia di protoni) venisse messo in un campo magnetico uniforme, i protoni potrebbero assumere due orientazioni di spin quasi ugualmente popolate a temperatura ambiente, dato che la differenza di energia tra i due stati è dell'ordine di 10−26 J, minore dell'energia termica a 25 °C (kT=4,11×10−21 J). Con l'impiego di un campo magnetico rotante (che può essere sostituito da una radiofrequenza opportuna) è possibile provocare la transizione di spin dal livello di energia inferiore a quello superiore. Le due popolazioni, quindi, tendono a eguagliarsi, ma esistono processi di rilassamento che hanno la tendenza a riportare il sistema nelle condizioni iniziali. Se, anziché considerare un insieme di protoni, consideriamo un insieme di molecole contenenti atomi di 1H, il campo magnetico effettivo esercitato sui protoni risulta inferiore a quello applicato, a causa dell'atmosfera elettronica che circonda i protoni, a sua volta influenzata dagli atomi degli altri elementi che costituiscono la molecola.
In conseguenza di questi effetti, la frequenza alla quale i protoni entrano in risonanza risulta diversa da quella calcolabile teoricamente; la differenza viene chiamata spostamento chimico (chemical shift). Se una molecola di un composto contiene vari atomi di 1H, ciascuno con diverso intorno chimico, siccome essi non sono equivalenti, la risonanza dei loro nuclei ha luogo a frequenze diverse. Per misurare gli spostamenti chimici si usa uno spettrometro a risonanza magnetica nucleare. Esso è costituito da un magnete capace di creare un campo molto uniforme, variabile entro un piccolo intervallo intorno al valore richiesto per la risonanza, e dal campo magnetico rotante, oltre che da un dispositivo di rilevamento. Facendo variare il campo magnetico, si possono registrare le frequenze alle quali i nuclei di idrogeno con diverso intorno chimico entrano in risonanza. Se si passa dalla risonanza protonica, la più comunemente studiata, a quella del nuclide 13C, assai meno abbondante in natura del nuclide 12C, il cui nucleo non possiede momento di spin, il problema delle prestazioni richieste allo spettrometro diventa ancora più difficile.
Spettrometria di massa
La spettrometria di massa è una tecnica analitica per la caratterizzazione di specie molecolari, basata sull'identificazione dei frammenti ionici che da esse si generano sotto l'azione di elettroni ad alta energia. Nello spettrometro di massa, le molecole del composto in esame, in quantità che può variare da pochi milligrammi a un nanogrammo, sono portate allo stato gassoso e sono sottoposte a bombardamento da parte di un fascio di elettroni, che dà luogo alla formazione di ioni positivi, ioni negativi e specie radicaliche. La semplice asportazione di un elettrone dalla molecola genera un radicale-ione, lo ione molecolare, che possiede una massa eguale a quella della molecola. Si ha, contemporaneamente, la frammentazione della molecola, con formazione di varie specie ioniche di cui viene misurato il rapporto massa/carica. Gli ioni possono essere rivelati con lo spettrometro, se la loro vita media è superiore a un valore minimo, che è dell'ordine di 10−5 s. Se la loro stabilità è minore, essi possono subire sia ulteriori processi di frammentazione spontanea sia processi di associazione dovuti a collisione con radicali.
Dopo la frammentazione, gli ioni positivi vengono accelerati sotto l'azione di un campo elettrico e separati in base al rapporto massa/carica con dispositivi che variano secondo il tipo di strumento impiegato. Gli ioni, dopo il processo di separazione, raggiungono il rivelatore, che dà origine a una corrente ionica proporzionale al numero di cariche elettriche che essi portano. L'identificazione di una data specie può essere eseguita mediante l'esame dello spettro di massa. Il peso molecolare può essere immediatamente determinato se è presente lo ione molecolare, e cioè lo ione positivo risultante dalla cessione di un elettrone da parte della molecola neutra, senza frammentazione. L'esperienza ottenuta esaminando gli spettri di massa di un gran numero di composti organici ha permesso di formulare una serie di regole di frammentazione delle molecole, cosicché dall'esame della serie di frammenti formati è possibile risalire alla struttura molecolare del composto in esame.
