
genetica
Ramo delle biologia che si occupa del materiale ereditario, cioè della sua struttura, del suo modo di funzionare, delle modalità della sua trasmissione, sia da una cellula alle sue discendenti (se si tratta di cellule dello stesso organismo si parla di g. somatica) sia da una generazione all’altra di organismi pluricellulari, e della sua storia evolutiva.
Biologia
La g. può essere suddivisa secondo modalità differenti in base al criterio o, più frequentemente, alla combinazione di criteri adottata nei singoli casi. Il primo criterio è costituito dall’approccio e dal livello di analisi; il secondo dal fine; il terzo dal materiale biologico in esame.
L’approccio e il livello di analisi
Le principali branche della g. che possono essere individuate su questa base sono: la g. formale, la citogenetica, la g. biochimica, la g. molecolare, la g. delle popolazioni.
La g. formale
Consiste essenzialmente nello studio delle proporzioni relative delle classi fenotipiche che si trovano nella progenie di determinati incroci, e anche, più recentemente, delle frequenze con cui sono assortiti non a caso, in singoli genomi, alleli di siti diversi, ma strettamente associati a formare i cosiddetti aplotipi (linkage disequilibrium). Questo approccio, che è quello con cui è nata la g., è stato da solo sufficiente a far scoprire che l’eredità di molti caratteri dipende da un singolo fattore (caratteri mendeliani unifattoriali), il quale può esistere in forme alternative diverse, dette alleli; questi, a seconda dei casi, si comportano l’uno rispetto all’altro come dominanti, o come codominanti o come recessivi. In molti casi si sono anche individuate relazioni non casuali tra le frequenze con cui ‘segregano’ gli alleli di un gene e quelle di un altro gene, ottenendo così la prova statistica che alcuni fattori (che nel frattempo avevano preso il nome di geni) sono associati fra loro più o meno strettamente.
Questo approccio, combinato con quello citogenetico, ha portato a una conoscenza esatta e rigorosa dei meccanismi della trasmissione dei caratteri ereditari e della sede materiale (i cromosomi) dei fattori che li determinano. Non ha, tuttavia, dato – né avrebbe potuto – alcuna notizia sulla natura di questi fattori (dimensioni, struttura chimica e modo di funzionare), né sui motivi per i quali certi alleli si comportano da dominanti, altri da codominanti e altri ancora da recessivi. Questo approccio è inoltre applicabile soltanto a geni che mostrino una certa variabilità fenotipica.
Citogenetica
È la parte della g. che indaga con approcci morfologici essenzialmente al microscopio ottico il materiale ereditario, cioè i cromosomi durante la mitosi e la meiosi e, in casi più rari, in interfase. Quanto si è scoperto sul comportamento dei cromosomi in questi due tipi di divisione cellulare costituisce la perfetta spiegazione delle leggi della g. formale e la prova conclusiva che i cromosomi sono la sede fisica dei geni. Lo studio di cromosomi particolari, come quelli giganti delle ghiandole salivari di Drosophila melanogaster, ha portato a costruire mappe citogenetiche (in cui numerosi geni sono stati allineati e localizzati lungo i rispettivi cromosomi), che si sono dimostrate colineari rispetto alle mappe formali ottenute stimando le frequenze di ricombinazione tra geni situati nello stesso cromosoma.
La g. biochimica
Differisce dalla g. formale dei primordi per il livello al quale si effettua l’analisi fenotipica. Nella g. formale erano esaminati gli aspetti morfologici (piselli gialli o verdi, oppure lisci o rugosi ecc.), funzionali (individui con normale visione dei colori oppure daltonici ecc.), clinici (soggetti normali o emofilici; normali o con idiozia fenilpiruvica ecc.), sierologici (soggetti di gruppo 0, o A o B o AB; oppure Rh+ o Rh− ecc.), cioè un insieme di caratteri a base biochimica ignota. Successivamente, mediante lo studio biochimico di determinati fenotipi collegabili in modo non soltanto formale al gene che ne era responsabile, si è passati all’esame dei caratteri proteici. Questo è stato il passo decisivo verso la nozione di gene strutturale (➔ gene) e una delle tappe del percorso che ha portato alla corretta impostazione del problema del codice genetico. Si può quindi affermare che la g. biochimica abbia fatto da ponte tra la g. formale e la g. molecolare.
La g. molecolare
Ha portato alla conoscenza della natura chimica e del modo di funzionare dei geni, o meglio del DNA nel suo complesso. In una prima fase, detta proteica (iniziata negli anni 1940), queste informazioni si sono ottenute indirettamente, cioè per inferenza dalle proprietà delle catene polipeptidiche, sfruttando le relazioni biunivoche tra queste e i geni strutturali corrispondenti (n residui amminoacidici corrispondono a n triplette; un certo residuo amminoacidico in una certa posizione della sequenza della catena polipeptidica corrisponde a un certo codone, oppure a uno dei codoni che codificano per quell’amminoacido, nella posizione della sezione codificante del suo gene strutturale ecc.). In una seconda fase, iniziata negli anni 1980, il DNA è stato studiato direttamente a livello sia strutturale sia funzionale.
La g. delle popolazioni
Ha per oggetto di studio la popolazione, cioè un insieme di individui della stessa specie che, oltre a essere potenzialmente interfecondi (per definizione), si incrociano effettivamente tra loro (del tutto casualmente nelle popolazioni ideali dal punto di vista matematico, dette anche popolazioni mendeliane). La g. delle popolazioni è la base della scienza che studia l’evoluzione genetica e, soprattutto, la speciazione o la microevoluzione. La parte di essa che si occupa della popolazione umana si è rivolta alla formulazione di ipotesi ragionevoli riguardo alla sede e all’età dell’origine dell’uomo, nonché alla sua successiva diversificazione in gruppi distinti, le cosiddette razze, e ha poi elaborato un drastico ridimensionamento dell’entità di questa diversificazione===
Il fine
Sulla base di questo criterio si possono distinguere due g., la g. applicata e la g. di base. Scopo di quest’ultima è la conoscenza della natura chimica e dei modi con cui funziona, è trasmesso ed evolve il materiale ereditario, prescindendo completamente dall’eventuale utilizzabilità di questa conoscenza. Si tratta comunque di distinzioni spesso vaghe e sfumate. La distinzione ha tuttavia una sua validità sul piano concettuale, in quanto permette di individuare sezioni della g. che, almeno in linea di principio, sono da considerare g. applicata e sezioni che invece costituiscono una parte della g. di base.
Rientrano nel primo gruppo la g. agraria, la g. forense e varie parti della g. medica (in particolare quelle che hanno lo scopo di migliorare l’efficienza della prevenzione delle malattie g., per es. con procedimenti di consulenza g.).
Fanno parte della g. di base la g. biochimica, la g. molecolare, la g. evoluzionistica e l’immunogenetica. In quest’ultimo ambito, il chiarimento della genesi dei geni delle immunoglobuline e dei T-cell receptors ha aperto un nuovo, e fondamentale, capitolo della g. generale.
Il materiale biologico in esame
L’utilità di studiare organismi diversi varia a seconda dei molteplici problemi che si devono affrontare nel campo della biologia e particolarmente della genetica. Si sono così sviluppati rami differenti di questa disciplina, più o meno indipendenti l’uno dall’altro e riguardanti diversi tipi di organismi (insetti, funghi e batteri), come la g. di Drosophila melanogaster, di Neurospora crassa, di Saccharomyces cerevisiae, di Escherichia coli e dei suoi fagi sia virulenti (della serie T) sia lisogeni (il fago λ), del mais, del topo e, in un secondo tempo e in misura sempre più accentuata, dell’uomo. Le peculiarità biologiche di questi organismi, che hanno costituito per decenni, e in molti casi costituiscono tuttora, i materiali di elezione della g., hanno determinato la scelta dei problemi di carattere generale da affrontare con ciascuno di essi.
