
ferro
Elemento chimico di simbolo Fe, numero atomico 26, peso atomico 55,85, densità 7,85 g/cm3, punto di fusione 1536 °C. È il metallo più abbondante della Terra (costituisce il 34,6% della massa della Terra, il 5% della crosta). In natura se ne cono;scono 4 isotopi stabili 2654Fe, 2656Fe (il più abbondante, 92% circa), 2657Fe, 2658Fe.
Chimica
Proprietà
Il f. è un metallo bianco-argenteo, lucente, tenace, duttile e malleabile. In natura raramente si trova il f. metallico, mentre sono abbondanti i suoi composti, specialmente ossidi, idrossido, carbonato, solfuro. I più importanti minerali del f. sono la magnetite, l’ematite, la limonite, la siderite, la pirite. Allo stato chimicamente puro il f. può essere ottenuto per riduzione degli ossidi in corrente di idrogeno, ovvero per elettrolisi a caldo di soluzioni di solfato o cloruro ferroso, con speciali processi siderurgici nel forno elettrico (ferro Armco: usato, per es., per le sue proprietà magnetiche, nella costruzione di nuclei di elettromagneti). Se ottenuto per riduzione, quando questa viene effettuata a bassa temperatura, la polvere che si ottiene è piroforica, ossia suscettibile di autocombustione. Il ferro elettrolitico ha la caratteristica di contenere, probabilmente allo stato di occlusione, un certo quantitativo di idrogeno catodico il cui allontanamento non è realizzabile in modo completo anche tramite una rifusione sotto vuoto; tuttavia questo prodotto ha notevoli caratteristiche magnetiche.
Il f. non subisce alterazioni se in contatto con aria secca o con acqua priva di anidride carbonica; in presenza di umidità viene attaccato con formazione di ruggine secondo un meccanismo di ossido-carbonatazione che può procedere nel tempo a causa delle caratteristiche fisiche del deposito. Allo stato di estrema purezza il f. non sembra suscettibile di arrugginirsi: nel processo di ossidazione ha infatti notevole importanza la presenza di impurezze o eterogeneità locali che potrebbero causare la formazione di microelementi galvanici (➔ corrosione).
Il f. si scioglie in acido cloridrico e solforico diluiti, con sviluppo di idrogeno e formazione di cloruro o di solfato ferroso. L’acido solforico concentrato e caldo agisce riducendosi in parte ad anidride solforosa e producendo solfato ferroso e ferrico; l’acido nitrico non molto concentrato scioglie il f. senza o con sviluppo di ossidi di azoto e formazione di nitrato ferroso e ammonico o di nitrato ferrico; l’acido nitrico concentrato passiva il f. in modo da renderlo insolubile nell’acido diluito e incapace di spostare altri elementi quali il rame o l’argento dalle loro soluzioni saline: questo stato anomalo del f., dovuto probabilmente a una formazione di uno strato protettivo di ossido, è caratteristico anche, in determinate condizioni, di numerosi metalli, e può essere determinato anche da altri agenti ossidanti.
Forme allotropiche del ferro
Il f. si presenta in diverse forme allotropiche che vengono usualmente indicate con i simboli α, γ e δ. A temperatura ordinaria il f. cristallizza nella forma α, con reticolo cubico a corpo centrato; per riscaldamento a 911 °C il f. passa dal reticolo cubico a corpo centrato a quello a facce centrate, nella forma γ, stabile fino a 1392 °C; fra 1392 °C e la temperatura di fusione (1536 °C) il f. esiste nella forma δ con reticolo cubico a corpo centrato. Si ammette l’esistenza di un’altra forma, β, stabile fra 769 °C e 911 °C, che differisce dalla forma α non per caratteristiche cristalline ma per le proprietà magnetiche. Le varie forme allotropiche del f. hanno la proprietà di sciogliere diversamente il carbonio; il f. α ne scioglie pochissimo: da 0,007% a temperatura ordinaria a un massimo di 0,025% a 723 °C; la forma γ scioglie il carbonio allo stato solido fino a un massimo del 2,06% a 1147 °C. Queste differenze riguardo la solubilità del carbonio nelle varie forme del f. sono di fondamentale importanza perché da esse dipendono le proprietà degli acciai. Le temperature alle quali avvengono trasformazioni strutturali nel raffreddamento o nel riscaldamento del f. prendono il nome di punti critici o punti di transizione: si indicano rispettivamente con Ar e Ac (dal francese refroidissement e chauffage); questa doppia formulazione deriva dal fatto che, specie laddove intervengono modificazioni reticolari, si nota inevitabilmente una notevole inerzia termica e pertanto le temperature di trasformazione possono essere diverse a seconda che si effettui un riscaldamento oppure un raffreddamento. Accanto al simbolo Ac o Ar vengono usualmente posti i numeri, da 1 a 4, per contrassegnare le diverse trasformazioni: così Ac2 indicherà la presunta trasformazione α→β (per il f. puro, in tal caso, Ac2 coincide con Ar2, ovvero non si manifesta alcun fenomeno d’inerzia); Ac3 può collocarsi fra 915 e 935 °C, a seconda della purezza del materiale di partenza, e si sposta verso valori più bassi della temperatura col crescere del tenore in carbonio: esso è rappresentativo della trasformazione β→γ. Nel raffreddamento, Ar3 si manifesta invece a temperature più basse e precisamente intorno a 906 °C, il che dimostra il verificarsi di un fenomeno d’inerzia. Ac4 rappresenta infine la trasformazione, al riscaldamento, della forma γ in quella δ.