Metodi basati sulla distribuzione tra fasi
Da lungo tempo il processo di estrazione, che si realizza dibattendo una soluzione con un solvente non miscibile con essa, è stato utilizzato per isolare un dato composto o un gruppo di composti dalla soluzione in esame. Tale processo si basa sul principio di ripartizione, secondo cui una specie si distribuisce fra due fasi in modo da rendere costante il rapporto delle sue concentrazioni nelle due fasi. Pertanto, per il soluto A che si distribuisce fra un solvente organico e una fase acquosa, si ha [A]o/[A]aq=KD, in cui [A]o e [A]aq sono le concentrazioni nelle due fasi e KD è il coefficiente di ripartizione. Per effettuare l'isolamento di una data specie da una soluzione o da un sistema solido, l'estrazione può essere eseguita in modo discontinuo, in modo continuo o in controcorrente; in quest'ultimo caso, si tratta di un processo estrattivo, in cui due solventi immiscibili vengono messi in contatto mentre fluiscono in direzione opposta. Prendiamo in esame un processo in controcorrente discontinuo, in cui una quantità costante di solvente puro (fase stazionaria) viene messa in contatto con un ugual volume di un altro solvente (fase mobile). Se quest'ultimo viene successivamente trasferito in recipienti contenenti la fase stazionaria e dibattuto con essa, si osserva che una qualunque specie si ripartisce nelle due fasi secondo una distribuzione gaussiana, governata dal valore di KD .
Il processo di ripartizione, e la conseguente distribuzione di una specie fra due solventi non miscibili, rappresenta uno dei numerosi metodi (cromatografici) di distribuzione tra fasi diverse (come, per es., solido-liquido). Il termine cromatografia è stato introdotto dal botanico russo Michail Tswett all'inizio del Novecento, per descrivere la separazione dei pigmenti delle foglie verdi ottenuta facendo percolare etere di petrolio su una colonna impaccata con carbonato di calcio in polvere, alla cui estremità superiore era posto un estratto di foglie. In tutti i processi cromatografici si utilizza lo stesso principio, e cioè la diversa velocità con cui i differenti componenti di una miscela migrano in una fase stazionaria sotto l'influenza di una fase mobile, la quale ha il compito di trascinare i componenti della miscela. La differente velocità è determinata dagli equilibri cui prendono parte le specie che debbono essere separate, e che dipendono dalla loro interazione con la fase stazionaria.
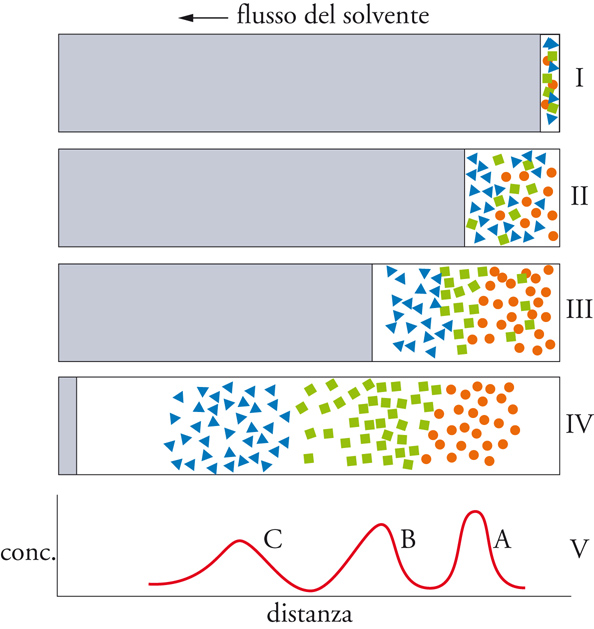
Durante un processo cromatografico, le molecole delle specie che devono essere separate e analizzate passano dalla fase stazionaria a quella mobile e da questa nuovamente nella fase stazionaria, e tale trasferimento si ripete molte volte secondo i vari dispositivi sperimentali. Di conseguenza, le molecole, allorché si trovano nella fase mobile, procedono nella direzione del flusso di quest'ultima, mentre sono praticamente ferme durante il tempo in cui permangono nella fase stazionaria. La velocità di migrazione di ciascun soluto è determinata dal rapporto tra i tempi che esso trascorre nelle due fasi e quindi dal suo rapporto di distribuzione. I processi che intervengono nel trasferimento da una fase mobile a una stazionaria sono quelli di adsorbimento, ripartizione, scambio ionico ed esclusione. Qualunque sia la natura dell'interazione fra i soluti di una miscela da analizzare e la fase stazionaria, il processo cromatografico che determina il loro frazionamento e la conseguente separazione dei costituenti è quello schematizzato nella fig. 4.

