
proteine
Macromolecole costituite da una, o più, lunghe catene polipeptidiche (dette anche protidi). Le p. costituiscono la classe di molecole organiche più abbondanti in tutti gli organismi viventi; si trovano in tutte le cellule e costituiscono il 50% o più del loro peso secco. Le p. sono essenziali per tutti i processi biologici legati alla vita, svolgendo un ruolo fondamentale per la struttura e la funzione cellulare, come nei processi di catalisi enzimatiche, di trasporto e deposito, di supporto meccanico, di protezione immunitaria, di generazione e trasmissione dell’impulso nervoso, di controllo della crescita e della differenziazione. La sintesi proteica da parte degli organismi viventi è detta protidopoiesi.
Struttura
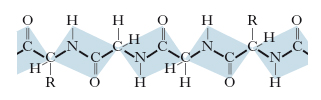
I primi studi fondamentali sulla struttura delle p. risalgono agli anni 1950. Le p. sono costituite da numerose molecole di α-amminoacidi (detti anche residui amminoacidici), legate fra loro mediante legami peptidici tra il gruppo carbossilico di una molecola e il gruppo amminico della successiva, costituendo nel loro insieme una catena polipeptidica (fig. 1). Il peso molecolare delle p. può variare da circa 5000-10.000 in quelle più piccole (40-80 amminoacidi) sino ad alcuni milioni per quelle più grandi e complesse. Tra tutti gli amminoacidi presenti in natura solo 20 sono i costituenti fondamentali delle p.; è importante notare che sono tutti esclusivamente L-isomeri. Ogni p. è caratterizzata da un punto isoelettrico (pI), cioè da un valore di pH al quale sono uguali il numero delle cariche negative (derivanti dalla dissociazione dei gruppi carbossilici) e il numero di quelle positive (derivanti dalla protonazione dei gruppi amminici) presenti nella molecola proteica; a tale valore di pH, la p. è globalmente neutra e presenta solubilità minima.
La struttura primaria. Detta anche struttura covalente, è determinata dalla sequenza dei diversi amminoacidi costituenti la catena polipeptidica, senza alcun riferimento al suo arrangiamento spaziale; questa struttura riguarda l’insieme dei legami covalenti della molecola, sia il legame peptidico che unisce i singoli residui amminoacidici, sia gli eventuali legami disolfuro che possono stabilirsi tra residui di cisteina, anche distanti, all’interno della stessa catena polipeptidica. La composizione amminoacidica di una data p. viene determinata idrolizzando la p. in esame, in modo da rompere i singoli legami peptidici e ottenere una miscela contenente tutti gli amminoacidi liberi. Più complessa è la determinazione della successione dei residui amminoacidici nella catena, anche se ormai esistono metodi sperimentali molto affidabili e precisi. Oggi sono note le sequenze primarie di numerose p.; questo ha permesso di evidenziare come p. funzionalmente omologhe, ma di specie diverse (per es., emoglobine e citocromi), possiedono gli stessi residui amminoacidici in alcune posizioni costanti della catena, mentre gli altri residui possono variare da una specie all’altra.
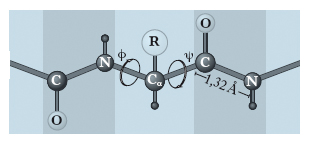
La struttura secondaria. È determinata dalla disposizione strutturale, cioè dalle relazioni steriche, degli amminoacidi che si trovano vicini nella sequenza lineare e permettono alla p. di ripiegarsi a formare una struttura ripetitiva regolare. Sono state determinate due forme conformazionali più comuni: l’α-elica (elica destrogira) e la struttura β (a foglietto ripiegato). A grandi linee, l’α-elica è una struttura che si riscontra più frequentemente nelle p. globulari, mentre la struttura β a foglietto ripiegato è caratteristica delle p. fibrose. Entrambi i tipi di struttura sono generalmente presenti solo per alcuni tratti della molecola proteica e possono essere presenti contemporaneamente in punti diversi della stessa proteina. La possibilità di assumere una di queste conformazioni è dovuta alle piccole rotazioni che possono avvenire nei legami dei carboni α adiacenti al legame peptidico (fig. 2). Infatti, mentre quest’ultimo costituisce una struttura rigidamente piana, possedendo in parte le caratteristiche di un doppio legame, i legami singoli che uniscono il carbonio e l’azoto del gruppo peptidico agli atomi di carbonio α adiacenti ne permettono una certa libertà di rotazione. L’angolo di rotazione corrispondente al legame Cα-C viene definito angolo ψ mentre quello riguardante Cα-N, angolo ϕ. Il valore effettivo degli angoli ψ e ϕ che può esistere nelle p. è però limitato, perché la libertà di rotazione è impedita dalla presenza di altri atomi vicini. Se si conoscono i valori di ψ e ϕ per ogni amminoacido costituente la catena polipeptidica, è possibile ottenere valide informazioni sull’intera conformazione della p., tracciando il grafico degli angoli ψ e ϕ permessi e non permessi (➔ Ramachandran, Gopalasamudram Narayana).
Per quanto riguarda l’α-elica, essa si potrebbe formare sia con D-, sia con L-amminoacidi, ma non potrebbe sicuramente crearsi in una catena polipeptidica che contenesse una miscela di residui D- e L-. Inoltre, a partire dagli L-amminoacidi si possono formare spirali elicoidali sia destrogire sia levogire, ma l’elica destrogira è molto più stabile. Generalmente, i tratti ad α-elica non superano i 4 nm di lunghezza, tranne che per p. particolari come la miosina, la cui struttura è interamente ad α-elica. Vi sono alcuni residui amminoacidici, come la prolina, la tirosina, la glicina e l’asparagina, che tendono a destabilizzare l’avvolgimento a spirale.
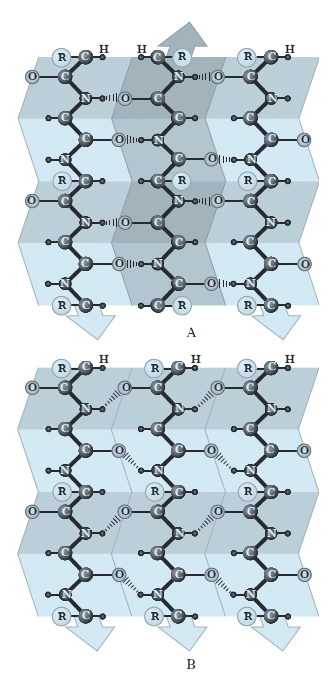
Come per l’α-elica, la conformazione del foglietto ripiegato β cade nelle regioni permesse dal diagramma di Ramachandran e utilizza completamente la capacità dello scheletro polipeptidico di formare legami idrogeno, che si generano tra catene polipeptidiche vicine o tra i residui amminoacidici appartenenti a tratti differenti della stessa catena. Esistono due varietà di strutture β: il foglietto ripiegato β antiparallelo, in cui i legami idrogeno si formano tra tratti di catene o tra catene vicine che vanno in direzioni opposte; il foglietto ripiegato β parallelo, nel quale i tratti di catena o le due catene vanno nella stessa direzione (fig. 3). Le conformazioni che consentono la formazione ottimale dei legami idrogeno necessari per la struttura β sono leggermente diverse da quelle completamente estese del polipeptide (cioè con angoli di 180°). I foglietti β sono unità strutturali comuni nelle p.; nelle p. globulari, i foglietti β che coinvolgono catene proteiche diverse sono costituiti da un minimo di 2 fino a un massimo di 15 catene polipeptidiche, con un valore medio intorno a 6, formando un aggregato dello spessore di circa 2,5 nm. Le catene polipeptidiche impegnate per la formazione di una struttura β coinvolgono fino a un massimo di 15 residui amminoacidici, anche se è più frequente che 6 amminoacidi siano coinvolti nel foglietto β (corrispondente cioè a una lunghezza media di 2,1 nm). È raro trovare foglietti ripiegati paralleli formati da meno di 5 amminoacidi; ciò suggerisce che la struttura β parallela sia meno stabile di quella antiparallela, forse a causa del fatto che i legami idrogeno della prima sono distorti se paragonati a quella della seconda.
