
metabolismo
Il complesso delle trasformazioni chimiche che avvengono nelle cellule degli organismi eucarioti e procarioti in modo coordinato e finalizzato, al quale cooperano molti enzimi e sistemi multienzimatici intracellulari. Il m. ha quattro funzioni principali e specifiche: ricavare energia chimica dalla degradazione di sostanze nutritive ricche di energia, dall’ambiente o dall’energia solare; convertire le molecole nutritive nei precursori di base delle macromolecole cellulari; utilizzare questi precursori di base per formare proteine, acidi nucleici, lipidi, polisaccaridi e altre sostanze; formare e degradare biomolecole necessarie a funzioni specializzate delle cellule.
Biologia
Per m. intermedio si intendono le sequenze specifiche dei prodotti successivi di trasformazione, i metabolici; esso presenta due fasi principali: l’anabolismo e il catabolismo. Nell’anabolismo (o biosintesi), che costituisce la fase costruttiva sintetica del m., piccoli precursori o molecole di base sono utilizzati per formare i grandi componenti macromolecolari cellulari, come le proteine e gli acidi nucleici. Il catabolismo è la fase degradativa del m. nella quale le molecole organiche di nutrimento, come i carboidrati, i lipidi e le proteine provenienti dall’ambiente extracellulare o da riserve accumulate nella cellula, sono degradate da reazioni a tappe successive in prodotti finali più semplici e a minore peso molecolare, come l’acido lattico, l’anidride carbonica e l’ammoniaca.
Il m. è attività di ogni singola cellula; in rapporto alle differenze citomorfologiche delle cellule e delle loro attività metaboliche, si distinguono un m. delle cellule eucariotiche e un m. delle cellule procariotiche.
M. delle cellule eucariotiche
Reazioni anaboliche e cataboliche
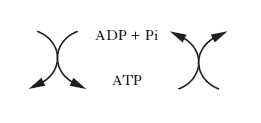
Le reazioni chimiche, siano esse anaboliche o cataboliche, possono avvenire spontaneamente solo se la variazione di energia libera è negativa, ovvero se l’energia libera dei prodotti (stato finale della reazione) è maggiore di quella dei reagenti (stato iniziale della reazione). Le reazioni anaboliche di sintesi sono prevalentemente endoergoniche (cioè richiedono un apporto di energia), al contrario di quelle cataboliche di degradazione che sono prevalentemente reazioni esoergoniche, nelle quali si liberano notevoli quantità di energia. Questa viene temporaneamente trasferita a composti intracellulari specifici ad alto potenziale energetico, che a loro volta trasportano e rendono disponibile l’energia nelle sedi dei vari processi anabolici. Il principale di questi composti è l’adenosintrifosfato; la demolizione delle molecole nutritive a prodotti di rifiuto determina, utilizzando ADP e Pi, la sintesi di ATP, il quale rende disponibile l’energia accumulata cedendo il gruppo fosforico per la sintesi di macromolecole o per lo svolgimento di varie forme di lavoro, secondo lo schema generale (v. fig.):
Per questo motivo, il sistema ATP/ADP costituisce una sorta di ‘moneta di scambio’ utilizzabile per la maggior parte delle reazioni che implicano trasferimenti di energia. Le reazioni chimiche generalmente deputate alla produzione di energia, da conservare sotto forma di composti altamente energetici, sono quelle ossidoriduttive associate al trasferimento degli elettroni. Esistono tre meccanismi fosforilativi fondamentali di sintesi dell’ATP associati ai tre tipi di vie ossidative esistenti in natura, e cioè: la glicolisi, la fosforilazione ossidativa e la fotosintesi clorofilliana.