Composti
I più importanti composti del f., il quale si può comportare da bivalente, trivalente ed esavalente sono: i ferrati, i composti ferrici e i composti ferrosi.
Ferrati
Denominazione dei sali di un ipotetico acido ferrico da f. esavalente, di formula generale Me2FeO4, con Me metallo monovalente. È noto, per es., il ferrato di sodio, Na2FeO4, le cui soluzioni (soltanto in soluzione il composto è dotato di una certa stabilità) sono intensamente colorate in rosso. Il ferrato di potassio, K2FeO4, di colore rosso violetto, si ottiene ossidando a caldo la limatura di f. con nitrato di potassio oppure ossidando l’idrossido ferrico con cloro e bromo in soluzione alcalina concentrata. I f. sono ossidanti più forti del permanganato.
Composti ferrici
Sono i composti che contengono ferro trivalente. In essi il f. ha deboli proprietà basiche e perciò non si conoscono sali stabili con acidi deboli. I sali con acidi forti in acqua si idrolizzano e la soluzione appare rossa per la presenza di idrossido ferrico colloidale.
Cloruro ferrico
Composto, FeCl3; si può preparare ossidando con cloro o con acido nitrico una soluzione di cloruro ferroso; il sale che si forma per via umida cristallizza con diverse quantità di acqua (di tali idrati il più noto è l’esaidrato, FeCl3 • 6H2O, in cristalli giallo-arancio assai deliquescenti) e non è suscettibile di disidratazione termica perché soggetto a decomposizione. Per ottenere cloruro ferrico anidro occorre far reagire il cloro secco con f.: si presenta in tal caso come un solido bruno scuro, solubile in acqua e alcol. Il cloruro ferrico trova impiego come mezzo clorurante nella manifattura di altri composti del f., come catalizzatore, mordente in tintoria, agente coagulante-flocculante nei trattamenti delle acque, nell’industria ceramica per produrre effetti decorativi superficiali, in medicina nella fabbricazione del cotone emostatico ecc.
Idrossido ferrico (anche ossido idrato ferrico)
Composto FeO(OH), cui tradizionalmente si attribuisce la formula Fe(OH)3, anche se la forma completamente idrata non esiste. Si origina per esposizione del f. all’aria umida, ovvero per precipitazione con alcali da una soluzione di un sale ferrico; ottenuto con il secondo metodo è suscettibile di dializzazione e pertanto se ne può preparare una soluzione colloidale di color rosso bruno che trova impiego per es., in medicina (ferro dializzato). Si ritrova in natura in alcuni minerali (per es. limonite, goethite, lepidokrokite). Si presenta in forma di precipitato bruno, flocculento, solubile negli acidi, negli alcali caustici (con formazione di ferriti di sodio e di potassio ecc.), insolubile in acqua, alcol ed etere; i suoi impieghi più importanti sono: come costituente di masse depuranti (di Laming) nell’industria chimica, nella purificazione delle acque, nella preparazione di pigmenti.
