
retina
Membrana nervosa che costituisce la tunica interna dell’occhio e si estende dal nervo ottico all’orifizio pupillare.
Anatomia
Anatomia umana. Nella r. si distinguono tre porzioni: una posteriore (porzione coroidea della r. o parte ottica della r. o solo r.), una media (porzione ciliare della r. o r. ciliare), e un segmento anteriore (porzione iridea della r. o r. iridea). Le porzioni media e anteriore non sono in grado di percepire stimoli visivi mentre la porzione posteriore è completamente sviluppata e funzionalmente efficiente.
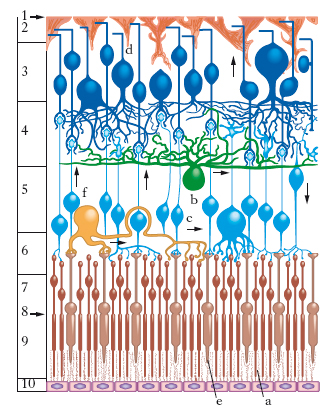

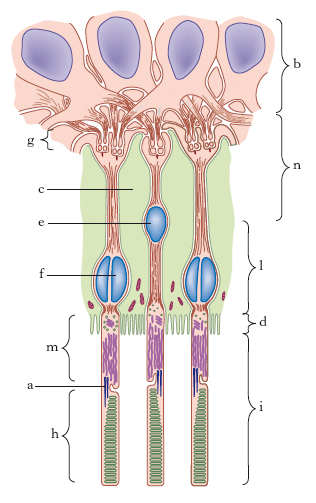
La r. propriamente detta presenta due speciali regioni: la papilla ottica e la macula lutea. La prima corrisponde al punto in cui il nervo ottico si espande a ventaglio continuandosi con la r. stessa; la macula lutea occupa invece il polo posteriore dell’occhio, si presenta come una macchia ovale e mostra nella sua parte centrale una depressione, detta fovea centralis. Dal punto di vista istologico la r. si compone essenzialmente, come il neurasse da cui deriva, di due ordini di elementi: elementi nervosi ed elementi di sostegno, comprendenti questi ultimi le cellule di nevroglia. Tali differenti elementi costitutivi si dividono in 10 strati sovrapposti (fig. 1). Fra i vari strati quello dei coni e dei bastoncelli è il più importante in quanto questi elementi rappresentano l’estremità terminale dell’elemento nervoso. Essi non sono distribuiti uniformemente sulla retina. Il numero dei coni diminuisce dal centro alla periferia, mentre quello dei bastoncelli ha comportamento inverso. Sicché, mentre alla fovea si trovano quasi esclusivamente dei coni, questi sono rarissimi nelle parti periferiche. Coni e bastoncelli mancano completamente in corrispondenza della papilla (fig. 2), per cui questa zona retinica (macchia cieca, o punto cieco, di Mariotte) si presenta non adatta alle percezioni luminose.
Le arterie della r. provengono dall’arteria centrale del nervo ottico, branca terminale dell’arteria oftalmica. Le vene seguono in senso inverso il decorso delle arterie sboccando nella vena centrale del nervo ottico.
Anatomia comparata. Le cellule visive, di cui i coni e i bastoncelli rappresentano la parte differenziata, si osservano in tutte le classi di Vertebrati; hanno uno sviluppo notevole nei Pesci, mostrano un prevalere dei coni nei Rettili e negli Uccelli, dei bastoncelli negli Anfibi e nei Mammiferi (fig. 3). Nella r. di tutti i Vertebrati, in corrispondenza dell’asse ottico dell’occhio, si distingue un’area dove le cellule visive si presentano più assottigliate e più stipate; di conseguenza anche le cellule bipolari e quelle gangliari sono assai più numerose: è la regione dell’area centrale, che ha più spiccate capacità percettive. Nell’Uomo e in alcuni Primati a quest’area corrisponde la fovea centralis. La fovea può essere unica o anche doppia, per es., negli Uccelli. Nei Vertebrati, in cui le cellule fotorecettrici della r. sono disposte in modo che i raggi luminosi, per raggiungerle e stimolarle, debbono attraversare i vari altri strati retinici, si ha la condizione della r. inversa, tipica degli occhi detti cerebrali. Negli Invertebrati, che hanno occhi di origine epiteliale, la r. si dice eversa. L’epitelio pigmentoso della r. è formato da grosse cellule cariche di pigmento, mostrando in tutte le classi di Vertebrati una certa uniformità di struttura.