Una miscela costituita da tre componenti, A, B e C (indicati rispettivamente con piccoli cerchi, quadrati e triangoli), è posta su un supporto (I stadio); allorché la fase mobile la investe, si ha una parziale separazione di questi costituenti (II stadio). Il frazionamento è più accentuato nel III stadio, mentre nel IV la separazione è pressoché completa, come risulta dal tracciato finale, che riporta la concentrazione delle tre specie nel sistema in cui è avvenuto il frazionamento. La cromatografia costituisce il procedimento più efficace per la separazione dei costituenti di una miscela e per la determinazione di una qualunque specie: dopo averla isolata, si può procedere allora alla sua identificazione e alla sua valutazione quantitativa. Le tecniche cromatografiche si eseguono con apparecchiature e modalità differenti a seconda della natura della fase mobile impiegata, che può essere un gas o un liquido, e a seconda della natura della fase stazionaria, che può essere un liquido o un solido con particolari proprietà. I vari procedimenti sono schematizzati nella tab. 2.
Gascromatografia
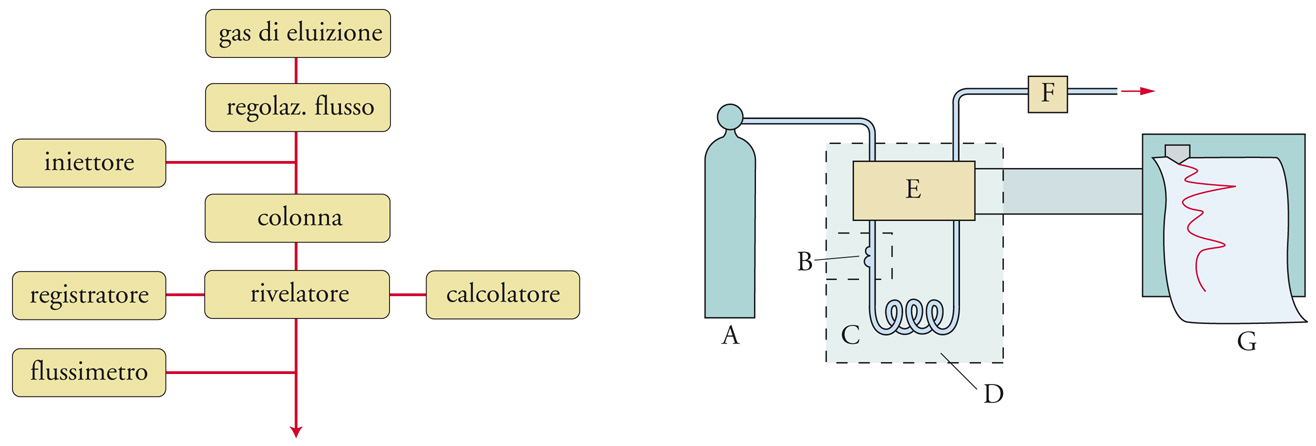
L'analisi cromatografica che si realizza usando come fase mobile un gas viene denominata gascromatografia. La fase stazionaria può essere un solido o un liquido non volatile trattenuto da un supporto inerte. Nel primo caso, si ha la cromatografia gas-solido o GSC (Gas-solid chromatography), nel secondo la cromatografia gas-liquido o GLC (Gas-liquid chromatography). Gli schemi a blocchi e quelli funzionali di un sistema cromatografico in fase gassosa sono riportati nella fig. 5. La fase mobile, detta anche gas di trasporto o gas vettore, costituita da un gas inerte quale elio, azoto o idrogeno, è fatta fluire con velocità costante attraverso una colonna cromatografica (il dispositivo in cui avviene il frazionamento della miscela in esame), mantenuta a temperatura costante per mezzo di una camera termostatica.
