
Aromaticità e composti aromatici
Aromaticità e composti aromatici
La classificazione delle sostanze organiche comprende un'ampia serie di molecole insature cicliche, strutturalmente affini al benzene e in origine note come aromatiche per l'odore caratteristico. I composti aromatici appartengono alla classe degli idrocarburi insaturi e sono per lo più caratterizzati da scarsa reattività chimica ed elevata stabilità termica. La definizione di aromatico, fin dalla scoperta del benzene da parte di Michael Faraday (1825), è stata attribuita agli idrocarburi benzenoidi che non mostrano la tipica reattività degli alcheni, ma hanno tendenza a subire le reazioni di sostituzione che spesso richiedono calore o catalizzatori. Le evidenze sperimentali rappresentarono ‒ negli anni successivi ai lavori di Friedrich August Kekulé (1865), James Dewar (1867) e Carl Ernst Claus (1867) ‒ i fondamenti della discussione sulla natura della aromaticità con riferimento alla configurazione elettronica-π del benzene.
formula
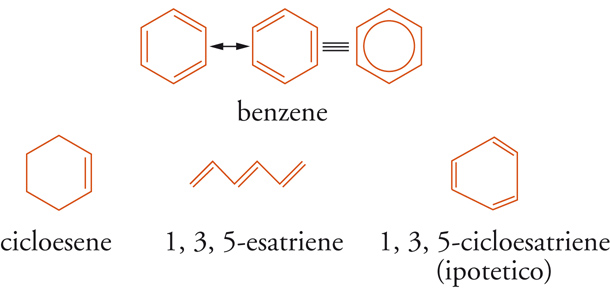
La rappresentazione elettronica π della struttura reale del benzene, definita risonanza aromatica (o risonanza di coniugazione), illustrata graficamente con un cerchio inscritto in un poligono regolare, è mediata da due (o più) forme di risonanza (forme canoniche) che coinvolgono gli orbitali p di 6 atomi di carbonio ibridati sp2. Ernst C. Crocker nel 1922, James W. Armit e Robert Robinson nel 1925, ipotizzarono che le proprietà aromatiche dei sistemi insaturi esagonali, come il benzene e la piridina, o pentagonali, come il pirrolo e il tiofene, fossero attribuibili alla presenza di elettroni detti π delocalizzati nell'anello e formanti due nubi a forma di ciambella generata dalla sovrapposizione dei 6 orbitali p residui sopra e sotto l'anello (sestetto aromatico). Erich Hückel nel 1931 eseguì calcoli quantomeccanici approssimati degli orbitali molecolari HMO (Hückel molecular orbital) di molecole insature e aromatiche, trattando separatamente gli elettroni-σ appartenenti ai legami che formano lo scheletro molecolare, dagli elettroni-π. Di particolare interesse sono quelli che occupano gli orbitali molecolari di frontiera FMO (Frontier molecular orbital): quelli a più alta energia, detti HOMO (Highest occupied molecular orbital), e quelli di antilegame π* a più bassa energia, detti LUMO (Lowest unfilled molecular orbital). Le illustrazioni delle forme di risonanza (tipiche strutture di Lewis, Kekulé, Dewar e Claus) e le consequenziali strutture reali di una molecola sono paradigmatiche e vincolate con una serie di postulati, da cui si può facilmente dedurre che la differenza tra l'energia del benzene nello stato fondamentale _(Ede_loc), con i legami π delocalizzati e quella (Eloc) di un ipotetico 1,3,5-cicloesatriene, con legami π localizzati dovrebbe corrispondere all'energia di coniugazione ΔEcon (o energia di risonanza aromatica), cioè al grado di stabilizzazione conferito al benzene dall'aromaticità.
