
Biotecnologie
BIOTECNOLOGIE
Biotecnologie di Alberto Albertini
sommario: 1. Introduzione generale. 2. Processi biotecnologici fondamentali: a) colture microbiche; b) attività enzimatiche; c) colture cellulari. 3. Processi biotecnologici avanzati: a) introduzione; b) immunizzazione; c) tecniche per la preselezione dei linfociti sensibilizzati; d) immortalizzazione; e) metodi di selezione cellulare; f) anticorpi modificati; g) produzione su larga scala di AcM. 4. Ingegneria genetica: a) tecniche di modificazione e trasferimento del DNA; b) vettori; c) espressione degli inserti; d) screening delle colonie batteriche; e) sequenziazione del DNA; f) polimorfismi del DNA. 5. Ingegneria proteica. 6. Applicazioni pratiche: a) impiego di anticorpi monoclonali in diagnostica e terapia; b) impiego di sonde genetiche per la rivelazione di agenti infettivi; c) settore agroalimentare; d) settore energetico ecologico; e) settore chimico farmaceutico. □ Bibliografia.
1. Introduzione generale
Nonostante che la pubblicazione della prima rivista specializzata in biotecnologie, il ‟Bullettin of the Bureau of Biotechnology", risalga al 1920, le biotecnologie si sono affermate solo qualche anno fa, quando cominciarono a essere commercializzati alcuni prodotti nati da sensazionali risultati ottenuti dall'ingegneria genetica.
Una definizione della biotecnologia, o meglio delle attività a essa inerenti, può essere quella formulata nel 1981 nell'ambito della Federazione Europea di Biotecnologia, secondo cui ‟la biotecnologia comprende l'uso integrato di microbiologia, biochimica e ingegneria al fine di realizzare l'applicazione industriale delle proprietà e delle capacità potenziali di microrganismi, cellule di tessuto coltivate o loro parti".
In pratica, ed è forse questa la cosa da notare, sono degli organismi viventi, o loro componenti opportunamente modificati o condizionati, a fungere da ‛macchine' o da sistemi di produzione su larga scala di sostanze di interesse commerciale e industriale.
Non è difficile quindi rendersi conto che, proprio perché mediate da esseri viventi, le attività biotecnologiche hanno accompagnato l'uomo fin dall'antichità e non deve sorprendere che i progressi nello sviluppo e nella ottimizzazione di sistemi biotecnologici vadano di pari passo con la conoscenza che l'uomo ha progressivamente accumulato in relazione all'ambiente che lo circonda.
Le prime attività biotecnologiche erano empiriche, anzi per molto tempo un alone magico o religioso ha circondato alcune attività tipicamente biotecnologiche, come la vinificazione dell'uva: il fenomeno era infatti ben conosciuto e controllato, fin dai tempi biblici, senza però che nessuno lo comprendesse.
In effetti si può senz'altro affermare che la trasformazione del cibo sia stata la prima attività biotecnologica intrapresa dall'uomo. Il trasformare un alimento di per sé deteriorabile, come il latte, in qualcosa di facilmente immagazzinabile, trasportabile e non deperibile, come il formaggio, doveva avere un tempo un'importanza strategica.
Probabilmente l'evoluzione dell'uomo agricoltore e pastore ha accelerato l'acquisizione di queste tecniche, diffondendole poi con un gran numero di varianti.
Molti alimenti e bevande abitualmente consumati sono il frutto di attività biotecnologiche che sfruttano spesso le risorse locali: ne sono un esempio il sakè giapponese e la birra bavarese. Questo aspetto è rilevante, in quanto le condizioni ambientali e l'ecologia della zona di residenza hanno orientato le produzioni agricole, l'allevamento e quindi la trasformazione dei prodotti, condizionando le culture dell'area abitata.
Tuttavia, nonostante che queste attività di trasformazione fossero molto fiorenti fin dall'antichità, è stato necessario attendere la seconda metà dell'Ottocento perché Pasteur le ponesse finalmente su rigorose basi razionali e scientifiche.
Tentativi per spiegare alcuni fenomeni, come per esempio la trasformazione del vino in aceto o la lievitazione del pane, erano stati affrontati da una nutrita schiera di studiosi di alchimia: a questi va senz'altro il merito di aver posto le basi - seppure in maniera disordinata - della chimica moderna, ma la loro comprensione dei fenomeni biologici non era andata al di là dell'ipotesi dell'esistenza di fantomatici ‛fermenti' a cui attribuire queste strane proprietà trasformanti.
Molto tempo dopo, nel 1871, Pasteur identificava e isolava l'agente responsabile della trasformazione del mosto in vino (fermentazione alcolica): un organismo vivente unicellulare chiamato ‛lievito'. Allo stesso modo sono stati identificati numerosi altri organismi, generalmente microscopici, capaci di trasformare il vino in aceto, il latte in yoghurt, ecc.
Più tardi sono state scoperte anche alcune sostanze, contenute in questi microrganismi, che da sole avevano le stesse proprietà trasformanti del microrganismo in toto. Nel 1878 W. Kühne chiama tali sostanze ‟enzimi". Nel 1897 i fratelli Buchner scoprono che gli enzimi riescono ad accelerare (catalizzare), a temperature e pressioni fisiologiche, molte reazioni che altrimenti abbisognerebbero di centinaia di gradi di temperatura e decine di atmosfere di pressione.
La seconda metà dell'Ottocento è comunque ricca di scoperte e intuizioni basilari per la moderna biologia: Mendel formula le leggi fondamentali della genetica classica e successivamente Miescher scopre l'esistenza di acidi nucleici all'interno delle cellule.
Il Novecento inizia con la produzione industriale di lievito; concettualmente il procedimento è semplice, perché si tratta di permettere a una colonia di cellule di lievito di crescere e suddividersi moltiplicandosi attivamente: è sufficiente fornire loro il nutrimento (zucchero) e una ventilazione adeguata. Questo procedimento è noto come fermentazione aerobica, in quanto l'ossigeno è necessario per le varie reazioni chimiche che gli enzimi del lievito catalizzano. Esistono anche dei microrganismi, detti anaerobi, che non hanno bisogno d'aria; anch'essi possono essere coltivati e possono operare trasformazioni mediante fenomeni di fermentazione analoghi a quelli del lievito.
Una delle prime applicazioni industriali di questo processo è rappresentata dalla produzione di acetone e butanolo, il primo utilizzato per la preparazione di esplosivi (siamo all'inizio del Novecento), il secondo per quella di vernici. L'uso della fermentazione e il miglioramento tecnico di questo processo subiranno però un'accelerazione notevole con la scoperta della penicillina (1928) e con il conseguente problema, negli anni quaranta, della crescita in coltura dei microrganismi che la sintetizzano: Penicillium notatum e, in seguito, Penicillium chrysogenum. In questo modo le biotecnologie classiche diventano parte integrante dell'industria farmaceutica.
Sempre nella prima metà del Novecento vengono formulate da L. Michaelis e M. L. Menten le leggi che regolano la catalisi enzimatica e viene purificato e cristallizzato il primo enzima (ureasi): è il primo passo per lo studio di queste straordinarie molecole proteiche dalle innumerevoli funzioni. Mediante gli enzimi, negli anni trenta, si scopre che con la fermentazione è possibile realizzare, su molecole complesse, modificazioni strutturali che non era possibile effettuare per mezzo della chimica classica di sintesi. In tal modo si riescono a produrre cortisone e suoi analoghi, amminoacidi, antibiotici modificati, il tutto a costi decisamente inferiori.
Parallelamente si assiste a uno sforzo notevole per il miglioramento degli impianti in cui avvengono i processi di fermentazione, oltre che degli strumenti di controllo per la misura di importanti parametri quali la temperatura, il pH, il volume, l'aria immessa. Si affinano anche i procedimenti per estrarre e purificare i prodotti che escono da tali fermentatori. Ormai la fermentazione sommersa, in cui le cellule sono mantenute totalmente immerse nel liquido di coltura in grandi recipienti metallici, è un procedimento affidabile che permette di coltivare enormi quantità di cellule o microrganismi e ottenere ingenti quantità di prodotto.
Tra gli anni cinquanta e sessanta J. D. Watson e F. H. C. Crick hanno una delle intuizioni più geniali della storia della biologia: concepiscono un modello di DNA a doppia elica che permette di ipotizzare il meccanismo della duplicazione del materiale genetico, ponendo così le basi molecolari dell'ereditarietà. Già nel 1941 si era scoperto che i cromosomi sono costituiti da DNA e che svolgono alcune attività di controllo sulla produzione degli enzimi. L'intuizione di Watson e Crick avviò nella giusta direzione una folta schiera di ricercatori occupati nella verifica sperimentale di tale teoria. Alla fine degli anni settanta era ormai perfettamente chiaro che l'informazione genetica è contenuta nel DNA codificata in sequenze di nucleotidi, che, attraverso la sintesi di vari tipi di acido ribonucleico (RNA), sono tradotte in proteine.
In questi anni nasce l'ingegneria genetica in seguito alla identificazione di particolari enzimi chiamati endonucleasi di restrizione, capaci di tagliare il DNA in punti prestabiliti, e di enzimi che, al contrario, possono ricucirlo (ligasi). Utilizzando questi e altri mezzi è possibile modificare in vitro tratti di DNA (geni) di un organismo e/o trasferirli in un altro. Ad esempio il tratto di DNA umano che codifica per la sintesi di insulina è stato inserito nel DNA di un batterio (Escherichia coli) rendendo quest'ultimo capace di sintetizzare insulina umana in grandi quantità. E ovvio che le applicazioni di una tale tecnologia possono rivelarsi rivoluzionarie.
In questo modo sono nate le biotecnologie avanzate, in cui la possibilità di intervento sul corredo genetico degli esseri viventi aumenta il potenziale di progettualità e di controllo delle attività biotecnologiche.
Nel 1975 G. Köhler e C. Milstein ottengono anticorpi coltivando in provetta le cellule del sistema immunitario che li producono (linfociti), opportunamente fuse con cellule tumorali di mieloma. Si ottiene così un ibrido che produce in gran quantità anticorpi con caratteristiche chimiche e funzionali ben definite, che potranno essere utilizzati in vari campi, non ultimo quello della diagnosi e della terapia delle neoplasie.
Attualmente un grosso sforzo per immagazzinare, catalogare ed elaborare l'enorme mole di risultati prodotti ogni giorno nei vari campi afferenti alle biotecnologie rappresenta una necessità irrinunciabile. Nasce così la bioinformatica, una branca delle biotecnologie avanzate che si occupa della creazione di banche dati per gestire informazioni che vanno dalle sequenze di DNA identificate ai brevetti internazionali, alla progettazione molecolare assistita dall'elaboratore.
Da tutto ciò appare chiara la dimensione tipicamente umana delle biotecnologie, in cui ciò che veramente conta è l'uomo con le sue idee e la sua creatività: molti hanno perciò paragonato lo sviluppo delle biotecnologie avanzate a quello dell'elettronica dopo la costruzione del microprocessore. In effetti le attività biotecnologiche richiedono un elevato grado di conoscenze, tali da produrre beni a elevato valore aggiunto.
Viste in questa ottica le biotecnologie sono divenute un enorme polo di interesse per investimenti di capitali pubblici e, cosa decisamente nuova per la ricerca biologica, privati.
Verso la fine degli anni settanta negli Stati Uniti sono nate una serie di piccole compagnie di biotecnologie fondate da prestigiosi ricercatori universitari e finanziate con capitali ad alto rischio (venture capitals) messi a disposizione da società finanziarie a cui spesso partecipano importanti gruppi industriali farmaceutici. Ultimamente anche queste grandi industrie, spesso acquistando piccole compagnie di ricerca biotecnologica, sono entrate in campo con tutta la loro potenza produttiva e commerciale. Buona parte dell'attività produttiva di queste aziende è rivolta al settore sanitario, dove attualmente si assiste a una notevole ricaduta in termini di prodotti commercializzati (v. tab. I).

Ciò nonostante nel prossimo futuro saranno i settori dell'agricoltura e dell'allevamento a subire il maggiore impatto delle biotecnologie innovative. La manipolazione genetica di piante può portare a specie resistenti a condizioni climatiche proibitive e a parassiti, e a migliorarne l'efficienza fotosintetica e il valore nutritivo. Lo stesso discorso si può applicare alla zootecnia, con vantaggi in termini di resistenza alle malattie e di resa nella conversione del foraggio in carne o latte.
Nel campo della produzione di sostanze chimiche si possono utilizzare reazioni biologiche per trasformare materie prime rinnovabili (come i rifiuti agricoli o urbani) in prodotti chimici intermedi o combustibili, attualmente prodotti dalla lavorazione del petrolio (d'altra parte il petrolio e le materie di origine vegetale sono due facce della stessa medaglia ma la tecnologia attuale ci ha consentito finora di utilizzarne solo una). Tutto ciò si rifletterebbe positivamente sulla diminuzione del carico inquinante proveniente dalle attività umane che si riversa sull'ambiente.
Non dimentichiamo peraltro che le attività biologiche non hanno bisogno di un'alta energia di attivazione (merito degli enzimi), per cui è necessario fornire poca energia per far funzionare un fermentatore; ciò contribuisce senza dubbio in modo favorevole alla limitazione del consumo di fonti di energia non rinnovabili.
Da queste considerazioni emerge il quadro di una società sempre più tesa all'utilizzo di tecnologie che sfruttano i meccanismi più profondi della vita. Questo significa un impatto più dolce e meno distruttivo delle attività umane sulla natura circostante: esigenza peraltro ormai non più prorogabile. Le biotecnologie innovative contribuiranno grandemente alla realizzazione di queste condizioni, e gran parte di questo articolo è dedicato proprio a esse. Tuttavia non sono state tralasciate quelle tecniche utilizzate da tempo in campo farmaceutico e alimentare che così grandemente hanno contribuito allo sfruttamento industriale di alcune delle principali proprietà degli organismi viventi. Da ultimo verranno considerati i campi di applicazione delle biotecnologie innovative, alcuni dei quali, come quelli agroalimentare ed ecologico, saranno i settori in cui, nel futuro a medio e lungo termine, si verificheranno le realizzazioni più interessanti e più utili per gran parte dell'umanità.
2. Processi biotecnologici fondamentali
a) Colture microbiche
I microrganismi. - I microrganismi di interesse biotecnologico appartengono a due gruppi tassonomici: Batteri e Miceti (v. microbiologia). I Batteri sono classificati come cellule procariote, prive cioè di membrana nucleare e fuso mitotico, di dimensioni comprese tra 0,5 e 2 μm; la loro struttura superficiale è caratterizzata dalla presenza di una parete cellulare rigida esterna alla membrana citoplasmatica, che ha lo scopo di proteggere la cellula dai traumi di tipo meccanico e osmotico. Dalla parete dipende la forma dei Batteri, che possono essere più o meno sferici (cocchi) oppure cilindrici (bacilli); alcuni batteri possono crescere in lunghi filamenti ramificati (Actinomiceti).
I Miceti sono cellule eucariote con più filamenti di DNA delimitati da una membrana nucleare. I miceti più semplici sono i lieviti, unicellulari, di dimensioni di circa 5 μm; le muffe sono invece miceti che danno origine a colonie filamentose multicellulari con morfologia complessa.
L'utilizzazione di batteri o miceti dipende dallo specifico campo d'azione biotecnologico: gli Actinomiceti sono tipici produttori di antibiotici; i lieviti sono utilizzati per produrre biomasse o metaboliti primari; batteri come Escherichia coli sono utilizzati come mezzo di espressione di geni di organismi superiori.
Colture microbiche. - A tutt'oggi è possibile separare e isolare in coltura pura tutti i microrganismi in grado di crescere in terreno di coltura; essi crescono formando delle colonie ognuna delle quali contiene organismi tutti uguali fra loro.
Sono stati istituiti dei centri specializzati nella raccolta, conservazione e distribuzione di colture microbiche; i processi biotecnologici, infatti, necessitano per il loro sviluppo di specie microbiche selezionate altamente specifiche.
Metodi di selezione. - I metodi di isolamento selettivo sfruttano le caratteristiche di un singolo ceppo; ad esempio i Batteri crescono a temperature più elevate (30-37 °C) rispetto ai Miceti (23-25 °C); valori bassi di pH permettono la crescita dei Miceti e non dei Batteri; l'aggiunta di un determinato antibiotico al terreno di coltura seleziona i microrganismi resistenti all'azione dell'antibiotico.
L'utilizzo di determinati substrati favorisce la crescita solo di quei microrganismi in grado di utilizzare quel substrato specifico. Se, per esempio, si vogliono isolare microrganismi capaci di utilizzare solo l'amido, quest'ultimo è introdotto nel terreno di coltura come unica fonte di carbonio. In queste condizioni possono crescere solo i microrganismi capaci di procurarsi energia degradando la molecola dell'amido, che possiedono cioè enzimi che lo attaccano, le amilasi. I microrganismi isolati in questo modo possono essere utilizzati per la produzione di amilasi impiegabili nell'industria alimentare.
Metabolismo microbico. - Il processo di demolizione degli zuccheri fino ad acqua, anidride carbonica ed energia implica una complessa serie di reazioni che portano alla produzione di metaboliti intermedi o finali di notevole utilità: acido piruvico, acido lattico, alcool etilico, ecc. (v. tab. II). Alcuni metaboliti, oltre a essere necessari per lo svolgersi del ciclo biologico del microrganismo che li produce, rivestono importanza biotecnologica; ad esempio un amminoacido come l'acido glutammico non solo serve al microrganismo per sintetizzare le proteine, ma trova impiego anche nell'industria alimentare per la produzione di surrogati di brodo di carne.
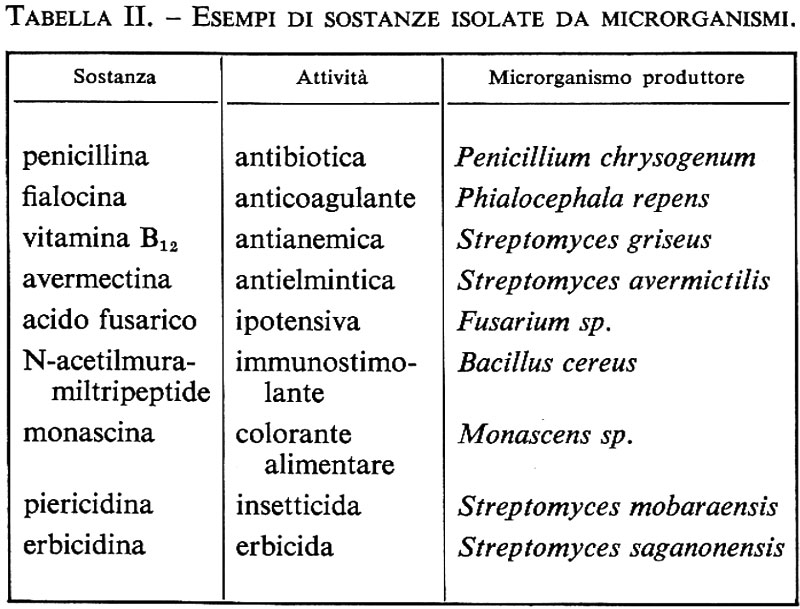
Altri metaboliti, come gli antibiotici, sono di non chiara utilità per il microrganismo che li produce, ma rivestono un importante ruolo terapeutico sia in campo umano (avendo permesso di fronteggiare quasi tutte le malattie infettive di origine batterica), sia in campo veterinario (favorendo un aumento della carne prodotta), sia in campo vegetale (contro i parassiti delle piante).
Colture di microrganismi per la produzione di metaboliti primari. - Sono definiti metaboliti primari quelle molecole, presenti nei microrganismi, essenziali per il metabolismo di base oppure necessarie per la sintesi di macromolecole essenziali o coenzimi.
Un microrganismo in condizioni normali di crescita tende a evitare una produzione di metaboliti che superi le proprie esigenze vitali, perché ciò rappresenterebbe un inutile consumo di energia. Esistono quindi dei meccanismi di regolazione che consentono di evitare consumi inopportuni. Tuttavia questi meccanismi possono essere modificati in laboratorio. I microrganismi hanno caratteristiche metaboliche che rendono facilmente aggredibili i loro meccanismi di controllo: crescono e si riproducono infatti in tempi assai brevi (dell'ordine di decine di minuti) e quindi replicano il loro DNA con frequenza maggiore rispetto agli organismi più complessi. Per quest'ultimo motivo è più probabile che il DNA vada incontro a errori di replicazione dando origine a cellule ‛mutate'; essendo spesso unicellulari è possibile isolare anche una singola cellula mutata. Le mutazioni avvengono frequentemente in natura, ma possono anche facilmente essere indotte sperimentalmente al fine di reprimere importanti enzimi regolatori.
Si può anche intervenire non influendo sul patrimonio genetico del microrganismo, semplicemente con l'uso di terreni di crescita sbilanciati, con forti eccessi di zuccheri e scarsità di sali. Quale che sia il metodo utilizzato, si induce il funzionamento preferenziale o in eccesso di date vie metaboliche, con conseguente accumulo di enormi quantità di sostanze che in condizioni normali sono prodotte in quantità più ridotte o non prodotte affatto.
Metabolismo primario aerobico. - I prodotti del metabolismo primario aerobico sono amminoacidi, vitamine, nucleotidi e alcuni acidi organici. In condizioni normali tutte queste sostanze sono prodotte in quantità controllate da meccanismi di regolazione e da barriere che impediscono la loro uscita dalla cellula. Esistono diversi livelli di complessità nella regolazione di una via metabolica. Il più semplice implica la regolazione da parte di tutti quei parametri che influenzano la velocità di una reazione enzimatica (pH, concentrazione dell'enzima, degli intermedi, degli ioni e dei coenzimi all'interno della cellula). Il secondo meccanismo si basa sull'azione di enzimi regolatori che vengono inibiti dal prodotto finale (inibizione da feed-back o retroinibizione). Se si allontana il prodotto finale dall'ambiente cellulare, l'enzima, non più inibito, può funzionare ininterrottamente portando alla produzione continua del prodotto finale. Alternativamente si può produrre una mutazione che renda l'enzima regolatore insensibile all'azione del prodotto finale. Il terzo meccanismo prevede il controllo genico della sintesi enzimatica.
Metabolismo primario anaerobico. - La complessa rete di reazioni che porta alla produzione di metaboliti in completa assenza di aria prende il nome di fermentazione. Pasteur definì la fermentazione come ‟vita in assenza di aria": aveva cioè intuito che nel processo di fermentazione l'energia metabolica è ottenuta dai microrganismi per mezzo della loro capacità di utilizzare l'ossigeno che è combinato negli zuccheri. La fermentazione è un meccanismo metabolico di tipo essenzialmente respiratorio, ma che si verifica in anaerobiosi e in cui l'accettore finale di elettroni è un composto inorganico diverso dall'ossigeno, in genere un nitrato o un solfato.
Le condizioni di anaerobiosi necessarie per la fermentazione si realizzano facilmente in ambienti liquidi. Nell'industria a tale scopo si utilizzano recipienti di notevoli dimensioni incubati staticamente, in cui l'ossigeno atmosferico non raggiunge gli strati profondi ed è in genere sostituito, anche nella fase gassosa sovrastante, dall'anidride carbonica (CO2). Nell'industria si usa spesso il termine ‛fermentazione' per indicare qualsiasi processo produttivo su larga scala che sia catalizzato da microrganismi, anche nei casi in cui, come per la produzione di antibiotici, sia necessaria una notevole aerazione della coltura. Il metabolismo anaerobico è poco favorevole al microrganismo, che deve disporre di grandi quantità di substrati organici; trova tuttavia largo impiego applicativo quando si voglia trasformare una qualsiasi sostanza organica, in genere uno zucchero, in un'altra con diversa struttura e diverso stato di ossidazione.
I coenzimi. - I coenzimi sono molecole termostabili non proteiche a basso peso molecolare. Sono state inizialmente distinte dagli enzimi perché potevano venire da essi separate mediante dialisi, con con seguente perdita di attività da parte dell'enzima stesso. Tuttavia l'attività dell'enzima poteva essere ripristinata ponendolo in presenza del sovranatante di un estratto cellulare in cui gli enzimi erano stati precedentemente inattivati. Le reazioni che avvengono durante la fermentazione e la respirazione dei microrganismi utilizzano coenzimi identici a quelli presenti nei Mammiferi. I coenzimi noti sono: il coenzima A, che agisce nel trasferimento di gruppi acilici; il tiamin-difosfato, per il trasferimento di gruppi derivati da un chetone; la biotina, per il trasferimento di anidride carbonica; l'acido lipoico, il NAD, il NADP, i derivati della riboflavina, l'ubichinone, i citocromi e altri, per il trasporto di idrogeno e di elettroni; l'ATP, per il trasferimento di fosfato e di energia.
Tipi di fermentazioni. - Molte fermentazioni, pur portando alla produzione di prodotti finali differenti, seguono la via glicolitica fino alla produzione di acido piruvico. La produzione di ATP in questa sequenza dipende dall'ossidazione del triosofosfato a spese del NAD+ che si riduce a NADH + H+. Poiché il contenuto cellulare di NAD+ è molto limitato, la fermentazione si interromperebbe rapidamente se il NADH non fosse continuamente riossidato durante il metabolismo dell'acido piruvico. I microrganismi eseguono questa reazione secondo modalità diverse.
Fermentazione lattica. È il tipo di fermentazione più semplice: una sola reazione catalizzata da una latticodeidrogenasi legata a NAD+ riduce l'acido piruvico ad acido lattico, senza formazione di gas. Questo tipo di fermentazione si realizza nei processi di acidificazione del latte e di altri alimenti. Per le modificazioni di aroma e di sapore che esse determinano, queste fermentazioni assumono un'importanza particolare dal punto di vista economico, ad esempio nel caso dell'industria casearia.
Fermentazione alcolica. L'acido piruvico attivato dal coenzima tiamin-difosfato viene trasformato in anidride carbonica (che viene trattenuta in alcune bevande) e in aldeide acetica; questa viene quindi ridotta ad alcol etilico mediante una reazione in cui interviene il NADH. Questa fermentazione è caratteristica dei lieviti. La sua importanza applicativa nella lievitazione del pane e nella produzione di bevande è nota da millenni.
Fermentazione propionica. L'acido piruvico è carbossilato ad acido ossalacetico, il quale subisce due riduzioni per diventare acido succinico e quindi, per decarbossilazione, acido propionico. I microrganismi che possono seguire questa via metabolica sono capaci di fermentare l'acido lattico, prodotto finale di un'altra fermentazione. L'acido lattico è dapprima ossidato ad acido piruvico, una parte del quale è ridotta ad acido propionico, mentre il resto è ossidato ad acido acetico e anidride carbonica. Nei formaggi tipo emmental la produzione di anidride carbonica da parte batterica, dopo che è stata completata la fermentazione del latte, provoca la formazione dei ‛buchi', mentre l'acido propionico prodotto contribuisce a determinarne l'aroma.
Fermentazione butirrica. È una via metabolica seguita da batteri anaerobi obbligati che possono attivare idrogeno molecolare tramite un coenzima: la ferrodossina. I prodotti finali sono: acetone, alcool isopropilico, acido butirrico e alcool butilico.
Altre fermentazioni. Di scarso interesse biotecnologico sono la fermentazione acido-mista e la butilen-glicolica.
Le fermentazioni lattica e butirrica sono impiegate per la produzione di polimeri dell'acido lattico e dell'acido butirrico, che presentano l'importante caratteristica di essere biodegradabili e quindi potenzialmente utilizzabili in sostituzione della comune plastica; tuttavia i costi elevati dei substrati hanno notevolmente limitato la loro produzione industriale.
b) Attività enzimatiche
Gli enzimi sono proteine specializzate nella catalisi di reazioni biologiche (v. enzimi). Il legame con il substrato si realizza in un'area particolare della superficie enzimatica, definita ‛sito attivo'. L'attività enzimatica è estremamente specifica e accelera di molto la reazione (catalisi). Gli enzimi non sono consumati al termine della reazione e possono quindi intervenire in una reazione successiva; inoltre svolgono la loro azione in condizioni assai moderate di temperatura e di pH e a pressione atmosferica.
L'elevata specificità rappresenta un chiaro vantaggio per l'utilizzazione extracellulare degli enzimi; tuttavia per garantire questa specificità è necessario poter disporre di preparazioni enzimatiche estremamente pure. Un limite al loro utilizzo a fini biotecnologici è dato dal fatto che gli enzimi vengono di solito ‛persi' al termine della reazione, in quanto il loro recupero dal mezzo acquoso in cui è avvenuta la reazione è assai costoso; per ovviare a tale inconveniente si è sviluppata la tecnica di immobilizzazione enzimatica.
Fonti di enzimi. - Gli enzimi si possono ottenere da cellule vegetali e da organi animali, ma la fonte principale è rappresentata dai microrganismi. Per la produzione di enzimi a partire da batteri o miceti si devono attuare precise condizioni di coltura e, naturalmente, è necessaria l'aggiunta dei substrati adeguati. Anche in questo caso la tecnologia del DNA ricombinante ha trovato vasta applicazione, in quanto può permettere la secrezione di enzimi altrimenti non secreti, oppure l'alterazione della struttura di un enzima per aumentarne la resistenza o modificarne leggermente l'attività.
