
emofilia
emofilia
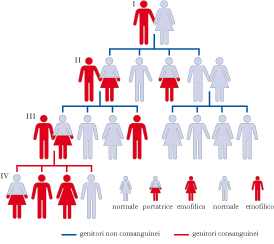
Disordine ereditario recessivo che causa una grave insufficienza nella coagulazione del sangue dovuta alla mancanza, totale o parziale, del fattore VIII (e. A), o del fattore IX (e. B) della coagulazione. Più rara è l’e. C, data dalla mancanza totale o parziale del fattore XI. Il soggetto emofilico può essere colpito da emorragie spontanee, oppure provocate da traumi, queste con esiti più gravi.
Emofilia A
Si tratta della forma più diffusa di e.: colpisce un individuo ogni 10.000. È caratterizzata da una carenza della subunità C del fattore VIII, una componente proteica che svolge un’importante azione nel processo di coagulazione. Il deficit è legato al cromosoma sessuale X. Questa patologia colpisce quindi esclusivamente i maschi e si trasmette dalla madre, portatrice sana, al figlio maschio. Nel 30% dei casi può verificarsi in una donna non per trasmissione ereditaria, ma come mutazione puntiforme del cromosoma X. Il deficit può essere quantitativamente variabile, così da indurre quadri clinici diversi: con livelli funzionali di fattore VIII fino al 2% (e. A grave) sono frequenti gli ematomi (o il rischio di gravi emorragie, come quelle cerebrali) e, in età infantile, gli emartri spontanei (emorragie delle cavità articolari); con livelli di fattore VIII dal 2 al 5% (e. A moderata) e inoltre pari al 5÷25% (e. A lieve), gli emartri sono rari e si hanno emorragie solo dopo interventi chirurgici. Le manifestazioni emorragiche più frequenti sono, oltre agli emartri, gli ematomi sottocutanei o muscolari, le emorragie del sistema gastro-enterico e di quello uro-genitale, le epistassi, le emorragie post-intervento chirurgico anche semplice (per es., le estrazioni dentarie). Inoltre, dal momento che la subunità C del fattore VIII è coinvolta nell’emostasi secondaria, l’emorragia può presentarsi in un momento successivo alla lesione: dapprima la perdita di sangue si arresta per la formazione regolare del tappo emostatico, ma in una seconda fase la carenza del coagulo fibrinico impedisce l’arresto dell’emorragia. Le analisi di laboratorio evidenziano allungamenti dei tempi di coagulazione, ma la diagnosi si ottiene solo dosando il fattore VIII. La terapia è incentrata sull’assunzione di preparazioni concentrate del fattore VIII.
Emofilia B
È trasmessa geneticamente attraverso il cromosoma X, ma è molto più rara della A, con un quadro clinico che le si sovrappone. L’anomalia genica determina una carenza di fattore IX funzionale, a cui corrisponde un deficit della parte antigenica nel 70% dei casi, e della sola parte funzionale nel 30%. La terapia è sostitutiva e prevede l’infusione di concentrati di complesso protrombinico.
Emofilia C
È ancora più rara e il difetto, che riguarda il fattore XI, è trasmesso come deficit autosomico recessivo. Le manifestazioni sono più rare e più lievi dei difetti precedenti: si hanno emorragie dopo interventi chirurgici e dopo traumi importanti.