
genètiche, malattìe

genètiche, malattìe Patologie ereditarie, dovute ad anomalie cromosomiche o a mutazioni di singoli geni (malattie da deficit di enzimi; errori congeniti del metabolismo). La frequenza delle m.g. nella popolazione mondiale dei neonati è di ca. il 3%. Esse incidono significativamente sulla frequenza delle morti neonatali e perinatali e sono responsabili di oltre il 20% dei ricoveri pediatrici nel mondo. Nonostante lo sviluppo di sofisticate strategie, che perseguono l'obiettivo del trattamento delle m.g., l'intervento più realistico ed efficace nel loro controllo resta tuttora affidato alla prevenzione, attraverso la diagnosi prenatale.
Abstract di approfondimento da Genetica medica di Antonio Pizzuti (Enciclopedia della Scienza e della Tecnica)
Malattie genetiche
Se consideriamo le cause di morbilità nella popolazione generale, più del 75% degli individui sarà nella propria vita colpito da patologie con importante componente genetica, il 5% prima dei 24 anni. Di queste patologie lo 0,4% saranno legate a disordini cromosomici, il 2% a patologie monogeniche, mentre le malattie cosiddette ‘complesse’ rappresenteranno il gruppo di gran lunga più importante.
Eredità mendeliana
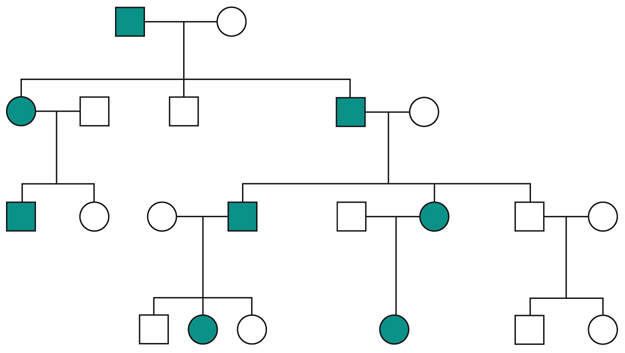
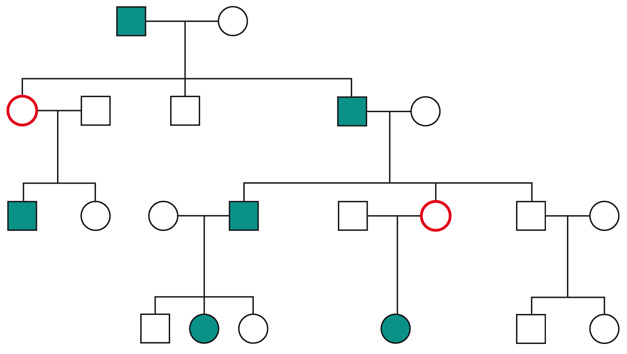
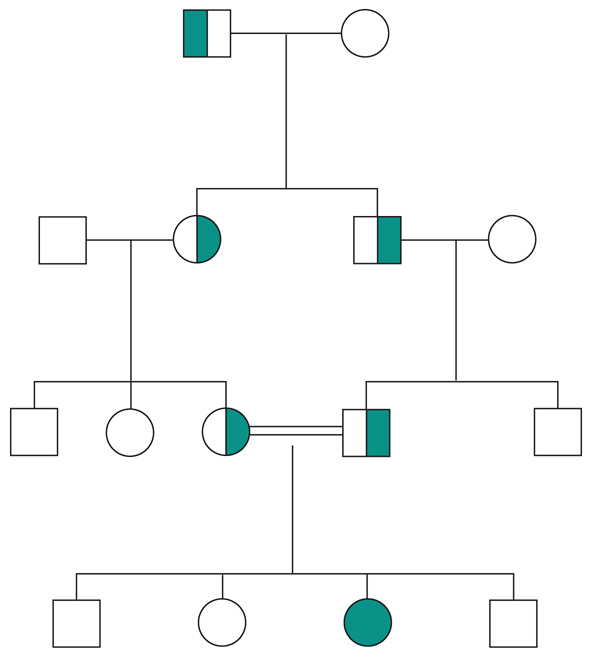
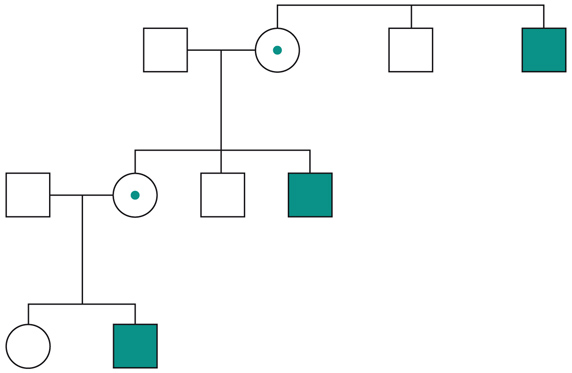
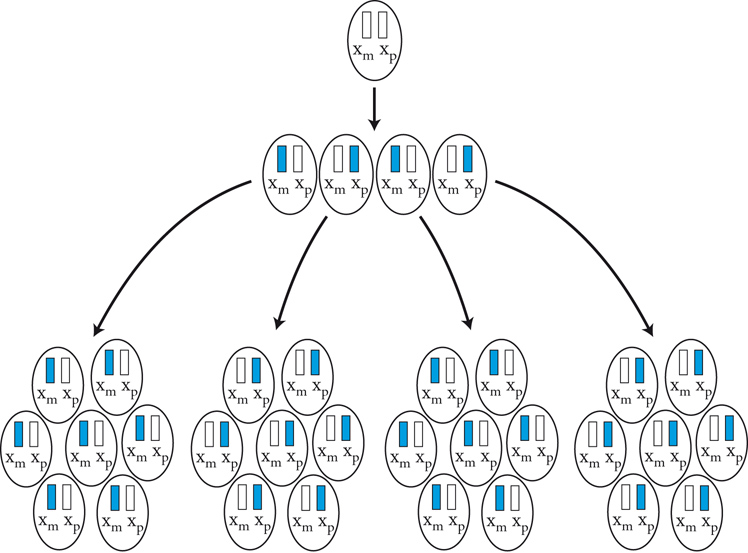
Le malattie causate in parte preponderante dalla mutazione di un solo gene, dette ‘malattie mendeliane’, vengono distinte in patologie autosomiche e patologie legate ai cromosomi del sesso a seconda che la posizione (locus) dei geni, le cui mutazioni sono responsabili della malattia, sia su un autosoma o su un cromosoma del sesso (in particolare il cromosoma X). A loro volta queste patologie vengono distinte in malattie recessive e malattie dominanti. In realtà la classica definizione delle patologie monogeniche, come patologie nelle quali tutti i fenomeni clinici sarebbero secondari alla mutazione di un singolo gene, è solamente una semplificazione. Esistono fattori genetici e non genetici in grado di variare gli effetti patologici delle singole mutazioni (geni e fattori modificatori), cosicché anche in una stessa famiglia, nella quale venga trasmessa la stessa mutazione genetica, i diversi portatori del difetto genetico possono mostrare caratteristiche cliniche differenti.Ogni gene posizionato sugli autosomi (geni autosomici) esiste nel genoma umano in duplice copia, una copia di origine materna e una di origine paterna. Le due copie del gene (alleli) possono essere uguali (e l’individuo è detto omozigote per quel gene) o differenti (eterozigote). La distribuzione degli alleli a un singolo locus è detta genotipo, ed è il substrato molecolare per la determinazione delle caratteristiche individuali (fenotipo). I principî di dominanza determinano il modo nel quale il rapporto tra i due alleli a un singolo locus determina il fenotipo. Un allele è detto dominante (A) su un altro