Produzione di enzimi. - Gli enzimi esplicano la loro azione all'interno della cellula o nel mezzo extracellulare. I metodi più utilizzati per estrarli sono: omogeneizzazione, uso di ultrasuoni, congelamento e successiva rottura della cellula, shock osmotico, uso di detergenti o di altri enzimi. Durante questi trattamenti bisogna controllare tutti quei fattori che possono inattivare anche parzialmente l'enzima, quali la temperatura e l'azione concomitante di altri enzimi.
Per concentrare le preparazioni enzimatiche così ottenute si ricorre usualmente alla precipitazione con una soluzione salina altamente concentrata (ad esempio una soluzione satura di solfato di ammonio). L'aggiunta del sale in concentrazioni elevate riduce la solubilità delle proteine. Si possono così separare frazioni proteiche con diverso grado di solubilità: in una di queste si troverà l'enzima desiderato. Tale metodo presenta l'inconveniente di denaturare facilmente l'enzima, rendendolo inattivo. Attualmente si utilizzano sistemi di ultrafiltrazione attraverso membrane porose o gelatinose, congelamento o liofilizzazione.
Con i metodi descritti si ottengono delle purificazioni parziali. Nel caso si presenti l'esigenza di preparazioni più pure si ricorre a metodi più sofisticati, come la cromatografia per adsorbimento. Tale tecnica prevede l'utilizzo di colonne di carbonato di calcio, in cui viene fatta percolare la soluzione contenente la miscela proteica. La separazione avviene grazie alla diversa velocità di migrazione delle molecole attraverso la colonna: le proteine che si adsorbono prima migrano più lentamente, separandosi lungo la colonna. Lavando poi la colonna con una soluzione appropriata, le varie frazioni potranno essere recuperate separatamente le une dalle altre. In sostituzione del carbonato di calcio si possono usare gel (materiale colloidale) o resine a scambio ionico. In quest'ultimo caso la separazione avviene poiché le proteine hanno diversa affinità per la resina a causa della loro diversa carica elettrica. Le varie frazioni proteiche possono essere eluite modificando il pH all'interno della colonna.
Un altro metodo è rappresentato dalla cromatografia per affinità e sfrutta la capacità dell'enzima di legarsi al substrato specifico o a un anticorpo monoclonale (v. cap. 3) diretto contro il sito attivo dell'enzima. Il substrato (o l'anticorpo monoclonale) posto nella colonna fissa l'enzima corrispondente, che viene poi recuperato mediante trattamento a pH acido.
Immobilizzazione degli enzimi. - Il problema della rigenerazione dell'enzima al termine della reazione è stato risolto con la tecnica di immobilizzazione enzimatica. Essa consiste nel fissare o adsorbire gli enzimi a matrici insolubili in acqua, cosicché possono essere separati dal mezzo acquoso contenente il substrato e il prodotto.
Havewala e Pitcher hanno presentato per primi nel 1973 un lavoro sulla immobilizzazione della glucosioisomerasi in cui si considerava la cinetica, l'effetto della temperatura sull'attività e sulla stabilità dell'enzima e altri problemi connessi con la produzione enzimatica su larga scala.
Nella selezione di un sistema con enzima immobilizzato si deve tener conto di vari parametri, come la fonte e il grado di purificazione dell'enzima, la natura del vettore e il tipo di enzima.
La stabilità è molto importante, in quanto l'attività dell'enzima deve essere duratura per fornire vantaggi economici. Poiché il pH a cui agisce un enzima immobilizzato può essere diverso da quello a cui opera l'enzima nativo, si devono studiare attentamente le condizioni di pH che influenzano altamente la stabilità e la scelta del supporto.
Nella scelta dell'enzima deve essere attentamente analizzata la cinetica; infatti, poiché la velocità delle reazioni di importanza industriale è molto elevata, si pone il problema dell'inibizione causata dal prodotto finale. Ad esempio la β-galattosidasi prodotta da Aspergillus oryzae subisce una minore inibizione dal prodotto finale rispetto a quella prodotta da A. niger.
Nel selezionare la natura del supporto bisogna considerare le condizioni di reazione (pH), il peso molecolare e la forma dell'enzima, il tipo di reattore che si intende utilizzare e il tipo di legame con l'enzima. Deve essere attentamente analizzata la struttura porosa del substrato: i pori devono essere abbastanza larghi per permettere l'immobilizzazione dell'enzima e il contatto dell'enzima con il substrato. La composizione del supporto determina il tipo e la durata del legame con l'enzima; è importante la scelta di un supporto chimicamente inerte sia per la sopravvivenza dell'enzima, sia per la purezza del prodotto.
Attualmente si lavora alla progettazione di reattori più idonei e alla messa a punto di supporti più duraturi e meno costosi che possano legare una maggiore quantità di enzima.
c) Colture cellulari
Solo all'inizio del XX secolo si prese in considerazione la possibilità di far crescere e replicare in vitro una cellula rimossa da un animale superiore. Successivamente si scoprì che gli agenti filtrabili, cioè i virus, potevano crescere e replicarsi nelle cellule isolate da un organismo. Ciò ha permesso la produzione di grandi quantità di virus per la messa a punto dei primi vaccini (v. Belin e Houillon, 1928). È stato successivamente possibile ottenere da una singola cellula una popolazione cellulare omogenea (v. Bryant e altri, 1958); oggi, tramite l'utilizzo della tecnologia del DNA ricombinante, è possibile ottenere l'espressione di geni esogeni introdotti nella cellula.
Stabilizzazione di cellule in coltura. - La sospensione cellulare ottenuta partendo da un tessuto costituisce una cosiddetta coltura primaria e può essere utilizzata come tale, per esempio per la produzione di virus, oppure può essere usata per generare una linea cellulare, cioè una coltura di cellule che crescono, si dividono e possono dare origine a nuove colture.
La scelta del metodo di dissociazione del tessuto dipende dalle caratteristiche di quest'ultimo: può essere di volta in volta un trattamento enzimatico, chimico o meccanico. Gli agenti più comunemente usati sono gli enzimi idrolitici che distruggono la matrice extracellulare; questo metodo, pur essendo molto efficiente, può danneggiare la membrana cellulare provocando la lisi della cellula, per cui il tempo di esposizione agli enzimi deve essere minimo (v. Norrby e altri, 1966). Gli enzimi maggiormente utilizzati sono la tripsina, le pronasi, le collagenasi, la elastasi, la dispasi, la ialuronidasi. Una miscela di enzimi risulta essere molto efficace, in quanto, mentre un enzima indebolisce lo stroma, l'altro dissocia rapidamente le cellule. Poiché i cationi del calcio e del magnesio rivestono un ruolo importante nell'adesione cellulare, agenti chelanti come l'EDTA rompono i legami tra cellula e cellula. I tessuti possono inoltre essere dissociati mediante omogeneizzazione oppure tramite spremitura attraverso una rete di nailon. È possibile anche ottenere un certo numero di cellule mediante perfusione di determinati organi (ad esempio epatociti e cellule delle isole pancreatiche).
Le fonti più comuni di cellule per colture primarie sono: rene di scimmia, vitello, cane, coniglio, embrione di pollo, polmone e rene fetale umano. È preferibile l'uso di tessuti embrionali per la minor quantità di tessuto connettivo in essi presente.
Stabilizzazione di cellule emopoietiche. - Le cellule emopoietiche sono molto diverse tra loro. Leucociti ed entrociti non presentano possibilità di crescita, i macrofagi hanno scarsa capacità di crescita in vitro, i linfociti possono crescere e dividersi solo se opportunamente stimolati. I linfociti possono essere separati mediante centrifugazione in gradiente di densità e successiva adesione su superfici plastiche. I linfociti sono stimolati con agenti mitogeni, come la fitoemoagglutinina (PHA), o con fattori di crescita specifici, come la interleuchina-2 (IL-2), oppure possono essere trasformati con il virus di Epstein-Barr.
Metodi di separazione cellulare. - Le tecniche di dissociazione descritte non sono specifiche e danno origine a popolazioni cellulari miste di cellule epiteliali, endoteliali e fibroblasti; questi ultimi possono predominare sugli altri tipi cellulari presenti nella stessa coltura. Si ricorre quindi a: a) trattamenti enzimatici selettivi: ad esempio l'uso di tripsina rimuove selettivamente i fibroblasti (v. Owens, 1976); b) uso di inibitori specifici: l'acido iodoacetico è più tossico per i fibroblasti che per le cellule epiteliali (v. Gilbert e Migeon, 1975); c) passaggi alternati in vivo e in vitro che selezionano cellule tumorali; d) clonaggio, che consiste in diluizioni successive sino a ottenere una sospensione contenente un'unica cellula, che cresce formando una colonia di cellule figlie identiche a sé; e) centrifugazione in gradiente di densità, che sfrutta la diversa densità cellulare, oppure centrifugazione differenziale, che si basa sulla diversa velocità di sedimentazione (v. Sheeter e Doolittle, 1980); f) cromatografia di affinità, che permette di separare cellule con diverse caratteristiche di membrana, mediante il legame con vari immunoadsorbenti; g) metodi fisici, come l'elettroforesi, che separano le cellule a seconda della loro capacità di migrare in un campo elettrico; h) uso di un citofluorimetro a flusso (v. Melamed e altri, 1979); questo strumento permette l'analisi della sospensione cellulare mediante l'utilizzo di un raggio laser: le cellule che passano attraverso il raggio generano dei segnali dovuti alla dispersione della luce o all'emissione di fluorescenza, che sono captati da fotorecettori e opportunamente amplificati.
Subcolture cellulari. - La subcoltivazione di una coltura di cellule primarie dà origine a una linea cellulare primaria. Tale linea ha una vita limitata (generalmente non più di 20 passaggi). In queste linee, per mutazioni naturali o per trattamenti con agenti mutageni, possono emergere linee cellulari stabilizzate, capaci di crescere indefinitamente in vitro, caratterizzate da un numero di cromosomi maggiore del normale (poliploidi).
Le colture cellulari primarie sono di solito preferibili per quanto riguarda la produzione di vaccini (soprattutto utilizzati in medicina umana), perché le cellule coltivate più volte tendono a perdere la sensibilità ai virus (v. Van Wezel e altri, 1980). Le linee cellulari sono tuttavia più convenienti, in quanto non richiedono la disponibilità continua di animali, il loro controllo e il loro mantenimento; il loro utilizzo è quindi preferibile quando possibile.
Valutazione della crescita cellulare. - Il metodo maggiormente utilizzato per la misura della vitalità cellulare si basa sulla colorazione con trypan blu (v. Sandford e altri, 1951); in genere le tecniche di colorazione sfruttano la capacità delle cellule di resistere all'incorporazione del colorante e sono quindi una misura di permeabilità.
Una misura indiretta della vitalità si basa sulla valutazione dell'attività metabolica. I parametri più impiegati sono la velocità di metabolizzazione del glucosio e dell'ossigeno, la produzione di acido lattico, di acido piruvico, di ATP e di anidride carbonica, la presenza di determinati enzimi e il grado di sintesi delle proteine e degli acidi nucleici.
Per ottenere un valore quantitativo della crescita cellulare si possono utilizzare metodi chimici o fisici. Il metodo chimico più usato è la determinazione della concentrazione delle proteine totali con il metodo di Lowry modificato (v. Oyama ed Eagle, 1956). I metodi fisici sono più sensibili: ad esempio il cromo radioattivo presenta una diversa capacità di legame con le proteine a seconda che sia in forma ridotta - in questo caso si lega alle proteine non vitali - oppure in forma ossidata - in questo caso si lega alle proteine vitali (v. Scaife e Brohee, 1967).
Il metodo di valutazione della crescita cellulare dovrà essere selezionato in base alle caratteristiche dello studio che si intende affrontare; la scelta dipende quindi dalla necessità di verificare l'aumento della massa cellulare totale, la capacità di divisione delle cellule oppure una determinata caratteristica metabolica.
Clonaggio. - Per clonaggio si intende la stabilizzazione di una popolazione cellulare originata da una singola cellula per ottenere una popolazione cellulare omogenea; è possibile perciò selezionare la popolazione cellulare maggiormente idonea per lo studio che si intende affrontare (v. Spier e Clarke, 1980).
Per produrre cloni da una sospensione cellulare devono essere separate le singole cellule, in modo che le colonie da esse formate possano essere facilmente isolate le une dalle altre. Il modo più facile per separare le singole cellule consiste nell'eseguire una serie di diluizioni scalari finché si arriva a ottenere una singola cellula per unità di volume di terreno di coltura. Le cellule così diluite sono poste in coltura e osservate al microscopio per seguire la comparsa delle prime colonie. È necessario sostituire a intervalli regolari il terreno di coltura con terreno fresco addizionato di siero e fornire cellule fresche, inibite nella loro capacità di riprodursi (ad esempio irradiate), che rappresentano una fonte di fattori di crescita. Quando le cellule sono sufficientemente cresciute, si isolano le singole colonie che possono venire direttamente utilizzate o congelate.
Sincronizzazione della crescita cellulare. - Normalmente all'intemo di una popolazione cellulare le cellule si trovano in varie fasi del ciclo cellulare (mitosi, G1, S, G2); molti studi richiedono l'uso di colture di cellule sincronizzate, che possono essere ottenute con metodi di blocco e di separazione. L'utilizzo di inibitori della mitosi induce un blocco delle cellule a un punto stabilito, con conseguente accumulo delle cellule a questo livello; la rimozione del blocco permette il proseguimento della crescita in sincronia. Le cellule in mitosi non si associano facilmente tra loro e al substrato (v. Tobey e altri, 1967): possono quindi essere staccate dal substrato con una forza minore rispetto alle cellule in fase di riposo. Applicando una forza minima si riesce a selezionare una popolazione cellulare nella stessa fase mitotica, ma con scarsa resa: è preferibile quindi associare il trattamento con l'uso di una sostanza bloccante per aumentare il numero di cellule mitotiche. Questa tecnica è naturalmente applicabile solo a cellule che crescono in monostrato su un supporto solido, e non a cellule che crescono in sospensione.
Immobilizzazione cellulare. - L'immobilizzazione delle cellule su matrici semisolide presenta numerose applicazioni pratiche, anche se la funzione fondamentale è quella di stabilizzare la cellula e di proteggerla da condizioni non ottimali. Le cellule possono essere immobilizzate tramite adsorbimento, legami covalenti, legami crociati o inclusione in matrici polimeriche. L'utilizzo di cellule immobilizzate nella tecnologia delle fermentazioni presenta molti vantaggi, in quanto il terreno può essere facilmente aggiunto o tolto, variando il rapporto tra le cellule e il volume di terreno; inoltre i prodotti cellulari riversati nel terreno possono essere recuperati senza pericolo di contaminazione con elementi cellulari (v. Nilsson e altri, 1983).
Conservazione delle cellule. - L'unico procedimento che assicura l'assoluta conservazione delle cellule è il congelamento a −196 °C. Le cellule mantenute in coltura tendono a perdere o a variare le loro proprietà: la conservazione di cellule di riferimento è fondamentale per alcuni programmi di ricerca, per la conservazione del patrimonio genetico delle linee cellulari utilizzabili nei processi biotecnologici, per mantenere stabili quelle linee già sottoposte a controllo di qualità, per prolungare la vitalità di cellule a vita breve. Le cellule sono congelate in presenza di un crioprotettore, di solito dimetilsolfossido (DMSO).
Collezioni di linee cellulari. - Uno dei maggiori problemi della manipolazione cellulare è rappresentato dal rischio di contaminazioni microbiche delle colture. È quindi opportuno controllare spesso le caratteristiche delle linee cellulari utilizzate in un laboratorio, oppure servirsi di linee provenienti da collezioni di colture cellulari. L'esistenza di tali collezioni permette a ogni laboratorio di disporre di linee cellulari sicuramente prive di contaminanti e di linee altrimenti non disponibili, ad esempio gli ibridomi (v. cap. 3). Inoltre, cautela contro il rischio di perdere materiale insostituibile e stimola lo sviluppo di metodi per la caratterizzazione, il controllo di qualità e la conservazione delle cellule. Si comprende quindi come le collezioni di colture cellulari rivestano enorme importanza per lo sviluppo futuro in biotecnologia.
Colture cellulari su larga scala. - Le cellule animali crescono in vitro in due modi diversi: aderenti a un supporto solido o in sospensione. È chiaro che dal tipo di cellule utilizzate dipende lo sviluppo di una particolare tecnologia di coltura.
A tutt'oggi, metodi per coltivazioni cellulari su larga scala sono stati messi a punto solo in pochi casi, quali la produzione di vaccini per l'afta epizootica (v. Nardelli e Panina, 1977; v. Radlett e altri, 1985) e dei vari tipi di interferoni (v. Finter e Fantes, 1980; v. Reuveny e altri, 1980). La produzione di vaccini virali umani si basa tuttora sull'allestimento di colture primarie o di colture di cellule trasformate. La necessità di produrre anticorpi monoclonali e altre proteine in cellule di mammifero manipolate geneticamente ha stimolato la messa a punto di nuove tecnologie per la coltivazione di cellule su larga scala. La produzione di sostanze di interesse farmacologico, quali antigeni virali, anticorpi ed enzimi, richiede specifiche glicosilazioni, non ottenibili in batteri o lieviti (v. Berman e altri, 1985; v. Patzer e Obijeski, 1985). Inoltre, tramite l'utilizzo della tecnica del DNA ricombinante, queste sostanze possono essere prodotte in grandi quantità. Poiché il pericolo maggiore nell'allestimento di colture cellulari su larga scala è indubbiamente identificabile nell'inquinamento microbico, è necessario fare riferimento a banche cellulari che forniscono cellule sicuramente prive di contaminanti (v. Telling e Radlett, 1970).
I recipienti in cui si effettuano le colture cellulari su larga scala sono progettati in modo tale da garantire una buona miscelazione della sospensione cellulare e offrono la possibilità di aggiungere terreni di coltura liquidi e di insufflare gas senza provocare danni alle cellule. Inoltre, l'agitazione e l'aerazione mantengono la coltura omogenea; la temperatura, la tensione di ossigeno e il pH possono essere controllati e possono essere prelevati campioni rappresentativi della coltura. Un sistema di coltura cellulare in sospensione utilizza i fermentatori a corrente d'aria ascensionale. Questo tipo di fermentatore è stato utilizzato per molti anni per le colture batteriche e solo recentemente il suo uso è stato esteso alle colture cellulari (v. Katinger e altri, 1979). Miscele di gas sono insufflate dalla base del fermentatore in un tubo immerso e, lungo tale condotto, risalgono fino alla superficie della coltura. La densità del liquido all'interno del condotto viene ridotta, rispetto a quella esterna, dall'insufflazione di gas, favorendo quindi la circolazione della coltura. Il gas introdotto ha, oltre allo scopo di mescolare la coltura, anche quello di fornire ossigeno. Il vantaggio maggiore di questo tipo di reattore è la semplicità, in quanto non necessita di agitatori.
Un altro metodo per colture cellulari su larga scala si basa sull'utilizzo di microvettori. Le linee cellulari diploidi necessitano per la loro crescita di un supporto solido cui ancorarsi: in laboratorio queste cellule sono coltivate in appositi contenitori (fiasche), ma possono anche essere coltivate in sospensione su sferette che fungano da vettori. Attualmente questo sembra il metodo più facilmente applicabile alle colture cellulari su larga scala, dal momento che fornisce una vasta superficie per unità di volume di terreno (v. Van Wezel, 1967; v. Griffiths, 1985; v. Reuveny e altri, 1983). Nelle colture cellulari su microvettori l'agitazione deve essere vigorosa per impedire l'aggregazione delle sferette vettrici che altrimenti tenderebbero ad agglomerarsi. L'utilizzo dei microvettori assume un ruolo fondamentale nella produzione di molecole ricombinanti, che vengono espresse in modo soddisfacente solo da cellule che crescono aderenti a un supporto solido.
3. Processi biotecnologici avanzati
a) Introduzione
Gli anticorpi monoclonali (AcM) sono prodotti da un singolo clone di linfociti B: tutte le molecole presentano la stessa sequenza amminoacidica e la stessa specificità di legame nei confronti dell'antigene (Ag). Negli anni settanta Köhler e Milstein hanno dimostrato la possibilità di ottenere linee cellulari continue, capaci di secernere anticorpi, mediante la tecnica dell'ibridazione cellulare. Essa consiste nella fusione di cellule neoplastiche (mieloma) con linfociti B presensibilizzati in vivo o in vitro verso un determinato antigene. La cellula ibrida che ne deriva (ibridoma) mantiene la capacità della cellula di mieloma di riprodursi per un tempo indefinito, mentre del linfocita conserva la capacità di secernere immunoglobuline specifiche. Gli AcM sono reagenti estremamente omogenei e specifici e, essendo producibili in elevate quantità, sono diventati strumenti efficaci in molti campi della ricerca biomedica e industriale. La tecnica di produzione degli ibridomi comporta diverse fasi: immunizzazione, immortalizzazione, selezione e produzione.
b) Immunizzazione
La somministrazione di sostanze estranee, antigeni (Ag), nell'organismo determina la produzione di anticorpi specifici (Ac) diretti contro l'Ag da parte delle cellule del sistema immunitario. La risposta anticorpale è detta policlonale, in quanto vengono stimolate più popolazioni linfocitarie: ognuna di queste produce Ac in grado di riconoscere e legare alcune porzioni antigeniche con diverso grado di affinità e specificità (v. Krause, 1970; v. Klinman, 1972).
Immunizzazione in vivo. - Scopo dell'immunizzazione è quello di ottenere linfociti B sensibilizzati e proliferanti. Le molecole impiegate come immunogeni presentano nella loro struttura alcune porzioni chiamate ‛epitopi' o ‛determinanti antigenici', in grado di stimolare la produzione di anticorpi specifici da parte dei linfociti B. Le condizioni critiche che influenzano il risultato dell'immunizzazione sono costituite dalla specie animale, dalla via scelta per la somministrazione dell'Ag, dal suo grado di purezza e dall'impiego di sostanze adiuvanti. Per quanto riguarda la specie animale, si impiegano generalmente topi del ceppo BALB/c per la loro perfetta caratterizzazione genotipica e per il grado di sviluppo del sistema reticoloendoteliale.
La purezza dell'antigene influenza direttamente la specificità della risposta anticorpale. Uno dei vantaggi offerti dalla tecnologia degli ibridomi consiste nella possibilità di ottenere anticorpi specifici anche nei confronti di molecole difficilmente purificabili od ottenibili in scarsa quantità. Nonostante i risultati positivi ottenuti da Secher e Burke (v., 1980) nella produzione di interferone, è comunque preferibile utilizzare molecole parzialmente purificate.
La quantità di antigene necessaria per l'immunizzazione dipende largamente dalle caratteristiche immunogeniche della molecola. Antigeni presenti sulla superficie cellulare sono fortemente immunogenici, mentre antigeni solubili come polipeptidi, carboidrati e steroidi lo sono solo scarsamente. La loro immunogenicità può essere aumentata mediante coniugazione con macromolecole come la keyhole limpet haemocyanin (KLH) o la sieroalbumina.
L'uso di sostanze adiuvanti, quali il Freund o l'idrossido di alluminio (eventualmente associato a microrganismi quale Bordetella pertussis), è necessario quando vengano impiegati antigeni proteici (v. Herbert, 1978; v. Munoz, 1964; v. Munoz e Bergman, 1977). Gli adiuvanti sono in grado di amplificare la risposta immunitaria e di condizionare la classe immunoglobulinica degli Ac prodotti (v. Handman e Remington, 1980; v. Stähli e altri, 1980; v. Hirashima, 1981). La quantità di antigene necessario, inoltre, può essere fortemente ridotta qualora esso venga incorporato in micelle o liposomi.
La prima inoculazione dell'Ag (miscelato ad adiuvante) viene effettuata nell'animale per via sottocutanea o intraperitoneale. Dopo alcune settimane viene somministrata un ulteriore dose in soluzione salina, per via endovenosa; a essa fa seguito la valutazione del titolo degli anticorpi circolanti, indice di attivazione del sistema immunitario. Un basso titolo anticorpale indica generalmente una risposta inadeguata da parte dell'animale. Quando l'antigene è costituito da proteine a basso peso molecolare e/o da molecole che non presentano nella loro struttura residui amminoacidici a cui poter legare molecole vettrici, si rendono necessarie vie di somministrazione diverse da quella intraperitoneale: in questo caso la sede di elezione è rappresentata dal cuscinetto popliteo, e i linfociti sensibilizzati per la fusione vengono prelevati dai linfonodi che drenano questo distretto.
Per produrre anticorpi della classe IgA è invece opportuno stimolare il tessuto linfoide del sistema gastroenterico somministrando l'antigene mediante sonda gastrica.
Una tecnica per incrementare notevolmente l'efficienza dell'immunizzazione e per ottenere quindi un'alta percentuale di ibridi secernenti consiste nel trasferimento di cellule spleniche di un animale immunizzato in un animale ‛singenico', reso immunodepresso mediante irradiamento con raggi X (v. Siraganian e altri, 1983). Rispetto alle tecniche di immunizzazione tradizionali questa tecnica consente di ottenere un numero di ibridi secernenti fino a 50 volte maggiore.
Immunizzazione in vitro. - La stimolazione dei linfociti in coltura rappresenta un'attraente alternativa rispetto a quella in vivo, soprattutto nella produzione di ibridomi umani. Esistono tuttavia molte altre situazioni in cui questa tecnica viene impiegata anche nella produzione di anticorpi murini, in particolare contro antigeni che normalmente non danno risposta anticorpale, antigeni labili e antigeni self (v. Borrebaek, 1983; v. Reading, 1982 e 1983). I vantaggi offerti da questa tecnica sono costituiti dal breve tempo necessario per la sensibilizzazione cellulare e dall'utilizzo di quantità di antigene fino a 1.000 volte inferiori rispetto a quelle utilizzate nelle procedure in vivo (v. Luben e altri, 1982). Il numero degli ibridi ottenibili è alto, ma gli anticorpi prodotti sono prevalentemente della classe IgM, fattore questo che ne limita alcuni impieghi (v. Buchman e altri, 1985).
Durante la stimolazione in vitro è necessario aggiungere al terreno di coltura alcuni fattori di crescita che stimolano la proliferazione linfocitaria. Per le cellule murine vengono utilizzati terreni condizionati con timociti, mentre per le cellule umane, generalmente rappresentate da linfociti periferici, la stimolazione viene favorita dai linfociti T e dai monociti presenti nella sospensione cellulare. Altre sostanze che agiscono sulle cellule B in coltura sono il lipopolisaccaride batterico (LPS) e il pokeweed mitogen (PWM) (v. Andersson e Melchers, 1978; v. Ilfeld e altri, 1981).
Per la produzione di ibridi umani secernenti anticorpi verso antigeni associati a neoplasie vengono impiegate cellule B di soggetti portatori della neoplasia, purificate dal sangue periferico o dai linfonodi circostanti la massa tumorale. In questo caso la frequenza degli ibridi secernenti risulta molto inferiore rispetto a quella ottenibile impiegando linfociti presensibilizzati (v. Olsson e Kaplan, 1980; v. Olsson e altri, 1983).
c) Tecniche per la preselezione dei linfociti sensibilizzati
Queste tecniche hanno lo scopo di selezionare da una popolazione eterogenea solo i linfociti sensibilizzati nei confronti dell'antigene somministrato. La selezione viene effettuata in base alla presenza sulla superficie dei linfociti B di recettori immunoglobulinici, che hanno nei confronti dell'antigene la stessa specificità degli anticorpi secreti. I sistemi impiegati sono il FACS (fluorescent activated cell sorter), la marcatura con avidina-biotina e la rosettazione.
Selezione mediante fluorescent activated cell sorter (FA CS). - Questo metodo consente di separare sottopopolazioni presenti in una sospensione cellulare attraverso l'impiego di un raggio laser. La separazione avviene in base sia alle dimensioni cellulari sia alla fluorescenza loro associata. Antigeni marcati con fluoresceina si legano alle immunoglobuline di superficie presenti sui linfociti B sensibilizzati (v. Herzenberg e Herzenberg, 1978). Parks e altri (v., 1979) hanno descritto l'uso combinato del FACS e di microsfere fluorescenti per la separazione e la selezione di cellule ibride. Le microsfere coniugate chimicamente all'antigene sono in grado di legarsi alle cellule ibride, che vengono poi separate dalle altre cellule mediante il FACS. Analogo procedimento può essere impiegato per la selezione di linfociti antigene-specifici da una popolazione di splenociti prima della fusione.
Marcatura con il sistema avidina-biotina. - La coniugazione di antigeni e/o anticorpi mediante il complesso avidina-biotina è in grado di trasformare un processo casuale e a bassa efficienza, quale la fusione cellulare, in un sistema controllato, specifico e con alte rese. L'avidina è una glicoproteina, estratta dall'albume d'uovo, che presenta un'elevatissima affinità per il coenzima biotina. Nella preselezione delle cellule immunocompetenti l'avidina viene legata in modo covalente all'antigene, mentre la biotina viene associata alle cellule di mieloma. Si incubano poi opportune concentrazioni di linfociti immunizzati con i complessi antigene-avidina e biotina-mieloma. I linfociti sulla cui superficie sono presenti immunoglobuline antigene-specifiche formeranno complessi stabili e altamente affini costituiti da linfocita B-Ag-avidina-biotina-mieloma (v. Guesdon e altri, 1979).