Ossido ferrico
In natura è uno dei più importanti minerali di f. (➔ ematite; limonite). Può prepararsi artificialmente per disidratazione dell’idrossido, per riscaldamento all’aria di sali ferrici volatili e di composti del f. (per es., dalla pirite per combustione). Costituisce uno dei principali componenti dei pigmenti del f. (➔ pigmento); si usa inoltre come abrasivo per vetri, metalli preziosi e diamanti, come catalizzatore nelle reazioni di ossidoriduzione, nella fabbricazione di semiconduttori.
Composti ferrosi
Sono i composti che contengono ferro bivalente. I sali ferrosi normali e le loro soluzioni sono colorati in verde chiaro; si formano facendo agire sul f. i corrispondenti acidi, anche quelli ossidanti (per es. nitrico) diluiti; a freddo danno il sale ferroso che si ossida più o meno lentamente a ferrico. Le soluzioni dei sali ferrosi hanno la proprietà di solubilizzare l’ossido d’azoto, NO, colorandosi in bruno, ciò che costituisce anche la base di metodi analitici di riconoscimento dell’acido nitrico, dell’acido nitroso e dei loro sali.
Acetato ferroso
Fe (CH3COO)2 • 4H2O; cristalli verdastri (se allo stato puro e fuori del contatto dell’aria), solubili in acqua e alcol. Si può preparare facendo reagire il f. con acido acetico; trova impiego in medicina, come colorante nell’industria tessile, per la conservazione del legno.
Carbonato ferroso
FeCO3, esistente in natura come minerale (siderite). Si può ottenere per azione di CO2 sotto pressione su idrossido ferroso, ovvero da una soluzione di solfato ferroso trattata con carbonati (o bicarbonati) alcalini; polvere biancastra che si ossida rapidamente all’aria dando idrossido ferrico; solubile negli acidi, insolubile in acqua, si usa in medicina e può servire per la preparazione di altri sali di ferro.
Cloruro ferroso
FeCl2 • 4H2O, si ottiene sciogliendo il f. in soluzioni acquose di acido cloridrico. Sotto forma di cristalli idrati si presenta di colore verde: in tali condizioni non è suscettibile di ulteriore disidratazione perché soggetto a decomposizione.
Fosfato ferroso
Fe3(PO4)2 • 8H2O, esiste anche in natura come minerale (vivianite). Si può preparare per elettrolisi di una soluzione di fosfato sodico usando anodi di f., ovvero per reazione fra solfato di f. e fosfato di ammonio; composto instabile, insolubile in acqua, solubile negli acidi. In commercio si trova allo stato di miscela costituita da ossido ferroso, fosfato ferroso e ferrico. Si usa come catalizzatore, nella preparazione di manufatti ceramici e in medicina nella cura della clorosi e dell’anemia.
Idrossido ferroso
Fe(OH)2, composto instabile; si può ottenere trattando con alcali una soluzione di un sale ferroso. La massa gelatinosa biancastra che in un primo tempo si forma tende rapidamente a mutare colore (prima bruno-verdastro e poi rosso-bruno) per effetto della trasformazione in idrossido ferrico; può presentarsi anche in forma di polvere amorfa bianca o verdastra insolubile in acqua.
Ossido ferroso
FeO; si può preparare per riduzione dell’ossido ferrico con ossido di carbonio o per azione dell’acqua sul f. rovente o anche per decomposizione termica dell’ossalato. Si presenta sotto forma di polvere nera, quasi mai allo stato puro, facilmente ossidabile, insolubile in acqua, solubile negli acidi. L’ossido ferroso costituisce un prodotto intermedio nella riduzione dei minerali di f. all’altoforno, per la fabbricazione della ghisa.
Solfuro ferroso
FeS; si può preparare trattando a caldo limatura di f. con zolfo; il prodotto di fusione viene frantumato o colato in forma di cilindri o lingotti. Si forma anche per precipitazione da una soluzione di sale ferroso o ferrico con aggiunta di un solfuro alcalino: ha aspetto e lucentezza metallica. Trova impiego nella preparazione d’idrogeno solforato poiché è capace di svolgere questo gas per trattamento con acidi diluiti; ha anche applicazioni nell’industria ceramica. Il solfuro ferroso ottenuto per via umida si usa in medicina come antidoto nell’avvelenamento da arsenico.