Insetti
Le caratteristiche dei cromosomi giganti delle ghiandole salivari delle larve e quelle delle uova hanno permesso di chiarire alcune proprietà biologiche importanti. I cromosomi giganti, oltre a essere molto lunghi (DNA relativamente poco spiralizzato) e molto spessi (cromosomi politenici), presentano il fenomeno dell’appaiamento degli omologhi (sinapsi somatica tra i cromosomi ‘fratelli’) che rende particolarmente evidenti eventuali aberrazioni cromosomiche allo stato eterozigote. A causa di queste caratteristiche, a partire dagli anni 1930, si sono potuti localizzare i geni mediante mappe citogenetiche estremamente dettagliate, ed è stato possibile constatare la loro corrispondenza con quelle basate sulla frequenza di ricombinazione (mappe statistiche o formali) e comprendere la natura delle aberrazioni cromosomiche, quali le inversioni, le traslocazioni, le delezioni e le duplicazioni.
Le grandi dimensioni delle uova rendono possibili manipolazioni in grado di fornire importanti informazioni riguardo alle prime fasi dello sviluppo embrionale e consentono anche la costruzione di animali transgenici. Si sono, inoltre, scoperte mutazioni con effetti su fasi anche precocissime della differenziazione embrionale che hanno chiarito la base genetica del controllo delle varie fasi dell’embriogenesi, comprese quelle pre- e postzigotica.
Funghi
Gli ascomiceti sono funghi, come quelli dei generi Neurospora e Saccharomyces, in cui i prodotti aploidi di ogni singolo evento meiotico (spore) rimangono racchiusi insieme in un unico involucro, detto asco. Nel caso di N. crassa, inoltre, l’asco è un sacchetto oblungo al cui interno le 8 spore (i 4 prodotti della meiosi vanno ciascuno incontro a una divisione mitotica supplementare) si dispongono in modo ordinato, per cui è possibile non solo esaminare le singole meiosi separatamente l’una dall’altra, ma addirittura sapere in quale delle divisioni meiotiche si è verificato l’evento in esame (per es. un evento ricombinatorio). L’analisi delle singole meiosi ha portato alla scoperta di un fenomeno del tutto inatteso, la gene conversion, che costituisce un’eccezione alla segregazione mendeliana (nella quale metà delle spore derivate dalla divisione meiotica di una cellula diploide eterozigote A1A2 ha l’allele A1 e l’altra metà ha l’allele A2): in alcuni aschi, derivati da un diploide eterozigote, il numero delle spore con un allele è diverso da quello delle spore con l’altro allele (5 a 3 oppure 6 a 4). Questo fenomeno – raro (ca. 1‰) per la maggior parte del genoma, ma molto frequente (fino a 50%) per alcuni marcatori genetici – è fondamentale per le sue numerose e generali implicazioni e non sarebbe mai stato scoperto in organismi di altre specie nelle quali i prodotti di meiosi diverse sono mescolati tutti assieme.
Batteri
Fra le caratteristiche di Escherichia coli e dei suoi batteriofagi, quella che si è rivelata più decisiva per la comprensione della struttura fine e del modo di funzionare e di evolvere del materiale genetico è la rapidità della loro moltiplicazione; quindi, la grandezza dei numeri di elementi in gioco, associata alla facilità di individuare e di isolare, per selezione, fenotipi anche rarissimi (perfino dell’ordine di 10−8-10−9), ha reso possibile uno studio accurato di eventi così eccezionali da non essere scopribili, e tanto meno analizzabili, nella grande maggioranza degli altri sistemi biologici.

Allo studio dei batteri e dei batteriofagi la g. deve alcuni concetti fondamentali: a) il materiale genetico è costituito dal DNA; b) la sua replicazione è semiconservativa; c) la regolazione dell’espressione dei geni per gli enzimi adattivi (inducibili o repressibili) è mediata da molecole diffusibili (repressori oppure attivatori) che, legandosi oppure staccandosi da una breve sequenza di DNA che costituisce il loro bersaglio specifico e che è detta operatore, inibiscono o promuovono, a seconda dei casi, la trascrizione dei geni strutturali a valle di quell’operatore (l’insieme di questi geni, dell’operatore e del promotore costituisce un cistrone regolativo, l’operone; attualmente le molecole diffusibili che regolano l’espressione dei geni sono chiamate fattori di trascrizione); d) il concetto di replicone (segmento discreto di DNA che si replica come un’unità) nella replicazione appunto del DNA; e) la struttura fine del gene e il concetto di cistrone, definiti a livello formale nei batteriofagi T di Escherichia coli; f) la dimostrazione che agenti letali ad azione immediata (come i fagi virulenti T) non inducono in una coltura di batteri sensibili la comparsa di mutanti resistenti, ma si limitano a selezionare quelli già presenti casualmente nella coltura; g) la dimostrazione che, invece, l’esposizione di batteri a condizioni non letali ma che non ne consentono la crescita (per es., la semina di batteri che richiedono triptofano e non sono in grado di utilizzare il lattosio in un terreno minimo contenente lattosio come unica fonte di carbonio organico) è seguita dopo tempi lunghi, cioè dopo molti giorni, dalla comparsa di mutanti evidentemente non pre-esistenti che formano colonie (o papille entro singole colonie), all’interno delle quali i doppi mutanti si formano nella coltura con una frequenza dello stesso ordine di grandezza di quella di una delle due mutazioni richieste per moltiplicarsi in quel terreno (invece che con una frequenza pari al prodotto delle due frequenze di mutazione: per es., con frequenza di 10−9 invece che di 10−16, che è come dire 0); h) il ciclo integrazione-escissione del fago temperato come modello di ricombinazione illegittima, accompagnata o meno da trasduzione di sequenze di DNA batterico adiacenti al sito di integrazione del fago nel cromosoma batterico; i) la coniugazione (fig. 1), cioè il passaggio da una cellula batterica donatrice, detta F+ o Hfr (high frequency of recombination), a una cellula ricevente, la F–, di una porzione più o meno lunga di una copia del suo genoma a partire da un punto di inizio fisso, a seconda della frequenza rispettivamente bassa o molto alta di trasferimento; il genoma trasmesso per coniugazione è sintetizzato via via che viene trasferito nell’F− (dato che la cellula donatrice conserva il suo genoma, che resta separato dalla F−, questo processo non è assimilabile alla formazione di uno zigote attraverso la fusione di due gameti); poiché il trasferimento è sequenziale e può essere interrotto in qualsiasi momento, questo sistema si presta a mappare i geni del cromosoma batterico registrando l’ordine temporale con cui essi vengono trasferiti nel batterio F− (si può cioè accertare quali geni vengono trasferiti per primi 1′, 2′, 3′ dopo l’inizio della coniugazione e quali in tempi successivi); l) la trasformazione, cioè il passaggio di DNA ‘nudo’ dal mezzo di coltura fino all’interno della cellula batterica, seguito dalla sua integrazione nel genoma batterico. L’osservazione di tale fenomeno ha permesso di dimostrare che il materiale ereditario è costituito da acidi nucleici, constatando che i caratteri ereditari possono essere trasferiti con DNA puro.