Le strutture secondarie regolari, le α-eliche e i β-foglietti, rappresentano solo la metà della struttura di una p. media. La rimanente parte dei segmenti della catena polipeptidica ha conformazioni a forma di «spirale» (coil) o di «ansa» (loop), sempre abbastanza ordinate, ma di difficile descrizione. Il «gomitolo casuale» (random coil) è un gruppo di conformazioni fluttuanti e totalmente disordinate assunte dalle p. denaturate e da altri polimeri in soluzione. Quasi tutte le p. con più di 60 amminoacidi contengono una o più anse, costituite da 6 a 16 residui che non fanno parte né di strutture α né β, e che occupano uno spazio inferiore ai 1 nm. Questa struttura è nota con il nome di ansa Ω (Ω loop); è molto compatta, in quanto le catene laterali dei residui che la compongono tendono a riempire la cavità interna. Dato che queste strutture sono sempre localizzate sulla superficie delle p., si pensa che abbiano un ruolo fondamentale nei processi di riconoscimento biologico.
La struttura terziaria. Riguarda la disposizione spaziale delle p. ed è il prodotto dell’interazione tra le catene laterali degli amminoacidi costituenti la p. che assume, così, una ben definita conformazione tridimensionale. La carica positiva o negativa posseduta dalle catene laterali permette l’attrazione o la repulsione fra loro. I legami tra le catene laterali possono essere sia legami idrogeno, sia i cosiddetti ponti salini, che si stabiliscono tra residui di acido aspartico, o glutammico, e lisina, o arginina. Un altro tipo di attrazione coinvolge i residui non polari come valina e fenilalanina; in questo caso, l’attrazione deriva dalla disposizione all’interno della p. di questi residui, al di fuori del contatto con molecole d’acqua, provocando interazioni idrofobiche tra questi amminoacidi.
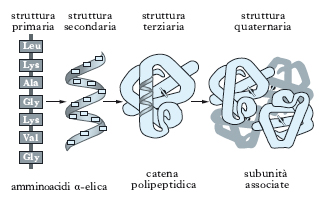
Nelle p. ricche di cisteina, la conformazione tridimensionale della catena polipeptidica è fortemente influenzata dai possibili ponti disolfuro (−S−S−) che si stabiliscono tra questi residui, a volte anche molto lontani tra loro; in questo caso, la catena si ripiega formando un’ansa chiusa dal ponte disolfuro stesso. La formazione di queste interazioni dipende esclusivamente dai residui amminoacidici presenti; quindi è la struttura primaria che determina la conformazione tridimensionale della p.; ovvero, l’informazione necessaria a specificare la complessa struttura spaziale della p. è contenuta nella sequenza dei suoi amminoacidi (fig. 4). La struttura terziaria definisce l’attività di una p.; infatti, se una p. viene denaturata, cioè viene trattata in modo da spezzare i vari legami che ne definiscono l’arrangiamento tridimensionale, essa perde non solo la sua conformazione (assumendo la forma a gomitolo casuale), ma anche la sua attività biologica.
La struttura quaternaria. Le p. che sono costituite da più di una catena polipeptidica possiedono un ulteriore livello di organizzazione strutturale, la struttura quaternaria, che si riferisce al modo in cui le catene si associano tramite interazioni non covalenti o legami covalenti trasversali. Ogni catena polipeptidica di queste p., dette oligomeriche o polimeriche a seconda del numero complessivo di catene che le compongono, costituisce una subunità. La p. oligomerica più conosciuta, e una tra le più semplici, è l’emoglobina, costituita da 4 subunità, uguali 2 a 2, legate tra loro da legami idrogeno e da interazioni idrofobiche. Sebbene sia un tetramero, le 4 subunità sono così strettamente legate tra loro da far definire l’emoglobina come una molecola, anche se non esistono legami covalenti tra le 4 catene polipeptidiche. In altre p., per es. l’insulina o le immunoglobuline, si stabiliscono invece legami covalenti, come i ponti disolfuro, che uniscono subunità differenti. Non è detto che le varie subunità siano identiche tra loro, così come che siano della stessa lunghezza; inoltre, la simmetria della struttura quaternaria non è solo tetraedrica, ma può essere anche ciclica, cubica, diedrica, icosaedrica e anche più complessa (fig. 5), come nel caso dell’enzima piruvatodeidrogenasi (➔ piruvico, acido) di Escherichia coli.
Classificazione
Le p. sono classificate tenendo conto fondamentalmente di quattro parametri: la composizione chimica, la forma delle molecole, la solubilità in acqua e la funzione biologica. La composizione chimica permette di distinguerle in p. semplici e p. coniugate e la forma delle molecole in p. globulari e p. fibrose. La solubilità in acqua costituisce un criterio che, anche se non del tutto caduto in disuso, è attualmente considerato poco soddisfacente, dato che p. con struttura e funzioni diverse possono presentare simile solubilità e, al contrario, p. simili per funzione e struttura presentano solubilità molto diversa. Infine, la classificazione in base alle funzioni biologiche è quella più utilizzata e anche la più soddisfacente, anche se si deve ricordare che molte p. possono avere più di una sola funzione. Per es., la p. contrattile miosina può anche agire come una adenosintrifosfatasi (ATPasi), cioè come un enzima in grado di idrolizzare l’ATP. Nonostante la funzione specifica di molte p. sia ancora sconosciuta, una tale classificazione funzionale è utilizzata per dimostrare la correlazione tra la struttura e la funzione di una proteina.
