
genoma
In biologia, il corredo aploide dei cromosomi di una cellula, con i geni in essa contenuti.
Il Progetto genoma
Nel 1990 negli Stati Uniti è iniziato ufficialmente il progetto di ricerca internazionale denominato Progetto genoma umano o HGP (Human genome project), coordinato dal Dipartimento per l’energia (DOE, Department Of Energy) e dal Centro nazionale di ricerca sul g. umano, che fa parte dei National Institutes of Health (NIH). Gli scienziati che hanno fatto parte del progetto si sono posti l’obiettivo di conoscere la sequenza dei geni della specie umana e di posizionarli sui vari cromosomi, costruendo così una mappa del genoma. L’annuncio del sequenziamento del g. umano a opera di una società privata, la Celera Genomics, è stato dato nel 2000 e i risultati raggiunti sia da questa società sia dal consorzio internazionale pubblico (International human genome sequencing consortium) sotto l’egida dei NIH sono stati pubblicati nel 2001. Insieme al sequenziamento del g. umano sono iniziati progetti di mappatura e sequenziamento di g. più piccoli di organismi diversi, che si sono rivelati essenziali per raggiungere l’obiettivo finale. Tutti questi progetti hanno portato al potenziamento di tecnologie quali la costruzione di mappe genetiche e fisiche ad alta risoluzione, alla ideazione di programmi per la gestione informatizzata della grande quantità di dati, alla creazione di apparecchi ad alte prestazioni per rendere più veloci, in termini di costi e di efficienza, le tecniche necessarie, quali, per es., la preparazione dei cloni, l’elettroforesi e il sequenziamento del DNA (➔ biotecnologie).

La storia del sequenziamento del DNA ebbe inizio quando, nel 1977, F. Sanger pubblicò il metodo per determinare l’ordine dei nucleotidi del DNA; nello stesso anno fu isolato e sequenziato il primo gene umano. Nel corso dei primi anni 1980 è stato anche messo a punto un metodo di clonazione non selettivo, chiamato shotgunning in quanto con esso si producevano frammenti di DNA a caso come se fossero colpiti dalla rosa di proiettili che si determina sparando con un fucile. Questo è rimasto il metodo di elezione per l’analisi del g. su larga scala. Il metodo è stato notevolmente esteso nei vent’anni successivi utilizzando vettori di clonazione nei quali si potevano inserire frammenti di DNA sempre più grandi (➔plasmide; YAC; BAC). In fig. 1 è rappresentato il metodo utilizzato per sequenziare grandi g. seguendo un ordine gerarchico di frammentazione: grandi frammenti di DNA genomico vengono clonati in BAC e successivamente assemblati nello stesso ordine nel quale erano nel g. integro. Ciascun frammento è suddiviso in tratti più piccoli che sono sequenziati avendo cura, nei successivi processi di assemblaggio, di non lasciare intervalli di sequenze ignote fra un segmento e l’altro. È inoltre apparso chiaro che le sequenze di cDNA (complementary DNA), che rappresentano le sequenze di geni trascritti nel g., sono essenziali per studiare i geni umani; è stato pertanto intrapreso dal TIGR (The Institute for Genomic Research, nel Maryland) un progetto di sequenziamento di cloni di genoteche di cDNA derivate da quasi tutti i tessuti e gli organi umani. Si è potuto così creare un catalogo di brevi sequenze corrispondenti agli RNAm provenienti da cloni di cDNA. Queste sequenze sono denominate EST (expressed sequence tag) e si possono utilizzare per stabilire l’estensione e la diversità dei geni espressi e lo schema dell’attività di trascrizione di ciascuno di essi.