Rosettazione. - La tecnica di elezione da impiegare nella preselezione, qualora sia possibile coniugare l'antigene agli eritrociti, è quella della rosettazione. Essa si basa sulla capacità delle cellule B di formare rosette con i globuli rossi di pecora a cui sia stato preventivamente associato l'antigene (v. Legrain e altri, 1983). I linfociti in grado di formare rosette sono quelli che esprimono immunoglobuline di superficie specifiche; essi possono essere individuati con il microscopio e separati mediante centrifugazione in gradiente di densità. Falsi positivi (rosette aspecifiche) possono formarsi con cellule non vitali per adsorbimento di globuli rossi sulla superficie. La velocità e semplicità di esecuzione rendono questo metodo estremamente vantaggioso: l'unico fattore limitante è costituito dalla notevole quantità di antigene necessaria.
Un altro metodo di preselezione consiste nell'arricchimento della sospensione di linfociti B antigene-specifici mediante rimozione di cellule B non sensibilizzate (v. Walker e altri, 1977; v. Kozbor e Roder, 1983). In molti casi le cellule B, dopo la reazione con l'antigene, perdono i recettori globulinici di superficie per fenomeni di capping, shedding, stripping (v. Ault e Unanue, 1974). Le cellule B che non legano l'antigene possono essere rimosse mediante rosettazione con globuli rossi, sulla cui superficie vengono fatte aderire immunoglobuline anti-Ig della specie e complemento. Questo metodo, detto ‛di selezione negativa', permette di incrementare notevolmente il numero dei linfociti specifici.
d) Immortalizzazione
Per ottenere l'immortalizzazione vengono utilizzate linee di tumori plasmacellulari (mielomi) indotti sperimentalmente, rese mutanti sia per la sintesi di immunoglobuline sia per l'assenza degli enzimi ipoxantina-guanina-fosforibosiltransferasi (HGPRT) e timidina chinasi (TK). La selezione, dopo la fusione cellulare, viene effettuata bloccando la sintesi di DNA, e quindi la replicazione cellulare, mediante l'impiego di veleni metabolici che inibiscono gli enzimi indispensabili a tale sintesi.
Nelle cellule la sintesi di DNA può avvenire attraverso due vie: la prima prevede la sintesi di nucleotidi partendo da zuccheri e amminoacidi, la seconda (impiegata in alternativa quando la via principale sia bloccata) utilizza ipoxantina e guanidina mediante gli enzimi HGPRT e TK. Il blocco della via principale avviene aggiungendo al terreno di coltura analoghi dell'acido folico, come l'amminopterina. Quando i linfociti B sensibilizzati (che posseggono gli enzimi HGPRT e TK) vengono fusi con cellule di mieloma (mancanti di HGPRT e TK), solo le cellule ibride sono in grado di crescere nel terreno di coltura che contiene amminopterina, ipoxantina e timidina (terreno HAT). Questo terreno, infatti, favorisce la crescita solo delle cellule HGPRT+ e TK+, mentre inibisce la crescita delle cellule di mieloma mancanti di questi enzimi.
Le linee cellulari di mieloma murino più impiegate sono derivate da un tumore, il MOPC 21, indotto sperimentalmente in animali del ceppo BALB/c mediante somministrazione intraperitoneale di olio minerale. Il tumore secerne molecole di immunoglobuline. Oltre alla capacità di crescere continuamente in vitro ed essere HGPRT- queste cellule non sono in grado di sintetizzare Ig né di esercitare un'azione soppressiva nei confronti dei geni preposti alla sintesi di immunoglobuline di derivazione linfocitaria. In particolare la linea P3-NS1-Ag 4.1 (NS1), la più usata, non e in grado di produrre catene pesanti, mentre la X63-Ag 8.653 (NSO/1) non produce catene né pesanti né leggere (v. Köhler e altri, 1976; v. Kearney e altri, 1979). Altre linee di mieloma murino non secernenti ampiamente utilizzate sono la SP2/0 Ag 14 (SP2) e una sua variante, la SP2F0 (v. Shulman e altri, 1978).
La linea 210-RCY3-Ag1, resistente all'azoguanina, quindi HGPRT-, descritta da Querineyean e Milstein nel 1972, deriva invece da un tumore ileocecale evocato in ratti del ceppo LOU/Ws 1, che presenta un'elevata incidenza di tumori plasmacellulari spontanei (v. Bazin, 1973). Gli ibridi ratto-ratto sono una valida alternativa agli ibridi topo-topo. Infatti nei ratti la milza è il maggior organo, se non il solo, che produce anticorpi quando si somministra l'antigene per via endovenosa; inoltre il numero di ibridi ottenibili, nonché la quantità di siero e ascite, sono molto maggiori rispetto a quando vengono impiegati topi (v. Milstein, 1980; v. Galfré e altri, 1979).
L'efficienza di ibridazione ottenuta impiegando linee di mieloma umano nella produzione di ibridi uomo-uomo è invece molto bassa, per la loro instabilità e scarsa attitudine a crescere in vitro; esse sono nella maggior parte dei casi derivate da cellule linfoblastoidi dopo trasformazione di linfociti B con il virus di Epstein-Barr (EBV).
Fusione. - La fusione delle membrane cellulari è un evento spontaneo che si osserva in vari fenomeni biologici quali la pinocitosi, la secrezione, la formazione di liposomi, la fecondazione. Nel caso specifico della produzione di AcM, la fusione si effettua mettendo a contatto i linfociti sensibilizzati con le cellule di mieloma in opportuni rapporti, in presenza di agenti in grado di modificare le membrane cellulari: il polietilenglicole (PEG) e virus inattivati quali il virus Sendai. Essi agiscono depolarizzando le membrane e favorendo la formazione di ponti citoplasmatici a cui fa seguito il rigonfiamento cellulare e la formazione dell'eterocarionte, nel cui citoplasma si possono riconoscere due o più strutture nucleari. Nel periodo immediatamente successivo avviene la fusione del materiale nucleare con ricombinazione e perdita di una parte di esso. L'ibrido che ne deriva, dopo la stabilizzazione, avrà caratteristiche genetiche e biochimiche di entrambe le cellule parentali.
Un'alternativa all'impiego di agenti chimici (PEG) o biologici (virus Sendai inattivati) è costituita dall'elettrofusione. Mediante l'applicazione di un campo elettrico è possibile modificare temporaneamente le proprietà elettriche delle membrane cellulari. Queste sono costituite da un doppio strato di fosfolipidi nel quale sono incorporate le molecole proteiche: i gruppi idrofobi, interni, delle molecole lipidiche agiscono da isolante, mentre i gruppi idrofili, posti all'esterno, fungono da portatori di carica elettrica. La cellula può quindi essere paragonata a un condensatore sferico. La sospensione cellulare costituita da linfociti sensibilizzati e da cellule di mieloma viene posta in una camera di fusione contenente due elettrodi collegati a un generatore di frequenza a impulsi. Applicando un campo elettrico si genera una diversa distribuzione delle cariche elettriche presenti sulla membrana cellulare, con formazione di dipoli, favorenti l'avvicinamento delle cellule. Il procedimento si può seguire al microscopio: quando le membrane sono in contatto si applica un impulso elettrico detto ‛impulso di fusione', che, superando la tensione critica della membrana citoplasmatica, provoca l'apertura temporanea di pori in corrispondenza dei quali le membrane si fondono (v. Zimmerman, 1983). Questo processo avviene con una frequenza elevata ma non è selettivo. La sua specificità può essere aumentata se si realizza un avvicinamento preliminare delle cellule sensibilizzate impiegando, ad esempio, il sistema avidina-biotina. I vantaggi sono dovuti alla natura fisica del metodo oltre che alla possibilità di seguire direttamente le varie fasi del processo al microscopio. I tempi di fusione sono minimi e le rese buone. Gli anticorpi prodotti da ibridi ottenuti con questo sistema presentano un'affinità maggiore rispetto a quelli ottenuti con metodi convenzionali (v. Lo e altri, 1984).
Immortalizzazione mediante virus trasformanti. - Oltre che con i metodi precedentemente descritti, l'attitudine a crescere e a moltiplicarsi in vitro può essere conferita alle cellule mediante l'impiego di virus trasformanti. A questo scopo il virus più impiegato per la trasformazione dei linfociti umani è il virus di Epstein-Barr (EBV), un Herpesvirus linfotropo che infetta selettivamente i linfociti B e che consente di ottenere linee cellulari stabili secernenti anticorpi (v. Rosen e altri, 1977). Le cellule trasformate mantengono tutte le caratteristiche delle cellule B originali ed esprimono un antigene nucleare EBV-specifico (EBNA). La trasformazione indotta dall'infezione dà luogo a una produzione anticorpale di tipo policlonale. Per ottenere linfociti B secernenti anticorpi della specificità desiderata è conveniente preselezionare le cellule con appropriati siti di combinazione per l'antigene. A questo scopo si impiega il test della rosettazione con eritrociti precedentemente descritto (v. Kozbor e Roder, 1983; v. Steinitz e altri, 1977). Dopo la preselezione i linfociti del sangue periferico di donatori vengono infettati mediante l'aggiunta del sovranatante di coltura delle cellule infettate con EBV. I linfociti vengono poi coltivati in condizioni opportune; dopo circa un mese si ottengono delle linee stabilizzate. Tutte le cellule vitali dovrebbero esprimere come risultato dell'infezione l'Ag EBNA. Le cellule vengono poi separate per isolare i doni di linfociti producenti anticorpi di interesse. Questa tecnica è stata impiegata per la produzione di AcM contro gli Ag del sistema Rh (v. Koskimies, 1979), contro il polisaccaride dello streptococco di gruppo A (v. Steinitz e altri, 1979), contro la tossina tetanica (v. Kozbor e Roder, 1981), contro il virus dell'epatite B e contro gli Herpesvirus di tipo 1 e 2 (v. Paire e Desgranges, 1985).
Transfezione. - La transfezione, che consiste nel trasferimento diretto e nell'incorporazione di DNA genomico nei cromosomi di cellule eucariote in replicazione, consente di stabilizzare in vitro linee cellulari (v. Davis e altri, 1985). L'immortalizzazione di cellule linfoidi mediante transfezione con DNA genomico, contenente sequenze oncogene, oltre che costituire un'importante applicazione delle tecnologie del DNA ricombinante, ha fornito nuovi approcci alla produzione di AcM. Il metodo è simile a quello impiegato per la produzione degli ibridomi. La principale differenza è costituita dall'uso di DNA in luogo di cellule intere durante la fusione. Il DNA utilizzato viene estratto, con le usuali tecniche biochimiche, da linee leucemiche umane Reh o da linee di mieloma murino SP2/0Ag 14 e X63Ag 8 653 in fase logaritmica di crescita (v. Jonak e altri, 1983 e 1984). Il trasferimento del DNA all'interno delle cellule ospiti viene mediato dal fosfato di calcio. Le cellule linfoidi sensibilizzate vengono messe a contatto con il DNA; dopo un periodo di incubazione, la sospensione cellulare viene trattata con PEG e quindi risospesa in terreno di coltura. Le condizioni per la crescita sono uguali a quelle adottate per gli ibridomi derivanti da fusioni.
e) Metodi di selezione cellulare
La selezione degli ibridi formati durante la fusione avviene in due fasi successive: 1) preselezione di ibridi mediante l'analisi dei sovranatanti di coltura (screening); 2) clonaggio degli ibridi al fine di ottenere una progenie cellulare omogenea e secernente stabilmente AcM.
Screening. - Una prima selezione viene effettuata già alcuni giorni dopo la fusione, analizzando i sovranatanti di coltura. I test più comunemente impiegati a questo scopo sono costituiti da metodi radioimmunometrici (IRMA) o enzimoimmunometrici (ELISA) (v. Catt e Tregear, 1967; v. Engvall e Perlmann, 1971). I requisiti necessari per un test di screening sono: sensibilità, applicabilità a un elevato numero di campioni, rapidità di esecuzione e buona specificità. Entrambi i sistemi prevedono l'impiego di fasi solide, generalmente micropiastre di polivinile, a cui viene legato mediante adsorbimento passivo l'antigene. In ogni pozzetto viene poi aggiunto il sovranatante di coltura contenente AcM e il legame fra Ag e AcM viene rivelato mediante l'aggiunta di un anticorpo antimmunoglobuline marcato. La differenza fra i due metodi è rappresentata dal tipo di molecole impiegato come tracciante: nell'IRMA il tracciante è costituito da isotopi radioattivi (125I), mentre nell'ELISA da enzimi quali perossidasi, fosfatasi, β-galattosidasi.
Altri metodi impiegati nello screening sono l'immunofluorescenza, la citotossicità e la rosettazione.
I test di immunofluorescenza consentono di selezionare gli ibridi secernenti sfruttando la presenza, sulla loro superficie cellulare, di anticorpi in grado di reagire specificamente con l'antigene precedentemente coniugato con molecole fluorescenti (v. Goding, 1976). La loro rivelazione viene effettuata mediante l'impiego del microscopio a fluorescenza o del FACS, il quale è anche in grado di separare gli ibridi positivi (v. Ledbetter e Herzenberg, 1976; v. Herzenberg e altri, 1976).
I test di citotossicità vengono impiegati per la selezione di ibridi secernenti AcM in grado di fissare il complemento, diretti contro antigeni presenti sulla superficie cellulare. Le cellule che presentano l'antigene vengono incubate con il sovranatante contenente AcM; una volta stabilitosi il legame, viene aggiunto il complemento, che determina la lisi cellulare. Il grado di citolisi può essere misurato impiegando coloranti vitali quali il trypan blu, l'arancio di acridina o l'etidio bromuro.
Clonaggio. - Il clonaggio è il procedimento attraverso cui vengono isolate le cellule ibride in modo da ottenere una progenie omogenea (clone) derivante da una singola cellula. A questo scopo la sospensione cellulare contenente ibridi positivi viene diluita scalarmente secondo la tecnica della limiting dilution (v. Lefkovits e Waldmann, 1979).
Un altro metodo impiegato consiste nel seminare la sospensione cellulare contenente gli ibridi su un terreno semisolido (soft agar) e nel trasferire poi le singole colonie sviluppate in pozzetti di coltura (v. Coffino e altri, 1972; v. Metcalf, 1977).
Entrambe le tecniche vanno ripetute più volte affinché la progenie cellulare ottenuta sia omogenea e geneticamente ben stabilizzata. Gli ibridi, infatti, durante le fasi di crescita e moltiplicazione vanno incontro a una perdita graduale di materiale genetico che può portare all'inibizione della capacità di produrre e secernere anticorpi.
Diversi fattori, quali il controllo genetico della mitosi, la replicazione asincrona del DNA, la condensazione prematura dei cromosomi (PCC), la disarmonia genica fra i genomi parentali e le alterazioni del metabolismo proteico, sono probabilmente le cause della perdita cromosomica nelle cellule ibride. Anche la mancanza di normali interazioni nella fase della divisione mitotica può comportare una perdita di materiale genetico o un'anomala distribuzione di cromosomi nelle cellule figlie, determinando l'arresto nella produzione di anticorpi.
f) Anticorpi modificati
Anticorpi chimerici uomo-topo. - Utilizzando tecniche di ingegneria genetica sono stati prodotti anticorpi ‛chimerici', cioè anticorpi il cui sito di legame con l'antigene deriva da AcM murini, mentre il resto della molecola è costituito da sequenze umane. L'importanza applicativa di queste molecole anticorpali è legata essenzialmente al loro uso in tecniche diagnostiche in vivo e nella terapia antineoplastica. Boulianne e altri (v., 1984) hanno prodotto e caratterizzato anticorpi in cui le regioni variabili presenti nelle catene pesanti e leggere erano di origine murina, mentre le regioni costanti erano di origine umana (v. Munro, 1984). Essi hanno sintetizzato dei geni costituiti da frammenti di DNA codificanti per le regioni variabili murine, dirette verso l'Ag di interesse, legati a segmenti di DNA codificanti per le regioni costanti umane. Questi geni ‛chimerici' sono stati poi trasferiti in un vettore (plasmide) con il quale sono state transfettate cellule di mieloma. Poiché le cellule transfettate secernono segmenti anticorpali privi del frammento cristallizzabile (Fc), si è resa possibile la produzione di anticorpi chimerici bifunzionali: essi infatti sono in grado di legarsi specificamente all'Ag oltre che di veicolare un enzima in sostituzione dell'Fc (v. Sharon e altri, 1984; v. Neurberger e altri, 1984).
Anticorpi bispecifici. - Vengono definiti anticorpi bispecifici le immunoglobuline che posseggono nella loro molecola due siti combinatori specifici per due diversi determinanti antigenici. Nel 1983 Milstein e Cuello hanno prodotto AcM bispecifici diretti contro la somatostatina e la perossidasi e impiegati con successo nelle tecniche immunoistochimiche. La tecnica di produzione si basa su una doppia fusione: viene prima prodotto un ibrido specifico per un determinato Ag, secondo il procedimento usuale, e questi ibridi vengono poi fusi con linfociti sensibilizzati verso un secondo Ag rappresentato da un enzima, come ad esempio la perossidasi. L'ibrido che ne risulta viene selezionato con tecniche immunoistochimiche in rapporto alla sua duplice attività. La molecola anticorpale risultante rappresenta solo una delle dieci possibili combinazioni fra catene pesanti e leggere prodotte dalle cellule parentali. Tali AcM sono una valida alternativa a quelli ottenuti mediante trattamento chimico, che spesso comporta una parziale perdita dell'attività anticorpale e la possibilità di legami aspecifici.
Commutazione di classe (switch di isotipo). - Talvolta gli AcM secreti dagli ibridomi ottenuti dopo la fusione possono non presentare caratteristiche idonee all'uso cui sono destinati (carenza legata alla classe o sottoclasse di appartenenza). È possibile selezionare varianti, dovute a mutazioni spontanee, tra gli ibridomi che producono anticorpi di isotipo diverso rispetto al clone parentale. Ad esempio, tra ibridomi che producono AcM IgG1 diretti verso glicoproteine di superficie di cellule tumorali, ma non in grado di causare la lisi delle cellule, sono stati selezionati mutanti switch, capaci di secernere IgG2, che lisano le cellule target in presenza di complemento. La selezione di queste varianti switch viene effettuata con l'impiego di reagenti antisotipo specifici, di tecniche avanzate come l'ELISA e di strumenti come il FACS (v. Bargellesi, 1985; v. Herzenberg, 1985). Gli ibridomi secernenti AcM switch esprimono ancora l'originale regione variabile, ma hanno ‛mutato' l'espressione di una regione costante, che risulta appartenere a un'altra classe o sottoclasse (v. Parham, 1983).
g) Produzione su larga scala di AcM
Produzione in vitro. - Dopo il clonaggio e la stabilizzazione le colture vengono espanse; il sovranatante di coltura viene recuperato per la purificazione degli AcM prodotti. Se le colture vengono espanse su larga scala è possibile ottenere quantità di AcM illimitate e perfettamente omogenee. Uno dei fenomeni che si osservano più frequentemente nella produzione in vitro è la perdita della capacità di secernere da parte degli ibridi. Questo è dovuto sia alla perdita di materiale genetico, sia all'alterazione delle condizioni fisico-chimiche del mezzo di crescita. Il pH ottimale per la crescita cellulare è compreso tra 7,5 e 7,6; valori superiori o inferiori comportano una diminuita moltiplicazione cellulare (v. Ceccarini e Eagle, 1971). I vantaggi offerti dalla produzione su larga scala degli AcM in vitro sono legati alla possibilità di condurre il processo in condizioni di sterilità e di usare, come unica sostanza estranea, il siero fetale bovino (FCS), necessario alla crescita cellulare.
Produzione in vivo. - La tecnica più diffusa per lo sviluppo degli ibridomi secernenti AcM impiegati per la ricerca è rappresentata dalla coltura in vivo. I doni secernenti vengono iniettati per via intraperitoneale in animali istocompatibili trattati preventivamente con pri stano (2,6,10,14-tetrametilpentadecano), un olio minerale che favorisce lo sviluppo del tumore e la produzione di liquido ascitico da cui vengono purificati gli AcM. Per lo sviluppo di ibridi tra specie diverse si impiegano animali immunodepressi. La tecnica di produzione in vivo, pur consentendo di ottenere una concentrazione di AcM per unità di volume assai superiore a quella ottenibile con colture in vitro, ha però alcuni limiti: non avviene in maniera sterile e vi può essere una contaminazione da parte di virus animali che rendono gli AcM non utilizzabili nella sperimentazione in vivo. Inoltre la presenza di grandi quantità di proteine animali nel liquido ascitico rende necessario l'impiego di complessi metodi di purificazione per poter successivamente utilizzare gli AcM.
4. Ingegneria genetica
a) Tecniche di modificazione e trasferimento del DNA
Il DNA è una molecola polimerica formata dalla ripetizione di monomeri costituiti da una base azotata (adenina, guanina, citosina, timina) unita a uno zucchero (desossiribosio) a sua volta legato a un gruppo fosforico; questa struttura è organizzata in doppia elica (v. acidi nucleici).
Il processo che porta dal DNA alla sintesi di proteine da esso codificate avviene attraverso diverse fasi. La doppia elica si svolge, permettendo all'enzima RNA-polimerasi di ancorarsi all'estremità di una delle due eliche e di trascrivere il messaggio contenuto nel DNA in una seconda molecola (RNA messaggero), che differisce dal DNA originario unicamente per la sostituzione di una base azotata (uracile anziché timina) e dello zucchero (ribosio). Attraverso un complesso meccanismo, non ancora del tutto chiarito, l'RNA messaggero viene tradotto nella corrispondente proteina.
Da più di dieci anni il DNA è oggetto di studio a livello molecolare mediante tecniche di manipolazione che competono a quella branca della biologia definita ora ‛ingegneria genetica' ora ‛tecnologia del DNA ricombinante' e che rientra nell'ambito della ‛biologia molecolare' (v. biologia molecolare, suppl.).
L'ingegneria genetica si basa principalmente sulla possibilità di donare un frammento di DNA e cioè di inserirlo, tramite un vettore (v. È b), in una cellula (generalmente un batterio), in modo che il DNA inserito sia conservato in tutte le discendenti di tale cellula. In questo caso il clonaggio corrisponde, in senso lato, a un metodo di purificazione: infatti, isolando il batterio contenente il frammento di DNA che interessa, si potranno ottenere innumerevoli copie di quel singolo frammento, separato dalle altre sequenze (analogamente, ottenere un anticorpo monoclonale significa, come si è visto, isolare uno solo dei molteplici anticorpi prodotti da un organismo).
Mentre, in linea di principio, si potrebbe pensare di donare una sola sequenza (quella che interessa), in pratica risulta più semplice donare un insieme di diversi frammenti e poi individuare, tra i molti cloni, quello desiderato, tramite una delle varie strategie possibili, descritte più avanti.
Una collezione di frammenti di DNA di un singolo organismo inseriti in opportuni vettori costituisce una ‛genoteca' o ‛libreria genomica'.
In determinate situazioni risulta più conveniente inserire nel vettore (cioè clonare) solo le sequenze codificanti del DNA. A tal fine si estrae l'RNA messaggero, da cui sono state excise le sequenze non codificanti definite ‛introni', e si sfrutta il fatto che, in presenza di un particolare oligonucleotide (oligo-dT), l'enzima transcrittasi inversa, prodotto da una famiglia di virus a RNA conosciuti come Retrovirus (v. virus), è in grado di retro-trascrivere l'RNA messaggero in DNA privato degli introni. Il DNA così ottenuto è definito cDNA poiché costituisce la copia complementare dell'RNA messaggero.
Esistono attualmente diverse procedure per costruire una libreria di cloni di DNA; tutte si basano sull'uso di vari enzimi, che modificano in qualche modo il DNA: enzimi capaci di frammentarlo (endonucleasi di restrizione), ligasi, che uniscono frammenti di DNA in modo covalente, fosfatasi, che rimuovono gruppi fosfato terminali, e molti altri.
In particolare le endonucleasi di restrizione sono enzimi estratti da microrganismi, la cui funzione biologica consiste nell'eliminare le molecole di acido nucleico esogeno che penetrano nei microrganismi. Le tecniche di biologia molecolare utilizzano questi enzimi sfruttando la loro capacità di riconoscere una data sequenza, costituita solitamente da 4-6 nucleotidi, e di tagliare in sua corrispondenza la doppia elica di DNA. I siti di taglio degli enzimi di restrizione sono costituiti da sequenze palindromiche, sequenze cioè dotate di simmetria bilaterale nella composizione nucleotidica, per cui leggendo da sinistra a destra un'elica si ottiene la composizione dell'elica complementare letta da destra a sinistra.
Esistono due tipi di enzimi di restrizione: alcuni creano tagli sfasati nella doppia elica dando luogo a polinucleotidi con ‛estremità coesive' (sticky ends), altri creano tagli netti e ne risultano delle ‛estremità spuntate' (blunt ends).
Attualmente si conoscono più di 150 enzimi di restrizione, ma solo alcuni di essi vengono utilizzati nella manipolazione del DNA.
b) Vettori
Il frammento o i frammenti di DNA ottenuti dalla digestione con enzimi di restrizione devono essere inseriti in un sistema che ne assicuri la conservazione e la replicazione: tale sistema si chiama ‛vettore'. Si utilizzano vettori diversi a seconda delle dimensioni del frammento di DNA da clonare e a seconda che si vogliano utilizzare cellule procariote o eucariote per l'espressione dell'inserto.
I vettori più utilizzati per donare frammenti di piccole dimensioni (dell'ordine di poche migliaia di basi) in cellule procariote sono i plasmidi, le più piccole unità autoreplicantisi conosciute in natura. I plasmidi sono molecole circolari di DNA che si trovano nei Batteri e contengono pochi geni, di cui uno o più responsabili della resistenza dei Batteri ad alcuni antibiotici.
I plasmidi vengono tagliati con un enzima di restrizione, in modo che assumano una forma linearizzata, e sono messi in presenza di frammenti di DNA digerito con lo stesso enzima di restrizione. Per l'intervento di un altro enzima, la ligasi, una buona parte dei frammenti di DNA si integra nei plasmidi che ritornano nella forma circolare. I plasmidi vengono quindi messi a contatto con un ceppo batterico, solitamente di Escherichia coli, esposto precedentemente a un'elevata concentrazione di cloruro di calcio, che ha la proprietà di permeabilizzare la membrana batterica e di favorire quindi l'ingresso dei plasmidi nel batterio, che a questo punto viene definito ‛competente'.
I plasmidi che integrano un frammento di DNA sono definiti ‛ricombinanti'. Per selezionare i batteri contenenti i plasmidi e poi quelli i cui plasmidi hanno raccolto un segmento di DNA esogeno, si sfrutta la proprietà dei plasmidi di conferire alla cellula ospite la resistenza a qualche antibiotico. Indichiamo genericamente con A e B due antibiotici e supponiamo che il plasmide da usare come vettore contenga i due geni responsabili della resistenza ai due antibiotici. Piastrando i batteri sull'antibiotico A (per esempio) si fa in modo che sopravvivano solo quei batteri che hanno assunto un plasmide. Se si è avuta l'accortezza di tagliare - per inserire il DNA esogeno - il gene del plasmide che conferisce la resistenza all'antibiotico B, piastrando sull'antibiotico B un certo numero di batteri derivati da quelli sopravvissuti all'azione dell'antibiotico A, si individuano i ceppi contenenti plasmidi modificati (perché, non essendo più B-resistenti, soccombono all'azione dell'antibiotico B).
Il fenomeno della penetrazione e replicazione di un plasmide ricombinante in una cellula batterica viene definito ‛trasformazione'.
Se il gene o i geni che si intendono donare superano le 15.000 basi (15 kb) si utilizzano come vettori i batteriofagi, cioè virus che infettano i Batteri. Fra i più conosciuti e utilizzati in biologia molecolare vi è il fago lambda, che infetta Escherichia coli. Oltre a consentire l'inserimento di frammenti di DNA di maggiori dimensioni, il batteriofago, o fago, costituisce un ottimo strumento di amplificazione. Infatti, una volta penetrato nel batterio ospite, il fago ricombinante si replica diverse migliaia di volte. Il DNA che contiene l'inserto viene impacchettato all'interno di nuove particelle virali complete che reinfettano i batteri circostanti amplificando in tale maniera il ciclo. L'infezione di un batterio da parte di un fago ricombinante viene definita ‛transfezione'.
Una terza classe di vettori è rappresentata da ibridi sintetici costruiti con plasmidi e fagi, noti come cosmidi. Tali vettori hanno la caratteristica di poter ospitare inserti di DNA delle dimensioni anche di 50 kb. I cosmidi contengono i cosiddetti ‛siti cos' del fago lambda, che assicurano l'imballaggio e la circolarizzazione del frammento di DNA esogeno nella testa del fago; sono inoltre presenti uno o più geni responsabili della resistenza agli antibiotici di derivazione plasmidica.