Funzioni
Gli enzimi rappresentano la classe più grande delle proteine. Alcuni enzimi, detti regolatori, sono in grado, attraverso la modulazione della loro attività catalitica, in risposta a vari tipi di segnali molecolari, di influenzare la velocità di un intero ciclo metabolico. Una simile funzione è svolta anche da varie p. che aiutano a regolare l’attività cellulare fisiologica, come, per es., gli ormoni. Tra questi sono compresi, per es.: l’adrenalina, che media la trasmissione dell’impulso nervoso; la tiroxina, che accelera le ossidazioni intracellulari delle p., dei carboidrati e dei lipidi; l’insulina, che regola il metabolismo glicidico; l’ormone somatotropo, che regola la crescita dell’organismo; l’ormone paratiroideo, che influisce direttamente sul trasporto del Ca++ e del fosfato. Inoltre p. regolatrici, dette repressori, regolano la biosintesi di enzimi nelle cellule batteriche.
Un’altra classe di p. ha la funzione di materiale di riserva, che serve da sostanza nutritiva e da materiale costruttivo per la crescita embrionale; ne sono un esempio l’albumina dell’uovo, la caseina del latte e la ferritina, che è una p. di deposito del ferro nella milza. Anche nei semi delle piante troviamo p. con identica funzione, come la gliadina e la zeina presenti rispettivamente nel seme di grano e di mais.
Alcune p. hanno una funzione di trasporto; infatti esse sono in grado di legare e trasportare nel sangue ioni o molecole più complesse, permettendone il trasferimento da un organo a un altro. Per es., l’emoglobina degli eritrociti trasporta ossigeno dai polmoni ai tessuti periferici, dove viene rilasciato e utilizzato dalle cellule per le ossidazioni di sostanze che producono energia. La mioglobina, presente nel tessuto muscolare, ha invece la funzione di legare l’O2 nei miociti. Negli invertebrati sono le emocianine, le clorocruorine e le emeritrine a svolgere la funzione di trasportatori di ossigeno. Il plasma sanguigno contiene lipoproteine che trasportano i lipidi fra intestino, fegato e tessuti adiposi, mentre gli acidi grassi liberi si legano all’albumina sierica che li veicola dal tessuto adiposo ai vari organi. Non meno importanti sono le p. di trasporto presenti nelle membrane cellulari, in grado di legare e trasportare, per es., glucosio, amminoacidi, ioni inorganici, con meccanismi diversi ma fondamentali per molti processi cellulari.
Esistono, poi, p. che sono elementi essenziali nei sistemi contrattili e motori; l’actina e la miosina, per es., sono i principali elementi proteici del sistema contrattile del muscolo scheletrico, ma si trovano anche in molte altre cellule non muscolari; un altro esempio è la tubulina, costituente dei microtubuli dei flagelli e delle ciglia che permettono alle cellule di compiere movimenti ameboidi.
Un’ulteriore classe di p. comprende quelle che servono da elementi strutturali, ovvero come filamenti di supporto, cavi o lamine per dare ai sistemi biologici robustezza e protezione. Per es., il maggiore componente dei tendini e delle cartilagini è il collagene, p. fibrosa con un’elevatissima capacità elastica. Nelle cartilagini sono presenti in notevole quantità anche glicoproteine che conferiscono proprietà lubrificanti alle secrezioni mucose e al liquido sinoviale delle articolazioni dei Vertebrati. I legamenti contengono elastina, una p. strutturale in grado di allungarsi in due dimensioni. Inoltre, i capelli, le unghie e le penne sono formati in gran parte di cheratina, p. fibrosa insolubile e resistente.
Alcune p. hanno una funzione protettiva o difensiva, come le p. del sistema di coagulazione del sangue, trombina e fibrinogeno, che impediscono la perdita di sangue in caso di danni al sistema vascolare; la catalasi, la glutationeperossidasi e la superossidodismutasi, che proteggono le cellule dalla tossicità delle specie radicaliche dell’ossigeno (➔ radicale); la glutationetransferasi e la NADPH-citocromo-P450-reduttasi, coinvolte nella detossificazione degli xenobiotici, sostanze naturali o sintetiche normalmente estranee alla nutrizione e al metabolismo dell’organismo. Tra le p. protettive più importanti bisogna ricordare le immunoglobuline, o anticorpi, prodotte dai linfociti; infatti, esse sono in grado di riconoscere e precipitare, o neutralizzare, batteri, virus, tossine o elementi estranei all’organismo. Anche le tossine batteriche, le p. tossiche delle piante, come la ricina, e i veleni dei serpenti possono essere considerati come p. di difesa degli organismi che li producono.
Metodi di studio
I metodi più comunemente utilizzati per l’isolamento, la purificazione e la caratterizzazione delle p. sono alla base degli studi biochimici e sono ora anche utilizzati per scopi clinici.
Isolamento. Non esiste una procedura standard per l’isolamento delle p. da organi e tessuti, anche se la prima tappa del procedimento è di portarle in soluzione, rompendo la struttura del tessuto stesso. Le tecniche utilizzate per questo scopo dipendono dalle caratteristiche meccaniche del tessuto considerato e dalla localizzazione nella cellula della p. in esame. Se la p. si trova libera nel citoplasma, si ricorre alla lisi cellulare, ottenuta con mezzi chimici o meccanici; in alternativa, se la p. è un componente di una struttura subcellulare, come, per es., membrane e mitocondri, la lisi cellulare deve essere accompagnata da una centrifugazione differenziale, che permette l’isolamento e la purificazione parziale delle stesse strutture subcellulari. Quando sono prive dell’ambiente protettivo della cellula, le p., così come altre macromolecole, devono essere trattate in modo da garantire la loro stabilità e da prevenirne il danneggiamento e la denaturazione. Questi ultimi processi sono provocati da valori estremi di pH, temperature elevate, presenza di sostanze ossidanti, degradazione enzimatica e chimica e drastici trattamenti meccanici.
Purificazione. Le p. vengono purificate con procedimenti di frazionamento successivo; infatti, in una serie di tappe indipendenti, vengono sfruttate le varie proprietà chimico-fisiche della p. di interesse, per separarla progressivamente dalle altre sostanze e dalle altre p. presenti. Le proprietà utilizzate sono: solubilità, carica ionica, massa molecolare, proprietà di adsorbimento e affinità di legame per altre biomolecole. Le tecniche analitiche che permettono la purificazione di p. in base alle loro proprietà chimico-fisiche sono la ultracentrifugazione, la cromatografia e l’elettroforesi.