I cromosomi facilmente osservabili del mais e il gran numero di semi di ogni singola pannocchia hanno consentito a B. McClintock di integrare la g. formale con la citogenetica in modo così geniale da portare alla scoperta degli elementi mobili (o trasposoni) già nel 1948, quando un simile concetto costituiva un’eresia. La g. di quell’epoca era del tutto impreparata a recepire l’idea che alcuni tratti del genoma, lungi dall’avere una posizione fissa nel genoma stesso, abbandonassero frequentemente la loro sede iniziale e ‘saltassero’ in un altro punto integrandosi in un nuovo sito (a quell’epoca non si sarebbe ancora detto ‘in una nuova sequenza di DNA’), influenzando infine l’espressione del gene adiacente. I dati di McClintock, essendo inoppugnabili, furono accettati, ma passò una ventina d’anni prima che i trasposoni, riscoperti in Escherichia coli, entrassero ufficialmente nella g. ortodossa e se ne comprendesse appieno il significato funzionale nella ‘ricombinazione illegittima’, cioè nella ricombinazione che si verifica senza un appaiamento esteso tra sequenze omologhe.
Topi
Il topo è, insieme all’uomo, l’unico Mammifero, anzi l’unico Vertebrato, tra gli organismi che hanno avuto un ruolo importante nello sviluppo della g., e ciò lo rende assolutamente insostituibile. Le caratteristiche che hanno fatto di questa specie un materiale biologico particolarmente adatto per la g. risiedono nel fatto che i topi sono molto piccoli, molto prolifici e con tempi di generazione molto brevi (donde l’alto potere di risoluzione della loro g. formale); il loro allevamento è, inoltre, facile ed economico.
Alla g. del topo si devono le conoscenze dell’immunogenetica, del fenomeno di Mary Lyon, dell’imprinting genomico. Inoltre i topi sono stati i primi fra i Vertebrati nei quali è stato possibile trasferire geni di altre specie per studiarne l’espressione (topi transgenici). Uno degli aspetti essenziali dell’immunogenetica consiste nello studio della struttura della regione genetica responsabile della sintesi delle molecole MHC (major histocompatibility complex), denominata regione H2 nel topo e regione HLA nell’uomo. Le molecole MHC si suddividono in due classi ben distinte tanto per struttura quanto per funzione: le MHC di classe I e le MHC di classe II. Mediante molecole MHC, le cellule esibiscono sulla loro superficie esterna, cioè accessibile alla sorveglianza immunologica, una serie di peptidi sia derivati dalla degradazione delle catene polipeptidiche sintetizzate nel loro citoplasma, sia formati nei lisosomi per degradazione di molecole peptidiche esogene (la presentazione dei due tipi di peptidi sulla superficie cellulare è svolta rispettivamente dalle MHC di classe I, che sono presenti in tutte le cellule dell’organismo, e dalle MHC di classe II, che sono presenti soltanto in determinate cellule, quali i macrofagi, le cellule dendritiche ecc.).
Il fenomeno di Mary Lyon (o compensazione della differenza di dosaggio genico) si riferisce a quei processi i quali fanno sì che nelle femmine di Mammifero i geni presenti nel cromosoma X abbiano lo stesso livello di espressione rispetto ai maschi, malgrado le femmine possiedano due cromosomi X e quelli maschili uno solo. Questo fenomeno è dovuto al fatto che, in una fase molto precoce dello sviluppo degli embrioni di femmine di Mammifero, in ognuna delle loro cellule somatiche viene inattivato a caso uno dei due cromosomi X, quello di origine paterna o quello di origine materna; la ‘decisione’ presa viene trasmessa inalterata a tutte le cellule discendenti, cioè si generano cloni nei quali tutte le cellule hanno come cromosoma X attivo quello paterno e cloni dove invece in tutte le cellule si esprime solo l’X materno.
L’imprinting genomico è una delle eccezioni più importanti ai profili di eredità mendeliana e consiste nella variazione nell’espressione di un gene o di un gruppo di geni, dipendente dal fatto che quel gene proviene dal genitore maschio o dal genitore femmina. Il fenomeno è stato scoperto nel topo, osservando che l’instaurarsi di un fenotipo normale richiede non solo l’esistenza di due copie dello stesso gene, ma anche che una di esse sia di origine paterna e che l’altra sia di origine materna. Ciò implica che quelle regioni di DNA, prima di essere trasmesse, hanno nel contesto maschile un destino epigenetico (che lascia cioè intatta la sequenza nucleotidica) diverso da quello che ha luogo nel contesto femminile e che entrambe le regioni (sia quella di origine paterna sia quella di origine materna) sono necessarie per la formazione di un fenotipo normale. Sono ormai note molte malattie umane dovute al fatto che l’embrione deriva da uno zigote che presenta un tratto del genoma diploide solo paterno o solo materno. Da questo si può dedurre che molte regioni genomiche sono soggette a imprinting. Condizioni patologiche legate a questo fenomeno sono due tipi di triploidie, la mola idatiforme (neoformazione nella quale sono presenti due assetti cromosomici paterni e un assetto cromosomico materno) e i teratomi (che contengono due assetti materni e uno paterno). Esse, insieme ad altre evidenze, suggeriscono che il genoma paterno favorisca la differenziazione degli annessi embrionali a scapito dell’embrione vero e proprio e il genoma materno eserciti un’influenza di segno opposto.
I topi transgenici sono l’esempio più noto di trasferimento e integrazione di un gene di una specie nel genoma di un organismo di un’altra specie. Si tratta di un tipo di procedimento ormai classico di ingegneria genetica, dal quale si sono ottenute informazioni sulle conseguenze fenotipiche sia dell’introduzione nel genoma murino di determinati geni (in certi casi dopo averli opportunamente modificati in vitro, per esempio con un sistema di mutagenesi sito-specifica, site directed mutation), sia dell’inattivazione di quelli pre-esistenti (quando un gene viene sostituito da una sua forma inattiva introdotta per transgenizzazione, il topo viene chiamato knock-out per quel determinato gene).
L’uomo
Ciò che ha reso l’uomo uno dei materiali biologici ottimali della g. generale è la nascita di g. nuove, che si sono andate via via ad aggiungere alla g. formale e alla citogenetica. Per la g. biochimica e quella molecolare, che rappresentano i due nuovi indirizzi della g., l’uomo costituisce un eccellente materiale biologico di ricerca per una serie di motivi. Il principale è che la g. biochimica è nata e si è sviluppata partendo da malattie ereditarie. Gli errori congeniti del metabolismo (inborn errors of metabolism), studiati da A.E. Garrod fin dagli inizi del 20° sec., sono ora denominati genetici, invece che congeniti, perché, in una malattia ereditaria del metabolismo, ciò che è senza eccezioni congenito è il genotipo, non il fenotipo patologico che, in molte malattie ereditarie, si manifesta solo tardivamente.
L’approfondito studio biochimico di molte malattie ereditarie ha condotto alla scoperta della proteina alterata di ciascuna determinata malattia ereditaria, alla scoperta cioè di un fenotipo proteico, collegabile al genotipo corrispondente in termini molecolari e non più solo formali. In un primo momento, sulla base delle conoscenze delle relazioni tra sequenze amminoacidiche e sequenze nucleotidiche, si è potuto dedurre la natura chimica delle mutazioni in esame; successivamente, è stato possibile studiarle direttamente a livello di DNA. Non c’è da stupirsi se, divenute le malattie ereditarie il punto di partenza, l’uomo sia diventato un materiale biologico di elezione: si hanno molte più conoscenze a riguardo delle malattie umane che di quelle di tutte le altre specie messe assieme.