Definizione magnetica di aromaticità

Già a partire dal 1912 Paul Pascal considerò la suscettività magnetica come una delle proprietà degli aromatici. La maggiore suscettività magnetica e lo spostamento a frequenze più alte del segnale degli idrogeni di un composto aromatico, rispetto all'omologo aciclico, erano alcuni fenomeni inizialmente utilizzati per stabilire se una molecola contenesse un sistema di elettroni-π ciclico. L'aromaticità fu allora definita come la capacità di generare una corrente di anello in una molecola sottoposta a un campo magnetico uniformemente applicato. L'introduzione in chimica organica di strumenti per l'analisi spettroscopica sempre più evoluti, in particolare la 1H-NMR, ha permesso una più specifica ricognizione sperimentale dei composti aromatici (sistemi diatropici). La circolazione degli elettroni-π nel perimetro molecolare sarebbe in grado di generare un campo magnetico indotto, che, al centro dell'anello, si oppone al campo magnetico applicato (effetto paramagnetico) mentre è concorde alla periferia (effetto diamagnetico) (fig. 2).
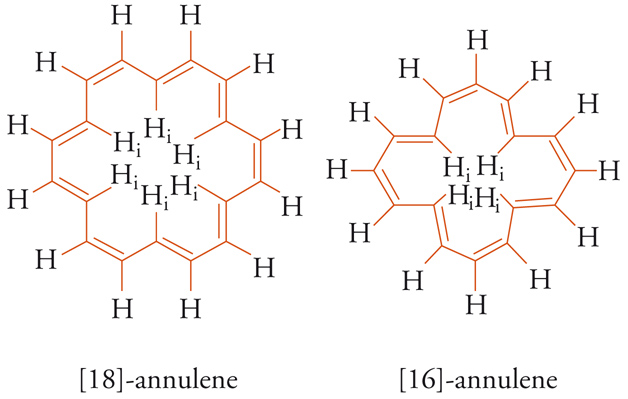
La frequenza di risonanza (o spostamento chimico δ) del segnale spettroscopico degli idrogeni (protoni) esterni all'anello, regione dello spazio in cui il campo magnetico applicato e quello indotto si sommano, 'risuona' a campo magnetico più basso (alti valori di δ), mentre il segnale degli idrogeni interni, dove i campi magnetici applicato e indotto sono opposti, 'risuona' a campo magnetico più alto (bassi valori di δ). Il modello descritto nel 1956 da John A. Pople per il benzene è utilizzato come criterio di aromaticità per qualsiasi molecola planare carbo- o eteroaromatica contenente 4N+2 elettroni-π ciclici (diatropici) e per i composti 4N antiaromatici (paratropici). Questi ultimi sono caratterizzati da una corrente d'anello paramagnetica, perciò gli effetti schermanti e deschermanti sono rovesciati in base allo sdoppiamento Jahn-Teller. Gli spettri NMR di due sistemi, diatropico e paratropico, dimostrano la dipendenza dell'aromaticità (o dell'antiaromaticità) dalla delocalizzazione elettronica-π. Nello spettro 1H-NMR del [18]-annulene (4N+2) i segnali degli idrogeni interni (Hi) risuonano a −2,99 δ e quelli esterni a 9,28 δ, mentre nel caso del [16]-annulene (4N) gli idrogeni interni risuonano a 10,32 δ e quelli esterni a 5,28 d [5].
[5] formula
Modelli sperimentali e numerici integrati, concernenti le proprietà magnetiche, energetiche e strutturali di classi definite di benzenoidi o di eteroaromatici, si riferiscono generalmente alla quantomeccanica. Lo studio di Robert C. Haddon, sulla relazione analitica tra l'energia di risonanza calcolata con il metodo di HMO e la corrente di anello per una serie di [N]annuleni (4N+2 con N=1÷5), rappresenta un valido criterio di aromaticità, ma limitatamente alla classe degli annuleni considerati. Il calcolo quantomeccanico e la spettroscopia NMR sono stati utilizzati per spiegare le profonde differenze strutturali ed energetiche osservate tra sistemi monociclici 4N, come l'elusivo ciclobutadiene e il cicloottatetraene, da un lato, e i sistemi benzenoidi medio- e macrociclici 4N+2, come gli [10]-, [14]-, [18]-annuleni (4N+2, con N = 2÷4 atomi di carbonio), dall'altro [6].