Caratterizzazione. Una volta che la p. in esame sia stata ottenuta pura e in quantità sufficiente, si possono intraprendere studi strutturali e funzionali, il cui scopo è quello di trovare le correlazioni esistenti tra la struttura e la funzione di quella data proteina. Si procede, quindi, con l’indagine analitica per determinare la sequenza primaria della p. pura in esame, ovvero la sequenza dei suoi residui amminoacidici. La prima tappa di quest’analisi è stabilire il contenuto della p. in catene polipeptidiche chimicamente diverse, mediante l’analisi dei gruppi carbossi- e amminoterminali. Vengono poi rotti gli eventuali ponti disolfuro presenti ed è determinata la composizione in amminoacidi, idrolizzando tutti i legami peptidici della p. e ottenendo, così, una miscela degli amminoacidi che la compongono. Tale miscela viene poi sottoposta ad analisi qualitativa e quantitativa per il contenuto in amminoacidi. In seguito, le catene polipeptidiche purificate vengono idrolizzate specificamente a livello di precisi residui amminoacidici, mediante metodi enzimatici o chimici, in peptidi più piccoli, che sono separati, purificati e di cui viene infine determinata la sequenza amminoacidica. Ripetendo questa procedura, e avendo l’accortezza di usare diversi sistemi di idrolisi delle p., si generano peptidi che presentano sovrapposizioni nelle loro sequenze amminoacidiche dei frammenti peptidici ottenuti con una prima idrolisi. Confrontando le sequenze delle diverse serie di frammenti, e tenendo presenti le sovrapposizioni dei diversi peptidi, è possibile ricostruire il loro ordine e quindi la sequenza della p. originale. La sequenza amminoacidica dei peptidi che si ottengono dalla digestione della p. viene eseguita con l’uso di strumenti automatizzati, detti sequenziatori di peptidi. La determinazione della struttura primaria viene completata stabilendo la posizione degli eventuali ponti disolfuro. A questo scopo è necessario degradare la p. lasciando tali ponti intatti; quindi, sequenziando le coppie di peptidi uniti dal ponte −S−S−, è possibile dedurre la loro posizione nella p. nativa.
L’analisi dei livelli successivi dell’organizzazione delle p., cioè la determinazione della struttura secondaria, terziaria e quaternaria, viene affrontata con l’ausilio di tecniche analitiche differenti da quelle usate per la determinazione della struttura primaria. Si ricorre, così, a metodi spettrofotometrici, spettrofluorimetrici, di dicroismo circolare, di risonanza magnetica nucleare e di risonanza paramagnetica di spin, di cristallografia a raggi X ecc. Quest’ultima tecnica richiede, però, che la p. in esame sia estremamente pura e che sia cristallizzabile, cosa quest’ultima che non sempre si verifica. La cristallografia a raggi X e l’uso di modelli matematici computerizzati rappresentano certamente il passo finale che determina il completamento degli studi strutturali di una proteina. Parallelamente, gli studi funzionali vengono condotti per comprendere il ruolo biologico di una p. nella complessa organizzazione cellulare e, più ancora, dell’intero organismo.
Nel caso di p. strutturali, cioè non dotate di attività enzimatica, si cerca di comprendere come l’organizzazione tridimensionale della p. allo studio si accordi con una funzione biologica da essa svolta. Ovviamente, se ne cerca la localizzazione tessutale e intracellulare e l’associazione con altre molecole, proteiche e non proteiche. Al fine di individuarne la distribuzione cellulare o tessutale, si ricorre spesso all’uso di anticorpi specifici contro quella p., ottenuti mediante tecniche di immunizzazione di animali da laboratorio.
Nel caso di p. dotate di attività enzimatica, si caratterizzano le proprietà cinetiche delle reazioni catalizzate, si determina l’affinità per il substrato (o i substrati), si cerca se la p. in esame abbia gruppi prospettici indispensabili per la sua attività catalitica, si valuta se essa necessiti di coenzimi o altri cofattori, come piccoli ioni inorganici, si studia l’effetto di inibitori della reazione da essa catalizzata. Se sono note la struttura primaria, secondaria e terziaria, si individua il meccanismo molecolare dell’interazione p.-substrato, arrivando a determinare il sito catalitico della p. e i residui amminoacidici direttamente coinvolti nella reazione enzimatica. L’importanza della conoscenza delle relazioni fra struttura e funzione delle p. si può comprendere meglio qualora si pensi al numero elevato di farmaci che inibiscono specifiche reazioni enzimatiche e la cui sintesi è stata spesso guidata dalle conoscenze strutturali dell’enzima coinvolto in quella data reazione. Anche per tali motivi gli studi strutturali e funzionali sulle p. rimangono senz’altro una delle aree principali della ricerca biochimica.
Sintesi delle proteine
Il processo in generale può essere schematizzato nella seguente formula, chiamata ‘dogma centrale’:

Il meccanismo della traduzione è assolutamente irreversibile, nel senso che non si ammette una trasmissione di informazioni in senso inverso. Invece si è osservato che in alcuni virus oncogeni il processo di trascrizione può essere invertito. H.M. Temin ipotizzò nel 1964 che l’RNA virale – per certi virus privi di DNA – venisse dapprima trascritto su un DNA a doppia elica e che successivamente questo DNA fosse integrato nel DNA nucleare della cellula ospite (➔ retrovirus). Alla base della duplicazione del DNA e della formazione dell’RNAm c’è un processo di riconoscimento tra le basi complementari. Però, mentre nella duplicazione cellulare il DNA si autoreplica completamente, nella sintesi proteica solo alcuni tratti di DNA (i geni propriamente detti) vengono trascritti e tradotti in proteine.
Trascrizione. La sintesi degli RNA è definita trascrizione perché il messaggio scritto nel DNA con un linguaggio a 4 lettere (A, G, C, T, cioè le basi azotate adenina, guanina, citosina, timina) è trascritto nell’RNAm con lo stesso linguaggio (A, G, C, U: l’uracile sostituisce la timina). I filamenti del DNA si separano parzialmente e su uno di essi (stampo) sono attratti i ribonucleotidi complementari, secondo il principio dell’accoppiamento di basi (A-U, G-C). Nelle cellule eucariotiche la trascrizione dell’RNAm, dell’RNAr e dell’RNAt avviene nel nucleo e, solo dopo una serie di modificazioni, gli RNA passano nel citoplasma dove avviene la traduzione mentre nei procarioti trascrizione e traduzione avvengono nel citoplasma senza soluzione di continuità.
Traduzione. In tutte le cellule comunque l’RNAm trasporta il messaggio genetico dal DNA ai ribosomi dove avviene il processo di sintesi proteica vera e propria, ossia di traduzione del messaggio. Questo termine si riferisce all’intero processo attraverso cui la sequenza di basi dell’RNAm viene utilizzata per ordinare e unire gli amminoacidi in una p., cioè per passare dal linguaggio polinucleotidico a 4 basi a quello polipeptidico a 20 amminoacidi. Sorge qui il problema del rapporto tra il numero di basi presenti in un gene e il numero di amminoacidi di cui è costituita la catena polipeptidica da esso codificata. Il calcolo combinatorio suggerisce che ciascun amminoacido può essere codificato da una sequenza di 3 basi (tripletta o codone); infatti, combinando a 3 a 3 le 4 basi, si ottengono 64 triplette, in largo eccesso rispetto ai 20 amminoacidi presenti nelle proteine.
Mediante alcuni brillanti esperimenti, condotti inizialmente da M.W. Nirenberg, è stato possibile pervenire a una decifrazione completa del codice genetico. La decifrazione del codice ha dimostrato che esistono 61 triplette (triplette di senso) che codificano i 20 amminoacidi presenti nella cellula e che più di una tripletta può pertanto codificare lo stesso amminoacido. Le 3 triplette rimaste (triplette non senso) non designano alcun amminoacido ma determinano il punto di arresto della formazione della catena polipeptidica.