Vettori particolari di cui si parlerà più avanti sono rappresentati da alcuni virus.
c) Espressione degli inserti
L'inserimento del DNA esogeno in un vettore non equivale alla sua espressione (trascrizione, modificazioni post-trascrizionali, traduzione e modificazioni post-traduzionali). Affinché uno o più geni si esprimano è necessario che si trovino sotto il controllo di sequenze regolatrici ('promotori') poste in determinate posizioni rispetto alla sequenza codificante del gene. La costruzione dei vettori di espressione deve tener conto di questa regolazione genica; pertanto vengono inserite determinate sequenze nucleotidiche in opportune posizioni perché l'inserto di DNA possa esprimersi.
Si hanno vettori per l'espressione genica nei Batteri e vettori per l'espressione genica in cellule eucariote; in quest'ultimo caso i promotori derivano di solito da virus oncogeni. Tali virus sono utilizzati per infettare cellule eucariote sfruttando la loro proprietà di integrarsi nel genoma cellulare direttamente (virus a DNA) o attraverso un passaggio intermedio che prevede la trascrizione da RNA a DNA mediata dalla transcrittasi inversa (virus a RNA). Un tratto del genoma virale non essenziale per la replicazione viene sostituito con il frammento di DNA da clonare; il virus così manipolato è in grado di infettare le cellule e di integrarsi nel loro genoma per venire poi espresso.
Il virus a DNA SV40 della famiglia dei Papovavirus è uno dei più studiati e utilizzati; insieme al virus HTLV-III, della famiglia dei Retrovirus, l'SV40 viene sfruttato anche per la costruzione di vettori di espressione, a causa della presenza nel suo genoma di sequenze regolatrici non specifiche che permettono la regolazione e in alcuni casi l'espressione amplificata di geni posti sotto il loro controllo.
L'SV40 possiede sequenze dette enhancer, costituite da 72 paia di basi, ripetute in tandem (a coppie). Quando tali sequenze vengono donate in un plasmide insieme al gene che codifica per la β-globina di coniglio e il plasmide viene poi introdotto in cellule umane, si assiste a un aumento di RNA messaggero della β-globina, indice di un'aumentata trascrizione del gene corrispondente; il plasmide privato delle sequenze enhancer non produce RNA messaggero per la β-globina. Con lo stesso fine viene utilizzato un gene del virus HTLV-III (gene tat) che è in grado di aumentare l'espressione di geni posti sotto il suo controllo.
d) Screening delle colonie batteriche
Dopo aver ottenuto il frammento di DNA da inserire nel vettore più opportuno e aver trasferito i vettori nelle cellule batteriche rese competenti, è necessario far crescere le colonie batteriche su piastre con terreno di agar incubate a 37 °C e individuare infine quali di queste colonie contengono il segmento di DNA esogeno. Un metodo utilizzato a tale scopo è quello della ibridazione delle colonie o procedura di Grunstein-Hogness. Dopo aver duplicato, per semplice apposizione, le colonie batteriche cresciute in agar su un filtro di nitrocellulosa e averle lisate con alcali, la nitrocellulosa viene posta a essiccare a 80 °C sotto vuoto; questo passaggio permette al DNA, precedentemente reso monoelica dagli alcali, di legarsi covalentemente al supporto solido. Avendo a disposizione una sequenza nucleotidica complementare al segmento di DNA che si intende clonare (sonda genetica a DNA o DNA probe) è possibile rivelare la presenza del segmento di DNA sulla nitrocellulosa; infatti la sonda, opportunamente marcata con sostanze radioattive o non radioattive, si ibrida con la sequenza complementare corrispondente al frammento clonato. L'autoradiografia della nitrocellulosa permette di svelare in quale colonia batterica è presente il segnale e da qui si potrà risalire alla corrispondente colonia batterica della piastra madre.
Una strategia complementare a questa viene utilizzata per evidenziare la produzione di una determinata proteina in colonie batteriche trasformate con vettori di espressione. I passaggi seguiti corrispondono a quelli utilizzati nella metodica con le sonde a DNA, ma in questo caso le colonie contenenti il segmento clonato e la corrispondente proteina, dopo essere state lisate, vengono identificate attraverso l'impiego di anticorpi monoclonali marcati diretti contro la proteina in questione.
e) Sequenziazione del DNA
La manipolazione del DNA non potrebbe avvenire senza il supporto costituito dalla conoscenza della sequenza nucleotidica dell'acido desossiribonucleico.
Le tecniche utilizzate per studiare la composizione nucleotidica del DNA sono quelle sviluppate da Maxam e Gilbert (v., 1977) e da Sanger e altri (v., 1977 e 1980).
La tecnica di Maxam e Gilbert si basa sull'impiego di un metodo chimico di degradazione del DNA. Il campione di DNA viene marcato a un'estremità con fosforo radioattivo e posto a contatto con quattro diversi agenti chimici in quattro reazioni separate, in determinate condizioni di temperatura e di forza ionica. Questi composti chimici interrompono l'elica del DNA in corrispondenza di determinate basi, creando una miscela di segmenti di DNA marcati, la lunghezza dei quali dipende dalla distanza esistente fra l'interruzione operata dall'agente chimico e l'estremità marcata dell'acido nucleico. I segmenti di DNA così ottenuti vengono prelevati e fatti migrare per elettroforesi su gel, dove formano diverse bande a seconda dei rispettivi pesi molecolari: i segmenti più piccoli migrano più velocemente e sono quelli da cui si inizia a leggere la sequenza nucleotidica, mentre nella parte alta del gel si trovano i segmenti a elevato peso molecolare.
Il metodo per sequenziare il DNA sviluppato da Sanger si basa su una reazione enzimatica che prevede l'utilizzo di alcuni nucleotidi trifosfati (2′,3′-didesossinucleotidi trifosfati-ddNTP) in grado di bloccare l'allungamento di una catena del DNA quando aggiunti in posizione 3′. La procedura prevede l'impiego di una catena di DNA che funge da stampo, di una seconda catena (primer) in accrescimento marcata all'estremità 5′ con fosforo radioattivo, dell'enzima DNA-polimerasi I, dei quattro nucleotidi e di un didesossinucleotide per ogni condizione. Il didesossinucleotide deve essere in eccesso rispetto al corrispondente desossinucleotide, in maniera da venire incorporato preferenzialmente. Tutte le volte che viene incorporato un didesossinucleotide la crescita del DNA primer viene arrestata; l'inserimento di uno o dell'altro didesossinucleotide dipende dalla base complementare presente sull'elica di DNA di cui si vuole conoscere la sequenza.
Alla fine della reazione vi saranno frammenti di DNA di diverse dimensioni e pesi molecolari; facendo migrare i quattro diversi campioni su gel di poliacrilammide si otterranno diverse bande e, come nella metodica di Maxam e Gilbert, si potrà leggere la sequenza nucleotidica del DNA in esame partendo dalle bande che sono migrate più velocemente e risalendo a quelle di dimensioni maggiori.
f) Polimorfismi del DNA
Si chiamano ‛alleli' le forme alternative di uno stesso gene. Un gene si definisce polimorfico quando un suo allele non dominante supera la frequenza dell'1% in una determinata popolazione.
Un metodo utile per riconoscere i polimorfismi si basa sul fatto che una differenza fra due alleli può consistere in una differenza di sequenza che non permette più il riconoscimento da parte di un enzima di restrizione. In questi casi il DNA genomico totale o di un determinato cromosoma viene digerito con una serie di enzimi di restrizione; i frammenti così ottenuti vengono separati per elettroforesi e trasferiti su un foglio di nitrocellulosa mediante una tecnica definita blotting.
Utilizzando il DNA estratto da un clone di una libreria genomica, dopo aver individuato la sua posizione sul cromosoma, è possibile impiegarlo per costruire una sonda molecolare opportunamente marcata con fosforo radioattivo.
Una volta ibridata la sonda con i frammenti di DNA immobilizzati sulla nitrocellulosa, l'autoradiografia evidenzierà uno o più segnali; ciò significa che il clone utilizzato come sonda riconosce almeno un frammento di DNA digerito con l'enzima di restrizione di volta in volta impiegato.
Supponiamo che il frammento individuato dalla sonda impiegata sia di 7 kb; la nostra sonda ha riconosciuto almeno una parte di frammento delimitato alle estremità da due siti di restrizione riconosciuti dall'enzima impiegato.
Molte malattie genetiche sono determinate da mutazioni che interessano uno o più geni; è verosimile che la mutazione si ripercuota anche sulla composizione nucleotidica dei siti di restrizione posti in vicinanza del gene mutato. La conseguenza immediata è che l'enzima non riconoscerà più la sequenza palindromica, taglierà il sito di restrizione successivo e quindi la digestione genererà un frammento di dimensioni superiori alle 7 kb. Conoscendo l'ubicazione dei siti di restrizione e disponendo di sonde che riconoscono frammenti di DNA compresi fra tali siti, è possibile diagnosticare le forme di eterozigosi, nonché di omozigosi, di alcune malattie genetiche. In un nucleo familiare con un membro affetto da malattia genetica ereditata con carattere autosomico recessivo la digestione del DNA con un determinato enzima darà luogo a due frammenti della stessa lunghezza, nel caso si tratti di un soggetto omozigote sano, a due frammenti di diversa lunghezza, nel caso il soggetto sia eterozigote per quell'allele, e a due frammenti della stessa lunghezza ma di dimensioni diverse da quelle osservate nel primo caso se il soggetto è omozigote e malato.
5. Ingegneria proteica
Gli enzimi impiegati in processi biotecnologici spesso non sono sufficientemente stabili in varie condizioni operative di pH, temperatura, pressione, in presenza di solventi organici, detergenti, ecc. Pertanto un'esigenza primaria nell'ambito della tecnologia degli enzimi riguarda la loro stabilizzazione, in modo da poter condurre bioconversioni in condizioni non fisiologiche. A questo scopo, negli ultimi anni, sono stati sviluppati vari procedimenti di stabilizzazione di enzimi, tra cui l'immobilizzazione su supporti solidi e la modifica chimica con reagenti bifunzionali.
Il recente sviluppo dell'ingegneria genetica sta avendo un notevole impatto anche nell'ambito della tecnologia degli enzimi, in quanto le tecniche di manipolazione del DNA attualmente disponibili permettono di alterare un gene clonato in vitro mediante sostituzione di una qualsiasi base nucleotidica con un'altra, eliminazione di un segmento del gene o inserimento di un nuovo segmento. Questa tecnica è comunemente detta ‛mutagenesi sito-specifica' (sitedirected mutagenesis) o ‛ingegneria proteica' e di recente è stata usata per modificare la struttura primaria (sequenza degli amminoacidi) di varie proteine, i prodotti dell'espressione genica (v. Breslow, 1982; v. Ulmer, 1983; v. Rastetter, 1983; v. Fersht e altri, 1984 e Binding..., 1986; v. Mutter, 1985; v. Winter e Fersht, 1984). È pertanto possibile variare in modo selettivo e predeterminato uno o più residui di amminoacido in un enzima e utilizzare questa modifica per impartire a esso proprietà di stabilità in varie condizioni operative, e nel contempo variare anche proprietà funzionali dell'enzima come ad esempio proprietà di binding per il substrato e di catalisi (v. tab. III).

Il termine ‛mutagenesi sito-specifica' si applica a qualsiasi tecnica che permetta di modificare in modo selettivo una o più basi del DNA. Sono disponibili varie procedure per ottenere questo scopo, ma quella che implica l'uso di oligonucleotidi sintetici sta trovando le applicazioni più sistematiche e promettenti. La tecnica consiste nella sintesi chimica di un oligonucleotide (12-16 nucleotidi) complementare al gene nelle vicinanze del sito che si desidera mutare, ma che contiene una o più basi variate in funzione del residuo di amminoacido che si vuole introdurre. L'oligonucleotide è appaiato a una copia a catena singola del gene ed è esteso con una DNA-polimerasi in modo da sintetizzare il rimanente della catena complementare del DNA. Si aggiunge quindi la DNA-ligasi in modo da unire le parti terminali del DNA così ottenuto. A questo punto il duplex della molecola del DNA contenente ora il desiderato scambio di basi viene introdotto, ad esempio, in E. coli. La replicazione del batterio comporta l'ottenimento di cloni originali e cloni mutati. Questi ultimi possono essere identificati mediante ibridizzazione del loro DNA con l'oligonucleotide sintetico radioattivo impiegato per il processo di mutazione.
L'impiego della mutagenesi per ottenere enzimi con variate proprietà funzionali e strutturali in verità non è nuovo, in quanto in passato la ricerca di enzimi più adatti per scopi industriali è stata fatta mediante mutazione di microrganismi in grado di produrre enzimi con caratteristiche ottimali per definiti scopi. Un classico esempio è costituito dalla produzione batterica della penicillina, la cui resa di produzione, dovuta all'apparato enzimatico del batterio, è stata migliorata di 10.000 volte negli ultimi 40 anni. Inoltre, la specificità di substrato di enzimi di origine batterica è stata variata anche mediante selezione di mutanti in grado di crescere in presenza di opportuni substrati. Evidentemente la ricerca di mutazioni random di enzimi implica notevole dispendio di tempo e non può essere di certo programmata in modo da alterare punti specifici della catena polipeptidica di un enzima, come mediante l'utilizzo della mutagenesi sito-specifica. A questo proposito, merita anche ricordare che in natura esistono varie famiglie di proteine e di enzimi omologhi (citocromi, emoglobine, deidrogenasi, ecc.), cioè con struttura primaria simile e con solo alcuni amminoacidi variati.
Bisogna ricordare che il processo di mutagenesi sopra descritto implica comunque il clonaggio del gene dell'enzima di interesse e il suo inserimento in un opportuno vettore (plasmide, batteriofago). Questo non costituisce però attualmente un problema, poiché le tecniche di ingegneria genetica hanno raggiunto un tale grado di sofisticazione da permettere la clonazione di qualsiasi gene delle proteine presenti in natura.
Con la disponibilità attuale di sintetizzatori automatici di oligonucleotidi, nonché di tutta una metodologia di clonaggio, le prospettive dell'ingegneria proteica appaiono pertanto eccezionali. Infatti appare possibile modificare sistematicamente importanti proprietà degli enzimi, come specificità di substrato, proprietà catalitiche, effetto del pH sulla catalisi, stabilità in varie condizioni operative, ecc. (v. tab. III). Inoltre sin d'ora è prevedibile la possibilità, sebbene ambiziosa, di disegnare nuove proteine con nuove e inusuali proprietà funzionali e catalitiche (de novo enzyme synthesis).
Sebbene le potenzialità sopra menzionate appaiano concrete, vari elementi limitano la generalità di impiego dell'ingegneria proteica. Queste limitazioni risiedono nella insufficiente conoscenza su basi quantitative delle relazioni fra struttura e attività di enzimi e dei loro meccanismi di azione, per cui non sempre è prevedibile con certezza e quantificabile in termini di proprietà chimico-fisiche e funzionali l'effetto di una modifica della sequenza degli amminoacidi. In tale ambito svolge un ruolo fondamentale la grafia molecolare computerizzata, sviluppata di recente, che permette di visualizzare e manipolare il modello tridimensionale di un enzima di struttura nota (computer-aided protein design: v. Langridge e altri, 1981; v. Blundell e Sternberg, 1985).
Poiché attualmente non è ancora possibile la predizione della struttura tridimensionale di un enzima di cui sia nota la struttura primaria, l'ingegneria proteica può essere utilmente impiegata solo con una proteina o un enzima di cui sia nota la struttura tridimensionale, o almeno la struttura di un corrispondente enzima omologo. In questi casi è possibile predire e quantizzare il binding di un substrato o inibitore all'enzima e le variazioni conformazionali e funzionali che conseguono a una modifica della sequenza degli amminoacidi. Pertanto, le informazioni ottenibili dagli studi di ingegneria proteica porranno su basi quantitative molti problemi di correlazione fra struttura e attività di proteine e nel contempo permetteranno di creare in laboratorio nuove proteine con proprietà funzionali e chimico-fisiche ottimali ai fini di applicazioni industriali.
L'ingegneria proteica, benché di recente introduzione, già conta numerose applicazioni; l'elenco dei mutanti di enzimi e proteine finora ottenuti e studiati è riportato nella tab. IV (v. Villafranca e altri, 1983; v. Mas e altri, 1986; v. Perry e Wetzel, 1984 e 1986; v. Hecht e altri, 1986; v. Shortle e Meeker, 1986). Attualmente lo studio più sistematico di ingegneria proteica è stato condotto sulla tirosil-tRNA-sintetasi (v. Winter e altri, 1982; v. Wilkinson e altri, 1984; v. Fersht e altri, 1985 e Quantitative..., 1986; v. Leatherbarrow e altri, 1985; v. Wells e Fersht, 1985 e 1986). Naturalmente, per le limitazioni sopra accennate, la tecnica è stata applicata soprattutto a enzimi di struttura tridimensionale nota, ottenendo risultati oltremodo incoraggianti e dimostrando chiaramente le eccezionali potenzialità della tecnica.

Le proteine finora studiate (v. tab. IV) sono essenzialmente di natura enzimatica, ma merita ricordare che le tecniche e le metodologie sviluppate sono applicabili per la modifica di qualsiasi proteina, ad esempio proteine di interesse clinico, farmaceutico o veterinario (interferone, ormone della crescita, anticorpi, ecc.).
Su queste basi appare comprensibile l'eccezionale e recentissimo sviluppo degli studi di ingegneria proteica presso laboratori di ricerca sia accademica che industriale, poiché appaiono ben evidenti le enormi potenzialità della tecnica nel modificare importanti proprietà di enzimi anche di interesse industriale. La realizzazione di alcune delle potenzialità sopra menzionate potrà permettere trasformazioni di varie metodologie di processo attualmente in uso, richiedenti alte pressioni e temperature, in processi biotecnologici implicanti l'uso di enzimi che richiedono invece condizioni operative di bassa temperatura e pressione e quindi consumi energetici molto ridotti.
6. Applicazioni pratiche
a) Impiego di anticorpi monoclonali in diagnostica e terapia
La tecnologia degli ibridomi ha senz'altro rivoluzionato le procedure diagnostiche e terapeutiche, che fino a poco tempo fa si avvalevano dell'uso di anticorpi policlonali.
Le caratteristiche intrinseche dell'anticorpo monoclonale (purezza, omogeneità di legame, specificità, disponibilità illimitata) contribuiscono a farne uno dei reagenti più versatili e innovativi e con più largo spettro di applicazioni.
Il settore diagnostico è stato il primo a beneficiarne. Fin dagli anni sessanta erano stati sviluppati molti metodi di analisi basati sulle proprietà di riconoscimento degli anticorpi, che però presentavano alcuni problemi legati all'eterogeneità delle miscele anticorpali impiegate. In altre parole, non si disponeva di un'unica specie molecolare, bensì di una miscela di anticorpi con diverse capacità di legarsi a sostanze da analizzare.
L'anticorpo monoclonale ha consentito di superare questi problemi e ha aperto nuove strade per quanto riguarda lo studio e la comprensione dei meccanismi che stanno alla base della risposta immunitaria.
In campi come l'oncologia si è cominciato a cogliere risultati promettenti proprio quando si sono resi disponibili dei reagenti che hanno permesso di distinguere in modo estremamente specifico le strutture tipiche prodotte da cellule tumorali. Tali sostanze potevano essere utilizzate come indicatori per la diagnosi e il monitoraggio delle neoplasie.
La stessa proprietà di riconoscimento può essere sfruttata impiegando gli anticorpi monoclonali come vettori di farmaci antitumorali a scopi terapeutici. In tal modo gli immunofarmaci giungerebbero sulla cellula bersaglio maligna ed esplicherebbero la loro azione nel modo più efficace e selettivo possibile, riducendo drasticamente il problema della loro elevata tossicità.
Finora sono stati prodotti anticorpi monoclonali contro molecole associate a melanoma, a tumore dell'ovaio, del tratto gastrointestinale, del polmone, del fegato.
Gli anticorpi monoclonali vengono impiegati anche in infettivologia, in sostituzione degli indaginosi metodi di coltura e isolamento utilizzati per l'identificazione di agenti patogeni (virus, batteri, miceti). Tali sistemi presentavano infatti alcuni problemi di sensibilità e soggettività.
La possibilità di riconoscere i vari microrganismi o parti di essi utilizzando anticorpi monoclonali diretti contro strutture peculiari di ogni ceppo ha costituito senz'altro un notevole passo avanti: ha infatti portato alla comprensione dell'epidemiologia di alcune malattie quali l'epatite virale A e B, l'infezione da Herpesvirus di tipo I e da Chlamydia, la mononucleosi.
Gli anticorpi monoclonali sono stati impiegati anche in immunologia, per studiare le varie sottopopolazioni linfocitarie e la cooperazione che si attua durante la risposta immunitaria, grazie al riconoscimento di strutture specifiche di ogni sottopopolazione. In tal modo sono state identificate le cause di alcune patologie a carico del sistema immunitario, come le immunodeficienze ereditarie e acquisite o le malattie autoimmuni, in cui le funzioni di difesa sono rivolte verso il proprio organismo.
Lo stesso discorso si può fare per quanto riguarda l'identificazione di alcuni tipi di leucemia, di linfomi T e B, e per la ricerca di linfociti immaturi in circolo.
Da ultimo, anche se questo settore ha storicamente beneficiato per primo dell'utilizzo degli anticorpi monoclonali, rimane il campo dello sviluppo e della standardizzazione di metodi immunometrici innovativi. In questo senso l'uso di un reagente chimicamente definito e altamente puro come l'anticorpo monoclonale ha consentito di migliorare le prestazioni di metodi di analisi di varie sostanze presenti nei liquidi biologici in concentrazioni minime, come ormoni, farmaci, vitamine, la cui misura è essenziale sia nella diagnosi sia nel monitoraggio degli schemi terapeutici.
Gli anticorpi monoclonali hanno consentito lo sviluppo di metodi di analisi basati sull'utilizzo della reazione antigene-anticorpo quale sistema per modulare la variazione di particolari condizioni chimico-fisiche, come la differenza di potenziale di un campo elettrico oppure le condizioni di rifrangenza della luce. In questo senso sono già in fase di studio sistemi con biosensori che, oltre che in campo diagnostico, potranno essere utilizzati come rilevatori per il controllo dell'inquinamento ambientale e degli alimenti.
b) Impiego di sonde genetiche per la rivelazione di agenti infettivi
La tecnologia del DNA ricombinante ha trovato finora applicazione solo in campo diagnostico, anche se attualmente nuove prospettive si presentano per un suo utilizzo in ambito terapeutico.
Sonde molecolari in diagnostica. - A tutt'oggi è disponibile una cospicua quantità di sonde genetiche che riconoscono sequenze specifiche degli acidi nucleici e che trovano il loro impiego soprattutto in campo infettivologico. Il meccanismo biochimico del riconoscimento della sequenza specifica di acido nucleico da parte della sonda è la complementarietà delle basi azotate. Nel caso particolare, la sonda è costituita da un segmento genomico del microrganismo da studiare ed è quindi in grado di complementare parte del genoma del microrganismo stesso.
L'uso diagnostico di sonde offre un elevato grado di sensibilità e specificità: nelle infezioni virali la sonda può rivelare la presenza del virus integrato nel genoma cellulare anche quando non si ha replicazione del virus ed espressione delle proteine virali. Il grado di specificità ottenibile con le sonde molecolari può essere variato a seconda che si intenda individuare uno specifico microrganismo (elevata specificità) o si intendano studiare microrganismi filogeneticamente correlati (bassa specificità). Il grado di specificità desiderato si ottiene variando le condizioni di ‛stringenza' del mezzo in cui avviene la reazione di ibridazione. Esse dipendono da diversi fattori: temperatura, forza ionica, presenza di formammide. Un'elevata stringenza permetterà solamente alle sequenze strettamente omologhe di appaiarsi, mentre una bassa stringenza darà la possibilità di ibridare anche a quelle non perfettamente complementari.
Sonde molecolari in oncologia clinica e sperimentale. - Dopo che Cooper, Weinberg e Wigler identificarono gli oncogeni del linfoma del pollo e un oncogene di cellule di carcinoma vescicale umano, sempre maggior attenzione è stata posta nello studio di questi geni.
Gli oncogeni sono sequenze nucleotidiche di origine animale che, integrate nel genoma di un virus, lo rendono capace di trasformare alcune linee cellulari, che assumono così il fenotipo neoplastico. La corrispondente sequenza normale viene definita protoncogene: si tratta di un gene che ha la funzione di codificare per importanti fattori e recettori di crescita necessari alla proliferazione e alla differenziazione cellulare.
I meccanismi attraverso i quali un protoncogene si trasforma in oncogene sono: mutazioni, traslocazioni e amplificazioni geniche. A seguito della trasformazione viene persa la capacità di regolazione controllata dall'attivazione delle sequenze geniche. Un esempio di mutazione puntiforme è quello che interessa l'oncogene c-k-ras attivamente trascritto in cellule di carcinoma del colon e del polmone umani. La traslocazione coinvolge invece l'oncogene c-myc, che dal cromosoma 8, sua sede abituale, viene traslocato al cromosoma 14 sotto il controllo del promotore del gene che codifica per le catene pesanti immunoglobuliniche; gli mRNA trascritti sono strutturalmente normali, ma la loro quantità e la loro emivita sono aumentate nelle cellule del linfoma di Burkitt.
Un'importante applicazione clinica delle attuali conoscenze sugli oncogeni è lo studio del neuroblastoma, tumore maligno che colpisce i gangli nervosi: in questo caso l'oncogene N-myc è presente in un gran numero di copie nelle cellule maligne come segmento genico ripetuto lungo il cromosoma o in frammenti extracromosomici. Studi clinici hanno dimostrato la correlazione positiva fra stadio del tumore e amplificazione dell'oncogene: le fasi più avanzate della neoplasia sono caratterizzate da un numero di copie di oncogeni sempre crescente. Avendo a disposizione la sequenza nucleotidica dell'oncogene, è possibile costruire una sonda molecolare corrispondente e, attraverso le tecniche di ibridazione degli acidi nucleici, utilizzarla per rivelare la presenza di oncogeni attivati nelle linee cellulari tumorali. Gli sviluppi di queste tecnologie troveranno un'applicazione diagnostica sempre maggiore in oncologia sperimentale e clinica. Le tecniche del DNA ricombinante potrebbero rendere possibile la conoscenza e il controllo dell'espressione dei geni responsabili della trasformazione neoplastica.
Sonde molecolari per la diagnosi di malattie genetiche. - La diagnosi prenatale di malattie genetiche si avvale ora anche dell'uso di sonde molecolari. Fino a poco tempo fa potevano venir rilevate solo le mutazioni genetiche che interessavano il fenotipo cellulare; l'impiego di sonde molecolari specifiche del genoma umano consente invece l'analisi e l'individuazione di mutazioni genetiche anche quando queste ultime non danno luogo a prodotti genici modificati. Questa peculiarità è utile nella rivelazione di alterazioni del patrimonio cromosomico che, nel caso di patologie di tipo autosomico recessivo, non danno luogo a malattia clinicamente manifesta.
Le strategie attuate nella diagnosi genetica di malattie ereditarie mediante l'uso di tecniche molecolari dipendono dalle conoscenze della struttura e della localizzazione cr0mosomica del gene responsabile della malattia. Nel caso sia conosciuta la sequenza nucleotidica del gene normale, l'analisi del gene alterato permetterà di svelare direttamente la localizzazione e il tipo di mutazione presente. In questo caso non è possibile la complementazione fra la sonda, costituita dal segmento normale, e il gene alterato, a causa della presenza di delezioni, sostituzioni o inserzioni in un sito riconosciuto da un enzima di restrizione non più in grado di tagliare il DNA.
Un diverso tipo di approccio alla rivelazione di mutazioni di una singola base è quello che utilizza sonde di oligonucleotidi sintetici: la sonda sintetica, appositamente costruita, è in grado di ibridare con il gene patologico e non con il gene normale. Tale metodica viene utilizzata soprattutto nella diagnosi delle emoglobinopatie e in particolare delle talassemie. Quando non è noto il gene responsabile della malattia (per cui non è possibile stabilirne la posizione cromosomica) si sfrutta il linkage del gene in questione con siti di restrizione limitrofi; è così che vengono studiate due gravissime malattie: la fibrosi cistica e la malattia di Duchenne. La stretta vicinanza fra il sito di restrizione considerato e il presunto gene patologico rende improbabile una diversa segregazione dei due segmenti durante il crossing-over. Affinché l'informazione sia utile il sito di restrizione e il gene non devono distare più di 10 kb. In questo caso è possibile diagnosticare un difetto genetico pur non avendo informazioni dettagliate sul gene alterato. Anche se meno attendibile dei metodi prima descritti, questa metodica è attualmente la più utilizzata e viene denominata RFLP (restriction fragment length polymorphism; v. genetica, suppl., cap. 4, È a).