recessivo (a), quando il fenotipo determinato dal genotipo omozigote AA e da quello eterozigote Aa sono identici. Nelle condizioni autosomiche dominanti, un individuo è affetto se è portatore di almeno uno dei due alleli della coppia genica mutato (eterozigote). Nelle singole famiglie, la malattia colpisce entrambi i sessi in proporzioni simili e può essere trasmessa alle generazioni successive da genitori di ambedue i sessi (fig.). Un paziente ha generalmente un genitore affetto e il 50% della prole rischia di essere affetta. Una patologia autosomico-dominante può tuttavia colpire un individuo i cui genitori non sono affetti. Ciò può avvenire più frequentemente per la comparsa di una nuova mutazione durante la formazione dei gameti in uno dei genitori, oppure perché un genitore che costitutivamente non reca la mutazione ha alcune delle cellule della linea germinale mutate (mosaico germinale). È anche possibile che uno dei genitori sia portatore della mutazione ma non esprima il fenotipo patologico, un fenomeno che è detto ‘difetto di penetranza’ della mutazione (fig.). Quando una mutazione è completamente penetrante, tutti i portatori della mutazione saranno affetti. Altre volte la patologia ha un’espressività variabile e alcuni individui esprimono il difetto genetico solo a livelli subclinici. Espressività variabile e difetto di penetranza sono i tipici fenomeni determinati da modificatori genetici o ambientali, e indicano come anche le malattie che generalmente sono considerate monogeniche sono in realtà il risultato dell’azione di diverse componenti, stante un contributo importante della singola mutazione genetica. Mentre alcune forme cliniche mendeliane sono invariabilmente dovute a mutazioni di un gene singolo, altre mostrano il fenomeno della eterogeneità genetica (di locus), dove la stessa malattia può essere causata da singole mutazioni ma in geni differenti. Per esempio, più di 40 geni possono essere responsabili del fenotipo clinico della retinite pigmentosa. Questo fenomeno, oltre a essere di impaccio nella scelta del test genetico a cui sottoporre i pazienti con queste forme patologiche, sta rivoluzionando la nosologia di molte specialità mediche. Quando mutazioni differenti dello stesso gene danno origine a quadri clinici simili si parla di eterogeneità allelica. Questo fenomeno è conosciuto da tempo, e alcuni geni possono mutare in migliaia di modi differenti, e anche questa è una difficoltà quando si prende in considerazione la possibilità di un test genetico. Il fenomeno opposto a quello dell’eterogeneità genetica di locus è quello della serie allelica. Qui mutazioni differenti dello stesso gene danno luogo a malattie diverse o perché regioni differenti dello stesso gene hanno distinte specificità funzionali, oppure perché il gene è espresso in diversi tessuti. Nonostante la diversità tra i Mammiferi e altre specie animali, come Vermi o Insetti, il numero di geni nei vari genomi non è molto differente. Il genoma umano contiene solo il doppio dei