RNAr e RNAt. Nella sintesi proteica, oltre all’RNAm vi sono altri due tipi di molecole di RNA che svolgono ruoli distinti in cooperazione fra loro. L’RNAr si combina con una serie di p. formando i ribosomi, i quali possiedono i siti di legame per tutte le molecole che interagiscono nella sintesi proteica. L’RNAt costituisce la chiave di lettura del codice genetico; non essendovi affinità stereochimica fra gli amminoacidi e le basi dell’RNAm non è possibile che gli amminoacidi si allineino su uno stampo di RNA, come avviene nella trascrizione. Gli amminoacidi, specificati da una sequenza di basi di una molecola di RNAm, vengono trasportati e depositati all’estremità in accrescimento di una catena polipeptidica da molecole di RNAt che hanno la funzione di molecole adattatrici. Le molecole di RNAt sono corte e diversificate. Si ritiene che ve ne siano di 30-40 tipi diversi nei batteri e circa 50 nelle cellule animali e vegetali. In soluzione le molecole di RNAt si ripiegano assumendo una struttura tridimensionale. Schematicamente la configurazione assunta è una struttura a steli e anse. La sequenza di una molecola di RNAt termina sempre, all’estremità 3′, con una tripletta citosina-citosina-adenina (CCA) che costituisce il sito di legame con l’amminoacido; 3 dei 4 steli hanno all’estremità un’ansa costituita da 7 o 8 basi. Al centro dell’ansa, portata dallo stelo centrale, vi sono 3 basi, che costituiscono l’anticodone e possono appaiarsi con 3 nucleotidi di un codone dell’RNAm. Un singolo RNAt è in grado di riconoscere più di una tripletta che corrisponde a un dato amminoacido, a causa della cosiddetta oscillazione del codice o appaiamento non standard delle basi tra il terzo nucleotide del codone e il suo corrispondente nucleotide dell’anticodone.
Il legame fra amminoacido e RNAt è catalizzato da uno dei 20 enzimi presenti nella cellula e denominati amminoacil-tRNA-sintetasi. Ogni enzima può attaccare un particolare amminoacido all’estremità 3′ di un RNAt corrispondente e può riconoscere i vari RNAt di uno stesso amminoacido. Il legame si forma nel corso di una reazione che richiede la scissione di una molecola di ATP. Enzima, amminoacido e ATP formano un complesso, liberando AMP e pirofosfato inorganico (PPi). La pirofosfatasi successivamente scinde il legame ad alta energia del pirofosfato e l’equilibrio della reazione è spostato verso la formazione di un amminoacido attivato o amminoacil-tRNA, che trattiene l’energia liberata dall’ATP mediante la scissione dei legami fosforici.
Ribosomi. L’anticodone dell’RNAt legato all’amminoacido corretto riconosce un codone dell’RNAm a livello dei ribosomi. Infatti i legami chimici altamente specifici della traduzione non hanno luogo liberamente all’interno della cellula in quanto questa funzione critica comporta più di un milione di legami peptidici al secondo e richiede l’interazione di numerosissimi gruppi chimici presenti negli RNA e nelle p. che servono alla sintesi. Se ognuno di questi componenti si trovasse libero in soluzione, le interazioni simultanee fra le varie molecole sarebbero rare e la velocità di polimerizzazione degli amminoacidi molto bassa. Al contrario, l’RNAm, con la sua informazione in codice, e i singoli RNAt caricati con i corretti amminoacidi si incontrano grazie al legame contemporaneo con i ribosomi. Il ribosoma è una particella citoplasmatica costituita da singole molecole di RNA e da più di 50 p. e organizzata in due subunità, una maggiore e una minore.
Fasi della sintesi proteica. La sintesi proteica a livello dei ribosomi si divide in 3 fasi: inizio, allungamento, terminazione.
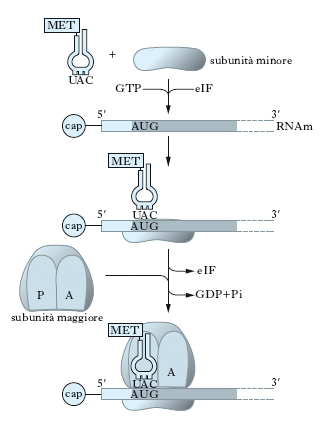
Il primo evento della fase di inizio è l’attacco di una molecola di metionina libera all’estremità di una molecola di RNAt da parte della specifica metionil-tRNA sintetasi (fig. 6). Vi sono almeno due tipi di tRNAmet ma solo uno, chiamato tRNAmeti, può iniziare la sintesi proteica. Nei Batteri il gruppo amminico della metionina viene modificato per l’aggiunta di un gruppo formile e questo fa sì che la sintesi parta dall’estremità carbossilica libera del primo amminoacido. Successivamente il tRNAmeti, insieme a una molecola di GTP (guanosina trifosfato) e alla subunità ribosomiale minore, si lega a un sito specifico dell’RNAm che, nella maggior parte dei casi, è collocato nelle immediate vicinanze del codone di inizio AUG (adenina uracile guanina): si costituisce così il complesso di inizio. L’energia richiesta nelle diverse tappe della sintesi proteica è fornita dall’idrolisi del GTP che diventa GDP e libera fosforo inorganico. Nei Batteri il ribosoma viene guidato nel sito di inizio da una corta sequenza nucleotidica, complementare a una sequenza dell’RNAm situata a circa 5-10 nucleotidi prima della tripletta di inizio AUG. Negli Eucarioti, l’inizio della traduzione avviene invece mediante il riconoscimento di una speciale modifica, il cappuccio (➔ cap), situata all’estremità 5′ dell’RNAm. Il complesso di inizio si può formare solo se sono presenti una serie di p., dette fattori di inizio (IF), che svolgono ognuna un ruolo preciso. Solo dopo che il codone AUG è stato collocato correttamente, la subunità ribosomiale maggiore si lega al complesso di inizio e comincia la seconda fase.
