
emoglobina
Proteina contenente ferro, dotata di funzione respiratoria, capace di combinarsi reversibilmente con l’ossigeno molecolare, indicata con la sigla Hb.
Biologia
Caratteristiche della molecola
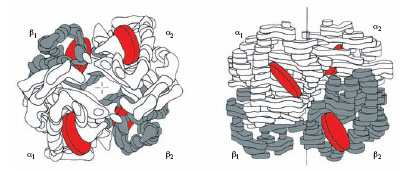
Nei Mammiferi l’e. è una molecola con peso molecolare di circa 68.000, costituita (v. fig.) da 4 catene polipeptidiche uguali a due a due, due catene α e due catene β. A ciascuna catena dell’e. è legata una molecola di eme, un complesso del ferro con la protoporfirina IX: ogni molecola di e. porta legate 4 molecole di eme, ciascuna in grado di trasportare una molecola di O2 . A seconda dello stato di ossidazione del ferro, le e. si possono suddividere in e. contenenti ferro ferroso (ridotto) ed e. contenenti ferro ferrico (ossidato). Le principali e. contenenti ferro ferroso sono la desossiemoglobina (Hb), nella quale le molecole di ossigeno sono rimpiazzate da molecole d’acqua, l’ ossiemoglobina (HbO2), che è la forma ossigenata dell’e., e la carbossiemoglobina (HbCO), nella quale l’ossigeno è sostituito da una molecola di monossido di carbonio (l’elevata tossicità del monossido di carbonio deriva dal fatto che per esso l’e. ha un’affinità circa 50 volte maggiore di quella per l’O2). L’e. contenente ferro ferrico è la metemoglobina (MetHb), che non si combina con l’ossigeno molecolare bensì con una serie di leganti anionici.
L’analisi della struttura tridimensionale ha rivelato che l’e. ha una conformazione spaziale quasi sferica con numerosi tratti a elica e che le 4 subunità presentano numerosi punti di contatto, che variano durante la reazione dell’e. con l’O2. In ciascuna catena sono presenti regioni caratteristiche, che consentono all’e. di svolgere la sua funzione di trasportatrice di ossigeno e permettono la regolazione della sua affinità per l’O2 da parte di una serie di effettori allosterici positivi e negativi. Infatti, a una determinata pressione parziale di ossigeno presente nel mezzo in cui è disciolta l’e., la frazione di e. ossigenata varia in funzione della temperatura, del pH, della concentrazione di alcuni ioni (K+, Cl−, PO43−, HCO3−), della pressione parziale di CO2 e della concentrazione di alcuni metaboliti fosforilati.
Il ruolo dell’e. nell’uomo
Nell’uomo, in particolare, il ruolo principale di regolatore dell’affinità dell’e. per l’O2 è svolto dall’acido 2,3-difosfoglicerico. Nella reazione di legame dell’e. con l’O2 bisogna ricordare il cosiddetto fenomeno dell’interazione fra emi (o effetto cooperativo dell’e.) che provoca cambiamenti di conformazione che avvengono in seguito al legame di una molecola di O2 con una delle 4 catene dell’e.; questi cambiamenti favoriscono e accelerano il legame delle successive 3 molecole di O2 ai siti di legame liberi sugli emi delle altre 3 catene. Le caratteristiche strutturali e funzionali consentono quindi che nell’uomo l’e. trovi negli alveoli polmonari le condizioni per essere totalmente ossigenata (elevata pressione parziale di O2, pH leggermente alcalino, bassa pressione parziale di CO2) e, una volta giunta ai tessuti, sia in grado di cedere l’O2 necessario per il metabolismo aerobio cellulare in seguito ad alcuni fattori che ne diminuiscono l’affinità (bassa pressione parziale di O2, pH leggermente acido, elevata pressione parziale di CO2). Nell’uomo adulto la componente principale dell’e. normale è la cosiddetta HbA1, che rappresenta circa il 95% del contenuto emoglobinico, formata da due catene α e due β (α2β2) costituite rispettivamente da 141 e 146 amminoacidi. Il rimanente 5% è formato dalla HbA1−c, forma glicosilata dell’HbA1 (un residuo glicosidico è legato all’estremità N-terminale di ciascuna catena β dell’e.), che riveste una particolare importanza poiché il suo contenuto aumenta fino a tre volte nei soggetti diabetici, dalla HbA2, costituita da due catene α e due δ, e dalla MetHb. Negli embrioni umani fino alla 12a settimana di vita intrauterina sono presenti, al posto delle catene β, le catene ε e ζ.
Nei feti umani di età superiore alle 12 settimane il componente emoglobinico principale è l’ e. fetale (HbF), costituita da due catene α e due γ. L’e. fetale è presente nel sangue neonatale anche dopo la nascita, ma viene rapidamente sostituita dall’HbA1 (nell’adulto sono presenti solo tracce di HbF). Negli eritrociti umani normali sono contenuti circa 31 g di e. per 100 ml di globuli rossi, corrispondenti a circa 13,5-15 g di e. per 100 ml di sangue.
La conoscenza della sequenza amminoacidica di un gran numero di e. (provenienti da specie molto distanti fra loro) ha permesso di stabilire l’esistenza di una notevole somiglianza nella struttura primaria delle varie e. e di costruire un albero filogenetico che indica la loro origine da una proteina ancestrale comune. Si pensa che i geni che codificano per le diverse catene si siano originati per duplicazione (e successivo differenziamento) da un gene ancestrale comune.
Medicina
Emoglobinemia e emoglobulinuria
L’ emoglobinemia è l’abnorme presenza di e. libera nel plasma, in seguito a emolisi; si determina quantitativamente mediante emoglobinometria, effettuata con l’ emoglobinometro e con metodi fotometrici.