Leghe ferro-carbonio
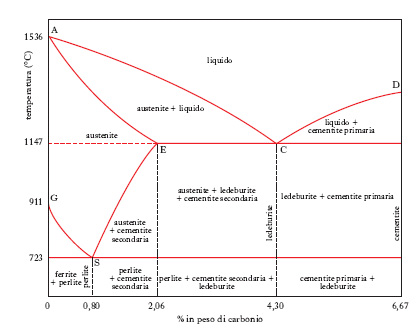
Le più importanti leghe del f. sono quelle che esso forma con il carbonio, giacché da tale unione scaturiscono i prodotti siderurgici, cioè gli acciai e le ghise. Le condizioni di formazione e di trasformazione dei vari costituenti di questi prodotti sono rappresentabili sul cosiddetto diagramma di stato f.-carbonio (fig. 1). La temperatura di fusione del f. puro (1536 °C) viene abbassata (curva AC) da tenori crescenti di carbonio fino a un valore minimo (1147 °C) che corrisponde all’eutettico (ledeburite), contenente il 4,30% di carbonio (punto C); per tenori più elevati di questo elemento la temperatura di fusione della lega si innalza (curva CD) fino a un massimo (punto D) che si raggiunge in corrispondenza del 6,67% di carbonio, cioè del composto noto con il nome di cementite (carburo di ferro, Fe3C); nel campo del diagramma binario al di sotto del 2,06% di carbonio si trovano le composizioni rappresentative degli acciai, in quello al di sopra di tale valore si trovano le ghise.
Nel campo degli acciai ha particolare importanza un altro punto singolare corrispondente a 0,80% di C, alla temperatura di 723 °C, rappresentativo di un eutettico tra fasi solide o, come si dice, di un eutettoide (perlite), dato che è il punto d’incontro delle linee GS e SE lungo le quali si ha separazione rispettivamente di f. (ferrite) e di cementite (secondaria, per distinguerla da quella primaria che si separa nelle leghe con più del 4,3% di C, per raffreddamento a partire dal liquido). Tra la curva del solidus AE (nel campo 0-2,06% di carbonio) e la curva GSE esiste il campo di stabilità dell’austenite, soluzione solida di carbonio nel f. γ che, come appare dal diagramma, può essere stabile fino a 723 °C se contiene esattamente un tenore di carbonio dello 0,80%; a temperatura inferiore l’austenite scompare per dare origine all’eutettoide perlite; per tenori in carbonio inferiori o superiori allo 0,80% si hanno, accanto alla perlite, quantitativi più o meno sensibili rispettivamente di ferrite o di cementite secondaria; però è possibile, in opportune condizioni, conservare anche a temperatura ordinaria la struttura austenitica. In effetti, esistendo un’inerzia alla trasformazione dell’austenite in perlite, tanto più sentita quanto più alta è la velocità di raffreddamento, si ha la possibilità di portare, a mezzo di un adeguato trattamento termico (tempra), la soluzione solida in un campo di temperature nel quale la velocità di trasformazione, nel senso e secondo le previsioni deducibili dal diagramma di stato, è assai esigua: ne deriva che l’austenite può restare tutta, o in parte, come tale anche a temperatura ambiente.
