
Colloidi
Colloidi
Nel 1907, Wolfang Ostwald definì i sistemi colloidali "il mondo delle dimensioni trascurate", ponendo l'accento sul fatto che le dimensioni dei componenti di tali sistemi non hanno né l'estensione tipica delle molecole, né quella dei materiali cristallini. La definizione generale di colloide poggia ancora oggi sulla dimensione lineare dei suoi componenti ‒ compresa tra 1 nm e 1 μm ‒ considerata la caratteristica fondamentale delle particelle che lo compongono. Per questa ragione, lo studio dei colloidi è fortemente interdisciplinare e abbraccia la chimica, la fisica, la biologia, la scienza dei materiali, e molte altre scienze. La chimica dei colloidi è dunque la scienza sia delle grandi molecole, sia dei sistemi multifase in stato di elevata suddivisione. Nello studio di tali sistemi si incontrano la scienza dei colloidi e quella delle interfasi: infatti, tanto più una certa fase è suddivisa, tanto maggiore è l'estensione di area superficiale a parità di peso del campione, e quindi tanto più rilevanti sono i fenomeni che si verificano al contatto tra la fase suddivisa e le altre fasi (interfase).
Non esiste ramo dell'industria chimica che non coinvolga proprietà o metodologie dei colloidi e delle interfasi. Inoltre, sistemi nell'intervallo dimensionale dei colloidi si incontrano regolarmente nelle scienze ambientali (aerosol, nebbie, schiume, ecc.), nella scienza dei materiali (leghe, cementi, fibre, ecc.) e nei prodotti per uso domestico e cosmetico (latte, maionese, ecc.). Molte sospensioni (vale a dire dispersioni di solidi nei liquidi), come le vernici, gli inchiostri, le lacche, gli anticrittogamici, ecc., sono di uso comune; altre, come per esempio le sospensioni di minerali in acqua, che si prestano al trasporto in condotta, lo diverranno presto. Altri tipi di sistemi, come le schiume strutturali e, in senso lato, i materiali compositi, sostituiranno tra breve i materiali ferrosi e le leghe di alluminio; altri ancora ‒ per esempio, i liposomi, le micelle o le emulsioni ‒ sono già entrati nella preparazione di alcuni farmaci, potenziandone enormemente le proprietà. A testimonianza della grande importanza dei colloidi potremmo citare numerosi altri esempi, come quelli biologici (sangue, cellule, lecitine o fosfolipidi di membrana).
Storicamente, la scienza dei colloidi nasce nel 1845 con l'opera di Francesco Selmi, il quale descrisse alcune "strane proprietà" di soluzioni in acqua di zolfo, cloruro di argento e blu di Prussia. Nel 1861, Thomas Graham coniò il termine colloide ‒ che deriva dalla parola greca kálla e significa proprio materiale colloso ‒ per indicare un insieme amorfo e mal definito. Graham riteneva che esistessero due stati diversi della materia con proprietà chimico-fisiche completamente differenti, i cristalloidi e appunto i colloidi, assimilabili i primi ai minerali e i secondi a una massa organizzata. In realtà, a seconda della specifica applicazione del colloide, le loro condizioni di preparazione possono essere modulate in modo da privilegiare alternativamente l'estensione del contatto interfasale o la crescita dei cristalliti. Tuttavia, le proprietà di maggiore interesse dei sistemi colloidali si individuano nel comportamento del sistema nel suo complesso (reologia, proprietà ottiche, stabilità) e, nel caso dei sistemi multifase, nello stato superficiale e nella reattività interfasale.
Classificazione dei sistemi colloidali
Possono essere considerati colloidi sia le grandi molecole, sia i materiali (i solidi, i liquidi e anche i gas) in stato di elevata suddivisione, a patto che le componenti del sistema (molecole o particelle) abbiano dimensioni comprese tra 1 nm e 1 μm. La differenza tra questi due tipi di colloidi è collegata alla relazione esistente tra la particella colloidale e il mezzo in cui essa si trova. Per esempio, i colloidi macromolecolari danno luogo a vere soluzioni ‒ nel senso termodinamico del termine, ossia attraverso un processo spontaneo ‒ con il mezzo che li circonda. Una fase suddivisa, d'altra parte, forma con il mezzo un sistema almeno bifasico, con la presenza di un confine di fase esteso. I sistemi colloidali monofasici sono definiti liofili, quelli bifasici liofobi. I colloidi liofili possono formare vere soluzioni, che si creano spontaneamente quando soluto e solvente sono messi in contatto. In assenza di cambiamenti chimici o di temperatura, un sistema colloidale liofilo è stabile indefinitamente. Viceversa, quando due fasi sono portate in contatto, esse non danno origine spontaneamente a una dispersione finemente suddivisa, anzi, se si lascia riposare il sistema, si instaura il processo opposto: per esempio, olio e acqua possono essere mescolati vigorosamente per produrre un fluido eterogeneo e non trasparente, ma, a riposo, il sistema evolverà verso due strati omogenei e trasparenti.