Un certo numero di molecole, comune a entrambi i processi, permette una interconnessione tra le vie anaboliche e quelle cataboliche. Si possono riconoscere, sia nel catabolismo sia nell’anabolismo, tre stadi diversi costituiti da una serie di reazioni enzimatiche successive. Nel catabolismo, il primo stadio è rappresentato dalla demolizione delle macromolecole (proteine, lipidi, polisaccaridi, acidi nucleici) nei loro blocchi costitutivi (amminoacidi, monosaccaridi, acidi grassi, nucleotidi); questo stadio è essenzialmente idrolitico e non libera energia utilizzabile dalle cellule. Nel secondo stadio, si ha la demolizione dei blocchi costitutivi in pochi intermedi fondamentali formati da 2-4 atomi di carbonio, come il piruvato, l’ossalacetato, l’acetato (sotto forma di acetil-CoA) e pochi altri, attraverso vie metaboliche specifiche per ogni tipo di composto (come la glicolisi, la β-ossidazione degli acidi grassi ecc.). A questo stadio è associata la liberazione di una parte relativamente piccola dell’energia totale racchiusa nei legami delle biomolecole. Infine, nel terzo stadio, i pochi intermedi ottenuti precedentemente sono incanalati in un unico processo ciclico terminale, il ciclo dell’acido citrico o ciclo di Krebs, che estrae tutta l’energia possibile dai legami chimici tramite reazioni ossidoriduttive. La riossidazione delle molecole ridotte nel ciclo dell’acido citrico è accoppiata alla produzione di energia sotto forma di ATP durante la fosforilazione ossidativa, nella quale l’accettore finale degli elettroni, provenienti si può dire da tutti i cicli metabolici precedenti, è l’ossigeno molecolare. È in quest’ultimo stadio comune che viene liberata e conservata la maggior quantità dell’energia chimica presente nelle molecole da metabolizzare.
Nel primo stadio dell’anabolismo, invece, piccole molecole entrano nel ciclo di Krebs, o si trasformano, comunque, in precursori a basso peso molecolare. Il ciclo dell’acido citrico, perciò, non può essere considerato esclusivamente una via catabolica, in quanto partecipa anche alla maggioranza delle vie biosintetiche, fornendo i precursori essenziali. Nel secondo stadio, questi piccoli precursori (piruvato, ossalacetato, acetil-CoA ecc.) vengono trasformati in molecole specifiche che rappresentano i blocchi costitutivi delle varie classi di molecole biologiche (monosaccaridi, acidi grassi, amminoacidi ecc.). In questo stadio è, in genere, necessaria più energia di quella che le corrispondenti vie cataboliche sono in grado di liberare. Nel terzo e ultimo stadio, i singoli blocchi costitutivi sono uniti tra loro con reazioni essenzialmente di disidratazione, che perciò richiedono una quantità notevole di energia sotto forma di ATP, per formare le 4 classi fondamentali di macromolecole biologiche: proteine, polisaccaridi, lipidi complessi, acidi nucleici.
Controllo del metabolismo
Il m. è sottoposto a una serie di fattori di regolazione che si differenziano a seconda del loro meccanismo d’azione, della loro localizzazione intracellulare e dell’ordine temporale della loro azione. Vi è una regolazione automatica, termodinamica e cinetica, che agisce in base alla concentrazione degli intermedi (substrati e prodotti delle singole reazioni enzimatiche) o dei coenzimi e del loro stato ossidoriduttivo, a livello del sito attivo degli enzimi interessati. Questo tipo di controllo è influenzato in modo determinante dall’organizzazione della via metabolica (lineare o ciclica) e dall’organizzazione strutturale degli enzimi (liberi, sotto forma di complessi multienzimatici, o legati a membrane). La regolazione, basata sulla compartimentazione intracellulare delle vie metaboliche, consiste nella precisa localizzazione dei processi anabolici e catabolici in diversi compartimenti cellulari ed è caratteristica degli organismi eucarioti. Per es., l’ossidazione degli acidi grassi ha luogo nei mitocondri, mentre la loro sintesi è localizzata nella frazione solubile del citoplasma, il citosol.