Mediante un opportuno rivelatore ‒ indicato con E nella fig. 5 ‒ che fornisce di solito un responso differenziale misurando una proprietà del gas di eluizione prima e dopo il passaggio nella colonna, è possibile mettere in evidenza i singoli componenti della miscela in esame, che sono stati frazionati dalla colonna e che fuoriescono da essa in tempi successivi. Il responso del rivelatore è amplificato e inviato a un registratore, da cui si ottiene il relativo tracciato, indicato come gascromatogramma.
La cromatografia consente di ricavare eccezionali informazioni sulla composizione di tutti i sistemi naturali o sintetici costituiti da specie gassose o vaporizzabili e, in virtù delle possibilità di frazionamento delle colonne cromatografiche e della sensibilità dei sistemi di rivelazione, grazie ai quali si possono rivelare quantità pari a 10−12÷10−13 g, è stato possibile risalire alla natura di sistemi complessi.
Cromatografia liquida ad alta pressione (HPLC)
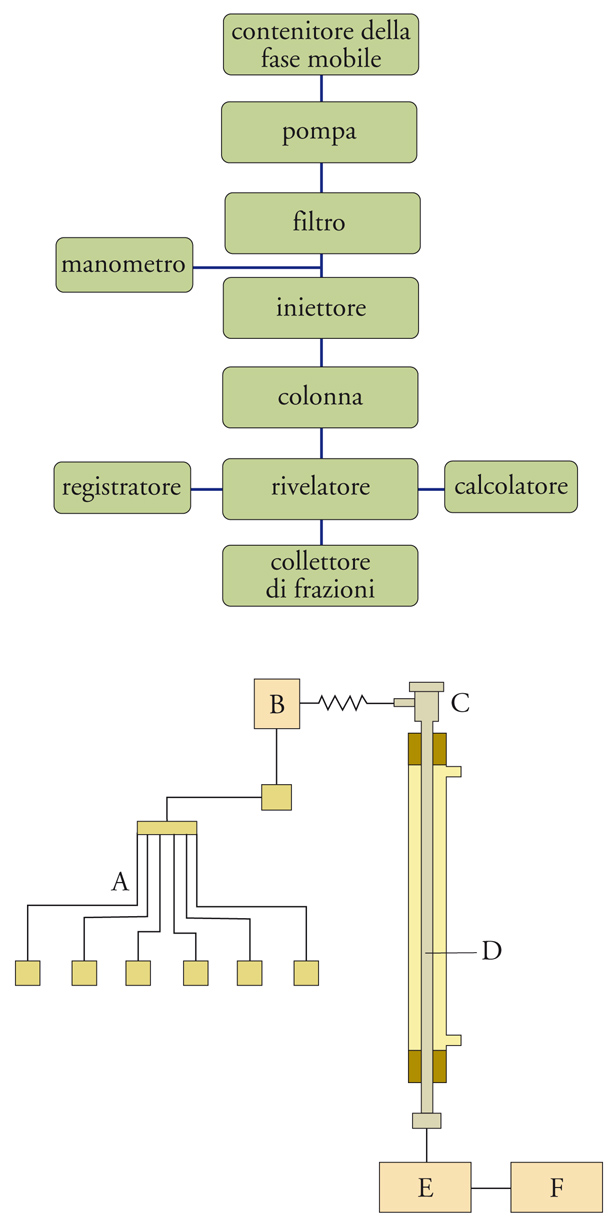
La cromatografia in fase gassosa è impiegabile solo per le sostanze gassose o vaporizzabili; tuttavia, una colonna cromatografica non può essere utilizzata a una temperatura superiore a circa 250 °C, perché la fase stazionaria si decompone. Questa limitazione non esiste nella cromatografia liquida, in cui si utilizza come fase mobile un liquido e si opera a temperatura ambiente. In queste condizioni, qualsiasi specie può essere separata, identificata e determinata quantitativamente. Utilizzando come fase mobile un liquido, in luogo di un gas, e operando con una fase stazionaria supportata da particelle solide di dimensioni molto piccole, per esaltare la capacità di separazione, la velocità di flusso si abbassa notevolmente; per ovviare a questo inconveniente è stato opportuno esercitare a monte della colonna una pressione molto alta sulla fase mobile. Utilizzando questi criteri è stata sviluppata la cromatografia liquida ad alta pressione, detta anche più propriamente cromatografia liquida ad alta risoluzione o HPLC (High performance liquid chromatography). Lo schema a blocchi di un sistema per HPLC è riportato nella fig. 6, insieme con lo schema funzionale. L'apparecchio è costituito da una pompa che convoglia alla colonna il liquido prescelto come fase mobile, dopo che esso ha attraversato il dispositivo mediante il quale la sostanza in esame viene iniettata nella colonna. All'uscita della colonna è disposto un rivelatore collegato con il registratore. La funzione della pompa è quella di assicurare un'elevata pressione di ingresso (3,5÷35 MPa) e una portata controllata della fase mobile con un flusso costante (1÷2 ml/min) attraverso la colonna in cui si trova la fase stazionaria. In genere, la colonna ha un diametro interno pari a 2÷4 mm e una lunghezza di 10÷30 cm, ed è riempita di materiali omogenei costituiti da particelle di diametro molto piccolo (250 μm).