Sistemi colloidali liofili

Per descrivere i sistemi colloidali liofili si può fare riferimento a una classe molto importante di composti: i tensioattivi, che sono formati da molecole anfifiliche. Queste sono costituite da una parte polare, affine all'acqua nella quale la molecola è solubile, e da una 'coda' idrocarburica apolare, poco compatibile con l'acqua e tendente a determinare una reattività particolare. Tale situazione fa sì che le molecole anfifiliche tendano ad andare incontro, da una parte, a fenomeni di adsorbimento (ossia a localizzarsi in corrispondenza dei contatti interfasali), dall'altra, a fenomeni di autoassociazione in soluzione acquosa. Nella fig. 2A è riportato l'andamento della tensione superficiale γ (misurata all'interfase aria-acqua) in funzione della concentrazione di tensioattivo in soluzione. Si osservi che, nella regione di concentrazioni più basse, la tensione superficiale diminuisce in seguito all'adsorbimento del monomero di tensioattivo nell'interfase aria-acqua, in accordo con l'equazione di adsorbimento di Gibbs a temperatura e pressione costanti,
[1] formula
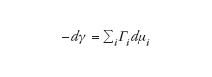
dove Γi sono gli eccessi superficiali e μi i potenziali chimici nella fase massiva. Al crescere della concentrazione del tensioattivo in soluzione, da un valore critico in poi la tensione superficiale diviene pressoché costante e nel sistema si verificano brusche variazioni di numerosi parametri chimico-fisici (fig. 2B), quali il potere di diffusione della luce, la conducibilità, la pressione osmotica.
In corrispondenza di questo specifico valore di concentrazione detto CMC (Critical micelle concentration), le forze autoassociative dei monomeri di tensioattivo portano alla formazione spontanea di aggregati tridimensionali (micelle) perfettamente stabili e compatibili con il solvente, organizzati con la parte polare verso la soluzione e la coda idrocarburica verso l'interno. Una soluzione micellare rappresenta un tipico esempio di colloide liofilo: esso, infatti, si forma spontaneamente, è omogeneo ed è stabile. Il numero di aggregazione dei monomeri, la dimensione e la forma delle micelle possono variare profondamente in funzione del tipo di sistema, nonché della natura e della concentrazione del tensioattivo. Le molecole di tensioattivi non ionici possono dare luogo ad aggregati costituiti da 1000 o più monomeri, mentre nel caso dei tensioattivi ionici, in conseguenza delle interazioni repulsive che si instaurano tra le teste polari, il numero dei monomeri negli aggregati oscilla tra 10 e 100.
La popolazione delle micelle è spesso polidispersa e la loro forma varia con la concentrazione. Sebbene le micelle possano anche essere perfettamente sferiche, è più frequente che nella regione della concentrazione critica di micellizzazione esse abbiano la forma di una sfera appiattita. A concentrazioni molto maggiori, alcuni sistemi danno origine a strutture costituite da strati molecolari paralleli, chiamate micelle lamellari; in questo caso, i monomeri giacciono perpendicolarmente agli strati, con le teste polari disposte verso l'esterno se si trovano in una soluzione acquosa, verso l'interno se in un mezzo apolare. Le micelle lamellari mostrano una stretta somiglianza con le membrane biologiche e sono spesso utilizzate come sistemi modello in ricerche di base. Per concentrazioni elevate di tensioattivo, le micelle possono subire un'ulteriore organizzazione in aggregati tridimensionali organizzati spazialmente, i cristalli liquidi isotropici. L'importanza applicativa e tecnologica dei sistemi micellari è enorme. Infatti, poiché il core della micella ha le caratteristiche di un idrocarburo liquido e, come mostrano i dati di risonanza magnetica nucleare, le code idrocarburiche al suo interno sono poco meno mobili che non nella fase massiva, le micelle possono fungere da solvente-serbatoio per sostanze altrimenti non solubili nel mezzo (esempi si hanno nella catalisi micellare, nei processi attraverso cui si veicolano i farmaci, nel recupero assistito del petrolio, ecc.).