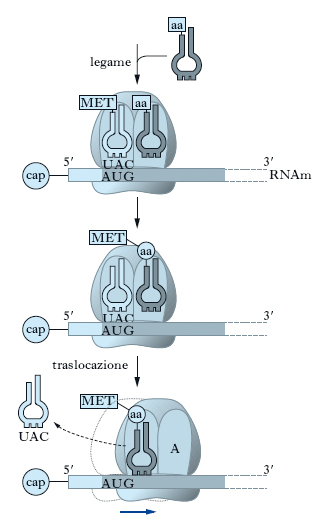
Nella fase di allungamento gli amminoacidi vengono aggiunti uno alla volta e legati in una catena polipeptidica (fig. 7). Le reazioni di allungamento coinvolgono l’appaiamento RNAm-RNAt a livello dei due principali siti attivi sul ribosoma, il sito A e il sito P. I ribosomi sono infatti caratterizzati da due principali stati conformazionali: una conformazione espone il sito A che lega l’RNAt legato a un solo amminoacido (amminoacil-tRNA); l’altra espone il sito P che ha affinità per un RNAt a cui è legata la catena polipeptidica (peptidil-tRNA). Nella fase di allungamento si osservano 3 sequenze di eventi che si ripetono in modo ciclico: l’amminoacil-tRNA si lega al sito A; si forma un legame peptidico; si procede alla traslocazione, in cui il peptidil-tRNA formatosi a livello del sito A si sposta sul sito P. Dopo la fase di inizio, il ribosoma, avendo legato un tRNAmeti a livello del sito P e un amminoacil-tRNA a livello del sito A, è pronto per la formazione del primo legame peptidico. Tale legame, catalizzato da un enzima (peptidiltransferasi), si forma tra la metionina, portata dall’RNAt iniziatore a livello del sito P, e l’amminoacido, portato dall’RNAt a livello del sito A. L’energia richiesta per la reazione deriva principalmente dall’idrolisi del legame tra la metionina e l’RNAt iniziatore. Al termine della reazione, entrambi gli amminoacidi associati in un dipeptide sono legati all’RNAt localizzato nel sito A. In virtù di questo legame, l’RNAt del sito A da amminoacil-tRNA diventa peptidil-tRNA. Un RNAt vuoto rimane pertanto a livello del sito P. Non sono richiesti fattori di allungamento per la formazione del legame peptidico. L’RNAt vuoto viene rilasciato dal ribosoma. Segue la traslocazione, durante la quale il ribosoma si muove lungo l’RNAm nella direzione 5′-3′ per una distanza equivalente a un codone. L’energia per la traslocazione è fornita dall’idrolisi di una molecola di GTP. Durante la traslocazione, infatti, il ribosoma funziona come trasduttore di energia, convertendo l’energia chimica rilasciata dall’idrolisi del GTP in energia meccanica per il movimento. Il movimento porta il codone precedentemente localizzato al sito A, con il suo peptidil-tRNA, sul sito P, ed espone un nuovo codone libero a livello del sito A. In tal modo il ciclo può ricominciare; durante ogni ciclo il ribosoma avanza di un codone e aggiunge un amminoacido alla catena polipeptidica nascente. La sintesi procede alla velocità di 2 o 3 cicli al secondo negli Eucarioti e si ripete fino a quando i ribosomi non raggiungono un codone non senso o di terminazione (UAA, UAG o UGA) alla fine della regione codificante dell’RNAm.
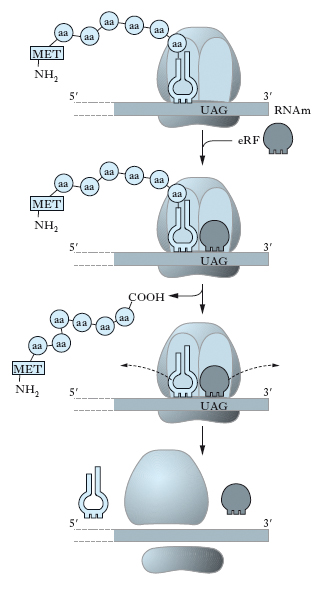
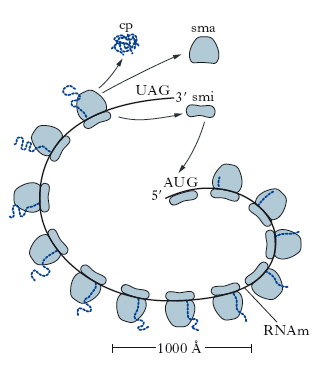
Nella fase di terminazione, le reazioni sono catalizzate da un singolo fattore di rilascio (eRF) (fig. 8). L’eRF si lega al sito A insieme a un’altra molecola di GTP non appena nel sito A compare una tripletta di terminazione (o non senso). Il legame di eRF stimola l’idrolisi, catalizzata dalla stessa peptidiltransferasi, del legame tra la catena peptidica e l’RNAt. In tal modo la catena polipeptidica completa viene rilasciata dal ribosoma, eRF viene rimosso dal sito A, l’RNAt vuoto viene rimosso dal sito P e le subunità ribosomiali si separano dall’RNAm. Più catene polipeptidiche possono venire sintetizzate contemporaneamente dallo stesso RNAm; molti ribosomi infatti possono essere coinvolti nella traduzione dello stesso messaggio. Essi sono distribuiti sulla molecola di RNAm come le perle di una collana e l’intera struttura è chiamata polisoma (fig. 9). Il numero dei ribosomi in un polisoma dipende dalla lunghezza della regione codificante della molecola di RNAm e va da un massimo di circa 100 ribosomi in lunghi RNAm a un minimo di 2 in un piccolo RNAm che codifica le protammine.
Inibizione della sintesi proteica. Gli antibiotici sono inibitori della sintesi proteica in quanto uccidono o danneggiano le cellule interferendo con le singole fasi dei processi di inizio, allungamento e terminazione. Molti antibiotici uccidono o danneggiano selettivamente le cellule batteriche, perché interagiscono con alcuni dei componenti coinvolti nella sintesi proteica batterica, che sono simili ma non uguali a quelli eucariotici.
Modificazioni postraduzionali delle proteine
Quasi tutte le p., dopo essere state rilasciate dal ribosoma, possono subire modificazioni postraduzionali, di tipo covalente, che possono essere irreversibili e reversibili. Le prime sono catalizzate da uno o più enzimi specifici e hanno come prodotto finale una p. modificata irreversibilmente in uno o più punti della sua struttura primaria. Le seconde sono modificazioni enzimatiche, operate su specifici amminoacidi, che possono essere rimosse da enzimi differenti, che ripristinano la molecola proteica originaria.
In genere, il gruppo delle modificazioni postraduzionali reversibili comprende quelle che fungono da sistema di attivazione e disattivazione della funzione biologica di una data proteina. Rientrano in questo gruppo le reazioni di fosforilazione-defosforilazione, di acetilazione-deacetilazione e di ADP-ribosilazione di molte proteine. Ne sono un esempio tipico le reazioni di fosforilazione-defosforilazione delle p. di trasporto di membrana che agiscono da pompe, trasportando attivamente alcune sostanze contro un gradiente elettrochimico (➔ membrana) come la pompa Na+-K+ e la pompa del Ca++. Anche le reazioni di polimerizzazione-depolimerizzazione sono modificazioni reversibili che operano come meccanismo di controllo della funzione di p. enzimatiche e non enzimatiche. Per es., sia nel caso della tubulina, che costituisce i microtubuli, sia in quello dell’actina, che con la miosina è coinvolta nel processo della contrazione muscolare, uno dei fattori regolanti la polimerizzazione-depolimerizzazione sembra essere lo stato di fosforilazione della p. stessa, cioè l’interazione della tubulina con GTP o GDP e dell’actina con ATP o ADP.