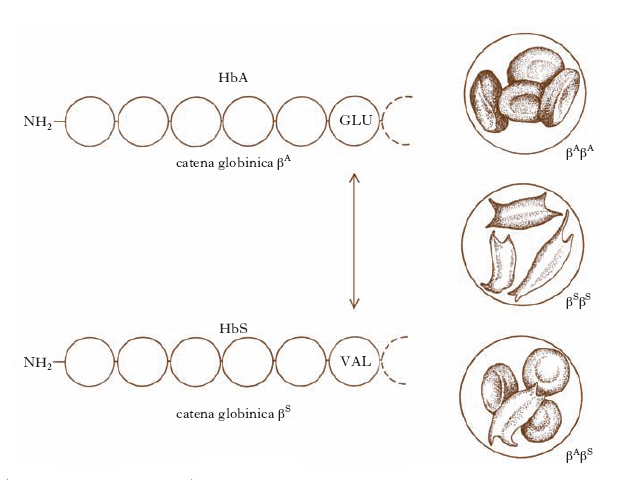
Il gene che più si presta a illustrare il nuovo ruolo assunto dall’uomo come strumento per lo sviluppo della g. di base è quello dell’emoglobina, Hb (ora diremmo il gene della catena globinica β). Nel 1956, per la prima volta nella storia della biologia, si scoprì un fenotipo a livello molecolare, cioè una molecola proteica anormale che fu chiamata HbS (i globuli rossi dei soggetti con l’allele anormale βS hanno una forma a falce ‒ in inglese sickle ‒ invece che la normale forma a disco biconcavo) e che è presente da sola negli omozigoti βSβS (i malati di anemia falciforme), oppure insieme alla HbA nei βAβS (i falcemici, che sono sani; fig. 2). Successivamente si è compresa la differenza tra la struttura della HbS e quella della HbA: il sesto residuo amminoacidico della catena βA è costituito dall’acido glutammico, mentre nella catena βS è costituito dalla valina. La scoperta della HbS ha segnato la nascita non solo della g. molecolare, ma anche, più in generale, della biologia molecolare. La delucidazione dell’anatomia strutturale della HbS ha dimostrato che i geni codificano la sequenza amminoacidica delle catene polipeptidiche amminoacido per amminoacido. A questo punto sorgeva il problema della decifrazione del codice genetico, che poteva ormai essere posto nella sua forma corretta: bisognava capire quali fossero le relazioni tra la sequenza nucleotidica del gene strutturale di una catena polipeptidica e la sequenza amminoacidica di quella catena polipeptidica; il problema sarebbe stato risolto negli anni 1960 (➔ codice).
Lo studio dei geni dell’emoglobulina umana ha permesso di effettuare un’altra scoperta fondamentale per la g. di base: le talassemie, anch’esse partite dallo studio di una malattia umana, il morbo di Cooley (o talassemia maior). L’indagine a livello molecolare dei geni talassemici ha portato a due importanti risultati: a) è stata studiata e raccolta una serie di mutazioni tutte risultanti nella compromissione più o meno completa dell’espressione di un gene strutturale, anche se diverse fra loro per i meccanismi e i livelli di tale compromissione funzionale (per es., delezione parziale o totale del gene, oppure alterazione funzionale a livello della trascrizione o della maturazione del suo prodotto, da pre-mRNA a mRNA, mediante alterazioni dello splicing, e/o a livello della traduzione – frameshift mutations, comparsa di un codone di stop, allungamento della catena globinica ecc. – e/o a livello delle proprietà della globina prodotta dal gene talassemico); b) HbS e talassemia costituiscono il modello di polimorfismo bilanciato meglio compreso, a livello molecolare, di tutta la biologia e rappresentano inoltre esempi di interazione tra le evoluzioni di due specie, quella di un organismo parassitato e quella del suo parassita (rispettivamente Homo sapiens e Plasmodium falciparum, l’agente eziologico delle forme più gravi di malaria). In molte delle popolazioni esposte a lungo a un’intensa endemia malarica si trova a frequenza elevata l’allele βthal (o il βS), cioè un allele letale recessivo (i βthal / βthal sono malati di morbo di Cooley), situazione assolutamente eccezionale perché gli alleli letali sono, di regola, rari. La frequenza elevata è dovuta alla particolare resistenza degli eterozigoti per la talassemia alla malaria: ciò costituisce, in un ambiente fortemente malarico, un vantaggio selettivo tale da rendere la loro idoneità biologica o fitness (valutata in base al numero medio di figli) maggiore di quella degli omozigoti per l’allele normale non talassemico. Polimorfismi come questi vengono detti bilanciati, perché sono mantenuti indefinitamente da un bilanciamento tra gli svantaggi dei due genotipi omozigoti rispetto al genotipo eterozigote, per cui nessuno dei due alleli, nemmeno quello letale, tende a scomparire dalla popolazione.
G. umana
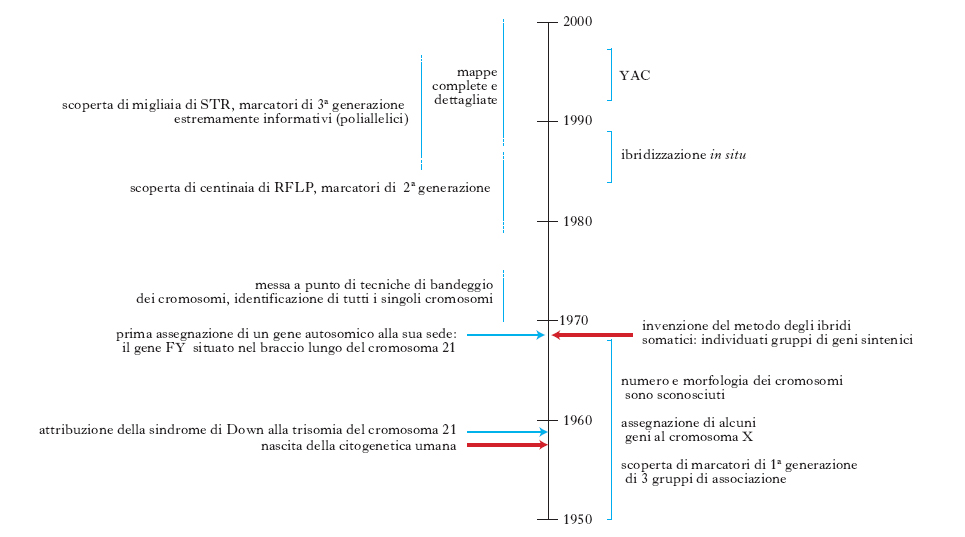
Due fondamentali capitoli della g. umana riguardano il primo lo studio dell’anatomia del genoma umano, il secondo il modo di procedere seguito da questa disciplina nella delucidazione della base genetica dei caratteri unifattoriali; la storia dei principali processi conoscitivi è sinteticamente riportata in fig. 3.
Anatomia del genoma umano
Fino agli anni 1950 e oltre, l’anatomia del genoma umano non era ancora in sostanza neppure agli albori: sul versante citogenetico non era nemmeno noto il numero dei cromosomi umani. La nascita della citogenetica umana si può datare al 1957, quando furono introdotte due innovazioni tecniche: l’uso della colchicina (che blocca le mitosi in metafase, facendone così aumentare il numero) e di soluzioni ipotoniche (che disperdono i cromosomi della piastra equatoriale metafasica, districandoli cioè l’uno dall’altro). È diventato così possibile ottenere molte metafasi con i cromosomi ben distinti l’uno dall’altro e, quindi, suscettibili di essere contati e suddivisi in gruppi diversi sulla base di caratteristiche morfologiche ben evidenti: lunghi e metacentrici, lunghi e submetacentrici ecc. (classificazione di Denver). I cromosomi di uno stesso gruppo sono rimasti, tuttavia, indistinguibili fra loro ancora per una quindicina d’anni. Subito dopo si è scoperto che la sindrome di Down è dovuta a una trisomia del cromosoma 21; in seguito si sono scoperte numerose altre anomalie di numero di cromosomi (X0, XYY, XXY, trisomia 13, trisomia 18 ecc.) o di struttura (delezione del braccio lungo del cromosoma 4 e varie altre). All’inizio degli anni 1970 la messa a punto di tecniche di bandeggio ha permesso di distinguere i singoli cromosomi, cioè anche quelli appartenenti allo stesso gruppo della classificazione di Denver.