Sistemi colloidali liofobi

La caratteristica comune a tutti i sistemi colloidali liofobi bifasici (tab. 1) è la presenza di un elevato contatto interfasale. Consideriamo una massa sferica di 1 g di peso e 1 cm di raggio. Supponiamo di suddividere questo materiale in sfere di raggio progressivamente più piccolo, assumendo che la densità non vari. La sfera iniziale ha una superficie di area 12,57 cm2; la superificie totale di 64 sfere con raggio 0,25 cm diviene di 50,26 cm2; se il raggio è di 10−5 cm ‒ le dimensioni tipiche dei sistemi colloidali ‒ l'area totale aumenta fino a 1,26×106 cm2, vale a dire 126 m2 per grammo di materiale. Sappiamo dalla termodinamica che i processi spontanei ‒ e quindi anche la separazione di un sistema disperso bifasico con formazione di due distinte regioni omogenee ‒ inducono una diminuzione dell'energia libera di Gibbs, il cui termine relativo alla presenza di un'interfase è dato dal prodotto γa tra la tensione superficiale γ e l'estensione del contatto interfasale a; pertanto, un sistema costituito da più fasi evolve spontaneamente verso stati con valori minori del termine γa.
A questo proposito si può menzionare, per esempio, che la polvere lunare portata sulla Terra nel 1969 dall'equipaggio dell'Apollo 11 conteneva numerose sferette di vetro, formatesi in conseguenza della tensione superficiale al confine tra due fasi. La tensione superficiale, infatti, fa sì che corpi isolati con superfici omogenee tendano ad assumere la forma sferica, ossia quella di area minima per un volume dato. A ciò si deve, per esempio, la forma delle bolle di sapone e (in assenza di campi gravitazionali) delle gocce di qualsiasi liquido. Dato che le particelle solide non possono cambiare la loro forma per effetto della tensione superficiale, è chiaro che, nel caso delle sferette lunari, il materiale deve essere stato fluido prima di solidificare tramite un raffreddamento pressoché istantaneo. Un sistema colloidale liofobo, quindi, tende spontaneamente all'aggregazione per diminuire l'estensione del contatto interfasale. Tuttavia, la termodinamica non fornisce informazioni sulla cinetica del processo e, infatti, molte dispersioni bifasiche apparentemente possono non trasformarsi per lunghi periodi di tempo, pur essendo termodinamicamente instabili e tendendo quindi a evolvere verso sistemi formati da particelle più grandi o aggregate. Per ottenere sistemi stabili, soprattutto nel caso di colloidi liofobi fluidi, cioè emulsioni e schiume, si può intervenire riducendo il valore della tensione superficiale tramite l'aggiunta di tensioattivi e co-tensioattivi, che inducono un processo di adsorbimento all'interfase liquido-liquido o liquido-gas, stabilizzando il sistema. In particolare, nella produzione di schiume ed emulsioni sono utilizzati tensioattivi ramificati o polielettroliti, che inducono la formazione di corone interfasali espanse, le quali, oltre a determinare una diminuzione della tensione superficiale γ, costituiscono anche un ostacolo sterico alla coalescenza.
4. Sistemi colloidali solido-liquido
Morfologia
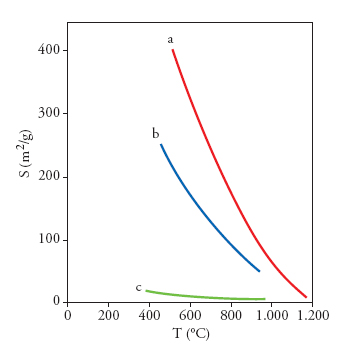
Il più evidente e vistoso aspetto connesso con un solido in stato di elevata suddivisione è l'ampiezza della sua area superficiale. In genere, l'estensione del contatto interfasale del solido è strettamente correlata con la sua applicazione e dipende in modo primario dal cammino seguito per ottenere il contatto stesso. La fig. 3 mostra la diminuzione dell'area superficiale di polveri di zirconia al crescere della temperatura utilizzata per calcinare il sale iniziale o il precursore idrato dell'ossido. Come si può vedere dalla rilevanza degli effetti prodotti, la temperatura di preparazione/calcinazione costituisce uno dei parametri principali che determinano l'estensione del contatto interfasale. L'andamento decrescente con la temperatura può essere spiegato con la crescita dei cristalliti nei solidi, spesso seguita da fenomeni di sinterizzazione tra i cristalliti per formare le particelle finali. Campioni ottenuti tramite calcinazione diretta di sali (fig. 3, curva c) mostrano aree superficiali meno estese rispetto a quelle dei campioni preparati seguendo percorsi che implichino stadi idrotermali (curva b) o sol-gel (curva a). Le differenze possono essere elevate perfino per lo stesso composto; la fig. 2 mostra, per esempio, che l'area superficiale della zirconia, ottenuta attraverso una calcinazione diretta di ZrCl4, è inferiore di circa 1 ordine di grandezza rispetto a quella dei campioni preparati per via umida. Se nella preparazione di una fase solida dispersa viene introdotto uno stadio in soluzione, generalmente si riesce a controllare in modo più accurato la morfologia delle polveri.