Terapia genica. - Per ‛terapia genica' si intende la manipolazione di materiale genetico al fine di introdurre nelle cellule geni che correggano le alterazioni geniche dovute a fenomeni di delezione o alterazione genica. Le affezioni per le quali viene attualmente contemplata l'ipotesi di tale terapia sono gravissime e quasi sempre letali. Ne sono esempi la sindrome di Lesh-Nyhan, caratterizzata da impulsi incontrollabili di automutilazione (dita delle mani e braccia), ritardo mentale, disordini del metabolismo dell'acido urico, e i deficit di adenosindeaminasi (ADA) e di nucleosidefosforilasi (PNP), che evidenziano una grave alterazione del sistema immunitario.
I geni responsabili di tali malattie sono stati a lungo studiati e attualmente è possibile inserire le loro varianti normali in virus a RNA (Retrovirus) opportunamente deleti in sequenze genomiche non necessarie all'integrazione nel genoma umano; i Retrovirus, infatti, hanno la proprietà di inserirsi stabilmente nel genoma delle cellule infettate.
Un prelievo di midollo osseo costituisce il primo passaggio della terapia genica; le cellule emopoietiche vengono infettate con il retrovirus manipolato geneticamente e contenente nel proprio genoma il gene normale. Il midollo osseo viene quindi reinfuso per via endovenosa al ricevente. Finora, tuttavia, la terapia genica è stata eseguita con alterna fortuna nei primati infenori.
I problemi che rimangono da risolvere sono la costruzione di opportuni vettori virali, la loro capacità infettante, la loro integrazione nel genoma e la percentuale di cellule integranti il virus. Anche la regolazione della trascrizione del gene inserito è un fattore critico, come pure lo è la possibilità di correggere il difetto a livello dell'organo bersaglio della malattia.
In questa prima fase delle ricerche la manipolazione genetica ha riguardato solamente le cellule somatiche; si sta ora dibattendo l'eventualità di intervenire anche su cellule germinali: in questo caso l'introduzione di materiale genetico verrebbe acquisita irreversibilmente dalla progenie, ponendo così dei problemi etici non trascurabili.
Appare chiaro che la terapia genica, quando verrà applicata, trasformerà radicalmente il decorso di alcune malattie genetiche, con la possibilità teorica di debellarle definitivamente.
c) Settore agroalimentare
Uno degli obiettivi che l'uomo si è sempre posto nella produzione di beni agroalimentari è rappresentato dall'incremento nella resa delle colture. Tale incremento riflette una maggiore fissazione di energia da parte delle piante; questa è a sua volta regolata da numerosi fattori interdipendenti, per cui qualunque strategia innovativa in campo agroalimentare deve prevedere obiettivi strategici articolati. Le principali fasi di intervento sono: a) fertilizzazione; b) irrigazione e drenaggio; c) sviluppo di bioinsetticidi; d) miglioramento del substrato genetico mediante incroci; e) riduzione delle perdite.
Fertilizzazione. - La fertilizzazione è il processo per cui si forniscono alle colture le sostanze nutritive di crescita. La principale categoria di fertilizzanti è costituita da sostanze azotate. Si stima che attualmente il consumo mondiale di azoto sia di circa 50 milioni di tonnellate, ma nel 2000 tale valore sarà almeno triplicato. Il consumo energetico per la produzione di questi fertilizzanti è elevato: per la fissazione di un kg di azoto è necessario infatti un metro cubo di gas naturale. Pertanto la richiesta di energia è pari a circa l'uno per cento del consumo mondiale.
Per rendere meno dispendioso il processo si è prospettato l'utilizzo di microrganismi in grado di fissare l'azoto atmosferico: molti di essi (Klebsiella, Azotobacter, Clostridium) vivono liberi nel terreno, altri (Rhizobium, Actinomiceti) vivono in simbiosi con le piante e particolarmente con le Leguminose.
Irrigazione e drenaggio. - È noto che alcune zone del nostro pianeta (Nordafrica, Asia Minore) non dispongono di sufficienti scorte idriche per la scarsità delle precipitazioni. Lo sviluppo biotecnologico di impianti di irrigazione e drenaggio (per ridurre l'acidità del terreno) è un obiettivo pratico e necessario per aumentare l'efficienza della fotosintesi e, in ultima analisi, la produzione agroalimentare.
Sviluppo di bioinsetticidi. - La lotta contro i parassiti vegetali e animali consentirà di migliorare la resa produttiva sia in agricoltura che in veterinaria. Lo sviluppo delle biotecnologie ha permesso (in parte) la produzione di grandi quantità di sostanze (antibiotici, insetticidi) fornite da microrganismi e l'individuazione e la selezione di batteri, virus e funghi patogeni per i parassiti.
Gli antibiotici insetticidi presentano lo svantaggio di essere troppo costosi e tossici per l'uomo: il loro impiego è stato quindi finora scarso. Recentemente, tuttavia, sono state prodotte due classi di sostanze poco tossiche e facilmente biodegradabili: le nikkomicine e le avermectine.
Fra i microrganismi patogeni per i parassiti Bacillus thuringiensis presenta una notevole efficacia e resistenza agli agenti atmosferici.
Miglioramento del substrato genetico mediante incroci. - L'efficienza della produttività agricola può essere perseguita mediante incroci genetici al fine di ottenere ceppi più resistenti alle variazioni di temperatura, allo stress idrico e alle malattie. È anche possibile ottimizzare il contenuto di componenti nobili, ad esempio aumentare il contenuto proteico del grano, o coltivare nuove piante in zone relativamente poco fertili.
Infine, mediante le tecniche di transfezione, si potranno introdurre nuove caratteristiche genetiche o potenziare quelle esistenti in ceppi già coltivati. Un esempio in tal senso è rappresentato dal plasmide Ti di Agrobacter tumefaciens che, opportunamente manipolato, conferisce alle cellule di tabacco la resistenza a un antibiotico; di per sé non è un fatto di grande rilevanza, ma lascia intravedere nuove prospettive applicative.
Le stesse tecniche di transfezione genica troveranno ampia applicazione per introdurre caratteristiche desiderate in animali da allevamento. Particolarmente importante e interessante è in questo senso la possibilità di inserire nuovi geni nelle cellule germinali, dando così origine a specie modificate.
Riduzione delle perdite. - Uno dei campi in cui le innovazioni biotecnologiche porteranno maggiori risultati è quello della riduzione delle perdite, a cui contribuiranno certamente sia la selezione di piante ricche in componenti nobili sia il miglioramento delle tecniche di degradazione e riutilizzo di residui, come la cellulosa.
Un altro campo suscettibile di ampie trasformazioni è quello della manipolazione e dell'impiego delle biomasse mediante l'individuazione di processi fermentativi e l'utilizzo di tecniche di sostituzione e conversione dei prodotti.
Nel settore zootecnico e veterinario le biotecnologie offrono importanti punti applicativi volti al miglioramento della produttività. In particolare, con le tecniche del DNA ricombinante sono stati già realizzati alcuni vaccini di interesse veterinario. Le ricerche in tal senso certamente approderanno a imponenti risultati nei prossimi anni, specie per le malattie da Herpesvirus, afta epizootica, ecc. Sono stati inoltre prodotti ormoni (come quello della crescita) che accrescono la produttività e sono stati sviluppati sistemi diagnostici che impiegano sonde molecolari o anticorpi monoclonali più sensibili e specifici di quelli usati precedentemente.
d) Settore energetico ecologico
Uno dei problemi più urgenti che si pongono in questi anni è rappresentato dalla necessità di reperire fonti energetiche sufficienti alle attività economico-produttive su scala mondiale, unitamente all'esigenza di ridurre il tasso di inquinamento derivante dalla produzione di sottoprodotti e materiali di scarico non biodegradabili nel corso delle trasformazioni industriali.
Anche se l'utilizzo del carbone risale a 4.000 anni fa, esso si è affermato solo negli ultimi 200 anni; da 100 anni viene utilizzato il petrolio e si fa uso dell'elettricità; l'impiego di gas naturale risale invece agli ultimi 50 anni e quello dell'energia nucleare a 30 anni fa. L'uso di energia di derivazione fossile (carbone, petrolio, gas naturale) è quindi relativamente recente, ma è prevedibile che in un prossimo futuro le risorse saranno comunque esaurite. D'altro canto sono ben noti i rischi inerenti all'impiego di energia nucleare.
Le biotecnologie offrono numerose possibilità di riutilizzo e riconversione di sostanze biologiche a scopo energetico, salvaguardando così l'integrità dell'ambiente.
In funzione del tipo di piante, è possibile selezionare tecniche di combustione e di conversione in gas o bioetanolo. Il bioetanolo è un liquido miscelabile con benzina che viene prodotto per fermentazione con un'efficienza energetica del 75%, il che significa che l'etanolo contiene il 75% dell'energia presente nella cellulosa, in zuccheri non degradabili e nei rifiuti biologici.
Poiché la combustione delle biomasse è in grado di fornire energia termica, sono stati proposti altri processi di conversione in grado di generare forme diverse di energia: l'idrolisi delle biomasse, in particolare della cellulosa e dell'emicellulosa, genera carboidrati del tipo degli esosi e dei pentosi, che possono essere utilizzati come tali o ulteriormente processati per generare etanolo, acetone, acido acetico, ecc. Questi materiali sono utilizzabili come carburanti per motori a combustione interna e possono sostituire prodotti simili derivanti dal petrolio. L'idrolisi delle biomasse può essere effettuata in soluzione acquosa a temperature di 240 °C e con l'aggiunta di acidi, o a temperature fra i 30-40 °C sotto l'effetto di fermenti prodotti da microrganismi (funghi e batteri). L'idrolisi enzimatica consuma meno energia, ma è altamente sensibile a sostanze tossiche presenti nel substrato; d'altro canto la purificazione e la produzione di enzimi su scala industriale non sono economicamente vantaggiose.
Un altro processo di trasformazione biologica è la fermentazione anaerobica delle biomasse, originariamente sviluppata per la trasformazione dei rifiuti biologici animali e urbani. In tale processo il materiale organico viene convertito in gas rappresentato per 2/3 da metano e per il resto essenzialmente da diossido di carbonio. Tale tecnologia presenta due importanti vantaggi: a) è possibile processare senza difficoltà materiali altamente diluiti, in quanto il gas si separa facilmente dal contenuto liquido rimanente; b) l'azoto rimane concentrato dopo la fermentazione nel materiale liquido, mantenendo così il suo potere fertilizzante. Queste tecniche consentono quindi la produzione di energia a partire dai rifiuti (generazione dei cosiddetti biogas), offrono importanti vantaggi ecologici e, nel contempo, consentono il riutilizzo delle sostanze fertilizzanti a favore della produttività agroalimentare.
La produzione di bioetanolo può essere accresciuta mediante lo sviluppo di monocolture idonee (che peraltro non sono vantaggiose in termini di produttività alimentare) o mediante processi integrati che si basano sull'impiego di materiali di rifiuto diversi (cellulosa, carboidrati, rifiuti biologici), degradati attraverso tecniche di fermentazione innovative. I vantaggi sono numerosi: a) preservazione ecologica dell'ambiente; b) basso consumo di energia per la preservazione di fibre vegetali; c) basso consumo energetico nelle tecniche di produzione del bioetanolo.
Relativamente a forme di energia più tradizionali, lo sviluppo delle biotecnologie ha reso ipotizzabile il miglioramento dell'estrazione del petrolio dai pozzi mediante l'impiego di agenti surfactanti di origine molecolare o di microrganismi opportunamente modificati con tecniche di ingegneria genetica.
Un altro capitolo interessante è rappresentato dal recupero di minerali con l'impiego di microrganismi e proteine (metallotioneine), atti a concentrarli da soluzioni acquose o capaci di esercitare la biolisciviazione dei minerali. A tali processi contribuisce soprattutto l'industria chimica, che per tradizione utilizza tecnologie (separazione, catalisi, purificazione) facilmente trasferibili in ambito biotecnologico.
Più strettamente inerente a tematiche di preservazione dell'ambiente è il problema della manipolazione dei rifiuti, reso più drammatico dalla formazione di sottoprodotti non biodegradabili nonché di prodotti finali (pur biodegradabili) in quantità tali da non poter essere smaltiti dai microrganismi dell'acqua e del suolo.
Le biotecnologie hanno un ruolo primario nella biodegradazione degli inquinanti delle acque di scarico; essa può realizzarsi mediante procedimenti aerobici impiegando impianti a filtrazione e a fanghi attivi. Peraltro il trattamento aerobico delle acque reflue presenta due svantaggi: genera elevate quantità di fanghi (non facilmente utilizzabili come tali perché spesso patogeni) e comporta un consumo di energia elevato. Un'alternativa è rappresentata dal trattamento anaerobico, che produce minori quantità di fanghi e, anziché consumarla, produce energia (sotto forma di biogas); i costi d'impianto sono tuttavia maggiori, e così pure i volumi di lavorazione. È presumibile comunque che attraverso opportune manipolazioni genetiche sia possibile aumentare la resa della degradazione aerobica e anaerobica esercitata dai microrganismi.
e) Settore chimico farmaceutico
La tecnica del DNA ricombinante e la produzione degli anticorpi monoclonali ha già influenzato in modo determinante le industrie farmaceutica e chimica. In particolare l'ingegneria genetica offre all'industria farmaceutica importanti vantaggi quali: a) la possibilità di produrre numerose sostanze biologiche in quantità sufficienti per verificarne l'efficacia clinica (per es. l'interleuchina 2 per il trattamento di neoplasie); b) costi di produzione assai minori rispetto alle tecniche di estrazione da tessuti; c) la possibilità di modificare e migliorare farmaci già in uso (antibiotici, chemioterapici); d) la produzione su larga scala di farmaci e vaccini specifici per diverse malattie; e) l'ottenimento di un prodotto altamente purificato e privo di contaminanti allergenici.
In base a tali presupposti, numerose industrie hanno reso disponibili prodotti ‛ricombinanti' in alternativa agli omologhi purificati o derivati da altre specie: è questo il caso dell'insulina umana e dell'ormone della crescita. Un capitolo particolarmente importante è quello attinente lo sviluppo di nuovi farmaci antineoplastici con proprietà immunomodulanti: è già disponibile l'alfa-interferone ricombinante ed è verosimile che in un prossimo futuro saranno largamente impiegati il gamma-interferone, l'interleuchina 2 e il fattore di necrosi tumorale (tumor necrosis factor). Un altro gruppo di prodotti è rappresentato dalle proteine della coagulazione (fattore VIII nella terapia dell'emofilia) e dagli anticoagulanti (TPA, attivatore tessutale del plasminogeno per la lisi dei trombi); tra le sostanze non proteiche vi sono gli ormoni steroidei, alcune vitamine (E, B12) e antibiotici (penicilline, tetracicline, cefalosporine).
La tecnologia del DNA ricombinante ha consentito inoltre lo sviluppo di nuovi vaccini specialmente per malattie virali (epatite B) o parassitarie (malaria, schistosomiasi): il vantaggio in questo caso è rappresentato dalla purezza e dalla sicurezza del prodotto rispetto ai vaccini tradizionali.
Anche la tecnologia degli ibridomi ha indotto una notevole trasformazione nell'industria farmaceutica. Le applicazioni più importanti al riguardo sono costituite dallo sviluppo di diagnostici in vitro, dalla produzione di traccianti per tecniche di radiolocalizzazione in vivo, dalla preparazione di immunotossine per terapia. L'impatto maggiore è stato riscontrato nello sviluppo di diagnostici utilizzabili per la diagnosi di malattie infettive e tumorali.
Anche l'ingegneria proteica, realizzando la modificazione strutturale e funzionale delle proteine, è suscettibile di applicazioni in campo farmaceutico. È possibile, infatti, attraverso tecniche di mutagenesi, attuate mediante la manipolazione del DNA, modificare la struttura primaria delle proteine; si possono ottenere in tal modo enzimi modificati, la cui attività risulta amplificata, e proteine di interesse farmacologico con maggiore attività biologica. Nel settore chimico le biotecnologie sono impiegate sia nella produzione di composti ad alto valore aggiunto (biopolimeri, biosurfactanti, enzimi, biocoloranti), sia nella trasformazione di biomasse.
bibliografia
Andersson, J., Melchers, F., The antibody repertoire of hydrib cell lines obtained by fusion of X63-Ag8 myeloma cells with mitogen activated B-cell blast, in ‟Current topics in microbiology and immunology", 1978, LXXXI, pp. 130-139.
Ault, K. A., Unanue, E. R., Events after the binding of antigen to lymphocytes removal and regeneration of the antigen receptor, in ‟Journal of experimental medicine", 1974, CXXXIX, pp. 1110-1112.
Barald, K. F., Wessells, N. K., Differential antigen adhesivity to select spleen cells for the production of monoclonal antibodies to embryonic neurons, in ‟Journal of immunological methods", 1984, LXXIII, pp. 1-15.
Bargellesi, A., Class switch of hybridomas as additional tools in monoclonal technology, in Biotech RIA 85, International Symposium on ‛The impact of biotechnology on diagnosis', Abs. 3, Roma 1985.
Bazin, H., Transplantable immunoglobulin-secreting tumors in rats. V. Monoclonal immunoglobulins secreted by 250 ileocecal immunocytomas in lou/WsI rats, in ‟Journal of the National Cancer Institute", 1973, LI, pp. 1359-1361.
Belin, M., Houillon, G., Vaccination antiaphteuse avec les complex vaccino-aphteuse stérilisés, in ‟Compte rendu des séances de la Société de Biologie", 1928, IC, pp. 1194-1196.
Benz, E. J. Jr., Clinical management of gene expression, in ‟New England journal of medicine", 1982, CCCVII, pp. 1515-1516.
Berman, P. W., Gregory, T., Crase, D., Laskey, L. A., Protection from genital Herpes simplex virus type 2 infection by vaccination with cloned type 1 glycoprotein D, in ‟Science", 1985, CCXXVII.
Bishop, J. M., Oncogenes, in ‟Scientific American", 1982, CCXLVI, pp. 80-82.
Blundell, T., Sternberg, M. J. E., Computer-aided design in protein engineering, in ‟Trends in biotechnology", 1985, III, pp. 229-235.
Borrebaek, C. A. K., In vitro immunization for the production of antigen specific lymphocyte hybridomas, in ‟Scandinavian journal of immunology", 1983, XVIII, 1, pp. 9-12.
Boulianne, G. L., Hozumi, N., Shulman, M. J., Production of functional chimaeric mouse-human antibody, in ‟Nature", 1984, CCCXII, pp. 643-646.
Boyer, P. D., Lardy, H., Myrbäck, K., The enzymes, New York 19703.
Breatnach, R., Chambon, P., Organization and expression of eucaryotic split genes coding for proteins, in ‟Annual review of biochemistry", 1981, L, pp. 349-383.
Breslow, R., Artificial enzymes, in ‟Science", 1982, CCXVIII, pp. 532-537.
Bryant, J. C., Shiling, E. L., Earle, W. L., Massive fluid suspension cultures of certain mammalian tissue cells, in ‟Journal of the National Cancer Institute", 1958, XXI, p. 331.
Buchman, D., Miller, D., Kock, G., Monoclonal antibodies to digoxin: comparison of in vitro and in vivo immunization, in ‟Hybridoma", 1985, IV, pp. 173-177.
Campbell, A. M., Monoclonal antibody technology, in Laboratory techniques in biochemistry and molecular biology (a cura di R. H. Burdon e P. H. von Knippenberg), vol. XIII, Amsterdam 1984, pp. 86-100.
Caskey, C. T., White, R. L., Recombinant DNA. Applications to human disease, Banburry report n. 14, Cold Spring Harbor, N. Y., 1983.
Catt, K. J., Treager, G., Solid-phase radioimmunoassay in antibodycoated tubes, in ‟Science", 1967, CLVIII, pp. 1570-1572.
Ceccarini, C., Eagle, H., pH as determinant of cellular growth and contact inhibition, in ‟Proceedings of the National Academy of Sciences", 1971, LXVIII, p. 229.
Chang, A. C. Y., Nunberg, J. H., Kaufman, R. J., Erlich, H. A., Schimke, R. T., Cohen, S. N., Phenotipic expression in E. coli of a DNA sequence coding for mouse dihydrofolate reductase, in ‟Nature", 1978, CCLXXV, pp. 617-624.
Cline, M. J., Stang, H., Mercola, K., Morse, L., Ruprecht, R., Browne, T., Salser, W., Gene transfer in intact animals, in ‟Nature", 1980, CCLXXXIV, pp. 422-425.
Coffino, P., Baumal, R., Laskov, I., Scharff, M. D., Cloning of mouse myeloma cells and detection of rare variants, in ‟Journal of cell physiology", 1972, LXXIX, pp. 429-439.
Crick, F. H. C., Split genes and RNA splicing in evolution of eucaryotic cells, in ‟Science", 1979, CCIV, pp. 264-271.
Davis, W. C., Yilma, T., Perryman, L. E., Mc Guire, T. C., Prospectives on the application of monoclonal antibody and transfection technology in veterinary microbiology, in Progress in veterinary microbiology and immunology, vol. I, Infection and immunity in farm animals, Basel 1985, pp. 1-24.
De Boer, H. A., Comstock, L. J., Vasser, M., The Tac promoter: a functional hybrid derived from the trp and lac promoters, in ‟Proceedings of the National Academy of Sciences", 1983, LXXX, pp. 21-25.
Demain, A. L., Industrial microbiology, in ‟Science", 1981, CCXIV, pp. 987-995.
Demain, A. L., New application of microbial products, in ‟Science", 1983, CCXIX, pp. 709-714.
Der, C. J., Krontiris, T. G., Cooper, G. M., Transforming genes of human bladder and lung carcinoma cell lines are homologous to the ras genes of Harvey and Kirsten sarcoma viruses, in ‟Proceedings of the National Academy of Sciences", 1982, LXXIX, pp. 3637-3640.
Doelle, H. W., Basic metabolic processes, in Biotechnology (a cura di H. J. Rehm e G. Reed), vol. I, Weinheim 1981, pp. 89-97.
Engvall, E., Perlmann, P., Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulins G, in ‟Immunochemistry", 1971, VIII, pp. 871-874.
Feder, J., Tolbert, W. R., The large scale cultivation of mammalian cells, in ‟Scientific American", 1983, CCXLVIII, pp. 24-31.
Fersht, A. R., Leatherbarrow, R. J., Wells, T. N. C., Binding energy and catalysis: a lesson from protein engineering of the tyrosyl-tRNA synthetase, in ‟Trends in biological sciences", 1986, XI, pp. 321-325.
Fersht, A. R., Leatherbarrow, R. J., Wells, T. N. C., Quantitative analysis of scructure-activity relationships in engineered proteins by linear free-energy relationships, in ‟Nature", 1986, CCCXXII, pp. 284-286.
Fersht, A. R., Shi, J. P., Knill-Jones, J., Lowe, D. M., Wilkinson, A. J., Blow, D. M., Brick, P., Carter, P., Waye, M. M. Y., Winter, G. P., Hydrogen bonding and biological specificity analysed by protein engineering, in ‟Nature", 1985, CCCXIV, pp. 235-238.
Fersht, A. R., Shi, J. P., Wilkinson, A. J., Blow, D. M., Carter, P., Waye, M. M. Y., Winter, G. P., Analysis of enzyme structure and activity by protein engineering, in ‟Angewandte Chemie" (international English edition), 1984, XXIII, pp. 467-473.
Finter, N. B., Fantes, K. H., The purity and safety of interferons prepared for clinical use: the care for lymphoblastoid interferon, in Interferon (a cura di I. Gresser), New York 1980, pp. 65-75.
Galfré, G., Howe, S. C., Milstein, C., Butcher, G. W., Haward, J. C., Antibodies to major histocompatibility antigens produced by hybrid cell lines, in ‟Nature", 1977, CCLXVI, pp. 550-552.
Galfré, G., Milstein, C., Wright, B., Rat × rat hybrid myelomas and a monoclonal anti-Fd portion of mouse IgG, in ‟Nature", 1979, CCLXXVII, pp. 131-133.
Gamper, H. B., Size of the unwond region of DNA in E. coli RNA polymerase and calf thymus RNA polymerase II ternary complex, in ‟Cold Spring Harbor symposia on quantitative biology", 1983, XLVII, pp. 447-453.
Gilbert, S. F., Migeon, B. R., D-Valine as a selective agent for normal human and rodent epithelial cells in culture, in ‟Cell", 1975, V, pp. 11-17.
Gilbert, W., Why genes in pieces?, in ‟Nature", 1978, CCLXXI, pp. 501-510.
Gilbert, W., Villa-Komaroff, L., Useful proteins from recombinant bacteria, in ‟Scientific American", 1980, CCXLII, pp. 68-82.
Goding, J. W., Conjugation of antibodies with fluorochromes: modification to the standard method, in ‟Journal of immunological methods", 1976, XIII, p. 215.
Griffiths, J. B., The production and properties of a tissue plasminogen activator from normal epithelial cells grown in microcarrier culture, in ‟Developments in biological standardization", 1985, LX, pp. 439-446.
Grunstein, M., Hoguess, D. S., Colony hybridization: a method for the isolation of cloned DNAs that contain a specific gene, in ‟Proceedings of the National Academy of Sciences", 1975, LXXII, pp. 3961-3965.
Guarantel, L., Laner, G., Roberts, T., Ptshane, M., Improved methods of maximizing expression of a cloned gene: a bacterium that synthesizes rabbit-globin, in ‟Cell", 1980, XX, pp. 543-553.
Guesdon, J. L., Ternynck, T., Avrameas, S., The use of avidin-biotin interaction in immunoenzymatic techniques, in ‟Journal of histochemistry and cytochemistry", 1979, XXVII, pp. 1131-1139.
Handman, E., Remington, S., Serological and immunochemical characterization of monoclonal antibodies to toxoplasma Gondii, in ‟Immunology", 1980, XL, pp. 579-588.
Hecht, M. H., Sturtevant, J. M., Sauer, R. T., Stabilization of repressor against thermal denaturation by site-directed Gly→Ala changes in α-helix 3, in ‟Proteins. Structure, function and genetics", 1986, I, pp. 43-46.
Helfman, D. M., Feramisco, J. R., Fiddes, J. C., Thomas, G. P., Hughes, S. H., Identification of clones that encode chicken tropomyosin by directed immunological screening of a cDNA expression library, in ‟Proceedings of the National Academy of Sciences", 1983, LXXX, pp. 31-35.
Herbert, W. J., Mineral-oil adjuvants and immunization of laboratory animals, in Handbook of expermental immunology (a cura di D. M. Weir), 3 voll., Oxford 1978.
Herzenberg, L. A., New approaches to hybridoma specificity and function, in Biotech RIA 85, International Symposium on ‛The impact of biotechnology on diagnosis', Abs. 2, Roma 1985.
Herzenberg, L. A., Herzenberg, L. A., Analysis and separation using the fluorescence activated cell sorter (FACS), in Handbook of experimental immunology (a cura di D. M. Weir), 3 voll., Oxford 1978, pp. 121-123.
Herzenberg, L. A., Sweet, R. G., Herzenberg, L.A., A new tool for isolating functional cell types, in ‟Scientific American", 1976, CCXXXIV, p. 108.
Hirashima, M., Regulatory role of IgE binding factors from rat T lymphocytes. V. Formation of IgE-potentiating factor by T lymphocytes from rats treated with Bordetella pertussis vaccine, in ‟Journal of immunology", 1981, CXXVI, pp. 838-842.
Hohn, B., Collins, J., A small cosmid for efficient cloning of large DNA fragments, in ‟Gene", 1980, XI, pp. 291-298.
Ilfeld, D. N., Cathcart, H. K., Krakamer, R. S., Blaese, M., Human splenic and peripheral blood lymphocyte response to lipopolysaccharide, in ‟Cellular immunology", 1981, XV, pp. 400-407.
Jonak, Z. L., Braman, V., Kennet, R. H., Transfection of primary lymphocytes with human tumor DNA: production of continuous cell lines producing monoclonal antibodies, in ‟Hybridoma", 1983, II, pp. 124-128.
Jonak, Z. L., Braman, V., Kennet, R. H., Production of continuous mouse plasma cell lines by transfection with human leukemia DNA, in ‟Hybridoma", 1984, III, pp. 107-118.
Kan, Y. W., Hemoglobin abnormalities: moleculary and evolutionary studies, in ‟Harvey lectures", 1982, LXXVI, pp. 75-93.
Katinger, H. W. D., Scheirer, W., Kromer, E., Bubble column reactor for mass propagation of animal cells in suspension culture, in ‟German chemical engineering", 1979, II, pp. 31-38.
Kearney, J. F., Radbruch, A., Liesegang, B., Rajewsky, K., A new mouse myeloma cell line that has lost immunoglobulin expression but permits the construction of antibody-secreting hybrid cell lines, in ‟Journal of immunology", 1979, CXXIII, pp. 1548-1550.
Klinman, N. R., The mechanism of antigenic stimulation of primary and secondary clonal precursor cells, in ‟Journal of experimental medicine", 1972, CXXXVI, pp. 241-260.
Köhler, G., Howe, C. S., Milstein, C., Derivation of specific antibody-producing tissue culture and tumor lines by cell fusion, in ‟European journal of immunlogy", 1976, VI, pp. 292-299.
