
emofilia
Malattia ereditaria, che colpisce quasi esclusivamente gli individui di sesso maschile della specie umana, ai quali viene trasmessa dalle femmine, che possono essere portatrici della malattia, senza essere clinicamente malate. Alcune sindromi emorragiche clinicamente simili all’e., ma non tutte ereditarie, si dicono emofiloidi.
Aspetti clinici
Il quadro morboso dell’e. si evidenzia sin dall’infanzia con manifestazioni emorragiche mucose, cutanee, viscerali e articolari (queste, ripetendosi, inducono processi degenerativi e deformanti: emoartrosi). I sintomi di solito si attenuano dopo i 30 o 40 anni. Il tempo di emorragia, la retrazione del coagulo e il numero delle piastrine sono normali. La diagnosi differenziale dell’ e. A ( e. classica), che è dovuta alla carenza del fattore VIII della coagulazione, consente di distinguerla da altre analoghe sindromi emorragiche, soprattutto la malattia di Christmas (detta anche e. B), che è connessa al deficit del fattore IX della coagulazione, e con la deficienza di fattore Rosenthal (o deficienza di PTA). La diagnosi di laboratorio si basa sulla normalità del tempo di emorragia e del tempo di protrombina e sull’alterazione del tempo di tromboplastina parziale, nonché sul dosaggio specifico del fattore carente.
Aspetti genetici
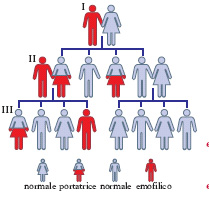
Lo schema ereditario. - L’e. è un carattere ereditario che si trasmette secondo lo schema della cosiddetta eredità legata al sesso (eredità diaginica). Il gene che determina questo carattere è cioè localizzato nei cromosomi sessuali, e propriamente nel cromosoma X. Nel maschio (che ha la coppia di cromosomi XY) la presenza della mutazione per l’e. nell’unico cromosoma X è sufficiente a determinare la manifestazione del carattere. Invece nella femmina (che ha la coppia di cromosomi XX), se il gene dell’e. è allo stato eterozigote, cioè presente in un solo cromosoma X, la malattia non si manifesta perché l’allele normale, presente nell’altro X, è dominante sulla mutazione. Perché una femmina sia emofilica è necessario quindi che sia figlia di un uomo emofilico e di una donna eterozigote o emofilica. Le femmine emofiliche sono quindi rare. Le femmine eterozigoti invece sono tutt’altro che rare (circa 1 su 200), e trasmettono la malattia alla metà dei figli maschi (v. fig.).
E. A. - Il gene del fattore VIII è localizzato all’estremità del braccio lungo del cromosoma X; è un gene grande, costituito da 186 chilobasi, che contiene 26 esoni. Le mutazioni del gene che codifica il fattore VIII sono molteplici; sono state identificate in questo gene 78 grandi delezioni e 223 mutazioni di tipo puntiforme, quali piccole delezioni o inserzioni. La grande varietà di mutazioni e la grandezza del gene da una parte hanno fornito molte informazioni sulla struttura e sulla funzione della proteina codificata ma, dall’altra, hanno reso difficile identificare il difetto specifico di ogni famiglia in cui sono presenti individui affetti.
Sono state messe a punto metodiche di screening che consentono la ricerca di specifiche mutazioni, e l’applicazione di queste metodiche allo studio degli esoni e delle giunzioni esone/introne ha consentito di identificare le mutazioni di quasi tutti quei pazienti che presentano un fattore VIII anomalo. In essi è presente un’inversione del cromosoma X che interrompe il gene che codifica il fattore VIII. L’inversione si verifica a causa di una ricombinazione omologa fra un piccolo segmento di DNA chiamato gene A, situato all’introne 22 del gene, e una delle due copie dello stesso gene, situata verso l’estremità del cromosoma X. L’inversione causa la perdita completa del fattore VIII e una forma grave di emofilia. Questa singolare ricombinazione omologa, mai descritta prima in altre malattie umane, si verifica soltanto durante la meiosi maschile, probabilmente a causa della presenza nel maschio di un solo cromosoma X non appaiato. Questa osservazione ha importanti implicazioni cliniche, perché si può assumere che un bambino affetto abbia una mamma portatrice anche in assenza di una storia familiare, in quanto la ricombinazione anomala è avvenuta durante la formazione dei gameti del nonno materno.
E. B. - La prevalenza di questa malattia è circa 1/10 rispetto a quella dell’e. A. Il gene è costituito da 34 chilobasi e contiene 8 esoni. Anche l’e. B si associa a un gran numero di mutazioni diverse del gene. Una variante particolare di e. B, denominata fattore IX Leiden, è particolarmente interessante. I pazienti affetti presentano e. grave, che migliora dopo la pubertà. L’analisi molecolare può fornire una spiegazione a questo fenomeno: le mutazioni identificate in questi individui sono localizzate nella sequenza che regola la trascrizione del fattore IX, il promotore. Fattori regolativi di questo gene sono gli androgeni, la cui concentrazione aumenta durante la pubertà e determina la produzione di fattore IX, anche se il promotore del gene è mutato.
La terapia
La terapia dell’e. A consiste nella sostituzione del gene che codifica il fattore VIII di coagulazione del sangue, assente negli individui affetti. La cura è stata a lungo solo sintomatica o, più raramente, profilattica, effettuata prevalentemente, se non esclusivamente, con trasfusioni di sangue intero o con concentrati purificati del fattore di coagulazione. Dagli inizi degli anni 1980 l’infezione da HIV ha avuto una inevitabile ricaduta sulla terapia dell’emofilia. Anche se dopo il 1985 lo screening dei donatori per l’infezione da HIV e il miglioramento delle procedure di preparazione del fattore VIII hanno efficacemente eliminato il rischio di infezioni, l’identificazione e il clonaggio del gene che codifica il fattore VIII da parte di società biotecnologiche hanno aperto la prospettiva di produrre tale fattore mediante la tecnologia del DNA ricombinante e hanno permesso una migliore comprensione della base molecolare della malattia. Nel 1994 l’uso di fattore VIII ricombinante privo di rischi di infezioni è stato approvato per fini terapeutici. Successivamente è stato preparato un fattore VIII ricombinante di seconda generazione, caratterizzato dalla delezione di una frazione centrale della catena singola; questo prodotto, denominato r-VIII SQ (S per serina, Q per acido glutammico, i due amminoacidi che si uniscono dopo la delezione, Ser 743-Glu 1638), è ugualmente attivo rispetto ai precedenti emoderivati, presenta una maggiore attività specifica ed è stabile pur essendo privo di albumina umana. Anche per il fattore IX è stato possibile preparare di recente un emoderivato ricombinante, prodotto in assenza di proteine umane, con attività indistinguibile da quella del fattore IX plasmatico. Il fattore VIIa ricombinante è stato ampiamente utilizzato nei pazienti affetti da e. A o e. B, nei quali si sia sviluppato un inibitore diretto rispettivamente contro il fattore VIII o contro il fattore IX, tale da rendere inefficace la classica terapia sostitutiva con emoderivati specifici. Il fattore VIIa ricombinante è risultato efficace nel controllo degli episodi emorragici e nella prevenzione delle emorragie postoperatorie di questi emofiliaci, e contemporaneamente meno trombogenico rispetto agli emoderivati contenenti i fattori attivati del complesso protrombinico (II, VII, IX, X), utilizzati nel medesimo tipo di pazienti.