geni di un semplice verme. Tuttavia negli animali più complessi ogni gene è in grado di codificare per più prodotti (per di più proteici). Ovvero, i genomi evolvono non solo moltiplicando e cambiando i geni presenti, ma anche utilizzandoli in modo alternativo. Nelle patologie a trasmissione autosomica recessiva gli individui sono affetti quando sono omozigoti per alleli mutati e sono generalmente figli di individui sani eterozigoti portatori di una mutazione genica. Anche in questo caso la patologia colpisce in modo identico i due sessi, e il 25% dei figli di due portatori sarà malato, il 50% sarà portatore sano e un altro 25% sarà di sani e non portatori. Quando nella popolazione generale lo stato di portatore è raro, l’occorrenza di un individuo malato per una malattia recessiva autosomica deve sempre fare considerare la possibilità che i genitori siano consanguinei. Maggiore è il grado di consanguineità, maggiore la condivisione del genoma e quindi la possibilità di generare figli omozigoti per mutazioni per i quali i genitori siano portatori sani, mutazioni che hanno ereditato da un avo comune (fig.). Questo è il motivo per il quale il rischio di patologia malformativa e di ritardo mentale è superiore nei matrimoni tra consanguinei, rispetto al rischio generale.Altre patologie monogeniche sono dovute a mutazioni di geni presenti sul cromosoma X (patologie legate alla X, fig.). Questi geni sono presenti in duplice copia nelle femmine (che recano nelle loro cellule due cromosomi X, uno di origine paterna e uno materna) e in singola copia nei maschi emizigoti, che hanno un solo cromosoma X di origine materna e un piccolo cromosoma Y di origine paterna (che determina il sesso maschile, tramite l’azione di un piccolo gene, SRY, che è il determinante umano del testicolo). Gli individui affetti da malattie recessive legate alla X sono quasi esclusivamente maschi, con la mutazione del gene sul cromosoma X ereditato da una madre eterozigote portatrice sana oppure il risultato di un evento de novo durante la gametogenesi materna. Una portatrice avrà un rischio del 50% di avere figli maschi affetti, e del 50% di avere figlie femmine portatrici. Se un padre è affetto, tutte le figlie femmine saranno portatrici e tutti i figli maschi saranno sani, dato che un figlio maschio eredita dal padre affetto il cromosoma Y e non quello X.Lo sbilanciamento genico tra i due sessi per quanto riguarda il cromosoma X (due X nella femmina e una nel maschio) viene aggiustato da un’inattivazione quasi completa di uno dei due cromosomi X nella femmina (inattivazione del cromosoma X). L’inattivazione funzionale di un cromosoma X nella femmina è casuale, in modo tale che in un individuo femmina alcune cellule inattivano la X di origine paterna e altre quella di origine materna (fig.). L’inattivazione è legata a un cambiamento chimico del DNA e delle proteine associate, ed è in qualche modo provocata dall’azione di un gene (Xist), che è attivo sul cromosoma X inattivo e inattivo su quello attivo.