Le modificazioni postraduzionali irreversibili delle p. spesso sono reazioni finalizzate a promuovere l’interazione della p. modificata con alcune specifiche strutture cellulari, a conferire alla p. modificata una specifica attività enzimatica o a segregarla in determinati compartimenti cellulari dove deve svolgere la sua funzione biologica. Fanno parte di queste modificazioni permanenti le trasformazioni o scissioni proteolitiche specifiche, le quali rappresentano la fase finale della biosintesi di molte proteine. Queste reazioni avvengono in molti modi diversi, ma hanno in comune il fatto che viene scisso un determinato legame peptidico nella catena polipeptidica del precursore della p. matura, che viene poi rilasciata come p. biologicamente attiva. Questo processo regolativo interessa molti enzimi digestivi, ormoni proteici, p. della crescita, p. vasoattive, p. del sistema del complemento e della coagulazione sanguigna, p. virali e fagiche, p. costituenti il collagene e p. dei processi di sviluppo. Anche la glicosilazione, che avviene nel reticolo endoplasmatico, fa parte delle trasformazioni permanenti; questa reazione aggiunge una sequenza saccaridica a una data p., sequenza che ricopre un ruolo di segnale di riconoscimento e d’innesco di una particolare funzione biologica. Infatti, la catena saccaridica conferisce a molte glicoproteine una notevole stabilità conformazionale, resistenza agli enzimi proteolitici e capacità di interagire con sostanze polari di varia natura. Gli scopi dei processi fisiologici di glicosilazione sono soprattutto la produzione di p. con funzione di recettori sulla superficie cellulare, la produzione di un segnale per il riconoscimento di particolari p. e, infine, la produzione di un sistema per l’interazione tra cellule.
Le modificazioni postraduzionali assumono particolare importanza nei processi di traslocazione delle p. attraverso le membrane, che riguardano tutte quelle p. destinate a espletare la loro funzione esclusivamente all’interno di organuli cellulari come, per es., mitocondri, cloroplasti, lisosomi, vacuoli ecc. Infatti, queste p. al termine della loro biosintesi fuoriescono dal reticolo endoplasmatico e vengono indirizzate e trasportate alla loro sede definitiva grazie a un meccanismo di trasporto unidirezionale che ne impedisce l’eventuale ritorno al citoplasma. In genere, tutte le p. da traslocare presentano una sequenza guida di amminoacidi-segnale all’estremità amminoterminale, che viene riconosciuta da un recettore di membrana il quale, agendo da pompa endoenergetica, spinge la p. all’interno dell’organulo e poi scinde proteoliticamente la sequenza segnale. Solo nel caso del trasferimento all’interno del nucleo, il passaggio avviene attraverso i pori nucleari (➔ nucleo) senza dispendio di energia e, inoltre, le sequenze segnale caratteristiche delle p. nucleari rimangono legate come parti integranti della p. stessa anche dopo che il trasporto di queste molecole è stato completato.
Oltre alle modificazioni postraduzionali tutte le complesse reazioni della sintesi proteica rappresentano l’obiettivo di meccanismi di controllo, che agiscono per accelerare o rallentare le varie fasi del processo e quindi la velocità dell’intero meccanismo (➔ regolazione).
Metabolismo proteico
Le p. alimentari sono degradate nello stomaco e nell’intestino dalle proteasi (o proteinasi), gruppo di enzimi appartenenti alla classe delle idrolasi, che idrolizzano le p. fino a trasformarle in amminoacidi; questi sono poi assorbiti dalla mucosa intestinale, per passare in circolo ed essere utilizzati dall’organismo. Le proteasi agiscono sul legame peptidico catalizzando la reazione
in cui R e R′ rappresentano le catene laterali degli amminoacidi. Le proteasi di cui si è più approfonditamente studiato il meccanismo d’azione sono le proteasi a serina, le quali posseggono un residuo di serina nel sito catalitico, indispensabile per la loro attività enzimatica. La tripsina e la chimotripsina appartengono entrambe a questo sottogruppo. Oltre che nei processi di digestione delle p. alimentari, l’attività delle proteasi è di vitale importanza per l’organismo; infatti, esse intervengono nei processi di maturazione di p., ormoni, enzimi, trasformando i rispettivi precursori nelle forme biologicamente attive, svolgono un ruolo primario nel processo della coagulazione del sangue ecc. Poiché l’attività delle proteasi deve essere sotto stretto controllo, al fine di non provocare l’idrolisi indesiderata di p., la maggior parte di esse è sintetizzata come proenzima (tripsinogeno, chimotripsinogeno, fibrinogeno), trasformato a sua volta nell’enzima attivo a opera di altre proteasi e successivamente per azione autocatalica; altre proteasi sono invece inibite allostericamente da inibitori di natura diversa.
All’inizio degli anni 1990 sono state identificate proteasi codificate dai geni del virus HIV che svolgono un’azione importante nell’assemblaggio delle molecole costituenti la particella virale matura. Utilizzando alcuni inibitori delle proteasi virali si sono ottenuti progressi sostanziali nella terapia dell’AIDS, consentendo ai pazienti un miglioramento della qualità di vita e una marcata riduzione dell’espansione virale, anche quando il sistema immunitario risultava gravemente compromesso.
Proteine G
Le p. G sono le p. integrali di membrana che si legano ai nucleotidi GDP e GTP e trasmettono il segnale ormonale all’interno della cellula. Le p. G sono quindi p. per l’accoppiamento di segnali; si distinguono in p. che stimolano (p. G stimolatrici o Gs) e p. che inibiscono (p. G inibitrici o Gi) un enzima che catalizza la sintesi di un secondo messaggero. Tutte le p. G sono trimeri costituiti da una subunità α (peso molecolare 39.000-52.000) che si lega sia al GDP sia al GTP, una subunità β (35.000) e una subunità γ (10.000). Le p. G sono un trimero (Gαβγ) solo nello stato inattivo, con la subunità legata al GDP. Quando un complesso costituito dall’ormone con il suo rispettivo recettore si lega al complesso p. G-GDP, si ha una reazione di scambio nucleotidico, cioè il GTP sostituisce il GDP. Il trimero si dissocia nel dimero βγ e nel complesso attivo Gα-GTP, che stimola o inibisce l’enzima responsabile della sintesi del secondo messaggero. Dato che la Gα idrolizza il GTP in GDP+Pi, questa sua attività fosfatasica (GTPasi) costituisce un meccanismo di autoregolazione che inattiva automaticamente la p. G. Dopo che il GTP è stato idrolizzato, il risultante complesso Gα-GDP si riassocia con il dimero βγ e ritorna allo stato originario inattivo. Nei Mammiferi una p. Gsα-GTP stimola l’adenilatociclasi a trasformare l’ATP in AMPciclico (AMPc), provocando in essa un cambiamento conformazionale.
Proteomica
Si dice proteoma l’insieme di tutti i possibili prodotti proteici espressi in una cellula, oggetto di studio della proteomica. Essa sviluppa nuove tecnologie non solo per studiare le singole p. codificate dai geni, ma soprattutto per comprendere le loro relazioni. Poter rivelare i cambiamenti nell’espressione del genoma sia durante lo sviluppo e il differenziamento sia in risposta a cambiamenti ambientali dovrebbe rivelarsi di grande importanza per la comprensione delle malattie umane e dei processi di invecchiamento.