Un altro tipo di controllo del m. è dato dalla regolazione allosterica (➔ allosterismo; enzima) basata sulla modulazione dell’attività catalitica di particolari enzimi oligomerici (detti enzimi allosterici o regolatori), situati in posizione chiave delle vie metaboliche, in risposta alle variazioni di concentrazioni di effettori che si legano a siti particolari dell’enzima, detti siti allosterici. Frequentemente, uno dei prodotti finali della catena di reazione in cui sono coinvolti enzimi allosterici è in grado di inibirne specificatamente l’attività: questo tipo di inibizione è noto come inibizione da prodotto finale o inibizione a feedback o retroinibizione (➔). Quando uno o più prodotti di una sequenza metabolica inattivano più enzimi allosterici di quella stessa sequenza, si ha la cosiddetta inibizione sequenziale.
Il controllo del m. è anche garantito dalla presenza di forme multiple di uno stesso enzima note come isoenzimi. Ne sono un esempio gli isoenzimi della latticodeidrogenasi o della creatinfosfochinasi che, nell’uomo, catalizzano ognuno la propria reazione con velocità e affinità per il substrato differenti a seconda delle diverse esigenze metaboliche del tessuto al quale appartengono. La regolazione del m. basata sulle modificazioni covalenti degli enzimi è un tipo di controllo più lento e permette di mantenere l’enzima in uno stato attivo, o inattivo, per parecchi eventi catalitici. In certi casi, la modificazione covalente ha lo scopo di rendere l’enzima attivo solo quando raggiunge la sua sede d’azione; spesso, essa ha lo scopo di promuovere eventi a cascata, in cui un enzima attivato utilizza come substrato un altro enzima da attivare, amplificando in tal modo la risposta a un determinato stimolo metabolico. Ne è un esempio l’attivazione degli zimogeni (➔). In questo tipo di controllo, sono comprese anche le modificazioni covalenti post-traduzionali che subiscono alcune proteine, una volta sintetizzate e rilasciate dai ribosomi. Lo scopo è di operare, prevalentemente in modo reversibile, sull’attività biologica della proteina che subisce la modificazione. L’esempio più comune è la fosforilazione proteica da parte di chinasi e la successiva defosforilazione a opera di fosfatasi (➔ glicogeno).
Alcune proteine subiscono modificazioni permanenti per svolgere la loro attività biologica: così molti enzimi vengono legati stabilmente a coenzimi, come la biotina, l’acido lipoico, il piridossalfosfato. Vi sono poi altre modificazioni come l’acetilazione dell’estremità N-terminale della proteina o la metilazione di specifici residui amminoacidici. Inoltre, esiste un meccanismo di controllo che si esercita attraverso la regolazione della concentrazione di un dato enzima nella cellula.
La velocità di sintesi di alcuni enzimi è molto accelerata in certe condizioni, cosicché la concentrazione reale dell’enzima nella cellula risulta sostanzialmente aumentata. Questo tipo di controllo del m. è più lento, perché agisce sul livello degli enzimi presenti all’interno di una cellula; il controllo può essere permanente (i livelli dei vari enzimi sono determinati geneticamente), o può agire in risposta a determinati stimoli metabolici. In quest’ultimo caso l’azione può avvenire a livello genico sulla sintesi degli enzimi (a livello della trascrizione o della traduzione), oppure sulla loro degradazione intracellulare.
In alcuni casi, il controllo dell’espressione genica di un determinato enzima dipende dall’interazione di substrati o prodotti dell’enzima stesso con la sequenza nucleotidica del DNA che codifica la traduzione per quel particolare enzima. Tali sequenze di DNA sono denominate operoni. Inoltre, esiste un controllo del m. che si manifesta per mezzo di una regolazione a distanza negli organismi multicellulari, sia attraverso l’uso di messaggeri chimici (ormoni, neuromediatori ecc.) sia attraverso connessioni fisse intercellulari. Questa regolazione risponde alla necessità di una coordinazione più generale fra cellule e tessuti diversi per garantire l’armonizzazione del m. negli organismi complessi e agisce spesso attraverso l’azione ulteriore di messaggeri intracellulari, che trasmettono e amplificano l’azione del segnale primario al sistema effettore finale.
Infine, si deve tener conto che anche le concentrazioni intracellulari relative di ATP, ADP e AMP e soprattutto il loro rapporto, definito come carica energetica cellulare, hanno un ruolo molto importante nella regolazione del m., nel senso che un’alta carica d’energia inibisce le vie di produzione di ATP (cataboliche), mentre stimola le vie di utilizzazione dell’ATP stesso (anaboliche).