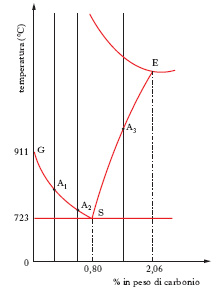
Trattamenti termici
Si limita l’esame alla porzione di sinistra, semplificata (fig. 2), del diagramma della fig. 1, assumendo come limite superiore di concentrazione in carbonio quello del 2,06%, e si considera brevemente l’influenza esercitata sulle proprietà della lega da un raffreddamento non lento. Per ottenere una struttura diversa da quella caratteristica delle leghe raffreddate lentamente, non occorre partire da un prodotto che si trovi allo stato fuso ma è sufficiente raggiungere, per es. a mezzo di opportuno riscaldamento, una temperatura che interessi il campo di stabilità dell’austenite, cioè è sufficiente superare la curva GSE. Realizzando un raffreddamento rapido si può riuscire a impedire, per l’esaltazione dei fenomeni d’inerzia alla trasformazione, il passaggio dalla struttura austenitica a quella perlitica; a seconda della drasticità dell’operazione è possibile ottenere tutta una serie di strutture intermedie fra le due strutture limite possibili e cioè l’austenitica (raffreddamento rapidissimo) e la perlite (raffreddamento assai lento). Per es., con un raffreddamento sufficientemente rapido, avendo l’austenite raggiunto la temperatura che corrisponde a un punto della curva (A1, A2 o A3, a seconda del contenuto di carbonio della lega in esame), il reticolo del f. γ si trasforma nel reticolo tetragonale di un nuovo costituente strutturale nel quale il carbonio resta disciolto allo stato di soluzione soprasatura e quindi in condizioni di metastabilità. È questa la cosiddetta struttura martensitica, che all’esame micrografico si presenta, a seconda del metodo d’esame, in forma di aghi bianchi, grigi o addirittura neri. Se il raffreddamento è eseguito meno rapidamente, si può realizzare una struttura, detta troostitica, nella quale il carbonio, combinato in forma di cementite con il f., comincia a separarsi; infine, agendo ancor meno drasticamente, la separazione della cementite raggiunge uno stadio più avanzato, dando luogo alla cosiddetta struttura sorbitica, senza tuttavia arrivare al completamento, come nel caso della perlite (➔ trattamento; tempra).
Elementi speciali nelle leghe ferro-carbonio
Quantitativi più o meno elevati di elementi speciali (Ni, Cr, Mn, Co, Si, V, Mo ecc.) esercitano un effetto sulle caratteristiche meccaniche, fisiche e chimiche delle leghe f.-carbonio con riferimento all’intervallo 0-2,06% di carbonio (➔ acciaio), escludendo tenori superiori (➔ ghisa). Gli elementi che presentano la stessa struttura reticolare del f. γ (nichel, rame, oro) sono in esso completamente solubili e pertanto tendono a rallentare la trasformazione γ → α, aumentando in definitiva il campo di stabilità dell’austenite verso le basse temperature. È perciò più facile ottenere una struttura austenitica (o prossima a questa) in acciai che contengono tenori sensibili, per es. di nichel, ovvero è possibile, con un raffreddamento più moderato, realizzare le stesse condizioni conseguenti a una tempra molto drastica. Al contrario sarà, in via generale, più difficile conservare la forma austenitica (o martensitica) in acciai nei quali sono presenti elementi speciali come cromo, tungsteno, molibdeno, vanadio che, essendo a reticolo cubico a corpo centrato, e quindi solubili nel f. α, aumentano il campo di esistenza di quest’ultimo e pertanto si oppongono alla trasformazione in austenite, riducendo sensibilmente, specie per elevati tenori, il campo di stabilità della soluzione solida γ. Ciò modifica ovviamente le modalità secondo cui deve essere eseguito il trattamento termico.
Ferroleghe
Le leghe del f. con altri elementi diversi dal carbonio vengono chiamate ferroleghe. Trovano impiego soprattutto in metallurgia allo scopo di introdurre nell’acciaio comune un certo quantitativo di uno o più elementi speciali, così da conferirgli determinate caratteristiche chimiche, fisiche e meccaniche. La funzione delle ferroleghe non è però limitata alla possibilità di ottenere un acciaio di opportuna composizione finale, ma comporta anche un’energica azione affinante che si esplica, per es., attraverso l’asportazione dell’ossigeno contenuto nel bagno liquido, con conseguente formazione di una scoria nella quale maggiormente si concentrano le impurezze non altrimenti eliminabili. Queste ferroleghe si preparano quasi esclusivamente al forno elettrico, riducendo miscugli naturali o artificiali dei rispettivi ossidi: così dalla cromite si ottiene un ferrocromo ecc. Nelle ferroleghe i diversi metalli, o non metalli, si possono trovare presenti come soluzioni solide o come carburi dei corrispondenti elementi.
Ferro-alluminio
Contiene fino al 20% di alluminio e piccole quantità di silicio, manganese, carbonio e rame; presenta struttura cristallina lucente ed è di colore grigio scuro; usata essenzialmente come disossidante.
Ferro-boro
Contiene in media il 16% di boro; è usato per la sua energica azione disossidante e soprattutto per introdurre boro nel bagno fuso per preparare acciai di particolare durezza.