Percorsi mirati alla preparazione di fasi con caratteristiche morfologiche controllate, spesso prevedono l'uso di agenti complessati organici o inorganici. In questi casi, generalmente vengono progettate reazioni idrotermali multistadio che permettano di effettuare un controllo rigoroso sulle varie fasi della reazione di precipitazione, in particolare sulla reazione di nucleazione del materiale solido, in modo da ottenere particelle che presentino tutte la stessa forma e la stessa dimensione (monodisperse). Egon Matijevic e i suoi collaboratori hanno messo a punto, nel corso degli ultimi decenni, percorsi preparativi complessi per ottenere una grande varietà di metalli, ossidi e composti poco solubili, perfettamente monodispersi e con forme e dimensioni rigorosamente controllate, utilizzando nella maggior parte dei casi una tecnica di idrolisi controllata, generalmente in condizioni debolmente acide e in presenza di specifiche specie complessate.
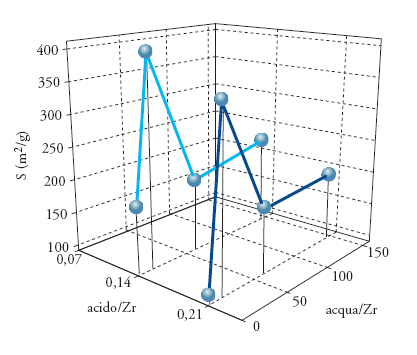
In anni recenti, sono state ampiamente utilizzate le sintesi sol-gel per ottenere fasi solide con caratteristiche morfologiche e di composizione di fase controllate. In queste reazioni, condotte generalmente in miscela idroalcolica, di norma si ottiene in un primo stadio una fase solida dispersa intermedia, il sol, che poi viene successivamente solubilizzata e policondensata per dare il gel. Le particolari caratteristiche reologiche del gel sono il risultato della presenza di un articolato network tridimensionale, nelle cui cavità si trovano molecole di solvente e specie ioniche. La rimozione finale di acqua e solvente dal gel può essere ottenuta per semplice evaporazione a pressione atmosferica (xerogel), o tramite liofilizzazione (criogel), oppure in condizioni supercritiche (aerogel). I parametri, sia chimici sia fisici, che possono essere modulati nella progettazione del percorso preparativo di queste sintesi sono molto numerosi e questo rende possibile ottenere prodotti con caratteristiche mirate e progettate a priori. La fig. 4 riporta la variazione dell'area superficiale di campioni di biossido di zirconio in funzione della composizione della miscela di reazione per una sintesi sol-gel.
Stato superficiale
Nel caso dei sistemi con elevata estensione di contatto interfasale, le proprietà della superficie solida sono estremamente rilevanti. All'interfase è presente una discontinuità nella distribuzione degli atomi o, più generalmente, delle molecole. In corrispondenza degli strati esterni del reticolo cristallino si verificano fenomeni di rilassamento e di ricostruzione, originati proprio dalla presenza di forze superficiali sbilanciate, che provocano modificazioni della posizione degli atomi sia in direzione perpendicolare al piano dell'interfase (rilassamento), sia in direzione parallela (ricostruzione). Per esempio, nel caso di sistemi a più componenti la composizione superficiale è in genere diversa da quella massiva. Inoltre, interazioni tra la superficie e l'intorno possono causare mutamenti importanti nello stato effettivo della superficie di un materiale rispetto alla fase massiva sottostante. Di conseguenza, la superficie di un solido risulta differente dalla fase massiva sottostante per quanto riguarda sia gli aspetti morfologici, sia quelli di composizione e di intorno chimico.