[6] formula
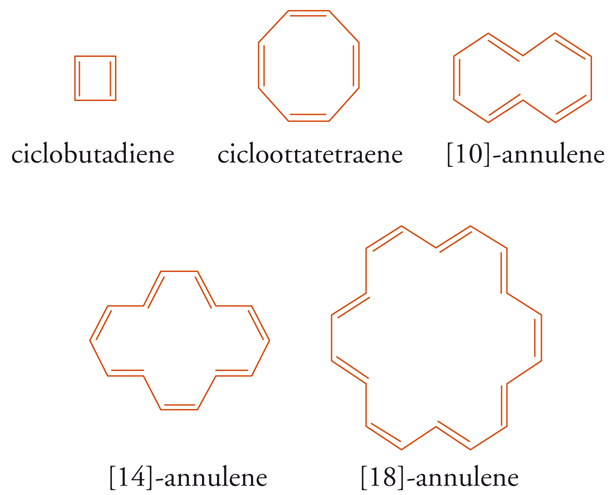
Le strutture chimiche del ciclobutadiene e del cicloottatetraene dovrebbero corrispondere a un certo grado di stabilità per coniugazione ciclica, fatta eccezione per la tensione d'anello dovuta agli angoli dei legami-σ (rispettivamente 90° e 135°), ma secondo la regola di Hückel le strutture quasi complanari, con lunghezze di legame C-C approssimativamente eguali e con solo 4N elettroni-π coniugati, sono antiaromatiche. In altre parole, i sistemi ciclici 4N, aventi strutture geometriche in tensione, sono caratterizzati da una minore stabilità rispetto ai corrispondenti composti aciclici, come dimostrano i calcoli delle rispettive energie.
La misura dell'antiaromaticità di un sistema 4N dipende dal grado di destabilizzazione prodotto nella configurazione elettronica di frontiera; talune molecole diminuiscono l'effetto destabilizzante assumendo forme asimmetriche, in cui la struttura è determinata sia dai legami-σ sia dai legami-π. La struttura complanare del ciclobutadiene assume preferibilmente una conformazione rettangolare (simmetria D2h) rispetto a quella di un quadrato regolare (simmetria D4h), caratterizzata da legami alternati i cui valori calcolati sono: singolo (1,565 Å) e doppio (1,344 Å; cfr. ciclobutano 1,543 Å). Le due forme isomere rettangolari sono state confermate mediante spettroscopia 1H-NMR del ciclobutadiene e degli addotti Diels-Alder dell'1,2-dideuteriociclobutadiene, relativamente più stabili. La struttura rettangolare è stata confermata con l'analisi ai raggi X a bassa temperatura [7].
[7] formula
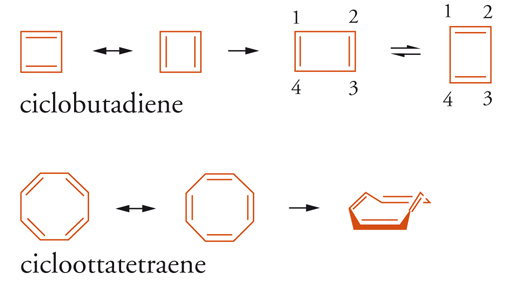
Dal punto di vista elettronico, il ciclobutadiene è quindi una molecola non delocalizzata ed è meglio rappresentato da due strutture rettangolari, una con asse orizzontale e l'altra verticale, che isomerizzano con barriera di attivazione di 1,6÷10 kcal mol-1 [7]. Le configurazioni elettroniche di queste strutture sono interpretate, a livello di calcolo MO, come una distorsione del tipo Jahn-Teller, che presenta una mescolanza tra stati elettronici degeneri: uno degli orbitali singolarmente occupati a energia più alta della forma simmetrica complanare (D4h) diminuisce in energia fino a uno stato di singoletto (1 elettrone spaiato migra nell'altro orbitale che diviene lo stato fondamentale), con formazione di due strutture equivalenti ma con geometrie meno simmetriche (D2h). La tensione angolare e la perdita della coniugazione ciclica sono le cause principali dell'instabilità dei sistemi antiaromatici e si può ipotizzare che eventuali conformazioni simmetriche siano caratterizzate da assenza di carattere aromatico e antiaromatico, perché entrambe queste condizioni richiederebbero la massima sovrapposizione degli orbitali-p. Analogamente, gli orbitali-p del cicloottatetraene non raggiungono la massima sovrapposizione; la molecola tende ad assumere una conformazione non delocalizzata, con i 4 doppi legami isolati, e chimicamente si comporta come un alchene. Gli esempi riportati confermano la marcata interdipendenza del criterio di aromaticità e della geometria molecolare.