Al 1968 risale l’invenzione del metodo degli ibridi somatici per l’identificazione di geni situati sullo stesso cromosoma, detti sintenici. In breve tempo si sono scoperte molte coppie, o addirittura gruppi, di geni sintenici. Con la messa a punto delle tecniche di bandeggio che hanno permesso di individuare singolarmente i cromosomi umani è diventato possibile assegnare i gruppi di geni sintenici ai rispettivi cromosomi. Si è trattato, semplicemente, di caratterizzare le sottocolture ibride in esame anche per i cromosomi: se un gruppo sintenico mostrava una concordanza perfetta di comportamento con un certo cromosoma umano era possibile assegnarlo a quel cromosoma. Per quanto questo approccio abbia costituito una pietra miliare nella delucidazione della topografia dei geni umani, esso tuttavia aveva un limite molto grave, derivante dal fatto che si poteva utilizzare solo per geni espressi e studiabili nelle cellule in coltura (si trattava quasi sempre, rispettivamente, di geni strutturali di enzimi e di fibroblasti). Anche questo limite è stato superato quando è diventato possibile identificare i geni non dai loro prodotti, ma direttamente, come sequenze di DNA, mediante sonde molecolari specifiche (probes). È stato possibile così accertare la presenza o meno di un determinato gene umano in una certa sottocoltura utilizzando un probe capace di identificare la sequenza di quel gene (in modo specifico rispetto non solo agli altri geni umani ma anche al corrispettivo gene murino). Questo metodo ha costituito un’innovazione significativa in quanto ha permesso il riconoscimento di numerosi gruppi sintenici, ognuno costituito da parecchi geni; esso però non è in grado di fornire alcuna informazione sulle distanze fisiche o di ricombinazione tra i geni di uno stesso gruppo sintenico.
Tappa successiva è stata la scoperta di centinaia di RFLP (restriction fragment length polymorphisms). Gli RFLP sono polimorfismi diallelici che riguardano una sequenza di 4-7 coppie di basi che può corrispondere a quella di un sito riconosciuto da un enzima di restrizione, allele+, oppure non corrispondere a tale sito, allele–. Fino agli inizi degli anni 1980, i limiti più seri nella costruzione di mappe di ricombinazione (dette anche mappe statistiche) del genoma umano sono stati rappresentati dal numero molto esiguo di marcatori genetici polimorfici noti e dal fatto che la maggioranza di essi fosse solo moderatamente polimorfica, cioè avesse un grado di eterozigosità (H) piuttosto piccolo. Un marcatore genetico è, infatti, un sito sul DNA identificato attraverso la sua variabilità nella popolazione: se questa variabilità è elevata, cioè se la frequenza H degli eterozigoti (o grado di eterozigosità) è almeno dell’1%, il marcatore è detto polimorfico. In una popolazione in stato di stabilità (equilibrio di Hardy-Weinberg) per un certo marcatore, condizione rispettata quasi senza eccezioni, la frequenza degli eterozigoti per quel marcatore è data dalla somma delle frequenze di tutti i possibili eterozigoti, essendo la frequenza di ogni eterozigote pari al doppio prodotto di quelle degli alleli di cui è costituito. L’informatività potenziale di un marcatore ai fini di una mappatura statistica è tanto maggiore quanto maggiore è H. La scarsezza di siti polimorfici rendeva molto difficile la mappatura statistica di un sito genetico rispetto a un altro, perché questo procedimento si basa necessariamente sull’analisi della progenie di individui eterozigoti per entrambi i siti. Fino agli inizi degli anni 1960, gli unici marcatori polimorfici (cioè gli unici utilizzabili come punto di riferimento per descrivere la topografia del genoma umano) sono stati i gruppi sanguigni che sono poco meno di una decina, cioè in media uno per ogni 2-3 autosomi: situazione che era paragonabile a quella di un geografo che disponga di una decina di punti trigonometrici di altitudine per descrivere l’orografia di tutte le catene montuose del mondo. A partire dagli anni 1960, con la scoperta dei polimorfismi enzimatici, i punti di riferimento sono diventati circa 30, cioè la situazione, anche se migliorata, è rimasta pressoché immutata: il magro raccolto di geni associati che erano stati individuati fino alla fine degli anni 1970 consisteva solo di 3 coppie di geni associati, senza tenere conto naturalmente dei geni di uno stesso gruppo, come quelli della famiglia genica delle globine. La vera scoperta determinante è stata quella degli RFLP: dai 30 punti di riferimento si è passati, nel breve volgere di pochi anni, a varie centinaia di marcatori polimorfici, molti dei quali sono stati assegnati prima al rispettivo cromosoma e successivamente localizzati in una sua parte specifica. Si era così giunti a disporre di varie centinaia di punti di riferimento, correttamente allineati e localizzati nei rispettivi cromosomi e separati l’uno dall’altro da distanze pari in media a una decina di centimorgan (un centimorgan corrisponde a una frequenza di ricombinazione dell’1% ed equivale in media a una megabase, cioè a un milione di coppie di desossiribonucleotidi).
Il progetto di sequenziare l’intero genoma è stato portato a termine nell’aprile 2003. La sua attuabilità pratica è diventata realistica grazie alla scoperta delle STR (simple tandem repeats) e all’invenzione di 3 tecnologie, due delle quali (la PCR, polymerase chain reaction, e le tecniche di sequenziamento del DNA) hanno letteralmente rivoluzionato la genetica (la terza tecnologia è quella degli YAC, yeast artificial chromosomes). Le STR, dette anche SSR (simple sequence repeats) o microsatelliti, sono brevi sequenze ripetute in tandem un numero n di volte, per esempio (CAG)n. Il loro elevato grado di polimorfismo riguarda il numero di ripetizioni, per cui di ognuna delle regioni costituita da una successione di ripetizioni (repeats) esistono di regola non due alleli (come nel caso degli RFLP), ma molti alleli, per es. 5: (CAG)6, (CAG)7, (CAG)8, (CAG)9 e (CAG)10. Esistono, sparsi nel genoma, moltissimi siti (CAG)n, moltissimi (CAT)n, (TGA)n ecc. È tuttavia possibile individuare ognuno di essi in modo specifico perché ciascuno è situato in un contesto proprio, cioè è fiancheggiato da due sequenze, una a monte e una a valle, che differiscono da quelle che fiancheggiano le altre STR dello stesso tipo. Per es. un certo sito (CAG)n, che denominiamo A, è fiancheggiato da 2 sequenze specifiche, 5′A e 3′A; un altro sito B, anch’esso del tipo (CAG)n, è fiancheggiato da due sequenze specifiche, 5′B e 3′B, diverse dalla 5′A e dalla 3′A e così via. Quindi, se in un esperimento di amplificazione mediante PCR effettuato sul DNA di un certo soggetto, si usano sonde complementari al 5′F e al 3′F, si amplificano specificamente i due alleli (CAG)nF di quel soggetto, che sono uguali oppure diversi fra loro per il numero n delle ripetizioni (CAG) a seconda che esso sia omo- o eterozigote per quel sito. Si possono così studiare separatamente l’una dall’altra tutte le STR.
I polimorfismi per le STR sono estremamente più utili degli RFLP ai fini della mappatura statistica del genoma umano perché, oltre a essere molto numerosi, il loro grado medio di eterozigosità H è di gran lunga maggiore (perché, a differenza degli RFLP che sono in genere diallelici, gli alleli delle STR sono in genere più di due). In aggiunta a queste proprietà biologiche così favorevoli, si deve notare che sono tecnicamente molto più facili da studiare degli RFLP e quindi molto più economici (queste considerazioni valgono anche per la loro utilizzazione nella medicina forense, tanto che sono diventati i marcatori medico-legali di elezione). Attualmente sono state localizzate lungo il genoma umano più di 5000 STR. Esse hanno costituito la piattaforma di partenza del progetto del suo sequenziamento completo.