Ferro-cromo
Se ne conoscono diversi tipi nei quali, oltre a variare la percentuale di cromo presente (55-70%), può variare anche il tenore di carbonio. Si ha un f.-cromo a basso tenore di carbonio (ferro-cromo affinato), contenente dallo 0,03 al 2% di carbonio e piccole percentuali di silicio e manganese, usato prevalentemente per la preparazione di acciai speciali e inossidabili a basso tenore di carbonio: è un prodotto compatto, di colore grigio-argento, che si può fabbricare al forno Héroult, con elettrodi di grafite assai pura, per riduzione della cromite con f.-silicio (90-95% di Si). Un altro tipo di f.-cromo, ad alto contenuto di carbonio (ferro-cromo comune), è impiegato per fabbricare acciai al cromo a tenori più elevati di carbonio, e contiene in media 60-65% di cromo, 4-9% di carbonio, quantitativi modesti di silicio, manganese e tracce di fosforo: si presenta alla frattura di colore argenteo brillante; si può preparare in forni elettrici monofase o trifase, partendo da minerali di cromo contenenti in media il 50% di cromo. Si preparano anche tipi di f.-cromo adatti per usi speciali: così, il f.-cromo da fonderia, usato in particolare per l’aggiunta in siviera del cromo alla ghisa liquida, contenente, oltre f. e cromo, 6-9% di silicio e 1-7% di carbonio; un f.-cromo a basso o a elevato tenore di carbonio, con il 4-6% di silicio e manganese, impiegato per le aggiunte in siviera, data la sua alta velocità di dissoluzione nell’acciaio liquido.
Ferro-fosforo
Contiene di solito dal 20 al 30% di fosforo; è usato per introdurre quest’ultimo in acciai speciali (meccanici) o in ghise da fonderia, specie per piccoli getti. Alla frattura presenta cristalli aghiformi poco saldati fra loro, talvolta anzi assai friabili; è di colore grigio f. con riflessi azzurrognoli.
Ferro-manganese
Quello comune contiene 75-80% di manganese accanto al 6-8% di carbonio e all’1% massimo di silicio ed è usato in acciaieria (convertitori, forno Martin, forni elettrici) come disossidante e per introdurre manganese nel bagno. Alla frattura è di colore grigio f. e si presenta in cristalli assai fini e compatti. Il f.-manganese a basso tenore in carbonio (ferro-manganese affinato), impiegato per arricchire di manganese acciai molto dolci e anche per prodotti speciali (acciai inossidabili ecc.), è internamente di colore grigio perla con grana serrata e contiene 80-90% di manganese, 0,6-0,75% di carbonio, 0,3-0,5% di fosforo e tracce di zolfo. Le leghe f.-manganese contenenti meno del 25% di manganese sono dette ghisa speculare o manganesifera (➔ ghisa).
Ferro-molibdeno
Contiene 55-75% di molibdeno e 0,1-2,5% di carbonio; usato per la preparazione di acciai speciali, specie acciai per utensili a elevata velocità di lavoro (autotempranti); è aggiunto a volte direttamente all’acciaio liquido. Può essere preparato o al forno elettrico o col sistema dell’alluminotermia. Il prodotto è assai duro, di color cenere, e presenta una grana molto serrata.
Ferro-nichel
Ferrolega caratterizzata da un basso coefficiente di dilatazione termica variabile con il contenuto di nichel. Con il 21-30% di nichel si hanno ferroleghe non ferromagnetiche, usate per costruire rotismi per macchine elettriche; con il 36% di nichel si ha la lega invar, col minimo coefficiente di dilatazione e la massima resistività elettrica; con il 46% di nichel si ha la lega platinite, con lo stesso coefficiente di dilatazione del vetro e del platino; con il 78% di nichel, infine, si ha la lega permalloy, con permeabilità magnetica molto elevata.
Ferro-niobio
Contiene 50-60% di niobio, 5% massimo di manganese, 8% di silicio, 0,4% di carbonio; è usato prevalentemente nella fabbricazione di acciai inossidabili ‘stabilizzati’, cioè a bassa fragilità intercristallina e maggiore resistenza alle fessurazioni ad alte temperature.