Taluni benzenoidi policiclici possiedono uno o più doppi legami carbonio-carbonio (C=C), i cui idrogeni sono meno deschermati (1H-NMR) rispetto a quelli del benzene e in genere favoriscono l'addizione di elettrofili piuttosto che la reazione di sostituzione; ciò dipende dal contributo che ciascuna forma di risonanza apporta alla struttura reale.
[8] formula
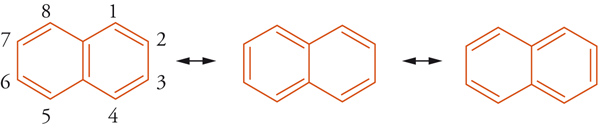
Se si assume che il contributo dato dalle 3 forme di risonanza, illustrate in [8], alla struttura reale del naftalene sia eguale, il legame 1,2 sarà di ordine maggiore rispetto al legame 2,3. Gli ordini di legame ottenuti con calcoli MO sono rispettivamente 1,724 e 1,603 (cfr. benzene 1,663), e, in accordo con questa previsione, le distan-ze di legame sono 1,360 e 1,415 Å. Generalmente, in un sistema aromatico esiste una correlazione lineare tra i valori dell'ordine di legame e le costanti di accoppiamento spin-spin (J) NMR fra gli idrogeni degli atomi di carbonio che formano quei legami [9].
[9] formula
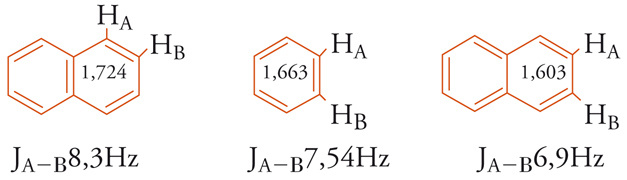
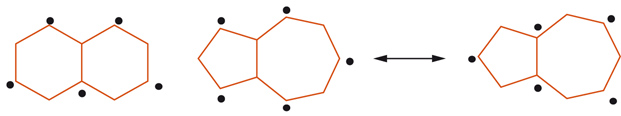
La non equivalenza dei legami (parziale localizzazione del legame) è comune ai sistemi aromatici condensati e le energie di risonanza aromatica aumentano con l'aumentare del numero delle forme di risonanza possibili. Sistemi come il benzene (2 forme), il naftalene (3 forme), l'antracene (4 forme) e il fenantrene (5 forme) hanno energia empirica di risonanza aromatica (RE)=36, 61, 84 e 92 kcal mol-1, calcolata dai rispettivi calori di combustione. La parziale localizzazione elettronica del fenantrene o dell'antracene (3 anelli benzenici condensati disposti angolarmente o linearmente) facilita le reazioni d'addizione elettrofila e le cicloaddizioni, con ossidazione dei legami-9,10 parzialmente localizzati, rilasciando 2 anelli benzenici con energia di risonanza di 36 kcal mol-1 ciascuno. L'alternanza tra doppio e singolo legame nei benzenoidi condensati è importante per la forma grafica, la topologia combinatoria e le proprietà chimiche. È conveniente suddividere questi composti in due categorie: aromatici alternanti e aromatici non-alternanti; come mostra l'esempio [10] in cui i gruppi degli atomi (evidenziati e non con un pallino) dell'alternante naftalina non sono direttamente legati tra loro, mentre il non alternante azulene possiede 2 atomi appartenenti allo stesso gruppo legati tra loro. La distinzione è meramente elettronica e geometrica e permette di stabilire: (a) la distribuzione della carica elettrica in funzione del numero di atomi della struttura (frazione di carica totale); (b) il numero di orbitali di legame occupati; (c) se gli elettroni-π sono uniformemente distribuiti sugli atomi insaturi (frazione di carica parziale).