Una delle tecnologie più fruttuose dell’ingegneria genetica ai suoi albori è consistita nel clonaggio di una sequenza in esame in un plasmide, seguito dalla raccolta del DNA così amplificato. Un limite molto serio di questa tecnologia è la brevità della sequenza (dell’ordine di poche kilobasi, kb), che può essere incorporata in un genoma piccolo come quello di un plasmide. Con gli YAC la lunghezza della sequenza clonabile è stata aumentata di un fattore dell’ordine di 100, arrivando quindi alla megabase (pari cioè a un centesimo, o poco meno, di un cromosoma umano). Uno YAC è una sequenza, derivata per es. da un genoma umano, che è stata trasformata in una sorta di cromosoma di lievito con procedimenti ormai standardizzati di ingegneria genetica. È stata creata una genoteca costituita da molte migliaia di YAC e, dato che ciascuno di essi comprende una sequenza umana pari in media a una megabase e il genoma umano è costituito da circa 3000 megabasi, si suppone che tale genoteca sia completa, cioè che tutte le sequenze umane siano veicolate in almeno uno degli YAC della genoteca.
La PCR è certamente per la genetica molecolare, anzi per tutta la biologia molecolare, l’innovazione tecnica più importante e più utilizzata degli ultimi 10-15 anni. Essa permette di amplificare specificamente anche di milioni di volte una sequenza di DNA lunga fino a 1-2 kb (anche di più opportuni accorgimenti sperimentali), se si conosce la sequenza delle due regioni che la fiancheggiano.

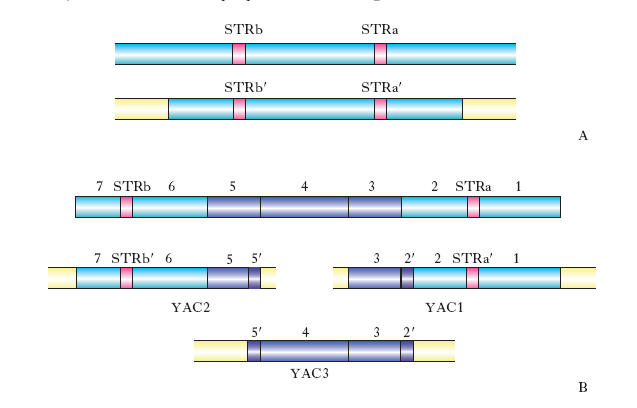
Nel corso degli anni sono stati messi a punto procedimenti sempre più rapidi (anche automatici), attendibili ed economici finalizzati al sequenziamento del DNA. La fase attuale ha avuto come punto di partenza la situazione presentata nella fig. 4, che mostra i 23 cromosomi del corredo aploide dell’uomo (22 autosomi + X; Y non è mostrato) con 2335 posizioni definite da 5264 marcatori (quasi tutti STR) localizzati lungo di essi. Il problema da risolvere è stato il sequenziamento dei tratti interposti tra questi e 2335 posizioni. Lo si è affrontato creando una raccolta ‘completa’ di YAC allo scopo di individuare quelli che comprendono una sequenza complementare a una delle 2335 posizioni di riferimento e soprattutto di trovarne alcuni che si sovrappongano a due STR adiacenti. Tenendo conto del modo con cui gli YAC sono stati ottenuti (ognuno di essi ha una sezione centrale derivata da una sequenza continua del genoma umano fiancheggiata da due sequenze, una al suo 5′ e una al suo 3′, che le conferiscono proprietà di YAC; fig. 5) si può essere conseguentemente certi che in ogni YAC che comprende una sequenza a′ uguale a quella a di un marcatore, e un’altra b′ uguale a quella b di un altro marcatore adiacente al precedente, la sequenza interposta tra a′ e b′ corrisponde necessariamente alla sequenza interposta nel genoma umano tra le due STR in questione.
Dal fenotipo al DNA
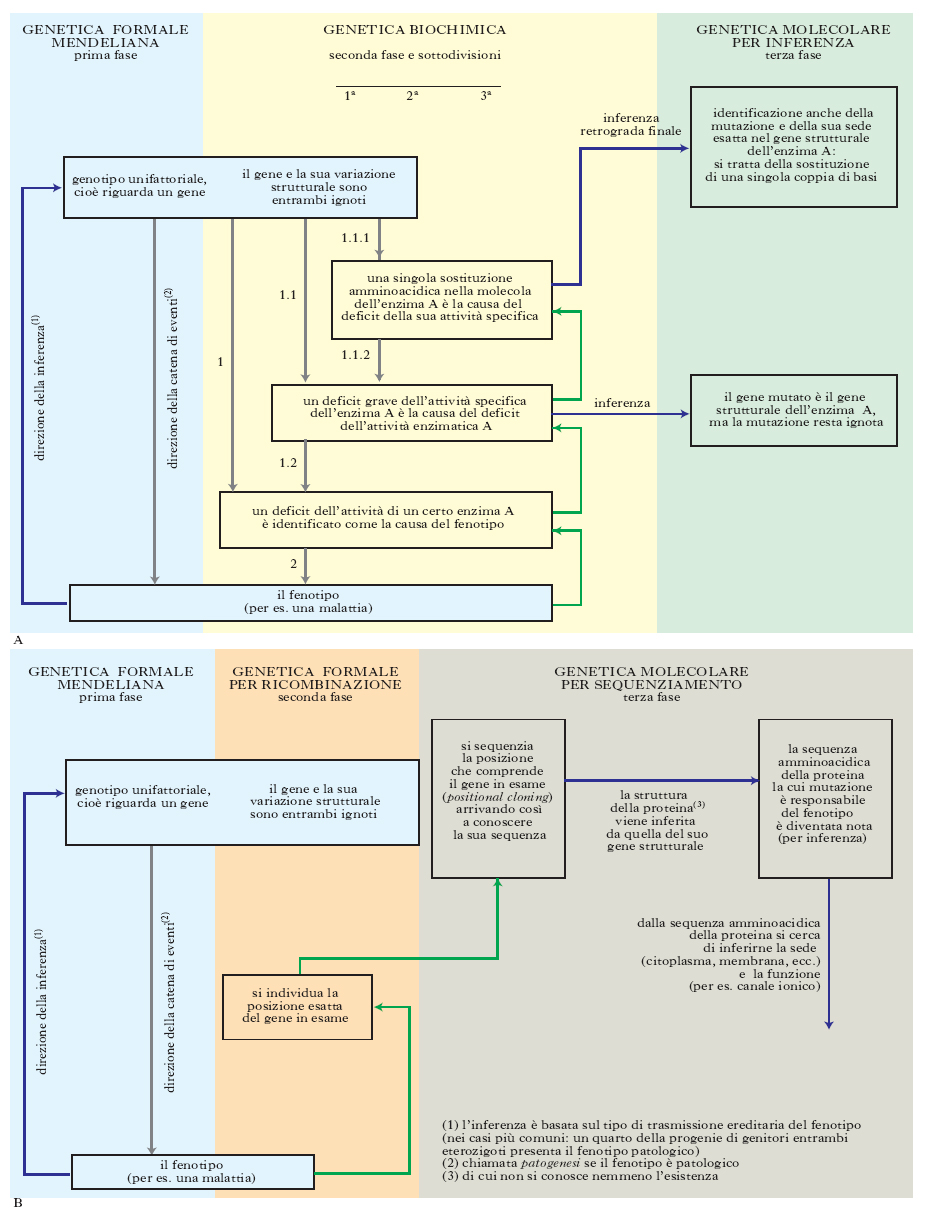
Per una novantina d’anni, dei circa cento del suo sviluppo, la g. ha proceduto risalendo, a ritroso e un passo alla volta, da un fenotipo unifattoriale verso il genotipo. Il problema che si è affrontato, e risolto con successo in migliaia di casi, è consistito quindi nell’individuare la natura molecolare della variazione genetica responsabile della variazione fenotipica, avendo come punto di partenza la certezza che la variazione genetica era trasmissibile come una singola entità secondo due possibili modalità: la prima come variazione di un singolo sito genetico (per es., la sostituzione di una singola coppia di basi con un’altra, oppure la variazione del numero di ripetizioni in una STR), la seconda come variazione di due o più siti così vicini da essere trasmessi in blocco, cioè senza mai ricombinare (se in una certa sequenza di DNA molto breve esistono due siti variabili A e B e un individuo ha il genotipo A1B2/A2B1, esso trasmette alla prole o A1B22 o A2B; non trasmette mai, o quasi mai, A1B1 o A2B2: cioè queste due coppie di alleli, pur riguardando siti diversi, si comportano come un unico fattore mendeliano). Come si mostra schematicamente nella fig. 6 A nei casi più tipici si risaliva a ritroso fino al genotipo responsabile del fenotipo unifattoriale in esame (per es., una malattia del metabolismo): innanzitutto scoprendo, attraverso la g. biochimica, che esso era da attribuire a una carenza di una determinata proteina (anzi, più precisamente, di una determinata catena polipeptidica), per es. dell’attività di un enzima; poi, scoprendo che questa carenza consisteva in una riduzione dell’attività specifica della molecola enzimatica; infine, individuando l’alterazione della struttura primaria della catena polipeptidica responsabile della riduzione della sua attività specifica come enzima. A questo punto si poteva dedurre l’alterazione genetica responsabile basandosi sulle conoscenze certe delle relazioni intercorrenti tra la sequenza amminoacidica di una catena polipeptidica e la sequenza nucleotidica che la codifica. Lo stesso tipo di procedimento può essere immaginato per i casi nei quali l’attività enzimatica era deficitaria perché le molecole di enzima erano troppo poche, e ciò poteva dipendere da una bassa stabilità della molecola enzimatica. Se alla base del fenotipo di una scarsa attività enzimatica non c’era un’alterazione strutturale della molecola dell’enzima, bensì una compromissione della sua sintesi, allora l’identificazione dell’alterazione genetica che ne era responsabile era risolvibile in modo altrettanto diretto solo in certi casi (nelle mutazioni frameshift), mentre in altri casi la sua soluzione era più difficile, come per le mutazioni che compromettono la trascrizione del gene (mutazioni del suo promotore), oppure la maturazione del suo prodotto, il pre-mRNA a mRNA, e/o la traduzione di quest’ultimo. Lo stesso tipo di processo è stato talora, e solo molto più di recente, compiuto in una sola tappa (fig. 6 B). Il punto di partenza è anche in questo caso il fatto di aver accertato, con metodi classici di g. formale, che il fenotipo in esame ha una base genetica unifattoriale.