M. basale e m. secondario
Il m. basale è il dispendio energetico dell’organismo in condizioni ‘di base’, ossia in completo riposo fisico e psichico, a digiuno da almeno 12-16 ore e in neutralità termica. Esso rappresenta il fabbisogno calorico necessario al mantenimento delle funzioni vitali (attività cardiocircolatoria, respirazione, termoregolazione ecc.) e può essere espresso in kilocalorie per ora e per m2 di superficie corporea (la quale si calcola, mediante apposite tabelle, dall’altezza e dalla massa del soggetto; 1 kcal = 4187 J). Nell’uomo adulto normale in condizione di base il dispendio calorico è valutato pari a 40 kilocalorie per m2 e per ora (un po’ meno nella donna; un po’ più nei bambini): ossia, per un individuo di media corporatura, intorno a 1700 kcal giornaliere. Fra le numerose condizioni patologiche che modificano, nell’uomo, il m. basale si possono ricordare lo stato febbrile, le malattie con dispnea o con tachicardia, l’acromegalia, affezioni tiroidee. Esistono malattie dipendenti direttamente da alterazioni delle vie metaboliche conosciute come malattie congenite del metabolismo.
Il m. secondario è costituito da vie metaboliche che portano alla formazione o alla degradazione di sostanze nell’ordine di appena alcuni milligrammi al giorno. Ne sono un esempio la biosintesi di coenzimi e ormoni, che sono prodotti e utilizzati solo in tracce, e la formazione di nucleotidi e pigmenti. Si tratta di biomolecole altamente specializzate molto importanti per la vita degli organismi che le sintetizzano. Le vie metaboliche del m. secondario sono generalmente interconnesse con le reazioni del m. centrale. Numerose malattie metaboliche dipendono da difetti ereditari che interessano enzimi appartenenti a vie del m. secondario, come quelle del m. fosfolipidico.
Particolarità del m. nelle piante superiori
Nelle piante, accanto al m. primario che si svolge secondo meccanismi pressoché universali volti a sopperire alle funzioni vitali (per es., sintesi proteica, metabolismo glucidico ecc.), è particolarmente intensa un’attività riferita al m. secondario, mediante la quale si forma un gran numero di sostanze, di diversa natura chimica, che non sono essenziali alle funzioni vitali, ma rappresentano peculiari specializzazioni delle cellule e sono atte a influenzare e regolare funzioni dell’organismo o anche di altri organismi determinando, per es., forme di attrazione o di repulsione. I metaboliti secondari sono spesso sostanze di notevole interesse farmacologico e industriale.
M. delle cellule procariotiche
Il m. dei procarioti si differenzia da quello degli organismi superiori soprattutto a livello di alcune vie cataboliche, mentre il loro anabolismo è fondamentalmente simile. Nei procarioti aerobi, per la mancanza di organelli intracellulari definiti, è assente il sistema di controllo del m. dovuto alla compartimentazione cellulare, e i sistemi enzimatici delle vie metaboliche si trovano sparsi nel citoplasma o associati alla membrana plasmatica, come nel caso degli enzimi della fotosintesi o della fosforilazione ossidativa.