[10] formula
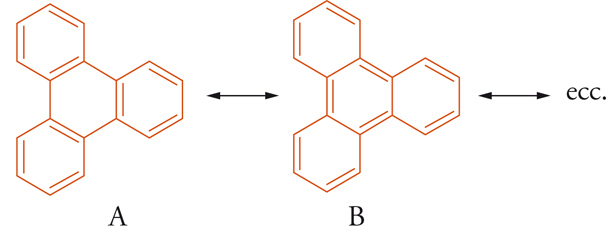
È possibile spiegare la maggiore ‒ e spesso anomala ‒ reattività degli idrocarburi benzenoidi condensati, rispetto ai monociclici, attribuendo a uno dei cicli la configurazione elettronica del benzene e all'altro (o agli altri) un parziale carattere d'idrocarburo insaturo. Un tipico esempio di distribuzione elettronica localizzata preferenziale è il trifenilene, in cui i 18 elettroni-π esterni generano 8 forme di risonanza principali di tipo (A) (in cui nessuno dei 3 legami che collegano gli anelli benzenici è un doppio legame), e solo una forma del tipo (B) con i 3 legami centrali aventi carattere di doppio legame. La configurazione elettronica risultante dal relativo contributo di queste forme impartisce al trifenilene elevata stabilità e reattività, comparabili a quelle del benzene [11].
[11] formula
A partire dagli anni Settanta del Novecento, il continuo incremento delle potenzialità computazionali e la corrispondente diminuzione del costo di calcolo hanno favorito lo sviluppo di nuovi (o la rielaborazione di noti) modelli molecolari basati sul calcolo quantomeccanico: (a) modello Hess-Scaad, modelli Gutman-Aihara o dell'energia di risonanza topologica TRE (Topological resonance energy) e modelli Herndon; essi predicono ottimamente la stabilità aromatica di sistemi semplici, ma non sono adatti per pronosticare la reattività; (b) modello del circuito coniugato integrato: l'energia di risonanza di una molecola policiclica coniugata è calcolata dal rapporto della sommatoria di tutti i circuiti 4N+2 e 4N coniugati con il numero delle forme di Kekulé possibili. I valori numerici degli anelli Rn (n=1÷3) sono calcolati per il benzene, il naftalene e l'antracene con una parametrizzazione più accurata rispetto ai valori DRE (Dewar resonance energy). Il modello è molto buono per la predizione della stabilità aromatica di grandi sistemi policiclici, ma non per la reattività; (c) modello della durezza: il metodo migliore per prevedere la stabilità cinetica molecolare. La durezza assoluta η è generalmente equiparata a 1/2 della differenza delle energie HOMO-LUMO calcolate con i metodi Hartree-Fock (HF-SCF), Hückel (HMO) o con la formula (I−A)/2, dove I è il potenziale di ionizzazione verticale e A è l'affinità elettronica.
L'approccio al calcolo numerico sempre più accurato e il crescente numero di sistemi aromatici conosciuti hanno introdotto una serie di criteri di aromaticità basati sull'ECM. I lavori più significativi consistono in misure sperimentali integrate con modelli teorici: (a) l'energia di risonanza e le energie di stabilizzazione aromatica, entrambe determinate mediante la valutazione di sistemi di riferimento (polieni e olefine) e la rielaborazione di equazioni isodesmiche ed equazioni omodesmotiche utilizzando il metodo PMO; (b) modello dell'oscillatore armonico dell'aromaticità HOMA (Harmonic oscillator model of aromaticity), basato sulle variazioni del legame (EN) e del fattore geometrico (GEO) in funzione del numero e della disposizione spaziale dei circuiti elettronici integrati; (c) modello basato sulla polarizzabilità dei legami, ottenuto mediante analisi CICL: un metodo ab initio MO basato sulla combinazione tra l'interazione di configurazione CI (Configuration interaction), l'orbitale molecolare localizzato LMO (Localization molecular orbital) e lo spazio di configurazioni in cui costruire la funzione d'onda CI (Complete active space-SCF); (d) incremento della suscettività magnetica (Λ) e lo spostamento chimico nucleo-indipendente NICS (Nucleus-indipendent chemical shift), ora basati su un tensore perpendicolare all'anello (parallelo al campo magnetico applicato) più adatto a fornire misure magnetiche relative alla corrente di anello indotta e la delocalizzazione degli elettroni-π ciclici; (e) l'indice di aromaticità (Ia), valutazione statistica dell'ordine di legame nell'anello.