Ferro-silicio
Si distinguono un f.-silicio al 5% di silicio, di larghissimo impiego per lamierini di trasformatori elettrici; un f.-silicio al 12-15% di silicio, usato in fonderia, che equivale praticamente a una ghisa siliciosa; un f.-silicio di uso analogo al precedente, ma col 25-30% di silicio; un f.-silicio al 45-50%, usato largamente, oltre che come disossidante, per apporto di silicio in acciaieria e fonderia; un f.-silicio al 75-80%, impiegato a preferenza per la fabbricazione di acciai ad alto contenuto di silicio; infine, un f.-silicio all’85% e uno al 90-95%, di uso e di preparazione analoga al precedente. Le leghe f.-silicio si preparano in genere al forno elettrico partendo da miscele di minerali di f. o da f. metallico e quarzo, o quarziti e carbone. I vari prodotti di cui sopra si presentano all’aspetto con colori sempre più tendenti al bianco quanto più elevato è il tenore di silicio; ciò vale fino a un tenore di 70% di silicio, oltre il quale la lega è di colore azzurrognolo. Parallelamente aumenta la durezza e la fragilità della lega. Leghe f.-silicio si usano, per la loro resistenza agli agenti chimici, per fabbricare recipienti, tubazioni.
Ferro-silicio-manganese
Contiene, oltre a silicio, anche manganese in quantità dal 5% al 60-65%; è usato specialmente nell’affinazione dell’acciaio.
Ferro-titanio
Può contenere tenori variabili sia di titanio (fino al 40%), sia di carbonio (da piccole percentuali sino al 6-8%), oltre che alluminio, silicio ecc. Ha colore grigio opaco scuro; è usato per aggiunte di titanio agli acciai.
Ferro-tungsteno
Contiene 70-80% di tungsteno e non più dello 0,6% di carbonio, è usato per la fabbricazione di acciai al tungsteno e acciai rapidi; è di colore cenere opaco e si prepara in genere con il sistema dell’alluminotermia.
Ferro-vanadio
Contiene tenori variabili di vanadio (45-50%) accanto a quantitativi molto bassi di carbonio (0,05-0,2%; prodotto non carburato), ovvero più elevati (9-10%; prodotto carburato); ha colore bianco argenteo e si prepara comunemente per mezzo dell’alluminotermia; si impiega nella fabbricazione di acciai speciali, come disossidante e deazotante.
Ferro delle paludi
Deposito di origine biochimica, costituito pressoché esclusivamente di ossidi di f. idrati. Si genera per azione di alghe unicellulari e di alcuni batteri sul carbonato ferroso disciolto nelle acque palustri o lacustri; i microrganismi, per ossidazione, liberano anidride carbonica e causano la precipitazione di ossido di f. idrato, che talvolta forma depositi che possono essere utilizzati per l’estrazione industriale del ferro.
Spugna di ferro
F. metallico ottenuto sotto forma di piccole masse spugnose per riduzione diretta (con carbone) o indiretta (con gas riducenti) di ossido di f., a temperature minori di quella di fusione del f. risultante; si usa, per es., per depurare il gas di cokeria dai composti solforati.
Produzione
Il f., fino dall’antichità, è il metallo più utilizzato (rappresenta il 95% della produzione mondiale di metalli). Alla metà del 20° sec. i quattro maggiori produttori di minerali di f. (Stati Uniti, URSS, Svezia, Francia) fornivano oltre i 4/5 della produzione mondiale. Il rilancio dell’industria siderurgica, a partire dagli anni 1950, ha agito nel senso di una forte diversificazione produttiva. Numerosi paesi dell’America Latina (Brasile, Cile, Messico, Perù), dell’Africa occidentale (Liberia, Mauritania), dell’Asia (India e Cina) e dell’Oceania (Australia), che prima producevano modestissimi quantitativi, hanno moltiplicato l’offerta a ritmi notevolissimi, acquistando posizioni di crescente rilievo nel commercio internazionale. Attualmente i primi produttori sono Cina, Brasile, Australia, India e Russia, che insieme coprono il 70% della produzione mondiale. Questa nel 2007 ammontava a 1,9 miliardi di t. L’Australia è il paese leader nell’esportazione.