Due tipi di m. prettamente procariotico sono la fermentazione e la respirazione anaerobia. La fermentazione è un’ossidazione anaerobia nella quale non interviene l’ossigeno come accettore finale di elettroni. Nei procarioti, l’acido piruvico derivante dalla via glicolitica è trasformato attraverso 4 tipi di fermentazione: a) fermentazione alcolica, caratteristica dei lieviti, di alcuni funghi e di pochi batteri; è un tipo di fermentazione utilizzata industrialmente per la produzione di alcol etilico; b) fermentazione lattica o omolattica, che porta solamente alla produzione di acido lattico dall’acido piruvico, per contemporanea ossidazione del NADH. È la stessa fermentazione che si osserva nel m. anaerobio delle cellule eucariotiche ed è caratteristica dei lattobacilli e di alcuni cocchi; c) fermentazione propionica, caratteristica del genere Propionibacterium, che produce anidride carbonica, acido propionico e acido acetico. L’associazione delle fermentazioni propionica e lattica è ampiamente utilizzata nell’industria casearia; d) fermentazione formica, propria dei batteri enterici, come Escherichia coli, Proteus, Salmonella ecc., che porta alla produzione di numerosi acidi organici quali acido acetico, formico, lattico e succinico. La respirazione anaerobia è caratteristica dei procarioti anaerobi sia facoltativi sia obbligati, i quali sono in grado di utilizzare come accettore finale di elettroni molecole inorganiche diverse dall’ossigeno molecolare, o molecole organiche che, a differenza di quanto avviene nella fermentazione, non derivano dal substrato utilizzato in partenza. In questo caso, i più comuni accettori di elettroni sono i nitrati, che vengono ridotti a nitriti, o a ossido d’azoto, o ad azoto molecolare. I procarioti aventi questo tipo di m. sono, però, aerobi facoltativi, a differenza di altri che utilizzano come accettori di elettroni i solfati, riducendoli a idrogeno solforato. Inoltre, per es. nei metanobatteri, è l’anidride carbonica che viene utilizzata e ridotta a metano; mentre in Streptococcus è il fumarato ad accettare gli elettroni riducendosi a succinato.
Una differenza importante riscontrabile nel m. dei procarioti riguarda la fotosintesi: la clorofilla presente nei batteri è diversa da quella delle piante superiori nella composizione delle catene laterali alifatiche, che permettono di distinguere quattro tipi di batterioclorofille identificate con le lettere a, b, c, d. Infine, va ricordato un aspetto importante del m. procariotico, e cioè la produzione degli antibiotici.
Medicina
Da quando (1908) A.E. Garrod coniò l’espressione ‘malattie congenite del m.’ (inborn errors of metabolism) a proposito dell’alcaptonuria, sono stati descritti molti altri quadri, accomunati da un meccanismo eziologico analogo, dato dal deficit parziale o totale di una specifica attività enzimatica oppure di una proteina di trasporto dei vari composti all’interno della cellula. Pertanto, la via metabolica può andare incontro a un rallentamento o a un blocco con tutte le ripercussioni del singolo caso. In particolare si può verificare: accumulo dei metaboliti a monte del blocco (singolo difetto enzimatico ecc.); impossibilità di giungere al prodotto finale della via metabolica che risulterà scarso o assente; attivazione di vie alternative finalizzate al superamento del blocco.
In base a quanto anticipato esistono difetti del m. a vari livelli, con specifici quadri patologici degni di una distinta collocazione nosologica. Una premessa recente riguarda l’identificazione di numerosi meccanismi patogenetici, basati spesso su alterazioni geniche; a ciò si associa la possibilità di effettuare diagnosi precoci e di identificare il trattamento più fisiologico, basato sulla terapia sostitutiva a base del prodotto enzimatico carente o sulla terapia genica.
Malattie del m. dei carboidrati
Il deficit di fruttochinasi causa una forma benigna nota come fruttosuria essenziale. Al contrario il deficit di 1,6-bifosfato-aldolasi porta alla forma nota come intolleranza ereditaria al fruttosio con sintomatologia grave fino alla morte per insufficienza epatica e renale, reversibili all’eliminazione totale dei carboidrati interessati.
Fra le affezioni caratterizzate da accumulo di glicogeno (glicogenosi), i disordini di più frequente riscontro durante l’infanzia sono: deficit di glucosio-6-fosfatasi (tipo I, malattia di E. von Gierke), autosomica recessiva con ipoglicemia, chetosi, iperlipemia ecc.; deficit di α-1,4-glucosidasi lisosomiale (tipo II, malattia di J.C. Pompe) con prognosi gravissima (evoluzione letale entro il secondo anno di vita); deficit dell’enzima deramificante (tipo III) con clinica simile al tipo I, in forma attenuata e il deficit di fosforilasi chinasi epatica (tipo IX). Il disordine di più frequente riscontro nell’adulto è il deficit della fosforilasi muscolare (tipo V, malattia di B. McArdle) con crampi muscolari dolorosi, possibile emoglobinuria ecc.