A monte della colonna vi è un dispositivo per l'introduzione del campione e all'uscita un rivelatore che segnala i vari componenti separati dalla colonna. La registrazione della risposta del rivelatore in funzione del tempo dà luogo a un tracciato cromatografico, il cromatogramma liquido, che viene utilizzato per la valutazione qualitativa e quantitativa della sostanza analizzata. I dati del cromatogramma possono essere elaborati con opportuni sistemi computerizzati. Oltre alla fase stazionaria, anche la fase mobile svolge nella HPLC un ruolo attivo: infatti, essa dà luogo a interazioni con i componenti della miscela da separare. Così, per variare l'entità di tali interazioni, la fase mobile, anziché consiste-re in un solo solvente, può essere una miscela di duesolventi, e la sua composizione può essere mantenutacostante (procedimento isocratico) o fatta variare nel corso dell'eluizione. Questo secondo procedimento viene utilizzato quando i componenti di un campione possiedono caratteristiche di polarità molto diverse, per migliorare la separazione e per ridurre i tempi di eluizione. Come nella gascromatografia, l'analisi qualitativa, intesa come identificazione del picco di ciascun componente, si esegue sulla base della misura dei tempi di eluizione, che vengono confrontati con quelli che si ottengono eluendo nelle stesse condizioni i singoli composti puri. L'analisi quantitativa è basata sulla misura delle aree dei picchi, previa appropriata calibrazione con miscele a contenuto noto.
Bibliografia
Bauer 1978: Instrumental analysis, edited by Henry H. Bauer, Boston, Allyn and Bacon, 1978 (trad. it.: Analisi strumentale, Padova, Piccin, 1986).
Christian 2004: Christian, Gary D., Analytical chemistry, 6. ed., Hoboken (N.J.), Wiley, 2004.
Harris 2007: Harris, Daniel C., Quantitative chemical ana-lysis, 7ed., New York, Freeman, 2007.
Kolthoff, Elving 1959-1980: Treatise on analytical chem-istry, edited by Izaak M. Kolthoff, Philip J. Elving, New York, Wiley, 1959-1980, 33 v.
Kolthoff 1969: Kolthoff, Izaak M. e altri, Quantitative chemical analysis, 4. ed., New York-London, Macmillan, 1969 (trad. it.: Analisi chimica quantitativa, Padova, Piccin, 1980).
Poole, Schouette 1984: Poole, Colin F. - Schouette, Sheila A., Contemporary practice of chromatography, Amsterdam-Oxford, Elsevier, 1984.
Saini, Liberti 1980: Saini, Guido - Liberti, Arnaldo, Chimica analitica, Torino, UTET, 1980.
Skoog, West 1971: Skoog, Douglas A. - West, Donald M., Principles of instrumental analysis, New York, Holt, Rinehart and Winston, 1971.
Vogel 1972: Vogel, Arthur I., A textbook of quantitative inorganic analysis including elementary instrumental analysis, 3. ed., London, Longman, 1972.
Wilson, Wilson 1959-2001: Comprehensive analytical chem-istry, edited by Cecil L. Wilson, David W. Wilson, Amsterdam, Elsevier, 1959-2001.