L’ emoglobinuria è l’abnorme presenza di emoglobina nelle urine, le quali appaiono di colore variabile dal rosa al bruno. Condizione necessaria è l’emolisi e il concomitante passaggio di e. nel plasma e quindi nell’urina. Può verificarsi in corso di malattie infettive (malaria, tetano, tifo ecc.), intossicazioni (da chinino, solfammidici, fosforo, veleni di serpenti, funghi velenosi ecc.), difetti enzimatici ereditari (per es. favismo), in seguito a trasfusioni di sangue, o a introduzione endovenosa di sostanze ipotoniche rispetto al siero di sangue. Una forma particolare è l’ emoglobinuria parossistica notturna, malattia emolitica caratterizzata da un’emolisi particolarmente accentuata nelle ore notturne.
Emoglobinopatie
Le alterazioni di origine genetica della struttura primaria della molecola dell’e. vanno sotto il nome di emoglobinopatie, termine generico designante uno stato patologico connesso con la presenza di una e. patologica. Alle emoglobinopatie vengono ascritte le talassemie, la falcemia e altre forme di più recente descrizione.
Uno dei moderni metodi per la diagnosi è l’elettroforesi, che permette di determinare eventuali diversità di carica elettrica dovuta a sostituzioni di amminoacidi nell’e. patologica. Per es., nell’e. S, responsabile della falcemia, l’acido glutammico è sostituito dalla valina; per l’e. C, spesso associata all’e. S, si tratta della sostituzione dell’acido glutammico con la lisina. Per le talassemie α e β l’alterazione dei rapporti quantitativi fra le catene di globina deriva da vari tipi di mutazioni a carico dei geni per la globina α o per la β. Tali mutazioni sono, per es., delezioni di parte del gene, crossing over asimmetrico (e. Lepore), sostituzioni di basi in punti determinanti per la funzione del gene.
Le emoglobinopatie si suddividono in due grandi categorie: per difetti quantitativi e per difetti qualitativi.
Emoglobinopatie per difetti quantitativi
L’e. normale ha una uguale quantità di α-globina e di β-globina, mentre gli individui affetti da α-talassemia o da β-talassemia presentano uno sbilancio rispettivamente nella quantità di globina α o β. Le α-talassemie derivano da anomalie nel dosaggio genico: le persone normali hanno 2 geni e quindi 4 alleli della α-globina (genotipo αα/αα). Le persone con 2 alleli dell’α-globina presentano sintomi lievi (genotipo α-/α- oppure αα/--); quelle che hanno solo un allele α hanno la malattia in forma grave (α -/-), mentre la mancanza di tutti i geni (genotipo --/--) è letale per l’insorgenza dell’idrope fetale. Alcune forme di α-talassemia sono causate da instabilità dell’RNAm o dell’emoglobina. Per es., la Hb Constant Spring presenta una mutazione in un codone di stop dell’α-globina, pertanto la traduzione della proteina continua per altri 30 codoni e determina un prodotto proteico instabile. In un altro caso la mutazione è nella sequenza segnale di poliadenilazione dell’RNAm. Una varietà rara di α-talassemia è causata da una mutazione associata al cromosoma X, il che dimostra l’esistenza sul cromosoma X di un fattore di controllo dell’espressione della α-globina. Le mutazioni identificate nel gene che codifica la β-globina e che danno luogo a β-talassemie sono molteplici: per es., mutazioni non-senso che determinano l’interruzione prematura della sintesi della globina; mutazioni nel promotore del gene; mutazioni che eliminano normali siti di splicing o attivano nuovi siti di splicing (detti siti criptici di splicing). In un tipo di β-talassemia, l’ e. Lepore, i prodotti genici sono codificati da un gene di fusione formato da sequenze del gene δ e del gene β. Questo gene di fusione conserva il promotore a bassa attività del gene δ e questo porta a β-talassemia. La gravità della β-talassemia è spesso ridotta dalla persistenza ereditaria dell’e. fetale (HPFH, hereditary persistence of fetal hemoglobin). Le cause della HPFH sono varie e complesse, ma possono includere delezioni di sequenze di DNA che interessano regioni di controllo e anche mutazioni nel promotore della γ-globina.
Emoglobinopatie per difetti qualitativi
Le sostituzioni di amminoacidi che alterano le proprietà ma non la produzione delle globine producono fenotipi la cui varietà ha fornito molte informazioni sulla funzione delle emoglobine. Oltre all’anemia falciforme (➔ falcemia), causata dalla sostituzione nella β-globina dell’acido glutammico con la valina, vi sono altre forme di anemia causate da diverse mutazioni del gene che codifica la globina. Nell’e. la transizione dalla forma ossigenata a quella non ossigenata è accompagnata da importanti cambiamenti nella struttura terziaria; la sostituzione di un amminoacido può modificare questo equilibrio e pertanto l’e. mutata, con alta affinità per l’ossigeno, non riesce a portare ossigeno ai tessuti, causando una eritrocitosi solitamente benigna, mentre quella con bassa affinità per l’ossigeno determina anemia e cianosi. Molte varianti emoglobiniche sono instabili a causa dell’indebolimento della loro struttura terziaria o quaternaria dovuto a variazioni nelle catene di amminoacidi. In alcuni casi l’e. instabile precipita formando aggregati intracellulari (corpi di Heinz). Questo causa l’anemia emolitica cronica, un’affezione spesso lieve, ma gravemente esacerbata da determinati farmaci e infezioni. Le e. M sono caratterizzate da cambiamenti nella conformazione tridimensionale della molecola nell’ansa che trattiene il gruppo eme; l’atomo di ferro in questo caso assume lo stato ferrico non fisiologico. Questo stato di metaemoglobinemia determina una cianosi cronica, solitamente benigna.