Malattie del m. lipidico
A proposito di dislipidemie familiari, la ricerca ha fornito oggi gli strumenti per andare oltre la vecchia classificazione di D.S. Fredrickson, basata sull’individuazione delle frazioni lipoproteiche aumentate e fornendo quindi un solo dato fenotipico della patologia. Le conoscenze attuali consentono di fornire una classificazione genotipica delle alterazioni, in grado di integrare sia gli aspetti metabolici sia quelli genetici. Alcuni esempi sono dati da: ipercolesterolemia familiare classica, conseguente ad alterazioni geniche a carico del recettore per le LDL, responsabili di un loro alterato catabolismo e conseguente accumulo; iperchilomicronemia familiare (deficit genetico a carico della lipoproteinlipasi, LPL, o di ApoC-II, suo cofattore) con prevalente ipertrigliceridemia (eruzioni xantomatose, rischio di pancreatite acuta) ecc.
Tra le alterazioni del m. lipidico sono incluse le lipidosi (o lipoidosi). Alcuni esempi sono dati da: malattia di Gaucher, disordine autosomico recessivo, conseguente al deficit dell’enzima lisosomiale β-glucosidasi, con esordio e presentazione clinica variabile, a seconda dell’età; malattia di Niemann-Pick (lipidosi sfingomielinica), da deficit, più o meno completo a seconda dei vari fenotipi (A, B, C ecc.), dell’enzima sfingomielinasi con accumulo di sfingomielina all’interno della cellula che va incontro a morte; malattia di Tay-Sachs da deficit enzimatico di β-esoaminidasi A con progressiva neurodegenerazione conseguente all’accumulo intracellulare di ganglioside GM2.
Malattie del m. dei mucopolisaccaridi
Costituiscono un’altra manifestazione di patologia lisosomiale coinvolgente il m. dei mucopolisaccaridi. Si distinguono diverse manifestazioni in base alla specifica attività enzimatica coinvolta: sindrome di G. Hurler (tipo I) da deficit di L-iduronidasi con gravi dimorfismi, epatosplenomegalia ecc.; sindrome di C.H. Hunter (tipo II) da deficit di iduronatosolfatasi con presentazione simile al tipo I ma in forma attenuata; sindrome di S.J. Sanfilippo (tipo III), di cui si conoscono quattro forme, contraddistinte con le lettere A, B, C e D, dovute rispettivamente a deficit di eparan-N-solfato-solfatasi, N-acetil-α-D-glucosaminidasi, acetil-CoA-α-glicosaminide-N-acetiltransferasi, N-acetilglicosamina-6-solfato-solfatasi, con dismorfismi, ancor meno accentuati; sindrome di Morquio (tipo IV), da deficit di N-acetil-galattosamina-solfatasi con nanismo, protrusione della mandibola o altri dismorfismi facciali ecc.; sindrome di H.G. Schede (tipo V) con deficit enzimatico uguale alla sindrome di Hurler, dalla quale però si distingue nettamente sul piano clinico: esordio nel soggetto adulto con opacità corneali, compromissioni tendinee e articolari ecc.; sindrome di P. Maroteaux - M. Lamy (tipo VI), da deficit di arilsofatasi B, con dismorfismi che ricordano anche in questo caso la sindrome di Hurler; sindrome di Sly (tipo VII), dovuta a deficit di β-glicuronidasi.
Attualmente non esiste una cura risolutiva per le mucopolisaccaridosi, che vengono gestite con terapie sintomatiche (logopedia, fisioterapia, ecc.); per alcune forme si è tentato il trapianto di midollo osseo per poter giungere alla produzione endogena dell’enzima mancante. Tale strategia, insieme alla terapia genica, rappresenta il target terapeutico più fisiologico e risolutivo.