Artigianato
Ferro battuto
La tecnica del f. battuto (battitura a caldo sull’incudine delle lamine, poi tagliate, traforate, cesellate o battute entro coni) ha avuto vastissima applicazione sul piano delle arti applicate. Per le sue caratteristiche di malleabilità e di resistenza, il f. è stato usato per serramenti, cancelli e grate, ringhiere, lampadari, mensole ecc., a volte con risultati altissimi nella decorazione lineare e bidimensionale. I primi capolavori risalgono al 12°-13° sec. (ferramenta delle cattedrali spagnole, francesi e inglesi). In Italia, nel 14° sec., si ebbero maestri come Conte di Lello Orlandi (cancellate del duomo di Orvieto, 1337-38). In Germania si elaborò un metodo di lavorazione a freddo; tra la fine del 14° sec. e il 15° fu notevole la produzione di cancelli,di ferrature di porte, armadi, cofani, scrigni, di elementi decorativi minori. Tra 15° e 16° sec. la perfezione tecnica dei fabbri italiani contribuì alla decorazione di edifici civili e religiosi, con cancelli (cappella Barbazza in S. Petronio a Bologna), balconi (Palazzo Bevilacqua a Bologna, convento di S. Maria della Quercia a Viterbo) lanterne e portastendardi (lanterne dei palazzi Strozzi, Riccardi e Guadagni a Firenze, attribuiti a N. Grosso). Tra 16° e 17° sec. in Germania con la cesellatura a freddo si realizzarono opere monumentali (trono di Rodolfo II). Nel 17° e 18° sec. l’evoluzione del gusto verso il rococò e il virtuosismo tecnico sfruttò tutte le possibilità del f. battuto, in Francia (Versailles, cattedrale di Nancy), Germania (Würzburg), e in tutta Europa; in Italia una certa classicità fu ricercata da artefici come G.B. Malagoli (1729-1797). Dopo una parentesi in periodo neoclassico l’arte del f. battuto risorse in Francia dalla metà del 19° sec. per iniziativa di E. Viollet-le-Duc e per l’attività di E. Robert e della sua scuola. Fiorente in Italia la bottega di A. Mazzucotelli. La tradizione italiana conobbe una nuova fioritura dopo l’esposizione di Parigi del 1925, confermata nelle esposizioni delle arti decorative di Monza.
Biologia
L’organismo umano adulto contiene 3,5-5 g di f. di cui il 65-75% in complessi con porfirine, mentre il restante è presente in complessi proteici e in forma inorganica. Tutte le proteine coniugate contenenti f. nel gruppo prostetico sono note come ferroproteine, composti di interesse biologico in quanto svolgono alcune funzioni fondamentali, quali il trasporto di ossigeno e la catalisi di numerose ossidoriduzioni biologiche. Il f. entra nella composizione dell’emoglobina, il pigmento respiratorio dei vertebrati e di molti invertebrati. È in virtù di questo f. che l’ossigeno può legarsi in forma reversibile per essere trasportato e ceduto ai tessuti. Una delle funzioni essenziali per gli organismi quale la respirazione si ricollega dunque a questo elemento. Il f. fa parte anche di altre proteine respiratorie come le eritrocruorine di molti invertebrati e l’emeritrina di certi vermi. Oltre che nei pigmenti deputati alla respirazione esogena, il f. entra nella composizione di altre cromoproteine interessate ai processi ossidoriduttivi che si svolgono nelle cellule e cioè alla respirazione endogena. Sono queste i citocromi, le perossidasi, la catalasi e altre ancora. In una particolare combinazione con una proteina forma la ferritina, che regola l’assorbimento del f. nell’organismo. Nel sangue si combina con una α-globulina dando origine alla transferrina e sotto questa forma è trasportato ai tessuti, dove è fissato all’emosiderina che funge da proteina-deposito.
Per lo studio del metabolismo del f. attualmente si fa uso di un isotopo radioattivo di questo elemento. Il f. ingerito è ridotto, assorbito (soprattutto nel duodeno) e quindi immagazzinato nella ferritina e nell’emosiderina sotto forma ossidata. Quando il tasso ematico del f. diminuisce, il f. passa dalla ferritina al sangue per essere trasportato ai tessuti o ai depositi sotto forma di transferrina. Non si conoscono forme di escrezione del f. in eccesso, per cui somministrazioni continuative di f. o ripetute trasfusioni possono provocare accumuli nei tessuti (➔ emocromatosi).
Il fabbisogno giornaliero di f. di un adulto è tra 10 e 20 mg; i costituenti della dieta più ricchi di f. sono la carne rossa, la verdura, il pane integrale, il torlo d’uovo, le carote e la frutta.