Nel 1995 è stato sequenziato il batterio Haemophilus influenzae di 1,8 megabasi (Mb); alla fine del 1998 si sono rese disponibili le sequenze complete di 18 g. batterici in banche dati pubbliche. I g. batterici hanno una lunghezza che varia da 580.070 coppie di basi (bp) di Mycoplasma genitalium fino a 4.411.529 bp di Mycobacterium tuberculosis. Nel 1997 ha suscitato grande entusiasmo nel mondo scientifico la pubblicazione della sequenza completa (4.639.221 bp) di Escherichia coli. La sequenza del g. del primo organismo eucariotico, il lievito Saccharomyces cerevisiae (12.068 kilobasi, o kb) venne completata nel 1996. Nel 2000 è stato sequenziato il primo g. eucariotico complesso, quello di Drosophila melanogaster, di 120 Mb. Sono stati anche sequenziati i g. del nematode Caenorhabditis elegans (97 Mb), della pianta Arabidopsis thaliana (100 Mb) e del topo Mus musculus (3000 Mb).
Numero dei geni
Gli studi sul g. umano hanno individuato un numero di geni minore rispetto a quanto si potesse prevedere (circa 30.000). Il numero dei geni è solo uno degli elementi, e forse non il più decisivo, che serve a misurare la complessità di un organismo. Importantissimo è invece il modo con cui i geni determinano la loro influenza sull’organismo mediante la funzione delle proteine costruite con le loro istruzioni: i geni sono formati da segmenti distinti e la proteina viene codificata solo da una parte di questi segmenti (➔ esone; introne); un gene spesso determina la formazione di più di una proteina, così come più geni possono concorrere alla formazione di una sola proteina funzionante. Una volta sintetizzate, le proteine possono essere modificate in vari modi, e non è noto quanto questo sia frequente in rapporto all’aumento della complessità degli organismi. Per codificare le proteine, i geni devono ricevere segnali da altri geni e tutti sono funzionalmente connessi in una rete di informazioni. In questa rete ogni gene può avere ruoli diversi, sia perché influenza altri gruppi di geni in modo vario a seconda delle circostanze, sia perché le proteine da esso codificate possono avere funzioni diverse in cellule appartenenti a tessuti diversi. La complessità funzionale del g. dipende quindi soprattutto dal numero, per lo più ignoto, delle proteine e dalla loro funzione (➔ proteomica).
Struttura del genoma
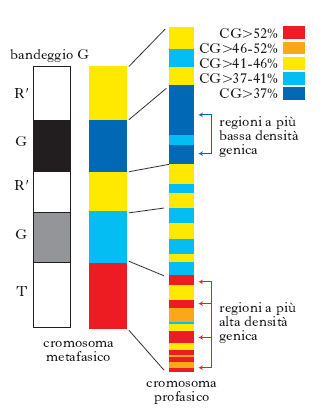
Gli studi sul bandeggio cromosomico hanno rivelato che circa il 17-20% del complemento cromosomico consiste di bande C, ossia di eterocromatina costitutiva. La maggior parte dell’eterocromatina è polimorfa e consiste di differenti famiglie di DNA (detto alfa-satellite), con sequenze ripetute di vario tipo localizzate per lo più nelle regioni del centromero dei cromosomi. Il rimanente 80% del g., la parte cioè che è stato possibile sequenziare con le tecniche a disposizione, è costituita dalla componente eucromatica identificata, nel bandeggio cromosomico, come bande G, R e T. Queste bande, osservabili a livello citogenetico, differiscono in composizione e densità di geni: la densità maggiore è presente nelle zone ad alto contenuto di sequenze CG (citosina-guanina), anche se il sequenziamento del g. ha messo in evidenza che la correlazione fra contenuto in CG e densità dei geni non è così determinante come si pensava (fig. 2). La più alta concentrazione di geni si osserva nei cromosomi 17, 19 e 22, la più bassa nei cromosomi 4, 18, 13 e X. Riassumendo, il g. eucariotico è costituito essenzialmente da due tipi di sequenze di DNA: classi di DNA a sequenze non ripetute o scarsamente ripetute e classi di DNA ripetute. Alla prima categoria appartengono i geni codificanti proteine, che costituiscono una piccola parte del g. e che possono essere singoli e localizzati in tutto il g. o raggruppati in famiglie geniche. Parecchi tipi di proteine sono codificati da famiglie di geni omologhi, che comprendono un numero variabile di geni: la famiglia delle globine ha 5 geni, quella delle actine comprende da 5 a 30 geni, quella