Köhler, G., Milstein, C., Continuous cultures of fused cells secreting antibody of predefined specificity, in ‟Nature", 1975, CCLVI, pp. 495-497.
Konarska, M., Filipowicz, W., Gross, H. J., RNA ligation via 2′-phosphomoester, 3′-5′-phosphodiester linkage: requirement of 2′-3′-cyclic-phosphate termini and involvement of a 5′-hydroxyl polinucleotide kinase, in ‟Proceedings of the National Academy of Sciences", 1982, LXXIX, pp. 1474-1478.
Konecki, D. S., Brennand, J., Fuscoe, J. C., Caskey, C. T., Chinault, A. C., Hypoxantine-guanine phosphoribosyltransferase genes of mouse and chinese hamster: construction and sequence analysis of cDNA recombinants, in ‟Nucleic acids research", 1982, X, pp. 6763-6775.
Kornberg, A., 1982 supplement to DNA replication, San Francisco 1982.
Koskimies, S., Human lymphoblastoid cell lines producing specific antibody against Rh-antigen D, in ‟Scandinavian journal of immunology", 1979, XII, pp. 355-358.
Kozbor, D., Roder, J. C., Requirements for the establishment of high-titered human monoclonal antibodies against tetanus toxoid using the Epstein-Barr virus technique, in ‟Journal of immunology", 1981, CXXVII, pp. 1275-1280.
Kozbor, D., Roder, J. C., The production of monoclonal antibodies from human lymphocytes, in ‟Immunology today", 1983, IV, pp. 72-79.
Krause, R. M., The search for antibodies with molecular uniformity, in ‟Advances in immunology", 1970, XII, pp. 1-56.
Kruger, K., Grabowski, P. J., Zang, A. J., Sands, J., Gottschling, D. E., Cech, T. R., Self splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena, in ‟Cell", 1982, XXXI, pp. 147-157.
Kurtz, D. T., Nicodemus, C. F., Cloning of alpha 2v globulin cDNA using a high efficiency technique for the cloning of trace messenger RNAs, in ‟Gene", 1981, XIII, pp. 145-152.
Lafferty, R. M. (a cura di), Enzyme technology, Berlin 1982.
Land, H., Schutz, G., Schmale, H., Richter, D., Nucleotide sequence of cloned cDNA encoding bovine arginine vasopressin-neurophysin II precursor, in ‟Nature", 1982, CCXCV, pp. 299-303.
Langridge, R., Ferrin, T. E., Kuntz, I. D., Connolly, M. L., Real-time color graphics in studies of molecular interactions, in ‟Science", 1981, CCXI, pp. 661-666.
Leatherbarrow, R. J., Fersht, A. R., Winter, G., Transition-state stabilization in the mechanism of tyrosyl-tRNA synthetase revealed by protein engineering, in ‟Proceedings of the National Academy of Sciences", 1985, LXXXII, pp. 7840-7844.
Ledbetter, J. A., Herzenberg, L. A., Xenogenic monoclonal antibodies to mouse lymphoid differentiation antigens, in ‟Immunological review", 1976, XLVII, p. 63.
Lefkovits, I., Waldmann, H., Limiting dilution analysis of cells in the immune system, Cambridge, Mass., 1979.
Legrain, P., Juy, D., Buttin, G., Rosette forming cell assay for detection of antibody-syntetizing hybridomas, in Methods in enzymology (a cura di S. P. Colowick e N. O. Kaplan), vol. XCII, New York 1983, pp. 175-182.
Levinson, B., Khoury, G., Woude, G. V., Gruss, P., Activation of SV40 genome by 72-base pair tandem repeats of Moloney sarcoma virus, in ‟Nature", 1982, CCXCV, pp. 568-572.
Lewin, R., On the origins of introns, in ‟Science", 1982, CCXVII, pp. 921-922.
Lo, M. M. S., Tsong, T. Y., Conrad, M. K., Strittmatter, S., Hester, L., Snyper, S., Monoclonal antibody production by receptor-mediated electrically induced cell fusion, in ‟Nature", 1984, CCCX, pp. 792-794.
Luben, R. A., Brazeau, P., Bohlen, P., Guillemin, R., Monoclonal antibodies to hypothalamic growth hormone-releasing factor with picomoles of antigen, in ‟Science", 1982, CCXVIII, pp. 887-889.
Mc Kearn, T. J., 51Cr-release cytotoxicity assay, in Monoclonal antibodies hybridoma: a new dimension in biological analyses (a cura di R. H. Kennett, T. J. Mc Kearn e K. B. Bechtol), New York 1980, pp. 393-394.
Maniatis, T., Fritsch, E. F., Sambrook, J., Molecular cloning: a laboratory manual, Cold Spring Harbor, N. Y., 1982.
Mas, M. T., Chen, C. Y., Hitzeman, R. A., Riggs, A. D., Active human-yeast chimeric phosphoglycerate kinases engineered by domain interchange, in ‟Science", 1986, CCXXXIII, pp. 788-790.
Maxam, A. M., Gilbert, W. A., A new method for sequencing DNA, in ‟Proceedings of the National Academy of Sciences", 1977, LXXIV, pp. 560-564.
Melamed, M. R., Mullaney, P. F., Mendelshon, M. (a cura di), Flow cytometry and sorting, New York 1979.
Mertz, J. E., Davis, R. V., Cleavage of DNA by RI restriction endonuclease generates cohesive ends, in ‟Proceedings of the National Academy of Sciences", 1972, LXIX, pp. 3370-3374.
Messing, J., Crea, R., Seeburg, P. H., A system for shotgun DNA sequencing, in ‟Nucleic acids research", 1980, IX, pp. 309-321.
Metcalf, D., Hemopoietic colonies. In vitro cloning of normal leukaemic cells, in Recent results in cancer research, vol. LXI, Heidelberg 1977, pp. 15-17.
Milstein, C., Monoclonal antibodies, in ‟Scientific American", 1980, CCXLIII, 4, pp. 56-64.
Milstein, C., Cuello, A. C., Hybrid hybridomas and their use in immunohistochemistry, in ‟Nature", 1983, CCCV, pp. 537-540.
Morandi, E., Comitti, R., Racchetti, G., Galante, Y. M., Immunizatoin protocols with low MW proteins for the production of murine monoclonal antibodies, in Biotech RIA 85, International Symposium on ‛The impact of biotechnology on diagnosis', Abs. 86, Roma 1985.
Munoz, J., Effect of bacteria and bacterial products on antibody response, in ‟Advances in immunology", 1964, IV, pp. 397-440.
Munoz, J., Bergman, A., Bordetella pertussis. Immunological and other biological activities, New York 1977.
Munro, A., Uses of chimaeric antibodies, in ‟Nature", 1984, CCCXII.
Murray, V., Holliday, R., Mechanism for RNA splicing of gene transcripts, in ‟FEBS letters. Federation of European Biochemical Societies", 1979, CVI, pp. 5-7.
Mutter, M., The construction of new proteins and enzymes: a prospect for the future?, in ‟Angewandte Chemie" (international English edition), 1985, XXIV, pp. 639-653.
Nardelli, L., Panina, G. F., 10 years experience with a 28,000 roller bottle plant for FMD vaccine production. Joint WHO/IABS Symposium on the standardization of cell substrates for the production of virus vaccines, Genève, dec. 1976, in ‟Developments in biological standardization", 1977, XXXVII, pp. 133-138.
Neel, G., Suresh, C., Chaganti, R. S., Harward, W., Two human c-oncogenes are located on the long arm of chromosome 8, in ‟Proceedings of the National Academy of Sciences", 1982, LXXIX, pp. 7842-7846.
Neurberger, M. S., Williams, G. T., Fox, R. O., Recombinant antibodies possessing novel effector functions, in ‟Nature", 1984, CCCXII, pp. 604-608.
Nilsson, K., Sheirer, W., Merten, O. W., Östberg, L., Liehl, E., Katinger, H. W. D., Mosbach, K., Entrapment of animal cells for production of monoclonal antibodies and other biomolecules, in ‟Nature", 1983, CCCII, pp. 629-630.
Norrby, K., Knutson, F., Lundin, P. M., On the single cell state in enzymatically produced tumor cell suspensions, in ‟Experimental cellular research", 1966, XLIV, pp. 421-428.
Okayama, H., Berg, P., High-efficiency cloning of full-length cDNA, in ‟Molecular and cellular biology", 1982, II, pp. 161-170.
Olsson, L., Kaplan, H. S., Human-human hybridomas producing monoclonal antibodies of predefined antigenic specificity, in ‟Proceedings of the National Academy of Sciences", 1980, LXXVII, pp. 5429-5431.
Olsson, L., Wonstroem, H., Cambon-De Mounoz, A., Honsik, C., Brodin, T., Jakobsen, B., Antibody producing human-human hybridomas. I. Technical aspects, in ‟Journal of immunological methods", 1983, LXI, pp. 17-32.
Owens, R. B., Selective cultivation of mammalian epithelial cells, in Methods in cell biology (a cura di D. M. Prescott), vol. XIV, New York 1976, pp. 341-354.
Oyama, V. I., Eagle, H., Measurement of cell growth in tissue culture with a phenol reagent, in ‟Proceedings of the Society for Experimental Biology and Medicine", 1956, XCI, pp. 305-307.
Paire, J., Desgranges, C., Production of human monoclonal antibodies by EBV immortalization, in Biotech RIA 85, International Symposium on ‛The impact of biotechnology on diagnosis', Abs. 74, Roma 1985.
Pardue, R. L., Brady, R. C., Perry, G. M., Demand, J. R., Production of monoclonal antibodies against calmodulin by in vitro immunization of spleen cells, in ‟Journal of cell biology", 1983, XCVI, pp. 1149-1154.
Parham, P., Isolation of heavy chain class switch variants of monoclonal anti DC-1 hybridoma cell line effective conversion of noncytotoxic IgG 1 antibodies to cytotoxic IgG 2 antibodies, in ‟Human immunology", 1983, VIII, pp. 141-151.
Parks, D. R., Bryan, V. M., Oi, V. T., Herzenberg, L. A., Antigenspecific identification and cloning of hybridomas with a fluorescence-activated cell sorter, in ‟Proceedings of the National Academy of Sciences", 1979, LXXVI, 4, pp. 1962-1966.
Patzer, E. J., Obijeski, J. F., Vaccines prepared from translation products of cloned viral genes, in Immunochemistry of viruses: the basis for serodiagnosis and vaccines (a cura di M. H. V. Van Regenmortel e A. R. Neurath), New York 1985, pp. 153-170.
Perry, L. J., Wetzel, R., Disulfide bond engineered into T4-lysozyme. Stabilization of the protein toward thermal inactivation, in ‟Science", 1984, CCXXVI, pp. 555-557.
Perry, L. J., Wetzel, R., Unpaired cystine-54 interferes with the ability of an engineered disulfide to stabilize T4-lysozyme, in ‟Biochemistry", 1986, XXV, pp. 733-739.
Radlett, P. J., Pay, T. W. F., Garland, A. J. M., The use of BHK suspension cells for commercial production of foot-and-mouth disease vaccines over a twenty year period, in ‟Developments in biological standardization", 1985, LX, pp. 163-170.
Rastetter, W. H., Enzyme engineering: applications and promises, in ‟Trends in biotechnology", 1983, I, pp. 1-5.
Reading, C. L., Theory and methods for immunization in culture and monoclonal antibody production, in ‟Journal of immunological methods", 1982, LIII, pp. 261-291.
Reading, C. L., Procedures for in vitro immunization and monoclonal antibody production, in Hybridoma and cellular immortality (a cura di H. T. Baldwing e J. P. Allison), New York 1983, pp. 235-248.
Reuveny, S., Bino, T., Rosenberg, H., Traub, A., Mizrahi, A., Pilot plant scale production of human lymphoblastoid interferon, in ‟Developments in biological standardization", 1980, XLVI, pp. 281-288.
Reuveny, S., Mizrahi, A., Kotler, M., Freeman, A., Mammalian cell propagation on derivatized polycrylamide microcarriers, in ‟Developments in biological standardization", 1983, LV, pp. 11-23.
Rosen, A., Gergely, R., Sondal, M., Klein, G., Policlonal Ig production after Epstein-Barr virus infection of human lymphocytes in vitro, in ‟Nature", 1977, CCLXVII, pp. 52-54.
Sandford, K. K., Earle, W. R., Evans, V. J., Waltz, H. K., Shannon, J. E., The measurement of proliferation in tissue cultures by enumeration of cell nuclei, in ‟Journal of the National Cancer Institute", 1951, XI, pp. 773-795.
Sanger, F., Coulson, A. R., A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase, in ‟Journal of molecular biology", 1975, XCIV, pp. 441-448.
Sanger, F., Coulson, A. R., Barrel, B. G., Smith, A. J., Roe, B. A., Cloning in single-strained bacteriophage as an aid to rapid DFNA sequencing, in ‟Journal of molecular biology", 1980, CXLIII, pp. 161-178.
Sanger, F., Nicklen, S., Coulson, A. R., DNA sequencing with chain-terminating inhibitors, in ‟Proceedings of the National Academy of Sciences", 1977, LXXIV, pp. 5463-5467.
Scaife, J. F., Brohee, H. A., A quantitative measurement of cell damage by means of 51Cr binding, in ‟Experimental cell research", 1967, XLVI, pp. 612-615.
Schonherr, O. T., Houwink, E. H., Antibody engineering, a strategy for the development of monoclonal antibodies, in ‟Antoine Van Leeuwenhoek journal of microbiology and serology", 1984, L, pp. 597-623.
Scoper, R., Protein purification, principles and practice, Berlin 1982.
Secher, D. S., Burke, D. C., A monoclonal antibody for large-scale purification of human leukocyte interferon, in ‟Nature", 1980, CCLXXXV, pp. 446-450.
Sharon, J., Gefter, M. L., Mausert, J. L., Oi, V. T., Ptashener, M., Expression of Vk Ck chimaeric protein in mouse myeloma cells, in ‟Nature", 1984, CCCIX, pp. 364-367.
Sheeter, P., Doolittle, M. H., Separation of mammalian cells by velocity sedimentation, in ‟International laboratory", March 1980, pp. 71-84.
Shimizu, K., Goldfarb, M., Perucho, N., Wigler, M., Isolation and preliminary characterization of the transforming gene of a human neuroblastoma cell line, in ‟Proceedings of the National Academy of Sciences", 1983, LXXX, pp. 383-387.
Shimizu, K., Takahashi, N., Yaoita, Y., Honjio, T., Organization of constant-region gene family of the mouse immunoglobulin heavy chain, in ‟Cell", 1982, XXVIII, pp. 499-506.
Shortle, D., Meeker, A. K., Mutant forms of staphylococcal nuclease with altered patterns of guanidine hydrochloride and urea denaturation, in ‟Proteins. Structure, function and genetics", 1986, I, pp. 81-89.
Shows, T. B., Sakaguchi, A. Y., Naylor, S. L., Mapping the human genoma, cloned genes, DNA polymorphisms and inherited disease, in ‟Advances in human genetics", 1982, XII, pp. 341-452.
Shulman, M., Wilde, C. D., Köhler, G., A better cell line for making hybridomas secreting specific antibodies, in ‟Nature", 1978, CCLXXVI, pp. 269-270.
Siraganian, R. P., Fox, P. C., Berenstein, E. H., Methods of enhancing the frequency of antigen-specific hybridomas, in Methods in enzymology (a cura di S. P. Colowick e N. O. Kaplan), vol. XCII, New York 1983, pp. 17-26.
Sothern, E. M., Detection of specific sequences among DNA fragments separated by gel electrophoresis, in ‟Journal of molecular biology", 1975, XCVIII, pp. 503-517.
Spier, R. E., Clarke, J. B., Variation in the susceptibility of BHK population and cloned cell lines to three strains of foot-and-mouth disease virus, in ‟Archives of virology", 1980, LXIII, pp. 1-9.
Stähli, C., Stähelin, T., Miggiano, V., Schmidt, J., Häring, P., High frequencies of antigen specific hybridomas. Dependence on immunizaton parameters and prediction by spleen cell analysis, in ‟Journal of the immunological methods", 1980, XXXII, pp. 297-304.
Steinitz, M., Klein, G., Koskimies, S., Makel, O., EB virus-induced B lymphocytes cell lines producing specific antibody, in ‟Nature", 1977, CCLXIX, pp. 420-422.
Steinitz, M., Seppola, I., Eichmann, K., Klein, G., Establishment of human lymphoblastoid cell line with specific antibody production against group A streptococcal carbohydrate, in ‟Immunobiology", 1979, CLVI, p. 41.
Steinmetz, M., Winoto, A., Minard, K., Hood, L., Cluster of genes encoding mouse transplantation antigenes, in ‟Cell", 1982, XXVIII, pp. 489-492.
Suggs, S. V., Wallace, B. R., Hirose, T., Kawashima, E. H., Itakura, K., Use of synthetic oligonucleotides as hybridization probes: isolation of cloned cDNA sequences for human B2-microglobulin, in ‟Proceedings of the National Academy of Sciences", 1981, LXXVIII, pp. 6613-6617.
Telling, R. C., Radlett, P. J., Large scale cultivation of mammalian cells, in ‟Advances in applied microbiology", 1970, XIII, pp. 91-119.
Temin, H. M., Function of the retrovirus long terminal repeat, in ‟Cell", 1982, XXVIII, pp. 3-5.
Tobey, R. A., Anderson, E. C., Petersen, D. F., Properties of mitotic cells prepared by mechanically shaking monolayer cultures of Chinese hamster cells, in ‟Journal of cellular physiology", 1967, LXX, pp. 63-68.
Tooze, J., DNA tumor viruses. Molecular biology of tumor viruses, Cold Spring Harbor, N. Y., 19812.
Treisman, R., Proudfoot, N. J., Shauder, M., Maniatis, T., A single-base change at a splice site in a β0-thalassemic gene causes abnormal RNA splicing, in ‟Cell", 1982, XXIX, pp. 903-911.
Trent, J. M., Olson, S., Lawn, R. M., Chromosomal localization of human leukocyte, fibroblast and immune interferon genes by means of in situ hybridizaton, in ‟Proceedings of the National Academy of Sciences", 1982, LXXIX, pp. 7809-7813.
Ulmer, K., Protein engineering, in ‟Science", 1983, CCXIX, pp. 666-671.
Van Wezel, A. L., Growth of cell-strains and primary cells on microcarriers in homogeneous culture, in ‟Nature", 1967, CCXVI, pp. 64-65.
Van Wezel, A. L., Van der Velden- de Groot, C. A. M., Van Herwaarden, J. A. M., The production of inactivated poliovaccine on serially cultivated kidney cells from captive-bred monkeys, in ‟Developments in biological standardization", 1980, XLVI, pp. 151-158.
Villafranca, J. E., Howell, E. E., Voet, D. H., Strobel, M. S., Ogden, R. C., Abelson, J. N., Kraut, J., Directed mutagenesis of dihydrofolate reductase, in ‟Science", 1983, CCXXII, pp. 782-788.
Wade, N., Gene therapy caught in more entanglements, in ‟Science", 1981, CCXII, pp. 24-25.
Walker, S. M., Meinke, G. C., Keigle, W. O., Enrichment of antigenspecific B lymphocytes by direct removal of B cell not bearing specificity for the antigen, in ‟Journal of experimental medicine", 1977, CXLVI, pp. 445-448.
Watson, J. D. (a cura di), The DNA story, San Francisco 1981.
Watson, J. D., Crick, F. H. C., Molecular structure of acids: a structure for deoxribose nucleic acid, in ‟Nature", 1953, CLXXI, pp. 737-738.
Wells, T. N. C., Fersht, A. R., Hydrogen bonding in enzymatic catalysis analysed by protein engineering, in ‟Nature", 1985, CCCXVI, pp. 656-657.
Wells, T. N. C., Fersht, A. R., Use of binding energy in catalysis analyzed by mutagenesis of the tyrosyl-tRNA synthetase, in ‟Biochemistry", 1986, XXV, pp. 1881-1886.
White, R. A. H., Mason, D. W., Williams, A. F., Galfré, G., Milstein, C., T lymphocyte heterogeneity in the rat separation of functional subpopulation using a monoclonal antibody, in ‟Journal of experimental medicine", 1978, CXLVIII, 3, pp. 664-673.
Wilkinson, A. J., Fersht, A. R., Blow, D. M., Carter, P., Winter, G., A large increase in enzyme-substrate affinity by protein engineering, in ‟Nature", 1984, CCCVII, pp. 187-188.
Williamson, B., Gene therapy, in ‟Nature", 1982, CCXCVIII, pp. 416-418.
Winter, G., Fersht, A. R., Engineering enzymes, in ‟Trends in biotechnology", 1984, II, pp. 115-119.
Winter, G., Fersht, A. R., Wilkinson, A. J., Zoller, M., Smith, M., Redesigning enzyme structure by site-directed mutagenesis: tyrosyl-tRNA synthetase and ATP binding, in ‟Nature", 1982, CCIC, pp. 756-758.
Wiseman, A. (a cura di), Enzyme biotechnology, Chichester 1975.
Wu, R. (a cura di), Recombinant DNA, in Methods in enzymology (a cura di S. P. Colowick e N. O. Kaplan), vol. C, New York 1983.
Wynn, C. H., Struktur und Funktionen von Enzymen, Stuttgart 1978.
Zimmerman, U., Electrofusion of cells: principles and industrial potential, in ‟Trends in biotechnology", 1983, I, pp. 149-155.
Processi enzimatici industriali di Walter Marconi
sommario: 1. Introduzione. □ 2. I catalizzatori biologici: a) enzimi di origine vegetale; b) enzimi di origine animale; c) enzimi microbici e cellule. □ 3. Tecnologia dei processi enzimatici: a) tipo di bioconversione; b) economicità di processo: uso di enzimi e cellule immobilizzati. □ 4. I bioreattori: a) reattore a batch; b) reattori in continuo. □ 5. Esempi di processi enzimatici industriali: a) liquefazione e saccarificazione dell'amido; b) isomerizzazione del glucosio; c) idrolisi della penicillina G; d) risoluzione ottica di amminoacidi; e) idrolisi del lattosio. □ 6. Prospettive. □ Bibliografia.
1. Introduzione
I fenomeni biologici sono resi possibili da particolari sostanze contenute in tutti gli organismi viventi, dal più piccolo microrganismo all'uomo, note sotto il nome di enzimi (v. enzimi). Queste sostanze sono dei veri e propri catalizzatori biologici, cioè degli acceleratori di quelle reazioni chimiche che sono caratteristiche di un organismo vivente (v.catalisi enzimatica). Già prima che se ne conoscesse la natura proteica (v. proteine) l'uomo imparò a sfruttarli artigianalmente per la produzione di alcuni cibi e bevande (pane, formaggi, vino, birra), in processi che con il progredire della tecnologia diventarono industriali e formarono la base dell'industria fermentativa. Ulteriori processi tecnologici permisero di ampliare la base di questa industria fino a comprendere i prodotti più svariati solventi (come l'acetone e il butanolo), acidi organici (come l'acido lattico e l'acido gluconico), amminoacidi e vitamine , per trovare un coronamento nella produzione di antibiotici e di enzimi.
La tecnologia degli enzimi, intesa come l'utilizzazione industriale di preparati più o meno purificati di uno o di pochi enzimi per eseguire trasformazioni chimiche, si può dire nata negli anni sessanta, quando si sostituì l'idrolisi acida dell'amido a glucosio con l'idrolisi enzimatica. Da allora la tecnologia degli enzimi ha subito un intenso sviluppo e ha raggiunto un alto grado di sofisticazione, trovando applicazione in questi ultimi decenni in alcuni settori dell'industria chimica (soprattutto farmaceutico e alimentare), come pure nella diagnostica clinica, nel trattamento di effluenti inquinanti e in campo biomedico.
Degli oltre 2.000 enzimi di cui si conoscono le principali proprietà funzionali, solo un numero molto ristretto trova impiego industriale (v. tab. I). Sono prevalentemente enzimi idrolitici utilizzati nella produzione di detersivi, nella ‛liquefazione' e saccarificazione dell'amido di mais, nella caseificazione del latte, nella produzione di succhi di frutta, nella panificazione, nella preparazione di idrolizzati proteici e nella produzione della birra. Tra i processi non idrolitici di importanza industriale citiamo l'isomerizzazione del glucosio a fruttosio e la modificazione di steroidi.
2.I catalizzatori biologici
Gli enzimi catalizzano all'interno degli organismi viventi una serie di reazioni chimiche volte al compimento di specifiche funzioni metaboliche. Anche se ogni organismo vivente può essere considerato una potenziale fonte di enzimi, in pratica solo poche piante e pochi organi animali sono utilizzati industrialmente per estrarre enzimi; la maggior parte degli enzimi industriali proviene invece dai microrganismi (v. tab. I).
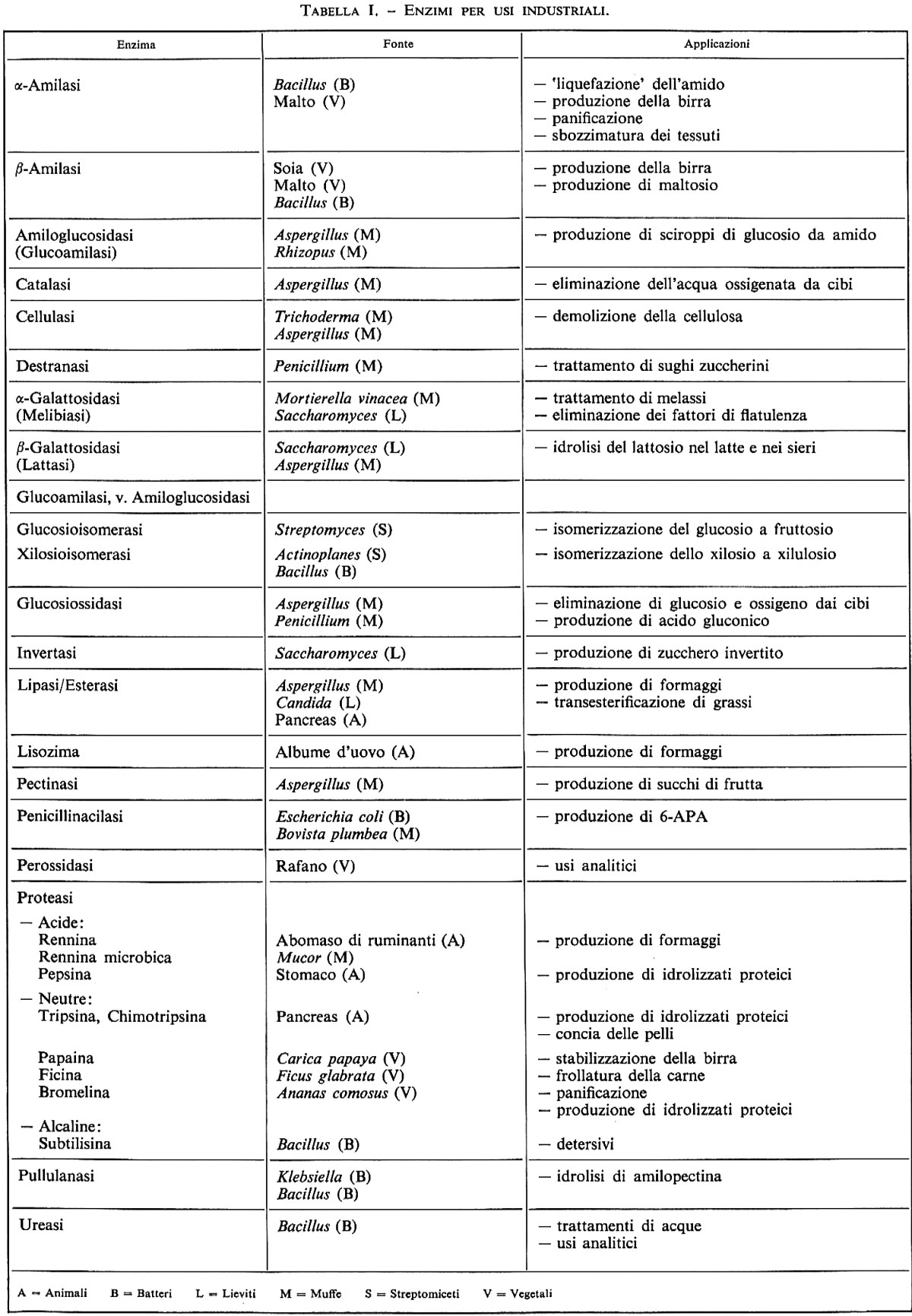
a) Enzimi di origine vegetale
Sono generalmente enzimi proteolitici (come la papaina, la bromelina, la ficina), gli enzimi amilolitici dei cereali e alcuni altri presenti nella buccia degli agrumi. Gran parte di questi enzimi, commercializzati sotto forma di preparati grezzi, sono estratti da piante coltivate in aree tropicali dell'Africa, del Sudamerica e dell'Asia. La precarietà delle piantagioni, soggette all'influenza di una serie di fattori naturali e geopolitici (lenta crescita, pericoli di siccità, di epidemie, lontananza dai luoghi d'uso, mutamenti nei governi), costituisce un grave vincolo per l'uso industriale di questi enzimi.
b) Enzimi di origine animale
Sono enzimi come la tripsina pancreatica, la lipasi e la rennina, prodotti sia in bulk per usi industriali, sia sotto forma di preparazioni pure. La forte richiesta di enzimi di origine animale (in particolare la rennina estratta dallo stomaco dei vitelli e usata nella caseificazione del latte), rispetto alla scarsa disponibilità di organi di animali macellati, ha fatto non solo lievitare i prezzi, ma anche orientare la produzione verso le fonti microbiche.
c) Enzimi microbici e cellule
La produzione di enzimi per uso industriale si sta sempre più orientando verso le fonti microbiche, data la facilità con cui si può disporre di microrganismi e produrli nella quantità voluta e a costi contenuti, inducendo in loro la sintesi degli enzimi desiderati. Gli enzimi microbici di importanza industriale sono prodotti da non più di 11 specie di funghi, 8 di batteri e 4 di lieviti (v. tab. I).