Geni e proteine. Il numero delle p. è senza dubbio minore rispetto alla capacità codificante del genoma di un dato organismo. I calcoli sulla quantità dei geni del genoma umano pubblicati nel 2001 hanno mostrato che esso contiene almeno 30.000 geni, ma solo una piccola parte di essi codifica proteine. Mentre in Saccharomyces cerevisiae quasi il 70% del genoma codifica p. ed è presente un gene ogni 2000 paia di basi, nell’uomo meno del 5% del genoma è codificante ed è presente circa un gene ogni 30.000 paia di basi. È tuttavia da rilevare che le p. formano fra di loro una rete intricatissima di interazioni; per es., nel lievito, usando un campione di 1004 p., si sono individuate ben 957 interazioni diverse. La comprensione di queste interazioni è uno degli obiettivi, complesso ma irrinunciabile, della proteomica.
Un gene eucariotico è costituito da sequenze codificanti, gli esoni, che sono frapposte a sequenze non codificanti, gli introni, i quali vengono generalmente rimossi attraverso il meccanismo dello splicing. Talvolta, per un meccanismo di regolazione cellulare, accade che un introne, invece di essere rimosso dal trascritto primario, costituisca parte integrante dell’RNAm citoplasmatico: questo fenomeno viene definito splicing alternativo. In questo caso lo stesso gene codifica p. diverse che però possiedono porzioni in comune. Una volta stabilito che nella stessa cellula sono presenti polipeptidi diversi ma prodotti dallo stesso gene, è importante analizzare quantitativamente i due prodotti, in quanto differenze di concentrazione di una determinata p. sono correlabili a diversi stadi di sviluppo o differenziamento della cellula. Queste informazioni costituiscono un importante contributo per lo studio di alcune patologie determinate da carenza o da eccesso di prodotto genico. Bisogna inoltre tenere presente che i geni codificano p. che possono subire modificazioni dopo la loro sintesi. Queste modificazioni permettono alle p. di divenire funzionali o inattive; è pertanto importante stabilire in quale momento della fisiologia cellulare una specifica p. è attiva o inattiva a causa delle modificazioni subite.
Tecniche. L’apparato tecnologico su cui si basa la proteomica è sicuramente meno sviluppato rispetto alle complesse e molteplici tecniche disponibili per l’analisi dei genomi. Tuttavia rispetto al passato, quando generalmente si procedeva all’identificazione di una p. dopo aver sottoposto un lisato cellulare a elettroforesi, vi sono nuove e sofisticate tecnologie e, avendo a disposizione la sequenza di un intero genoma, lo studio della sua espressione può essere facilitato. L’utilizzazione di sonde legate su membrane secondo specifiche disposizioni (array) permette di osservare i livelli di trascrizione dei geni (➔ chip), anche se questo metodo non fornisce informazioni sulle p. tradotte. A questo scopo, tradizionalmente, vengono utilizzati western blots (➔ blotting) per rivelare p. separate per elettroforesi o anticorpi accoppiati a sostanze fluorescenti per rivelare la localizzazione delle p. in vivo. Per seguire la sintesi e la localizzazione di specifiche p. in una grande varietà di cellule viventi è utilizzata una p. fluorescente naturale, scoperta nella medusa Aequorea victoria: la GFP (green fluorescent protein; fig. 10).
Gli studi della seconda metà degli anni 1990 hanno portato alla costruzione di geni di fusione che contengono la sequenza nucleotidica codificante la GFP, legata con quella codificante la p. di interesse; il gene così modificato viene poi introdotto nelle cellule, dove codifica una p. di fusione fluorescente che si può osservare dopo l’esposizione alla luce blu o ultravioletta. A differenza di altre p. bioluminescenti, la GFP non richiede l’aggiunta di substrati o cofattori per emettere la fluorescenza; è inoltre molto piccola e non interferisce con l’attività delle p. alle quali è legata. Mediante mutagenesi del gene per la GFP si sono anche prodotte forme varianti che producono luce blu o gialla, permettendo in questo modo lo studio di due o più p. che si esprimono o simultaneamente o in momenti differenti. Si stanno infatti utilizzando le fusioni con GFP per studiare i cambiamenti nell’espressione di tutti i geni codificanti p. coinvolte in una particolare via metabolica, in risposta ai trattamenti delle cellule o dei tessuti con una specifica sostanza o con un agente potenzialmente terapeutico. Nuove tecnologie basate su elettroforesi effettuata in piccoli tubi capillari e monitorate da sensibili microfotorivelatori e da sofisticati programmi informatici permetteranno di osservare cambiamenti nei livelli di p. di fusione in grandi quantità di campioni simultaneamente.
Un altro modo per affrontare gli studi riguardanti la p. si basa sul fatto che la funzione delle p., come quella delle altre macromolecole, dipende dalla loro struttura. Per determinare la struttura tridimensionale delle macromolecole si usano principalmente tecniche biofisiche, quali la cristallografia a raggi X e la risonanza magnetica nucleare, metodi che sono tecnicamente difficili e che richiedono tempi lunghi. Riuscire a prevedere la struttura e, quindi, la funzione delle p. in base alla sequenza dei loro elementi di base renderebbe questi tempi più brevi. A tale scopo sono stati messi a punto vari programmi informatici che mirano a prevedere la struttura secondaria delle p. in base alla sequenza amminoacidica. I parametri su cui si basano questi programmi sono sia le proprietà fisiche ed energetiche degli amminoacidi e dello scheletro polipeptidico, sia la frequenza statistica degli amminoacidi all’interno delle strutture secondarie.
Per prevedere la struttura secondaria delle p. è stata anche applicata la metodologia informatica delle reti neurali. Le reti neurali, costituite da un grande numero di unità di calcolo organizzate in strati interconnessi e nelle quali lo strato di ingresso (input) riceve i dati e può o meno trasmettere queste informazioni allo strato successivo, sono capaci di adeguare, in risposta ai dati, i parametri che definiscono le interconnessioni tra le sue subunità e pertanto possono essere addestrate a identificare quadri molto complessi insiti nei dati di ingresso. Per es., si possono usare le sequenze amminoacidiche di p. di cui si conosce la struttura cristallina per addestrare una rete, cioè per adeguare i suoi parametri a prevedere la struttura secondaria di nuove sequenze amminoacidiche. Per prevedere invece la struttura terziaria della p. in base alla struttura primaria viene usato un metodo comparativo, detto modellazione per omologia o modellazione in base a forme note di omologia, che sembra dare buoni risultati. Questo metodo si basa sul fatto che la p. da studiare è omologa a un’altra p., o a una famiglia di p., la cui struttura terziaria sia già stata risolta mediante cristallografia a raggi X. Il primo successo di questo metodo è stato la previsione della struttura di una p. codificata dall’HIV, omologa a proteasi già note, dato confermato dalla determinazione della struttura cristallina effettiva della proteasi di HIV.