Per alcuni fenotipi unifattoriali, tuttavia, la g. biochimica non riesce a individuare la proteina la cui alterazione ha causato il fenotipo in esame, quindi non si può attuare il procedimento graduale a ritroso (dal fenotipo al genotipo) appena descritto. L’esempio più noto di una situazione di questo genere è quello della fibrosi cistica. In molti di questi casi l’identificazione molecolare del gene responsabile è ugualmente riuscita mediante un approccio alternativo. Identificato il gene responsabile, dalla sequenza codificante si è risaliti alla sequenza amminoacidica della catena polipeptidica ricercata e da essa si è potuto spesso dedurre la sua localizzazione cellulare e alcuni aspetti del suo funzionamento. Nel caso paradigmatico della fibrosi cistica si è compreso che la proteina anomala è una proteina di membrana adibita al trasporto di ioni. Questo tipo di procedimento è stato indicato talora come reverse genetics (perché si è arrivati alla proteina deducendone la struttura da quella del suo gene codificante e non procedendo a ritroso come nella genetica classica) e talora come positional cloning (perché si è arrivati al gene ‘clonando la posizione’ del genoma nel quale è situato).
G. medica
Metodiche e strumenti
Il progresso della g. medica è stato fortemente influenzato dallo sviluppo delle tecniche di biologia molecolare. L’analisi sistematica delle malattie umane ha dimostrato che la maggior parte di esse ha una base o una larga componente genetica (malattie poligeniche e multifattoriali). L’identificazione dei geni responsabili delle malattie ha un impatto importante sui programmi dedicati alla loro assistenza e al loro controllo: in primo luogo, sulla diagnosi dei pazienti e delle persone potenzialmente a rischio; in secondo luogo, sulla qualità del trattamento, che viene razionalizzato dalla conoscenza della funzione del prodotto genico mutato nella malattia; infine, e in una dimensione a più lungo termine, sulla terapia genica, la quale fonda i suoi presupposti sulla conoscenza dell’espressione del gene che, se mutato, determina la malattia. Le strategie generali della terapia genica perseguono 4 principali obiettivi: aumentare la dose genica, attraverso l’introduzione di copie aggiuntive del gene normale nelle cellule dei pazienti affetti da malattie causate dalla perdita della funzione di un gene e nelle quali la patogenesi è reversibile; uccidere in maniera mirata specifiche popolazioni cellulari, in particolare nei processi neoplastici; correggere una mutazione; inibire in maniera mirata l’espressione genica (➔ terapia).
Le ricerche di g. molecolare hanno sviluppato efficaci strumenti per la diagnosi di malattie ereditarie. A livello citogenetico, al bandeggio, che ha permesso di identificare i cromosomi e ha definito una nuova categoria di sindromi cromosomiche, sono subentrate l’alta risoluzione cromosomica e la citogenetica molecolare. La comprensione delle basi biologiche delle malattie ha ricevuto uno straordinario impulso dallo sviluppo di strategie che consentono di localizzare sul cromosoma (mappare) e di isolare (clonare) i geni di interesse.
Malattie ereditarie
Queste analisi molecolari hanno rivelato la grande complessità delle malattie ereditarie, che possono essere distinte in malattie mendeliane monogeniche e poligeniche o multifattoriali. Nelle malattie monogeniche lo stesso gene può essere mutato in domini funzionali diversi, e quindi causare quadri clinici distinti. Conoscere le diverse mutazioni dello stesso gene (eterogeneità allelica) migliora la comprensione delle correlazioni tra il genotipo e il fenotipo. Un fenomeno relativamente comune è inoltre quello dell’eterogeneità genetica, che definisce la presenza di mutazioni in geni diversi, associate a fenotipi simili (➔ eterogeneità). È illustrativo l’esempio della retinite pigmentosa, una malattia degenerativa della retina che porta progressivamente alla cecità. Nel caso delle malattie multifattoriali il fenotipo è causato dall’interazione tra geni localizzati su cromosomi diversi, che interagiscono con un ambiente sfavorevole. Sono esemplificativi gli oltre 20 geni della suscettibilità al diabete mellito di tipo 1, o i numerosi polimorfismi nei geni che conferiscono circa il 50% della suscettibilità verso l’ictus o l’infarto giovanile. Sebbene l’analisi molecolare renda possibile decifrare molte malattie a livello delle loro basi biologiche, il rapporto che intercorre tra il progresso scientifico e il suo impatto a livello clinico spesso non è diretto e immediato.