Eredità mitocondriale
Non tutti i geni nell’uomo sono presenti nel nucleo delle cellule. Un organulo cellulare, il mitocondrio, possiede un proprio piccolo genoma. I mitocondri originano probabilmente come batteri a metabolismo ossidativo endosimbionti di cellule eucariotiche primordiali, inglobati dagli eucarioti quando l’atmosfera del pianeta, in origine riducente, divenne ossidante a causa dell’accumulo di ossigeno. Questo rimasuglio del genoma originario del batterio inglobato nelle cellule contiene 37 geni, che hanno a che fare con il metabolismo respiratorio e con le funzioni di replicazione del DNA, trascrizione e traduzione all’interno del mitocondrio. Mutazioni a carico dei geni presenti nel genoma mitocondriale sono responsabili di alcune patologie umane. L’eredità di patologie dovute a mutazioni del genoma mitocondriale hanno alcune caratteristiche particolari. Un individuo eredita i mitocondri quasi esclusivamente dalla madre. Pertanto la trasmissione di queste patologie avviene sempre in modo matrilineare e i maschi affetti non trasmetteranno la malattia alla prole. Eredità multifattoriale. La gran parte delle malattie genetiche ha modelli di trasmissione più complessi (caratteri multifattoriali) ed è determinata dall’interazione di più fattori genetici e di fattori ambientali. In queste patologie viene spesso riscontrata una certa aggregazione familiare, che tuttavia non può essere spiegata dai modelli mendeliani. Molte di queste patologie hanno una predilezione di sesso (per es., il sesso femminile nell’artrite reumatoide) e i rischi di ricorrenza in una famiglia decrescono in modo deciso con il diminuire del grado di parentela. Gran parte della ricerca genetica molecolare si sta occupando dello studio dei geni responsabili dell’aumentata o diminuita suscettibilità a queste patologie, tramite l’analisi della distribuzione nelle popolazioni di varianti genetiche comuni e la loro possibile associazione alle patologie complesse. Accanto ai classici modelli di patologie monogeniche e patologie multifattoriali stanno emergendo altre modalità di trasmissione ereditaria come quella digenica o trigenica, dove due o tre mutazioni in geni differenti sono necessarie per determinare un effetto patologico.
La consulenza genetica e i test genetici
Nell’attività del genetista medico il momento fondamentale è la consulenza genetica. La consulenza genetica è il processo per il quale pazienti o parenti, a rischio per una malattia che possa essere ereditaria, sono informati sulle conseguenze del disturbo, la probabilità di svilupparlo o trasmetterlo e sui mezzi per prevenirlo, evitarlo o migliorare la condizione clinica. Obiettivo della consulenza genetica è quello di aiutare il consultante, la coppia o la famiglia a comprendere le informazioni mediche, inclusa la diagnosi, la prognosi e le terapie disponibili; rendersi conto del contributo ereditario alla malattia e del rischio di ricorrenza; prendere le decisioni che sembrano appropriate in rapporto al rischio di ricorrenza, ai progetti familiari, agli standard etici e religiosi e agire in accordo con queste decisioni; ottenere il miglior possibile adattamento alla malattia in un soggetto affetto o a rischio di ricorrenza. Durante la consulenza il genetista medico traccia un albero genealogico della famiglia (pedigree), pone una diagnosi clinica, stima i rischi di occorrenza e di ricorrenza della patologia, informa sulle caratteristiche clinico-genetiche della stessa e consiglia eventuali test genetici, atti a verificare o scartare l’ipotesi clinica di patologie ereditarie.Il test genetico è detto ‘test diagnostico’ quando viene fatto su persone che hanno o possono avere un particolare disordine. Un test è presintomatico, se la sua positività in un individuo sano indica il futuro sviluppo quasi inevitabile della malattia. Un terzo tipo di test è quello predittivo, che copre un ampio range di situazioni, il risultato del quale determina un aumentato o ridotto rischio di una malattia, ma con un grado di certezza non completo. I test genetici possono essere eseguiti prima della nascita (test prenatali) su tessuto o materiale fetale, come i villi coriali, le cellule del liquido amniotico, il liquido stesso, o il sangue, o dopo la nascita (in generale sul sangue o su altri liquidi organici). Vi sono test genetici che vengono eseguiti su particolari gruppi di individui (screening genetici). Un test di screening può essere applicato a tutta la popolazione (per es., i test neonatali), a sottopopolazioni particolari (per es., lo screening per la malattia di Tay-Sachs negli ebrei aschenaziti, dove ha un’incidenza notevolmente superiore alle altre etnie), o come test a cascata sui parenti di un paziente affetto da una patologia genetica, che fossero essi stessi a rischio di sviluppare o trasmettere la malattia. Test particolarmente utili sono quelli di citogenetica.