In chimica organica l'aromaticità ha assunto sempre di più una fondamentale importanza nella teoria e nella pratica, per l'insegnamento e la ricerca. Essa è importante anche nella chimica degli eterocicli, che costituiscono la maggior parte dei composti organici conosciuti, sia perché formano le basi della vita sia per la loro massiccia presenza in medicina e nello sviluppo tecnologico. Tutte le importanti monografie sulla chimica degli eterocicli fanno riferimento all'aromaticità non solo dal punto di vista qualitativo, ma riportano i progressi effettuati allo scopo di ottenere un criterio quantitativo di valutazione di questa proprietà. La discussione sul criterio di aromaticità come misura universale, basata sulla relazione lineare tra le proprietà energetiche, geometriche e magnetiche, ed eventualmente estesa ai sistemi antiaromatici, è molto sviluppata, ma l'aromaticità è stata ipotizzata anche come un fenomeno multidimensionale. Studi integrati dell'energia aromatica di stabilizzazione ASE (Aromatic stabilization energy), dell'incremento della suscettività magnetica (Λ) e della componente anisotropa χanis del benzene, del ciclopentadiene anione e di una serie di composti eteroaromatici, hanno dimostrato che vi è buona correlazione lineare dei valori energetici e geometrici tra loro, ma non vi è linearità quando sono correlati alle misure magnetiche.
La maggior parte delle teorie elaborate, originariamente valide quando applicate a benzenoidi o eteroaromatici semplici, sono meno efficaci nel definire il criterio di aromaticità di sistemi più complessi, come gli eteroaromatici condensati (benzofurano, benzotiofene, dibenzotiofene) o benzenoidi policlici complanari e non (pirene, crisene, trifenilene, fullerene, corannulene). Molte ricerche sono orientate a determinare le eccezioni che originano l'incertezza ed eventualmente a formulare o rivedere ipotesi in cui queste siano interpretate e considerate in modo più approfondito. L'accesso al calcolo quantomeccanico, ora disponibile mediante programmi commerciali (Wavefunction Spartan, Gamess, MOPAC e Gaussian) funzionanti su vari tipi di elaboratori da tavolo, permette di ottenere una serie di dati ‒ strutturali (geometria, angolo e distanza di legame), termochimici (calore di formazione) ed energetici (energia di ionizzazione, energia degli stati di equilibrio e di transizione), unitamente ad alcune proprietà fisiche (funzione d'onda, momento di dipolo, frequenza vibrazionale, spettri IR, UV-vis, 1H- e 13C-NMR e strutture conformazionali) ‒ che possono essere utilmente integrati (o comparati) con i dati ottenuti mediante tecniche spettroscopiche avanzate: NMR ad alto campo, dello stato solido, bidimensionale o altre.
Bibliografia
Aihara 1976: Aihara, Jun-ichi, A new definition of Dewar-type resonance energies, "Journal of the American Chemical Society", 98, 1976, pp. 2750-2758.
Bernardi 1988: Bernardi, Fernando - Bottoni, Andrea - Venturini, Alessandro, An ab initio SCF-MO study of the aromaticity of some compounds, "Journal of molecular struc-tures. Theochem", 163, 1988, pp. 173-189.
Bird 1997: Bird, Clive W., Absolute hardness as a convenient criterion of heteroaromaticity, "Tetrahedron", 41, 1997, pp. 3319-3324.
Breslow 1968: Breslow, Roland, Small antiaromatic rings, "Angewandte Chemie. International edition in english", 7, 1968, pp. 565.
Breslow 1973: Breslow, Roland, The antiaromaticity, ‟Accounts of chemical research", 6, 1973, pp. 393-398.
Bühl, Hirsch 2001: Bühl, Michael - Hirsch, Andreas, Spherical aromaticity of phullerenes, ‟Chemical reviews", 101, 2001, pp. 1153-1183.