Recentemente, sono state sviluppate tecniche molto avanzate per la caratterizzazione spettroscopica delle superfici al fine di ottenere una superficie pulita. In condizioni normali, una superficie è costantemente bombardata da particelle di gas che la ricoprono molto rapidamente non appena essa viene esposta. La velocità di ricoprimento può essere valutata tramite la teoria cinetica dei gas e l'espressione che si ottiene per il numero di collisioni per unità di area e per unità di tempo, Zw, è la seguente:
[2] formula
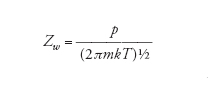
dove m è la massa, k è la costante di Boltzmann, T è la temperatura e p è la pressione. Dalla relazione si ottiene che a 1 atm e 25 °C la frequenza di collisione è pari a 3×1027m−2s−1, e poiché in 1 m2 di superficie sono contenuti circa 1019 atomi (per es., nel caso di un metallo), si può calcolare che ciascun atomo in superficie venga colpito circa 108 volte in 1 s. Anche se solo poche collisioni portano all'adsorbimento di una molecola di gas alla superficie, il tempo di vita di una superficie non modificata è molto breve. Dato che il metodo più ovvio per mantenere la superficie pulita è quello di ridurre la pressione, sono state sviluppate tecniche molto sofisticate di ultra alto vuoto o UHV (Ultra high vacuum), che permettono di portare le pressioni a valori estremamente bassi, compresi tra 0,1 μPa e 1 nPa. Per questi valori di pressione, ogni atomo della superficie viene colpito in media ogni 104÷106 s, quindi circa 1 volta al giorno.
Valori di pressione così bassi, ottenuti grazie al progresso tecnologico degli ultimi 10-15 anni, hanno permesso di sviluppare l'analisi spettroscopica di superficie con metodi molto avanzati, il cui numero è divenuto enorme al variare della sorgente di eccitazione (calore, campi elettrici e magnetici, particelle neutre, specie ioniche, fotoni, elettroni) e del relativo fenomeno di emissione. Inoltre, usando sorgenti di eccitazione della stessa natura ma in intervalli di energia differenti, si riesce a ottenere anche informazioni di altro tipo. Le tecniche di ionizzazione ‒ che si differenziano fra loro per la profondità di fuga degli elettroni, cioè la massima profondità da cui provengono gli elettroni emessi che non abbiano subito urti anelastici ‒ permettono di ottenere informazioni riguardo alla composizione superficiale, quando la profondità di fuga è tra 0,1 e 1,0 nm, e, quindi, quando solo le specie superficiali contribuiscono al segnale.
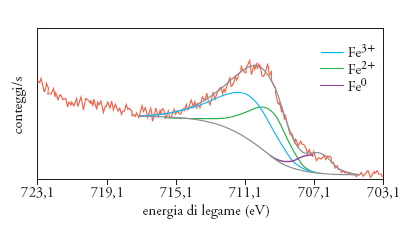
La spettroscopia di fotoelettroni o XPS (X-ray photoelectron spectroscopy) è una delle tecniche più utilizzate per ottenere informazioni sullo stato superficiale di polveri e materiali policristallini; essa si basa sull'analisi dell'energia cinetica degli elettroni emessi da un materiale solido, liquido o gassoso, colpito da una radiazione elettromagnetica di energia opportuna. La fig. 5 mostra lo spettro XPS della linea spettrale 2p del ferro (Fe 2p), per una polvere di ferro metallico utilizzata in studi riguardanti gli inibitori dei processi di corrosione: la larghezza e la deconvoluzione del picco indicano la presenza di più componenti, e cioè specie relative a ferro metallico (Fe0), specie ferrose (Fe2+) e ferriche (Fe3+). Di conseguenza, ci si può attendere che la reattività di questo materiale sia più confrontabile con quella di un ossido-idrossido che con quella di un metallo.
Per valutare la composizione e l'intorno chimico dei materiali solidi suddivisi, in alcuni casi vengono utilizzate, oltre all'analisi XPS, anche le spettroscopie Auger e SIMS (Secondary ion mass spectroscopy). Gli aspetti morfologico-strutturali dei sistemi dispersi, generalmente policristallini, non risultano chiaramente evidenziati da spettroscopie tipicamente mirate allo studio di superfici di cristalli singoli, come, per esempio, la spettroscopia LEED (Low energy electron diffraction); le microscopie elettroniche a scansione e trasmissione, invece, offrono molte informazioni sulla forma, sulla dimensione e sulla tessitura superficiale delle particelle. La microscopia a effetto tunnel o STM (Scanning-tunneling microscopy), sviluppata negli ultimi anni, è una tecnica estremamente potente e versatile, capace di fornire immagini tridimensionali di superfici con risoluzione che arriva al livello atomico; essa ha permesso di ottenere immagini di superfici e di adsorbati oltremodo dettagliate. Inoltre, la mappatura ad alta risoluzione di una superficie può essere fatta non solo in ambiente di UHV, ma anche all'aria o in liquidi isolanti, ossia in condizioni sperimentali nelle quali non sono utilizzabili le tecniche convenzionali di microscopia elettronica, come SEM (Scanning electron microscopy) e TEM (Transmission electron microscopy), che operano in ambienti di alto vuoto (HV, High vacuum) o di ultra alto vuoto (UHV). Il componente centrale del microscopio a effetto tunnel è la punta di platino-rodio della sonda, che opera una scansione del campione. Essa è connessa a un cilindro in materiale ceramico piezoelettrico, che si espande e contrae in direzione della lunghezza in risposta a una corrente elettrica. Quando la parte estrema della punta è portata molto vicino alla superficie, gli elettroni passano per effetto tunnel nello spazio tra la punta e il campione. Dato che la probabilità di avere effetto tunnel è in funzione molto sensibile della distanza, il microscopio può determinare piccole variazioni in scala atomica nel profilo della superficie.