Malattie da alterato m. degli amminoacidi
Sono numerose e comprendono turbe sia del trasporto degli amminoacidi sia del loro m. intermedio. La loro base biochimica è individuabile in un deficit enzimatico specifico; la caratteristica chimico-clinica che generalmente le accomuna è l’elevato contenuto nelle urine di un determinato amminoacido (amminoaciduria). Da tempo note sono l’alcaptonuria, connessa a un deficit di omogentisinico-ossidasi, e la fenilchetonuria, in cui è implicato un difetto di fenilalaninaidrossilasi. Altri esempi sono dati dalle turbe del m. di tirosina, triptofano, istidina, prolina, e vari altri amminoacidi per deficit enzimatici specifici e altrettanto caratteristica presentazione clinica. Anche gli amminoacidi a catena ramificata – valina, leucina, isoleucina – sono interessati da alterazioni congenite metaboliche, la cui incidenza nella popolazione generale è estremamente ridotta.
Difetti ereditari del ciclo della sintesi dell’urea
Si accompagnano ad aumento dell’ammonio ematico che agirebbe come agente tossico. Le forme che interessano le prime tappe del ciclo dell’urea e che presentano anche ammoniemia elevata sono: il deficit di carbamilfosfatosintetasi e il deficit di ornitina-carbamil-transferasi, detti anche rispettivamente iperammoniemia di tipo I e di tipo II. I quadri clinici comprendono in generale episodi di vomito, gravi manifestazioni morbose fino al coma, ritardo mentale, avversione per i cibi proteici. Il principale obiettivo della terapia nelle iperammoniemie ereditarie punta a mantenere un compenso biochimico in grado di contrastare l’insorgenza dei danni neurologici permanenti. A tal fine un ruolo decisivo è svolto dalla dieta ipoproteica associata a farma;ci che favoriscono l’escrezione dell’azoto mediante vie alternative al ciclo dell’urea.
Altri quadri dismetabolici
Altre forme di patologia del m. sono: la citrullinemia (aumento di citrullina nel sangue per difetto di arginino-succinico-sintetasi); l’arginino-succinato-aciduria (eliminazione urinaria di arginino-succinato da difetto di arginino-succinasi); il deficit di arginasi. Tutte queste forme sono trasmesse in via recessiva. Sono stati individuati altri quadri dismetabolici clinicamente dominati dai fenomeni chetosici: isovalericoacidemia causata da un difetto di isovaleril-CoA-deidrogenasi con ricorrenti episodi di scompenso metabolico acuto, vomito, chetoacidosi e coma; acidemia propionica, da difetto enzimatico di propionil-CoA-carbossilasi che blocca la trasformazione del propionil-CoA in metilmalonil-CoA, inizia nel neonato con grave chetoacidosi, disordini respiratori ecc.; metilmalonico-acidemia, da blocco nella conversione del metilmalonil-CoA a succinil-CoA, catalizzata dalla metilmalonil-CoA-mutasi ecc. Per la vicinanza clinica con le forme caratterizzate da chetoacidosi merita di essere qui ricordato il deficit di succinil-CoA-transferasi, che è causa di blocco del trasferimento del coenzima A dal succinil-CoA all’acido aceto-acetico. Le conseguenze sono chetonemia persistente con gravi episodi di chetoacidosi, per difetto di metabolizzazione dei corpi chetonici.
Malattie da alterato m. purinico e pirimidico
Le più note malattie conseguenti ad alterazioni del m. purinico sono rappresentate da: gotta; xantinuria, dovuta a deficit di xantino-ossidasi con eliminazione urinaria di xantina e ipoxantina, anziché di acido urico, il cui contenuto nel sangue è molto basso; sindrome di M. Lesch-W. L. Nyhan da deficit di ipoxantina-guanosina-fosforibosil-transferasi, caratterizzata da iperuricemia, ritardo mentale, spasticità ecc.
La più nota sindrome connessa a turbe del m. pirimidinico è l’orotico-aciduria, malattia a trasmissione autosomica recessiva la cui base enzimatica è costituita dai deficit di orotidina-5-fosfato-pirofosforilasi e di orotidina-5-fosfato-decarbossilasi. Il quadro clinico, scarsamente significativo (ritardo della crescita, lieve deficit mentale, ipotonia muscolare, anemia) è favorevolmente influenzato dalla somministrazione di uridina.