degli istoni da 100 a 1000 geni. Esistono anche sequenze ripetute corte e sparse in tutto il g. che costituiscono i minisatelliti (➔ impronta; VNTR), i microsatelliti (➔) e gli elementi trasponibili di vario tipo (➔ trasposone). Un tipo di elementi trasponibili è costituito dai retrotrasposoni, sequenze che sono state disperse in tutto il g. dopo trascrizione inversa dell’RNA, quali le sequenze Alu. Le ripetizioni corte (circa 200 nucleotidi) disperse in varie zone del g., come le sequenze Alu, sono chiamate collettivamente SINE (short interspersed elements). Gli elementi sparsi lunghi, chiamati LINE (long interspersed elements), sono anch’essi un esempio di elementi trasponibili nei Mammiferi. Sono sequenze omologhe ai retrovirus, lunghe 1000 o 2000 bp e presenti in 20.000-40.000 copie per g., nelle quali sono presenti anche sequenze che vengono tradotte. I processi che regolano l’espressione dei geni sono estremamente complessi e non ancora del tutto chiariti. Molto importanti sono la struttura della cromatina (➔ epigenesi) e anche la posizione relativa dei geni e degli altri segmenti di DNA del genoma.
La genomica
La genomica è la branca della genetica che studia la caratterizzazione molecolare e l’espressione di interi g., specie per specie. Il termine, coniato alla fine degli anni 1990, definisce nuove branche della conoscenza genetica nate dal progetto di sequenziamento del g. umano e di altri organismi rappresentativi (batteri, lieviti e Drosophila melanogaster). Genomica strutturale Studio tendente all’identificazione dell’intero insieme dei geni in un genoma. Genomica funzionale Studio delle diverse modalità di espressione dei geni e delle loro interazioni nel g. considerato come un’unica entità. Malgrado si conosca la sequenza delle basi del DNA sia per l’uomo sia per molti altri organismi, rimane ancora una notevole incertezza circa il numero dei geni presenti nei vari g. analizzati.
Tra le cause per le quali la sequenza di un gene non è sufficiente per identificarne con certezza la funzione, ricordiamo le modalità con cui si può identificare un gene. Frequentemente viene usato il prodotto della trascrizione, l’RNAm; da questa molecola si produce successivamente il cDNA. Dato che l’RNAm e quindi il cDNA non presentano gli introni che sono stati eliminati durante lo splicing, non si può conoscere con questo metodo la completa struttura del gene. Inoltre, dato che non tutti i geni sono espressi allo stesso livello, vi sono RNAm più abbondanti di altri e le specie meno rappresentate rischiano di non essere presenti tra i cDNA prodotti. L’identificazione di un gene a partire dal cDNA, pur tenendo conto dei limiti descritti, ha dato notevoli risultati e sono state messe a punto tecniche sempre più raffinate che usano cDNA per il sequenziamento del g. umano. Un’altra condizione per attribuire la valenza di gene a una sequenza di DNA è che non possieda all’interno di una sequenza di almeno 300 basi una tripletta di termine (➔ codice). Questa sequenza, detta ORF (open reading frame), non viene tuttavia considerata un vero e proprio gene sino a quando non si riesca a identificare una mutazione che determini una variazione osservabile nel fenotipo, oppure non si riesca a isolare una molecola di RNAm da essa trascritta. Questo approccio permette di conoscere la completa sequenza di un gene, introni compresi. Se, avendo una nuova sequenza da analizzare, si confrontano poi le numerose ORF di diverse specie ormai disponibili nelle banche dati e risulta una elevata omologia con una di esse, è ragionevole pensare che la nuova sequenza e quella già nota svolgano la stessa funzione. I limiti della genomica strutturale possono venire superati dalla genomica funzionale e dalla più recente branca di questa, la proteomica. Alla fine degli anni 1990 è stata messa a punto una nuova tecnica, detta DNA-array o chip a DNA (➔ chip), attraverso la quale è possibile accertare se una sequenza di DNA viene espressa e l’ordine temporale della sua espressione.