La neurofibromatosi di tipo 1 esemplifica una malattia a elevata variabilità fenotipica, della quale è stato clonato da tempo il gene (nf1). Si tratta di un gene a organizzazione complessa, in quanto contiene al suo interno altri geni, e che si estende per una regione genomica di circa 350 kb. Mentre nei casi familiari (almeno 2 soggetti affetti) è possibile seguire la segregazione della sequenza mutata, utilizzando polimorfismi intragenici, nei casi sporadici le grandi dimensioni del gene e la variabilità delle mutazioni rendono poco probabile la possibilità di caratterizzare la mutazione. Questo si riflette, in pratica, nell’impossibilità di monitorizzare con la diagnosi prenatale una parte delle gravidanze a rischio. In altri casi mappare e clonare il gene che, se mutato, determina una malattia ereditaria può incidere significativamente sulla malattia. Per es., i pazienti affetti da rene policistico di tipo adulto sviluppano cisti renali in media nel 22% dei casi già all’età di 10 anni, nel 68% dei casi a 20 anni, nell’86% a 30 anni; a 40 anni presentano i primi sintomi di insufficienza renale, a 50 anni necessitano di trattamento dialitico e attorno ai 55 anni richiedono il trapianto renale. Le analisi molecolari consentono di identificare in tutte le famiglie le persone a rischio in epoca presintomatica e addirittura nella vita fetale.
Consulenza genetica
Il controllo più efficace delle malattie ereditarie resta affidato alla prevenzione, che ha i suoi fondamenti nella diagnosi. Questo obiettivo viene raggiunto attraverso la consulenza genetica, un complesso servizio medico rivolto ai pazienti e alle famiglie in cui è presente una patologia che può essere o non essere ereditaria, per la quale si tentano di definire l’origine e l’eventuale ricorrenza familiare, e nei cui confronti vengono avviati interventi di prevenzione volti a migliorare la qualità di vita dei pazienti (➔ dismorfologia).
Medicina predittiva e genetica
La possibilità di intervenire predittivamente sulle malattie costituisce uno degli aspetti più rivoluzionari della moderna g. medica, che ha la sua espressione più evidente nelle cosiddette malattie da mutazioni dinamiche. Si tratta di affezioni, prevalentemente di interesse neurologico, nelle quali la malattia origina dall’espansione di una sequenza di 3 lettere (triplette), normalmente presenti in un basso numero di ripetizioni. È dimostrata una correlazione tra le dimensioni dell’espansione, l’età di esordio e l’espressività della malattia, che spiega l’anticipazione della malattia stessa, cioè la sua comparsa in età sempre più precoce con il passare delle generazioni. In conseguenza di questo meccanismo, una malattia tipicamente dell’adulto può presentarsi anche in forma congenita nella stessa famiglia.
Test e screening genetici
Poiché il rapporto tra sequenze nucleotidiche e manifestazioni cliniche non ha un margine di prevedibilità controllabile, è necessario che le tecniche molecolari entrino nel bagaglio della medicina moderna, ma non prendano il sopravvento sulla clinica. Questo rischio è potenziale, in considerazione della tipologia dei test genetici, che li classifica come: diagnostici, che pongono o confermano la diagnosi di una malattia genetica; predittivi, che identificano un fattore di rischio per lo sviluppo di una malattia, quando non venga controllata la predisposizione (per es., fenilchetonuria ed effetto della dieta a basso contenuto di fenilalanina); prognostici, che prevedono l’insorgenza futura di una malattia genetica (per es., corea o malattia di Huntington); che conferiscono suscettibilità, ovvero stabiliscono la vulnerabilità a un fattore ambientale (per es., deficit di α-1-antitripsina ed enfisema polmonare); che conferiscono resistenza, cioè proteggono dallo sviluppo di certe malattie, in presenza di un gene mutato; probabilistici, che individuano un genotipo a rischio con una probabilità significativamente maggiore rispetto a quella della popolazione; di profilo genetico, che identificano un’associazione empirica tra una mutazione e l’aumento dell’incidenza della malattia. Queste ampie possibilità diagnostiche e la documentata presenza di mutazioni nel genoma di ogni persona, nella quale queste ultime costituiscono potenziali fattori di malattia o di suscettibilità, propongono l’avvio di screening genetici finalizzati all’identificazione delle persone a rischio. I programmi di questo tipo, che si rivolgono a varie categorie di utenti, come i neonati o gli adulti in età riproduttiva, possono in teoria avere importanti ripercussioni sia sull’individuo sia sull’intera società. I test genetici che si rivolgono ad ampie fasce di popolazione devono inoltre tenere conto degli aspetti tecnici, dell’efficienza, dell’accuratezza, dei costi e delle priorità. L’aspetto tecnico appare quello più facilmente risolvibile, come sembra suggerire l’immissione sul mercato dei cosiddetti MASDA (multiple allele-specific diagnostic assay), che consentono di analizzare contemporaneamente i campioni di 500 persone per 106 mutazioni in 7 geni-malattia. La tecnica dei chip a DNA (➔ chip) è in grado di fissare 16.000 nucleotidi su un’area di 1,5 cm2. Appositi lettori ottici interpretano i risultati dell’analisi, che può riguardare interi tratti di genoma ed essere applicata a bassi costi ad ampie fasce di popolazione.
Questioni bioetiche
La manipolazione genetica sull’uomo – termine con il quale si designa qualunque forma di modificazione indotta nel materiale genetico con tecniche molecolari – comporta alcuni problemi etici, per comprendere i quali occorre fare riferimento ai vari livelli di intervento sui geni e alle finalità di tali interventi. Per quanto riguarda i livelli di intervento, le modificazioni genetiche possono avvenire sulle cellule somatiche, sulle cellule germinali o sugli embrioni. Nel primo caso, si tratta di interventi particolarmente promettenti nell’ambito della ricerca terapeutica, volti a modificare degenerazioni o difetti cellulari, nell’ambito dei quali l’eventuale dispersione di cellule non comporta danni per l’individuo, non ponendo, pertanto, particolari problemi di natura etica. Le modifiche sulle cellule germinali restano di fatto escluse per l’attuale impossibilità di guidare l’inserimento dei geni corretti. In ogni caso, potrebbero presentare rischi per le generazioni future, non potendosi prevedere le conseguenze di tali interventi, soprattutto sulla prole. Gli interventi sull’embrione umano presentano l’alto rischio di compromettere la vita dell’embrione e il suo avvenire genetico. Laddove si trattasse di mera sperimentazione scientifica, dunque, non terapeutica per il singolo soggetto sul quale si interviene, si solleverebbero gravi problemi di illiceità, a prescindere dalle finalità perseguite (alterazione e/o miglioramento del patrimonio genetico e delle possibilità future della scienza). La preventiva programmazione dell’utilizzo e della selezione degli embrioni a scopi meramente sperimentali costituisce, infatti, uno sfruttamento discriminatorio di vite umane. Le finalità d’impiego di alcuni interventi genetici possono determinarne la liceità o illiceità etica: finalità diagnostiche, terapeutiche, produttive, alterative, sperimentali (distruttive).
In generale, tuttavia, i principi dai quali non si può prescindere nella valutazione etica degli interventi genetici sull’uomo sono: a) salvaguardia della vita e dell’identità genetica di ogni individuo umano; b) principio terapeutico, ossia di intangibilità del patrimonio genetico dell’individuo, a meno che l’intervento sia benefico in riferimento al soggetto sul quale si interviene per correggere un difetto o una malattia non altrimenti curabile, e non in previsione di sacrificare qualcuno per portare vantaggio ad altri; c) salvaguardia dell’ecosistema e dell’ambiente; d) competenza e responsabilità della comunità scientifica.
Agraria
Il miglioramento delle razze delle piante e degli animali utili all’uomo è stato per il passato il prodotto della selezione, che ha favorito la scoperta e l’utilizzazione di molte mutazioni spontanee, ulteriormente perfezionata, con il contributo della citogenetica, sfruttando anche mutazioni sperimentali. La g. agraria ha contribuito, insieme con la migliore tecnica colturale, al miglioramento qualitativo e quantitativo della produzione nei diversi settori.