Fisiologia
Per le sue caratteristiche strutturali, e in particolare per la maggiore densità dei coni nella zona centrale, l’acuità visiva nella r. è massima nella fovea centralis, sempre notevole al centro e diminuisce a mano a mano che ci si avvicina alla periferia. Gli elementi sensibili, cioè i recettori specifici, sono rappresentati dai coni (preposti alla visione diurna) e dai bastoncelli (per la visione crepuscolare). Quando i raggi luminosi colpiscono la r., determinano nel pigmento contenuto nei coni e bastoncelli un cambiamento della struttura e dei rapporti tra i due elementi costitutivi, il gruppo cromoforo e la proteina. Questi mutamenti del pigmento visivo sono alla base dell’insorgenza del potenziale d’azione del recettore che da questo si trasmette come impulso nervoso al neurone con cui è articolato.
La struttura nervosa della r. comprende neuroni disposti in catena (cellule bipolari e cellule gangliari) e neuroni di associazione (cellule orizzontali e cellule amacrine). La r. comunque non è un semplice organo di raccordo per la trasmissione degli impulsi visivi, ma ha una funzione di vero e proprio centro visivo, poiché già a questo livello si svolgono un’analisi e un’integrazione degli stimoli visivi.
Patologia
La r. può andare incontro ad alterazioni di diversa natura: infiammatoria, degenerativa, vascolare, tumorale ecc.
Relativamente frequente è il distacco della r. dalla coroide, dovuto a cause traumatiche o morbose (miopia di grado elevato, retinite, iridociclite, iridocoroidite, retinopatie), che per lo più provocano accumulo di liquido sieroso, essudato o sangue. Nella zona della r. corrispondente al distacco vi è compromissione più o meno completa della facoltà visiva. Il distacco può essere primitivo o secondario. La forma primitiva, che colpisce prevalentemente i soggetti in età presenile con marcata miopia e con alterata vascolarizzazione della periferia retinica, è dovuta a una rottura della r. attraverso cui il vitreo si insinua distaccando progressivamente la r. dalla coroide (distacco retinico totale). Le forme secondarie sono legate a malattie della coroide quali infiammazioni, emorragie, tumori. La cura è chirurgica (criochirurgia, vitrectomia e, a scopo preventivo, la laserterapia). La prognosi è molto buona, soprattutto se l’intervento è tempestivo, tanto da consentire oltre il 90% di guarigioni.
Il processo infiammatorio della r. (retinite) si manifesta con emorragie e alterazioni vascolari, opacità più o meno diffuse e chiazze biancastre circoscritte dovute all’edema, all’infiltrazione parvicellulare e a essudazione fibrinosa. Le lesioni si presentano sempre più accentuate in vicinanza della papilla ottica. Clinicamente la retinite si manifesta con una diminuzione della funzione proporzionata alla gravità del processo. La diminuzione del visus è anche in rapporto con la sede della flogosi; una retinite centrale dà in genere un abbassamento di vista più notevole di una retinite periferica. Il decorso è sempre lungo; i casi gravi e recidivanti conducono spesso all’atrofia della membrana, con più o meno grave compromissione del potere visivo. Le forme cliniche della retinite assumono fisionomie diverse a seconda della causa determinante (ipertensione arteriosa grave, diabete, glomerulonefrite cronica), contro la quale deve tendere precocemente ogni terapia. Il processo morboso, consistente nella contemporanea presenza di retinite e coroidite, è detto retino-coroidite o coroidoretinite.
La retinite pigmentosa (o degenerazione pigmentosa della r.) è una retinopatia degenerativa trasmessa con carattere autosomico o legata al sesso, caratterizzata da una progressiva atrofia della retina. La lesione ha inizio con la scomparsa dei coni e dei bastoncelli e si continua con una progressiva atrofia dello strato pigmentato e degli elementi nervosi e con un’altrettanto progressiva ipertrofia degli elementi connettivali. L’affezione, sempre bilaterale, si manifesta con debolezza di vista, specie a luce scarsa (emeralopia), che va gradatamente accentuandosi mentre il campo visivo progressivamente si restringe. Il decorso è assai lungo e i sintomi vanno man mano aggravandosi finché, dopo anni, la funzionalità visiva scompare. Studi di genetica molecolare hanno consentito di identificare alcuni loci cromosomici associati alle numerose varianti della malattia. La forma degenerativa della macula è propriamente detta maculopatia (➔ macula lutea).
Il retinoblastoma è un tumore primitivo della r., geneticamente determinato da mutazioni cromosomiche, spesso ereditario; ha tendenza a invadere l’orbita e a dare metastasi, e solo raramente regredisce spontaneamente. Il gene responsabile, RB1, mappato sul braccio lungo del cromosoma 13 (13q14.2), è stato il primo gene oncosoppressore a essere clonato, nel 1986. Nelle forme ereditarie e sporadiche, i tumori retinici sono generalmente le uniche neoplasie, anche se gli individui affetti da retinoblastoma ereditario vanno incontro a un rischio maggiore di sviluppare un osteosarcoma.