Elettrificazione interfasale
Lo stato superficiale effettivo e la stechiometria del solido si riflettono nella reattività interfasale della polvere in sospensione acquosa. Il doppio strato elettrico che si forma al contatto tra un solido e la soluzione è responsabile di tutta la reattività della sospensione, includendo sia le interazioni al contatto solido-solido (stabilità-flocculazione, eterocoagulazione, adesione), sia interazioni solido-soluto (adsorbimento). Per una corretta comprensione della stabilità, della reologia e di molte altre proprietà dei solidi dispersi nei liquidi, è essenziale un'indagine quantitativa della distribuzione delle cariche e dei potenziali intorno alla particella. Vari processi possono essere alla base della localizzazione di una carica elettrica alla superficie delle particelle in sospensione: l'adsorbimento preferenziale di particolari specie ioniche, la sostituzione isomorfa reticolare o l'adsorbimento di polielettroliti.
Riguardo al primo caso, è necessario tenere presente che alcune specie ioniche ‒ per esempio gli ioni H+ e OH− per gli ossidi e, per molti solidi poco solubili, gli stessi ioni reticolari ‒ hanno un'affinità così elevata per la superficie da poterle ritenere parte di essa. Queste particolari specie ioniche vengono dette ioni potential determining. Per ragioni connesse con l'elettroneutralità, la localizzazione sulla parete del solido disperso di una carica fissa provoca il riarrangiamento delle specie ioniche in soluzione, con formazione di una regione spaziale, denominata doppio strato elettrico, con caratteristiche differenti da quelle della soluzione massiva. Il doppio strato è formato da una regione interna e una esterna di compensazione, detta doppio strato diffuso.
Nel caso di un ossido, la carica superficiale, σ0, è definita come:
[3] formula,


dove F è la costante di Faraday e Γ sono gli eccessi superficiali degli ioni potential determining; pertanto, la carica superficiale dipende dal pH. In corrispondenza del valore del pH della soluzione a cui gli eccessi di H+ eguagliano gli eccessi di OH−, la carica netta superficiale è nulla; questo valore viene definito punto di carica zero. La tab. 2 riporta alcuni valori di punto di carica zero (pristini), non modificati da effetti di adsorbimento specifico, che, come si può vedere, sono tipici per ciascun materiale: passando dalla silice alla gibbsite, la superficie diventa più basica. Sperimentalmente, il punto di carica zero viene determinato tramite curve di carica ottenute al variare del pH. In condizioni pristine, cioè in assenza di adsorbimento specifico, il valore del punto di carica zero diviene identico al valore del punto isoelettrico. Le due grandezze si riferiscono a due regioni diverse del doppio strato elettrico: il punto di carica zero alla regione più interna, cioè alla parete dell'ossido, e il punto isoelettrico alla regione più esterna, cioè al piano di scorrimento, che può essere considerato rappresentativo del doppio strato diffuso.
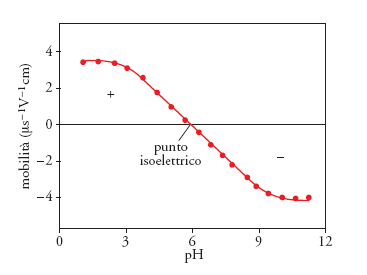
Se non si verificano fenomeni di chemisorbimento dell'elettrolita, i due parametri necessariamente coincidono, in quanto descrivono le caratteristiche di elettrificazione intrinseche del materiale. In anni recenti, è diventato più facile avere accesso a dati di punto isoelettrico utilizzando metodologie molto avanzate, che sfruttano la luce laser o l'accoppiamento di altri fenomeni; gli strumenti DELSA (Doppler electrophoretic light scattering analysis), per esempio, determinano la mobilità elettroforetica delle particelle (fig. 6), misurando lo spostamento Doppler della luce laser diffusa al variare dell'angolo di scattering e, poiché l'effetto di diffusione della luce dipende dalla dimensione della particella, la tecnica fornisce anche dati sulla distribuzione dimensionale delle particelle analizzate. Negli ultimi anni sono state sviluppate metodologie per l'analisi delle sospensioni concentrate: gli strumenti ESA (Electrokinetic sonic amplitude), molto utilizzati per la caratterizzazione degli slurry precursori dei materiali ceramici, misurano l'ampiezza sonora elettrocinetica delle onde di pressione generate quando un campo elettrico a frequenza alternata (1 MHz) viene applicato a una dispersione colloidale.