Un'importante suddivisione degli enzimi microbici è basata sui diversi meccanismi che ne regolano la biosintesi. Si distinguono al riguardo gli enzimi induttivi e quelli costitutivi. La produzione dei primi è sostanzialmente collegata alla presenza nel terreno di coltura di una particolare sostanza, detta induttore, mentre la produzione dei secondi è relativamente indipendente dalla composizione del terreno di coltura. L'induttore può essere il substrato stesso su cui l'enzima deve agire, ma spesso è una sostanza strutturalmente simile al substrato, che tuttavia l'enzima è incapace di modificare. Con l'uso di induttori è possibile aumentare la produzione di un enzima anche fino a 100 volte.
La biosintesi degli enzimi costitutivi non richiede la presenza di particolari sostanze (e ciò è un notevole vantaggio economico, perché spesso gli induttori sono composti molto costosi), ma anche nella produzione degli enzimi costitutivi - come del resto in quella degli enzimi induttivi - bisogna tener conto di alcuni meccanismi di regolazione della biosintesi, che sono l'inibizione a retroazione (feedback repression) e l'inibizione da catabolita (catabolite repression). L'inibizione a retroazione è esercitata dal prodotto della reazione enzimatica e viene evitata impedendo l'accumulo nella cellula del prodotto stesso. L'inibizione da catabolita, che si manifesta quando la cellula si moltiplica rapidamente a spese di fonti di carbonio e d'azoto facilmente utilizzabili, viene evitata utilizzando altre sostanze nutritive consumate più lentamente.
Anche sulla base della loro localizzazione rispetto alla cellula, gli enzimi microbici possono essere distinti in due gruppi - gli endocellulari e gli esocellulari - e ciò ha rilevanza, come vedremo subito, nei processi di purificazione. I primi sono sintetizzati, utilizzati e trattenuti all'interno della cellula, mentre i secondi sono secreti nel terreno di coltura. Gli enzimi esocellulari tendono generalmente a catalizzare reazioni di tipo idrolitico, mentre quelli endocellulari, spesso presenti sotto forma di sistemi multienzimatici altamente organizzati, sono di solito associati a reazioni di sintesi. Le peculiari proprietà catalitiche degli enzimi esocellulari rispecchiano la loro funzione metabolica, che è quella di fornire alla cellula i necessari nutrienti, demolendo, con reazioni idrolitiche, composti di alto peso molecolare presenti nel terreno di coltura. Questi enzimi (generalmente proteasi e carboidrasi) hanno trovato larga applicazione industriale per una serie di motivazioni: innanzitutto perché sono generalmente stabili come conseguenza della necessità di operare, senza alcuna protezione, in un ambiente esterno alla cellula, soggetto a variazioni di natura chimico-fisica; in secondo luogo perché possono essere prodotti in grandi quantità, e infine perché sono facilmente isolabili. Le tecniche e le procedure per l'isolamento degli enzimi esocellulari sono molto semplici: occorre rimuovere dal brodo di fermentazione, mediante filtrazione o centrifugazione, le cellule, e separare l'enzima utilizzando una o più delle tecniche riportate nella tab. II (v. anche enzimi).

Nel caso degli enzimi endocellulari le tecniche di isolamento e purificazione sono più complesse e laboriose, soprattutto se gli enzimi sono legati a strutture cellulari. Occorre, nella maggior parte dei casi, procedere alla rottura delle cellule, che viene ottenuta sia con mezzi fisici (omogeneizzatori ad alta pressione) sia con mezzi chimici o enzimatici (lisi). Solo in alcuni casi è possibile utilizzare tecniche più blande, come lo shock osmotico o l'estrazione con tensioattivi, che liberano l'enzima lasciando pressoché intatta la struttura cellulare. Un omogenato o un lisato di cellule contiene, oltre agli enzimi, anche gli acidi nucleici, e quindi la procedura di purificazione con le tecniche riportate nella tab. II deve essere preceduta da opportuni trattamenti (precipitazione, idrolisi enzimatica) atti a eliminare gli acidi nucleici. Nella pratica industriale non sempre si utilizzano enzimi, più o meno purificati, ma, quando è possibile, anche le cellule intere, e ciò per evitare le costose operazioni di estrazione e di purificazione.
3. Tecnologia dei processi enzimatici
Da un'analisi dei diversi tipi di catalizzatore enzimatico e dei relativi sistemi reattoristici si configura per la biocatalisi la possibilità di un'ampia scelta di sistemi operativi. La scelta del biocatalizzatore (enzimi liberi o cellule intere) può essere fatta sulla base di una serie di considerazioni inerenti al tipo e alla durata della bioconversione, alla qualità del prodotto, alla stabilità e al riutilizzo del catalizzatore, all'economicità dell'intero processo.
a) Tipo di bioconversione
La desiderata conversione biochimica può richiedere la partecipazione di un singolo enzima, di più enzimi operanti in successione, o di enzimi forniti di coenzima. Questi ultimi rivestono notevole importanza, dal momento che circa un terzo degli enzimi noti necessitano, per svolgere la loro attività, di cofattori quali NAD+, NADP+, FAD, ATP, ecc., presenti all'interno di ogni cellula vivente come parte del patrimonio enzimatico. Dovendo essere i coenzimi costantemente presenti in forma attiva nel mezzo di reazione perché la reazione proceda, una loro utilizzazione può risultare possibile a condizione che essi siano rigenerati e utilizzati più volte. Anche se in alcuni laboratori di ricerca si è tentato con qualche successo il riutilizzo dei coenzimi, non esiste ancora un processo industriale che sfrutti questo sistema complesso. Si deduce da ciò che, in tutte quelle reazioni di bioconversione in cui siano implicati un certo numero di enzimi oppure un singolo enzima richiedente un coenzima, l'uso delle cellule intere risulta favorito. Infatti, grazie proprio alla struttura ben organizzata delle cellule, capace di operare trasduzioni energetiche e rigenerazione di cofattori, si ha, con il loro impiego, la possibilità di catalizzare reazioni generalmente molto complesse, come quelle di biosintesi. Le cellule possono essere sia morte che vitali. Nel primo caso la cellula è assimilabile a un piccolo involucro contenente l'enzima o l'insieme di enzimi attivi. Nel secondo caso la cellula dispone di tutti i suoi componenti metabolici intatti, ed è quindi in grado di riprodursi quando introdotta in un mezzo nutriente (che è poi il normale terreno di coltura di una fermentazione). Queste cellule possono essere utilizzate indifferentemente sia nello stadio di crescita sia in quello di stato stazionario (resting cells).
b) Economicità di processo: uso di enzimi e cellule immobilizzati
Lo sviluppo applicativo di un processo enzimatico è condizionato da un certo numero di fattori che influenzano l'economicità dell'intero processo. I più determinanti sono: il costo del catalizzatore, le rese di bioconversione, la purificazione del prodotto di reazione, il sistema reattoristico. Per ogni processo esistono condizioni ottimali per l'uso del catalizzatore, fissate da alcuni parametri che correlano attività iniziale, concentrazione e quantità di substrato, stabilità operativa e tempi di reazione. Le condizioni, oltre a determinare la velocità di reazione e la quantità di prodotto che si forma per unità di biocatalizzatore, contribuiscono a valutare l'incidenza del costo del catalizzatore per unità di prodotto. Questo costo risente anche di una serie di fattori connessi: a) col processo fermentativo usato per ottenere la cellula o l'enzima microbico; b) con le rese di enzima attivo per unità di massa di fermentazione; c) con il tipo di catalizzatore (cellula intera o enzima libero); d) con il livello di purificazione del preparato enzimatico; e) con la stabilità operativa; f) con le possibilità di recupero e di riutilizzo. Allo scopo di ridurre l'incidenza economica del catalizzatore enzimatico in un processo industriale è nata, a metà di questo secolo, la tecnica di immobilizzazione degli enzimi.
Il termine ‛enzima immobilizzato' sta a indicare un sistema o una preparazione in cui l'enzima o la cellula sono confinati o localizzati in una regione di spazio appartenente a un supporto insolubile in acqua. Questa definizione abbastanza generica vuole indicare un sistema capace di essere separato dal mezzo di reazione, recuperato e utilizzato ripetutamente sia in processi a batch che in quelli in continuo. L'immobilizzazione si presenta quindi vantaggiosa, perché elimina le laboriose operazioni di separazione del catalizzatore dal mezzo di reazione, conferisce al catalizzatore una maggiore stabilità strutturale, consente l'utilizzo del catalizzatore per più cicli, riducendo in ultima analisi l'incidenza economica del catalizzatore nel processo. I metodi di immobilizzazione di enzimi o cellule rientrano sostanzialmente in quattro classi: a) attacco covalente a un supporto organico o inorganico insolubile in acqua e reso reattivo mediante attivazione con gruppi funzionali; b) reticolazione dell'enzima con macromolecole insolubili in acqua mediante reagenti reticolanti multifunzionali; c) adsorbimento su un supporto insolubile; d) inglobamento fisico in matrici polimeriche permeabili a composti chimici di basso peso molecolare.
L'attacco covalente a polimeri o matrici attivati è il metodo d'immobilizzazione più comunemente usato in laboratorio, ma non su scala industriale. Questo metodo ha il vantaggio di fissare il biocatalizzatore alla matrice solida con legami chimici, che rendono l'aggancio irreversibile rispetto a forza ionica, pH, substrati, ecc. Il complesso biologico coniugato al supporto può essere considerato come un derivato di quello in soluzione, anche se può avere proprietà fisiche e chimiche (stabilità termica, attività, ecc.) molto diverse e talora superiori. Un aspetto negativo di questo metodo è costituito dal fatto che durante la reazione di legame la molecola biologica può subire denaturazione o inattivazione da parte dei reagenti chimici. I migliori metodi sono quelli che fanno uso di condizioni di reazione (temperatura, pH, ecc.) blande e di reattivi poco aggressivi.
Numerosi sono i reagenti bifunzionali utilizzati per reticolare enzimi e cellule a macromolecole sintetiche e naturali. Tra questi la glutaraldeide è il più diffuso. Le metodiche di preparazione del complesso attivo immobilizzato possono distinguersi in tre tipi: a) semplice reticolazione intra- e intermolecolare tra le stesse molecole bioattive; b) adsorbimento della molecola bioattiva a supporti (tipo collageno, albumina, gelatina, chitina) e successiva reticolazione; c) introduzione del reagente bifunzionale in polimeri sintetici preformati e contemporaneo attacco covalente della molecola biologica.
L'adsorbimento rappresenta la procedura più semplice per immobilizzare molecole biologiche. Infatti la metodica consiste nel porre a contatto la soluzione biologica con la superficie attiva dell'adsorbente e in un successivo lavaggio per rimuovere le molecole non trattenute. L'adsorbimento può essere attribuito a un meccanismo di scambio ionico e quindi a interazioni elettrostatiche, oppure a legami fisico-chimici dovuti a interazioni idrofobiche, forze di van der Waals, legami a idrogeno, ecc. L'immobilizzazione per adsorbimento è preferita quando la molecola biologica è sensibile a modificazioni chimiche ed è inattivata da legami covalenti. Il meccanismo di adsorbimento dipende principalmente dalla natura della sostanza adsorbente. Poiché l'adsorbimento implica solo deboli interazioni, si verifica sovente un rilascio della molecola immobilizzata. Questo fenomeno è accentuato da variazioni di pH, forza ionica e temperatura.
L'ultimo metodo di immobilizzazione si basa sull'intrappolamento di enzimi e di cellule all'interno di un materiale poroso, le cui cavità sono munite di pareti che non lasciano passare le molecole bioattive a causa delle loro grosse dimensioni, mentre consentono la diffusione all'interno e all'esterno di molecole di basso peso molecolare.
4. I bioreattori
Nel bioreattore uno o più enzimi - sia liberi, sia contenuti in cellule o singolarmente immobilizzati - sono utilizzati per la produzione di uno o più composti di interesse commerciale. La funzione del reattore è quella di fornire un ambiente controllato per conseguire il miglior sfruttamento del sistema catalitico, svolgendo in questo modo un ruolo critico nell'economia di un processo. L'obiettivo principale nella progettazione di un bioreattore è di minimizzare i costi di produzione operando su una serie di parametri, quali l'attività specifica del catalizzatore, la sua vita operativa, i tempi di contatto, ecc. (v. tab. III), con lo scopo di ottenere elevate rese di conversione ed elevata produttività, cioè massima concentrazione di prodotto associata a minima formazione di sottoprodotti. Per ottenere l'insieme di questi obiettivi occorre generalmente ricercare un compromesso tra tempo di vita operativa del catalizzatore e produttività volumetrica. Nel caso specifico di catalizzatori immobilizzati la produttività volumetrica dipende, oltreché dalla percentuale del volume di reattore occupato dal catalizzatore, anche da una serie di altri fattori (v. tab. IV). In genere l'attività delle cellule immobilizzate non può eccedere quella delle cellule libere, a causa della barriera diffusiva costituita dalla matrice, a meno che altri fattori non contribuiscano a un incremento dell'attività specifica. Per aumentare la densità del catalizzatore, e quindi la produttività, occorre isolare l'enzima o gli enzimi che, validamente immobilizzati, possono fornire catalizzatori a elevata attività specifica, soprattutto se confrontati con le rispettive cellule immobilizzate (v. tab. V).



L'attività specifica degli enzimi può variare da 1 a 10.000 unità per milligrammo di proteina (una unità corrisponde a 1 μmole di substrato trasformato per minuto nelle condizioni ottimali di reazione). Dal momento che alcuni reattori possono contenere 50 grammi di enzimi immobilizzati per litro, è facile calcolare che la produttività volumetrica può risultare superiore a 100 moli per ora e per litro. Tali risultati sono tuttavia difficili da ottenersi per una serie di limitazioni di natura fisica e ingegneristica. Fenomeni di trasporto di materia condizionano sovente l'efficienza di un catalizzatore. Nel caso di catalizzatori immobilizzati, le cui particelle (a forma di sferette, di granuli, di filamenti, ecc.) hanno dimensioni che per gli enzimi si aggirano tra 0,15 e 0,8 mm, mentre per le cellule tra 1,5 e 4 mm, l'efficienza è condizionata da fenomeni diffusivi tanto più sentiti quanto più grandi sono le dimensioni delle particelle e meno porose le matrici di supporto. Nel caso di cellule un'ulteriore limitazione è costituita dalla membrana cellulare. Ma accanto a questi fenomeni di trasporto di materia all'interno del catalizzatore, occorre evitare anche le limitazioni connesse col trasporto di materia dalla massa di soluzione alla superficie del catalizzatore. A questo proposito la scelta del tipo di reattore risulta determinante.
Differenti tipi di reattori possono essere utilizzati con enzimi o cellule sia liberi che immobilizzati e possono operare sia a batch (in discontinuo) sia a flusso continuo.
a) Reattore a batch
Il reattore a batch è molto semplice, non necessita di particolari strumentazioni ed è generalmente utilizzato quando devono essere preparate piccole quantità di prodotto. La catalisi con enzimi in forma libera viene generalmente effettuata con questo tipo di reattore. Le operazioni sono semplici: l'enzima viene caricato nel reattore insieme col materiale di partenza e la reazione termina quando si è raggiunto il voluto grado di conversione. Generalmente non si cerca di recuperare l'enzima dal prodotto di reazione, perché il costo di recupero risulta eccessivo e in molti casi l'enzima al termine della reazione può essere anche poco attivo; teazioni di questo tipo, con enzimi a perdere, sono per lo più utilizzate nel settore alimentare. Nel caso di enzimi o cellule immobilizzate, la separazione del catalizzatore al termine della reazione avviene facilmente. Questo tipo di reattore offre numerosi vantaggi: può essere, ad esempio, utilizzato per una varietà di reazioni, sostituendo catalizzatore, reagenti e condizioni operative (pH, temperatura, ecc.); se poi uno dei reagenti inibisce il processo, tale reagente può essere aggiunto gradualmente con il procedere della reazione. Se il prodotto di reazione è un forte inibitore, il reattore a batch è da preferire rispetto a quello in continuo, perché la massima concentrazione di prodotto si ha solo alla fine della reazione. Tra i processi industriali più noti operanti con questo tipo di sistema reattoristico sono: la saccarificazione dell'amido (idrolisi dell'amido a glucosio), l'idrolisi del lattosio nel latte, l'idrolisi della penicillina G, l'idrossilazione degli steroidi.
b) Reattori in continuo
Due sono i tipi di reattori operanti in continuo: il reattore tubolare a letto fisso e il reattore agitato alimentato in continuo (Continuous Stirred Tank Reactor o CSTR). Un ibrido di questi due tipi è il reattore a letto fluido. L'uso di questi sistemi reattoristici è subordinato alla disponibilità di catalizzatori in forma immobilizzata. I principali vantaggi dei processi in continuo sono costituiti dal minor costo di gestione dell'impianto, da minori costi di investimento e da una maggiore produttività, soprattutto se confrontata con i processi a batch. I principali inconvenienti sono dovuti a una minore flessibilità nella produzione e a una maggiore complessità della tecnologia reattoristica.
La scelta tra i due tipi di reattori dipende dalle caratteristiche cinetiche e operative dell'intero processo di bioconversione, e in particolare dai seguenti fattori: a) forma del catalizzatore immobilizzato; b) natura del substrato (completamente solubile o con particelle in sospensione); c) richiesta di controlli cinetici; d) capacità di impaccamento; e) rapporto tra superficie del catalizzatore e volume del reattore; f) caratteristiche del trasporto di materia; g) facilità di riempimento del reattore e di sostituzione del catalizzatore nel reattore.
Nel reattore a letto fisso il substrato che fluisce attraverso il letto del catalizzatore in una determinata direzione si trova ovunque in condizioni di stato stazionario. Se il profilo della velocità del fluido è uniforme, il reattore assume le caratteristiche di un Plug-Flow Reactor (PFR), con valori di conversione del substrato crescenti nella direzione di flusso e uguali per tutti i punti della soluzione che si trovino sulle stesse sezioni del reattore. Deviazioni da queste condizioni ideali possono essere dovute a: a) gradienti di velocità in direzione normale al flusso; b) diffusione di substrato in direzione assiale; c) gradienti di temperatura in direzione normale al flusso. Si tratta, quindi, di fenomeni legati al trasporto di materia e alla distribuzione del calore di reazione, la cui influenza è molto avvertita nei reattori industriali. Nonostante ciò, questo sistema risulta il più diffuso tra i processi catalitici utilizzati per produrre quantità massive di prodotto. Tra i catalizzatori più usati sono: enzimi legati chimicamente a sferette di vetro; enzimi legati a particelle di polisaccaridi artificiali quali la dietilamminoetilcellulosa (DEAE-cellulosa) e il dietilamminoetildestrano (DEAE-Sephadex); cellule reticolate con glutaraldeide, ottenute sotto forma di particelle cilindriche; cellule inglobate in materiali cellulosici in forma di sferette.
Esempi di processi industriali biocatalitici che utilizzano reattori a letto fisso sono: l'isomerizzazione del glucosio a fruttosio con glucosioisomerasi; la produzione di acido 6-amminopenicillanico con penicillinacilasi; la risoluzione di isomeri ottici degli amminoacidi con amminoacilasi; la produzione di alcool etilico con cellule di lievito immobilizzate.
Più di recente sono stati sviluppati reattori a letto fisso contenenti membrane in configurazione di fibre cave, i cui pori consentono il passaggio di substrati e prodotti, ma non di enzimi e di cellule, che risultano così immobilizzati, generalmente all'interno della membrana. Poiché la molecola enzimatica inglobata non è chimicamente modificata, il suo comportamento cinetico all'interno della fibra cava dovrebbe risultare pressoché identico a quello dell'enzima libero. Fenomeni diffusivi legati al passaggio del substrato all'interno della membrana hanno, al contrario, dimostrato di influenzare notevolmente la velocità catalitica del processo. Variazioni notevoli (di un fattore 1.700) sono state riscontrate, per esempio, nella Km (Km costante di Michaelis, ovvero concentrazione di substrato cui corrisponde una velocità di reazione metà di quella massima: v. catalisi enzimatica) della fosfatasi alcalina, come pure nei valori dell'energia di attivazione per l'ureasi (diminuzione da 8,6 a 5,4 kcal mol-1) e nell'optimun di pH.
Nel reattore agitato alimentato in continuo (CSTR) la composizione del mezzo di reazione è quella stessa della soluzione che esce alla fine della reazione; questo aspetto può fortemente condizionare la cinetica della reazione, riducendone la velocità, soprattutto nel caso di inibizione da prodotto. A questo inconveniente si aggiunge la bassa produttività volumetrica del catalizzatore. I principali vantaggi sono: a) la facilità nel controllare le condizioni di reazione; b) la facilità nel caricare e scaricare il catalizzatore immobilizzato; c) la possibilità di utilizzare substrati contenenti anche particelle insolubili in sospensione; d) l'ottimo controllo delle condizioni di agitazione e del contatto fra substrato e catalizzatore, con riduzione al minimo dei fenomeni connessi col trasporto di materia.
In un reattore a letto fluido le condizioni fluidodinamiche di alimentazione si avvicinano sia a quelle di un PlugFlow Reactor sia a quelle di un CSTR. In questo sistema il substrato è alimentato attraverso il letto del catalizzatore enzimatico dal basso in alto, a una velocità sufficiente a mantenere in sospensione le particelle (sfere, dischi, cilindri, ecc.), senza che queste, tuttavia, fuoriescano dall'estremità superiore del reattore. Questo sistema non permette una elevata densità di impaccamento del catalizzatore, ma consente di utilizzare soluzioni viscose di substrato, contenenti anche particelle in sospensione, senza provocare cadute di pressione.
5. Esempi di processi enzimatici industriali
Gli enzimi vengono usati industrialmente sia in processi produttivi sia nella formulazione di prodotti di largo consumo. Un terzo circa della produzione totale di enzimi microbici è costituito dalle proteasi alcaline utilizzate nei detersivi biologici. Le amilasi vengono utilizzate per togliere l'appretto ai tessuti o nella produzione di preparati per favorire il distacco delle carte da parati dai muri. Altri enzimi trovano applicazioni in campo analitico. Una metà circa degli enzimi prodotti trova applicazioni dirette nei processi produttivi, soprattutto delle industrie agroalimentari. Gli enzimi vengono utilizzati in questi processi per una o più delle seguenti ragioni: a) come componenti essenziali del processo produttivo (per es., l'α-amilasi nella liquefazione dell'amido); b) per migliorare l'economicità di un processo produttivo, che può essere condotto anche senza enzimi (le pectinasi vengono utilizzate per migliorare la resa in mosto durante la vinificazione o, più generalmente, la resa nella produzione di succhi di frutta); c) per migliorare la qualità del prodotto (con la lattasi si rende il latte più digeribile).
Descriveremo ora i principali processi produttivi che utilizzano enzimi.
a) Liquefazione e saccarificazione dell'amido
I primi sciroppi di glucosio (destrosio) sono stati ottenuti per idrolisi acida dell'amido, ma la tecnologia enzimatica ha fortemente contribuito allo sviluppo dell'industria amidiera, consentendo la produzione con rese più elevate di una grande varietà di sciroppi zuccherini con specifiche proprietà chimico-fisiche, che hanno trovato larga applicazione nell'industria alimentare. I principali enzimi utilizzati a questo scopo sono l'α-amilasi e la glucoamilasi (o amiloglucosidasi). Il primo ha la proprietà di idrolizzare i legami α-1-4-glucosidici delle molecole di amilosio e di amilopectina di cui è costituito l'amido. Il secondo catalizza l'idrolisi dei legami α-1-4-glucosidici e, più lentamente, degli α-1-6-glucosidici degli oligosaccaridi ottenuti per azione dell'α-amilasi, liberando singole unità di glucosio.
L'α-amilasi trova applicazione nel processo di liquefazione dell'amido, che avviene in reattori operanti sia a batch sia in continuo. Perché l'azione enzimatica possa essere efficace occorre che l'amido sia completamente gelatinizzato, la qual cosa si ottiene per riscaldamento a temperature superiori ai 60 °C. Al di sopra di questa temperatura, infatti, quei granuli dalla struttura interna molto complessa che costituiscono l'amido rigonfiano e si rompono, consentendo la dispersione in soluzione delle molecole di amilosio e di amilopectina. L'amido gelatinizzato ha un'elevata viscosità, che viene rapidamente ridotta sfruttando la capacità dell'α-amilasi di attaccare le catene poliglucosidiche all'interno della catena (per questo si dice che è un endoenzima) e non a un'estremità (come fanno gli esoenzimi, quali la glucoamilasi). Conseguentemente, l'attacco da parte dell'enzima, oltre che ridurre la viscosità della soluzione, ha l'effetto di prevenire la precipitazione dell'amilosio (retrogradazione) e di fornire più catene terminali non riducenti per la successiva operazione di saccarificazione enzimatica. Per effettuare il processo si impiega una α-amilasi batterica da Bacillus subtilis o da Bacillus amyloliquefaciens addizionata a una sospensione di amido al 30-40% (peso secco) gelatinizzata a 60-90 °C, cui si aggiungono ioni calcio nella misura di 100-150 ppm per stabilizzare l'enzima. L'azione idrolitica avviene in un reattore a tino ben agitato, tenuto per 60 minuti a 85-90 °C, ed è quindi necessario impiegare un enzima in grado di sopportare questa temperatura. Segue poi un riscaldamento (con vapore sotto pressione) a 140 °C per cinque minuti, che serve a coagulare le mucillagini e a favorire la filtrazione dello sciroppo. Si ottengono migliori risultati se dopo il riscaldamento a 140 °C si fa seguire un secondo trattamento con α-amilasi per 30-60 minuti a 85 °C. Si ottiene uno sciroppo a DE (Dextrose Equivalent) 12-20, che viene inviato alla saccarificazione. Il DE, o ‛valore equivalente di destrosio', viene usato per caratterizzare il grado di degradazione dell'amido; più speficamente rappresenta il potere riducente dell'idrolizzato amidaceo quando confrontato con destrosio puro, considerato come riferimento al 100% di potere riducente.
Il processo di liquefazione in continuo rappresenta un'evoluzione rispetto a quello a batch, sia sul piano economico, sia su quello tecnico. Questa possibilità è stata offerta dopo la scoperta di una α-amilasi da Bacillus licheniformis, termostabile per alcuni minuti alla temperatura di 105 °C. Anche questo enzima è una metallo-proteina, che però ha lo ione calcio inserito stabilmente nella struttura proteica e quindi richiede l'aggiunta di quantità molto più modeste (10 ppm) di ioni calcio, caratteristica che rappresenta un notevole vantaggio tecnologico se il glucosio deve successivamente essere isomerizzato a fruttosio (v. sotto). Il processo consiste nell'aggiungere l'enzima a una sospensione di amido al 30-40%, prima che passi nel cosiddetto jet-cooker, dove rimane a 105 °C per 5-10 minuti. Dopo di che l'idrolizzato viene raccolto in un recipiente a 95 °C e tenuto a questa temperatura per 1-2 ore, fino ad avere il desiderato DE. Il jet-cooker è generalmente un reattore tubolare in cui il vapore è introdotto nella sospensione amidacea con un'azione di mescolamento tale da consentire il riscaldamento immediato della massa alla temperatura prefissata e quindi una rapida gelatinizzazione. Il vantaggio principale di questo metodo è che consente l'utilizzazione di impianti precedentemente progettati per l'idrolisi acida, senza eccessivi investimenti addizionali; lo svantaggio è costituito dall'elevato consumo di vapore. La saccarificazione dell'amido, che segue la liquefazione, avviene in condizioni operative che differiscono soprattutto per il pH. Due sono gli enzimi utilizzati nella saccarificazione: l'amiloglucosidasi da Aspergillus niger e l'amilasi fungina da Aspergillus oryzae. Questi due enzimi, se usati separatamente o in combinazione, sono in grado di produrre una grande varietà di prodotti (v. tab. VI).