Cohen, Benson 1993: Cohen, N. - Benson, S.W., Estimation of heats of formation of organic compounds by additivity methods, ‟Chemical reviews", 93, 1993, pp. 2419-2438.
Cook 1974: Cook, Michael J. - Katritzky, Alan R. - Linda, Paolo, Aromaticity of heterocycles, "Advances in heterocyclic chemistry", 17, 1974, pp. 255-356.
Dewar 1969: Dewar, Michael J.S., The molecular orbital theory of organic chemistry, New York, McGraw-Hill, 1969.
Dewar, De Llano 1969: Dewar, Michael J.S. - De Llano, Carlos, Ground states of conjugated molecules XI. Improved treatment of hydrocarbons, "Journal of the American Chemical Society", 91, 1969, pp. 789-795.
Gutman 1977: Gutman, Ivan - Milun, Milorad - Trinajstic, Nenad, Graph theory and molecular orbitals XI. Nonparametric resonance energies of arbitrary conjugated systems, "Journal of the American Chemical Society", 99, 1977, pp. 1692-1704.
Haddon 1979: Haddon, Robert C., Unified theory of resonance energies, ring currents, and aromatic character in the (4n +2) pi-electron annulenes, "Journal of the American Chemical Society", 101, 1979, pp. 1722-1728.
Herndon 1973: Herndon, William C., Enumeration of res-onance structures, "Tetrahedron", 29, 1973, pp. 3-12.
Hess jr, Schaad 1971: Hess jr, B. Annes - Schaad, Larry J., Hückel molecular orbital pi resonance energies. New approach, "Journal of the American Chemical Society", 93, 1971, pp. 305-310.
Katritzky 1996: Comprehensive heterocyclic chemistry II, edited by Alan R. Katritzky, Charles W. Rees, Eric F.V. Scriven, Oxford, Pergamon, 1996.
Katritzky 1998: Katritzky, Alan R. e altri, Aromaticity as a quantitative concept. 7. Aromaticity reaffirmed as a multidimensional characteristic, "Journal of physical organic chemistry", 63, 1998, pp. 5228-5231.
Krygowski, Cyranski 2004: Krygowski, Tadeusz - Cyranski, Michael M., Two faces of the structural aspects of aromaticity, ‟Physical chemistry chemical physics", 6, 2004, pp. 249-255.
Lazzeretti 2004: Lazzeretti, Paolo, Assessment of aromaticy via molecular response properties,‟Physical chemistry chemical physics", 6, 2004, pp. 217-223.
Randic 1976: Randic, Milan, Conjugated circuits and resonance energies of benzenoid hydrocarbons, "Chemical physics letters", 38, 1976, pp. 68-70.
Randic 2003: Randic, Milan, Aromaticity of polycyclic conjugated hydrocabons, "Chemical reviews", 103, 2003, pp. 3449-3605.
Sakai 2002: Sakai, Shogo, New criterion on the aromaticity of six-membered rings, "Journal of physical chemistry", 106, 2002, pp. 10370-10377.
Sondheimer 1972: Sondheimer, Franz, The annulenes, ‟Accounts of chemical research", 5, 1972, pp. 81-91.
Von Ragué Schleyer 1996: von Ragué Schleyer, Paul e altri, Nucleus independent chemical shifts: a simple and efficient aromaticity probe, "Journal of the American Chemical Society", 118, 1996, pp. 6317-6318.
Von Ragué Schleyer 2001: Aromaticity, edited by Paul von Ragué Schleyer, "Chemical reviews", 101, 2001.
Von Ragué Schleyer 2005: Delocalization Pi and Sigma, edited by Paul von Ragué Schleyer, "Chemical reviews", 105, 2005.
Zhou, Navangult 1990 : Zhou, Zhongxiang - Navangult, Himanshoo V., Absolute hardness and aromaticity: MNDO stady of benzenoid hydrocarbons, "Journal of physical organic chemistry", 3, 1990, pp. 784-788.
Zhou, Parr 1989: Zhou, Zhongxiang - Parr, Robert G., New measures of aromaticity: absolute hardness and relative hardness, "Journal of the American Chemical Society", 111, 1989, pp. 7371-7379.