Stabilità elettrostatica
La stabilità di un sistema disperso costituisce il punto centrale della chimica colloidale, mentre la coagulazione o la flocculazione (cioè l'aggregazione tra le particelle) rappresentano i più importanti meccanismi di transizione di un sistema colloidale a uno stato più stabile. In un sistema colloidale costituito da un solo componente disperso e da una fase disperdente omogenea, le particelle si attraggono tra loro a causa delle forze di van der Waals (dette anche forze di dispersione di London), che si manifestano anche attraverso il mezzo condensato, cioè il mezzo disperdente, anche se quest'ultimo ne diminuisce l'intensità. Le più importanti interazioni sono di tipo dipolo-dipolo (forze di Keesom), dipolo-dipolo indotto (forze di Debye) e di dispersione (forze di London). Tranne che nel caso dei materiali fortemente polari, le forze di London sono il contributo più importante delle interazioni di van der Waals.
Le forze di London tra due molecole variano inversamente alla sesta potenza della distanza intermolecolare (V=a/r6) e pertanto la loro azione si esplica a corto raggio. Per un insieme di molecole, le forze di dispersione sono, in prima approssimazione, additive, cosicché l'energia di interazione di van der Waals tra due particelle (Gatt) deve essere calcolata sommando le interazioni tra tutte le coppie di molecole. Il risultato è che, in un sistema costituito da molte particelle, l'energia attrattiva di London decade con la distanza molto più lentamente di quanto non faccia in una coppia di molecole isolate. La repulsione di tipo elettrostatico (per una sostanza dispersa in mezzo acquoso) è provocata dalla presenza di una discontinuità dielettrica, caratteristica dell'interfase, dovuta alla formazione del doppio strato elettrico. Quando due particelle che presentano un doppio strato si incontrano (per es. a causa del moto browniano) si verifica una repulsione dovuta alla sovrapposizione del doppio strato diffuso. Per particelle sferiche, Boris Derjaguin ha derivato un'espressione approssimata, valida quando il raggio della particella è molto maggiore rispetto alla distanza di interazione. La forza repulsiva, Grep, dipende sia dal valore del potenziale alla parete, Φsia dalla distanza di Debye, kd, a sua volta funzione diretta della temperatura e inversa della forza ionica.
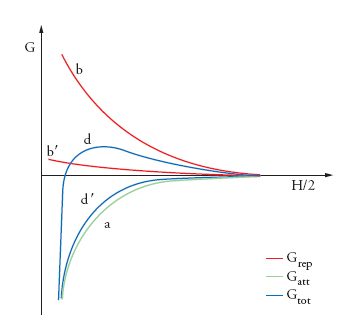
Le due relazioni, che rappresentano la variazione con la distanza del termine attrattivo e repulsivo, rispettivamente, possono essere combinate per fornire la dipendenza dalla distanza dell'energia totale di interazione, Gtot=Gatt+Grep. Un esempio schematico valido per due particelle piane e parallele è presentato nella fig. 7, dove la curva a rappresenta la componente attrattiva, Gatt, e la curva b il termine repulsivo, Grep. È evidente che la curva d, che risulta dalla combinazione di a e b, cresce via via che le particelle si avvicinano. Solo quando l'energia termica di queste ultime è abbastanza elevata da superare il massimo in d, esse possono avvicinarsi a distanze minori di quella corrispondente al massimo. In questo caso, le particelle giungono infine in contatto tra loro, cosa che è equivalente alla coagulazione. Se il massimo in d è sufficientemente più alto dell'energia del moto browniano, le particelle rimangono a distanze di avvicinamento superiori a questo valore, e, di conseguenza, la dispersione è stabile.