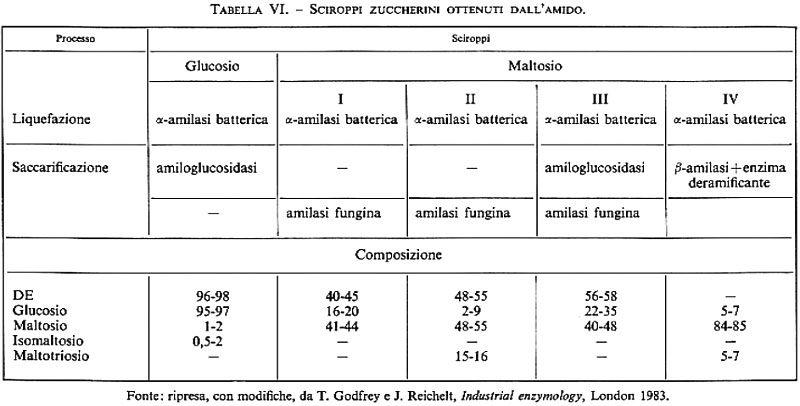
L'amiloglucosidasi è un enzima esocellulare che produce sciroppi di glucosio a 96-98 DE. L'amilasi fungina produce, per idrolisi dei legami α-1-4-glucosidici, sciroppi di maltosio a basso contenuto in destrosio (il maltosio è un disaccaride formato da due molecole di glucosio). Questo enzima differisce dalle α-amilasi batteriche per avere una specificità più ampia di substrato, capace di agire sia come enzima ‛destrinizzante' (le destrine sono il prodotto della iniziale liquefazione dell'amido), sia come enzima ‛saccarificante'.
Il processo di saccarificazione delle destrine a glucosio richiede tempi lunghi di reazione (48-96 ore) e viene eseguito in grandi tini muniti di agitatore. Questi tini sono utilizzati generalmente come reattori a batch. Tentativi di disporre in serie reattori alimentati in continuo non hanno dato risultati positivi, per la difficoltà di ottenere prodotti a DE elevato e costante. Il processo consiste nell'abbassare con acido cloridrico fino a 3,8-4,2 il pH del liquido proveniente dalla liquefazione, e quindi aggiungere l'amiloglucosidasi. La temperatura deve essere accuratamente mantenuta a 60 °C; temperature più alte disattivano l'enzima, più basse rallentano la velocità di idrolisi e aumentano il rischio di contaminazioni microbiche. L'amiloglucosidasi è anche capace di legare due molecole di glucosio per formare maltosio (legame α-1-4) o isomaltosio (legame α-1-6). Per questo motivo, appena raggiunto il DE desiderato (per esempio 98), si riscalda il liquido saccarificato a 80 °C per inattivare l'amiloglucosidasi.
Con l'amilasi fungina da Aspergillus oryzae si ottengono sciroppi contenenti fino al 55% di maltosio. Per ottenere sciroppi a più elevato tenore in maltosio, si ferma la liquefazione con α-amilasi quando il DE raggiunge il valore di 5-10, e la saccarificazione viene fatta con β-amilasi (vegetale o microbica). Poiché questo esoenzima stacca molecole di maltosio finché non incontra una ramificazione costituita da un legame α-1-6-glucosidico, le rese sarebbero molto basse: per questo motivo si aggiunge anche un apposito enzima deramificante (tipo pullulanasi o isoamilasi), che permette di ottenere sino all'85% di maltosio.
b) Isomerizzazione del glucosio
Il processo basato su enzimi immobilizzati che più di ogni altro ha avuto successo commerciale è quello della produzione di isomerosio tramite l'enzima glucosioisomerasi. La glucosioisomerasi è un enzima intracellulare generalmente stabile anche a temperature piuttosto elevate (60 °C) e quindi ideale per il suo impiego allo stato immobilizzato. L'enzima catalizza l'isomerizzazione parziale del glucosio a fruttosio, producendo una miscela (isomerosio) - molto simile allo zucchero invertito, composto di glucosio e fruttosio in parti uguali, ottenuto per idrolisi acida o enzimatica del saccarosio - che ha trovato larga applicazione in molti settori dell'industria alimentare. La storia dello sviluppo di questo processo catalitico, che ha portato sul mercato un nuovo prodotto in competizione con il saccarosio, è particolarmente indicativa di come i fattori economici risultino determinanti per il successo di un processo. La tecnologia dell'isomerizzazione enzimatica del glucosio era pronta per una sua applicazione negli Stati Uniti sin dal 1970, ma in quel periodo il saccarosio aveva prezzi molto bassi. Quando nel 1974, per diverse cause anche di natura politica, il prezzo dello zucchero sui mercati mondiali subì un'impennata, alcune società americane operanti nell'industria di trasformazione dell'amido di mais trovarono conveniente sperimentare la tecnologia enzimatica nei loro impianti produttivi. Da allora, l'isomerosio ha avuto una continua espansione di mercato (v. tab. VII), favorita dai continui miglioramenti apportati alla tecnologia enzimatica.
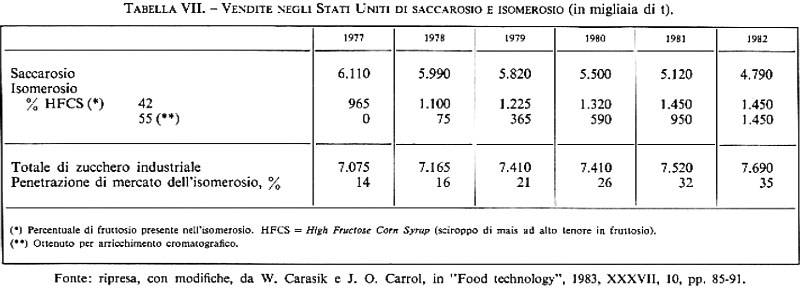
Il processo di isomerizzazione enzimatica del glucosio è oggi molto diffuso negli Stati Uniti, in Canada, nella Corea del Sud e in Giappone, mentre lo è poco in Europa, perché la Comunità Economica Europea (CEE), per proteggere la produzione di saccarosio, ha imposto all'isomerosio delle penalizzazioni fiscali.
Il primo impianto industriale per l'isomerizzazione del glucosio è stato realizzato dalla Clinton Corn Processing Co. (Stati Uniti); l'enzima, estratto da Streptomyces sp., era immobilizzato mediante adsorbimento su un supporto di DEAE-cellulosa. L'impianto, alimentato in continuo da uno sciroppo di glucosio al 30-50% di peso secco, consisteva in alcuni reattori a letto fisso disposti in serie, ciascuno dei quali conteneva il catalizzatore enzimatico, la cui attività era tanto più bassa quanto più a lungo esso era stato utilizzato nel processo. Quando l'attività dell'ultimo reattore della serie scendeva al disotto di un valore prefissato sulla base di alcuni parametri economici, il reattore veniva eliminato e sostituito all'inizio della serie con un altro contenente il catalizzatore al massimo dell'attività: si garantiva in tal modo una produttività costante nel tempo. La glucosioisomerasi da Streptomyces è inibita dagli ioni Ca2+ (che devono essere quindi allontanati dallo sciroppo da isomerizzare), mentre richiede la presenza di ioni Co2+ (utilizzando questo enzima sono quindi necessari due trattamenti di deionizzazione, dato che gli ioni Co2+ devono essere assenti dal prodotto finito perché tossici).
Gli attuali impianti industriali utilizzano in prevalenza la tecnologia sviluppata dall'industria danese NOVO. Il successo di questa tecnologia è legato alla scoperta di un microrganismo (Bacillus coagulans) alto produttore di enzima, ottenuto per fermentazione continua e senza necessità di usare induttori nel terreno di coltura. L'enzima è poi immobilizzato con una tecnica molto semplice, che sfrutta la proprietà della glutaraldeide di reticolare le cellule intere e gli omogenati cellulari, per ottenere, dopo vari trattamenti (macinazione, estrusione ed essiccamento della massa cellulare), il catalizzatore immobilizzato sotto forma di piccoli cilindri. La tecnologia NOVO, sia di preparazione sia di utilizzo del catalizzatore, ha subito numerose modifiche a partire dalla prima introduzione del procedimento sul mercato. L'applicazione del catalizzatore è passata così dai primi reattori a batch a quelli a letto fisso alimentati in continuo. Ciò è stato reso possibile apportando considerevoli miglioramenti alle proprietà meccaniche del catalizzatore: in particolare ne è stata aumentata la resistenza alla compressione, riducendo quei fenomeni di compattazione del letto catalitico che portavano a cadute di pressione nel reattore e a conseguenti fermate dell'impianto.
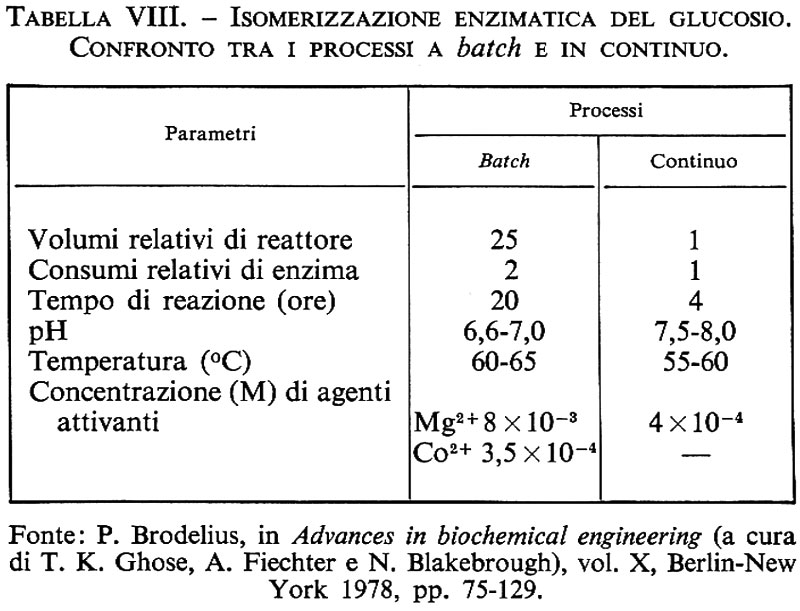
I vantaggi derivanti dal processo in continuo (v. tab. VIII) rispetto a quello a batch possono essere così riassunti: minore formazione di sottoprodotti colorati, nessuna aggiunta di ioni Co2+, ridotti volumi di impianto, minore consumo di enzima. Il conseguimento di questi risultati positivi è condizionato tuttavia a un maggior controllo del processo e più in particolare di alcuni parametri operativi (pH, temperatura, purezza, concentrazione e velocità di flusso dello sciroppo di alimentazione).
c) Idrolisi della penicillina G
La catalisi enzimatica sembra rispondere particolarmente bene alle esigenze produttive dell'industria farmaceutica, perché le reazioni di sintesi di composti farmaceutici richiedono generalmente alcune proprietà tipiche dei catalizzatori biologici, quali: elevata attività associata a specificità, stereoselettività e capacità di agire in condizioni molto blande di reazione.
L'idrolisi della benzilpenicillina (penicillina G) ad acido 6-amminopenicillanico (6-APA) e acido fenilacetico è catalizzata dall'enzima penicillinacilasi. Il 6-APA è il prodotto di partenza per la sintesi delle penicilline semisintetiche, composti nei quali l'acido fenilacetico è sostituito da altri sostituenti che conferiscono alla molecola una maggiore resistenza alla disattivazione nelle condizioni di impiego in vivo (resistenza alla penicillinasi) e un più ampio spettro di azione terapeutica.
L'importanza commerciale del 6-APA come intermedio per la sintesi di penicilline semisintetiche, quali l'ampicillina, l'amossicillina, ecc., ha fatto concentrare molti sforzi sull'ottimizzazione dell'idrolisi sia chimica sia enzimatica della penicillina G prodotta su larga scala per via fermentativa. Per molti anni il metodo chimico di idrolisi è prevalso, a livello industriale, su quello enzimatico. La scoperta di ceppi microbici alto-produttori di penicillinacilasi e lo sviluppo della tecnologia di immobilizzazione dei biocatalizzatori hanno reso poi altamente competitivo il processo enzimatico, che gradualmente ha soppiantato quello chimico. Oggi numerose industrie farmaceutiche hanno sviluppato in proprio un loro processo enzimatico, di cui ovviamente mantengono segrete le caratteristiche operative. Alcune società di ingegneria hanno, al contrario, pubblicizzato il proprio processo, dovendolo collocare sul mercato.
d) Risoluzione ottica di amminoacidi
L'uso degli amminoacidi come additivi (fortificanti proteici) per l'industria alimentare e come agenti terapeutici o dietetici è andato crescendo in questi ultimi anni. Con l'eccezione della glicina, tutti gli altri amminoacidi sono otticamente attivi, ma dei due isomeri solo quello a configurazione L risulta fisiologicamente attivo.
Gli amminoacidi sono prodotti sia con metodi fermentativi che con metodi di sintesi chimica. Questi ultimi portano però alla formazione di miscele racemiche di isomeri L e D, la cui separazione risulta generalmente difficile e costosa. Tra i metodi di risoluzione ottica, quello enzimatico con amminoacilasi risulta molto vantaggioso. Gli amminoacidi racemici sono prima trasformati in acetilderivati e poi mediante azione enzimatica il solo N-acetil-L-amminoacido viene deacetilato a L-amminoacido, mentre rimane inalterato l'N-acetil-D-amminoacido. L'isomero L dell'amminoacido viene separato per cristallizzazione, mentre l'N-acetil-D-amminoacido viene racemizzato e riciclato. Questo processo è stato applicato per la prima volta nel 1954 in Giappone dall'industria farmaceutica Tanabe Seiyaku Co. Ltd. e utilizzato per la produzione di vari amminoacidi fino al 1969. La reazione era ottenuta a batch incubando una miscela contenente il substrato e l'enzima in soluzione estratto da Aspergillus oryzae. La procedura presentava l'inconveniente che si dovevano rimuovere, al termine della reazione, sia l'enzima, mediante trattamenti termici o variazioni di pH, sia le altre proteine contaminanti, per poter alla fine isolare l'amminoacido puro. Per ovviare a questi problemi e ridurre i costi del catalizzatore, la Tanabe realizzò alla fine degli anni sessanta il primo impianto industriale basato su enzimi immobilizzati.
La procedura di immobilizzazione consiste nel far adsorbire mediante legami ionici l'amminoacilasi al polimero DEAE-Sephadex. Il polimero va poi introdotto all'interno di un reattore a colonna da 1 m3 di volume alimentato in continuo dalla soluzione di substrato.
Una volta avvenuta la reazione enzimatica, la soluzione effluente dalla colonna è concentrata e l'L-amminoacido separato per cristallizzazione, mentre l'acetil-D-amminoacido rimasto nelle acque madri è racemizzato per riscaldamento e riciclato all'impianto di separazione. L'impianto ha raggiunto una produttività mensile, per i vari amminoacidi, compresa tra le 2,1 e le 6,4 tonnellate.
Con questo processo sono stati prodotti vari amminoacidi, quali L-valina, L-metionina, L-fenilalanina, con un risparmio del 60% dei costi rispetto al precedente sistema operante a batch con l'enzima (non riutilizzabile) impiegato in soluzione.
e) Idrolisi del lattosio
L'industria lattiero-casearia utilizza gli enzimi in più processi: le proteasi (rennina animale e microbica, pepsina) nella caseificazione del latte per la produzione di formaggi; le lipasi per conferire al formaggio alcuni sapori caratteristici; la catalasi per ridurre gli effetti nocivi della ‛pastorizzazione a freddo' del latte con acqua ossigenata; il lisozima per evitare reazioni indesiderate durante la stagionatura di alcuni formaggi; la β-galattosidasi (lattasi) per idrolizzare il lattosio contenuto sia nel latte che nel siero. Solo quest'ultimo processo utilizza l'enzima in forma immobilizzata. L'interesse commerciale per l'applicazione della β-galattosidasi deriva dalla constatazione che una certa percentuale della popolazione mondiale è intollerante al lattosio e non può nutrirsi di latte per mancanza, dovuta a una carenza congenita, di lattasi nella mucosa intestinale. Questa deficienza enzimatica, presente in alcuni gruppi etnici, crea problemi non solo nutrizionali, ma anche di patologia generale, soprattutto negli individui in età neonatale e negli anziani. L'idrolisi preventiva del lattosio a glucosio e galattosio elimina questo inconveniente, lasciando inalterate le proprietà organolettiche e nutrizionali dell'alimento. Lo stesso lattosio è presente in quantità rilevanti nei sieri ottenuti come sottoprodotto della produzione di formaggio. L'idrolisi del lattosio valorizza i sieri, rendendo possibile la loro utilizzazione nell'industria gelatiera, sotto forma di sciroppi di glucosio e galattosio. Gelati ottimi, per struttura fisica e qualità organolettiche, sono preparati aggiungendo sciroppo dolce di siero fino al 20% del peso totale dei solidi.
La β-galattosidasi viene generalmente estratta dal lievito Saccharomyces lactis e dalla muffa Aspergillus niger. La scelta tra le due fonti enzimatiche è dettata dal tipo di applicazione: l'idrolisi dei sieri acidi viene eseguita solo con l'enzima da muffa, che ha un optimum di pH coincidente con quello dei sieri, mentre per il latte si usa l'enzima da lievito, che ha un optimum di pH intorno a 7. Entrambi gli enzimi sono stati immobilizzati su supporti sia inorganici sia organici, mentre cellule intere di lievito sono state immobilizzate per inglobamento in sferette di acetato di cellulosa.
Il primo impianto industriale per la delattosizzazione del latte è stato realizzato, in Italia, alla Centrale del latte di Milano, utilizzando la tecnologia Snamprogetti di immobilizzazione di enzimi in fibre di triacetato di cellulosa.
L'impianto risulta costituito da un reattore a batch contenente, oltre al latte scremato e sterilizzato, anche le fibre enzimatiche sotto forma di piccoli spezzoni della lunghezza di circa 3 cm. Dopo circa 20 ore di agitazione alla temperatura di 5-8 °C, quando l'80% del lattosio iniziale è stato idrolizzato, sia il latte sia le fibre sono rimossi dal reattore e inviati a una centrifuga che ha la funzione di recuperare tutto il latte e separarlo dal catalizzatore enzimatico. Le fibre, dopo accurato lavaggio con acqua tamponata, sono rinviate nel reattore per la successiva idrolisi, mentre il latte, prima di essere confezionato, viene riaddizionato della parte grassa e pastorizzato. Un originale sistema reattoristico è stato di recente sviluppato alla Centrale del latte di Milano con lo scopo di semplificare il processo e di ridurre le dimensioni d'impianto. Il nuovo reattore, formato da un recipiente cilindrico forato, contenente le fibre a elevata densità (60-70 kg/m3), è alimentato in continuo, attraverso dei fori situati nella parte interna del cilindro, con latte che, dopo aver attraversato gli strati di fibra enzimatica in cui avviene la reazione, viene riciclato al serbatoio di raccolta. Il rapporto tra il volume di latte da trattare e la quantità di fibra enzimatica è regolato in modo da ottenere in 20 ore l'idrolisi desiderata di lattosio e disporre di un sufficiente intervallo di tempo (4 ore) per il lavaggio sia della fibra sia dei recipienti prima della successiva idrolisi; i lavaggi si rendono generalmente necessari per mantenere sotto controllo la contaminazione batterica.
L'idrolisi su scala commerciale del lattosio dei sieri è stata realizzata dalla Corning Glass, sfruttando la tecnologia di immobilizzazione chimica dell'enzima su un supporto di vetro poroso. L'impianto risulta formato, oltre che da un reattore enzimatico a letto fisso, anche da una serie di apparecchiature ancillari per il pretrattamento (pastorizzazione, ultrafiltrazione, demineralizzazione) del siero destinato all'idrolisi enzimatica. Le condizioni operative del processo sono molto simili a quelle per l'idrolisi del lattosio nel latte.
6. Prospettive
Una nuova fase dell'enzimologia è iniziata quando la tecnologia del DNA ricombinante ha fornito nuovi concetti e nuovi strumenti per fare produrre ai batteri nella quantità voluta ogni enzima presente in natura. Ma le maggiori speranze per lo sviluppo della tecnologia enzimatica in campo industriale provengono dalla prospettiva di fabbricare nuovi enzimi con proprietà non più rispondenti alle esigenze metaboliche degli organismi viventi, bensì alle esigenze operative della catalisi industriale. Questa opportunità è offerta dalle nuove tecniche di sintesi chimica di frammenti di DNA, che permettono, con l'ausilio di specifici enzimi, di costruire geni sintetici completamente nuovi o di modificare quelli naturali mediante inserzioni, delezioni e sostituzioni di oligonucleotidi in singoli siti specifici della struttura genica.
Con i nuovi geni, donati e fatti esprimere attraverso le tecniche di ingegneria genetica, si potrebbero produrre enzimi completamente nuovi, sia per sequenza amminoacidica sia per proprietà strutturali, conferendo al catalizzatore le caratteristiche richieste dalle applicazioni industriali. Questo approccio totalmente nuovo per costruire i futuri enzimi si basa sulla possibilità di correlare sintesi genica con sequenza amminoacidica e conseguente struttura conformazionale. In teoria sarebbe sufficiente conoscere le caratteristiche strutturali associate a ogni specifica proprietà funzionale per poter creare, avvalendosi di metodi rapidi (computer graphics for model building) in grado di predire la struttura tridimensionale di una proteina a partire dalla sua sequenza amminoacidica, un enzima capace di associare tutte le proprietà richieste da un processo catalitico industriale. Questa prospettiva ha portato a un forte ritorno di interesse per le tecnologie enzimatiche, generando anche un'intensa attività di ricerca nel campo della catalisi biomimetica, che ha come scopo la riproduzione per via chimica, attraverso strutture artificiali non proteiche, del sito attivo di un enzima e del suo meccanismo di azione. In questo contesto, dopo i primi lavori di F. Cramer sulle ciclodestrine naturali o chimicamente modificate per facilitarvi l'inserimento di gruppi catalitici funzionali, la ricerca si è orientata su catalizzatori interamente artificiali, alcuni dei quali hanno mostrato specificità e velocità catalitiche dell'ordine di quelle proprie degli enzimi naturali. Ma sia l'ingegneria genetica applicata all'enzimologia sia la catalisi mimetico-enzimatica prospettano un futuro per le tecnologie enzimatiche tanto affascinante quanto ancora lontano da una pratica realizzazione, data la complessità dei problemi ancora da risolvere. Nel primo caso infatti si è visto che non è sufficiente sintetizzare un polipeptide contenente il solo sito attivo per avere un enzima, perché la selettività e la stabilità conformazionale sono fornite dal resto della molecola proteica, una struttura così complessa che non si è ancora in grado di riprodurla artificialmente. Nel caso invece della mimesi enzimatica, mentre sono state eguagliate con alcuni derivati delle ciclodestrine sia la velocità di reazione sia la specificità caratteristica degli enzimi, non si è ancora riusciti a preparare, per sintesi totale, molecole non polipeptidiche capaci di mimare completamente l'azione enzimatica, e non solo alcune fasi di essa.
Al contrario, i reattori enzimatici detti di seconda generazione, in quanto capaci di catalizzare complesse reazioni di sintesi o semplici biotrasformazioni in sistemi bifasici, sono una realtà di oggi, anche se ancora a livello di laboratorio. L'interesse per questi sistemi catalitici formati da acqua e da un solvente organico immiscibile con l'acqua deriva dalla prospettiva di estendere la catalisi enzimatica a composti insolubili o poco solubili in acqua, come gli steroidi, gli oli, i grassi, gli idrocarburi, ecc. La reazione catalitica avviene quando, per agitazione o mescolamento, il substrato idrofobico sciolto nel solvente organico viene a contatto con l'enzima o la cellula presenti nella fase acquosa. Un metodo che permette di ottenere elevata superficie interfacciale, tale da consentire rapide velocità di trasferimento, sia per il substrato sia per il prodotto, tra le due fasi, è quello delle micelle inverse: piccole goccioline di acqua disperse nella fase organica e contenenti l'enzima stabilizzato da un contorno di agenti tensioattivi. I vantaggi dei sistemi catalitici bifasici possono essere così riassunti: a) riduzione dei volumi di reazione per l'elevata solubilità dei reagenti; b) facilità di separazione del biocatalizzatore dal mezzo di reazione anche quando questo non è in forma immobilizzata; c) riduzione dei fenomeni di inibizione da substrato o da prodotto per la loro bassa concentrazione nella fase acquosa; d) possibilità, mediante enzimi idrolitici, di sintesi di peptidi e di esteri, grazie allo spostamento dell'equilibrio termodinamico.
Anche se la tecnologia della catalisi bifasica non è ancora perfezionata e restano da eliminare alcuni inconvenienti, sembra certo che entro pochi anni i reattori biocatalitici polifasici entreranno nei processi commerciali e contribuiranno a rendere competitivi molti processi enzimatici valutati oggi troppo costosi e sofisticati per competere con quelli convenzionali.
bibliografia
Breslow, R., Artificial enzymes, in ‟Science", 1982, CCXVIII, pp. 532-537.
Carrea, G., Biocatalysis in water-organic solvent two-phase systems, in ‟Trends in biotechnology", 1984, II, pp. 102-106.
Chibata, I., Immobilized enzymes, New York 1978.
Chibata, I., Development of enzyme engineering-application of immobilized cell system, in Food process engineering (a cura di P. Linko), vol. II, London 1979, pp. 1-26.
Cooney, C. L., Bioreactors. Design and operation, in ‟Science", 1983, CCXIX, pp. 728-733.
Cramer, F., Einschlussverbindungen, Berlin 1954.
Drexler, K. E., Molecular engineering. An approach to the development of general capabilities for molecular manipulation, in ‟Proceedings of the National Academy of Sciences", 1981, CXXVII, 9, pp. 5275-5278.
Fukui, S., Tanabe, A., Application of bio-catalysis immobilized by prepolymer methods, in Advances in biochemical engineering/biotechnology (a cura di A. Fiechter), vol. XXIX, Heidelberg 1984, pp. 1-34.
Hilhorst, R., Laane, C., Veeger, C., Reversed micelles as a medium for enzyme-catalyzed synthesis of apolar compounds, in Innovations in biotechnology (a cura di E. H. Houwkin), vol. XX, Amsterdam 1984, pp. 81-83.
Jensen, B. F., Norman, B. E., Bacillus acidopullulytriens pullulanase: application and regulatory aspects for use in the food industry, in ‟Process biochemistry", 1984, XIX, 4, pp. 129-134.
Klibanov, A. M., Immobilized enzymes and cells as practical catalysts, in ‟Science", 1983, CCXIX, pp. 722-727.
Linko, P., Linko, Y., Application of immobilized microbial cells, in Applied biochemistry and bioengineering (a cura di I. Chibata e L. Wingard), vol. IV, New York 1983, pp. 54-136.
Marconi, W., Bartoli, F., Cecere, F., Galli, G., Morisi, F., Synthesis of penicillins and cephalosporins by penicillin acylase entrapped in fibres, in ‟Agricultural and biological chemistry", 1975, XXXIX, 1, pp. 277-279.
Marconi, W., Bartoli, F., Morisi, F., Improved whey treatment by immobilized lactase, in Enzyme engineering (a cura di L. Weetell), vol. V, New York-London 1980, pp. 269-278.
Marconi, W., Morisi, F., Industrial applications of fiber-entrapped enzymes, in Applied biochemistry and bioengineering (a cura di L. Wingard, E. Katchalski-Katzir e L. Goldstein), vol. II, New York 1979, pp. 219-258.
Meier, P., Luisi, P. L., Micellar solubilization of biopolymers in hydrocarbon solvents. II. The case of horse liver alcohol dehydrogenase, in ‟Journal of solid-phase biochemistry", 1980, V, 4, pp. 269-282.
Messing, R. A., Carriers for immobilized biologically active systems, in Advances in biochemical engineering (a cura di A. Fletcher), vol. X, Heidelberg 1978, pp. 51-73.
Mosbach, K., Immobilized enzymes, in Methods in enzymology (a cura di S. P. Colowick e N. O. Kaplan), vol. XLIV, New York 1976.
Ostergaad, J., Knudsen, S. L., Use of Sweetzyme in industrial continuous isomerization, in ‟Die Stacke", 1976, XXVIII, pp. 350-356.
Oyama, K., Kihara, K., A new horizon for enzyme technology, in ‟Chemtech", 1984, XIV, pp. 100-105.
Pastore, M., Morisi, F., Reduction of lactose in milk by entrapped α-galactosidase. IV. Results of long experments with a pilot plant, in Enzyme engineering (a cura di E. K. Pye e H. Weetall), vol. III, New York-London 1978, pp. 537-542.
Solomon, B., Starch hydrolysis by immobilized enzymes. Industrial applications, in Advances in biochemical engineering (a cura di A. Fiechter), vol. X, Heidelberg 1978, pp. 131-177.
Ulmer, K., Protein engineering, in ‟Science", 1983, CCXIX, pp. 666-671.
Venkatasubramanian, K., Karkare, S. B., Vieth, W. R., Chemical engineering analysis of immobilized-cell systems, in Applied biochemistry and bioengineering (a cura di I. Chibata e L. Wingard), vol. IV, New York 1983, pp. 312-351.
Winter, G., Fersth, A. R., Wilkinson, A. J., Zoller, M., Smith, M., Redesigning enzyme structure by site-directed mutagenesis: tyrosyl tRNA synthetase and ATP binding, in ‟Nature", 1983, CCIC, pp. 756-758.