Ovviamente, l'instabilità può essere prodotta abbassando l'energia di attivazione, per esempio intervenendo sul termine kd, cioè comprimendo lo spessore del doppio strato diffuso tramite un aumento della forza ionica dell'elettrolita di fondo in soluzione. La curva b′ nella fig. 7 rappresenta il termine elettrostatico repulsivo modificato dalla compressione del doppio strato e la curva d′ è la nuova funzione risultante. È importante osservare che le curve di interazione indicano che le sospensioni stabilizzate elettrostaticamente sono sempre instabili dal punto di vista termodinamico. Solo perché le energie di attivazione possono essere rese così elevate da rendere il processo di coagulazione lentissimo, si riesce a ottenere una stabilità colloidale di tipo cinetico. Nelle sospensioni concentrate, per esempio quelle dei precursori dei materiali ceramici, la stabilità indotta dall'elettrificazione generalmente non è sufficiente; pertanto è necessario indurre anche una stabilità di tipo sterico, aggiungendo polielettroliti o polimeri.
Prospettive di impiego dei materiali colloidali
Le enormi potenzialità dei materiali di tipo colloidale riguardano sia settori applicativi, tra loro anche molto diversi, sia ricerche di carattere fondamentale. Da un punto di vista generale, diventa sempre più pressante l'esigenza di ottenere materiali nanostrutturati con particolari caratteristiche. Il controllo di queste ultime, per quanto riguarda sia la composizione di fase, sia gli aspetti morfologico-superficiali, si ottiene intervenendo in maniera mirata sull'iter sintetico del materiale. Attualmente, vi è un grandissimo interesse nei confronti delle sintesi 'template'. La ricerca di nuovi catalizzatori eterogenei, che possano sostituire i catalizzatori in fase omogenea nell'industria chimica e petrolchimica per questioni di interesse ambientale, ha portato alla produzione di materiali porosi nanostrutturati, che hanno attratto considerevole attenzione per le loro potenziali applicazioni in campi come l'elettronica, l'ottica, l'accumulo/conversione di energia e la catalisi avanzata. In questi ultimi anni viene profuso molto impegno per produrre materiali cataliticamente attivi, caratterizzati non solo da un'elevata area superficiale, ma anche porosi, con diametro dei canali interni progettato in modo da permettere l'accesso anche a molecole organiche di grandi dimensioni.
In queste sintesi, la morfologia del solido è determinata dalla mesostruttura tridimensionale di un tensioattivo, che funge da nucleante e che viene rimosso dopo la sintesi del materiale inorganico. Attualmente, la ricerca, inizialmente mirata a ottenere materiali silicei, viene estesa a un numero elevato di altri materiali, quali allumino-fosfati o allumino-silicati e numerosi ossidi di grande interesse applicativo (MnO2, Al2O3, TiO2, ZrO2, ecc.), per la cui sintesi è stato proposto un meccanismo di stampo a cristalli liquidi o LCT (Liquid crystal template), nel quale un tensioattivo organico, formando strutture tridimensionali, funge da templante per la formazione dei canali e delle cavità. Modulando le con-dizioni operative e le caratteristiche del tensioattivo utilizzato, è possibile indirizzare la sintesi verso fasi liquido-cristalline con diversa organizzazione, e conseguentemente verso strutture con pori di dimensioni più elevate.
Bibliografia
Adamson, Gast 1997: Adamson, Arthur W. - Gast, Alice P., Physical chemistry of surfaces, 6. ed., New York-Chichester, Wiley, 1997.
Ardizzone 1999: Ardizzone, Silvia, Colloidal solid-liquid systems, "La chimica e l'industria", 81, 1999, pp. 599-604.
Hunter 2000: Hunter, Robert J., Foundations of colloid science, 2. ed., New York, Oxford University Press, 2000.
Israelachvili 1998: Israelachvili, Jacob, Intermolecular and surface forces, 2. ed., London, Academic Press, 1998.
Masi, Carrà 1999: Masi, Maurizio - Carrà, Sergio, From sand to integrated circuits and optical wave-guides. An overview of not traditional applications of chemical reaction engineering, "La chimica e l'industria", 81, 1999, pp. 845-852.
Matijevich 2001: Matijevich, Egon, The past and the future of monodispersed colloids, New York, American Chemical Society, 2001.
Mittal, Shah 1991: Mittal, Kashmiri L. - Shah, Dinesh O., Surfactants in solution. Proceedings of the Eighth International Symposium held in Gainesville, Florida, june 10-15, 1990, New York, Plenum, 1991.
Moroi 1992: Moroi, Yoshikiyo, Micelles: theoretical and applied aspects, New York, Plenum, 1992.
Moser 1996: Advanced catalysts and nanostructured materials: modern synthetic methods, edited by William R. Moser, London-San Diego, Academic Press, 1996.
Pugh, Bergström 1994: Surface and colloid chemistry in advanced ceramic processing, edited by Robert J. Pugh, Lennart Bergström, New York, Dekker, 1994.
Somorjai 1994: Somorjai, Gabor A., Introduction to surface chemistry and catalysis, New York, Wiley, 1994.